

Abstract

We have recently reported a series of piano-stool ruthenium(II) complexes of the general formula [RuCl2(η6-arene)(P(1-pyrenyl)R2R3)] showing excellent cytotoxic activities (particularly when R2 = R3 = methyl). In the present study, new members of this family of compounds have been prepared with the objective to investigate the effect of the steric hindrance of a bulky phosphane ligand, namely diisopropyl(1-pyrenyl)phosphane (L), on exchange reactions involving the coordinated halides (X = Cl, I). Two η6-arene rings were used, i.e. η6-methyl benzoate (mba) and η6-p-cymene (p-cym), and four complexes were synthesized, namely [RuCl2(mba)(L)] (1Cl2iPr), [RuI2(mba)(L)] (1I2), [RuCl2(p-cym)(L)] (2Cl2iPr), and [RuI2(p-cym)(L)] (2I2). Unexpectedly, all of the complexes exhibited poor cytotoxic activities after 24 h of incubation with cells, in contrast to the related compounds previously reported. However, it was observed that aged DMSO solutions of 2I2iPr (from 2 to 7 days) exhibited better activities in comparison to freshly prepared solutions and that the activity improved over “aging” time. Thorough studies were therefore performed to uncover the origin of this lag time in the cytotoxicity efficiency. The data achieved clearly demonstrated that compounds 2I2 and 2Cl2iPr were undergoing a series of transformation reactions in DMSO (with higher rates for the iodido complex 2I2), ultimately generating cyclometalated species through a mechanism involving DMSO as a coordinated proton abstractor. The cyclometalated complexes detected in solution were subsequently prepared; hence, pure [RuCl(p-cym)(κ2C-diisopropyl(1-pyrenyl)phosphane)] (3CliPr), [RuI(p-cym)(κ2C-diisopropyl(1-pyrenyl)phosphane)] (3I), and [Ru(p-cym)(κS-dmso)(κ2C-diisopropyl(1-pyrenyl)phosphane)]PF6 (3dmsoiPr) were synthesized and fully characterized. Remarkably, 3Cl, 3IiPr, and 3dmso are all very efficient cytotoxic agents, exhibiting slightly better activities in comparison to the chlorido noncyclometalated complexes [RuCl2(η6-arene)(P(1-pyrenyl)R2R3)] described in an earlier report. For comparison purposes, the iodido compounds [RuI2(mba)(dimethyl(1-pyrenyl)phosphane)] (1I2Me) and [RuI2(p-cym)(dimethyl(1-pyrenyl)phosphane)] (2I2), bearing the less hindered dimethyl(1-pyrenyl)phosphane ligand, have also been prepared. The cytotoxic and chemical behaviors of 1I2Me and 1I2 were comparable to those of their chlorido counterparts reported previously.
Short abstract
DMSO gradually converts half-sandwich, 1-pyrenyl-containing ruthenium(II) complexes into cyclometalated species showing notable cytotoxic properties.
Introduction
Cancer has a major impact on society worldwide, as it represents one of the leading causes of death.1,2 Cisplatin is one of the most used drugs to treat various types of cancer.3 The remarkable chemotherapeutic properties of cisplatin have instigated tremendous research efforts in the area of platinum drugs.4−6 Nevertheless, cisplatin suffers from some severe side effects,7 and a decrease in its effectiveness may be observed with platinum-resistant tumors.8 Therefore, the development of more efficient and less toxic therapeutic agents is essential in this area of investigation. In that context, alternative transition metals have been used to generate new compounds.9−14 For instance, some ruthenium complexes have been reported that exhibit remarkable anticancer properties,15 making them potential drug candidates.16−22 Actually, two ruthenium compounds are currently undergoing clinical trials, namely BOLD-100 (Na[trans-RuCl4(Ind)2], Ind = indazole)23−25 and TLD1433 ([Ru(bpy)(IP-TT)]2+ (IP-TT = 2-(2′,2″:5′′,2‴-terthiophene)imidazo[4,5-f][1,10]phenanthroline).26,27 To date, there are no efficient molecules capable of targeting most types of disseminated tumor cells. NAMI-A shows interesting antimetastatic properties,28 as do some Ru(II)-arene complexes from the RAPTA family;29,30 for instance, RAPTA-T exerts antimetastatic activities.31,32 Hence, Ru-based compounds are increasingly being seen as potential next-generation anticancer metallodrugs.33,34
Recently, we have reported a series of half-sandwich ruthenium(II) complexes of the general formula [RuX2(η6-arene)(P(1-pyrenyl)R2R3)] (with η6-arene = p-cymene, methyl benzoate, R2 = methyl, phenyl, and R3 = methyl, phenyl) displaying valuable cytotoxic behaviors.35 In that previous study, the effect of the nature of the η6-arene on the cytotoxic activity was examined (viz. p-cymene vs. methyl benzoate), as well as that of different R groups on the monophosphane P(1-pyrenyl)R2R3 ligand; a significant effect of these two parameters on cell toxicity was observed.35 Therefore, we decided to investigate the role played by the halide, i.e. X, on the biological activity of [RuX2(η6-arene)(P(1-pyrenyl)R2R3)] compounds and thus study and compare the cytotoxic properties of the four complexes, depicted in Scheme 1: viz., 1Cl2iPr, 1I2, 2Cl2iPr, and 2I2.
Scheme 1. Representation of the Structure of the Piano-Stool Ruthenium Complexes Designed and Synthesized in the Present Study to Evaluate the Effect of Chloride and Iodide on the Cytotoxic Activity.

These Ru compounds all contain the ligand diisopropyl(1-pyrenyl)phosphane (L), purposely chosen for its steric hindrance that would favor the displacement of the halide ligands, for instance by water molecules.36,37 Two different arene rings were used, namely methyl benzoate and p-cymene, which have produced piano-stool ruthenium complexes with drastically distinct cytotoxic properties in a previous study.35
Surprisingly, cell viability assays with various cancer lines revealed that 1Cl2iPr, 1I2, 2Cl2iPr, and 2I2 were poorly active (mostly inactive) compounds after an incubation time of 24 h. However, complex 2I2iPr, namely [RuI2(η6-p-cymene)(diisopropyl(1-pyrenyl)phosphane)] (Scheme 1), exhibited a drastic improvement in its activity with time; for instance, 2I2 was 4 times more active against lung adenocarcinoma cells after 7 days (in comparison to its activity after 1 day). This highly interesting behavior was thoroughly investigated to elucidate the origin of this time-dependent enhancement of the cytotoxic properties; the mechanistic studies carried out showed that 2I2iPr (as well as its corresponding chlorido complex 2Cl2) was gradually converted into a highly cytotoxic, cyclometalated species, through a three-step process (whose rate was halide-dependent: i.e., the conversion process was faster with iodido complex 2I2iPr than with chlorido complex 2Cl2).
Results and Discussion
Preparation of the Ru Compounds 1Cl2iPr, 1I2, 2Cl2iPr, and 2I2
The ligand, namely diisopropyl(1-pyrenyl)phosphane (L), was prepared by the reaction of lithiated 1-bromopyrene with chlorodiisopropylphosphane in THF at −78 °C (Scheme S1). Ligand L is unstable in air (an oxide of the phosphane is produced); therefore, L was protected by the formation of its borane adduct. L·BH3 can be deprotected, just before use (to prepare the Ru compounds), by reaction with the tetrafluoroboric acid diethyl ether adduct in dichloromethane (Scheme S1).
The chlorido complexes [RuCl2(η6-methyl benzoate)(L)] (1Cl2iPr) and [RuCl2(η6-p-cymene)(L)] (2Cl2) were obtained in good yields by the reaction of ligand L with the corresponding ruthenium dimeric precursors: namely, [RuCl(μ-Cl)(η6-methyl benzoate)]2 for 1Cl2iPr and [RuCl(μ-Cl)(η6-p-cymene)]2 for 2Cl2 (Figure 1). The iodido compounds [RuI2(η6-methyl benzoate)(L)] (1I2iPr) and [RuI2(η6-p-cymene)(L)] (2I2) can be generated in good yields from 1Cl2iPr and 2Cl2 in the presence of an excess of sodium iodide in refluxing technical acetone (Figure 1).38 It should be pointed out that the conversion of 2Cl2iPr to 2I2 was achieved in 1 h, whereas 16 h was required for the chloride to iodide exchange that generated 1I2iPr from 1Cl2 (see the Experimental Section). Thus, it appears that the bulkier and more electron donating p-cymene ring significantly favors the chloride to iodide substitution. This substitution can easily be monitored by 31P NMR spectroscopy. For instance, the 31P{1H} NMR spectrum of 1Cl2iPr shows a singlet at +38.9 ppm, whereas the corresponding singlet is observed at +34.5 ppm for 1I2 (Δδ −4.4 ppm). Similarly, the 31P chemical shift is observed at +36.3 ppm for chlorido 2Cl2iPr and at higher field for the iodido complex 2I2: viz., +31.3 ppm (Δδ −5.0 ppm). The lower electronegativity of iodine, in comparison to that of chlorine, may explain why the P atoms of the iodido complexes are more shielded.
Figure 1.

Synthetic procedures used to prepare the half-sandwich Ru(II) chlorido 1Cl2iPr and 2Cl2 and iodido complexes 1I2iPr and 2I2.
All of the ruthenium compounds were characterized by common techniques, including X-ray diffraction, which confirmed their identity (see the Experimental Section for details).
Crystal Structures of Ru Compounds 1Cl2iPr, 1I2, 2Cl2iPr, and 2I2
Single crystals of the four compounds, suitable for X-ray diffraction studies, were obtained (see Experimental Section). Compounds 1Cl2iPr and 1I2 crystallize in the monoclinic space group P21/c, compound 2Cl2iPr crystallizes in the monoclinic space group C2/c, and 2I2 crystallizes in the triclinic space group P1̅ (see Tables S1 and S2). The solid-state structures of the four complexes are shown in Figure 2; selected (coordination) bonds and angles are given in Table S3 (see also Figure S1).
Figure 2.

Representation of the crystal structures of complexes 1Cl2iPr, 1I2, 2Cl2iPr, and 2I2. The atoms bonded to the metal center are labeled, and C represents the centroid of the η6-arene ring. Hydrogen atoms are omitted for clarity.
The four ruthenium compounds exhibit the expected, typical “three-legged piano-stool” geometry for such systems. The centroid to metal distance varies from 1.69 to 1.72 Å, and the Ru–P length is in the range 2.39–2.41 Å (Table S3). The Ru–Cl bond distances are 2.41 and 2.42 Å for 1Cl2iPr and 2.40 and 2.43 Å for 2Cl2. As could be predicted, the Ru–I bonds are longer (of about 0.3 Å), with values at around 2.72 Å for 1I2iPr, and 2.72 and 2.73 Å for 2I2 (Table S3). The coordination angles are similar for all four complexes (Table S3), and they are in the range expected for such molecules.35,39,40
Effects of Compounds 1Cl2iPr, 1I2, 2Cl2iPr, and 2I2 on Cell Viability
The ability of the compounds to inhibit cell growth was evaluated next. Hence, their cytotoxic properties were first assessed in A549 cells (lung adenocarcinoma) at a fixed complex concentration of 10 μM. The corresponding cell viabilities (in percent) are given in Table 1.
Table 1. Cell Viability Values (%) of Complexes 1Cl2iPr, 1I2, 2Cl2iPr and 2I2 (Fixed Concentration of 10 μM) for A549 (Lung Adenocarcinoma) Human Cells, after Incubation for 24 h.
After 24 h incubation at 37.5 °C.
The results are expressed as mean values ± SD out of three independent experiments.
Surprisingly, the cytotoxic activities of the Ru compounds are mostly poor; for instance, 1Cl2iPr and 1I2 are inactive (cell viability of around 100%; Table 1), and 2Cl2iPr only gives rise to some cell growth inhibition (73% cell viability; Table 1). Only compound 2I2 is efficiently capable of eradicating A549 cells (37% cell viability; Table 1). An unusual phenomenon was observed during the replication experiments with 2I2iPr. Indeed, clearly different cell viability values after 24 h of incubation were obtained for the same stock solution of 2I2 in DMSO, used after 1, 5 and 7 days after preparation. A significant improvement in the cytotoxic properties of 2I2iPr was indeed noticed from day 0 of the preparation of the stock solution to, for instance, days 5 and 7; actually, the value of 37 ± 17% (Table 1) corresponds to the average value from three replicates carried out with a stock solution of 2I2 used after 0, 5, and 7 days. The large deviations observed among the replicates are due to the significantly different activities of the aging solution. It was also noted that the aging DMSO solution of 2I2iPr became slightly darker after a few days (see the graphical abstract). It thus appears that 2I2 is gradually converted into a new and more active, “unknown” species.
Subsequently, half-maximum inhibitory concentrations (IC50) were determined for compounds 2Cl2iPr and 2I2, considering this feature (viz. the observed time-dependent evolution of the biological properties of 2I2iPr). IC50 values were not determined for 1Cl2 and 1I2iPr, which did not show any cytotoxic behavior (see Table 1). The IC50 data obtained are given in Table 2.
Table 2. Half-Maximum Inhibitory Concentrations (IC50, μM) of Compounds 2Cl2iPr and 2I2 for A549 (Lung Adenocarcinoma) Human Cells, after Incubation of 24 h, Using Freshly Prepared Stock Solutions of the Complexes (Day 0), and 5- and 7-Day Aged Solutions of the Complexesa.
| cell line | compound | day 0 | day 5 | day 7 |
|---|---|---|---|---|
| A549 | 2Cl2iPr | 24 ± 1 | 26 ± 10 | 27 ± 12 |
| A549 | 2I2iPr | 48 ± 8 | 26 ± 8 | 9.5 ± 1.5 |
| MCF7 | 2I2iPr | 12 ± 2 |
The IC50 value (μM) of a 7-day-old DMSO solution of 2I2iPr for MCF7 (breast carcinoma) human cells, after incubation for 24 h, is also included. The results are expressed as mean values ± SD out of three independent experiments.
Compound 2Cl2iPr gives an IC50 value of 24 μM after 24 h incubation with A549 (lung adenocarcinoma) human cells (Table 2), using a stock solution of Ru compound (in DMSO) prepared the same day (viz. the day in which the compound was incubated with the cells, day 0). As already noticed with the cell viability studies, the use of a 5-day-old or 7-day-old stock solution of 2Cl2 did not lead to different IC50 values (Table 2), suggesting that the integrity of the Ru compound is most likely maintained (in DMSO solution).
