

Abstract
Traumatic brain injury (TBI) results in disrupted brain function following impact from an external force and is a risk factor for sporadic Alzheimer's disease (AD). Although neurologic symptoms triggered by mild traumatic brain injuries (mTBI), the most common form of TBI, typically resolve rapidly, even an isolated mTBI event can increase the risk to develop AD. Aberrant accumulation of amyloid β peptide (Aβ), a cleaved fragment of amyloid precursor protein (APP), is a key pathologic outcome designating the progression of AD following mTBI and has also been linked to impaired axonal transport. However, relationships among mTBI, amyloidogenesis, and axonal transport remain unclear, in part because of the dearth of human models to study the neuronal response following mTBI. Here, we implemented a custom-microfabricated device to deform neurons derived from human-induced pluripotent stem cells, derived from a cognitively unimpaired male individual, to mimic the mild stretch experienced by neurons during mTBI. Although no cell lethality or cytoskeletal disruptions were observed, mild stretch was sufficient to stimulate rapid amyloidogenic processing of APP. This processing led to abrupt cessation of APP axonal transport and progressive formation of aberrant axonal accumulations that contained APP, its processing machinery, and amyloidogenic fragments. Consistent with this sequence of events, stretch-induced defects were abrogated by reducing amyloidogenesis either pharmacologically or genetically. In sum, we have uncovered a novel and manipulable stretch-induced amyloidogenic pathway directly responsible for APP axonal transport dysregulation. Our findings may help to understand and ultimately mitigate the risk of developing AD following mTBI.
SIGNIFICANCE STATEMENT Mild traumatic brain injury is a risk factor for sporadic Alzheimer's disease (AD). Increased amyloid β peptide generation after injury may drive this risk. Here, by using a custom-built device to impose mild stretch to human neurons, we found that stretch triggers amyloid precursor protein (APP) cleavage, and thus amyloid β peptide generation, consequently disrupting APP axonal transport. Compellingly, protecting APP from cleavage was sufficient to spare axonal transport dysregulation and the consequent aberrant axonal accumulation of APP. Supporting such protective mechanism, the expression of the AD-protective APPA673T genetic variant conferred protection against stretch-induced APP axonal transport phenotypes. Our data reveal potential subcellular pathways contributing to the development of AD-associated phenotypes following mild traumatic brain injury, and putative strategies for intervening in these pathways.
Keywords: Alzheimer's disease, amyloid beta peptide, amyloid precursor protein, axonal transport, biomechanics, mild traumatic brain injury
Introduction
In addition to aging, traumatic brain injury (TBI) is the strongest environmental risk factor for developing sporadic Alzheimer's disease (AD) (Schofield et al., 1997; Guo et al., 2000; Plassman et al., 2000; Fleminger et al., 2003; Gardner et al., 2014). Across the broad spectrum of injury severities, mild TBI (mTBI) is the most common subtype, affecting at least 42 million people worldwide annually (Greenwald et al., 2012; Gardner and Yaffe, 2015). Despite most clinical symptoms resolving rapidly following mTBI (Carroll et al., 2004; Ruff et al., 2009), even an isolated mTBI event significantly increases the risk to develop AD and related dementias (Guo et al., 2000; Kiraly and Kiraly, 2007; Bazarian et al., 2009; Gavett et al., 2010; Barnes et al., 2018; Fann et al., 2018; Schaffert et al., 2018). The linkage between mTBI and AD suggests that mild injury induces subclinical pathophysiological changes in brain cells, which may increase neurodegenerative susceptibility. Pathways underlying such changes are not fully understood.
Aberrant accumulation of amyloid β peptide (Aβ), which results from the amyloidogenic processing of amyloid precursor protein (APP) by β-site APP cleaving enzyme 1 (BACE1) and γ-secretase, is a key outcome that distinguishes AD from other injury-associated neurodegenerative sequelae (Johnson et al., 2010, 2012). In addition to rapidly emerging and prevalent amyloid pathology in patients subject to severe head injury, such pathology has also been observed following a single mTBI event and after repetitive mTBI (Emmerling et al., 2000; Ikonomovic et al., 2004; DeKosky et al., 2013; Gatson et al., 2013; D. H. Smith et al., 2013; Marklund et al., 2014; Stein et al., 2015; Shahim et al., 2016; Abu Hamdeh et al., 2018; Takahashi et al., 2019). Hence, sublethally impacted neurons that survive following mild focal or diffuse axonal injury might contribute to AD pathogenesis (Browne et al., 2011; Johnson et al., 2013; Vascak et al., 2018). We posit that identifying early molecular changes in human neurons following sublethal (i.e., mild) mechanical trauma will provide insights into pathways related to amyloid pathogenesis after mTBI.
Previous studies have used animal models to investigate AD-related phenotypes under controlled conditions of severe and mild injury (Pierce et al., 1996; Bramlett et al., 1997; Stone et al., 2000; Tran et al., 2011a,b; Greer et al., 2013; Tajiri et al., 2013; Hånell et al., 2015; Lou et al., 2018). However, with a few exceptions, most of these studies have been performed using transgenic mice models for AD that are inherently predisposed to develop amyloid pathology, even without injury (Loane et al., 2011; Tran et al., 2011a, 2012; Winston et al., 2013; Cartagena et al., 2016; Levy Nogueira et al., 2018; Lou et al., 2018). Collectively, despite supporting a link between brain injury and amyloidogenesis, animal models cannot simulate the spectrum of human response to injury, which is influenced by diverse and human-specific variations in genetic and epigenetic factors. In addition, early outcomes in neurons cannot be readily decoupled from glial and systemic contributions. The ability to capture neuronal outcomes longitudinally and with high temporal and spatial resolution also remains challenging.
Several in vitro models have been also generated to investigate neuronal response to injury by imposing stretch. These platforms largely deliver supraphysiological strains (i.e., simulating severe neuronal trauma) that often result in immediate cytoskeletal disruption and rapid cell death (D. H. Smith et al., 1999; Morrison et al., 2003; LaPlaca et al., 2005; Cullen and LaPlaca, 2006; Monnerie et al., 2010; Tang-Schomer et al., 2012; Dollé et al., 2014; Wang et al., 2014; Salvador et al., 2015; Sherman et al., 2016; Abdul-Muneer et al., 2017). Only a few studies have investigated mild deformations in vitro, providing some insight into neuronal response to sublethal injuries (Chung et al., 2005; Yuen et al., 2009; Staal et al., 2010; Yap et al., 2014, 2017; Li et al., 2019). However, none has explored AD-associated outcomes after injury, and have only rarely used human neurons (Sherman et al., 2016; Dollé et al., 2018; Bianchi et al., 2019).
In this study, toward filling these knowledge gaps, we developed a custom-built device to impose rapid, sublethal, unidirectional deformations on human neurons, simulating those experienced by neurons during TBI (Kimpara and Iwamoto, 2012; Sahoo et al., 2016). We exploited the high-resolution imaging and biochemical capabilities of our system to evaluate two key cellular pathways hypothesized to contribute to AD pathophysiology: APP amyloidogenic processing and APP axonal transport. Our work provides new insight into early molecular changes that may link mTBI to increased susceptibility for AD progression.
Materials and Methods
hiPSC lines
We used a human induced pluripotent stem cell (hiPSC) line derived from a cognitively unimpaired male individual previously established and characterized (CVB) (RRID:CVCL_1N86, GM25430) (Gore et al., 2011). All hIPSCs were cultured as previously described (Israel et al., 2012; Woodruff et al., 2013; Young et al., 2015), on a γ-irradiated mouse embryonic fibroblast feeder layer in HUES media: KO-DMEM (Invitrogen), 10% plasmanate (Talecris Biotherapeutics), 10% KOSR (Invitrogen), 1× nonessential amino acids (Invitrogen), 20 mm glutamax (Invitrogen), 1× penicillin/streptomycin (Invitrogen), with 20 ng/ml fibroblast growth factor (FGF) (Millipore) and passaged with Accutase (Innovative Cell Technologies). APP Icelandic (APPA673T) hiPSC line was generated using CRISPR-based genome editing as previously described (Woodruff et al., 2016; Fong et al., 2018). Briefly, CVB hiPSCs were pretreated with 10 μm Rock Inhibitor (Y-27632 dihydrochloride, Abcam) before nucleofection. To obtain single cells, hiPSCs were dissociated with Accutase and filtered through 100 μm filters; 8 × 105 cells were nucleofected using the Amaxa Human Stem Cell Nucleofector Kit I (Lonza) with 5 µg of modified pSpCas9(BB)-2A-GFP (PX458, expression under EF-1α promoter) vector (Ran et al., 2013), containing guide RNA (5′-GGAGATCTCTGAAGTGAAGA-3′) targeting APP exon 16, and 50 nmol (4 ng) of repair single-stranded oligonucleotide (5′-TGTAATACAGGTTCTGGGTTGACAAATATCAAGACGGAGGAGATCTCTGAAGTCAAGATGGATACAGAATTCCGACATGACTCAGGATATGAAGTTCATCATCAAAAATTGGTACGT-3′). After culturing the hiPSCs in the presence of Rock Inhibitor for 24 h, 1 × 104 GFP+ IPSCs were FACS sorted (FACS Aria IIu, BD Biosciences) and plated on 10 cm plates preseeded with mouse embryonic fibroblasts in the presence of Rock Inhibitor. After a week, visible single colonies were manually picked, transferred to 96-well plates, and expanded. DNA from each single-cell derived clone was harvested using QuickExtract DNA Extraction Solution (Epicentre), APP exon 16 was PCR-amplified using JumpStart REDTaq ReadyMix Reaction Mix (Sigma) and primers (forward, AGG CCT AGA AAG AAG TTT TGG GT; reverse, CAG GCT GGT CTC GAA TTT CT), purified using Exo-SAP-IT PCR Product Cleanup Reagent (Thermo Fisher Scientific) and Sanger sequenced (using forward primer). Sequencing results were aligned against the APP WT sequence to determine the presence of disruption in the vicinity of guide RNA/Cas9 predicted cutting site. Likely edited clones were expanded re-sequenced, and amplified PCR fragments from non-WT colonies were selected, cloned using the Zero Blunt TOPO TA PCR Cloning Kit (Invitrogen), and sequenced to analyze the (edited) genomic DNA sequence on both alleles. Selected clones were karyotyped by hybridization to the Infinium CoreExome-24 BeadChip (Illumina). One clone was identified to carry the A673T mutation in heterozygosis; this line was expanded and used for subsequent experiments.
Generation of hiPSC-derived NPCs and neurons
Here we used two NPC lines previously derived and characterized: CV4a (derived from CVB iPSC line) (Young et al., 2015) and APP-KO Clone IA1 (CRISPR-based genome edited from CVB parental iPSC line) (Fong et al., 2018). NPC line carrying the APPA673T genetic variant was generated and differentiated as reported (Gore et al., 2011; S. H. Yuan et al., 2011; Israel et al., 2012; Fong et al., 2018). Briefly, 2 × 105 FACS-purified iPSC TRA1-81+ cells were seeded onto two 10 cm plates pre-seeded with 5 × 105 PA6 cells the day before and were cultured in PA6 differentiation media: Glasgow DMEM, 10% KO serum replacement, 1 mm sodium pyruvate, 0.1 mm nonessential amino acids, and 0.1 mm β-mercaptoethanol (all Invitrogen), complemented with 10 μm SB431542 (Stemgent) and 0.5 µg/ml Noggin (R&D Systems). After 6 d, SB431542 and Noggin were removed from the media. At day 11, cells were dissociated with Accutase and ∼5 × 105 CD184+CD24+CD44−CD271− NPCs were FACS-purified and plated onto 20 mg/ml poly-L-ornithine (Sigma) and 5 mg/ml laminin (Sigma) precoated plates and cultured in NPC media: DMEM:F12 supplemented with Glutamax, 0.5 × N2, 0.5× B27, Pen/Strep (all Invitrogen), and 20 ng/ml FGF-2 (Millipore), and passaged with Accutase (Innovative Cell Technologies). For neuronal differentiation, NPCs were cultured until confluent in 10 cm plates, after which FGF-2 was withdrawn from the NPC media. Media was changed every other day. After 3 weeks, neurons were purified by FACS using a CD184-APC, CD44-PE−, CD24-PECy7+ (BD Biosciences) neuronal surface signature (S. H. Yuan et al., 2011; Israel et al., 2012; Woodruff et al., 2013); 250,000 sorted neurons (unless indicated) were plated per microfabricated device in NPC+++ media: NPC media supplemented with 20 ng/µl BDNF (PeproTech), 20 ng/µl GDNF (PeproTech), and 0.5 mm dbcAMP (Sigma) and kept in a humidified incubator with 5% CO2 at 37°C for 7 d without media change.
Microfluidic device fabrication and assessment
We modified a previous device demonstrated to stretch rodent dorsal root ganglia neurons seeded on a thin film of polydimethylsiloxane (PDMS) at a low strain rate by slowly increasing vacuum pressure to deform the PDMS surface (Bober et al., 2015). To closely simulate the higher and rapid strain that neurons might experience during mild traumatic injury, we modified our device by a step-change in vacuum pressure. Our PDMS-based device consists of two vacuum chambers (2 × 6 mm) connected by a 200-µm-wide channel that is firmly attached to a thin flexible PDMS membrane on which cells adhere (see Fig. 1A). Additional details of device fabrication methodology will be published elsewhere.
Figure 1.

A new microfluidic device to impose sublethal mechanical-induced stretch to hiPSC-derived neurons. A, Schematic depicting the vacuum-activated mechanism of unidirectional mechanical loading and representative images illustrating the cell culture chamber (without cells) in the prestretch condition and during stretch. Briefly, after the vacuum line is activated, suction displaces the walls flanking the cell culture chamber (in pink), which pulls the PDMS membrane bilaterally and attached cells located between them. The width between chamber walls in both conditions is described in the images and was used to calculate the substrate strain. Red double-headed arrow indicates the direction of the imposed stretch. B, Representative images of human neurons transfected with soluble GFP, in the prestretch condition (top image) and during holding stretch (bottom image). Yellow dotted vertical lines are placed on morphologic marks and used as reference to visualize neurite lengthening during stretch. Neurite strain was calculated as the percentage of increase in original length during stretch. C, Pearson's correlation between neurite initial length before stretch (μm) and the percentage of increase in neurite length during stretch (%) (n = 17). D, Differential interference contrast representative images depicting neuron morphology before and up to 4 min after stretch. Neurites oriented parallel to the direction of stretch were affected (white arrows), whereas neurites oriented perpendicular were not (black arrows). E, Representative images of axons stretched and fixed immediately after stretch, labeled with antibodies against α-tubulin, phosphorylated neurofilament H (NF-H-p), and MAP2. Following stretch, waves were induced (yellow arrows) without breaks or enlargements in axons. Axons oriented vertically to the stretch direction were not affected (white arrows). F, Representative images of neurites stained with silicon-rhodamine (SiR)-tubulin, in the prestretch condition, immediately (0 h), and at indicated times after stretch. Waves (yellow arrows) were induced immediately after stretch (0 h). Vertical axons initially seen before stretch and 0 h after stretch shifted to other focus planes over time. G, Quantitative analysis of ptau/Total tau ratio measured from neuronal lysate harvested at different time points after stretch. H, Pseudo-colored representative images depicting purified neurons stained with Fluo4 before and after stretch. Axonal wave was induced after stretch (white arrow) without triggering massive Ca2+ influx, illustrated by unchanged Fluo4 intensity after stretch (n = 30). I, Heat map of normalized gene expression ratio (stretch/control) at different time points after stretch. Changes in FOS expression were significant according with differential expression call error model (n = 3 or more). J, Quantitative analysis of pJNK/JNK ratio measured from neuronal lysate harvested at different time points after stretch (n = 3 or more). K, Quantification of extracellular LDH levels in neuron's media (relative to LDH levels before stretch) (n = 15) and ATP content in neuronal lysate (n = 9) 24 h (h) after stretch. a.u, Arbitrary units. Further characterization of microfluidic device and standardization of MSD-based assay to quantitatively assess JNK and pJNK levels in neuron's cell lysates are displayed in Extended Data Figure 1-1. Data are mean ± SEM; means were compared by unpaired Student's t test. Scale bars: D, H, 50 μm; E, 11 μm; F, 12 μm.
Briefly, an SU8 mold was made by pouring 15 g of SU8-2100 resist (Microchem) onto a 5-inch-diameter silicon wafer, which was dried for 2 h at 95°C to form a ∼0.8-mm-thick coating. The SU8 mold was then exposed through a mask, patterning multiple devices, each containing two vacuum chambers connected to vacuum input channels, for 20 min at 2 mW/cm2 (365 nm LED array). The mold was then baked for 1 min at 65°C and 15 min at 95°C, followed by development in propylene glycol monomethyl ether acetate (Sigma).
The device was made by bonding two layers of PDMS: (1) a 1-mm-thick bottom layer casted on the patterned SU8 mold with 15 g of silicone (Dow Corning 184, 1:10 ratio) and cured for 10 min at 85°C; and (2) a 5-mm-thick PDMS (1:10 ratio) layer casted on a separate clean wafer, with an aluminum block (1.5 × 6 × 10 mm) used to exclude polymer from regions in which cells were to be seeded. Each layer was air plasma-treated for 15 s (PlasmaPrene II oven) and then aligned and baked for 1 h at 85°C. The well for the cell culture chamber and vacuum port was then cored out, and each device was attached onto a plasma-treated membrane made by spin-coating PDMS (1:20 ratio) to 150 µm thickness (∼750 rpm for 30 s) and curing for 30 min at 85°C.
