

Abstract
PURPOSE
Synovial sarcoma (SS) is the second most common malignant soft tissue tumor in children. ARST0332 evaluated a risk-based treatment strategy for young patients with soft tissue sarcoma designed to limit therapy for low-risk (LR) disease and to test neoadjuvant chemoradiotherapy for unresected higher-risk disease.
METHODS
Newly diagnosed patients with SS age < 30 years were assigned to four treatment arms based on disease features: A (surgery only), B (55.8 Gy radiotherapy [RT]), C (ifosfamide and doxorubicin [ID] chemotherapy plus 55.8 Gy RT), and D (neoadjuvant ID and 45 Gy RT, then surgery and RT boost based on margins followed by adjuvant ID). Patients treated in Arms A and B were considered LR, arms C and D without metastases as intermediate-risk (IR), and those with metastases as high-risk (HR).
RESULTS
Of the 146 patients with SS enrolled, 138 were eligible and evaluable: LR (46), IR (71), and HR (21). Tumors were 80% extremity, 70% > 5 cm, 70% high-grade, 62% invasive, 95% deep, and 15% metastatic. Treatment was on arm A (29.7%), B (3.6%), C (16.7%), and D (50%). There were no toxic deaths and four unexpected grade 4 adverse events. By risk group, at a median follow-up of 6.8 years, estimated 5-year event-free survival was LR 82%, IR 70%, and HR 8%, and overall survival was LR 98%, IR 89%, and HR 13%. After accounting for the features that defined risk category, none of the other patient or disease characteristics (age, sex, tumor site, tumor invasiveness, and depth) improved the risk stratification model.
CONCLUSION
The risk-based treatment strategy used in ARST0332 produced favorable outcomes in patients with nonmetastatic SS relative to historical controls despite using RT less frequently and at lower doses. The outcome for metastatic SS remains unsatisfactory and new therapies are urgently needed.
INTRODUCTION
Synovial sarcoma (SS) is the second most common malignant soft tissue tumor in children and is characterized by SYT gene rearrangement. SS is considered more chemosensitive than other nonrhabdomyosarcoma soft tissue sarcomas (NRSTS).1 Until recently, the outcomes for children with SS were described only in small patient series. A retrospective review on 37 children with SS published in 1994 revealed a 5-year event-free survival (EFS) and overall survival (OS) of 54% and 65%, respectively.2 Another report on 31 children treated on CWS-81 protocol showed a 5-year EFS of 84% for patients with group I and II tumors and 58% for those with group III or IV tumors.3 Children with SS have better prognosis when compared with adults, and the outcome decreases with age.4 Previous clinical trials on NRSTS in North America included very few children with SS.5-7 Data from these clinical trials and other large retrospective series revealed that extent of disease (local v metastatic), extent of tumor resection, maximal tumor diameter, and tumor grade were the most important prognostic factors.8-10 The Children's Oncology Group (COG) study ARST0332 evaluated a risk-based treatment strategy built on these prognostic factors for young patients with soft tissue sarcoma designed to limit therapy for low-risk (LR) disease and to test neoadjuvant chemoradiotherapy to increase likelihood of resection in unresected high-risk (HR) disease.11 Here, we report the results of patients with SS enrolled on ARST0332.
CONTEXT
Key Objective
This is the first risk-based prospective clinical trial in children and young adults with synovial sarcoma (SS) in North America. The risk stratification was based on metastasis, tumor grade, tumor size, and extent of surgical resection.
Knowledge Generated
The outcome for patients with localized disease was excellent, whereas those with metastatic disease fared very poorly. Radiation and chemotherapy were avoided in nearly a third of patients. Neoadjuvant radiation therapy was feasible and safe in children.
Relevance
This study sets a new standard for care of children and young adults with SS in North America. Future clinical trials on SS will build upon this backbone.
METHODS
Study Design
The COG ARST0332 trial (Protocol, online only) enrolled newly diagnosed patients with NRSTS including SS who were age < 30 years and had an expected life expectancy exceeding 3 months. Primary tumor gross total resection was required before enrollment except for (1) tumor > 5 cm in maximal diameter and gross or microscopic residual disease anticipated following resection, (2) inability to grossly resect the tumor without unacceptable morbidity, and (3) metastatic disease. Patients with clinical or radiologic evidence of regional lymph node involvement were required to undergo lymph node sampling to confirm lymph node status before enrollment, and those with lymph node involvement underwent lymph node dissection either at study entry or at week 13 (at the time of definitive surgery) if receiving neoadjuvant therapy. Patients with metastatic disease in whom all metastatic lesions were < 1 cm in maximal diameter were required to undergo a biopsy to confirm the presence of metastatic tumor. Adequate performance status was defined as Lansky performance status score ≥ 50 for patients age ≤ 16 years and Karnofsky performance status score of ≥ 50 for patients age > 16 years. Patients should not have received prior anthracycline or ifosfamide chemotherapy if they were enrolling in the chemotherapy arms (see below) or have received prior radiotherapy (RT) to tumor sites. Adequate bone marrow function was defined as absolute neutrophil count ≥ 1,000/μL and platelet count ≥ 100,000/μL. Adequate renal function was defined as creatinine clearance or radioisotope glomerular filtration rate ≥ 70 mL/min/1.73 m2 or normal serum creatinine for age and sex. Adequate liver function was defined as total bilirubin ≤ 1.5 times the upper limit of normal for age. Adequate cardiac function was defined as shortening fraction ≥ 24% or ejection fraction ≥ 50%. Female patients of childbearing age required a negative pregnancy test. Sexually active patients agreed to use contraception and lactating females receiving chemotherapy were required not to breastfeed during treatment. Written informed consent was obtained from patients (or assent, for those younger than age 18 years) and/or their parents or legal guardians before enrollment. The study was approved by the institutional review boards of treating institutions.