A completely different behavior was observed for 2I2iPr. Indeed, a freshly prepared solution of 2I2 in DMSO (day 0) gave an IC50 value of 48 μM; hence, 2I2iPr was 2 times less cytotoxic than 2Cl2. However, if the stock solution of 2I2iPr is used 5 days after its preparation, then an IC50 value of 26 μM is obtained; the cytotoxic behavior of 2I2 is comparable with that of 2Cl2iPr(day 5, Table 1). Even more interestingly, after 7 days in DMSO, 2I2 becomes very active, as illustrated by the IC50 value of 9.5 μM (day 7, Table 2). Clearly, the initial compound 2I2iPr is slowly converted into a significantly more active species (the new “unknown” species is indeed 5 times more cytotoxic than the original Ru complex against A549 cells). The cytotoxicity of 2I2 against MCF7 (breast carcinoma) human cells was evaluated as well, which gave an interesting IC50 value of 12 μM with a 7-day-old solution of the complex (Table 2).
The remarkable behavior of 2I2iPr in DMSO, namely its progressive activation (i.e., its increased cytotoxicity), was subsequently investigated with the objective of elucidating the nature of the formed, more cytotoxic species.
NMR Studies
Since the stock solutions of the complexes used for the cytotoxicity assays were prepared in DMSO, the potential modification/alteration of 2I2iPr in this solvent was monitored by 31P{1H} NMR spectroscopy. The time-dependent corresponding spectra obtained after a period of 48 h are shown in Figure 3.
Figure 3.

31P{1H} NMR spectra of complex 2I2iPr in DMSO-d6 recorded during a period of 48 h, illustrating the progressive formation of new species.
Already after 5 min, two new peaks, in addition to that corresponding to 2I2iPr(+30.7 ppm), are found at −8.3 and +80.0 ppm. The chemical shift at −8.3 ppm is due to the free ligand diisopropyl(1-pyrenyl)phosphane L that is released from the complex (in CDCl3 δ is found at −8.8 ppm; see the Experimental Section). The second new peak at +80.0 ppm arises from the development of a new species, denoted N1 (Figure 3; X = I). On the basis of the chemical shift, a cyclometalation reaction involving the pyrenyl ring may be considered to explain the observed low-field value; indeed, a number of related cyclometalated (phosphane)ruthenium(II) complexes have been reported with chemical shifts in the range +70–90 ppm.41−44 After 30 min, the intensity of the peak corresponding to N1 increases, whereas those of 2I2 and L slightly decrease. After 24 h, two new peaks are detected at +55.7 and +86.6 ppm (Figure 3). The chemical shift at +55.7 ppm is attributable to the oxide of the ligand, i.e. diisopropyl(1-pyrenyl)phosphane oxide (L=O). Actually, L=O was purposely synthesized by oxidation of L with dihydrogen peroxide (see Figures S2 and S3 and Table S4 in the Supporting Information), and its 31P{1H} NMR spectrum gave a single signal at δ +56.9 ppm in CDCl3 (Figure S4). The second new peak at δ +86.6 ppm can be ascribed to another cyclometalated species, denoted N2 (Figure 3). After 48 h in DMSO, the signals corresponding to L and 2I2iPr disappeared completely, while traces of L=O could still be seen. The peak due to species N1 slightly decreased, whereas that of N2 clearly increased, hence suggesting that N2 may be formed from N1.
The same time-course study was carried out for the chlorido complex 2Cl2iPr, for comparison. The corresponding NMR spectra in DMSO-d6 are shown in Figure S5. The same behavior is observed for this compound. An intense broad peak at −8.3 ppm, corresponding to free phosphine L, is detected instantly; such peak broadness can be explained by rapid exchange processes between different species in solution. The chemical shift of 2Cl2 is found at +36.3 ppm and the oxidized ligand, viz. the phosphane oxide L=O, is detected at +55.7 ppm. As for 2I2iPr, two signals are observed above +80 ppm, in the “cyclometalated region”. By analogy with 2I2 (see above), the peak observed at +80.8 ppm is attributed to N1 (with X = Cl) and that at +86.6 ppm to N2. It can be stressed here that the N2 species is formed in significantly lower amounts for 2Cl2iPr in comparison to 2I2 (see Figure 3 and Figure S5) for the same aging time. Also, in contrast to 2I2iPr, the development of a third new species, labeled N3, is observed at δ +45.7 ppm (Figure S5). On the basis of its chemical shift, N3 may be associated with the cationic [RuCl(η6-p-cymene)(diisopropyl(1-pyrenyl)phosphane)(dmso)]+ species resulting from the substitution of one of the chlorido ligands of 2Cl2 by a DMSO molecule. Actually, when the time-dependent NMR experiments are carried out for 2Cl2iPr in pure CDCl3, the peaks corresponding to N1 (X = Cl), N2 and N3 (X = Cl) are not observed (Figure S6), suggesting that DMSO is actively involved in the formation of these species. Similarly, the time-dependent study for 2I2 in pure CDCl3 only shows the presence of the starting ruthenium(II) complex together with oxidized ligand L=O (Figure S7), in higher amounts than for 2Cl2iPr (Figure S6). It thus appears that stable DMSO-containing species (Scheme 2, right) are generated from the cyclometalated complexes [RuX(η6-p-cymene)(κ2C-diisopropyl(1-pyrenyl)phosphane)] (X = Cl, I; Scheme 2, left) in the presence of DMSO and that the process is slower with the chlorido complex (see Study of the Solvation of the Cyclometalated Complexes).
Scheme 2. Possible Cyclometalated N1 and N2 Species Generated from 2Cl2iPr (X = Cl) and 2I2 (X = I).

For comparison purposes, 31P{1H} NMR time-resolved studies in DMSO-d6 were performed for η6-methyl benzoate containing complexes 1Cl2iPr and 1I2. The corresponding spectra are shown in Figures S8 and S9. After 24 h, 1Cl2iPr, free ligand L and L=O are observed together with two new compounds that are most likely cyclometalated species (Figure S8). After 48 h, the same species are present in solution. For 1I2, the formation of a cyclometalated species is immediately observed (Figure S9). After 24 h, 1I2iPr has completely disappeared; as for the η6-p-cymene-containing compounds, it appears that the cyclometalation reaction is favored for the iodido complex.
Subsequently, the effect of water on DMSO stock solutions of 2Cl2iPr and 2I2 was investigated by 31P{1H} NMR, as biological studies are performed in aqueous media. Therefore, 24 h aged, concentrated solutions of complexes 2Cl2iPr and 2I2 in DMSO-d6 were mixed with D2O, using a DMSO-d6 to D2O ratio of 25:75. Under these conditions, precipitation was observed; the solids were filtered off, and the NMR spectra of the filtrates were recorded. For both samples (i.e. prepared from 2Cl2iPr and 2I2), the same single peak was detected in the cyclometalated region, corresponding to that obtained for the N2 intermediate (see above). Using 48 h aged, concentrated solutions of complexes 2Cl2iPr and 2I2, comparable data were obtained; an increase in dissolved sample over time was observed (Figure S10), hence suggesting both the polar nature of the new complex [Ru(η6-p-cymene)(κ2C-diisopropyl(1-pyrenyl)phosphane)(dmso)]+ in solution and its relatively slow formation process. The formation of an aquo complex of the type [Ru(η6-p-cymene)(κ2C-diisopropyl(1-pyrenyl)phosphane)(H2O)]+ could not be detected under the conditions applied for these NMR experiments.
Cyclometalated Compounds
The cyclometalated complexes [RuX(η6-p-cymene)(κ2C-diisopropyl(1-pyrenyl)phosphane)] (X = Cl, I) and [Ru(η6-p-cymene)(κ2C-diisopropyl(1-pyrenyl)phosphane)(dmso)]+ (depicted in Scheme 2) are thus clearly important end species formed in solution from 2Cl2iPr and 2I2. Therefore, these compounds were purposely prepared in their pure form.
The reaction of dichloro(p-cymene)ruthenium(II) dimer with diisopropyl(1-pyrenyl)phosphane (L) in methanol in the presence of sodium acetate42,45 produces the cycloruthenated complex [RuCl(η6-p-cymene)(κ2C-diisopropyl(1- pyrenyl)phosphane)] (3CliPr) with a yield of 52% (Figure 4). The iodido complex [RuI(η6-p-cymene)(κ2C-diisopropyl(1-pyrenyl)phosphane)] (3I) is obtained from 3CliPr with a yield of 82%, by halide exchange in the presence of an excess of sodium iodide in refluxing technical acetone38 (Figure 4). Finally, the cationic cyclometalated complex [Ru(η6-p-cymene)(κS-dmso)(κ2C-diisopropyl(1-pyrenyl)phosphane)]PF6 (3dmso) can be prepared in nearly quantitative yield, viz. 95%, by reaction of an excess of DMSO with complex 3CliPr in the presence of thallium hexafluorophosphate in dichloromethane solution at room temperature (Figure 4).
Figure 4.

Synthetic procedures to prepare cyclometalated complexes 3CliPr, 3I, and 3dmsoiPr.
It can be pointed out here that the 31P{1H} chemical shifts of 3CliPr, 3I, and 3dmsoiPr in CDCl3, respectively +80.8, +79.3, and +86.6 ppm (Experimental Section), are in the range of those mentioned in NMR Studies (see Figures 3 and 4), therefore corroborating the assumptions thus made regarding the potential nature of the “unknown” species labeled N1 (X = Cl, I) and N2.
Single crystals of the three cyclometalated compounds, suitable for X-ray diffraction analyses, were obtained (see the Experimental Section); all three complexes crystallize in the monoclinic space group P21/c (Tables S5 and S7). The crystal structures of the cyclometalated compounds are shown in Figure 5; selected (coordination) bonds and angles are given in Table S6 for 3CliPr and 3I and Table S8 for 3dmsoiPr. The pseudo-octahedral geometry of the Ru center in the three compounds is significantly distorted due to the cyclometalation; for instance, the C–Ru–P angles for all complexes are close to 80° (Tables S5 and S7), while the corresponding X–Ru–P angles in 2Cl2 and 2I2iPr are closer to the ideal value, namely 90° (Table S3). The coordination bond lengths are in the range of those found for similar complexes.34,42
Figure 5.

Representation of the crystal structures of cyclometalated 3CliPr, 3I, and 3dmsoiPr. The atoms bonded to the Ru center are labeled, and C stands for the centroid of the p-cymene ring. Hydrogen atoms are omitted for clarity.
Study of the Solvation of the Cyclometalated Complexes
Kinetic studies of the solvation of 3CliPr, 3I, and 3dmsoiPr by different solvents were carried out by UV–vis spectroscopy at distinct temperatures and pressures. Saturated solutions of 3Cl and 3IiPr were prepared in the solvents to be investigated: namely, DMSO, DMSO saturated with NaCl, n-butanol (chosen for comparative purposes), and water.
Generally, suspensions were obtained, which were sonicated for 5–6 min and filtered over glass wool (to eliminate any remaining insolubilized compound); the filtrates were then transferred into UV–vis cells that were subsequently placed in a thermostated UV–vis spectrophotometer. After temperature equilibration, time-resolved spectra were collected at different time scales and intervals to warrant total conversion (4–6 half-lives) to the expected solvato complexes.
The first-order rate constants obtained for all solvation experiments carried out are given in Tables S9 and S10. These constants were determined by fitting the time-resolved spectral data with Specfit46 or ReactLab,47 considering an A → B process. It should be noted that, upon dilution of the initial solution (1:1 and 1:5 dilutions), no variations in rate constants were observed, therefore indicating the nonactuation of multinuclear species during the process. Moreover, when DMSO saturated with NaCl was used (Figure 6a), no noticeable differences in kobs values were found, hence suggesting the nonequilibrium nature of the solvation process under the conditions applied. Examples of the temperature and pressure dependence of the kobs values for representative systems are shown in Figure 6b,c, respectively. The values determined for the thermal and pressure activation parameters for a series of solvation experiments with 3CliPr, 3I, and 3dmsoiPr are given in Table 3; the extrapolated kobs and 6 × t1/2 values at 37 °C (310 K) are also given in Table 3. It should be noted that the data provided for 3Cl in water were obtained from the time-resolved appearance of definite UV–vis spectra, resulting from the progressive aquation of the chlorido ligand producing polar ionic species, most likely [Ru(η6-p-cymene)(κ2C-diisopropyl(1-pyrenyl)phosphane)(H2O)]+, exhibiting higher (but still limited) water solubility in comparison to 3CliPr (see below).
Figure 6.

(a) Representative set of time-resolved spectral changes observed for a solution of 3CliPr in NaCl-saturated DMSO at 70 °C for 10 h. (b) Selected Eyring plots of the temperature dependence of kobs for 3Cl in n-butanol (black ■) and 3IiPr in DMSO (red ●). (c) Selected plots of the pressure dependence of ln kobs for 3Cl in DMSO (black ■) and 3CliPr in n-butanol (red ●).
Table 3. Summary of the Kinetic, Thermal, and Pressure Activation Parameters for the Solvation of 3CliPr, 3I, and 3dmsoiPr with Various Solventsa.
| compound | solvolysis by | 105 × 310kobs/s–1 (6 × t1/2/days) | ΔH⧧/kJ mol–1 | ΔS⧧/J K–1 mol–1 | ΔV⧧/cm3 mol–1 |
|---|---|---|---|---|---|
| 3CliPr | DMSOd | 0.35 (330) | 85 ± 5 | –78 ± 14 | 6.2 ± 0.6 |
| n-butanol | 2.4 (48) | 63 ± 1 | –133 ± 1 | 12 ± 2 | |
| H2Ob | 5.2 (22) | 52 ± 2 | –162 ± 5 | ndc | |
| 3IiPre | DMSOd | 1.6 (72) | 113 ± 3 | 25 ± 8 | 13 ± 0.5 |
| n-butanol | 12 (9.6) | 62 ± 2 | –123 ± 5 | 20 ± 1 | |
| 3dmsoiPr | H2Of | 2.0 (58) | 71 ± 4 | –110 ± 13 | –14 ± 2 |
[Complex] = 10–50 μM.
3CliPr is poorly soluble in water; the values are derived from the rate of appearance of definite UV–vis spectra.
Not determined.
DMSO containing 0.005% of water was used ([H2O] = 2.77 × 10–3 M).
3IiPr is completely insoluble in water.
Experiments performed using a saturated aqueous solution; [3dmsoiPr] ≈ 10 μM (such a low concentration had to be used due to the very low solubility of this compound in water).