Completed devices were sonicated for 20 min sequentially in water and isopropanol and then dried and sterilized using a UV lamp in a laminar flow hood continuously for 30 min. To ensure consistency in substrate deformation, sterilized devices were vacuum-tested before starting the coating procedure. Devices were then amine functionalized with 4% (3-aminopropyl) trimethoxysilane (Alfa Aesar) for 20 min, extensively washed using PBS, and overnight coated with N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride 0.1%/ml (Sigma) and 25 mg/ml laminin before cell seeding.
To assess imaging resolution of the PDMS surface for a variety of immersion fluids (ddH2O, Standard Leica immersion oil, and 41% or 80% glycerol in water; Table 1), the point spread function of 200 nm fluorescein-coated beads (which served as a point source) was characterized. Briefly, beads were seeded within the cell imaging chamber (i.e., PDMS thin film) or on glass coverslips (positive control) and visualized using a 63×/1.4 NA objective on a Leica Microsystems SP5 confocal microscopy system, using filter sets appropriate for fluorescein/FITC. Beads were imaged with substrates unstretched and held briefly at maximum deformation. The FWHM value was used to specify resolution. Resolutions were measured and averaged from at least five beads per imaging condition.
Table 1.
PDMS optical properties under different immersion fluidsa
| Immersion fluid | Resolution (µm) | Average maximum intensity (a.u) |
|---|---|---|
| Glass water | 0.3065 ± 0.01 (5) | 200.16 ± 39.5 (617) |
| PDMS water | 0.4321 ± 0.02 (5) | 68.052 ± 50.0 (374) |
| PDMS Glyc41 water | 0.3149 ± 0.01 (5) | 121.61 ± 58.6 (315) |
| PDMS Glyc41 water vacuum | 0.3078 ± 0.01 (5) | 132.68 ± 61.6 (397) |
| Glass oil | 0.2934 ± 0.01 (5) | 220.14 ± 28.4 (533) |
| PDMS oil | 0.3874 ± 0.03 (5) | 114.39 ± 58.9 (355) |
| PDMS oil+PC | 0.3001 ± 0.01 (5) | 157.20 ± 59.5 (556) |
| PDMS oil+PC vacuum | 0.2900 ± 0.00 (5) | 171.31 ± 57.1 (281) |
| Glass Glyc80 lens | 0.3043 ± 0.01 (5) | — |
| PDMS Glyc80 lens | 0.3595 ± 0.03 (5) | — |
aData are mean ± SD (n).
Substrate and neuronal stretch
Controlled unidirectional mechanical stretch on cultured neurons was achieved by using a vacuum pump. Briefly, devices were placed in a live cell imaging chamber integrated with a Carl Zeiss LSM780 inverted confocal microscope. This chamber allowed the maintenance of 5% CO2 and 37°C environment, and concurrent brightfield and fluorescence image acquisition during stretch. To facilitate preconditioning, including detachment of poorly adherent cells that could confound subsequent image analysis, each injury bout consisted of 40 consecutive deformations of the flexible substrate, each constituted of a step-stretch held for 3 s followed by a step-release held for 3 s; the “step”-strain and release rate was 30%/s. Given the timeline for restoring cell tension (see Fig. 1D), additional strain on cells was negligible for each successive stretch (i.e., cells and axons were not additionally elongated with each stretch, as they were tension-free.)
The degree of substrate strain was calculated as the percentage increase in the distance between both chamber walls following device activation compared with the original length. Images of device walls in unstretched and stretched configurations were acquired using a 10×/NA 0.45 objective, and the distance between chamber walls measured. To quantify strain experienced by neuronal projections during stretch, neurons were first transfected with soluble pEGFP-C1 (Clontech, 6084-1; 1500 ng) using Lipofectamine 2000, according to the manufacturer's protocol, to identify neuronal boundaries. Then, images of individual adherent neurons during prestretched, holding stretch, and post-stretch release were acquired using a 10× objective; any weakly adherent neurons that happened to detach during the process were not included in this or other image analysis. Strain was calculated as the percentage increase in neurite length between two static reference points after stretch compared with the original length in the prestretch condition. ImageJ (National Institutes of Health) software was used for all measurements.
Immunocytochemistry assays
Neurons were fixed with 4% PFA (Pierce) and 4% glucose (Sigma) in PBS for 15 min at room temperature, rinsed with PBS, and permeabilized with 0.2% Triton X-100 (Sigma) in PBS for 30 min. Nonspecific binding was blocked with 10% NGS (Vector Laboratories) and 4% BSA (Sigma) in PBS for 30 min and then incubated overnight with primary antibodies against MAP2 (1:1000, Abcam, ab5392, raised in chicken), α-tubulin (1:1000, Abcam, YOL1/34, ab6161, raised in rat), neurofilament H phosphorylated (1:500, Biolegend, 801602, raised in mouse), tau (1:1000, Sigma, T9450, raised in mouse), APP (1:100, Abcam, Y188, ab32136, raised in rabbit), APP (1:100, Sigma, clone 1D1, MABN2278, raised in rat), Aβ42 (1:1000, Oncogene, Ab-1, p150-25µg, raised in rabbit), BACE-1 (1:1000, Thermo Fisher Scientific, PA1-757, raised in rabbit), Presenilin-1 (PS1) (1:250, Millipore, clone PS1-loop, MAB5232, raised in mouse), and C99 fragment (C99; an APP fragment generated following BACE-1 cleavage) (1:1000, Millipore, clone 6C3, MABN254, raised in mouse); all diluted in 1% NGS, 2% BSA, and 0.2% Triton X-100. Devices were washed with cold PBS followed by incubation with AlexaFluor-488, -550, -594, or 649-conjugated secondary antibody (1/200, Abcam) for 1 h at room temperature and protected from light. Cells were mounted with mounting media containing DAPI (Vector Laboratories) into the cell chamber. Images were acquired with an inverted 63×/NA1.4 objective using confocal microscope (Carl Zeiss LSM780, as above). At the earliest time point, to preserve the transient waves in neurites after stretch, cells were immediately fixed for 15 min within the live cell imaging chamber, by adding 4% PFA/4% glucose as above. To calculate puncta intensity, puncta area, and puncta number in axons, quantitative analysis of immunofluorescence images was performed using the ImageJ plugin Puncta Analyzer (Ippolito and Eroglu, 2010).
Live cell imaging
All live-cell imaging assays were performed at 37°C and under 5% CO2 using the live cell confocal imaging chamber noted above and NPC+++ media without Phenol Red (Invitrogen) as imaging media. Real-time evaluation of stretch-induced morphologic alterations in FACS-sorted neurons was performed with differential interference contrast microscopy of the same neuron during stretch and after release over time, using a 20×/NA 0.8 objective at a rate of 1 frame per second. To evaluate whether trauma-induced waviness in neurites was dependent on neurite's orientation related to mechanical loading direction, we used ImageJ to determine neurite angles in at least three randomly chosen fields of view from control and stretched devices.
Cytosolic Ca2+ levels following mechanical trauma were monitored using Fluo-4 AM (Invitrogen). Briefly, FACS-sorted cultured neurons were incubated with 1 μm Fluo-4 AM (reconstituted in DMSO) diluted in freshly made NPC+++ media for 30 min at 5% CO2 and 37°C, washed twice with NPC+++ media, and again placed in the incubator for 20 min before imaging. Cytosolic Ca2+ levels were analyzed using a 20×/NA 0.8 objective, and image sequences were acquired during stretch at a rate of 1 frame per second with 3 s intervals; 488 nm Argon laser was used for excitation, and emission was captured in the 490-600 nm range. Images were processed using ImageJ, Fluo-4 intensity measured within an ROI over time (Barreto-Chang and Dolmetsch, 2009).
To visualize tubulin, purified neurons seeded in devices for 7 d were labeled with 100 nm of silicon-rhodamine-tubulin (Spirochrome) diluted in NPC+++ media during 12 h in 5% CO2 at 37°C. Following incubation, cells were washed twice with NPC+++ media. Images from the same imaging field were acquired before, immediately after, and 3, 6, 12, and 24 h after stretch with the 20×/NA 0.8 objective. Following image acquisition, cells were returned to the CO2 incubator until the next imaging session. Cells were imaged using 647 nm laser for excitation, and emission was captured in the 690-750 nm range. Intensities within a given ROI were measured using ImageJ.
To quantify APP transport parameters, 600,000 sorted neurons were grown in microfluidic devices for 7 d and transfected with 2.5 µg of pcDNA3-APP695-YFP (Kaether et al., 2000), pcDNA3-APPM596V-YFP (Rodrigues et al., 2012), or APPIce-GFP (APP695 isoform carrying the APPA673T mutation) (Das et al., 2016) plasmids using Lipofectamine 2000, as previously described (Stokin et al., 2005; Falzone et al., 2009; Rodrigues et al., 2012; Almenar-Queralt et al., 2014). Sixteen hours after transfection, cells were imaged using an inverted epifluorescent microscope (TE-2000U, Nikon) coupled to incubator chamber at 37°C, 5% CO2 and a 63×/NA 1.4 oil immersion objective connected to a CCD camera (Roper Scientific). Imaged neurons were selected based on their morphology being consistent with published data (Ramón y Cajal, 1909), specifically, possessing a small cell body with a single, long, thin primary axon that extended for at least 1 mm, and secondary, short, thick dendrites. Neurites with axonal morphology and oriented within 30 degrees of the direction of stretch direction containing YFP/GFP puncta were imaged before and after stretch for 15 s (150 frames with 100 ms exposure per frame) using MetaMorph 7.0 software (Universal Imaging). To assess axonal transport at 3 and 24 h after stretch, transfected neurons were stretched within the imaging chamber but only imaged at 3 or 24 h after stretch (i.e., no prestretch imaging) to avoid fluorophore bleaching. Mock cells were manipulated and imaged identically, but not submitted to stretch.
Directionality of moving puncta was determined based on trajectory relative to location of cell body and neuronal terminal. Axonal transport parameters were evaluated by kymography using the ImageJ plugin KymoAnalyzer (Neumann et al., 2017). The axonal transport of lysosomes was evaluated using a fluorescent fusion protein, lysosomal-associated membrane protein 2-green fluorescent protein (LAMP2-GFP), introduced by transfecting cells with 3000 ng of human LAMP-2-GFP.
To measure APP amyloidogenic processing immediately after stretch in axons, we transfected nonpurified APP-KO neurons with pcDNA3-APP-YFP, and fixed neurons immediately after stretch as described above. Neurons were stained with APP (1:100, Sigma, clone 1D1, MABN2278, raised in rat) and Aβ42 (1:1000, Oncogene, Ab-1, p150, 25 µg, raised in rabbit) antibodies, and images were acquired with 63×/NA1.4 objective using confocal microscopy (Carl Zeiss LSM780). Aβ42 puncta intensity and puncta number were obtained using ImageJ plugin Puncta Analyzer (Ippolito and Eroglu, 2010).
Meso scale discovery (MSD) measurements
Intracellular levels of phosphorylated c-Jun N-terminal kinase (p-JNK) and phosphorylated tau (p-tau) were measured in cell lysates obtained using MSD lysis buffer supplemented with 1× protease (Millipore) and phosphatase (Invitrogen) inhibitors cocktail according to the manufacturer's instructions and as previously described (Israel et al., 2012; Woodruff et al., 2013). Extracellular levels of Aβ40 and Aβ42 were measured in cell culture media samples (matched with cell lysate samples when possible). Collected media was immediately supplemented with 1× protease and phosphatase inhibitors, centrifuged at 14,000 rpm for 10 min at 4°C to remove cell debris, and stored at −80°C until use as described previously (Israel et al., 2012; Woodruff et al., 2013). MSD Phospho(Thr231)/Total Tau, Phospho (Thr183/Tyr185)/Total JNK Whole Cell Lysate and V-PLEX Aβ Peptide Panel 1 (6E10) MSD kits were used according to the manufacturer's instructions and analyzed in MSD QuickPlex SQ 120 plate reader. Positive controls (Neurodegeneration Controls set [MSD]) were used in each run for Aβ and p-tau assays.
All measurements were in the detection range based on standard curves generated using MSD-provided calibrators (Aβ and p-tau assays). For the p-JNK assay, the detection range was assessed measuring undiluted cell lysate (control neurons) plotted against dilution rates to determine assay linear detection range. Furthermore, to determine p-JNK assay specificity, sorted neurons were exposed to anisomycin (2.5 μm) or JNK inhibitor II (9 μm) for 30 min as positive (Fosbrink et al., 2010) and negatives controls, respectively (Besirli and Johnson, 2003; Wu et al., 2014). Exposure to geranylgeranyl pyrophosphate (GGPP) during 24 h was used to modulate extracellular Aβ levels in APPWT/Ice neurons (Zhou et al., 2003; Kukar et al., 2005).
Immunoblotting analysis
Jess Simple Western system (ProteinSimple) was used to quantify full-length APP, BACE-1, PS1, and β-actin protein levels according to the manufacturer's standard method for 12-230 kDa Jess separation module (SM-W004). Briefly, neuronal lysates were separated in capillaries as they migrated through a separation matrix at 475 V and then exposed to blocking reagent (Antibody diluent, ProteinSimple) during 5 min followed by exposure to primary and secondary antibody both for 30 min. APP (1:25, Abcam, Y188, ab32136, raised in rabbit), BACE-1 (1:50, Thermo Fisher Scientific, PA1-757, raised in rabbit), PS1 (1:25, Millipore, clone PS1-loop (PS1-CTF), MAB5232, raised in mouse), β-actin (1:150, Millipore, clone C4, MAB1501, raised in mouse), and HRP-conjugated secondary (ProteinSimple) antibodies were all diluted in antibody diluent (ProteinSimple). The chemiluminescent detection was performed using peroxide/luminol-S (ProteinSimple). Using Compass Simple Western software (version 4.1.0, ProteinSimple), a digital image of chemiluminescence in each capillary was captured, and peak heights and peak area were automatically calculated per sample. Quantification was performed using peak area values obtained for each sample, normalized to β-actin values for the same sample.
NanoString gene expression analysis
Gene expression profiling was performed using nCounter Human Neuropathology Panel (NanoString Technologies). Total RNA from a single device with 600,000 neurons was extracted using miRNeasy Micro Kit (QIAGEN). RNA from at least three devices obtained from at least three individual neuronal differentiations was pooled and concentrated using GeneJET RNA Cleanup and Concentration Micro Kit (Thermo Fisher Scientific) according to the manufacturer's protocol. Samples were quantified using Qubit 4 fluorometer (Invitrogen), and RNA Integrity Number was determined using Agilent 2100 Bioanalyzer. All measured RNA Integrity Numbers were between 5 and 9. mRNA expression analysis was performed using Nanostring nCounter platform according to the manufacturer's instructions. The raw Nanostring nCounter counts were normalized to housekeeping genes included on the chip and background level using nSolver analysis system (NanoString Technologies). Genes with ≥100 counts and fold-change expression ≥1.35 were displayed and considered for statistical analysis using the differential expression call error model to determine ratio (stretch/control) confidence.
Cell viability assessment
Cell viability after stretch was evaluated assessing intracellular ATP and extracellular lactate dehydrogenase (LDH) levels 24 h after stretch. ATP was measured using CellTiter-Glo Luminescent Cell Viability Assay (Promega) in cell lysates, obtained as described above, according to the manufacturer's instructions and as previously described (van der Kant et al., 2019). Luminescent signal generated was recorded using Odyssey CLx microplate reader (LI-COR). LDH content was measured in cell culture media samples (matched with cell lysates samples) using the LDH-Cytotoxicity Assay Kit II (BioVision) according to the manufacturer's instructions. Sample absorbance was assessed using Infinite M200 pro (Tecan) at 450 nm (using 440-490 nm filter) and 650 nm, as reference wavelength. LDH levels after stretch were normalized by matched measurements before stretch. Positive controls (purified ATP and LDH) were used in each run.
β- and γ-secretase inhibition
To validate APP secretase activity attenuation, purified neurons were grown in 96-well plates (200,000 purified neurons per well) and exposed to inhibitors. Briefly, half of the cell culture media was removed (100 µl) and replaced with 100 µl of fresh NPC+++ media with β-secretase inhibitor IV (βSi-IV), Compound E (CE) (both from EMD Chemicals), or vehicle (DMSO) (Sigma) during 16 h in CO2 incubator at 37°C. To evaluate whether CE-induced γ-secretase inhibition was reversible in our system, following initial exposure to CE cell culture, media was collected (200 µl) and cells were washed and incubated with PBS for 5 min at 5% CO2 and 37°C before being re-exposed to CE or vehicle for 3, 6, 12, 24, and 36 h. Importantly, CE exposure and release or re-exposure did not induce changes in intracellular Aβ levels or Aβx-42/Aβx-40 ratio (Extended Data Fig. 2-1F).