Treatment
Patients were required to submit tumor tissue for central review by enrolling on COG soft tissue sarcoma biology study D9902. The tissue was evaluated by two study pathologists to confirm the diagnosis and tumor grade both by Pediatric Oncology Group (POG)12 and Fédération Nationale des Centers de Lutte Contre le Cancer (FNCLCC) criteria.13 Tumors were classified as low (POG grade 1 or 2) and high grade (POG grade 3) for treatment assignment purposes. Extent of surgical resection was defined as R0 if the tumor was gross totally resected with surgical margins negative for tumor cells, R1 if tumor cells were present at surgical margins by microscopy following gross resection, and R2 if there was gross residual disease after surgery. Adequate margin was defined as resected tumor surrounded by a cuff of normal tissue at least 0.5-cm thick or intact fascia.
Patients were classified into LR, intermediate-risk (IR), and HR groups based on metastatic status, extent of surgical resection, POG grade, and size of the primary tumor (Fig 1). LR: Nonmetastatic R0 or R1 low-grade tumors were observed without further therapy and nonmetastatic R0 high-grade tumors ≤ 5 cm were observed without further therapy (arm A). Nonmetastatic R1 high-grade tumors ≤ 5 cm received adjuvant RT (55.8 Gy; arm B). IR: Nonmetastatic R0 or R1 high-grade tumor > 5 cm received adjuvant ifosfamide and doxorubicin chemotherapy and RT (55.8 Gy; arm C). Tumors that were unresectable or > 5 cm, high-grade, and anticipated to have R1 margins after resection were treated with neoadjuvant ifosfamide and doxorubicin chemotherapy and RT (arm D). RT was administered at week 4 (45 Gy) and surgical resection was attempted at week 13. An additional RT boost was given postoperatively for R1 resection (10.8 Gy) or R2 or no resection (19.8 Gy). HR: Patients with metastatic disease were HR. If the primary tumor was grossly resected, adjuvant chemotherapy and radiation therapy (arm C) was given. If the primary tumor was not resected at diagnosis, neoadjuvant chemoradiotherapy (arm D) was administered. Resection of metastases was performed at week 13 or end of therapy depending on sites involved. If feasible, radiation therapy to metastatic sites remaining after surgery was administered at the end of treatment. Patients with low-grade tumor and metastases could be observed on arm A if gross total resection of primary tumor and metastatic disease was achieved at study entry but none of the enrolled patients qualified for this arm.
FIG 1.

ARST0332 risk-based treatment assignment schema. HR, high-risk; IR, intermediate-risk; LR, low-risk; RT, radiotherapy.
The cumulative doses and dose per cycle of ifosfamide (54 g/m2; 9 g/m2/cycle × six cycles) and doxorubicin (375 mg/m2; 75 mg/m2/cycle × five cycles) were identical on arms C and D. Each 3-week cycle consisted of ifosfamide 3 g/m2/d administered intravenously over 3 h on days 1, 2, and 3, and doxorubicin 37.5 mg/m2/d administered intravenously over 24 hours on days 1 and 2. Doxorubicin was not administered at weeks 7 and 10 during RT and was deferred to week 25 in those requiring an RT boost after surgery. Toxicities were graded using National Cancer Institute Common Terminology for Adverse events version 4.0; unexpected grade 4 and all grade 5 events required reporting. Primary tumor response was evaluated using an elliptical model (0.5 times the product of the three largest perpendicular diameters) to measure tumor volume; nodal and distant metastases were assessed by using the RECIST 1.1. All imaging studies and measurements were centrally reviewed by two pediatric radiologists and one nuclear medicine physician.
Statistical Methods
Patient characteristics and clinical features were summarized using frequency and percentage. EFS was defined as the time from study enrollment to disease progression or recurrence, second malignant neoplasm, or death from any cause, whichever occurred first. OS was defined as the time from study enrollment to death from any cause. EFS and OS were censored at the patient's last contact date. Patient follow-up was current through June 30, 2018, when the data set was frozen for publication. EFS and OS by patient characteristics and clinical features were estimated using the Kaplan-Meier method. 95% CIs were estimated by the Peto-Peto method. The log-rank test was used to compare the EFS and OS distribution by patient characteristics and clinical features. A proportional hazards model was used to determine if there were any patient or disease characteristics that predicted outcome, after accounting for the factors that defined risk group. All statistical analyses were conducted using SAS 9.4 (Cary, NC).