Wide ranges of enthalpies (52–113 kJ mol–1) and entropies (−162 to +25 J K–1 mol–1) of activation are observed for these solvation processes (Table 3; compounds 3CliPr and 3I). These data suggest the operation of a rather uniform substitution mechanism showing a complete compensation between the activation entropies and enthalpies (Figure 7, black squares), with a clear isokinetic temperature, i.e. 79 °C, outside of the range experimentally used (3CliPr, Table S9; 3I, Table S6).48−50 The process is therefore explained by a rather pure interchange mechanism with a certain degree of dissociation and ordering, as indicated by the general trend of the set of ΔV⧧ (>0) and ΔS⧧ (<0) (see Table 3). On consideration that a neat charge separation is taking place during the process and that DMSO is less polar than n-butanol,51 the larger negative values of ΔS⧧ for the latter can be explained by solvent ordering of charge formation. Similarly, the more positive values of ΔV⧧ for n-butanol can be justified by a higher number of solvent molecules involved in the process. Furthermore, the more positive ΔV⧧ values for iodido complex 3IiPr in comparison with chlorido complex 3Cl (Table 3) suggest that higher volume changes on charge separation occur with the less polar compound, as one would expect.52 The results obtained are rather surprising, given the accepted associativeness of substitution mechanisms observed with ruthenium(II) complexes,53,54 which is normally accepted as a positive factor for biological applications. The important covalent character of the M–L bonds in organometallic 3CliPr and 3I most likely decreases the Lewis acidity of the metal center, by comparison with classical Werner-type complexes; consequently, the associative demand in the substitution process is reduced. Similar features were observed for substitution reactions with organometallic platinum(IV)55−57 and platinum(II) compounds.58,59
Figure 7.

Isokinetic (79 °C) compensation plot (black squares) for the thermal activation parameters obtained for the solvolysis reactions of 3CliPr and 3I (Table 3). The red square corresponds to the aquation of 3dmsoiPr.
The behavior of 3dmsoiPr in water was investigated next. As 3dmso is very poorly soluble in water, 10 μm solutions of this compound were prepared by sonication and the higher temperatures given in Table S9, i.e. 40–80 °C, were used (since the compound was hardly water soluble at room temperature). Despite these solubility issues, the large values of the extinction coefficients of the absorptions in the range of 350–400 nm allowed us to record satisfactory time-resolved spectra, even at variable pressures. The values of the kinetic and activation parameters obtained (with larger associated errors, compared with those of 3CliPr and 3I) are given in Table 3. The data point shown in Figure 7 (red square) suggests that a similar substitution mechanism for the DMSO/water exchange is taking place in 3dmsoiPr. However, the values of both the volume and entropy of activation are negative, hence suggesting that the ordering in this reaction is associated with a volume decrease; for 3dmso, no charge variation is occurring during the DMSO/water exchange, in contrast to the halide/DMSO exchanges in 3CliPr and 3I. As a result, the ordering and contraction go in the same direction in the case of 3dmsoiPr. Furthermore, from the data obtained, a very interesting shift to the associative activation side of the interchange process seems to apply for these cationic ruthenium(II) species.
In summary, once in solution, and especially in the presence of DMSO, compounds 2CI2iPr and 2I2 undergo a series of transformations, which are illustrated in Figure 8. Partial degradation of the compounds is observed, as the free phosphane ligand L is detected together with its oxidized form, namely phosphane oxide L=O (Figure 9, degradation pathway). In the absence of DMSO, cyclometalated species are not generated; therefore, this solvent clearly plays an important role in the transformation pathway. It is believed that the first step consists of the substitution of an halido ligand by a DMSO molecule (k1), converting 2CI2iPr or 2I2 into intermediate A. This intermediate is readily converted into 3CliPr or 3I(k2; Figure 8), through a cyclometalation reaction with DMSO acting as a Lewis base, in the so-called concerted metalation–deprotonation (CMD) mechanism.60,61 It is indeed proposed that coordinated DMSO makes an intramolecular hydrogen bond with the pyrenyl group, hence fostering the cyclometalation (Figure S11). 3CliPr and 3I then undergo substitution of the second halido ligand by a DMSO molecule, producing the cationic cyclometalated compound 3dmsoiPr (k3), which is finally aquated (i.e. DMSO is replaced with water, k4; Figure 8).
Figure 8.

Representation of the different species generated in solution upon dissolution of 2CI2iPr or 2I2 in the presence of DMSO.
Figure 9.
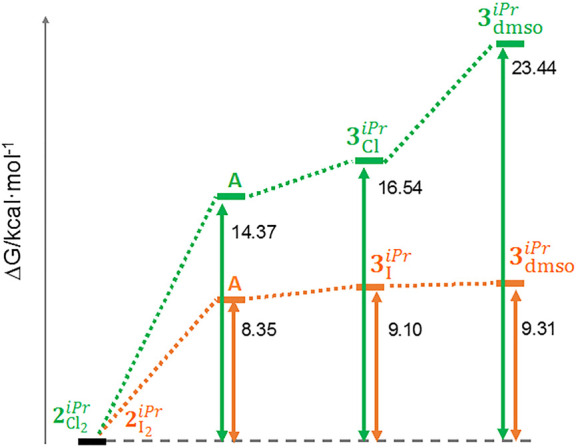
Energy profiles (in kcal mol–1) for intermediate A → 3XiPr → 3dmso processes: orange, X = I; green, X = Cl.
It should be noted that the k1 and k2 rate constants could not be determined. First, the degradation path masks the determination of the value of k1. Second, as k2 is clearly significantly higher than k1 (as indicated by the 31P{1H} NMR experiments), once 2Cl2iPr and 2I2 are converted into their respective intermediates A, the subsequent cyclometalation reaction to yield 3CliPr and 3I is immediate (k2 ≫ k1), not allowing a determination of the value of k2 under the experimental conditions applied in the present study. It can be pointed out here that k2I is most likely superior to k2Cl; indeed, NMR studies with 2Cl2iPr have shown (see above) that a species labeled N3 was forming in DMSO (δ +45.7 ppm; Figure S5), which may be ascribed to the chlorido intermediate A (X = Cl; Figure 8). In the case of 2I2, such a species was not observed by NMR (Figure 3), suggesting that when it is formed it is very rapidly converted into compound 3IiPr (hence k2I > k2Cl). The rate constants for the conversion of 3Cl and 3IiPr into 3dmso, respectively k3Cl and k3I, could be obtained (see Table 3), which showed that the iodido to DMSO substitution was almost 4.6 times faster than the chlorido to DMSO substitution (k3I > k3Cl; Table 3). It should be stressed here that one may expect an analogous trend for the first halido to DMSO substitution, namely k1I > k1Cl (2Cl2iPr or 2I2 → intermediate A; Figure 8); though additional in-depth studies are required to confirm this. Finally, the rate constant for the aquation of 3dmsoiPr, namely k4 (3dmso → aquo complex B; Figure 8), was also determined (Table 3), showing that it was a relatively slow process.
The changes in free energy (ΔG°) have been calculated for intermediates A (for X = Cl and X = I), 3CliPr and 3I, and 3dmsoiPr (Figure 8), with respect to the corresponding starting compounds 2Cl2 and 2I2iPr; these are shown in Figure 9. The energy profile is clearly more favorable when X = I (orange profile in Figure 9), corroborating the experimental data. For instance, the cyclometalation step (intermediate A → 3X) costs only 0.75 kcal mol–1 for X = I, whereas it is 2.17 kcal mol–1 for X = Cl. However, it can be noted that in both cases the cyclometalation is energetically inexpensive (although more feasible for X = I). The step 3XiPr → 3dmso is costless for X = I (energy difference of only 0.21 kcal mol–1), while a difference in energy of 6.9 kcal mol–1 is found between 3CliPr and 3dmso (Figure 9).
It is noteworthy to stress that the geometry optimization of intermediate A (Figure 8) shows a clear orientation of the oxygen atom of the DMSO ligand toward the hydrogen atom involved in the cyclometalation of the pyrenyl group (the O···H distances being 2.18 and 2.38 Å for X = Cl, I, respectively), as proposed in Figure S11, thus confirming the crucial involvement of the solvent in the cyclometalation pathway.
Cytotoxicity Behaviors of Cyclometalated Compounds 3CliPr, 3I, and 3dmsoiPr
IC50 values were then determined for compounds 3CliPr, 3I, and 3dmsoiPr at increasing “aging times”, using A549 (lung adenocarcinoma) human cells for comparison with the time-dependent IC50 values obtained for 2Cl2 and 2I2iPr for the same cell line (see Table 2). The results achieved are given in Table 4. It can be pointed out that a concentration range of 0.4–50 μM was used for these assays, because precipitation of the cyclometalated ruthenium compounds was observed at concentrations above 100 μM.
Table 4. Half-Maximum Inhibitory Concentrationsa (IC50, μM) of Compounds 3CliPr, 3I, and 3dmsoiPr for A549 (Lung Adenocarcinoma) Human Cells, after Incubation for 24 h (Day 0) and after 1, 2, and 7 days of Aging (before IC50 Determination).
| compound | day 0b | day 1 | day 2 | day 7 |
|---|---|---|---|---|
| 3CliPr | 2.26 ± 0.34 | 2.77 ± 0.38 | 4.92 ± 0.59 | 2.67 ± 1.61 |
| 3IiPr | 5.82 ± 1.89 | 5.58 ± 0.47 | 5.61 ± 2.16 | 4.32 ± 2.54 |
| 3dmsoiPr | 1.72 ± 0.67 | 2.47 ± 0.57 | 2.56 ± 0.44 | 2.34 ± 0.61 |
The results are expressed as mean values ± SD out of three independent experiments.
Day 0 corresponds to the first determination of IC50, after 24 h of incubation with freshly prepared stock solutions of the compounds.
Remarkably, in each case, comparable IC50 values were obtained for the samples with different “aging times”. These data indicate that the cyclometalated complexes remain unchanged in solution (in contrast to 2Cl2iPr and 2I2). The cytotoxic activities of compounds 3CliPr, 3I, and 3dmsoiPr are clearly better than those of the “parent compounds” 2Cl2 and 2I2iPr. It is interesting to note that the cytotoxic activities of 3dmso are comparable to those of 3CliPr and 3I, suggesting that replacement of the halide by DMSO has no effect on the biological properties. Surprisingly, compound 3IiPr is less cytotoxic than 3Cl; from the IC50 data obtained with 2Cl2iPr and 2I2 (see Table 2), one would have expected compound 3IiPr to be more active than 3Cl. This can be justified by the significantly higher conversion rates for 2I2iPr to form 3I and ultimately 3dmsoiPr (see above, Figure 8; k1, k2, and k3). Hence, the formation of the active cyclometalated species is significantly faster with 2I2; thus, the little amounts of 3IiPr (and of 3dmso) progressively generated in solution are enough to give the increasing cytotoxicity observed for 2I2iPr at days 5 and 7 (Table 2). Conversion of 2CI2 to a cyclometalated species is much slower; therefore, evolution of the IC50 value is not observed within a period of 7 days (see Table 2). It can be pointed out that the observed IC50 values, ranging from 1.72 to 5.82 μM, are comparable to data reported in the literature for various types of piano-stool ruthenium(II) complexes.62−65 However, it can be stressed that the IC50 values for 3CliPr, 3I and 3dmsoiPr, given in Table 2, were determined after 24 h of incubation with cells, whereas most of the IC50 values found in the literature were obtained after a drug-exposure time of 48, 72, or 96 h;66−68 hence, the low-micromolar IC50 values achieved after 24 h of incubation with 3CI, 3IiPr, and 3dmso indicate that they are highly cytotoxic. Notable activities after 24 h of incubation have been described for tethered,69 acylpyrazolonato-containing,70 or amino-oxime-based half-sandwich ruthenium(II) complexes,71 but 3CliPr, 3I, and 3iPrdmso are comparatively more efficient.
Dimethyl(1-pyrenyl)phosphane versus Diisopropyl(1-pyrenyl)phosphane
As mentioned in the Introduction, the use of the new ligand diisopropyl(1-pyrenyl)phosphane (L) originates from previous studies, which have shown that P-ligands of the type PR1R2(1-pyrenyl), which give rise to [RuX2(η6-arene)(PR1R2(1-pyrenyl))] compounds, exhibit remarkable cytotoxic properties; in particular, the chlorido Ru complex with the ligand dimethyl(1-pyrenyl)phosphane and η6-methyl benzoate, namely [RuCl2(η6-methyl benzoate)(dimethyl(1-pyrenyl)phosphane)], exhibited IC50 values in the low micromolar range for various cancer cell lines.35 The aim of the investigation presented herein was to examine the effect of the halide, viz. iodide versus chloride, on the cytotoxic properties of the corresponding Ru complexes bearing a sterically hindered P-ligand (for instance, to favor aquation, namely replacement of the halides by water molecules). Surprisingly, the cytotoxicity data obtained for compounds 1Cl2iPr, 1I2, 2Cl2iPr, and 2I2 were low (Table 1), especially for 1Cl2iPr and 1I2 (which are not active at all), which include the η6-methyl benzoate ligand. Indeed, in the earlier study, this η6-arene moiety generated the most cytotoxic agent of the series described therein, namely [RuCl2(η6-methyl benzoate)(dimethyl(1-pyrenyl)phosphane)] (1Cl2Me).35 For this reason, we decided to synthesize the iodido complexes of the ligand dimethyl(1-pyrenyl)phosphane,35 with both η6-methyl benzoate and η6-p-cymene, for comparison purposes. Hence, the iodide complexes [RuI2(η6-methyl benzoate)(dimethyl(1-pyrenyl)phosphane)] (1I2) and [RuI2(η6-p-cymene)(dimethyl(1-pyrenyl)phosphane)] (2I2Me) (Scheme S2) were prepared by halide exchange of their chlorido counterparts (chlorido Ru complexes are reported in ref (35)).