To inhibit β- and γ-secretase activity during and after stretch in stretch devices, cells were exposed to βSi-IV, CE, or vehicle control for 3 h before stretch and remained exposed to inhibitors or vehicle for further 24 h after injury. To evaluate stretch effects following γ-secretase reversible inhibition, cells were exposed to CE for 16 h and then washed and incubated with PBS for 5 min. Following incubation, fresh media was added, and stretch was imposed as previously described. Media was collected 24 h after injury. To inhibit β- and γ-secretase in APP axonal transport experiments, nonpurified neurons were exposed to βsi-IV, CE, or vehicle (DMSO) for 1 h and imaged in the presence of indicated inhibitor or corresponding vehicle control. In experiments probing for the formation of APP accumulation in neurons exposed to CE, nonpurified neuronal cultures were stretched in the presence of CE or vehicle, immediately washed, and submerged in fresh media before incubation after stretch.
Experimental design and statistical analysis
Statistics were assessed through GraphPad Prism (GraphPad Software, version 7.00). A minimum of n = 3 biological samples (different microfluidic devices) were obtained from at least three individual NPC differentiations per group. A p value ≤0.05 indicated statistically significant differences. Sample sizes and the statistical test applied for a given experiment are specified in the corresponding figure legend.
Results
A new device to impose sublethal stretch on hiPSC-derived neurons
To simulate a <15% axonal strain that neurons might experience under a sublethal traumatic injury (Kimpara and Iwamoto, 2012; Sahoo et al., 2016), we developed a PDMS-based microfluidic device capable of imposing unidirectional stretch on human neurons (Fig. 1A). Briefly, a vacuum system, via pressurization of the cell culture chamber walls, rapidly and reproducibly stretched the substrate by 24 ± 0.85% (mean ± SEM, n = 9), and attached neurons by 12 ± 1.26% (mean ± SEM, n = 21) (Fig. 1A,B). Strains achieved in neuronal projections were independent of their length, and no neurite breakage was observed even at maximal strains of ∼22% (Fig. 1C).
Microfluidic device further characterization and standardization of MSD-based assay to quantitatively assess JNK and pJNK levels in neuron's cell lysates. A) Representative images of neurons (with neurites oriented within 30 degrees of stretch direction) stretched and fixed immediately after to preserve waves in axon (Tau positive, white arrows) and dendrites (MAP2, yellow arrows). B) Quantification of the percent of neurites oriented parallel (0°-30°), inclined (30°-60°) and perpendicular (60°-90°) to the direction of the stretch in non-stretched microfluidic devices and the distribution of wavy neurites per angle following stretch (compared to 60°-90°, *p = 0.0119 for 0°-30° and *p = 0.0385 for 0°-30°) (n = 3). C) Quantitative analysis of Total tau and p-tau, measured from neuronal lysate harvested at different time points after stretch (n = 3 or more). D) Quantitative analysis of Fluo4 intensity in neurons cell body before and immediately after stretch. E) Assessment of assay linear detection range for Total JNK and pJNK (Thr183/Tyr185), measured using undiluted neuronal lysate (control neurons) plotted against dilution ranges (n = 4). 2.5X dilution range was chosen to perform further measurements. Quantitative analysis of Total JNK and pJNK (Thr183/Tyr185) levels and resultant pJNK/JNK ratio, following thirty minutes of exposure to F) Anisomycin (2.5 µM) (*p = 0.0306 for JNK, *p = 0.0435 for p-JNK and *p = 0.0444 p-JNK/JNK ratio) (n = 8) and G) JNK inhibitor II (9 µM) (*p = 0.0123 for p-JNK and *p = 0.0494 p-JNK/JNK ratio) (n = 8) (Vehicle = DMSO 0.01%). H) Quantitative analysis of Total JNK and pJNK, measured from neuronal lysate harvested at different time points up to 24 h (h) post-stretch (n = 3 or more). Data are mean ± SEM; means were compared by One-way ANOVA (B) unpaired Student's t test (C-H). Download Figure 1-1, TIF file (108.7MB, tif) .
Live imaging showed that immediately after release from stretching, neurites oriented parallel to the direction of the stretch, regardless of neurite identity (i.e., axonal or dendritic; Extended Data Fig. 1-1A) (Caceres et al., 1986), displayed a wavy phenotype, not observed before stretch (Fig. 1D, white arrow). In support, a wavy phenotype was reproducibly induced in 97 ± 2.18% (mean ± SEM, n = 21) in neurites oriented within 0-30 degrees of (i.e., parallel to) the direction of stretch and not in those oriented perpendicularly (Fig. 1D, black arrow; Extended Data Fig. 1-1B). This wavy phenotype is consistent with observations from previous in vitro models of stretch-based axonal injury and pathology of brains subject to severe injury (D. H. Smith et al., 1999; D. H. Smith and Meaney, 2000; Maas et al., 2008; Hayashi et al., 2009; Tang-Schomer et al., 2010, 2012; Johnson et al., 2013; Dollé et al., 2014, 2018). Waves suggest that neurites experienced tension, elongated because of tension, and retained their added length immediately after releasing tension. Upon release of stretch, neurites reverted to their original nonwavy morphology within minutes, indicating restoration of neurite tension (Fig. 1D).
Neurite waviness following supra-physiological strains of mechanical trauma have been previously associated with microtubule buckling, breakage, and ultimately the formation of focal swellings in these sites (Tang-Schomer et al., 2010, 2012). Exploiting the high-resolution imaging capabilities of our device (Table 1), we visualized microtubule network behavior in neurons by staining cells fixed immediately after stretch (Fig. 1E) or via live imaging over time using cell-permeable silicon-rhodamine-tubulin (Fig. 1F) (Lukinavičius et al., 2014). Together, they revealed neither formation of focal neurite swellings nor apparent breaks in microtubule network continuity in stretched neurites at any point within 24 h of injury, despite displaying clear neurite deformation after stretch (Fig. 1D–F). Similarly, consistent with restored neurite morphology, major discontinuities of phosphorylated neurofilament network immediately after stretch along wavy axons were not observed (Fig. 1E). Similarly, increased tau phosphorylation at Threonine 231(Thr231), which correlates with unstable microtubule-based cytoskeleton, was not observed up to 24 h after stretch (Fig. 1G; Extended Data Fig. 1-1C) (Vickers et al., 1994; C. Smith et al., 2003; Cho and Johnson, 2004; Anderson et al., 2008; Johnson et al., 2012; Li et al., 2015; Shibahashi et al., 2016; Yang et al., 2017; A. Yuan et al., 2017; Zanier et al., 2018).
Also consistent with sublethal stretch, increased calcium influx associated with plasma membrane rupture or mechanoporation (Yuen et al., 2009; Staal et al., 2010; Dollé et al., 2014; Patel et al., 2014; Abdul-Muneer et al., 2017) was not detected in the cell body or neurites of stretched neurons (Fig. 1H; Extended Data Fig. 1-1D). Similarly, expression of genes encoding proteins involved in injury/stress intracellular signaling or increased phosphorylation of JNK (p-JNK), which is part of a cellular stress-induced response pathway (Middlemas et al., 2003; Cavalli et al., 2005; Falzone et al., 2009; Yoshimura et al., 2011; Tran et al., 2012; Farley and Watkins, 2018; Liu et al., 2018), was unchanged by mild stretch (Fig. 1I,J; Extended Data Fig. 1-1E–H). Expression of the immediate-early response gene FOS (Duman et al., 2005; Bahrami and Drabløs, 2016), however, was significantly upregulated 3 h after stretch (Fig. 1I), suggesting a neuronal mechanotransduction response to mild stretch. Finally, unaffected levels of LDH release into media and intracellularly metabolized ATP (indicators of cell death and viability, respectively) (Severson et al., 2007; Kumar et al., 2018) 24 h after stretch support the nonlethality of the imposed stretch (Fig. 1K). Cumulatively, our analysis of mechanical and biological indicators of damage/injury establishes that our approach induces a sublethal neuronal stretch without major disruptions of cytoskeleton components.
Sublethal stretch is sufficient to stimulate Aβ generation in human neurons
To examine whether sublethal mechanical-induced stretch of neurons is sufficient to stimulate amyloidogenesis, we assessed changes in extracellular levels of Aβ accumulated in media after stretch. While Aβ extracellular levels were unchanged immediately after stretch or at earlier time points (Extended Data Fig. 2-1A), 24 h after stretch, Aβ extracellular levels were elevated compared with nonstretched neuronal cultures (Fig. 2A). As Aβ is generated by consecutive cleavage of APP by BACE-1 (the rate-limiting step) and γ-secretase (Citron et al., 1995) and in light of previous studies reporting increased expression of APP, BACE1, and PS1 following TBI (Iwata et al., 2002; Blasko et al., 2004; Nadler et al., 2008; Loane et al., 2009; Walker et al., 2012; Yu et al., 2012; Mannix et al., 2013; Thangavelu et al., 2020), we evaluated protein levels of APP, BACE1, and PS1 24 h after stretch. In agreement with our mRNA results (Fig. 1I), no significant changes were detected at the protein level in any of these proteins (Extended Data Fig. 2-1B).
Figure 2.

Sublethal stretch increases extracellular Aβ generation in human neurons. A, Quantitative analysis of extracellular Aβ40 and Aβ42 and resultant Aβ42/Aβ40 ratio, measured in neuronal media collected 24 h after stretch. Neuronal cultures were washed with PBS, and fresh media was added before stretch was imposed (*p = 0.0418 for Aβ40; *p = 0.0278 for Aβ42) (n = 7). Quantification of extracellular Aβ levels 24 h after stretch in neurons stretched in the presence of (B) βsi-IV (4 μm) (***p = 0.0005 for Aβ40; **p = 0.0040 for Aβ42) (n = 9) or (C) CE (200 nm) (*p = 0.0493 for Aβ40; *p = 0.0041 for Aβ42). Data are percent of change in extracellular Aβ levels after stretch relative to extracellular Aβ levels before stretch (n = 6) (vehicle = DMSO 0.001%). D, Diagram showing experimental design used to measure stretch effects on Aβ generation after CE reversible inhibition and quantitative analysis of extracellular Aβ40 and Aβ42 levels 24 h after stretch in neurons previously exposed to CE (200 nm) during 16 h (**p = 0.0035 for Aβ40; **p = 0.0042 for Aβ42) (n = 13). Measurements of intracellular Aβ levels after stretch are displayed in Extended Data Figure 2-1. Data are mean ± SEM; means were compared by unpaired Student's t test.
Stretch does not induce intracellular Aβ accumulation in human neurons. A) Quantitative analysis of extracellular Aβ40 and Aβ42 and resultant Aβ42/Aβ40 ratio, measured in neuronal media collected immediately and at different time points after stretch relative to matched Aβ levels before stretch (**p = 0.0078 for Aβ42 and *p = 0.0110 for Aβ42/Aβ40 ratio) (n = 3 or more). B) Representative bands and quantitative analysis of full-length APP, BACE1 and PS1 (PS1-CTF) protein levels measured in neuronal lysates 24 h after stretch (n = 7 or more) (C=control and S=stretched). Extracellular Aβ levels in neuronal media from naïve neurons exposed to C) CE (200 nM) (***p = 0.0001 for Aβ40 and Aβ42) (n = 4) or D) βsi-IV (4 µM) (***p = 0.0001 for Aβ40 and **p = 0.0046 for Aβ42) (n = 3) during sixteen hours (h) (Vehicle = DMSO 0.001%). E) Levels of extracellular Aβ peptides in media from neurons re-exposed to CE (200 nM) or vehicle (DMSO 0.001%) for different time points after been initially exposed to CE during sixteen hours, to demonstrate that CE inhibition is reversible (CE 16h+Vehicle, ***p = 0.0001 for Aβ40 and Aβ42) (n = 4). F) Quantitative analysis of intracellular Aβ peptides in cell lysates from neurons re-exposed to CE (200 nM) or exposed to vehicle (DMSO 0.001%) during twenty-four or thirty-six hours after been initially exposed to CE during sixteen hours (Vehicle 16h + Vehicle 24h ***p = 0.0001 for Aβ40 and Vehicle 16h + Vehicle 36h ***p = 0.0001 for Aβ40) (n = 4). ND = Non-detectable. Data are mean ± SEM; means were compared by unpaired Student's t test. Download Figure 2-1, TIF file (109.6MB, tif) .
Together, our results suggest that sublethal stretch actively stimulates new generation of Aβ. Accordingly, βSi-IV or the γ-secretase inhibitor CE, which efficiently suppressed accumulation of extracellular Aβ in naive neurons (Extended Data Fig. 2-1C,D), also attenuated the stretch-induced accumulation of extracellular Aβ (Fig. 2B,C). To further confirm the de novo production of Aβ after stretch, we took advantage of the reversibility of the γ-secretase inhibitor CE (Seiffert et al., 2000; Zhao et al., 2007). Before stretch, we applied CE to neurons for 16 h to eliminate pre-accumulation of extracellular Aβ in the system (Extended Data Fig. 2-1C,E,F); once stretched and on removal of CE and consequent resumption of γ-secretase activity, we found that levels of newly generated extracellular Aβ were higher in stretched neurons compared with non-stretched controls 24 h after stretch (Fig. 2D). These data confirmed that sublethal stretch is sufficient to enhance Aβ generation in human neurons by stimulating APP amyloidogenic processing.
Sublethal stretch interrupts APP axonal transport, leading to aberrant APP accumulations
APP axonal accumulations have been defined as a highly sensitive biomarker for axonal injury (Stone et al., 2000; Hoshino et al., 2003; Johnson et al., 2016). In support, we detected APP accumulations in sublethally stretched axons 24 h after stretch (Fig. 3A, arrows; Extended Data Fig. 3-1A). Injury-induced APP accumulations have been proposed to result from axonal transport impairment because of cytoskeletal disruptions and to contribute to enhanced Aβ generation after TBI (D. H. Smith et al., 1999; Uryu et al., 2007; X. H. Chen et al., 2009). The presence of APP accumulations in our system, despite the absence of major cytoskeletal disruptions or morphologic abnormalities (Figs. 1D-G and 3A), suggested that sublethal stretch may be sufficient to alter APP axonal transport. Indeed, using live imaging to assess the axonal transport of APP-YFP, we observed an immediate and severe reduction in the percentage of APP-YFP puncta moving in the anterograde direction (i.e., from the cell body toward the axonal terminal) and bidirectional reductions in run length and velocity after stretch compared with the prestretch conditions (Fig. 3B,C; Movie 1). The density of APP-YFP puncta in axons was unchanged immediately after stretch, ruling out the possibility that transport impairment was a consequence of fewer detected APP-YFP puncta at this time point (Fig. 3C). Supporting a causal relationship between axonal stretch and transport impairment, APP-YFP transport was not affected in neighboring axons oriented perpendicularly to the direction of stretch (i.e., no signs of stretched neurites; Figs. 1D and 3D; Extended Data Fig. 1-1B; Movie 2). Interestingly, the axonal transport of lysosomes, another vesicular cargo rapidly transported along the axon (Tsukita and Ishikawa, 1980; Ferguson, 2018), measured using GFP-labeled LAMP2 (a lysosomal protein), was not affected under these stretch conditions (Extended Data Fig. 3-1B; Movie 3), suggesting that stretch-induced axonal transport defects are cargo-specific.
Figure 3.

Stretch triggers APP transport impairment in neurons. A, Representative images of axons (within 30 degrees of stretch direction) fixed 24 h after stretch and labeled with antibodies against APP, α-tubulin, and neurofilament H phosphorylated (NF-H-p). Arrows indicate accumulated APP puncta along stretched axons in A. Quantitative analysis of APP-puncta intensity and puncta area are displayed in Extended Data Figure 3-1. B, Representative images depicting APP-YFP puncta in a transfected axon before and after stretch; the respective kymograph is displayed below the photomicrograph. Kymographs represent APP-YFP transport from 15 s, 10 Hz movies (Movie 1); right or left descending puncta represent anterograde and retrograde moving vesicles, respectively. Vertical lines indicate stationary puncta. C, Quantitative analysis of the APP-YFP movement parameters in axons immediately (n = 7), 3 h (n = 7 or more), and 24 h after stretch (n = 9 or more). Data are presented relative to control, moving vesicles (anterograde 0 h: *p = 0.0122; and 24 h: *p = 0.0186), net velocities (anterograde 0 h: **p = 0.0013; 3 h: *p = 0.0244; and 24 h: *p = 0.0413; retrograde 0 h: *p = 0.0100; 3 h: *p = 0.0235; and 24 h: **p = 0.0017), run length (anterograde 0 h: ***p = 0.0001; 3 h: *p = 0.0494; and 24 h: **p = 0.0013; retrograde 0 h: **p = 0.0055; 3 h: **p = 0.0018; and 24 h: ***p = 0.0001), and puncta density (*p = 0.0459 for 3 h and *p = 0.0455 for 24 h). D, Representative images and kymograph of an axon oriented perpendicular to the direction of stretch immediately after stretch (corresponds to Movie 2). E, Representative images and respective kymographs depicting stretched axons 3 and 24 h after stretch (corresponds to Movies 4 and 5). F, Representative images of axons transfected with APP-YFP, stretched and fixed 24 h after stretch, labeled with antibodies against APP, C99, Aβ42, BACE1, and PS1. Arrows indicate accumulated puncta along stretched axons in E and F. G, Quantitative analysis of APP-, C99-, Aβ42-, BACE1-, and PS1-puncta intensity within APP accumulation compared with respective puncta intensity in control axons (APP: **p = 0.0007; C99: ***p = 0.0001; Aβ42: ***p = 0.0001; BACE1: *p = 0.0462; PS1: ***p = 0.0001) (n = 7 or more). Data are mean ± SEM; means were compared by unpaired Student's t test. Scale bars: A, F, 5 μm; B, D, E, 10 μm.