RESULTS
A total of 146 patients with SS were enrolled between 2007 and 2012 (Fig 2). After excluding eight ineligible patients, 138 were eligible for analysis (Table 1). Approximately 80% of tumors arose in the extremities, with the remainder nearly evenly distributed into visceral, body wall, and head and neck sites. The majority of patients had POG grade 3 (70.3%) or FNCLCC grade 2 tumors (67.4%). Most tumors were > 5 cm in size at diagnosis (69.6%) and were classified as invasive (62.3%). Twenty-one (15.2%) patients had metastatic disease at diagnosis; 15 with isolated lung, 2 with isolated lymph node, and 4 with lung and lymph node metastases.
FIG 2.
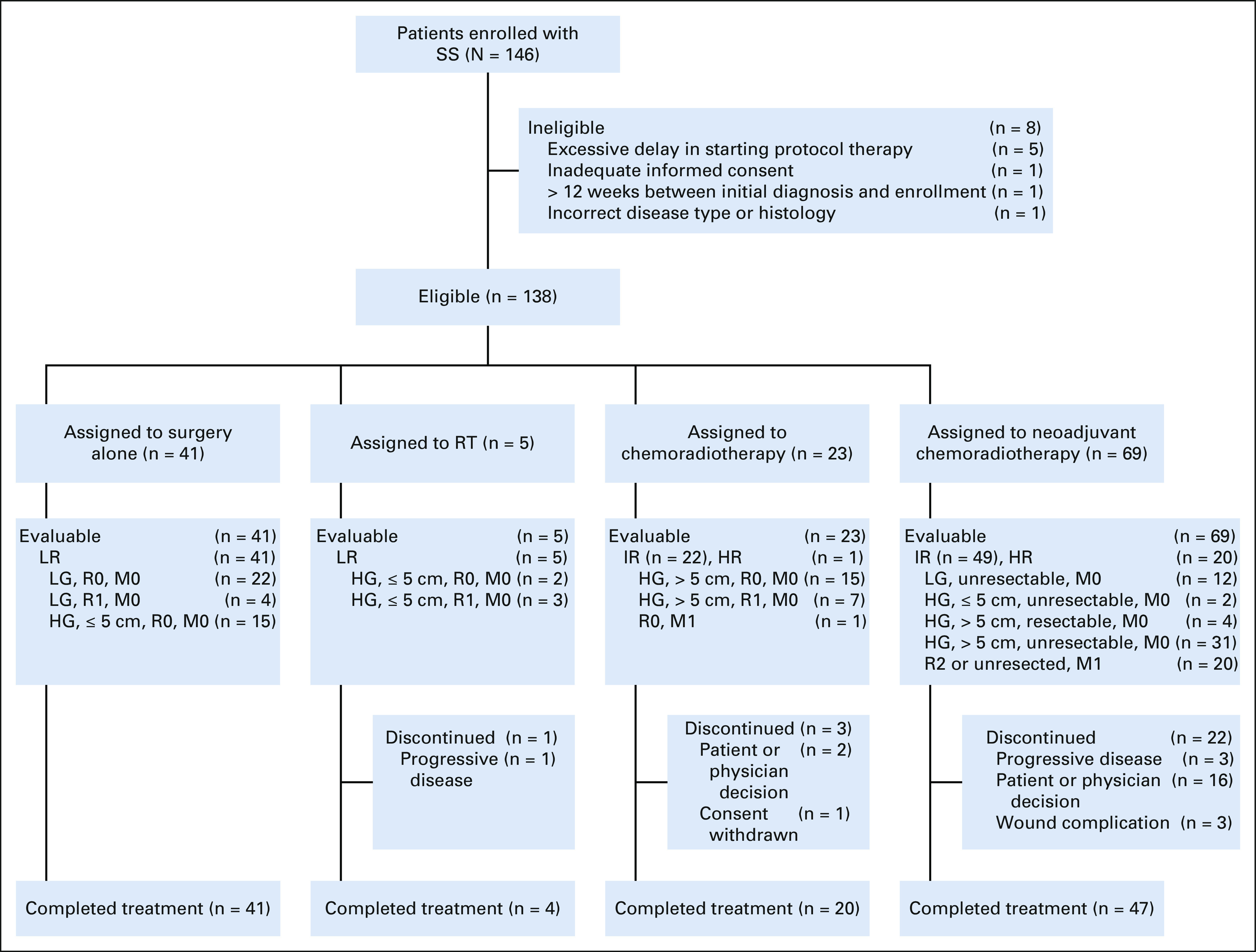
CONSORT diagram. HG, high-grade; HR, high-risk; IR, intermediate-risk; LG, low-grade; LR, low-risk; RT, radiotherapy; SS, synovial sarcoma.
TABLE 1.