The solid-state structures of 1I2Me and 2I2 could be obtained (Figure S12). 1I2Me crystallizes in the monoclinic space group P21/c and 2I2 in the orthorhombic space group P212121 (Table S11). Selected coordination bond lengths and angles are given in Table S12. The structural data of both compounds are comparable with those of 1Cl2iPr, 1I2, 2Cl2iPr, and 2I2, and are typical for such coordination geometry.35,39,40
IC50 values were then determined for 1I2Me and 2I2 using three different cell lines: namely, A549 (lung adenocarcinoma), SW620 (colorectal adenocarcinoma) and MCF7 (breast carcinoma). The data are given in Table 5. 1I2Me and 2I2 are significantly more cytotoxic than 1Cl2iPr, 1I2, 2Cl2iPr, and 2I2; moreover, no delay in their biological activity was observed (data not shown), in deep contrast to 2I2iPr. In fact, 1I2 and 2I2Me are behaving as their chlorido counterparts, i.e. [RuCl2(η6-methyl benzoate)(dimethyl(1-pyrenyl)phosphane)] (1Cl2) and [RuCl2(η6-p-cymene)(dimethyl(1-pyrenyl)phosphane)] (2Cl2Me), although less efficiently.35 Therefore, replacement of the chlorides by iodides does not seem to produce drastic changes regarding the biological properties of the corresponding Ru complexes; only for cell line A549 are iodido 1I2 and 1I2Me respectively 2 and 3 times more active than their chlorido counterparts (Table 5). It is important to note as well that, as observed in the earlier study,35 η6-methyl benzoate containing 1I2 is clearly more effective than η6-p-cymene containing 2I2Me (Table 5). In contrast, with diisopropyl(1-pyrenyl)phosphane as the P-ligand, η6-p-cymene containing 2Cl2 and 2I2iPr affect the viability of the cells, whereas η6-methyl benzoate containing 1Cl2 and 1I2iPr are not active at all (Table 1).
Table 5. Half-Maximum Inhibitory Concentrationsa (IC50, μM) of Compounds 1I2Me and 2I2 and their Chlorido Counterparts, Respectively [RuCl2(η6-methyl benzoate)(dimethyl(1-pyrenyl)phosphane)] (1Cl2Me) and [RuCl2(η6-p-cymene)(dimethyl(1-pyrenyl)phosphane)] (2Cl2),35 for A549 (Lung Adenocarcinoma), SW620 (Colorectal Adenocarcinoma) and MCF7 (Breast Carcinoma) Human Cells, after Incubation for 24 h.
| compound | A549 | SW620 | MCF7 |
|---|---|---|---|
| 1I2Me | 2.6 ± 0.4 | 9 ± 2 | 6 ± 1 |
| 1Cl2Meb | 5.0 ± 0.6 | 1.9 ± 0.1 | 5.1 ± 1.6 |
| 2I2Me | 6.3 ± 0.4 | 16 ± 4 | 17 ± 6 |
| 2Cl2Meb | 17.2 ± 0.5 | 6.5 ± 0.8 | 9.7 ± 0.1 |
The results are expressed as mean values ± SD out of three independent experiments.
Compounds described in ref (35).
These data again demonstrate the significance of the nature of the P-ligand on changing from dimethyl(1-pyrenyl)phosphane to the bulkier diisopropyl(1-pyrenyl)phosphane (that is from methyl to isopropyl substituents); the mechanism of action of the resulting Ru complexes is completely modified. When the bulkier ligand, namely ligand L (bearing two isopropyl groups), is combined with a bulkier η6 ring, namely p-cymene (1-methyl-4-isopropylbenzene), the corresponding complexes undergo a series of transformations in DMSO, leading to the formation of cytotoxic cyclometalated compounds. With the less sterically hindered ligand dimethyl(1-pyrenyl)phosphane, this cyclometalation pathway is minor, the noncyclometalated complexes obtained also being very cytotoxic, especially if the η6 ring is the less bulky η6-methyl benzoate. It can also be mentioned that 31P{1H} NMR studies revealed that cyclometalated species do not seem to be formed in DMSO with complex 1I2Me, which is mostly preserved in this solvent (Figure S13). Remarkably, for p-cymene containing 2I2, a slow cyclometalation reaction seems to take place; 2I2Me, however, remains the main species present in solution, even after 48 h (Figure S14). It can be pointed out that the cyclometalation is again favored in the presence of the bulkier p-cymene ring. This cyclometalation reaction with 2I2 will be investigated in the future.
In summary, the combination (i) of nonbulky ligands (phosphane + η6 ring) leads to typical piano-stool complexes and (ii) of bulky ligands generates cyclometalated complexes, both families of ruthenium(II) compounds exhibiting high cytotoxic activities.
Conclusions
Following recent studies with ruthenium(II) complexes of the type [Ru(η6-arene)X2(P(1-pyrenyl)R2R3)] (with η6-arene = benzoate, p-cymene and R2, R3 = methyl, phenyl) that showed interesting cytotoxic behaviors,35 four new members of this family of piano-stool complexes have been prepared, namely 1Cl2iPr, 1I2, 2Cl2iPr, and 2I2, with the objective of investigating the role played by the bulkiness of the phosphane ligand (R2 = R3 = isopropyl) as well as the nature of the coordinated anions (X = Cl, I) on the cytotoxic properties. Unexpectedly, complexes 1Cl2iPr, 1I2, 2Cl2iPr, and 2I2 were not as biologically active as the previously reported compounds 1Cl2Me and 2Cl2,35 bearing a less bulky ligand (i.e., R2 = R3 = methyl instead of isopropyl). However, compound 2I2iPr exhibited a striking behavior, since it was observed that its cytotoxic activity was increasing over time (viz. aged DMSO solutions of 2I2 gave lower IC50 values in comparison to freshly prepared solutions).
This surprising and very interesting feature should be due to a transformation of 2I2iPr in solution. Comprehensive studies were therefore carried out to try to elucidate the origin of the observed lag time in activity. It was found that 2I2 and 2CI2iPr were undergoing a series of transformations in DMSO (not in CHCl3), ultimately producing stable cyclometalated species (involving the pyrene ring): i.e. compounds 3Cl, 3IiPr , and 3dmso. The cyclometalation reaction was significantly faster with iodido 2I2iPe in comparison to chlorido 2Cl2. Notably, compounds 1Cl2iPr and 1I2, containing η6-methyl benzoate (whereas 2Cl2iPr and 2I2 have η6-p-cymene), only showed the formation of traces of cyclometalated complexes. Hence, cyclometalation is favored when both the η6-arene ring and the phosphane ligand are bulky; moreover, the cyclometalation rate is higher for the iodido complex. The cyclometalated compounds exhibited good IC50 values, in the range of 1.72–5.82 μM, showing slightly better activities (for the same cell line, i.e. A549) than the ruthenium complexes with much lower cyclometalation rates, namely iodido compounds 1I2Me and 2I2 (2.6 and 6.3 μM, respectively), as well as the previously reported compounds 1Cl2Me and 2Cl2 (5.0 and 17.2 μM, respectively).35
All of the data obtained evidenced that steric hindrance provided by the phosphane ligand is the key with regard to cyclometalation. Therefore, future studies will be dedicated to the detailed investigation of the cyclometalation reaction, for instance using P(1-pyrenyl)R2R3 phosphane ligands bearing mixed R2 and R3 groups of various sizes (e.g., R2 = methyl and R3 = isopropyl).
Experimental Section
Materials and Methods
The ligands and ruthenium complexes were synthesized using standard Schlenk and vacuum-line techniques, under a purified dinitrogen atmosphere. All solvents were purified by using a solvent purification system or by applying standard procedures.72 DMSO containing 0.005% of water (Acros Organics, ref 34844) was used for the solvation studies. 1H, 13C{1H}, and 31P{1H}, and HSQC 1H–13C NMR spectra were recorded at 298 K in CDCl3 unless otherwise stated, using 400 MHz spectrometers. The chemical shifts (δ) are reported in parts per million (ppm) and are referenced to the nondeuterated solvent peak (usually CHCl3: 7.26 ppm for 1H spectra). IR spectra were recorded using a FT-IR spectrometer (in the range 4000–400 cm–1) equipped with an ATR unit, and the main absorption bands are reported in cm–1. High-resolution mass analyses (HRMS) were carried out at the Centres Cientfics i Tecnòlogics de la Universitat de Barcelona, with a time-of-flight instrument using electrospray ionization. Elemental analyses were carried out at the Centres Cientfics i Tecnòlogics de la Universitat de Barcelona; satisfactory elemental analyses were obtained for most of the organometallic compounds described. In the case of í3dmsoiPr the 1H NMR spectrum (Figure S43) revealed the presence of two molecules of uncoordinated dmso, and hence the analysis of the solvato complex is reported. It can be stressed here that the difficulty sometimes encountered in characterizing organometallic compounds with this technique has recently been commented in an Editorial Note of an ACS journal.73
Preparation of Diisopropyl(1-pyrenyl)phosphane
Borane–diisopropyl(1-pyrenyl)phosphane Complex
1-Bromopyrene (1.12 g, 4.0 mmol) was dissolved in 20 mL of THF, and the resulting solution was cooled to −78 °C. A 1.6 M n-BuLi solution (2.4 mL, 3.8 mmol) was subsequently added using a syringe, and the mixture was stirred for 1 h. Chlorodiisopropylphosphane (0.51 mL, 534 mg, 3.5 mmol) was then added, and the reaction mixture was warmed to room temperature for 14 h. A 1 M borane·THF solution (7 mL, 7.0 mmol) was added, and the resulting solution was stirred for 1 h. Water (10 mL) was carefully added, and THF was removed under reduced pressure. The mixture was extracted with dichloromethane (3 × 10 mL), and the combined organic phases were washed with 20 mL of water. The final organic phase was dried over solid anhydrous sodium sulfate and filtered, and the solvent was removed under reduced pressure to give the crude product, which was purified by column chromatography (flash SiO2, hexane/ethyl acetate 95/5). The title product was obtained as a white solid. Yield: 419 mg (36%). 1H NMR (400 MHz, CDCl3): δ 8.86 (d, J = 9.2 Hz, 1H, Ar), 8.58 (dd, J = 11.2 Hz, 8.0 Hz, 1H, Ar), 8.29–8.17 (m, 5H, Ar), 8.11–8.05 (m, 2H, Ar), 2.94–2.85 (m, 2H, i-Pr), 1.40 (dd, 3JHH + 3JHP = 15.2 Hz, 7.2 Hz, 6H, i-Pr), 1.01 (dd, 3JHH + 3JHP = 14.8 Hz, 7.2 Hz, 6H, i-Pr) ppm (Figure S15). 31P{1H} NMR (162 MHz, CDCl3): δ +36.7 (s, br) ppm (Figure S16). HRMS (ESI): m/z calcd for C22H30BNP [M + NH4]+, 350.2203; found, 350.2207.
Diisopropyl(1-pyrenyl)phosphane (L)
The borane–diisopropyl(1-pyrenyl)phosphane complex (202 mg, 0.61 mmol) was dissolved in 20 mL of dichloromethane, and the resulting solution was cooled to 0 °C. HBF4·Et2O (0.43 mL, 3.1 mmol) was added, and the mixture was stirred for 1 h. A thoroughly deoxygenated solution of saturated aqueous NaHCO3 (10 mL) was carefully added to the mixture containing the formed phosphonium salt. The organic layer was then transferred to another flask, washed with thoroughly deoxygenated water, dried over sodium sulfate, filtered, and brought to dryness under reduced pressure. The title product was obtained as an air-sensitive solid. Yield: 181 mg (93%). 1H NMR (400 MHz, CDCl3): δ 9.19 (dd, J = 9.2 Hz, 6.0 Hz, 1H, Ar), 8.22–8.00 (m, 8H, Ar), 2.43 (s, br, 2H, i-Pr), 1.22 (dd, 3JHH + 3JHP = 15.6 Hz, 7.2 Hz, 6H, i-Pr), 0.98 (dd, 3JHH + 3JHP = 12.0 Hz, 6.8 Hz, 6H, i-Pr) ppm (Figure S17). 31P{1H} NMR (162 MHz, CDCl3): δ −8.8 (s) ppm (Figure S18).
Preparation of Ruthenium Compounds
[RuCl2(η6-methyl benzoate)(diisopropyl(1-pyrenyl)phosphane)] (1Cl2iPr)
Diisopropyl(1-pyrenyl)phosphane (180 mg, 0.57 mmol) was dissolved in 10 mL of dichloromethane, and [Ru(η6-methyl benzoate)Cl2]2 (184 mg, 0.30 mmol) was added. The resulting red solution was stirred for 1 h protected from light and filtered, and the solvent was removed under reduced pressure. The residue was recrystallized from dichloromethane/hexane to give the title product as a dark red solid. Yield: 250 mg (70%). IR: ν̅ 3056, 2960, 2927, 2872, 1732 (νC=O), 1428, 1382, 1287, 1266, 1101, 1041, 850, 767, 735, 645, 605, 595 cm–1. 1H NMR (400 MHz, CDCl3): δ 8.85 (d, J = 9.6 Hz, 1H, Ar), 8.56 (t, J = 8.0 Hz, 1H, Ar), 8.33–8.28 (m, 4H, Ar), 8.22 (d, J = 8.8 Hz, 1H, Ar), 8.16–8.11 (m, 2H, Ar), 6.41 (s, br, 1H, PhCOOMe), 6.28 (s, br, 1H, PhCOOMe), 5.09 (s, br, 1H, PhCOOMe), 4.53 (s, br, 1H, PhCOOMe), 4.27 (s, br, 1H, PhCOOMe), 4.01 (s, 3H, PhCOOMe), 3.92 (s, br, 1H, i-Pr), 3.41 (s, br, 1H, i-Pr), 1.81 (d, J = 9.2 Hz, 3H, i-Pr), 1.74 (m, br 3H, i-Pr), 1.36 (d, J = 8.8 Hz, 6H, i-Pr), 0.66 (s, br, 3H, i-Pr) ppm (Figures S19 and S20). 13C{1H} NMR(101 MHz, CDCl3): δ 164.1 (C, PhCOOMe), 133.2–123.4 (C, CH, Ar), 94.2 (s, 2CH, PhCOOMe), 91.1 (d, 2JCP = 10.2 Hz, C, PhCOOMe), 87.9 (s, CH, PhCOOMe), 84.8 (s, br, CH, PhCOOMe), 80.9 (s, br, CH, PhCOOMe), 53.4 (s, br, CH3, PhCOOMe), 23.4 (s, CH, i-Pr), 23.2 (s, CH, i-Pr), 22.0 (s, CH3,i-Pr), 19.0 (s, 3CH3, i-Pr), 17.6 (s, 2CH3, i-Pr) ppm (Figures S20 and S21). 31P{1H} NMR (162 MHz, CDCl3): δ +38.9 (s) ppm (Figure S22). HRMS (ESI): m/z calcd for [M – 2Cl – H]+, 555.1021; found, 553.1025. Anal. Calcd for C30H31Cl2O2PRu: C 57.51; H 4.99. Found: C, 57.08; H 4.97. Single crystals of 1Cl2iPr were obtained from CH2Cl2/n-hexane.