Stretch-induced inhibition of axonal transport is cargo-specific. A) Quantitative analysis of APP-puncta intensity and puncta area in stretched axons fixed 24 h after stretch, compared to control axons (puncta intensity **p = 0.033, puncta size **p = 0.0044) (n = 7 or more axons). B) Quantitative analysis of LAMP2-GFP axonal transport in axons before and after stretch (n = 5). Data are mean ± SEM; means were compared by unpaired Student's t test. Download Figure 3-1, TIF file (2MB, tif) .
In order to account for stretch-induced axonal APP accumulations that we observed at 24 h after stretch (Fig. 3A), we predict that APP axonal transport must recover, to at least some extent. In support of this prediction, the proportion of anterogradely moving APP-YFP puncta was restored to control levels 3 h after stretch, whereas net velocities and run lengths remained reduced (Fig. 3C). Accordingly, the density of APP-YFP puncta in the imaging field was also increased (Fig. 3C,E; Movies 4 and 5). At 24 h after stretch, bidirectional APP-YFP net velocities and run lengths remained suppressed, and the proportion of anterogradely moving APP-YFP puncta was again reduced (Fig. 3C,E).
Finally, consistent with human postmortem studies and with the hypothesis that APP accumulations can be loci of APP amyloidogenic processing (D. H. Smith et al., 1999; Uryu et al., 2007; X. H. Chen et al., 2009), we observed that APP C99, Aβ42, BACE-1, and PS1 accumulated within APP accumulations (Fig. 3F,G), as observed after TBI (Uryu et al., 2007; X. H. Chen et al., 2009). Together, our experiments demonstrated that a sublethal mechanical-induced deformation in human neurons is sufficient to impair APP axonal transport acutely and persistently, ultimately leading to aberrant axonal accumulations containing APP and indicators of its amyloidogenic processing.
Stretch-triggered APP axonal transport defects depend on APP amyloidogenic processing
Previous studies have suggested that enhanced APP amyloidogenic processing by its secretases can impact its own axonal transport (Kamal et al., 2001; Rodrigues et al., 2012; Almenar-Queralt et al., 2014). Thus, to test whether stretch-induced APP axonal transport defects are dependent on APP amyloidogenic processing, we assessed APP axonal transport dynamics before and immediately after stretch in neurons exposed to β- or γ-secretase inhibitors (βSi-IV and CE, respectively). While the percentage of APP-YFP puncta moving in the anterograde direction was similarly reduced following stretch in neurons exposed to DMSO (vehicle control for βSi-IV and CE) (Fig. 4A; Movie 6) and in untreated neurons (Fig. 3B), this effect was attenuated in neurons exposed to βSi-IV or CE (Fig. 4A; Movies 7 and 8). Importantly, exposure to inhibitors alone did not affect proportions of directionally moving APP-YFP puncta or their density (Extended Data Fig. 4-1). Together, these data indicate that stretch-induced impairment of APP axonal transport requires amyloidogenic processing of APP.
Figure 4.

Reduced APP processing before stretch abrogates stretch-induced APP transport defects. A, Kymographs and quantitative analysis of the percentage of APP-YFP puncta moving anterogradely and retrogradely immediately after stretch in neurons exposed to βsi-IV (40 μm), CE (5 μm), or vehicle (DMSO 0.01%) during 1 h before being stretched. Quantitative analysis of APP axonal transport parameters in nonstretched neurons exposed to βsi-IV or CE compared with vehicle are displayed in Extended Data Figure 4-1. Data are shown as relative to the prestretch condition for each experimental group (dashed red line). *p = 0.0506, anterograde compared to pre-stretch condition. n = 4 or more. B, Percentage of APPMV-YFP puncta moving before and immediately after stretch, and respective kymographs (n = 6). C, Representative images of neurites transfected with APP-YFP, fixed immediately after stretch, labeled with antibodies against APP and Aβ42, and quantitative analysis of Aβ42-puncta number and puncta intensity compared with control (puncta number: *p = 0.041; puncta intensity: *p = 0.0436). Data are mean ± SEM (A,B) and median and quartiles (C). Data were compared by unpaired Student's t test. Scale bars: A, B, 10 µm. Scale C, 5 µm.
APP axonal transport in neurons exposed to β- and γ-secretase inhibitors. Quantitative analysis of A) the percentage of APP-YFP puncta moving anterogradely and retrogradely, and B) APP-YFP particle density prestretch in neurons exposed to βsi-IV (40 µM), CE (5 µM) and vehicle (DMSO 0.01%) during 1 h (n = 4 or more). Data are mean ± SEM; means were compared by unpaired Student's t test. Download Figure 4-1, TIF file (1.7MB, tif) .
To further evaluate this possibility, we assessed axonal transport of a previously reported APP mutant (APPM596V) that abrogates BACE1-dependent cleavage of APP (Citron et al., 1995; Rodrigues et al., 2012). To avoid confounding effects caused by the presence of endogenous WT APP, we transiently transfected APPM596V-YFP (APPMV) into APP-KO neurons derived from genome-edited hiPSC lacking APP (Fong et al., 2018). As was also the case for secretase inhibition, axonal anterograde transport of APPMV-YFP puncta was not immediately disrupted by stretch (Fig. 4B; Movie 9). In support of amyloidogenic cleavage of APP occurring immediately after stretch, we observed that Aβ42 puncta number and intensity were increased in stretched compared with control axons (Fig. 4C), suggesting that stretch indeed actively stimulates the new generation of Aβ42 peptide in axons.
Together, our results demonstrate that APP axonal transport impairment is a direct and immediate consequence of stretch-induced amyloidogenic cleavage and that prestretch suppression of APP processing, either pharmacologically or genetically, prevents APP axonal transport defects caused by stretch.
Inhibition of APP amyloidogenic processing before stretch abrogates the formation of APP axonal accumulations
We demonstrated that stretch-induced defects in APP axonal transport can be abrogated by inhibiting APP amyloidogenic processing before stretch (Fig. 4A). Thus, it follows that by abrogating APP processing before stretch could also influence the formation of aberrant axonal APP accumulations after stretch. To test this hypothesis, we stretched neurons expressing APP-YFP in the presence of the γ-secretase inhibitor CE. Twenty-four hours after stretch, we found that APP-YFP density (puncta/µm) was lower in stretched neurons pre-exposed to CE than stretched neurons pre-exposed to DMSO (vehicle control), suggesting that exposure to CE abrogated the formation of APP accumulations (Fig. 5A). Corroborating these data, we also observed an increase in the percentage of APP-YFP puncta moving in the anterograde direction 24 h after stretch in axons pre-exposed to CE compared with stretched axons pre-exposed to vehicle control (Fig. 5A; Movie 10). Moreover, APPMV-YFP (i.e., unprocessed APP) did not show axonal transport defects or accumulation formation (i.e., increased puncta density) at 24 h after stretch (Fig. 5B; Movie 11).Cumulatively, our data demonstrate that APP processing is required for the formation of stretch-induced APP axonal accumulations and support the view that stretch-induced APP axonal transport defects precede formation of APP axonal accumulation.
Figure 5.

Reduced APP processing before stretch abrogates stretch-induced formation of APP axonal accumulations. A, Representative images and quantitative analysis of APP-YFP axonal transport 24 h after stretch, in axons exposed to CE (5 μm) or vehicle (DMSO 0.01%) during 1 h before being stretched (*p = 0.0251 for anterograde and *p = 0.0491 for density, both compared with DMSO) (n = 9 or more). B, Quantitative analysis of APPMV-YFP puncta movement and density 24 h after stretch (n = 5). Arrows indicate accumulated puncta along stretched axons in A. Data are mean ± SEM; means were compared by unpaired Student's t test. Scale bars: A, 10 μm.
AD-protective APPIce genetic variant prevents stretch-induced Aβ generation
The APPA673T genetic variant, also known as Icelandic (APPIce), is the first associated with a reduced risk to develop AD when carried in heterozygosity (Jonsson et al., 2012). Molecularly, it has been proposed that this protective variant reduces amyloidogenic BACE1-dependent processing of APP (Citron et al., 1995; Jonsson et al., 2012; Rodrigues et al., 2012; Benilova et al., 2014; Maloney et al., 2014). Thus, we tested the possibility that APPIce variant may also attenuate stretch-induced Aβ generation. To that end, we generated an APPIce//WT hiPSC using CRISPR/Cas9 genome editing technology (Fig. 6A). We confirmed that expression of a single copy of APPIce in neurons is sufficient to lower the basal levels of extracellular Aβ accumulated over time compared with control neurons (APPWT/WT) (Fig. 6B). Under our stretch paradigm, APPIce/WTneurons failed to accumulate Aβ 24 h after stretch compared with APPWT/WT (Fig. 6C). In contrast, both APPWT/WT and APPIce/WT were able to similarly stimulate Aβ production after exposure to GGPP, a previously reported stimulant of amyloidogenesis (Zhou et al., 2003; Kukar et al., 2005) (Extended Data Fig. 6-1), indicating that failure to stimulate Aβ after stretch in APPIce/WT was not related to a lack of inherent capacity to produce Aβ by these genetically engineered neurons.
Figure 6.

APP Icelandic variant abrogates stretch-induced increase in Aβ generation. A, Sequencing of genome-edited single-cell derived hiPSC lines confirms the generation of isogenic hiPSC line carrying the APP Icelandic mutation in heterozygosis. B, Quantitative analysis of extracellular Aβ40, Aβ42, and resultant Aβ42/Aβ40 ratio, accumulated over 7 d of culture in APPWT/Ice neurons compared with APPWT/WT neurons. Aβ levels are shown as relative to total tau levels in matched cell lysates (**p = 0.0048 for Aβ40; **p = 0.0033 for Aβ42) (n = 9). C, Schematic diagram illustrating the experimental design used for the quantification of extracellular Aβ levels in APPWT/Ice and APPWT/WT neurons 24 h after stretch (APPWT/WT, *p = 0.0418 for Aβ40; and *p = 0.0278 for Aβ42) (n = 7 or more). Exposure to GGPP increases Aβ generation in APPWT/Ice neurons (Extended Data Fig. 6-1). D, Quantitative analysis of the APPIce-GFP movement parameters in axons immediately after stretch (n = 5). Data are presented relative to pre-stretch condition. Data are mean ± SEM; means were compared by unpaired Student's t test. Scale D, 10 µm.
Exposure to GGPP increase Aβ generation in APPWT/Ice neurons. A) Extracellular Aβ40 and Aβ42 levels in APP WT/WT and APPWT/Ice neurons exposed to vehicle (Methanol 0.015% (Sigma)) or GGPP (15 µg/ml) during 24 h (APP WT/WT ***p = 0.001 for Aβ40 and *p = 0.0136 for Aβ42, and APPWT/Ice ***p = 0.001 for Aβ40 and *p = 0.0102 for Aβ42 (n = 4). Data are mean ± SEM; means were compared by unpaired Student's t test. Download Figure 6-1, TIF file (1.6MB, tif) .
Next, we tested whether stretch-induced APP axonal transport defects could also be attenuated by APPIce. To test this hypothesis, we transiently expressed APPIce-GFP (Das et al., 2016) in APP-KO neurons and measured APP transport dynamics before and immediately after stretch. In support of our hypothesis, APPIce-GFP was not significantly affected after stretch (Fig. 6D; Movie 12). In sum, consistent with protective impacts of pharmacological and genetic inhibition of APP processing on axonal transport and amyloid production (Figs. 2–5), these findings suggest that stretch-induced amyloidogenesis and APP axonal transport defects are blunted by a naturally occurring AD protective APP variant.
Discussion
In this study, to probe mechanisms potentially linking mTBI and increased risk for AD, we developed a new experimental approach to simulate mild stretch injury to human neurons. We demonstrated that sublethal stretch induced immediate amyloidogenic cleavage of APP. This cleavage directly results in impaired APP axonal transport and subsequently triggers the formation of axonal APP accumulations, the emergence of which correlated with elevated levels of extracellular Aβ after stretch. Pharmacological or genetic manipulation of APP amyloidogenic processing pathways before stretch effectively prevented these adverse stretch-associated outcomes. Consistent with these findings, the Icelandic AD protective variant of APP (APPA673T), which is less prone to amyloidogenic cleavage, also prevented stretch-induced amyloidogenesis and consequent APP axonal transport defects. These findings place APP amyloidogenic cleavage as an immediate upstream regulator of APP transport impairment and aberrant accumulations following mild neuronal injury (Fig. 7).
Figure 7.
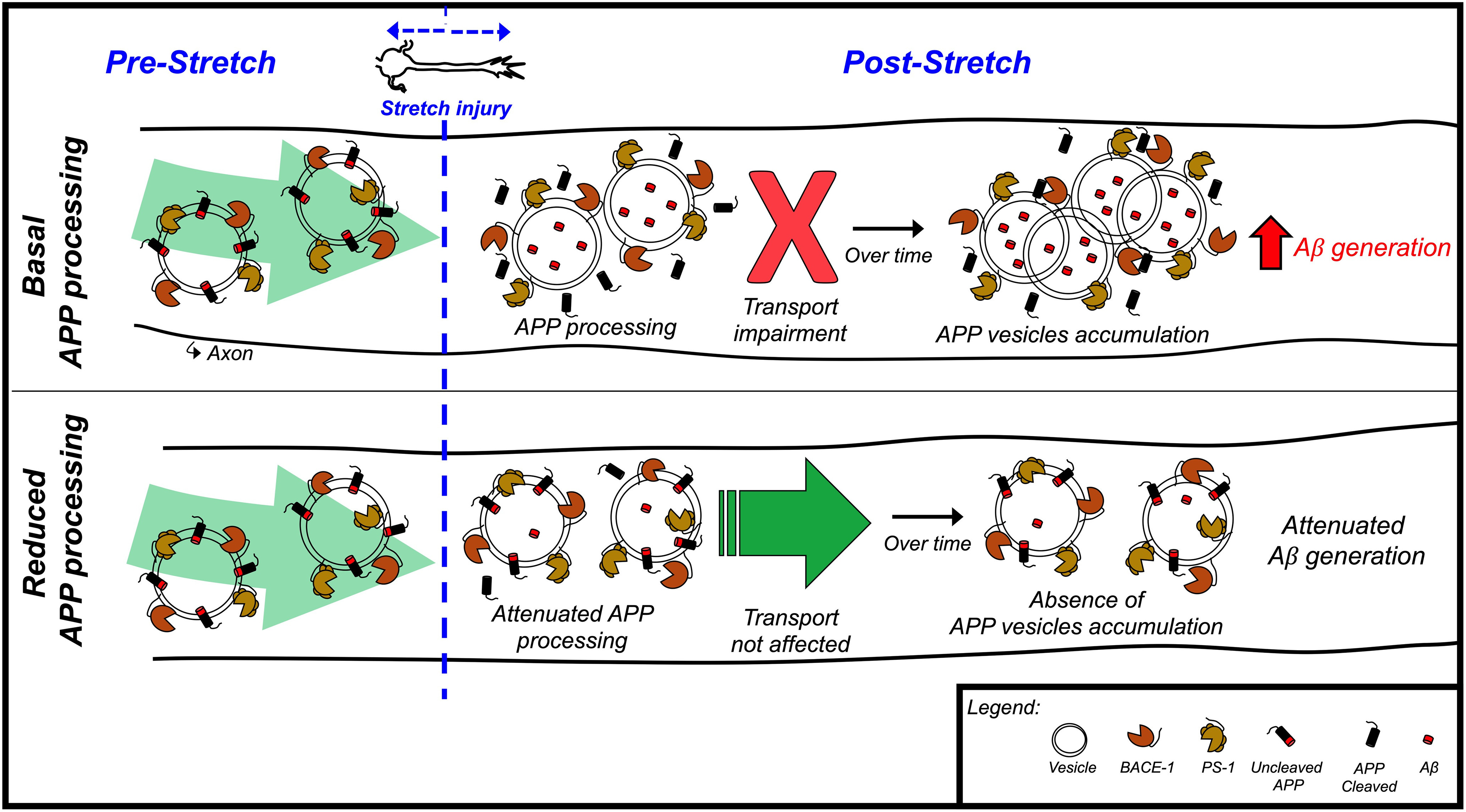
Schematic diagram illustrating the role of APP amyloidogenic cleavage as an immediate upstream regulator of APP transport impairment and aberrant accumulations following neuronal stretch.
Stretch impairs APP axonal transport. Movie showing APP-YFP puncta movement in a transfected axon before and after stretch (same axons as shown in Fig. 3B). Time is shown in milliseconds. Scale bar, 10 μm.
APP axonal transport is not affected in axons oriented perpendicular to the direction of stretch. Movie showing APP-YFP puncta movement in a transfected axon, oriented perpendicular to the stretch direction, after stretch (same axons as shown in Fig. 3D). Time is shown in milliseconds. Scale bar, 10 μm.
Lysosome axonal transport is not significantly altered after stretch. Movie showing LAMP-2-GFP puncta moving through axonal waves (white arrows) immediately after stretch. Time is shown in milliseconds. Scale bar, 10 μm.
APP axonal transport 3 h after stretch. Movie showing APP-YFP puncta movement in control and stretched axons 3 h after stretch (same axons as shown in Fig. 3E). Time is shown in milliseconds. Scale bar, 10 μm.