Clinical Features and Treatment by Risk Group and Treatment Arm

Treatment
Forty-one (29.7%) patients were treated with surgery alone (arm A), 5 (3.6%) with adjuvant RT (arm B), 23 (16.7%) with adjuvant chemoradiotherapy (arm C), and 69 (50%) with neoadjuvant chemoradiotherapy (arm D). Of the 41 LR patients assigned to arm A, 26 had POG grade 2 (low-grade) tumors (seven were > 5 cm in size), and 15 had localized POG grade 3 (high-grade) tumors ≤ 5 cm in size that were completely resected. Of the five LR patients assigned to arm B and treated with 55.8 cGy adjuvant radiation therapy, three had R1 resection and two had R0 resection with < 5-mm margins (considered R1 per protocol guidelines). Of the 23 patients assigned to arm C, 22 had localized high-grade tumors > 5 cm in size (15 R0 resection and seven R1 resection) and one patient had metastatic disease with upfront R0 resection of primary tumor. Twenty patients in arm C received a median radiation dose of 55.8 Gy; two patients did not receive radiation therapy because of amputation and one patient was removed from protocol therapy before radiation was due. Of the 69 patients assigned to arm D, 49 had IR (nonmetastatic) and 20 had HR (metastatic) disease. Of the 49 IR patients, 45 were unresectable (12 low-grade, two high-grade ≤ 5 cm, and 31 high-grade > 5 cm) and four were high-grade, > 5 cm, and anticipated to have R1 margins at the time of surgery. Forty-six of the 49 patients received neoadjuvant radiation therapy with a median dose of 45 Gy. Three patients were removed from protocol therapy before radiation therapy was due. Of these 46 patients, 43 underwent surgical resection following radiation therapy (35 R0 and 8 R1). Eighteen patients (11 R0 with < 5-mm margin, six R1, and one R2) received postoperative RT boost.
Overall, R0 or R1 resection of the primary tumor was achieved in 129 patients (93.5%): at study entry in 69 (53.5%) and after neoadjuvant chemotherapy in 60 (46.5%). Of these, 104 patients (80.6%) had an R0 resection: at study entry in 55 (53%) and after neoadjuvant chemotherapy in 49 (47%).
In the 69 patients who received neoadjuvant chemotherapy, response was evaluable in 55 patients. Two (3.6%) patients had a complete response, 9 (16.4%) had partial response (PR), 41 (74.6%) had stable disease, and 3 (5.5%) had progressive disease. The tumor tissue from 57 tumors were centrally reviewed after definitive resection. Forty-one (72%) tumors had < 90% necrosis, and 16 tumors (28%) had ≥ 90% necrosis.
Outcome
One-hundred seven of the 138 patients were alive at the time of this analysis with a median follow-up of 6.8 years (range, 0.01-10.98 years). The 5-year EFS and OS for the entire group were 64.9% (95% CI, 55.7 to 74.1) and 80.8% (73.3 to 88.3), respectively. EFS and OS differed significantly by risk group and treatment arm (Figs 3 and 4, Table 2). The 5-year EFS and OS for the 46 patients in the LR group were 81.9% (69 to 94.8) and 97.7% (92.7 to 100), respectively. The 5-year EFS and OS for the 23 patients in the IR group treated with adjuvant chemoradiotherapy (arm C) were 64% (42.4 to 85.8) and 89.5% (75.3 to 100), respectively. The 5-year EFS and OS for the 49 patients in the IR group treated with neoadjuvant chemoradiotherapy (arm D) were 71.16% (56.5 to 85.9) and 86.5% (75.6 to 97.3), respectively. The 5-year EFS and OS for the 37 patients with high-grade tumors in the IR group treated with neoadjuvant chemoradiotherapy (arm D) were 71% (54.3 to 87.8) and 84.9% (72 to 97.8), respectively. The 5-year EFS and OS for the 21 patients with metastatic disease were 7.6% (0 to 22) and 12.5% (0 to 28.7), respectively.
FIG 3.

EFS and OS by risk group. Kaplan-Meier curves representing (A) EFS and (B) OS for patients in the LR, IR, and HR groups. EFS, event-free survival; HR, high-risk; IR, intermediate-risk; LR, low-risk; OS, overall survival.
FIG 4.