[RuI2(η6-methyl benzoate)(diisopropyl(1-pyrenyl)phosphane)] (1I2iPr)
Compound 1I2iPr was prepared from 1Cl2 through halide exchange. Hence, a suspension of 1Cl2iPr (470 mg, 0.75 mmol) and excess NaI (1.5 g, 10 mmol) in 40 mL of technical acetone was refluxed, protected from light. Complete halide substitution was achieved after 16 h of reflux. The solvent was removed under reduced pressure, and the crude solid obtained was extracted in CH2Cl2/water. After separation, the organic phase was dried with anhydrous Na2SO4 and filtered and dichloromethane was evaporated under reduced pressure. The powder obtained was recrystallized from dichloromethane/hexane, filtered, and washed with pentane. Compound 1I2 was obtained as a brown crystalline solid with a yield of 72% (435 mg). Single crystals of 1I2iPr could be obtained that were suitable for X-diffraction analysis. IR: ν̅ 3052, 2961, 2926, 2865, 1735 (νC=O), 1430, 1296, 1265, 1104, 848, 761, 604 cm–1. 1H NMR (400 MHz, CDCl3): δ 9.03 (d, J = 9.2 Hz, 1H, Ar), 8.52 (t, J = 8.4 Hz, 1H, Ar), 8.34–8.20 (m, 5H, Ar), 8.15–8.10 (m, 2H, Ar), 6.49 (t, J = 5.6 Hz, 1H, PhCOOMe), 6.37 (d, J = 5.6 Hz, 1H, PhCOOMe), 5.24 (t, J = 5.6 Hz, 1H, PhCOOMe), 4.55 (dd, J = 9.6 Hz, J = 5.2 Hz, 1H, PhCOOMe), 4.39 (t, J = 5.2 Hz, 1H, PhCOOMe), 4.29–4.23 (m, 1H, i-Pr), 3.98 (s, 3H, PhCOOMe), 3.61–3.52 (m, 1H, i-Pr), 1.85 (dd, J = 16.4 Hz, J = 7.6 Hz, 3H, i-Pr), 1.71 (dd, J = 11.2 Hz, J = 7.2 Hz, 3H, i-Pr), 1.46 (dd, J = 16.0 Hz, J = 7.2 Hz, 3H, i-Pr), 0.66 (dd, J = 13.2 Hz, J = 7.2 Hz, 3H, i-Pr) ppm (Figures S23 and S24). 13C{1H} NMR (101 MHz, CDCl3): δ 164.1 (C, PhCOOMe), 133.1–123.3 (C, CH, Ar), 93.8 (CH, PhCOOMe), 93.3 (CH, PhCOOMe), 90.5 (CH, PhCOOMe), 89.2 (d, J = 9.4 Hz, C, PhCOOMe), 87.1 (CH, PhCOOMe), 83.8 (CH, PhCOOMe), 53.3 (CH3, PhCOOMe), 30.5 (d, J = 25.7 Hz, CH, i-Pr), 29.6 (d, J = 25.3 Hz, CH, i-Pr), 23.4 (CH3, i-Pr), 20.3 (d, J = 7.0 Hz, CH3, i-Pr), 20.0 (d, J = 7.2 Hz, CH3, i-Pr), 18.4 (CH3, i-Pr) ppm (Figures S24 and S25). 31P{1H} NMR (162 MHz, CDCl3): δ +34.5 (s) ppm (Figure S26). HRMS (ESI): m/z calcd for [M – I]+, 683.0144; found, 683.0142. Anal. Calcd for C30H31I2O2PRu: C, 44.52; H, 3.86. Found: C, 43.26; H, 3.83. Single crystals of 1I2 were obtained from CH2Cl2/n-hexane.
[RuCl2(η6-p-cymene)(diisopropyl(1-pyrenyl)phosphane)] (2Cl2iPr)
Diisopropyl(1-pyrenyl)phosphane (180 mg, 0.57 mmol) was dissolved in 10 mL of dichloromethane, and [Ru(η6-p-cymene)Cl2]2 (139 mg, 0.23 mmol) was subsequently added. The resulting red solution was stirred for 1 h, protected from light, and the solvent was removed under reduced pressure. The residue was recrystallized from dichloromethane/hexane to give the title product as a dark red solid. Yield: 210 mg (74%). IR: ν̅ 3043, 2960, 2926, 2870, 1581, 1460, 1382, 1204, 1083, 1053, 1039, 852, 722, 645, 609, 508 cm–1. 1H NMR (400 MHz, CDCl3): δ 8.85 (d, J = 9.2 Hz, 1H, Ar), 8.56 (t, J = 8.0 Hz, 1H, Ar), 8.31–8.25 (m, 3H, Ar), 8.22 (d, J = 5.6 Hz, 1H, Ar), 8.19–8.08 (m, 3H, Ar), 5.49 (s, br, 1H, p-cym), 4.97 (s, br, 1H, p-cym), 4.67 (s, br, 1H, p-cym), 4.17 (s, br, 1H, p-cym), 3.92 (s, br, 1H, i-Pr), 3.42 (s, br, 1H, i-Pr), 3.02 (h, 3JHH = 6.8 Hz, 1H, p-cym), 1.83 (s, br, 3H, i-Pr), 1.68 (s, br, 3H, i-Pr), 1.43 (s, 3H, p-cym), 1.32 (s, br, 6H, i-Pr), 1.17 (s, br, 3H, i-Pr), 0.66 (s, br, 3H, i-Pr) ppm (Figures S27 and S28). 13C{1H} NMR (101 MHz, CDCl3): δ 133.6–123.2 (C, CH, Ar), 111.6 (d, 2JCP = 5.2 Hz, C, Ar), 98.2 (s, C, p-cym), 88.6 (s, br, CH, p-cym), 87.8 (s, br, CH, p-cym), 86.6 (s, br, CH, p-cym), 85.1 (s, br, CH, p-cym), 30.5 (s, 2CH, i-Pr), 23.5–18.1 (m, 2CH, 4CH3, i-Pr), 17.7 (s, CH3, p-cym) ppm (Figures S28 and S29). 31P{1H} NMR (162 MHz, CDCl3): δ +36.3 (s) ppm (Figure S30). HRMS (ESI): m/z calcd for [M – 2Cl – H]+, 553.1592; found, 553.1593. Anal. Calcd for C32H37Cl2PRu: C, 61.54; H, 5.97. Found: C, 61.76; H, 6.11. Single crystals of 2Cl2iPr were obtained from CH2Cl2/n-hexane.
[RuI2(η6-p-cymene)(diisopropyl(1-pyrenyl)phosphane)] (2I2iPr)
Compound 2I2iPr was obtained from 2Cl2, by applying the procedure described above for the preparation of 1I2iPr. With 423 mg (0.68 mmol) of 2CI2 as the starting material and after 4 h of reflux, compound 2I2iPr was obtained as a brown crystalline solid with a yield of 75% (410 mg). Single crystals of 2I2 could be obtained that were suitable for X-ray diffraction analysis. IR: ν̅ 2952, 2926, 2870, 1448, 1200, 1026, 848, 609, 509 cm–1. 1H NMR (400 MHz, CDCl3): δ 8.96 (d, J = 9.6 Hz, 1H, Ar), 8.51 (t, J = 8.4 Hz, 1H, Ar), 8.32–8.23 (m, 4H, Ar), 8.20 (d, J = 8.8 Hz, 1H, Ar), 8.13 (d, J = 10.0 Hz, 1H, Ar), 8.09 (d, J = 7.6 Hz, 1H, Ar), 5.50 (s, br, 1H, p-cym), 4.86 (s, br, 1H, p-cym), 4.70 (s, br, 1H, p-cym), 4.40 (s, br, 1H, p-cym), 4.27 (s, br, 1H, i-Pr), 3.53 (s, br, 1H, i-Pr), 3.34 (sept, J = 6.8 Hz, 1H, p-cym), 1.85 (s, br, 3H, i-Pr), 1.69 (s, 3H, p-cym), 1.43 (s, br, 3H, i-Pr), 0.96 (s, br, 3H, i-Pr), 0.63 (s, br, 3H, i-Pr) ppm (Figures S31 and S32). 13C{1H} NMR (101 MHz, CDCl3): δ 133.9–122.8 (C, CH, Ar), 110.0 (C, p-cym), 101.3 (C, p-cym), 87.6–86.4 (4CH, p-cym), 31.4 (CH, i-Pr), 23.9 (br, 2CH3, i-Pr), 22.2 (br, CH3, i-Pr), 20.4 (br, CH3, i-Pr), 20.0 (br, CH3, i-Pr), 19.0 (CH3, i-Pr), 18.9 (br, CH3, p-cym) ppm (Figures S32 and S33). 31P{1H} NMR (162 MHz, CDCl3): δ +31.3 (s) ppm (Figure S34). HRMS (ESI): m/z calcd for [M – I]+, 681.0715; found, 681.0720. Anal. Calcd for C32H37I2PRu: C, 47.60; H, 4.62. Found: C, 46.44; H, 4.69. Single crystals of 2I2iPr were obtained from CH2Cl2/n-hexane.
[RuCl(η6-p-cymene)(κ2C-diisopropyl(1-pyrenyl)phosphane)] (3CliPr)
A suspension of [RuCl(μ-Cl)(η6-p-cymene)]2 (643 mg, 1.05 mmol), ligand L (716 mg, 2.25 mmol), and NaOAc (492 mg, 5.94 mmol) in 160 mL of methanol was stirred for 4 h at room temperature, protected from light. The solvent was removed under reduced pressure, and the residue was extracted with dichloromethane/water. The combined organic phase was dried with anhydrous Na2SO4 and filtered. After removal of the solvent, the crude product was purified by column chromatography (SiO2, dichloromethane/ethyl acetate 99.5/0.5). The solvent was removed under reduced pressure to give the title product as an orange solid. Yield: 646 mg (52%). IR: ν̅ 3029, 2954, 2923, 2867, 1667, 1437, 1384, 1301, 1246, 1184, 1111, 1032, 836, 739, 615 cm–1. 1H NMR (CDCl3, 400 MHz): δ 8.93 (s, 1H, Ar), 8.11–7.93 (m, 7H, Ar), 6.23 (d, J = 4.8 Hz, 1H, p-cym), 6.22 (d, J = 5.2 Hz, 1H, p-cym), 5.33 (d, J = 6.4 Hz, 1H, p-cym), 4.94 (d, J = 6.0 Hz, 1H, p-cym), 3.05–2.94 (m, 1H, i-Pr), 2.94–2.77 (m, 2H, i-Pr), 1.97 (s, 3H, p-cym), 1.59 (dd, J = 14.4 Hz, 7.2 Hz, 3H, i-Pr), 1.43 (dd, J = 15.6 Hz, 7.2 Hz, 3H, i-Pr), 1.23 (d, J = 6.8 Hz, 3H, i-Pr), 1.12 (d, J = 6.8 Hz, 3H, i-Pr), 1.10 (dd, J = 14.4 Hz, 7.2 Hz, 3H, i-Pr), 1.06 (dd, J = 13.6 Hz, 6.8 Hz, 3H, i-Pr) ppm (Figures S35 and S36). 13C{1H} NMR (CDCl3, 101 MHz): δ 169.2–122.4 (C, CH, Ar), 110.9 (C, p-cym), 97.6 (d, JCP = 6.1 Hz, CH, p-cym), 96.0 (C, p-cym), 92.0 (CH, p-cym), 88.6 (CH, p-cym), 84.9 (CH, p-cym), 30.8 (CH, i-Pr), 28.6 (d, JCP = 25.1 Hz, CH, i-Pr), 26.7 (d, JCP = 24.9 Hz, CH, i-Pr), 23.2 (CH3, i-Pr), 22.6 (CH3, i-Pr), 21.4 (d, JCP = 1.8, CH3, i-Pr), 19.7 (d, JCP = 2.5 Hz, CH3, i-Pr), 19.21 (CH3, i-Pr), 19.16 (CH3, i-Pr), 18.3 (CH3, p-cym) ppm (Figures S36 and S37). 31P{1H} NMR (CDCl3, 162 MHz): δ +80.8 (s) ppm (Figure S38). HRMS (ESI): m/z calcd for [M]+, 588.1281; found, 588.1305; calcd for [M – Cl]+, 533.1592; found, 533.1600. Anal. Calcd for C32H36ClPRu: C, 65.35; H, 6.17. Found: C, 65.35; H, 6.24. Single crystals of 3CliPr were obtained from CH2Cl2/n-hexane.
[RuI(η6-p-cymene)(κ2C-diisopropyl(1-pyrenyl)phosphane)] (3IiPr)
A suspension of 3CliPr (140 mg, 0.24 mmol) and NaI (893 mg, 6.00 mmol) in 20 mL of technical acetone was refluxed for 4 h, protected from light. The solvent was removed under reduced pressure, and the residue was extracted with dichloromethane/water. The combined organic phase was dried with anhydrous Na2SO4 and filtered. After evaporation of the solvent, the title product was obtained as a brown solid. Yield 133 mg (82%). IR: ν̅ 3033, 2960, 2921, 2866, 1707, 1568, 1436, 1382, 1259, 1079, 1017, 795, 737, 687, 653, 605 cm–1. 1H NMR (CDCl3, 400 MHz): δ 8.76 (s, 1H, Ar), 8.09–7.92 (m, 7H, Ar), 6.04 (d, J = 6.0 Hz, 2H, p-cym), 5.59 (d, J = 6.8 Hz, 1H, p-cym), 5.17 (d, J = 6.0 Hz, 1H, p-cym), 3.23–3.12 (m, 1H, i-Pr), 3.05 (sept, J = 6.8 Hz, 1H, i-Pr), 2.83–2.70 (m, 1H, i-Pr), 2.10 (s, 3H, p-cym), 1.64 (dd, J = 14.4 Hz, 7.2 Hz, 3H, i-Pr), 1.53 (dd, J = 16.0 Hz, 7.6 Hz, 3H, i-Pr), 1.29 (d, J = 6.8 Hz, 3H, i-Pr), 1.11 (d, J = 6.8 Hz, 3H, i-Pr), 1.01 (dd, J = 14.8 Hz, 7.2 Hz, 3H, i-Pr), 0.93 (dd, J = 13.2 Hz, 6.8 Hz, 3H, i-Pr) ppm (Figures S39 and S40). 13C{1H} NMR (CDCl3, 101 MHz): δ 165.1–122.3 (C, CH, Ar), 112.1 (C, p-cym), 98.0 (C, p-cym), 96.3 (d, JCP = 5.5 Hz, CH, p-cym), 90.0 (d, JCP = 3.5 Hz, CH, p-cym), 88.7 (CH, p-cym), 86.3 (CH, p-cym), 31.3 (CH, i-Pr), 30.2 (d, JCP = 26.4 Hz, CH, i-Pr), 29.8 (d, JCP = 24.5 Hz, CH, i-Pr), 23.7 (CH3, i-Pr), 23.1 (d, JCP = 2.2 Hz, CH3, i-Pr), 22.6 (s, CH3, i-Pr), 19.8 (CH3, i-Pr), 19.7 (d, JCP = 2.6 Hz, CH3, i-Pr), 19.0 (CH3, i-Pr), 18.7 (CH3, p-cym) ppm (Figures S40 and S41). 31P{1H} NMR (CDCl3, 162 MHz): δ +79.3 (s) ppm (Figure S42). HRMS (ESI): m/z calcd for [M + H]+, 681.0721; found, 681.0728; calcd for [M – I]+, 553.1592; found, 553.1593. Anal. Calcd for C32H36IPRu: C, 56.56; H, 5.34. Found: C 55.44; H 5.78. Single crystals of 3I were obtained from CH2Cl2/n-hexane.