APP axonal transport 24 h after stretch. Movie showing APP-YFP puncta movement in control and stretched axons 24 h after stretch (same axons as shown in Fig. 3E). Time is shown in milliseconds. Scale bar, 10 μm.
APP axonal transport in stretched neurons exposed to vehicle. Movie showing APP-YFP puncta movement before and after stretch in neurons exposed to vehicle (DMSO) during 1 h (same axons as shown in Fig. 4A). Time is shown in milliseconds. Scale bar, 10 μm.
APP axonal transport in stretched neurons exposed to CE. Movie showing APP-YFP puncta movement before and after stretch in neurons exposed to CE during 1 h (same axons as shown in Fig. 4A). Time is shown in milliseconds. Scale bar, 10 μm.
APP axonal transport in stretched neurons exposed to βsi-IV. Movie showing APP-YFP puncta movement before and after stretch in neurons exposed to βsi-IV during 1 h (same axons as shown in Fig. 4A). Time is shown in milliseconds. Scale bar, 10 μm.
APPMV axonal transport after stretch in isogenic APP-KO neurons. Movie showing APPMV-YFP puncta movement in APP-KO-transfected axons before and after stretch (same axons as shown in Fig. 4B). Time is shown in milliseconds. Scale bar, 10 μm.
APP axonal transport 24 h after stretch in neurons exposed to CE. Movie showing APP-YFP axonal transport 24 h after stretch, in axons exposed to CE during 1 h before being stretched (same axons as shown in Fig. 5A). Time is shown in milliseconds. Scale bar, 10 μm.
APPMV axonal transport 24 h after stretch in isogenic APP-KO neurons. Movie showing APPMV-YFP puncta movement in APP-KO-transfected axons 24 h after stretch. Time is shown in milliseconds. Scale bar, 10 μm.
APPIce axonal transport is not affected in stretched axons. Movie showing APPIce-GFP puncta movement in a transfected axon, before and immediately after stretch (kymograph shown in Fig. 6D). Time is shown in milliseconds. Scale bar, 10 μm.
A new experimental platform to examine early human neuronal response to sublethal mechanical-induced stretch
Investigation of the earliest pathophysiological events occurring following neuronal injury is imperative to understand mechanisms underlying mTBI-induced neurodegeneration. Our device, in comparison to others, uniquely enabled us to visualize early phases of the morphologic and intracellular response to stretch in live, adherent cells at high spatial and temporal resolution, using brightfield and fluorescence microscopy (Geddes et al., 2003; Patel et al., 2012, 2014; Di Pietro et al., 2013; Nakadate et al., 2014, 2017; Aomura et al., 2016; Gangoda et al., 2018; Rosas-Hernandez et al., 2018). This capability was driven by the optical compatibility of the thin PDMS-based substrate with high-magnification and high-resolution immersion objectives, in combination with rapid, vacuum-driven deformation of the thin-film substrate. We integrated our system with validated hiPSC models, allowing evaluation of outcomes in the axons of networked human neurons, free of non-neuronal or systemic influences. Our observation of axonal waviness, indicative of rapid stretch and release, has been observed in numerous postmortem and in vitro studies after TBI, suggesting stretch as a common feature in TBI pathology (Peerless and Rewcastle, 1967; Blumbergs et al., 1989; Gentleman et al., 1995; Niess et al., 2002; Tang-Schomer et al., 2010; Dollé et al., 2014). At higher strains, axons stretched in vitro typically returned to their original morphology within ∼40 min after stretch, if at all (D. H. Smith et al., 1999; Tang-Schomer et al., 2010; Dollé et al., 2013). In our system, at lower strains, waviness was rapidly reversed within 5 min after stretch. This reversibility is consistent with less severe stretch and a functional cytoskeleton. Indeed, unlike previous high-strain models, we did not see any apparent breaks in the continuity of microtubules or neurofilaments networks nor did we observe the formation of axonal swellings after injury (Tang-Schomer et al., 2010, 2012; Dollé et al., 2014). Moreover, we did not detect the activation of early axonal stress-induced response pathways in stretched neurons (Middlemas et al., 2003; Cavalli et al., 2005; Falzone et al., 2009; Yoshimura et al., 2011; Tran et al., 2012; Liu et al., 2018). These observations reinforce the validity of our system to study AD-related outcomes in neurons, free of other cellular or systemic influences, following a rapid, mild stretch, simulating those strains experienced by neurons during mTBI (Kimpara and Iwamoto, 2012; Sahoo et al., 2016).
Influence of sublethal stretch on APP amyloidogenic processing and axonal transport pathways
APP processing and Aβ generation are essential for normal physiological function (von Koch et al., 1997; Steinbach et al., 1998; Kamenetz et al., 2003; Plant et al., 2003; van der Kant and Goldstein, 2015). However, aberrant amyloidogenic processing is a response to generalized neuronal stress, and a key factor underlying plaque formation in AD (Masters et al., 1985; G. J. Chen et al., 2003; Almenar-Queralt et al., 2014). Altered amyloidogenesis is thus also posited to be a key outcome that distinguishes TBI-associated AD from other neurodegenerative sequelae. Although some have challenged this idea (Brody et al., 2008; Schwetye et al., 2010), rapid increases in amyloidogenesis have been frequently reported following TBI, regardless of severity (D. H. Smith et al., 1998; Emmerling et al., 2000; X. H. Chen et al., 2004; Abrahamson et al., 2006, 2009; Loane et al., 2009; Gatson et al., 2013; Marklund et al., 2014; Estrada-Rojo et al., 2018). Whether amyloidogenesis can result directly from mild neuronal stretch has not been explored. We observed the generation of Aβ42 peptide in axons immediately after stretch, as well as increased levels of accumulated extracellular Aβ within 24 h of stretch. That APP processing was enhanced, as opposed to increased secretion of existing Aβ after stretch or increased expression of APP, was supported by the following: (1) the lack of detectable increases in levels of APP and its cleaving enzymes after stretch; (2) an increase in stretch-induced Aβ after “zeroing-out” pre-accumulated Aβ by reversibly inhibiting γ-secretase before stretch; and (3) suppressed Aβ generation when neurons were exposed to APP secretase inhibitors before stretch. Together, to our knowledge, these findings provide the first direct evidence demonstrating that rapid sublethal neuronal stretch stimulates Aβ generation. The idea that cell deformation activates amyloidogenic cleavage of APP is supported by recent evidence that pulsatile stretch of brain vascular endothelial cells increases amyloidogenic cleavage (Gangoda et al., 2018).
It has been hypothesized that APP axonal transport defects can trigger amyloidogenic cleavage of APP following TBI, based on observations that APP accumulations colocalize with Aβ, β-, and γ-secretases, and stalled kinesin (Uryu et al., 2007; X. H. Chen et al., 2009). This idea places impaired axonal transport of APP as a key mechanistic link between neuronal deformation and Aβ generation (and by extension, TBI and AD). However, this hypothesis has not been directly tested, in large part because of limitations of existing model systems. So, we are left with fundamental knowledge gaps: (1) whether and to what extent rapid stretch can impair APP transport; (2) what the consequences are of a potential stretch-induced APP transport impairment; and (3) what the underlying causes are of any observed stretch-induced APP transport impairment. Our data provide insight into several of these gaps.
First, our data provide direct quantitative evidence that stretch immediately and robustly inhibits APP transport. This finding is novel. Importantly, axonal transport defects occurred only in adherent axons positioned in the direction of stretch, but not perpendicular to the direction of stretch (i.e., in unstretched neurons), confirming a mechanical influence on acute transport impairment. Unlike previous studies, in which microtubules were damaged at high strains (Tang-Schomer et al., 2010, 2012; Dollé et al., 2014), the fact that stretch-induced axonal transport defects did not disrupt the transport of all rapidly transported vesicular cargoes supports immunofluorescence-based observations that the cytoskeletal/microtubule lattice was not severely disrupted under mild axonal stretch.
Second, our analyses support the view that APP accumulations are triggered by impaired axonal transport. Indeed, we observed increased axonal density of APP(-YFP) within 3 h after stretch, and both kymography and immunocytochemistry assays indicated persistent accumulations 24 h after stretch. These may represent precursors or milder versions of APP-filled swellings observed pathologically following more severe TBI or more severely stretched neurons in vitro, which suggest sustained disruption of transport (D. H. Smith et al., 1999; Uryu et al., 2007; X. H. Chen et al., 2009). The development of accumulations is additionally supported by potential contributions of APP vesicles that continued to move after stretch, but with marked reductions in run length and velocity (Johnson et al., 2010). Stretch-induced transport defects may also enhance Aβ generation because of increased APP processing at sites of accumulation (D. H. Smith et al., 1999; Uryu et al., 2007; X. H. Chen et al., 2009); this consequence is consistent with detection of Aβ42, APPC99, BACE-1, and PS-1 in APP-positive accumulations.
Third, data from this study provided novel and unexpected insight into mechanisms underlying transport impairment. In contrast to predictions from more severe injuries, cytoskeletal disruption does not appear to explain immediate transport disruption following milder injury. On the other hand, given considerable prior evidence that APP processing can influence axonal transport (Kamal et al., 2001; Rodrigues et al., 2012; Almenar-Queralt et al., 2014), we deployed a number of complementary pharmacological and genetic approaches to test the hypothesis that reducing APP processing before stretch could prevent stretch-induced axonal transport defects and subsequent amyloidogenesis. Outcomes from each of several tested perturbations aligned with this prediction: (1) stretch-induced immediate amyloidogenic processing of APP; (2) stretch-induced transport defects, including the formation of axonal APP accumulations, were prevented by exposing neurons acutely to β- and γ-secretase inhibitors before imposing stretch; and (3) similar suppression of transport impairment and APP accumulation formation were observed with expression of APPMV and APPIce genetic variants (each abrogating β-secretase cleavage through different mechanisms). Together, our data demonstrate that amyloidogenic processing of APP is directly responsible for stretch-induced APP axonal transport defects. Further, by reducing APP processing before stretch, APP axonal transport may be shielded from stretch-induced impairment; this could in turn mitigate further Aβ generation in stretched neurons. These conclusions have substantial translational implications for suppressing stretch-associated transport defects and amyloid production, as it follows that intervention at any stage in this cascade could influence longer-term impacts. For example, pharmacological inhibition of APP processing pathways may represent a prophylactic or acute treatment strategy for mild neuronal injury. Indeed, blocking either β- or γ-secretase can ameliorate motor and cognitive deficits and reduce cell loss after experimental TBI in mice (Loane et al., 2009). In addition, the protective capability of the naturally occurring APP-Icelandic variant as well as engineered reduction of APP processing through the APPMV mutation raise the possibility that an individual's genomic profile could predict their early symptomatic response to mild injury as well as their eventual propensity to develop AD after TBI.
In conclusion, our findings validate a new system to study mechanisms underlying neurologic dysfunction following mild injury of human neurons. We demonstrated that sublethal neuronal stretch, simulating deformation levels experienced by neurons during mTBI, is sufficient to induce AD-related phenotypes. In particular, APP axonal transport and APP amyloidogenic processing pathways are adversely impacted by stretch. Further, given the dependence of transport impairment on APP amyloidogenic processing as well as the widely hypothesized dependence of APP accumulation and amyloidogenic processing on axonal transport, our data support the idea that these pathways interact in a feedforward manner, perturbing amyloidogenic homeostasis and physiological transport. Such a model may contribute to understanding of the emergence of early symptoms and the susceptibility for development of AD following mTBI.
Footnotes
This work was supported by U.S. Department of Defense Peer Reviewed Alzheimer's Research Program W81XWH-19-1-0315 and W81XWH-15-1-0561 to S.B.S. and A.A.-Q.; and National Institutes of Health/National Institute on Aging 1RF1AG048083-01 and 5P50AG005131-34 to L.S.B.G. We thank Dr. Ivan Garcia-Bassets, Dr. Rik Van der Kant, Dr. Utpal Das, and members of the L.S.B.G. laboratory for helpful comments on data interpretation and presentation; and Dr. Elsa Molina and University of California-San Diego Stem Cell Genomics Core for the technical assistance using the Nanostring nCounter platform (supported by a CIRM Major Facilities Grant FA1-00607 to the Sanford Consortium for Regenerative Medicine).
The authors declare no competing financial interests.
References
- Abdul-Muneer PM, Long M, Conte AA, Santhakumar V, Pfister BJ (2017) High Ca2+ influx during traumatic brain injury leads to caspase-1-dependent neuroinflammation and cell death. Mol Neurobiol 54:3964–3975. 10.1007/s12035-016-9949-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abrahamson EE, Ikonomovic MD, Ciallella JR, Hope CE, Paljug WR, Isanski BA, Flood DG, Clark RS, DeKosky ST (2006) Caspase inhibition therapy abolishes brain trauma-induced increases in Aβ peptide: implications for clinical outcome. Exp Neurol 197:437–450. 10.1016/j.expneurol.2005.10.011 [DOI] [PubMed] [Google Scholar]
- Abrahamson EE, Ikonomovic MD, Dixon CE, DeKosky ST (2009) Simvastatin therapy prevents brain trauma-induced increases in β-amyloid peptide levels. Ann Neurol 66:407–414. 10.1002/ana.21731 [DOI] [PubMed] [Google Scholar]
- Abu Hamdeh S, Waara ER, Moller C, Soderberg L, Basun H, Alafuzoff I, Hillered L, Lannfelt L, Ingelsson M, Marklund N (2018) Rapid amyloid-beta oligomer and protofibril accumulation in traumatic brain injury. Brain Pathol 28:451–462. 10.1111/bpa.12532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almenar-Queralt A, Falzone TL, Shen Z, Lillo C, Killian RL, Arreola AS, Niederst ED, Ng KS, Kim SN, Briggs SP, Williams DS, Goldstein LS (2014) UV irradiation accelerates amyloid precursor protein (APP) processing and disrupts APP axonal transport. J Neurosci 34:3320–3339. 10.1523/JNEUROSCI.1503-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson KJ, Scheff SW, Miller KM, Roberts KN, Gilmer LK, Yang C, Shaw G (2008) The phosphorylated axonal form of the neurofilament subunit NF-H (pNF-H) as a blood biomarker of traumatic brain injury. J Neurotrauma 25:1079–1085. 10.1089/neu.2007.0488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aomura S, Nakadate H, Kaneko Y, Nishimura A, Willinger R (2016) Stretch-induced functional disorder of axonal transport in the cultured rat cortex neuron. Integr Mol Med 3:654–660. 10.15761/IMM.1000218 [DOI] [Google Scholar]
- Bahrami S, Drabløs F (2016) Gene regulation in the immediate-early response process. Adv Biol Regul 62:37–49. 10.1016/j.jbior.2016.05.001 [DOI] [PubMed] [Google Scholar]
- Barnes DE, Byers AL, Gardner RC, Seal KH, Boscardin WJ, Yaffe K (2018) Association of mild traumatic brain injury with and without loss of consciousness with dementia in US military veterans. JAMA Neurol 75:1055–1061. 10.1001/jamaneurol.2018.0815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barreto-Chang OL, Dolmetsch RE (2009) Calcium imaging of cortical neurons using Fura-2 AM. J Vis Exp 23:1067. 10.3791/1067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazarian JJ, Cernak I, Noble-Haeusslein L, Potolicchio S, Temkin N (2009) Long-term neurologic outcomes after traumatic brain injury. J Head Trauma Rehabil 24:439–451. 10.1097/HTR.0b013e3181c15600 [DOI] [PubMed] [Google Scholar]