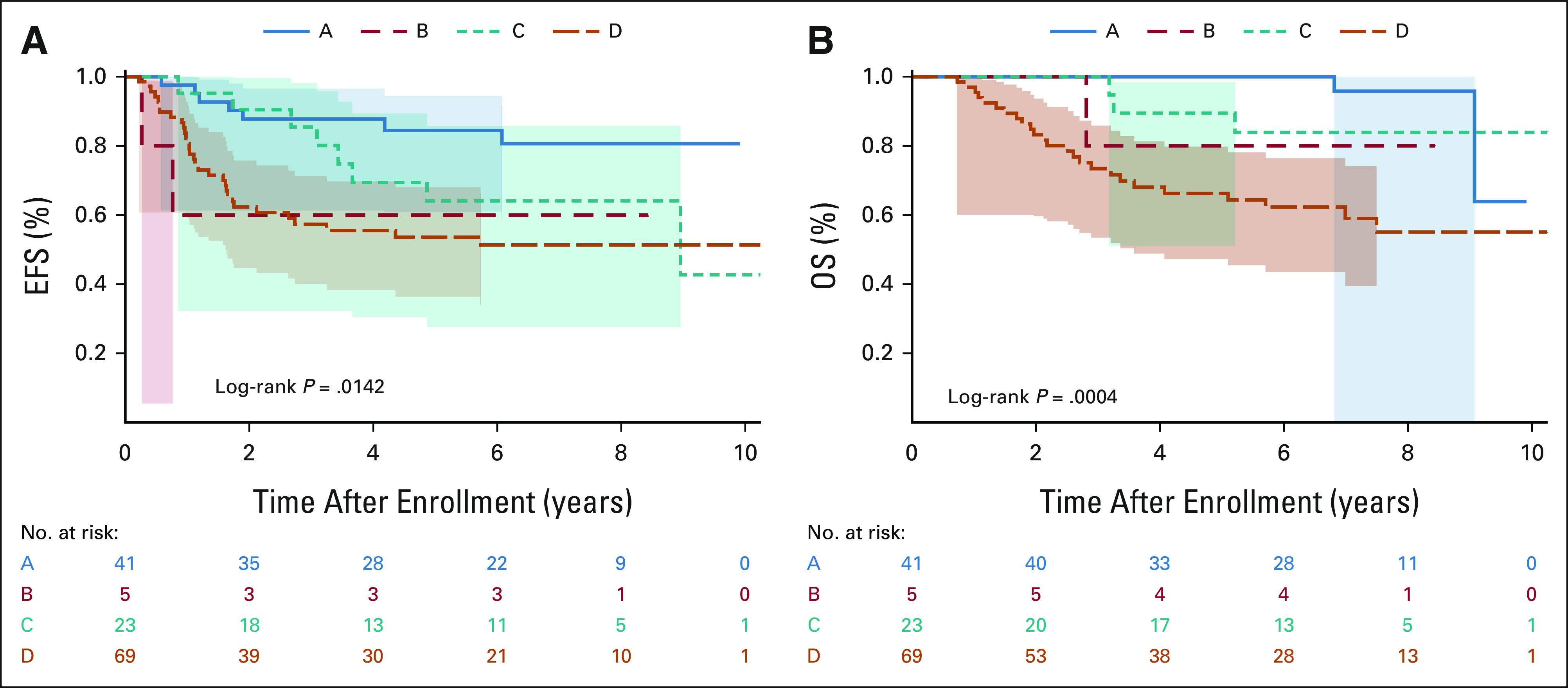
EFS and OS by treatment arm. Kaplan-Meier curves representing (A) EFS and (B) OS for patients treated on arm A (surgery alone), arm B (RT), arm C (adjuvant chemoradiotherapy), and arm D (neoadjuvant chemoradiotherapy). EFS, event-free survival; OS, overall survival; RT, radiotherapy.
TABLE 2.
Univariate Analysis of Outcome by Clinical Features, Risk Group, Treatment, and Response to Therapy

The following factors were significantly associated with inferior EFS and OS in univariate analysis (Table 2): maximal tumor diameter > 5 cm, invasive tumor, FNCLCC grade 3, presence of metastases, grossly incomplete resection, and positive microscopic surgical margin. Neither tumor invasiveness nor FNCLCC grade improves the risk stratification model generated using tumor grade, metastatic status, tumor size, and extent of resection (see Appendix Tables A1 and A2, online only).
There were no deaths related to treatment in the study. There were four unexpected grade 4 adverse events, all of which were observed in the neoadjuvant chemoradiotherapy arm (arm D). Three of those events were wound complications; one patient developed wound dehiscence over the biopsy site and required above-knee amputation, and two others developed wound complications following definitive surgery and required further surgical intervention. Overall, six patients (10.7%) out of 56 patients who underwent neoadjuvant chemoradiotherapy reported wound complications. Of these, two were removed from protocol therapy because of delay in resumption of chemoradiotherapy. Three (2.1%) second malignant neoplasms were reported: two were acute myeloid leukemia and one patient developed papillary thyroid carcinoma (outside the field treated with radiation).
Overall, 46 patients experienced a recurrence or progression of disease; the majority of the events (n = 36) were isolated metastatic recurrence, one was local and metastatic recurrence, and only nine events were isolated local recurrence. Thirty five of the 36 patients with metastatic failure had recurrence or progression in the lung (33 had lung only relapse). Other sites of recurrence included bone (n = 1), regional lymph node (one), posterior mediastinum (n = 1), and chest wall (n = 1). Of the nine patients with isolated local recurrence, seven were treated on arm A with surgery alone and two were treated on arm D with neoadjuvant chemoradiotherapy and delayed surgical resection. Ten recurrences were observed beyond 3 years (eight metastatic and two local) from diagnosis and three of them were observed after 5 years.
DISCUSSION
This is the largest prospective clinical trial conducted in young patients with SS in North America. The risk stratification based on metastasis, tumor grade, tumor size, and extent of surgical resection resulted in LR, IR, and HR groups with significantly different EFS and OS. The outcome for patients with localized disease was excellent, whereas those with metastatic disease fared very poorly. We avoided radiation and chemotherapy in nearly a third of patients (arm A). All recurrences in this group were local (7 of 41, 17%) and the 5-year OS was 100%. This approach was further confirmed by a pooled analysis with the European Pediatric Soft Tissue Sarcoma Study Group (EpSSG) NRSTS 2005 study.14 Based on these results, we do not recommend adjuvant therapy for patients with completely resected SS with tumors < 5 cm in size, and completely resected POG grade 2 tumors of any size. The treatment details at relapse for the seven patients with local relapse were not collected as part of this study. Therefore, we do not know the total burden of therapy for these patients.