[Ru(η6-p-cymene)(κS-dmso)(κ2C-diisopropyl(1-pyrenyl)phosphane)]PF6 (3dmsoiPr)
Complex 3CliPr (300 mg, 0.51 mmol) was dissolved in dichloromethane (30 mL) and dmso (0.35 mL, 4.93 mmol). Thallium hexafluorophosphate (196 mg, 0.54 mmol) was added, and the mixture was stirred overnight, protected from light. The suspension was filtered, and the solvent was removed under reduced pressure. The crude product was recrystallized in dichloromethane/diethyl ether at −20 °C, yielding the title product as a fine yellow solid. Yield 375 mg (95%). IR: ν̅ 2963, 1440, 1384, 1293, 1242, 1106, 1013, 832, 740, 668, 597, 574 cm–1. 1H NMR (acetone-d6, 400 MHz): δ 8.88 (s, 1H, Ar), 8.43–8.35 (m, 2H, Ar), 8.31–8.23 (m, 4H, Ar), 8.09 (t, J = 7.06 Hz, 1H, Ar), 7.11 (d, J = 6.4 Hz, 1H, p-cym), 7.08 (d, J = 6.8 Hz, 1H, p-cym), 6.35 (d, J = 5.6 Hz, 1H, p-cym), 6.10 (d, J = 6.4 Hz, 1H, p-cym), 3.56–3.43 (m, 1H, i-Pr), 3.45 (s, 3H, dmso), 3.29 (sept, J = 6.8 Hz, 1H, i-Pr), 2.90–2.81 (m, 1H, i-Pr), 2.46 (s, 3H, p-cym), 1.76 (dd, J = 16.8 Hz, 7.2 Hz, 3H, i-Pr), 1.66 (s, 3H, dmso), 1.53 (dd, J = 17.2 Hz, 7.2 Hz, 3H, i-Pr), 1.45 (d, J = 6.8 Hz, 3H, i-Pr), 1.38 (dd, J = 13.2 Hz, 6.8 Hz, 3H, i-Pr), 1.18 (d, J = 6.8 Hz, 3H, i-Pr), 0.29 (dd, J = 16.4 Hz, 6.8 Hz, 3H, i-Pr) ppm (Figures S43 and S44). 13C{1H} NMR (CD3COCD3, 101 MHz): δ 160.3–123.4 (C, CH, Ar), 114.4 (C, p-cym), 98.6 (CH, p-cym), 97.3 (br, CH, p-cym), 95.5 (CH, p-cym), 93.9 (br, CH, p-cym), 53.7 (CH3, dmso), 47.2 (CH3, dmso), 32.1 (CH, i-Pr), 31.2 (d, JCP = 27.7 Hz, CH, i-Pr), 26.4 (d, JCP = 27.0 Hz, CH, i-Pr), 24.8 (CH3, i-Pr), 21.4 (CH3, i-Pr), 19.6 (CH3, i-Pr), 19.4 (CH3, i-Pr), 19.2 (d, JCP = 2.4, CH3, i-Pr), 19.0 (CH3, p-cym), 18.1 (d, JCP = 6.1, CH3, i-Pr) ppm (Figures S44 and S45). 31P{1H} NMR (CD3COCD3, 162 MHz): δ +86.6 (s), −144.2 (sept, JPF = 708.8 Hz) ppm (Figure S46). 19F{1H} NMR (CD3COCD3, 377 MHz): δ −72.5 (d, JFP = 708.8 Hz) ppm (Figure S47). HRMS (ESI): m/z calcd for [M – PF6]+, 631.1732; found, 631.1741; calcd for [M – PF6 – dmso]+, 553.1592, found, 553.1604. Anal. Calcd for C38H54F6O3P2RuS3 (3dmso·2dmso): C, 48.97; H, 5.84. Found: C 48.89; H, 5.71. The presence of two molecules of dmso is observed as well in the 1H NMR spectrum of this compound (see Figure S43). Single crystals of 3dmsoiPr were obtained from CH2Cl2/n-hexane.
[RuI2(η6-methyl benzoate)(dimethyl(1-pyrenyl)phosphane)] (1I2Me)
Compound 1I2Me was prepared from [RuCl2(η6-methyl benzoate)(dimethyl(1-pyrenyl)phosphane)] (1Cl2), which was described previously.35 With 240 mg (0.42 mmol) of the chloro complex as the starting material, iodo complex 1I2Me was obtained as a brown crystalline solid with a yield of 37% (116 mg), after 11 days of reflux in technical acetone. The conversion of 1I2 into 1I2Me could be followed by 31P NMR (see Figure S48). Single crystals of 1I2 could be obtained that were suitable for X-ray diffraction analysis. IR: ν̅ 3065, 3035, 1935, 1733 (νC=O), 1288, 1270, 913, 849 cm–1. 1H NMR (400 MHz, CDCl3): δ 9.31 (d, J = 9.6 Hz, 1H, Ar), 8.51–8.10 (m, 8H, Ar), 6.25 (s, br, 2H, PhCOOMe), 5.70 (t, J = 5.6 Hz, 1H, PhCOOMe), 4.91 (s, br, 2H, PhCOOMe), 3.90 (s, 3H, PhCOOMe), 2.49 (d, J = 9.6 Hz, 6H, i-Pr) ppm (Figure S49). 13C{1H} NMR (101 MHz, CDCl3): δ 165.4 (C, PhCOOMe), 130.3–123.6 (C, CH, Ar), 95.0 (CH, PhCOOMe), 53.2 (CH3, PhCOOMe), 20.4 (CH3, i-Pr) ppm (Figure S50). 31P{1H} NMR (162 MHz, CDCl3): δ −5.2 (s) ppm (Figure S51). HRMS (ESI): m/z calcd for [M – I]+, 626.9518; found, 626.9518. Anal. Calcd for C26H23I2O2PRu: C, 41.45; H, 3.08. Found: C, 41.25; H, 3.11.
[RuI2(η6-p-cymene)(dimethyl(1-pyrenyl)phosphane)] (2I2Me)
Compound 2I2Me was prepared from [RuCl2(η6-p-cymene)(dimethyl(1-pyrenyl)phosphane)] (2Cl2), which was described previously.35 Using 230 mg (0.40 mmol) of the chloro precursor, compound 2I2Me was obtained with a yield of 68% (206 mg), after 24 h of reflux in technical acetone. Single crystals of 2I2 could be obtained that were suitable for X-ray diffraction analysis. IR: ν̅ 2957, 2917, 1624, 1430, 1383, 913, 845, 719, 698, 600, 532 cm–1. 1H NMR (101 MHz, CDCl3): δ 9.32 (d, J = 9.6 Hz, 1H, Ar), 8.39–8.26 (m, 5H, Ar), 8.21 (d, J = 8.0 Hz, 1H, Ar), 8.14 (d, J = 9.2 Hz, 1H, Ar), 8.11 (t, J = 8.0 Hz, 1H, Ar), 5.06 (d, J = 6.0 Hz, 2H, p-cym), 4.78 (s, br, 2H, p-cym), 3.14 (sept, J = 6.8 Hz, 1H, p-cym), 2.46 (d, J = 9.2 Hz, 6H, PMe), 1.98 (s, 3H, p-cym), 1.06 (d, J = 7.2 Hz, 6H, p-cym) ppm (Figures S52 and S53). 13C{1H} NMR (101 MHz, CDCl3): δ 133.3–121.1 (C, CH, Ar), 108.1 (C, p-cym) 96.2 (C, p-cym), 91.3 (d, J = 15.6 Hz, 2CH, p-cym), 86.2 (br, 2CH, p-cym), 31.9 (CH, p-cym), 22.6 (br, 2CH3, PMe), 20.0 (CH3, p-cym) ppm (Figures S53 and S54). 31P{1H} NMR (162 MHz, CDCl3): δ −5.8 (s) ppm (Figure S55). HRMS (ESI): m/z calcd for [M – I]+, 625.0089; found, 625.0090. Anal. Calcd for C28H29I2PRu: C, 44.76; H 3.89. Found: C, 44.78; H, 3.98.
X-ray Crystallography
Data for compounds 1Cl2iPr, 1I2, 2I2iPr, 3Cl, 3IiPr, 3dmso, 1I2Me, and 2I2 (see the Supporting Information) were collected on a Bruker APEX II QUAZAR diffractometer equipped with a microfocus multilayer monochromator with Mo Kα radiation (λ = 0.71073 Å). Data for compound 2Cl2iPr were collected using a Bruker D8 diffractometer with a Photon 100 detector on the Advanced Light Source beamline 11.3.1 at Lawrence Berkeley National Laboratory, from a silicon 111 monochromator (λ = 0.7749 Å). Data reduction and absorption corrections were performed by using SAINT and SADABS, respectively.74 The structures were solved using SHELXT75 and refined with full-matrix least squares on F2 by using SHELXL.76 Low-quality data for compound 3dmso could not be improved due to small crystal dimensions (very thin plates of 80 μm). In this case, a void containing only diffuse electron density was analyzed and taken into account with Olex2/Solvent Mask.77 An estimated content of five diffuse lattice CH2Cl2 molecules per asymmetric unit cell was deduced and included in the formula. All details can be found in CCDC 2054649–2054657, which contain the supplementary crystallographic data for the present paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Center via https://summary.ccdc.cam.ac.uk/structure.summary.form.
Computational Details
All calculations were performed using the Gaussian 09 (revision D01)78 electronic structure package using a 10–8 convergence criterion for the density matrix elements. The PBE functional was used for both exchange and correlation functionals.79,80 The fully optimized contracted triple-ζ all-electron Gaussian basis set with added polarization functions developed by Ahlrichs and co-workers was used for all the elements in all molecules,81 except for the ruthenium and iodine atoms, for which the Stuttgart/Dresden effective core potential (SDD)82−84 basis set was used. Full system optimization was carried out for all compounds investigated, followed by the corresponding vibrational analysis to calculate the thermochemical properties. The dimethyl sulfoxide solvent properties were modeled using a polarizable continuum model (self-consistent reaction field approximation) with Truhlar’s SMD variation.85
Cell Culture and Viability Assays
Human lung adenocarcinoma A549, colorectal adenocarcinoma SW620, and breast adenocarcinoma MCF7 cell lines were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). A549 and SW620 cells were cultured in DMEM medium with 10% heat-inactivated fetal bovine serum (FBS; Life Technologies, Carlsbad, CA, USA), 100 U mL–1 penicillin, 100 μg mL–1 streptomycin, and 2 mM glutamine. The MCF-7 cell line was cultured in DMEM–F12 (HAM) media (1/1) with 10% FBS, 50 μM sodium pyruvate, 10 μg mL–1 insulin (Sigma-Aldrich Chemical Co., St. Louis, MO, USA), 100 U mL–1 penicillin, 100 μg mL–1 streptomycin, and 2 mM glutamine. All reagents not specified above were obtained from Biological Industries, Beit Haemek, Israel. Cells were grown at 37 °C under a 5% CO2 atmosphere.
Cell viability assays were conducted using the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay. Amounts of 104 cells were seeded in 96-well plates (105 cells mL–1) and allowed to grow for 24 h. For single-point experiments, the cells were treated with compounds 1Cl2iPr, 1I2, 2Cl2iPr, and 2I2 at 10 μM for 24 h. For dose–response assays, the cells were treated with different concentrations of compounds 2Cl2iPr, 2I2, 1I2Me, and 2I2 (from 0.8 to 100 μM), and 3CliPr, 3I, and 3dmsoiPr (from 0.4 to 50 μM) for 24 h, using DMSO complex solutions of different “aging times”, Day 0 corresponds to a freshly prepared solution and days 1, 2, 5, and 7 correspond to 1-, 2-, 5- and 7-day-old solution, respectively. After the 24 h treatment, a 10 μM solution of MTT was added to each well and the plates were incubated for an additional 2 h at 37 °C. The medium was removed, and the purple formazan crystals were dissolved in 100 μL of DMSO. The absorbance was measured at 570 nm using a multiwell plate reader (Multiskan FC, Thermo Scientific). The cell viability was calculated according to the relation viability (%) = [(absorbance of treated wells)/(absorbance of control wells)] × 100. The IC50 values (corresponding to the compound concentrations that produce 50% reduction in cell viability) were obtained from the dose–response curves using GraphPad Prism V5.0 for Windows (GraphPad Software, San Diego, CA, USA). All data are shown as the mean value ± SD of at least three independent experiments for single-point assays and for the dose–response curves.
Acknowledgments
Financial support from the Spanish Ministerio de Ciencia Innovación, y Universidades (Project Nos. CTQ2015-65040-P, RED2018-102471-T, PID2019-107006GB-C21, PGC2018-098630-B-I00, and CTQ2017-88446-R AEI/FEDER, UE) and from the Instituto de Salud Carlos III (ISCIIIFIS PI18/00441, FEDER) is acknowledged. A.G. thanks the Royal Society of Chemistry for financial support (RSC Research Fund grant RF19-7147). J.C. thanks the Spanish MICINN for a Ramoń y Cajal research contract (RYC2018-024692-I) and the Spanish Structures of Excellence María de Maeztu program (MDM-2017-0767). P.G. acknowledges the Institució Catalana de Recerca i Estudis Avançats (ICREA).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.inorgchem.1c00507.