- Benilova I, Gallardo R, Ungureanu AA, Castillo Cano V, Snellinx A, Ramakers M, Bartic C, Rousseau F, Schymkowitz J, De Strooper B (2014) The Alzheimer disease protective mutation A2t modulates kinetic and thermodynamic properties of amyloid-β (Aβ) aggregation. J Biol Chem 289:30977–30989. 10.1074/jbc.M114.599027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besirli CG, Johnson EM (2003) JNK-independent activation of c-Jun during neuronal apoptosis induced by multiple DNA-damaging agents. J Biol Chem 278:22357–22366. 10.1074/jbc.M300742200 [DOI] [PubMed] [Google Scholar]
- Bianchi F, Pereno V, George JH, Thompson MS, Ye H (2019) Membrane mechanical properties regulate the effect of strain on spontaneous electrophysiology in human iPSC-derived neurons. Neuroscience 404:165–174. 10.1016/j.neuroscience.2019.02.014 [DOI] [PubMed] [Google Scholar]
- Blasko I, Beer R, Bigl M, Apelt J, Franz G, Rudzki D, Ransmayr G, Kampfl A, Schliebs R (2004) Experimental traumatic brain injury in rats stimulates the expression, production and activity of Alzheimer's disease β-secretase (BACE-1). J Neural Transm 111:523–536. 10.1007/s00702-003-0095-6 [DOI] [PubMed] [Google Scholar]
- Blumbergs PC, Jones NR, North JB (1989) Diffuse axonal injury in head trauma. J Neurol Neurosurg Psychiatry 52:838–841. 10.1136/jnnp.52.7.838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bober BG, Gutierrez E, Plaxe S, Groisman A, Shah SB (2015) Combinatorial influences of paclitaxel and strain on axonal transport. Exp Neurol 271:358–367. 10.1016/j.expneurol.2015.06.023 [DOI] [PubMed] [Google Scholar]
- Bramlett HM, Kraydieh S, Green EJ, Dietrich WD (1997) Temporal and regional patterns of axonal damage following traumatic brain injury: a beta-amyloid precursor protein immunocytochemical study in rats. J Neuropathol Exp Neurol 56:1132–1141. 10.1097/00005072-199710000-00007 [DOI] [PubMed] [Google Scholar]
- Brody DL, Magnoni S, Schwetye KE, Spinner ML, Esparza TJ, Stocchetti N, Zipfel GJ, Holtzman DM (2008) Amyloid-beta dynamics correlate with neurological status in the injured human brain. Science 321:1221–1224. 10.1126/science.1161591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browne KD, Chen XH, Meaney DF, Smith DH (2011) Mild traumatic brain injury and diffuse axonal injury in swine. J Neurotrauma 28:1747–1755. 10.1089/neu.2011.1913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caceres A, Banker GA, Binder L (1986) Immunocytochemical localization of tubulin and microtubule-associated protein 2 during the development of hippocampal neurons in culture. J Neurosci 6:714–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll LJ, Cassidy JD, Peloso PM, Garritty C, Giles-Smith L (2004) Systematic search and review procedures: results of the WHO Collaborating Centre Task Force on Mild Traumatic Brain Injury. J Rehabil Med 36:11–14. 10.1080/16501960410023660 [DOI] [PubMed] [Google Scholar]
- Cartagena CM, Mountney A, Hwang H, Swiercz A, Rammelkamp Z, Boutte AM, Shear DA, Tortella FC, Schmid KE (2016) Subacute changes in cleavage processing of amyloid precursor protein and tau following penetrating traumatic brain injury. PLoS One 11:e0158576. 10.1371/journal.pone.0158576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalli V, Kujala P, Klumperman J, Goldstein LS (2005) Sunday Driver links axonal transport to damage signaling. J Cell Biol 168:775–787. 10.1083/jcb.200410136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen GJ, Xu J, Lahousse SA, Caggiano NL, de la Monte SM (2003) Transient hypoxia causes Alzheimer-type molecular and biochemical abnormalities in cortical neurons: potential strategies for neuroprotection 1. J Alzheimers Dis 5:209–228. 10.3233/jad-2003-5305 [DOI] [PubMed] [Google Scholar]
- Chen XH, Siman R, Iwata A, Meaney DF, Trojanowski JQ, Smith DH (2004) Long-term accumulation of Amyloid-β, β-secretase, Presenilin-1, and Caspase-3 in damaged axons following brain trauma. Am J Pathol 165:357–371. 10.1016/s0002-9440(10)63303-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen XH, Johnson VE, Uryu K, Trojanowski JQ, Smith DH (2009) A lack of amyloid beta plaques despite persistent accumulation of amyloid beta in axons of long-term survivors of traumatic brain injury. Brain Pathol 19:214–223. 10.1111/j.1750-3639.2008.00176.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho JH, Johnson GV (2004) Primed phosphorylation of tau at Thr231 by glycogen synthase kinase 3β (GSK3β) plays a critical role in regulating tau's ability to bind and stabilize microtubules. J Neurochem 88:349–358. 10.1111/j.1471-4159.2004.02155.x [DOI] [PubMed] [Google Scholar]
- Chung RS, Staal JA, McCormack GH, Dickson TC, Cozens MA, Chuckowree JA, Quilty MC, Vickers JC (2005) Mild axonal stretch injury in vitro induces a progressive series of neurofilament alterations ultimately leading to delayed axotomy. J Neurotrauma 22:1081–1091. 10.1089/neu.2005.22.1081 [DOI] [PubMed] [Google Scholar]
- Citron M, Teplow DB, Selkoe DJ (1995) Generation of amyloid β protein from its precursor is sequence specific. Neuron 14:661–670. 10.1016/0896-6273(95)90323-2 [DOI] [PubMed] [Google Scholar]
- Cullen DK, LaPlaca MC (2006) Neuronal response to high rate shear deformation depends on heterogeneity of the local strain field. J Neurotrauma 23:1304–1319. 10.1089/neu.2006.23.1304 [DOI] [PubMed] [Google Scholar]
- Das U, Wang L, Ganguly A, Saikia JM, Wagner SL, Koo EH, Roy S (2016) Visualizing APP and BACE-1 approximation in neurons yields insight into the amyloidogenic pathway. Nat Neurosci 19:55–64. 10.1038/nn.4188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeKosky ST, Blennow K, Ikonomovic MD, Gandy S (2013) Acute and chronic traumatic encephalopathies: pathogenesis and biomarkers. Nat Rev Neurol 9:192–200. 10.1038/nrneurol.2013.36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Pietro V, Amorini AM, Tavazzi B, Hovda DA, Signoretti S, Giza CC, Lazzarino G, Vagnozzi R, Lazzarino G, Belli A (2013) Potentially neuroprotective gene modulation in an in vitro model of mild traumatic brain injury. Mol Cell Biochem 375:185–198. 10.1007/s11010-012-1541-2 [DOI] [PubMed] [Google Scholar]
- Dollé JP, Morrison B 3rd, Schloss RS, Yarmush ML (2013) An organotypic uniaxial strain model using microfluidics. Lab Chip 13:432–442. 10.1039/c2lc41063j [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dollé JP, Morrison B, Schloss RS, Yarmush ML (2014) Brain-on-a-chip microsystem for investigating traumatic brain injury: axon diameter and mitochondrial membrane changes play a significant role in axonal response to strain injuries. Technology (Singap World Sci) 2:106. 10.1142/S2339547814500095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dollé JP, Jaye A, Anderson SA, Ahmadzadeh H, Shenoy VB, Smith DH (2018) Newfound sex differences in axonal structure underlie differential outcomes from in vitro traumatic axonal injury. Exp Neurol 300:121–134. 10.1016/j.expneurol.2017.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duman RS, Adams DH, Simen BB (2005) Transcription factors as modulators of stress responsivity. In: Techniques in the behavioral and neural sciences (Steckler T, Kalin NH, Reul JM, eds), pp 679–698. Amsterdam: Elsevier. [Google Scholar]
- Emmerling MR, Morganti-Kossmann MC, Kossmann T, Stahel PF, Watson MD, Evans LM, Mehta PD, Spiegel K, Kuo YM, Roher AE, Raby CA (2000) Traumatic brain injury elevates the Alzheimer's amyloid peptide A beta 42 in human CSF: a possible role for nerve cell injury. Ann NY Acad Sci 903:118–122. 10.1111/j.1749-6632.2000.tb06357.x [DOI] [PubMed] [Google Scholar]
- Estrada-Rojo F, Martinez-Tapia RJ, Estrada-Bernal F, Martinez-Vargas M, Perez-Arredondo A, Flores-Avalos L, Navarro L (2018) Models used in the study of traumatic brain injury. Rev Neurosci 29:139–149. [DOI] [PubMed] [Google Scholar]
- Falzone TL, Stokin GB, Lillo C, Rodrigues EM, Westerman EL, Williams DS, Goldstein LS (2009) Axonal stress kinase activation and tau misbehavior induced by kinesin-1 transport defects. J Neurosci 29:5758–5767. 10.1523/JNEUROSCI.0780-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fann JR, Ribe AR, Pedersen HS, Fenger-Grøn M, Christensen J, Benros ME, Vestergaard M (2018) Long-term risk of dementia among people with traumatic brain injury in Denmark: a population-based observational cohort study. Lancet Psychiatry 5:424–431. 10.1016/S2215-0366(18)30065-8 [DOI] [PubMed] [Google Scholar]
- Farley MM, Watkins TA (2018) Intrinsic neuronal stress response pathways in injury and disease. Annu Rev Pathol 13:93–116. 10.1146/annurev-pathol-012414-040354 [DOI] [PubMed] [Google Scholar]
- Ferguson SM (2018) Axonal transport and maturation of lysosomes. Curr Opin Neurobiol 51:45–51. 10.1016/j.conb.2018.02.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleminger S, Oliver DL, Lovestone S, Rabe-Hesketh S, Giora A (2003) Head injury as a risk factor for Alzheimer's disease: the evidence 10 years on; a partial replication. J Neurol Neurosurg Psychiatry 74:857–862. 10.1136/jnnp.74.7.857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong LK, Yang MM, dos Santos Chaves R, Reyna SM, Langness VF, Woodruff G, Roberts EA, Young JE, Goldstein LS (2018) Full-length amyloid precursor protein regulates lipoprotein metabolism and amyloid-β clearance in human astrocytes. J Biol Chem 293:11341–11357. 10.1074/jbc.RA117.000441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fosbrink M, Aye-Han NN, Cheong R, Levchenko A, Zhang J (2010) Visualization of JNK activity dynamics with a genetically encoded fluorescent biosensor. Proc Natl Acad Sci USA 107:5459–5464. 10.1073/pnas.0909671107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangoda SV, Avadhanam B, Jufri NF, Sohn EH, Butlin M, Gupta V, Chung R, Avolio AP (2018) Pulsatile stretch as a novel modulator of amyloid precursor protein processing and associated inflammatory markers in human cerebral endothelial cells. Sci Rep 8:1689. 10.1038/s41598-018-20117-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner RC, Yaffe K (2015) Epidemiology of mild traumatic brain injury and neurodegenerative disease. Mol Cell Neurosci 66:75–80. 10.1016/j.mcn.2015.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner RC, Burke JF, Nettiksimmons J, Kaup A, Barnes DE, Yaffe K (2014) Dementia risk after traumatic brain injury vs nonbrain trauma: the role of age and severity. JAMA Neurol 71:1490–1497. 10.1001/jamaneurol.2014.2668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatson JW, Warren V, Abdelfattah K, Wolf S, Hynan LS, Moore C, Diaz-Arrastia R, Minei JP, Madden C, Wigginton JG (2013) Detection of beta-amyloid oligomers as a predictor of neurological outcome after brain injury. J Neurosurg 118:1336–1342. 10.3171/2013.2.JNS121771 [DOI] [PubMed] [Google Scholar]
- Gavett BE, Stern RA, Cantu RC, Nowinski CJ, McKee AC (2010) Mild traumatic brain injury: a risk factor for neurodegeneration. Alzheimers Res Ther 2:18. 10.1186/alzrt42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geddes DM, Cargill RS, LaPlaca MC (2003) Mechanical stretch to neurons results in a strain rate and magnitude-dependent increase in plasma membrane permeability. J Neurotrauma 20:1039–1049. 10.1089/089771503770195885 [DOI] [PubMed] [Google Scholar]
- Gentleman SM, Roberts GW, Gennarelli TA, Maxwell WL, Adams JH, Kerr S, Graham DI (1995) Axonal injury: a universal consequence of fatal closed head injury? Acta Neuropathol 89:537–543. 10.1007/BF00571509 [DOI] [PubMed] [Google Scholar]
- Gore A, Li Z, Fung HL, Young JE, Agarwal S, Antosiewicz-Bourget J, Canto I, Giorgetti A, Israel MA, Kiskinis E, Lee JH, Loh YH, Manos PD, Montserrat N, Panopoulos AD, Ruiz S, Wilbert ML, Yu J, Kirkness EF, Izpisua Belmonte JC, et al. (2011) Somatic coding mutations in human induced pluripotent stem cells. Nature 471:63–67. 10.1038/nature09805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwald BD, Ambrose AF, Armstrong GP (2012) Mild brain injury. Rehabil Res Pract 2012:469475. 10.1155/2012/469475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer JE, Hånell A, McGinn MJ, Povlishock JT (2013) Mild traumatic brain injury in the mouse induces axotomy primarily within the axon initial segment. Acta Neuropathol 126:59–74. 10.1007/s00401-013-1119-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Z, Cupples LA, Kurz A, Auerbach SH, Volicer L, Chui H, Green RC, Sadovnick AD, Duara R, DeCarli C, Johnson K, Go RC, Growdon JH, Haines JL, Kukull WA, Farrer LA (2000) Head injury and the risk of AD in the MIRAGE study. Neurology 54:1316–1323. 10.1212/wnl.54.6.1316 [DOI] [PubMed] [Google Scholar]
- Hånell A, Greer JE, McGinn MJ, Povlishock JT (2015) Traumatic brain injury-induced axonal phenotypes react differently to treatment. Acta Neuropathol 129:317–332. 10.1007/s00401-014-1376-x [DOI] [PubMed] [Google Scholar]
- Hayashi T, Ago K, Ago M, Ogata M (2009) Two patterns of β-amyloid precursor protein (APP) immunoreactivity in cases of blunt head injury. Legal Med 11:S171–S173. 10.1016/j.legalmed.2009.01.076 [DOI] [PubMed] [Google Scholar]
- Hoshino S, Kobayashi S, Furukawa T, Asakura T, Teramoto A (2003) Multiple immunostaining methods to detect traumatic axonal injury in the rat fluid-percussion brain injury model. Neurol Med Chir (Tokyo) 43:165–174. 10.2176/nmc.43.165 [DOI] [PubMed] [Google Scholar]
- Ikonomovic MD, Uryu K, Abrahamson EE, Ciallella JR, Trojanowski JQ, Lee VM, Clark RS, Marion DW, Wisniewski SR, DeKosky ST (2004) Alzheimer's pathology in human temporal cortex surgically excised after severe brain injury. Exp Neurol 190:192–203. 10.1016/j.expneurol.2004.06.011 [DOI] [PubMed] [Google Scholar]
- Ippolito DM, Eroglu C (2010) Quantifying synapses: an immunocytochemistry-based assay to quantify synapse number. J Vis Exp 45:2270. 10.3791/2270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Israel MA, Yuan SH, Bardy C, Reyna SM, Mu Y, Herrera C, Hefferan MP, Van Gorp S, Nazor KL, Boscolo FS, Carson CT, Laurent LC, Marsala M, Gage FH, Remes AM, Koo EH, Goldstein LS (2012) Probing sporadic and familial Alzheimer's disease using induced pluripotent stem cells. Nature 482:216–220. 10.1038/nature10821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwata A, Chen XH, McIntosh TK, Browne KD, Smith DH (2002) Long-term accumulation of amyloid-β in axons following brain trauma without persistent upregulation of amyloid precursor protein genes. J Neuropathol Exp Neurol 61:1056–1068. 10.1093/jnen/61.12.1056 [DOI] [PubMed] [Google Scholar]
- Johnson VE, Stewart W, Smith DH (2010) Traumatic brain injury and amyloid-β pathology: a link to Alzheimer's disease? Nat Rev Neurosci 11:361–370. 10.1038/nrn2808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson VE, Stewart W, Smith DH (2012) Widespread tau and amyloid-beta pathology many years after a single traumatic brain injury in humans. Brain Pathol 22:142–149. 10.1111/j.1750-3639.2011.00513.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson VE, Stewart W, Smith DH (2013) Axonal pathology in traumatic brain injury. Exp Neurol 246:35–43. 10.1016/j.expneurol.2012.01.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson VE, Stewart W, Weber MT, Cullen DK, Siman R, Smith DH (2016) SNTF immunostaining reveals previously undetected axonal pathology in traumatic brain injury. Acta Neuropathol 131:115–135. 10.1007/s00401-015-1506-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsson T, Atwal JK, Steinberg S, Snaedal J, Jonsson PV, Bjornsson S, Stefansson H, Sulem P, Gudbjartsson D, Maloney J, Hoyte K, Gustafson A, Liu Y, Lu Y, Bhangale T, Graham RR, Huttenlocher J, Bjornsdottir G, Andreassen OA, Jönsson EG, et al. (2012) A mutation in APP protects against Alzheimer's disease and age-related cognitive decline. Nature 488:96–99. 10.1038/nature11283 [DOI] [PubMed] [Google Scholar]
- Kaether C, Skehel P, Dotti CG, Lippincott-Schwartz J (2000) Axonal membrane proteins are transported in distinct carriers: a two-color video microscopy study in cultured hippocampal neurons. Mol Biol Cell 11:1213–1224. 10.1091/mbc.11.4.1213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamal A, Almenar-Queralt A, LeBlanc JF, Roberts EA, Goldstein LS (2001) Kinesin-mediated axonal transport of a membrane compartment containing β-secretase and presenilin-1 requires APP. Nature 414:643–648. 10.1038/414643a [DOI] [PubMed] [Google Scholar]
- Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, Sisodia S, Malinow R (2003) APP processing and synaptic function. Neuron 37:925–937. 10.1016/s0896-6273(03)00124-7 [DOI] [PubMed] [Google Scholar]
- Kimpara H, Iwamoto M (2012) Mild traumatic brain injury predictors based on angular accelerations during impacts. Ann Biomed Eng 40:114–126. 10.1007/s10439-011-0414-2 [DOI] [PubMed] [Google Scholar]
- Kiraly M, Kiraly SJ (2007) Traumatic brain injury and delayed sequelae: a review–traumatic brain injury and mild traumatic brain injury (concussion) are precursors to later-onset brain disorders, including early-onset dementia. Sci World J 7:1768–1776. 10.1100/tsw.2007.269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kukar T, Murphy MP, Eriksen JL, Sagi SA, Weggen S, Smith TE, Ladd T, Khan MA, Kache R, Beard J, Dodson M, Merit S, Ozols VV, Anastasiadis PZ, Das P, Fauq A, Koo EH, Golde TE (2005) Diverse compounds mimic Alzheimer disease–causing mutations by augmenting Aβ42 production. Nat Med 11:545–550. 10.1038/nm1235 [DOI] [PubMed] [Google Scholar]
- Kumar P, Nagarajan A, Uchil PD (2018) Analysis of cell viability by the lactate dehydrogenase assay. Cold Spring Harb Protoc 2018:pdb.prot095497. 10.1101/pdb.prot095497 [DOI] [PubMed] [Google Scholar]
- LaPlaca MC, Cullen DK, McLoughlin JJ, Cargill RS 2nd (2005) High rate shear strain of three-dimensional neural cell cultures: a new in vitro traumatic brain injury model. J Biomech 38:1093–1105. 10.1016/j.jbiomech.2004.05.032 [DOI] [PubMed] [Google Scholar]
- Levy Nogueira M, Hamraz M, Abolhassani M, Bigan E, Lafitte O, Steyaert JM, Dubois B, Schwartz L (2018) Mechanical stress increases brain amyloid β, tau, and α-synuclein concentrations in wild-type mice. Alzheimers Dement 14:444–453. 10.1016/j.jalz.2017.11.003 [DOI] [PubMed] [Google Scholar]
- Li Y, Zhang L, Kallakuri S, Cohen A, Cavanaugh JM (2015) Correlation of mechanical impact responses and biomarker levels: a new model for biomarker evaluation in TBI. J Neurol Sci 359:280–286. 10.1016/j.jns.2015.08.035 [DOI] [PubMed] [Google Scholar]
- Li Y, Li C, Gan C, Zhao K, Chen J, Song J, Lei T (2019) A precise, controllable in vitro model for diffuse axonal injury through uniaxial stretch injury. Front Neurosci 13:1063. 10.3389/fnins.2019.01063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Zhang CW, Zhou Y, Wong WQ, Lee LC, Ong WY, Yoon SO, Hong W, Fu XY, Soong TW, Koo EH, Stanton LW, Lim KL, Xiao ZC, Dawe GS (2018) APP upregulation contributes to retinal ganglion cell degeneration via JNK3. Cell Death Differ 25:663–676. 10.1038/s41418-017-0005-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loane DJ, Pocivavsek A, Moussa CE, Thompson R, Matsuoka Y, Faden AI, Rebeck GW, Burns MP (2009) Amyloid precursor protein secretases as therapeutic targets for traumatic brain injury. Nat Med 15:377–379. 10.1038/nm.1940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loane DJ, Washington PM, Vardanian L, Pocivavsek A, Hoe HS, Duff KE, Cernak I, Rebeck GW, Faden AI, Burns MP (2011) Modulation of ABCA1 by an LXR agonist reduces beta-amyloid levels and improves outcome after traumatic brain injury. J Neurotrauma 28:225–236. 10.1089/neu.2010.1595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou D, Du Y, Huang D, Cai F, Zhang Y, Li T, Zhou W, Gao H, Song W (2018) Traumatic brain injury alters the metabolism and facilitates Alzheimer's disease in a murine model. Mol Neurobiol 55:4928–4939. 10.1007/s12035-017-0687-z [DOI] [PubMed] [Google Scholar]
- Lukinavičius G, Reymond L, D'Este E, Masharina A, Göttfert F, Ta H, Güther A, Fournier M, Rizzo S, Waldmann H, Blaukopf C, Sommer C, Gerlich DW, Arndt HD, Hell SW, Johnsson K (2014) Fluorogenic probes for live-cell imaging of the cytoskeleton. Nat Methods 11:731–733. 10.1038/nmeth.2972 [DOI] [PubMed] [Google Scholar]
- Maas AI, Stocchetti N, Bullock R (2008) Moderate and severe traumatic brain injury in adults. Lancet Neurol 7:728–741. 10.1016/S1474-4422(08)70164-9 [DOI] [PubMed] [Google Scholar]
- Maloney JA, Bainbridge T, Gustafson A, Zhang S, Kyauk R, Steiner P, van der Brug M, Liu Y, Ernst JA, Watts RJ, Atwal JK (2014) Molecular mechanisms of Alzheimer disease protection by the a673t allele of amyloid precursor protein. J Biol Chem 289:30990–31000. 10.1074/jbc.M114.589069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannix RC, Zhang J, Berglass J, Qui J, Whalen MJ (2013) Beneficial effect of amyloid beta after controlled cortical impact. Brain Inj 27:743–748. 10.3109/02699052.2013.771797 [DOI] [PubMed] [Google Scholar]
- Marklund N, Farrokhnia N, Hanell A, Vanmechelen E, Enblad P, Zetterberg H, Blennow K, Hillered L (2014) Monitoring of beta-amyloid dynamics after human traumatic brain injury. J Neurotrauma 31:42–55. 10.1089/neu.2013.2964 [DOI] [PubMed] [Google Scholar]
- Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K (1985) Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci USA 82:4245–4249. 10.1073/pnas.82.12.4245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middlemas A, Delcroix JD, Sayers NM, Tomlinson DR, Fernyhough P (2003) Enhanced activation of axonally transported stress-activated protein kinases in peripheral nerve in diabetic neuropathy is prevented by neurotrophin-3. Brain 126:1671–1682. 10.1093/brain/awg150 [DOI] [PubMed] [Google Scholar]
- Monnerie H, Tang-Schomer MD, Iwata A, Smith DH, Kim HA, Le Roux PD (2010) Dendritic alterations after dynamic axonal stretch injury in vitro. Exp Neurol 224:415–423. 10.1016/j.expneurol.2010.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison B 3rd, Cater HL, Wang CC, Thomas FC, Hung CT, Ateshian GA, Sundstrom LE (2003) A tissue level tolerance criterion for living brain developed with an in vitro model of traumatic mechanical loading. Stapp Car Crash J 47:93–105. [DOI] [PubMed] [Google Scholar]
- Nadler Y, Alexandrovich A, Grigoriadis N, Hartmann T, Rao KS, Shohami E, Stein R (2008) Increased expression of the γ-secretase components presenilin-1 and nicastrin in activated astrocytes and microglia following traumatic brain injury. Glia 56:552–567. 10.1002/glia.20638 [DOI] [PubMed] [Google Scholar]
- Nakadate H, Fukumura Y, Kaneko Y, Kakuta A, Furukawa H, Aomura S (2014) In vitro uniaxial stretch model for evaluating the effect of strain along axon on damage to neurons. J Biomech Eng 9:14-00136. 10.1299/jbse.14-00136 [DOI] [Google Scholar]
- Nakadate H, Kurtoglu E, Furukawa H, Oikawa S, Aomura S, Kakuta A, Matsui Y (2017) Strain-rate dependency of axonal tolerance for uniaxial stretching. Stapp Car Crash J 61:53–65. [DOI] [PubMed] [Google Scholar]
- Neumann S, Chassefeyre R, Campbell GE, Encalada SE (2017) KymoAnalyzer: a software tool for the quantitative analysis of intracellular transport in neurons. Traffic 18:71–88. 10.1111/tra.12456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niess C, Grauel U, Toennes S, Bratzke H (2002) Incidence of axonal injury in human brain tissue. Acta Neuropathol 104:79–84. 10.1007/s00401-002-0525-9 [DOI] [PubMed] [Google Scholar]
- Patel TP, Ventre SC, Meaney DF (2012) Dynamic changes in neural circuit topology following mild mechanical injury in vitro. Ann Biomed Eng 40:23–36. 10.1007/s10439-011-0390-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel TP, Ventre SC, Geddes-Klein D, Singh PK, Meaney DF (2014) Single-neuron NMDA receptor phenotype influences neuronal rewiring and reintegration following traumatic injury. J Neurosci 34:4200–4213. 10.1523/JNEUROSCI.4172-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peerless SJ, Rewcastle NB (1967) Shear injuries of the brain. Can Med Assoc J 96:577–582. [PMC free article] [PubMed] [Google Scholar]
- Pierce JE, Trojanowski JQ, Graham DI, Smith DH, McIntosh TK (1996) Immunohistochemical characterization of alterations in the distribution of amyloid precursor proteins and beta-amyloid peptide after experimental brain injury in the rat. J Neurosci 16:1083–1090. 10.1523/JNEUROSCI.16-03-01083.1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plant LD, Boyle JP, Smith IF, Peers C, Pearson HA (2003) The production of amyloid β peptide is a critical requirement for the viability of central neurons. J Neurosci 23:5531–5535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plassman BL, Havlik RJ, Steffens DC, Helms MJ, Newman TN, Drosdick D, Phillips C, Gau BA, Welsh-Bohmer KA, Burke JR, Guralnik JM, Breitner JC (2000) Documented head injury in early adulthood and risk of Alzheimer's disease and other dementias. Neurology 55:1158–1166. 10.1212/wnl.55.8.1158 [DOI] [PubMed] [Google Scholar]
- Ramón y Cajal S (1909) Histologie du système nerveux de l'homme and des vertébrés, Paris: Maloine. [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F (2013) Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8:2281–2308. 10.1038/nprot.2013.143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues EM, Weissmiller AM, Goldstein LS (2012) Enhanced β-secretase processing alters APP axonal transport and leads to axonal defects. Hum Mol Genet 21:4587–4601. 10.1093/hmg/dds297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosas-Hernandez H, Cuevas E, Escudero-Lourdes C, Lantz SM, Gomez-Crisostomo NP, Sturdivant NM, Balachandran K, Imam SZ, Slikker W, Paule MG, Ali SF (2018) Characterization of biaxial stretch as an in vitro model of traumatic brain injury to the blood-brain barrier. Mol Neurobiol 55:258–266. 10.1007/s12035-017-0738-5 [DOI] [PubMed] [Google Scholar]
- Ruff RM, Iverson GL, Barth JT, Bush SS, Broshek DK; NAN Policy and Planning Committee (2009) Recommendations for diagnosing a mild traumatic brain injury: a National Academy of Neuropsychology Education Paper. Arch Clin Neuropsychol 24:3–10. 10.1093/arclin/acp006 [DOI] [PubMed] [Google Scholar]
- Sahoo D, Deck C, Willinger R (2016) Brain injury tolerance limit based on computation of axonal strain. Accid Anal Prev 92:53–70. 10.1016/j.aap.2016.03.013 [DOI] [PubMed] [Google Scholar]
- Salvador E, Burek M, Förster CY (2015) Stretch and/or oxygen glucose deprivation (OGD) in an in vitro traumatic brain injury (TBI) model induces calcium alteration and inflammatory cascade. Front Cell Neurosci 9:323. 10.3389/fncel.2015.00323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaffert J, LoBue C, White CL, Chiang HS, Didehbani N, Lacritz L, Rossetti H, Dieppa M, Hart J, Cullum CM (2018) Traumatic brain injury history is associated with an earlier age of dementia onset in autopsy-confirmed Alzheimer's disease. Neuropsychology 32:410–416. 10.1037/neu0000423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schofield PW, Tang M, Marder K, Bell K, Dooneief G, Chun M, Sano M, Stern Y, Mayeux R (1997) Alzheimer's disease after remote head injury: an incidence study. J Neurol Neurosurg Psychiatry 62:119–124. 10.1136/jnnp.62.2.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwetye KE, Cirrito JR, Esparza TJ, MacDonald CL, Holtzman DM, Brody DL (2010) Traumatic brain injury reduces soluble extracellular amyloid-β in mice: a methodologically novel combined microdialysis-controlled cortical impact study. Neurobiol Dis 40:555–564. 10.1016/j.nbd.2010.06.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seiffert D, Bradley JD, Rominger CM, Rominger DH, Yang F, Meredith JE, Wang Q, Roach AH, Thompson LA, Spitz SM, Higaki JN, Prakash SR, Combs AP, Copeland RA, Arneric SP, Hartig PR, Robertson DW, Cordell B, Stern AM, Olson RE, et al. (2000) Presenilin-1 and -2 are molecular targets for γ-secretase inhibitors. J Biol Chem 275:34086–34091. 10.1074/jbc.M005430200 [DOI] [PubMed] [Google Scholar]
- Severson WE, Shindo N, Sosa M, Fletcher T, White EL, Ananthan S, Jonsson CB (2007) Development and validation of a high-throughput screen for inhibitors of SARS CoV and its application in screening of a 100,000-compound library. J Biomol Screen 12:33–40. 10.1177/1087057106296688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahim P, Tegner Y, Gustafsson B, Gren M, Arlig J, Olsson M, Lehto N, Engstrom A, Hoglund K, Portelius E, Zetterberg H, Blennow K (2016) Neurochemical aftermath of repetitive mild traumatic brain injury. JAMA Neurol 73:1308–1315. 10.1001/jamaneurol.2016.2038 [DOI] [PubMed] [Google Scholar]
- Sherman SA, Phillips JK, Costa JT, Cho FS, Oungoulian SR, Finan JD (2016) Stretch injury of human induced pluripotent stem cell derived neurons in a 96 well format. Sci Rep 6:34097. 10.1038/srep34097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibahashi K, Doi T, Tanaka S, Hoda H, Chikuda H, Sawada Y, Takasu Y, Chiba K, Nozaki T, Hamabe Y, Ogata T (2016) The serum phosphorylated neurofilament heavy subunit as a predictive marker for outcome in adult patients after traumatic brain injury. J Neurotrauma 33:1826–1833. 10.1089/neu.2015.4237 [DOI] [PubMed] [Google Scholar]
- Smith C, Graham DI, Murray LS, Nicoll JA (2003) Tau immunohistochemistry in acute brain injury. Neuropathol Appl Neurobiol 29:496–502. 10.1046/j.1365-2990.2003.00488.x [DOI] [PubMed] [Google Scholar]
- Smith DH, Meaney DF (2000) Axonal damage in traumatic brain injury. Neuroscientist 6:483–495. 10.1177/107385840000600611 [DOI] [Google Scholar]
- Smith DH, Nakamura M, McIntosh TK, Wang J, Rodríguez A, Chen XH, Raghupathi R, Saatman KE, Clemens J, Schmidt ML, Lee VM, Trojanowski JQ (1998) Brain trauma induces massive hippocampal neuron death linked to a surge in beta-amyloid levels in mice overexpressing mutant amyloid precursor protein. Am J Pathol 153:1005–1010. 10.1016/S0002-9440(10)65643-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DH, Wolf JA, Lusardi TA, Lee VM, Meaney DF (1999) High tolerance and delayed elastic response of cultured axons to dynamic stretch injury. J Neurosci 19:4263–4269. 10.1523/JNEUROSCI.19-11-04263.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DH, Johnson VE, Stewart W (2013) Chronic neuropathologies of single and repetitive TBI: substrates of dementia? Nat Rev Neurol 9:211–221. 10.1038/nrneurol.2013.29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staal JA, Dickson TC, Gasperini R, Liu Y, Foa L, Vickers JC (2010) Initial calcium release from intracellular stores followed by calcium dysregulation is linked to secondary axotomy following transient axonal stretch injury. J Neurochem 112:1147–1155. 10.1111/j.1471-4159.2009.06531.x [DOI] [PubMed] [Google Scholar]
- Stein TD, Montenigro PH, Alvarez VE, Xia W, Crary JF, Tripodis Y, Daneshvar DH, Mez J, Solomon T, Meng G, Kubilus CA, Cormier KA, Meng S, Babcock K, Kiernan P, Murphy L, Nowinski CJ, Martin B, Dixon D, Stern RA, et al. (2015) Beta-amyloid deposition in chronic traumatic encephalopathy. Acta Neuropathol 130:21–34. 10.1007/s00401-015-1435-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinbach JP, Müller U, Leist M, Li ZW, Nicotera P, Aguzzi A (1998) Hypersensitivity to seizures in β-amyloid precursor protein deficient mice. Cell Death Differ 5:858–866. 10.1038/sj.cdd.4400391 [DOI] [PubMed] [Google Scholar]
- Stokin GB, Lillo C, Falzone TL, Brusch RG, Rockenstein E, Mount SL, Raman R, Davies P, Masliah E, Williams DS, Goldstein LS (2005) Axonopathy and transport deficits early in the pathogenesis of Alzheimer's disease. Science 307:1282–1288. 10.1126/science.1105681 [DOI] [PubMed] [Google Scholar]
- Stone JR, Singleton RH, Povlishock JT (2000) Antibodies to the C-terminus of the β-amyloid precursor protein (APP): a site specific marker for the detection of traumatic axonal injury. Brain Res 871:288–302. 10.1016/s0006-8993(00)02485-9 [DOI] [PubMed] [Google Scholar]
- Tajiri N, Kellogg SL, Shimizu T, Arendash GW, Borlongan CV (2013) Traumatic brain injury precipitates cognitive impairment and extracellular Aβ aggregation in Alzheimer's disease transgenic mice. PLoS One 8:e78851. 10.1371/journal.pone.0078851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi M, Yasuno F, Yamamuro K, Matsuoka K, Kitamura S, Yoshikawa H, Yamamoto A, Iida H, Fukuda T, Ihara M, Nagatsuka K, Kishimoto T (2019) Detection of brain amyloid-β deposits due to the repetitive head trauma in a former karate player. Psychogeriatrics 19:276–281. 10.1111/psyg.12383 [DOI] [PubMed] [Google Scholar]