Gross total resection of primary tumor was achieved in 94% of patients and R0 resection in 80% of patients. Gross total resection of primary tumor was achieved in a high proportion of patients (87%) who received neoadjuvant chemotherapy and 45 Gy RT, and fewer than 20% of these patients had microscopic residual disease after surgery requiring a radiation boost to 55.8 Gy. Therefore, the neoadjuvant chemoradiotherapy strategy produced a high rate of complete resection and high rates of local tumor control with lower doses of RT than were used previously. Overall, the therapy was well tolerated with no deaths because of protocol therapy. The incidence of wound complications in the neoadjuvant chemoradiotherapy group was lower than that reported in adult studies, although only wound infections that delayed postoperative therapy by > 2 weeks or were unexpected grade 4 events were tracked.15 Neoadjuvant chemoradiotherapy failed to improve outcomes for patients with metastatic disease, and the dismal outcomes in metastatic patients are similar to those reported by others.6,16
The EpSSG NRSTS 2005 clinical trial included 138 patients with SS.17 Although both trials used a similar approach, there were some significant differences, including the fact that the EpSSG trial did not include patients with metastases. Criteria used for LR, IR, and HR patients were different, and axial site was designated as HR irrespective of presence of other risk factors. The 5-year EFS and OS of patients with localized disease were 74.7% and 92.4%, respectively, in our study and are comparable to the EFS of 80.7% and OS of 90.7% reported in the NRSTS 2005 study. Approximately 20% of patients who received neoadjuvant chemoradiotherapy had an objective response in our study. This is much lower than the 55% of patients with objective response reported by EpSSG NRSTS 2005 clinical trial. The EpSSG trial divided PR into major (> 66% volume reduction) and minor (34%-65% volume reduction), whereas we defined PR as ≥ 64% decrease in volume. The number of patients achieving CR or major PR (22%) in the EpSSG study was similar to the patients achieving objective response (20%) in our trial. Moreover, the proportion of patients who did not experience progressive disease on treatment was similar in the COG (95%) and EpSSG trials (97%). Although patients with axial sites were designated and treated as HR in EpSSG trial, we did not find significant differences in outcome based on site.
Pathologic response to neoadjuvant therapy is a well-established prognostic factor in osteosarcoma. There is evidence that pathologic response may predict outcome in high-grade soft tissue sarcomas as well.18,19 In a large retrospective series in adults with extremity high-grade soft tissue sarcoma, the local recurrence rate was significantly lower and survival rate was higher in patients whose tumor had ≥ 95% tumor necrosis when compared with those with tumors with < 95% necrosis.18 Similar results were reported in a meta-analysis of 1,663 patients from 21 studies when comparing patients with < 90% necrosis with those who had ≥ 90% necrosis.19 Surprisingly, patients with ≥ 90% necrosis in our series seem to have worse EFS and OS when compared with patients with < 90% tumor necrosis, although the results were not statistically significant (Table 2). Necrosis may reflect inherent aggressiveness of the tumor rather than response to therapy and this may explain our finding. Outcome data from the study ARST1321 (when available), which was terminated early because of significant difference in tumor necrosis in the pazopanib plus chemotherapy arm compared with chemotherapy only arm, may help answer this question.20
All factors used in the risk stratification were significant predictors of outcome in univariate analysis except for POG grade. By contrast, FNCLCC grade was significant in univariate analysis. The exact reason for this is not known, and use of POG grade in risk stratification confounds this analysis. All patients with POG grade 2 tumors (n = 41) were classified as FNCLCC grade 2 but nearly half of the patients with POG grade 3 (n = 97) were classified as FNCLCC grade 2 and only 45 patients had FNCLCC grade 3 tumors. Recently, genomic complexity as measured by a genomic index (GI) tool using array comparative genomic hybridization has been described as an independent prognostic factor in SS.21 In 61 patients enrolled on the EpSSG NRSTS 2005 protocol, patients with no copy-number alterations (GI = 0) had a better EFS than those with one or more copy-number alterations (GI ≥ 1). Copy-number alteration data were not available for our cohort and as such we could not verify this finding. The prognostic role of specific fusion subtype in SS is conflicting with most studies concluding that it is not significant.22,23 We attempted to study the fusion subtype in our patients but the test could not be performed because of the poor quality of stored specimens.
The novel risk-based treatment strategy used in ARST0332 produced favorable outcomes in patients with localized SS relative to historical controls and sets a new standard for care of children and young adults with SS in North America. We were able to avoid radiation therapy in up to a third of patients and reduce the dose of radiation in a significant number of patients while maintaining excellent outcomes in those without metastatic disease. The outcome for patients with metastatic SS remains poor and new therapies are urgently needed.