Synthetic procedures for L and L=O, X-ray crystallographic data, 1H, 13C{1H}, 31P{1H}, and 1H–13C HSQC NMR spectra, time-dependent NMR studies, schematic representation of the DMSO-mediated cyclometalation reaction, rate constants for the solvation of 3CliPr, and Cartesian coordinates (computational calculations) (PDF)
Accession Codes
CCDC 2054649–2054657 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Siegel R. L.; Miller K. D.; Jemal A. Cancer Statistics, 2017. Ca-Cancer J. Clin. 2017, 67 (1), 7–30. 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- Ferlay J.; Soerjomataram I.; Dikshit R.; Eser S.; Mathers C.; Rebelo M.; Parkin D. M.; Forman D.; Bray F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136 (5), E359–E386. 10.1002/ijc.29210. [DOI] [PubMed] [Google Scholar]
- Alderden R. A.; Hall M. D.; Hambley T. W. The discovery and development of cisplatin. J. Chem. Educ. 2006, 83 (5), 728–734. 10.1021/ed083p728. [DOI] [Google Scholar]
- Kelland L. The resurgence of platinum-based cancer chemotherapy. Nat. Rev. Cancer 2007, 7 (8), 573–584. 10.1038/nrc2167. [DOI] [PubMed] [Google Scholar]
- Shaili E. Platinum anticancer drugs and photochemotherapeutic agents: recent advances and future developments. Sci. Prog. 2014, 97 (1), 20–40. 10.3184/003685014X13904811808460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galanski M. Recent developments in the field of anticancer platinum complexes. Recent Pat. Anti-Cancer Drug Discovery 2006, 1 (2), 285–295. 10.2174/157489206777442287. [DOI] [PubMed] [Google Scholar]
- Rothenberg M. L.; Carbone D. R.; Johnson D. H. Improving the evaluation of new cancer treatments: challenges and opportunities. Nat. Rev. Cancer 2003, 3 (4), 303–309. 10.1038/nrc1047. [DOI] [PubMed] [Google Scholar]
- Ohmichi M.; Hayakawa J.; Tasaka K.; Kurachi H.; Murata Y. Mechanisms of platinum drug resistance. Trends Pharmacol. Sci. 2005, 26 (3), 113–116. 10.1016/j.tips.2005.01.002. [DOI] [PubMed] [Google Scholar]
- Marzano C.; Pellei M.; Tisato F.; Santini C. Copper Complexes as Anticancer Agents. Anti-Cancer Agents Med. Chem. 2009, 9 (2), 185–211. 10.2174/187152009787313837. [DOI] [PubMed] [Google Scholar]
- Ott I. On the medicinal chemistry of gold complexes as anticancer drugs. Coord. Chem. Rev. 2009, 253, 1670–1681. 10.1016/j.ccr.2009.02.019. [DOI] [Google Scholar]
- Alessio E.; Guo Z. J. Metal Anticancer Complexes - Activity, Mechanism of Action, Future Perspectives. Eur. J. Inorg. Chem. 2017, 2017 (12), 1539–1540. 10.1002/ejic.201700196. [DOI] [Google Scholar]
- Medici S.; Peana M.; Nurchi V. M.; Lachowicz J. I.; Crisponi G.; Zoroddu M. A. Noble metals in medicine: Latest advances. Coord. Chem. Rev. 2015, 284, 329–350. 10.1016/j.ccr.2014.08.002. [DOI] [Google Scholar]
- Cuello-Garibo J.-A.; James C. C.; Siegler M. A.; Bonnet S. Ruthenium-based PACT compounds based on an N,S non-toxic ligand: a delicate balance between photoactivation and thermal stability. Chem. Sq. 2017, 1, 2. 10.28954/2017.csq.12.002. [DOI] [Google Scholar]
- Brissos R. F.; Korrodi-Gregório L.; Pérez-Tomás R.; Roubeau O.; Gamez P. Antiproliferative properties of iron supramolecular cylinders. Chem. Sq. 2018, 2, 4. 10.28954/2018.csq.11.001. [DOI] [Google Scholar]
- Hager L. A.; Mokesch S.; Kieler C.; Alonso-De Castro S.; Baier D.; Roller A.; Kandioller W.; Keppler B. K.; Berger W.; Salassa L.; Terenzi A. Ruthenium arene complexes bearing naphthyl-substituted 1,3-dioxoindan-2-carboxamides ligands for G-quadruplex DNA recognition. Dalton Trans. 2019, 48 (32), 12040–12049. 10.1039/C9DT02078K. [DOI] [PubMed] [Google Scholar]
- Poynton F. E.; Bright S. A.; Blasco S.; Williams D. C.; Kelly J. M.; Gunnlaugsson T. The development of ruthenium(II) polypyridyl complexes and conjugates for in vitro cellular and in vivo applications. Chem. Soc. Rev. 2017, 46 (24), 7706–7756. 10.1039/C7CS00680B. [DOI] [PubMed] [Google Scholar]
- Notaro A.; Gasser G. Monomeric and dimeric coordinatively saturated and substitutionally inert Ru(II) polypyridyl complexes as anticancer drug candidates. Chem. Soc. Rev. 2017, 46 (23), 7317–7337. 10.1039/C7CS00356K. [DOI] [PubMed] [Google Scholar]
- Zeng L. L.; Gupta P.; Chen Y. L.; Wang E. J.; Ji L. N.; Chao H.; Chen Z. S. The development of anticancer ruthenium(II) complexes: from single molecule compounds to nanomaterials. Chem. Soc. Rev. 2017, 46 (19), 5771–5804. 10.1039/C7CS00195A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergamo A.; Messori L.; Piccioli F.; Cocchietto M.; Sava G. Biological role of adduct formation of the ruthenium(III) complex NAMI-A with serum albumin and serum transferrin. Invest. New Drugs 2003, 21 (4), 401–411. 10.1023/A:1026243000320. [DOI] [PubMed] [Google Scholar]
- Alessio E.; Mestroni G.; Bergamo A.; Sava G. Ruthenium antimetastatic agents. Curr. Top. Med. Chem. 2004, 4 (15), 1525–1535. 10.2174/1568026043387421. [DOI] [PubMed] [Google Scholar]
- Hartinger C. G.; Zorbas-Seifried S.; Jakupec M. A.; Kynast B.; Zorbas H.; Keppler B. K. From bench to bedside - preclinical and early clinical development of the anticancer agent indazolium trans-[tetrachlorobis(1H-indazole)ruthenate(III)] (KP1019 or FFC14A). J. Inorg. Biochem. 2006, 100 (5–6), 891–904. 10.1016/j.jinorgbio.2006.02.013. [DOI] [PubMed] [Google Scholar]
- Hartinger C. G.; Jakupec M. A.; Zorbas-Seifried S.; Groessl M.; Egger A.; Berger W.; Zorbas H.; Dyson P. J.; Keppler B. K. KP1019, A New Redox-Active Anticancer Agent - Preclinical Development and Results of a Clinical Phase I Study in Tumor Patients. Chem. Biodiversity 2008, 5 (10), 2140–2155. 10.1002/cbdv.200890195. [DOI] [PubMed] [Google Scholar]
- Wernitznig D.; Kiakos K.; Del Favero G.; Harrer N.; Machat H.; Osswald A.; Jakupec M. A.; Wernitznig A.; Sommergruber W.; Keppler B. K. First-in-class ruthenium anticancer drug (KP1339/IT-139) induces an immunogenic cell death signature in colorectal spheroids in vitro. Metallomics 2019, 11 (6), 1044–1048. 10.1039/C9MT00051H. [DOI] [PubMed] [Google Scholar]
- Domotor O.; Hartinger C. G.; Bytzek A. K.; Kiss T.; Keppler B. K.; Enyedy E. A. Characterization of the binding sites of the anticancer ruthenium(III) complexes KP1019 and KP1339 on human serum albumin via competition studies. JBIC, J. Biol. Inorg. Chem. 2013, 18 (1), 9–17. 10.1007/s00775-012-0944-6. [DOI] [PubMed] [Google Scholar]
- Neuditschko B.; Legin A. A.; Baier D.; Schintlmeister A.; Reipert S.; Wagner M.; Keppler B. K.; Berger W.; Meier-Menches S. M.; Gerner C. Interaction with Ribosomal Proteins Accompanies Stress Induction of the Anticancer Metallodrug BOLD-100/KP1339 in the Endoplasmic Reticulum. Angew. Chem., Int. Ed. 2021, 60 (10), 5063–5068. 10.1002/anie.202015962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monro S.; Colon K. L.; Yin H. M.; Roque J.; Konda P.; Gujar S.; Thummel R. P.; Lilge L.; Cameron C. G.; McFarland S. A. Transition Metal Complexes and Photodynamic Therapy from a Tumor-Centered Approach: Challenges, Opportunities, and Highlights from the Development of TLD1433. Chem. Rev. 2019, 119 (2), 797–828. 10.1021/acs.chemrev.8b00211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smithen D. A.; Yin H. M.; Beh M. H. R.; Hetu M.; Cameron T. S.; McFarland S. A.; Thompson A. Synthesis and Photobiological Activity of Ru(II) Dyads Derived from Pyrrole-2-carboxylate Thionoesters. Inorg. Chem. 2017, 56 (7), 4121–4132. 10.1021/acs.inorgchem.7b00072. [DOI] [PubMed] [Google Scholar]
- Bergamo A.; Sava G. Linking the future of anticancer metal-complexes to the therapy of tumour metastases. Chem. Soc. Rev. 2015, 44 (24), 8818–8835. 10.1039/C5CS00134J. [DOI] [PubMed] [Google Scholar]
- Murray B. S.; Babak M. V.; Hartinger C. G.; Dyson P. J. The development of RAPTA compounds for the treatment of tumors. Coord. Chem. Rev. 2016, 306, 86–114. 10.1016/j.ccr.2015.06.014. [DOI] [Google Scholar]
- Su W.; Li Y. C.; Li P. Y. Design of Ru-arene Complexes for Antitumor Drugs. Mini-Rev. Med. Chem. 2018, 18 (2), 184–193. 10.2174/1389557517666170510113453. [DOI] [PubMed] [Google Scholar]
- Nhukeaw T.; Hongthong K.; Dyson P. J.; Ratanaphan A. Cellular responses of BRCA1-defective HCC1937 breast cancer cells induced by the antimetastasis ruthenium(II) arene compound RAPTA-T. Apoptosis 2019, 24 (7–8), 612–622. 10.1007/s10495-019-01544-w. [DOI] [PubMed] [Google Scholar]
- Lee R. F. S.; Escrig S.; Maclachlan C.; Knott G. W.; Meibom A.; Sava G.; Dyson P. J. The Differential Distribution of RAPTA-T in Non-Invasive and Invasive Breast Cancer Cells Correlates with Its Anti-Invasive and Anti-Metastatic Effects. Int. J. Mol. Sci. 2017, 18 (9), 1869. 10.3390/ijms18091869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thota S.; Rodrigues D. A.; Crans D. C.; Barreiro E. J. Ru(II) Compounds: Next-Generation Anticancer Metallotherapeutics?. J. Med. Chem. 2018, 61 (14), 5805–5821. 10.1021/acs.jmedchem.7b01689. [DOI] [PubMed] [Google Scholar]
- Rafols L.; Torrente S.; Aguila D.; Soto-Cerrato V.; Perez-Tomas R.; Gamez P.; Grabulosa A. Expanding the Range of Pyrenylphosphines and Their Derived Ru(II)-Arene Complexes. Organometallics 2020, 39 (16), 2959–2971. 10.1021/acs.organomet.0c00302. [DOI] [Google Scholar]
- Brissos R. F.; Clavero P.; Gallen A.; Grabulosa A.; Barrios L. A.; Caballero A. B.; Korrodi-Gregorio L.; Perez-Tomas R.; Muller G.; Soto-Cerrato V.; Gamez P. Highly Cytotoxic Ruthenium(II)-Arene Complexes from Bulky 1-Pyrenylphosphane Ligands. Inorg. Chem. 2018, 57 (23), 14786–14797. 10.1021/acs.inorgchem.8b02541. [DOI] [PubMed] [Google Scholar]
- Wang F. Y.; Habtemariam A.; van der Geer E. P. L.; Fernandez R.; Melchart M.; Deeth R. J.; Aird R.; Guichard S.; Fabbiani F. P. A.; Lozano-Casal P.; Oswald I. D. H.; Jodrell D. I.; Parsons S.; Sadler P. J. Controlling ligand substitution reactions of organometallic complexes: Tuning cancer cell cytotoxicity. Proc. Natl. Acad. Sci. U. S. A. 2005, 102 (51), 18269–18274. 10.1073/pnas.0505798102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H. M.; Parkinson J. A.; Morris R. E.; Sadler P. J. Highly selective binding of organometallic ruthenium ethylenediamine complexes to nucleic acids: Novel recognition mechanisms. J. Am. Chem. Soc. 2003, 125 (1), 173–186. 10.1021/ja027719m. [DOI] [PubMed] [Google Scholar]
- Hodson E.; Simpson S. J. Synthesis and characterisation of (eta(6)-cymene)Ru(L)X-2 compounds: single crystal X-ray structure of (eta(6)-cymene)Ru(P{OPh}(3))Cl-2 at 203 K. Polyhedron 2004, 23 (17), 2695–2707. 10.1016/j.poly.2004.06.016. [DOI] [Google Scholar]
- Pettinari R.; Marchetti F.; Condello F.; Pettinari C.; Lupidi G.; Scopelliti R.; Mukhopadhyay S.; Riedel T.; Dyson P. J. Ruthenium(II)-Arene RAPTA Type Complexes Containing Curcunnin and Bisdemethoxycurcumin Display Potent and Selective Anticancer Activity. Organometallics 2014, 33 (14), 3709–3715. 10.1021/om500317b. [DOI] [Google Scholar]
- Babak M. V.; Meier S. M.; Huber K. V. M.; Reynisson J.; Legin A. A.; Jakupec M. A.; Roller A.; Stukalov A.; Gridling M.; Bennett K. L.; Colinge J.; Berger W.; Dyson P. J.; Superti-Furga G.; Keppler B. K.; Hartinger C. G. Target profiling of an antimetastatic RAPTA agent by chemical proteomics: relevance to the mode of action. Chem. Sci. 2015, 6 (4), 2449–2456. 10.1039/C4SC03905J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun R. C.; Chu X. D.; Zhang S. W.; Li T. Y.; Wang Z.; Zhu B. L. Synthesis, Structure, Reactivity, and Catalytic Activity of Cyclometalated (Phosphine)- and (Phosphinite)ruthenium Complexes. Eur. J. Inorg. Chem. 2017, 2017 (25), 3174–3183. 10.1002/ejic.201700479. [DOI] [Google Scholar]
- Grabulosa A.; Granell J.; Font-Bardia M. Cyclometallated ruthenium complexes with P-stereogenic monophosphines containing a polycyclic aromatic substituent. J. Organomet. Chem. 2019, 896, 51–58. 10.1016/j.jorganchem.2019.05.015. [DOI] [Google Scholar]
- Kuwabara T.; Kato T.; Takano K.; Kodama S.; Manabe Y.; Tsuchida N.; Minami Y.; Hiyama T.; Ishii Y. P-C reductive elimination in Ru(ii) complexes to convert triarylphosphine ligands into five- or six-membered phosphacycles. Chem. Commun. 2018, 54 (42), 5357–5360. 10.1039/c8cc02539h. [DOI] [PubMed] [Google Scholar]
- Zhang S. W.; Chu X. D.; Li T. Y.; Wang Z.; Zhu B. L. Synthesis, Structures, and Reactivity of Single and Double Cyclometalated Complexes Formed by Reactions of [Cp*MCl2](2) (M = Ir and Rh) with Dinaphthyl Phosphines. ACS Omega 2018, 3 (4), 4522–4533. 10.1021/acsomega.8b00193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun R.; Chu X.; Zhang S.; Li T.; Wang Z.; Zhu B. Synthesis, Structure, Reactivity, and Catalytic Activity of Cyclometalated (Phosphine)- and (Phosphinite)ruthenium Complexes. Eur. J. Inorg. Chem. 2017, 2017 (25), 3174–3183. 10.1002/ejic.201700479. [DOI] [Google Scholar]
- Binstead R. A.; Jung B.; Zuberbühler A. D.. Specfit/32; Spectrum Software Associates: 2000.