- Tang-Schomer MD, Patel AR, Baas PW, Smith DH (2010) Mechanical breaking of microtubules in axons during dynamic stretch injury underlies delayed elasticity, microtubule disassembly, and axon degeneration. FASEB J 24:1401–1410. 10.1096/fj.09-142844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang-Schomer MD, Johnson VE, Baas PW, Stewart W, Smith DH (2012) Partial interruption of axonal transport due to microtubule breakage accounts for the formation of periodic varicosities after traumatic axonal injury. Exp Neurol 233:364–372. 10.1016/j.expneurol.2011.10.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thangavelu B, Wilfred BS, Johnson D, Gilsdorf JS, Shear DA, Boutté AM (2020) Penetrating ballistic-like brain injury leads to microRNA dysregulation, BACE1 upregulation, and amyloid precursor protein loss in lesioned rat brain tissues. Front Neurosci 14:915. 10.3389/fnins.2020.00915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran HT, LaFerla FM, Holtzman DM, Brody DL (2011a) Controlled cortical impact traumatic brain injury in 3xTg-AD mice causes acute intra-axonal amyloid-beta accumulation and independently accelerates the development of tau abnormalities. J Neurosci 31:9513–9525. 10.1523/JNEUROSCI.0858-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran HT, Sanchez L, Esparza TJ, Brody DL (2011b) Distinct temporal and anatomical distributions of amyloid-β and tau abnormalities following controlled cortical impact in transgenic mice. PLoS One 6:e25475. 10.1371/journal.pone.0025475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran HT, Sanchez L, Brody DL (2012) Inhibition of JNK by a peptide inhibitor reduces traumatic brain injury–induced tauopathy in transgenic mice. J Neuropathol Exp Neurol 71:116–129. 10.1097/NEN.0b013e3182456aed [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukita S, Ishikawa H (1980) The movement of membranous organelles in axons: electron microscopic identification of anterogradely and retrogradely transported organelles. J Cell Biol 84:513–530. 10.1083/jcb.84.3.513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uryu K, Chen XH, Martinez D, Browne KD, Johnson VE, Graham DI, Lee VM, Trojanowski JQ, Smith DH (2007) Multiple proteins implicated in neurodegenerative diseases accumulate in axons after brain trauma in humans. Exp Neurol 208:185–192. 10.1016/j.expneurol.2007.06.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Kant R, Goldstein LS (2015) Cellular functions of the amyloid precursor protein from development to dementia. Dev Cell 32:502–515. 10.1016/j.devcel.2015.01.022 [DOI] [PubMed] [Google Scholar]
- van der Kant R, Langness VF, Herrera CM, Williams DA, Fong LK, Leestemaker Y, Steenvoorden E, Rynearson KD, Brouwers JF, Helms JB, Ovaa H, Giera M, Wagner SL, Bang AG, Goldstein LS (2019) Cholesterol metabolism is a druggable axis that independently regulates tau and amyloid-β in iPSC-derived Alzheimer's disease neurons. Cell Stem Cell 24:363–375.5369. 10.1016/j.stem.2018.12.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vascak M, Jin X, Jacobs KM, Povlishock JT (2018) Mild traumatic brain injury induces structural and functional disconnection of local neocortical inhibitory networks via parvalbumin interneuron diffuse axonal injury. Cereb Cortex 28:1625–1644. 10.1093/cercor/bhx058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vickers JC, Riederer BM, Marugg RA, Buée-Scherrer V, Buée L, Delacourte A, Morrison JH (1994) Alterations in neurofilament protein immunoreactivity in human hippocampal neurons related to normal aging and Alzheimer's disease. Neuroscience 62:1–13. 10.1016/0306-4522(94)90310-7 [DOI] [PubMed] [Google Scholar]
- von Koch CS, Zheng H, Chen H, Trumbauer M, Thinakaran G, van der Ploeg LH, Price DL, Sisodia SS (1997) Generation of APLP2 KO mice and early postnatal lethality in APLP2/APP double KO mice. Neurobiol Aging 18:661–669. 10.1016/S0197-4580(97)00151-6 [DOI] [PubMed] [Google Scholar]
- Walker KR, Kang EL, Whalen MJ, Shen Y, Tesco G (2012) Depletion of GGA1 and GGA3 mediates postinjury elevation of BACE1. J Neurosci 32:10423–10437. 10.1523/JNEUROSCI.5491-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Franco R, Skotak M, Hu G, Chandra N (2014) Mechanical stretch exacerbates the cell death in SH-SY5Y cells exposed to paraquat: mitochondrial dysfunction and oxidative stress. Neurotoxicology 41:54–63. 10.1016/j.neuro.2014.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winston CN, Chellappa D, Wilkins T, Barton DJ, Washington PM, Loane DJ, Zapple DN, Burns MP (2013) Controlled cortical impact results in an extensive loss of dendritic spines that is not mediated by injury-induced amyloid-beta accumulation. J Neurotrauma 30:1966–1972. 10.1089/neu.2013.2960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodruff G, Young JE, Martinez FJ, Buen F, Gore A, Kinaga J, Li Z, Yuan SH, Zhang K, Goldstein LS (2013) The presenilin-1 DeltaE9 mutation results in reduced gamma-secretase activity, but not total loss of PS1 function, in isogenic human stem cells. Cell Rep 5:974–985. 10.1016/j.celrep.2013.10.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodruff G, Reyna SM, Dunlap M, Van Der Kant R, Callender JA, Young JE, Roberts EA, Goldstein LS (2016) Defective transcytosis of APP and lipoproteins in human iPSC-derived neurons with familial Alzheimer's disease mutations. Cell Rep 17:759–773. 10.1016/j.celrep.2016.09.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu ML, Chen CH, Lin YT, Jheng YJ, Ho YC, Yang LT, Chen L, Layne MD, Yet SF (2014) Divergent signaling pathways cooperatively regulate TGFβ induction of cysteine-rich protein 2 in vascular smooth muscle cells. Cell Commun Signal 12:22. 10.1186/1478-811X-12-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WJ, Chen W, Chen L, Guo YJ, Zeng JS, Li GY, Tong WS (2017) Involvement of tau phosphorylation in traumatic brain injury patients. Acta Neurol Scand 135:622–627. 10.1111/ane.12644 [DOI] [PubMed] [Google Scholar]
- Yap YC, Dickson TC, King AE, Breadmore MC, Guijt RM (2014) Microfluidic culture platform for studying neuronal response to mild to very mild axonal stretch injury. Biomicrofluidics 8:044110. 10.1063/1.4891098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap YC, King AE, Guijt RM, Jiang T, Blizzard CA, Breadmore MC, Dickson TC (2017) Mild and repetitive very mild axonal stretch injury triggers cystoskeletal mislocalization and growth cone collapse. PLoS One 12:e0176997. 10.1371/journal.pone.0176997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimura K, Ueno M, Lee S, Nakamura Y, Sato A, Yoshimura K, Kishima H, Yoshimine T, Yamashita T (2011) C-Jun N-terminal kinase induces axonal degeneration and limits motor recovery after spinal cord injury in mice. Neurosci Res 71:266–277. 10.1016/j.neures.2011.07.1830 [DOI] [PubMed] [Google Scholar]
- Young JE, Boulanger-Weill J, Williams DA, Woodruff G, Buen F, Revilla AC, Herrera C, Israel MA, Yuan SH, Edland SD, Goldstein LS (2015) Elucidating molecular phenotypes caused by the SORL1 Alzheimer's disease genetic risk factor using human induced pluripotent stem cells. Cell Stem Cell 16:373–385. 10.1016/j.stem.2015.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu F, Zhang Y, Chuang DM (2012) Lithium reduces BACE1 overexpression, beta amyloid accumulation, and spatial learning deficits in mice with traumatic brain injury. J Neurotrauma 29:2342–2351. 10.1089/neu.2012.2449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan A, Rao MV, Nixon RA (2017) Neurofilaments and neurofilament proteins in health and disease. Cold Spring Harb Perspect Biol 9:a018309. 10.1101/cshperspect.a018309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan SH, Martin J, Elia J, Flippin J, Paramban RI, Hefferan MP, Vidal JG, Mu Y, Killian RL, Israel MA, Emre N, Marsala S, Marsala M, Gage FH, Goldstein LS, Carson CT (2011) Cell-surface marker signatures for the isolation of neural stem cells, glia and neurons derived from human pluripotent stem cells. PLoS One 6:e17540. 10.1371/journal.pone.0017540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen TJ, Browne KD, Iwata A, Smith DH (2009) Sodium channelopathy induced by mild axonal trauma worsens outcome after a repeat injury. J Neurosci Res 87:3620–3625. 10.1002/jnr.22161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanier ER, Bertani I, Sammali E, Pischiutta F, Chiaravalloti MA, Vegliante G, Masone A, Corbelli A, Smith DH, Menon DK, Stocchetti N, Fiordaliso F, De Simoni MG, Stewart W, Chiesa R (2018) Induction of a transmissible tau pathology by traumatic brain injury. Brain 141:2685–2699. 10.1093/brain/awy193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao G, Tan J, Mao G, Cui MZ, Xu X (2007) The same γ-secretase accounts for the multiple intramembrane cleavages of APP. J Neurochem 100:1234–1246. 10.1111/j.1471-4159.2006.04302.x [DOI] [PubMed] [Google Scholar]
- Zhou Y, Su Y, Li B, Liu F, Ryder JW, Wu X, Gonzalez-DeWhitt PA, Gelfanova V, Hale JE, May PC, Paul SM, Ni B (2003) Nonsteroidal anti-inflammatory drugs can lower amyloidogenic Abeta42 by inhibiting Rho. Science 302:1215–1217. 10.1126/science.1090154 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Microfluidic device further characterization and standardization of MSD-based assay to quantitatively assess JNK and pJNK levels in neuron's cell lysates. A) Representative images of neurons (with neurites oriented within 30 degrees of stretch direction) stretched and fixed immediately after to preserve waves in axon (Tau positive, white arrows) and dendrites (MAP2, yellow arrows). B) Quantification of the percent of neurites oriented parallel (0°-30°), inclined (30°-60°) and perpendicular (60°-90°) to the direction of the stretch in non-stretched microfluidic devices and the distribution of wavy neurites per angle following stretch (compared to 60°-90°, *p = 0.0119 for 0°-30° and *p = 0.0385 for 0°-30°) (n = 3). C) Quantitative analysis of Total tau and p-tau, measured from neuronal lysate harvested at different time points after stretch (n = 3 or more). D) Quantitative analysis of Fluo4 intensity in neurons cell body before and immediately after stretch. E) Assessment of assay linear detection range for Total JNK and pJNK (Thr183/Tyr185), measured using undiluted neuronal lysate (control neurons) plotted against dilution ranges (n = 4). 2.5X dilution range was chosen to perform further measurements. Quantitative analysis of Total JNK and pJNK (Thr183/Tyr185) levels and resultant pJNK/JNK ratio, following thirty minutes of exposure to F) Anisomycin (2.5 µM) (*p = 0.0306 for JNK, *p = 0.0435 for p-JNK and *p = 0.0444 p-JNK/JNK ratio) (n = 8) and G) JNK inhibitor II (9 µM) (*p = 0.0123 for p-JNK and *p = 0.0494 p-JNK/JNK ratio) (n = 8) (Vehicle = DMSO 0.01%). H) Quantitative analysis of Total JNK and pJNK, measured from neuronal lysate harvested at different time points up to 24 h (h) post-stretch (n = 3 or more). Data are mean ± SEM; means were compared by One-way ANOVA (B) unpaired Student's t test (C-H). Download Figure 1-1, TIF file (108.7MB, tif) .
Stretch does not induce intracellular Aβ accumulation in human neurons. A) Quantitative analysis of extracellular Aβ40 and Aβ42 and resultant Aβ42/Aβ40 ratio, measured in neuronal media collected immediately and at different time points after stretch relative to matched Aβ levels before stretch (**p = 0.0078 for Aβ42 and *p = 0.0110 for Aβ42/Aβ40 ratio) (n = 3 or more). B) Representative bands and quantitative analysis of full-length APP, BACE1 and PS1 (PS1-CTF) protein levels measured in neuronal lysates 24 h after stretch (n = 7 or more) (C=control and S=stretched). Extracellular Aβ levels in neuronal media from naïve neurons exposed to C) CE (200 nM) (***p = 0.0001 for Aβ40 and Aβ42) (n = 4) or D) βsi-IV (4 µM) (***p = 0.0001 for Aβ40 and **p = 0.0046 for Aβ42) (n = 3) during sixteen hours (h) (Vehicle = DMSO 0.001%). E) Levels of extracellular Aβ peptides in media from neurons re-exposed to CE (200 nM) or vehicle (DMSO 0.001%) for different time points after been initially exposed to CE during sixteen hours, to demonstrate that CE inhibition is reversible (CE 16h+Vehicle, ***p = 0.0001 for Aβ40 and Aβ42) (n = 4). F) Quantitative analysis of intracellular Aβ peptides in cell lysates from neurons re-exposed to CE (200 nM) or exposed to vehicle (DMSO 0.001%) during twenty-four or thirty-six hours after been initially exposed to CE during sixteen hours (Vehicle 16h + Vehicle 24h ***p = 0.0001 for Aβ40 and Vehicle 16h + Vehicle 36h ***p = 0.0001 for Aβ40) (n = 4). ND = Non-detectable. Data are mean ± SEM; means were compared by unpaired Student's t test. Download Figure 2-1, TIF file (109.6MB, tif) .
Stretch-induced inhibition of axonal transport is cargo-specific. A) Quantitative analysis of APP-puncta intensity and puncta area in stretched axons fixed 24 h after stretch, compared to control axons (puncta intensity **p = 0.033, puncta size **p = 0.0044) (n = 7 or more axons). B) Quantitative analysis of LAMP2-GFP axonal transport in axons before and after stretch (n = 5). Data are mean ± SEM; means were compared by unpaired Student's t test. Download Figure 3-1, TIF file (2MB, tif) .
APP axonal transport in neurons exposed to β- and γ-secretase inhibitors. Quantitative analysis of A) the percentage of APP-YFP puncta moving anterogradely and retrogradely, and B) APP-YFP particle density prestretch in neurons exposed to βsi-IV (40 µM), CE (5 µM) and vehicle (DMSO 0.01%) during 1 h (n = 4 or more). Data are mean ± SEM; means were compared by unpaired Student's t test. Download Figure 4-1, TIF file (1.7MB, tif) .
Exposure to GGPP increase Aβ generation in APPWT/Ice neurons. A) Extracellular Aβ40 and Aβ42 levels in APP WT/WT and APPWT/Ice neurons exposed to vehicle (Methanol 0.015% (Sigma)) or GGPP (15 µg/ml) during 24 h (APP WT/WT ***p = 0.001 for Aβ40 and *p = 0.0136 for Aβ42, and APPWT/Ice ***p = 0.001 for Aβ40 and *p = 0.0102 for Aβ42 (n = 4). Data are mean ± SEM; means were compared by unpaired Student's t test. Download Figure 6-1, TIF file (1.6MB, tif) .
Stretch impairs APP axonal transport. Movie showing APP-YFP puncta movement in a transfected axon before and after stretch (same axons as shown in Fig. 3B). Time is shown in milliseconds. Scale bar, 10 μm.
APP axonal transport is not affected in axons oriented perpendicular to the direction of stretch. Movie showing APP-YFP puncta movement in a transfected axon, oriented perpendicular to the stretch direction, after stretch (same axons as shown in Fig. 3D). Time is shown in milliseconds. Scale bar, 10 μm.
Lysosome axonal transport is not significantly altered after stretch. Movie showing LAMP-2-GFP puncta moving through axonal waves (white arrows) immediately after stretch. Time is shown in milliseconds. Scale bar, 10 μm.
APP axonal transport 3 h after stretch. Movie showing APP-YFP puncta movement in control and stretched axons 3 h after stretch (same axons as shown in Fig. 3E). Time is shown in milliseconds. Scale bar, 10 μm.
APP axonal transport 24 h after stretch. Movie showing APP-YFP puncta movement in control and stretched axons 24 h after stretch (same axons as shown in Fig. 3E). Time is shown in milliseconds. Scale bar, 10 μm.
APP axonal transport in stretched neurons exposed to vehicle. Movie showing APP-YFP puncta movement before and after stretch in neurons exposed to vehicle (DMSO) during 1 h (same axons as shown in Fig. 4A). Time is shown in milliseconds. Scale bar, 10 μm.
APP axonal transport in stretched neurons exposed to CE. Movie showing APP-YFP puncta movement before and after stretch in neurons exposed to CE during 1 h (same axons as shown in Fig. 4A). Time is shown in milliseconds. Scale bar, 10 μm.
APP axonal transport in stretched neurons exposed to βsi-IV. Movie showing APP-YFP puncta movement before and after stretch in neurons exposed to βsi-IV during 1 h (same axons as shown in Fig. 4A). Time is shown in milliseconds. Scale bar, 10 μm.
APPMV axonal transport after stretch in isogenic APP-KO neurons. Movie showing APPMV-YFP puncta movement in APP-KO-transfected axons before and after stretch (same axons as shown in Fig. 4B). Time is shown in milliseconds. Scale bar, 10 μm.
APP axonal transport 24 h after stretch in neurons exposed to CE. Movie showing APP-YFP axonal transport 24 h after stretch, in axons exposed to CE during 1 h before being stretched (same axons as shown in Fig. 5A). Time is shown in milliseconds. Scale bar, 10 μm.
APPMV axonal transport 24 h after stretch in isogenic APP-KO neurons. Movie showing APPMV-YFP puncta movement in APP-KO-transfected axons 24 h after stretch. Time is shown in milliseconds. Scale bar, 10 μm.
APPIce axonal transport is not affected in stretched axons. Movie showing APPIce-GFP puncta movement in a transfected axon, before and immediately after stretch (kymograph shown in Fig. 6D). Time is shown in milliseconds. Scale bar, 10 μm.