APPENDIX
TABLE A1.
Cox Proportional Hazards Model for Factors Included in Risk Group

TABLE A2.
Model Fit Statistics for Addition of Tumor Invasiveness and FNCLCC Grade

R. Lor Randall
Honoraria: Biomet, Daiichi Sankyo, Onkos Surgical, Onclive
Travel, Accommodations, Expenses: Biomet, Daiichi Sankyo/Lilly
James Anderson
Employment: Merck
Stock and Other Ownership Interests: Merck Sharp & Dohme
Travel, Accommodations, Expenses: Merck Sharp & Dohme
Andrew Ostrenga
Honoraria: Amgen
Alberto Pappo
Honoraria: Bayer, Roche
Consulting or Advisory Role: Merck, Loxo/Bayer, EUSA Pharma, Debbio
Sheri L. Spunt
Research Funding: LOXO Oncology/Bayer AG/MedPace Inc, Bayer Healthcare Pharmaceuticals, Inc - US
No other potential conflicts of interest were reported.
PRIOR PRESENTATION
Presented in part at the American Society of Clinical Oncology Annual Meeting, May 29-June 2, 2015, Chicago, IL (abstr 10012); the Connective Tissue Oncology Society Annual Meeting, November 14-17, 2018, Rome, Italy.
SUPPORT
Supported by NCTN Operations Center Grant U10CA180886, NCTN Statistics & Data Center Grant U10CA180899, and St Baldrick's Foundation.
CLINICAL TRIAL INFORMATION
NCT00346164 (ARST0332)
DATA SHARING STATEMENT
The clinical trial data will be available publicly through the NCI NCTN data archive at https://nctn-data-archive.nci.nih.gov/.
AUTHOR CONTRIBUTIONS
Conception and design: Rajkumar Venkatramani, R. Lor Randall, Suzanne Wolden, James Anderson, Simon C. Kao, Andrew Ostrenga, Sheri L. Spunt
Administrative support: Simon C. Kao
Provision of study materials or patients: Suzanne Wolden, Jennifer Black
Collection and assembly of data: Rajkumar Venkatramani, Wei Xue, Suzanne Wolden, James Anderson, Jennifer Black, Simon C. Kao, Alberto Pappo, Sheri L. Spunt
Data analysis and interpretation: Rajkumar Venkatramani, Wei Xue, Suzanne Wolden, Dolores Lopez-Terrada, Jennifer Black, Simon C. Kao, Barry Shulkin, Andrew Ostrenga, Alberto Pappo, Sheri L. Spunt
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Synovial Sarcoma in Children, Adolescents, and Young Adults: A Report From the Children's Oncology Group ARST0332 Study
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/jco/authors/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
R. Lor Randall
Honoraria: Biomet, Daiichi Sankyo, Onkos Surgical, Onclive
Travel, Accommodations, Expenses: Biomet, Daiichi Sankyo/Lilly
James Anderson
Employment: Merck
Stock and Other Ownership Interests: Merck Sharp & Dohme
Travel, Accommodations, Expenses: Merck Sharp & Dohme
Andrew Ostrenga
Honoraria: Amgen
Alberto Pappo
Honoraria: Bayer, Roche
Consulting or Advisory Role: Merck, Loxo/Bayer, EUSA Pharma, Debbio
Sheri L. Spunt
Research Funding: LOXO Oncology/Bayer AG/MedPace Inc, Bayer Healthcare Pharmaceuticals, Inc - US
No other potential conflicts of interest were reported.
REFERENCES
- 1.Vlenterie M, Litière S, Rizzo E, et al. Outcome of chemotherapy in advanced synovial sarcoma patients: Review of 15 clinical trials from the European Organisation for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group; setting a new landmark for studies in this entity Eur J Cancer 5862–722016 [DOI] [PubMed] [Google Scholar]
- 2.Pappo AS, Fontanesi J, Luo X, et al. Synovial sarcoma in children and adolescents: The St Jude Children's Research Hospital experience J Clin Oncol 122360–23661994 [DOI] [PubMed] [Google Scholar]
- 3.Ladenstein R, Treuner J, Koscielniak E, et al. Synovial sarcoma of childhood and adolescence. Report of the German CWS-81 study Cancer 713647–36551993 [DOI] [PubMed] [Google Scholar]
- 4.Vlenterie M, Ho VKY, Kaal SEJ, et al. Age as an independent prognostic factor for survival of localised synovial sarcoma patients Br J Cancer 1131602–16062015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pratt CB, Pappo AS, Gieser P, et al. Role of adjuvant chemotherapy in the treatment of surgically resected pediatric nonrhabdomyosarcomatous soft tissue sarcomas: A Pediatric Oncology Group Study. J Clin Oncol. 1999;17:1219. doi: 10.1200/JCO.1999.17.4.1219. [DOI] [PubMed] [Google Scholar]
- 6.Pratt CB, Maurer HM, Gieser P, et al. Treatment of unresectable or metastatic pediatric soft tissue sarcomas with surgery, irradiation, and chemotherapy: A Pediatric Oncology Group Study Med Pediatr Oncol 30201–2091998 [DOI] [PubMed] [Google Scholar]