- King P.; Maeder M.. Reactlab Kinetics, 1.1; J PlusConsulting Pty Ltd: Karawarra, Western Australia, 2009.
- Tobe M. L.; Burgess J.. Inorganic Reaction Mechanisms; Longman: New York, 1999. [Google Scholar]
- Wilkins R. G.Kinetics and Mechanisms of Reactions of Transition Metal Complexes; VCH: 1991. [Google Scholar]
- Crespo M.; Martinez M.; Sales J. Effect of Fluorine Substituents in Intramolecular Activation of C–F and C–H Bonds by Platinum(II). Organometallics 1993, 12 (11), 4297–4304. 10.1021/om00035a014. [DOI] [Google Scholar]
- Reichardt C.Solvents and Solvent Effects in Organic Chemistry, 3rd ed.; Wiley-VCH: 2003. [Google Scholar]
- Burgess J.Ions in Solution; Woodhead Publishing: 1999. [Google Scholar]
- Merbach A. E. In High Pressure Chemistry and Biochemistry; van Eldik R., Jonas J., Eds.; Reidel: 1987; p 311. [Google Scholar]
- Vazquez M.; Martinez M. Kinetico-mechanistic Studies on the Substitution Reactivity on the {Ru-II(bpy)(2)} Core with Nucleosides and Nucleotides at Physiological pH. Inorg. Chem. 2016, 55 (13), 6731–6738. 10.1021/acs.inorgchem.6b01003. [DOI] [PubMed] [Google Scholar]
- Bernhardt P. V.; Gallego C.; Martinez M. Mechanisms of substitution reactions on cyclometallated platinum(IV) complexes: “Quasi-labile” systems. Organometallics 2000, 19 (23), 4862–4869. 10.1021/om000505y. [DOI] [Google Scholar]
- Bernhardt P. V.; Gallego C.; Martinez M.; Parella T. Substitution reactions on cyclometalated Pt(IV) complexes. Associative tuning by fluoro Ligands and fluorinated substituents. Inorg. Chem. 2002, 41 (7), 1747–1754. 10.1021/ic0108179. [DOI] [PubMed] [Google Scholar]
- Gallego C.; Gonzalez G.; Martinez M.; Merbach A. E. Activation volumes of substitution reactions on neutral and cationic organometallic platinum(IV) complexes: Definite proof of selective associative activation. Organometallics 2004, 23 (10), 2434–2438. 10.1021/om0499409. [DOI] [Google Scholar]
- Plutino M. R.; Scolaro L. M.; Albinati A.; Romeo R. Atropisomerization, C-H activation, and dissociative substitution at some biphenyl platinum(II) complexes. J. Am. Chem. Soc. 2004, 126 (20), 6470–6484. 10.1021/ja030486u. [DOI] [PubMed] [Google Scholar]
- Frey U.; Helm L.; Merbach A. E.; Romeo R. Dissociative Substitution in Four-Coordinated Planar Platinum(II) Complexes As Evidenced by Variable-Pressure High-Resolution Proton NMR Magnetization Transfer Experiments. J. Am. Chem. Soc. 1989, 111 (21), 8161–8165. 10.1021/ja00203a016. [DOI] [Google Scholar]
- Yuan B. F.; Wan J. W.; Guo X. G.; Gong Y. M.; Zhang F. L.; Li Q.; Wang G. Z.; Chen J. Y.; He R. X. Theoretical investigation on the Cu(i)-catalyzed N-carboxamidation of indoles with isocyanates to form indole-1-carboxamides: effects of solvents. New J. Chem. 2020, 44 (23), 9878–9887. 10.1039/D0NJ01116A. [DOI] [Google Scholar]
- Bjelopetrovic A.; Barisic D.; Duvnjak Z.; Dzajic I.; Juribasic Kulcsar M.; Halasz I.; Martinez M.; Budimir A.; Babic D.; Curic M. A Detailed Kinetico-Mechanistic Investigation on the Palladium C-H Bond Activation in Azobenzenes and Their Monopalladated Derivatives. Inorg. Chem. 2020, 59 (23), 17123–17133. 10.1021/acs.inorgchem.0c02418. [DOI] [PubMed] [Google Scholar]
- Harringer S.; Wernitznig D.; Gajic N.; Diridl A.; Wenisch D.; Hejl M.; Jakupec M. A.; Theiner S.; Koellensperger G.; Kandioller W.; Keppler B. K. Introducing N-, P-, and S-donor leaving groups: an investigation of the chemical and biological properties of ruthenium, rhodium and iridium thiopyridone piano stool complexes. Dalton Trans. 2020, 49 (44), 15693–15711. 10.1039/D0DT03165H. [DOI] [PubMed] [Google Scholar]
- Ballester F. J.; Ortega E.; Porto V.; Kostrhunova H.; Davila-Ferreira N.; Bautista D.; Brabec V.; Dominguez F.; Santana M. D.; Ruiz J. New half-sandwich ruthenium(ii) complexes as proteosynthesis inhibitors in cancer cells. Chem. Commun. 2019, 55 (8), 1140–1143. 10.1039/C8CC09211G. [DOI] [PubMed] [Google Scholar]
- Gatti A.; Habtemariam A.; Romero-Canelon I.; Song J. I.; Heer B.; Clarkson G. J.; Rogolino D.; Sadler P. J.; Carcelli M. Half-Sandwich Arene Ruthenium(II) and Osmium(II) Thiosemicarbazone Complexes: Solution Behavior and Antiproliferative Activity. Organometallics 2018, 37 (6), 891–899. 10.1021/acs.organomet.7b00875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid W. F.; John R. O.; Arion V. B.; Jakupec M. A.; Keppler B. K. Highly antiproliferative ruthenium(II) and osmium(II) arene complexes with paullone-derived ligands. Organometallics 2007, 26 (26), 6643–6652. 10.1021/om700813c. [DOI] [Google Scholar]
- Clavel C. M.; Paunescu E.; Nowak-Sliwinska P.; Griffioen A. W.; Scopelliti R.; Dyson P. J. Modulating the Anticancer Activity of Ruthenium(II)-Arene Complexes. J. Med. Chem. 2015, 58 (8), 3356–3365. 10.1021/jm501655t. [DOI] [PubMed] [Google Scholar]
- Marchetti F.; Pettinari R.; Di Nicola C.; Pettinari C.; Palmucci J.; Scopelliti R.; Riedel T.; Therrien B.; Galindo A.; Dyson P. J. Synthesis, characterization and cytotoxicity of arene-ruthenium(II) complexes with acylpyrazolones functionalized with aromatic groups in the acyl moiety. Dalton Trans. 2018, 47 (3), 868–878. 10.1039/C7DT04249C. [DOI] [PubMed] [Google Scholar]
- Tabrizi L.; Olasunkanmi L. O.; Fadare O. A. Experimental and theoretical investigations of cyclometalated ruthenium(ii) complex containing CCC-pincer and anti-inflammatory drugs as ligands: synthesis, characterization, inhibition of cyclooxygenase and in vitro cytotoxicity activities in various cancer cell lines. Dalton Trans. 2019, 48 (2), 728–740. 10.1039/C8DT03266A. [DOI] [PubMed] [Google Scholar]
- Chen F.; Romero-Canelon I.; Soldevila-Barreda J. J.; Song J. I.; Coverdale J. P. C.; Clarkson G. J.; Kasparkova J.; Habtemariam A.; Wills M.; Brabec V.; Sadler P. J. Transfer Hydrogenation and Antiproliferative Activity of Tethered Half-Sandwich Organoruthenium Catalysts. Organometallics 2018, 37 (10), 1555–1566. 10.1021/acs.organomet.8b00132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettinari R.; Pettinari C.; Marchetti F.; Skelton B. W.; White A. H.; Bonfili L.; Cuccioloni M.; Mozzicafreddo M.; Cecarini V.; Angeletti M.; Nabissi M.; Eleuteri A. M. Arene-Ruthenium(II) Acylpyrazolonato Complexes: Apoptosis-Promoting Effects on Human Cancer Cells. J. Med. Chem. 2014, 57 (11), 4532–4542. 10.1021/jm500458c. [DOI] [PubMed] [Google Scholar]
- de la Cueva-Alique I.; Sierra S.; Munoz-Moreno L.; Perez-Redondo A.; Bajo A. M.; Marzo I.; Gude L.; Cuenca T.; Royo E. Biological evaluation of water soluble arene Ru(II) enantiomers with amino-oxime ligands. J. Inorg. Biochem. 2018, 183, 32–42. 10.1016/j.jinorgbio.2018.02.018. [DOI] [PubMed] [Google Scholar]
- Armarego W. L. F.; Chai C. L. L.. Purification of Laboratory Chemicals., 7th ed.; Butterworth Heinemann: 2013. [Google Scholar]
- Gabbai F. P.; Chirik P. J.; Fogg D. E.; Meyer K.; Mindiola D. J.; Schafer L. L.; You S.-L. An Editorial About Elemental Analysis. Organometallics 2016, 35 (19), 3255–3256. 10.1021/acs.organomet.6b00720. [DOI] [Google Scholar]
- SAINT and SADABS; Bruker AXS Inc.: Madison, WI, USA.
- Sheldrick G. M. SHELXT - Integrated space-group and crystal-structure determination. Acta Crystallogr., Sect. A: Found. Adv. 2015, 71, 3–8. 10.1107/S2053273314026370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheldrick G. M. Crystal structure refinement with SHELXL. Acta Crystallogr., Sect. C: Struct. Chem. 2015, 71, 3–8. 10.1107/S2053229614024218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolomanov O. V.; Bourhis L. J.; Gildea R. J.; Howard J. A. K.; Puschmann H. OLEX2: a complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. 10.1107/S0021889808042726. [DOI] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A.; Nakatsuji H.; Caricato M.; Li X.; Hratchian H. P.; Izmaylov A. F.; Bloino J.; Zheng G.; Sonnenberg J. L.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Montgomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M. J.; Heyd J.; Brothers E. N.; Kudin K. N.; Staroverov V. N.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A. P.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Rega N.; Millam N. J.; Klene M.; Knox J. E.; Cross J. B.; Bakken V.; Adamo C.; Jaramillo J.; Gomperts R.; Stratmann R. E.; Yazyev O.; Austin A. J.; Cammi R.; Pomelli C.; Ochterski J. W.; Martin R. L.; Morokuma K.; Zakrzewski V. G.; Voth G. A.; Salvador P.; Dannenberg J. J.; Dapprich S.; Daniels A. D.; Farkas Ö.; Foresman J. B.; Ortiz J. V.; Cioslowski J.; Fox D. J.. Gaussian 09, Rev. D01; Gaussian Inc.: 2009.
- Perdew J. P.; Burke K.; Ernzerhof M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77 (18), 3865–3868. 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- Perdew J. P.; Burke K.; Ernzerhof M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1997, 78 (7), 1396–1396. 10.1103/PhysRevLett.78.1396. [DOI] [PubMed] [Google Scholar]
- Schafer A.; Huber C.; Ahlrichs R. Fully optimized contracted Gaussian basis sets of triple zeta valence quality for atoms Li to Kr. J. Chem. Phys. 1994, 100 (8), 5829–5835. 10.1063/1.467146. [DOI] [Google Scholar]
- Haussermann U.; Dolg M.; Stoll H.; Preuss H.; Schwerdtfeger P.; Pitzer R. M. Accuracy of energy-adjusted quasirelativistic ab initio pseudopotentials. Mol. Phys. 1993, 78 (5), 1211–1224. 10.1080/00268979300100801. [DOI] [Google Scholar]
- Kuchle W.; Dolg M.; Stoll H.; Preuss H. Energy-adjusted pseudopotentials for the actinides. Parameter sets and test calculations for thorium and thorium monoxide. J. Chem. Phys. 1994, 100 (10), 7535–7542. 10.1063/1.466847. [DOI] [Google Scholar]
- Leininger T.; Nicklass A.; Stoll H.; Dolg M.; Schwerdtfeger P. The accuracy of the pseudopotential approximation.2. A comparison of various core sizes for indium pseudopotentials in calculations for spectroscopic constants of InH, InF, and InCl. J. Chem. Phys. 1996, 105 (3), 1052–1059. 10.1063/1.471950. [DOI] [Google Scholar]
- Marenich A. V.; Cramer C. J.; Truhlar D. G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113 (18), 6378–6396. 10.1021/jp810292n. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.