- 7.Pappo AS, Devidas M, Jenkins J, et al. Phase II trial of neoadjuvant vincristine, ifosfamide, and doxorubicin with granulocyte colony-stimulating factor support in children and adolescents with advanced-stage nonrhabdomyosarcomatous soft tissue sarcomas: A Pediatric Oncology Group Study J Clin Oncol 234031–40382005 [DOI] [PubMed] [Google Scholar]
- 8.Spunt SL, Poquette CA, Hurt YS, et al. Prognostic factors for children and adolescents with surgically resected nonrhabdomyosarcoma soft tissue sarcoma: An analysis of 121 patients treated at St Jude Children's Research Hospital J Clin Oncol 173697–37051999 [DOI] [PubMed] [Google Scholar]
- 9.Okcu MF, Munsell M, Treuner J, et al. Synovial sarcoma of childhood and adolescence: A multicenter, multivariate analysis of outcome J Clin Oncol 211602–16112003 [DOI] [PubMed] [Google Scholar]
- 10.Brecht IB, Ferrari A, Int-Veen C, et al. Grossly-resected synovial sarcoma treated by the German and Italian Pediatric Soft Tissue Sarcoma Cooperative Groups: Discussion on the role of adjuvant therapies Pediatr Blood Cancer 4611–172006 [DOI] [PubMed] [Google Scholar]
- 11.Spunt SL, Million L, Chi Y-Y, et al. A risk-based treatment strategy for non-rhabdomyosarcoma soft-tissue sarcomas in patients younger than 30 years (ARST0332): A Children's Oncology Group prospective study Lancet Oncol 21145–1612020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Parham DM, Webber BL, Jenkins JJ, et al. Nonrhabdomyosarcomatous soft tissue sarcomas of childhood: Formulation of a simplified system for grading Mod Pathol 8705–7101995 [PubMed] [Google Scholar]
- 13.Coindre JM, Trojani M, Contesso G, et al. Reproducibility of a histopathologic grading system for adult soft tissue sarcoma Cancer 58306–3091986 [DOI] [PubMed] [Google Scholar]
- 14.Ferrari A, Chi Y-Y, De Salvo GL, et al. Surgery alone is sufficient therapy for children and adolescents with low-risk synovial sarcoma: A joint analysis from the European paediatric soft tissue sarcoma Study Group and the Children's Oncology Group Eur J Cancer 781–62017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.DeLaney TF, Spiro IJ, Suit HD, et al. Neoadjuvant chemotherapy and radiotherapy for large extremity soft-tissue sarcomas Int J Radiat Oncol Biol Phys 561117–11272003 [DOI] [PubMed] [Google Scholar]
- 16.Scheer M, Dantonello T, Hallmen E, et al. Primary metastatic synovial sarcoma: Experience of the CWS Study Group Pediatr Blood Cancer 631198–12062016 [DOI] [PubMed] [Google Scholar]
- 17.Ferrari A, De Salvo GL, Brennan B, et al. Synovial sarcoma in children and adolescents: The European Pediatric Soft Tissue Sarcoma Study Group prospective trial (EpSSG NRSTS 2005) Ann Oncol 26567–5722015 [DOI] [PubMed] [Google Scholar]
- 18.Eilber FC, Rosen G, Eckardt J, et al. Treatment-induced pathologic necrosis: A predictor of local recurrence and survival in patients receiving neoadjuvant therapy for high-grade extremity soft tissue sarcomas J Clin Oncol 193203–32092001 [DOI] [PubMed] [Google Scholar]
- 19.Salah S, Lewin J, Amir E, et al. Tumor necrosis and clinical outcomes following neoadjuvant therapy in soft tissue sarcoma: A systematic review and meta-analysis Cancer Treat Rev 691–102018 [DOI] [PubMed] [Google Scholar]
- 20.Weiss AR, Chen Y-L, Scharschmidt TJ, et al. Pathological response in children and adults with large unresected intermediate-grade or high-grade soft tissue sarcoma receiving preoperative chemoradiotherapy with or without pazopanib (ARST1321): A multicentre, randomised, open-label, phase 2 trial Lancet Oncol 211110–11222020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Orbach D, Mosseri V, Pissaloux D, et al. Genomic complexity in pediatric synovial sarcomas (Synobio study): The European pediatric soft tissue sarcoma group (EpSSG) experience Cancer Med 71384–13932018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stegmaier S, Leuschner I, Poremba C, et al. The prognostic impact of SYT-SSX fusion type and histological grade in pediatric patients with synovial sarcoma treated according to the CWS (Cooperative Weichteilsarkom Studie) trials Pediatr Blood Cancer 6489–952017 [DOI] [PubMed] [Google Scholar]
- 23.Takenaka S, Ueda T, Naka N, et al. Prognostic implication of SYT-SSX fusion type in synovial sarcoma: A multi-institutional retrospective analysis in Japan Oncol Rep 19467–4762008 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The clinical trial data will be available publicly through the NCI NCTN data archive at https://nctn-data-archive.nci.nih.gov/.