

Abstract
The unintended neurologic sequelae of chemotherapy contribute to significant patient morbidity. Chemotherapy-related cognitive impairment (CRCI) is observed in up to 80% of cancer patients treated with chemotherapy and involves multiple cognitive domains including executive functioning. The pathophysiology underlying CRCI and the neurotoxicity of chemotherapy is incompletely understood, but oxidative stress and DNA damage are highly plausible mechanisms based on preclinical data. Unfortunately, validating pathways relevant to CRCI in humans is limited by an absence of relevant neuropathologic studies of patient brain tissue. In the present study, we stained sections of frontal lobe autopsy tissue from cancer patients treated with chemotherapy (n = 15), cancer patients not treated with chemotherapy (n = 10), and patients without history of cancer (n = 10) for markers of oxidative stress (nitrotyrosine, 4-hydroxynonenal) and DNA damage (pH2AX, pATM). Cancer patients treated with chemotherapy had increased staining for markers of oxidative stress and DNA damage in frontal lobe cortical neurons compared to controls. We detected no statistically significant difference in oxidative stress and DNA damage by the duration between last administration of chemotherapy and death. The study highlights the potential relevance of oxidative stress and DNA damage in the pathophysiology of CRCI and the neurotoxicity of chemotherapy.
Keywords: Chemotherapy, Cognition, DNA damage, Neuron, Neurotoxicity, Oxidative stress
INTRODUCTION
There are an estimated 17 million cancer survivors in the United States, and this number is projected to exceed 22 million by 2030 (1). Despite advances in immunomodulatory and targeted anticancer therapies, chemotherapy remains a standard of treatment. Chemotherapy is associated with a number of serious neurologic adverse effects involving the musculoskeletal system and central and peripheral nervous systems (2).
Chemotherapy-related cognitive impairment (CRCI), colloquially termed “chemobrain” or “chemofog,” is becoming an increasingly clinically recognized adverse effect of chemotherapy that contributes to significant patient morbidity. CRCI involves multiple cognitive domains including memory, executive functioning, attention, and speed processing, and is detected in approximately 20% of cancer patients treated with chemotherapy (3), though the prevalence may approach 80% (4). In accordance with these cognitive deficits, radiologic studies have shown volumetric losses, changes to white matter integrity, and functional alterations in the brains of chemotherapy patients (5–8). The deficits associated with CRCI may improve over time (9), but patients can have persistent changes to cognitive function (10, 11) that last 20 years after cessation of treatment (12). The severity of CRCI is generally mild-to-moderate when patients undergo formal neurocognitive testing (13). Nevertheless, the impact of CRCI on patients’ quality of life, daily functioning, social relationships, and ability to work is nontrivial (14–16).
Unfortunately, development of disease-modifying interventions to reduce the burden of CRCI is hindered by an incomplete understanding of its underlying pathophysiology. Pathways involving oxidative stress (17–23), DNA damage (23–26), neuroinflammation/proinflammatory cytokines (23, 27–30), direct chemotherapy-induced injury to vulnerable central nervous system (CNS) cell populations (31), hippocampal neurogenesis (32–34), and dysmyelination (27, 29), among others (35, 36), may contribute to the pathogenesis of CRCI.
However, with rare exceptions (27), studies of CRCI have not included neuropathologic analysis of brain tissue from chemotherapy patients, limiting our understanding of potential mechanisms shown to be directly relevant to human CRCI and chemotherapy-induced neurotoxicity more generally. To address this limitation, we used immunohistochemistry and immunofluorescence to stain for markers of oxidative stress and DNA damage in frontal lobe cortical neurons of chemotherapy patients and control patients. The frontal lobe was selected as our region of interest given the reported deficits in executive functioning (37–39) and radiologic alterations involving frontal lobe (6, 40, 41) in cancer patients treated with chemotherapy.
MATERIALS AND METHODS
The neuropathology autopsy archives at Brigham and Women’s Hospital (Boston, MA) were reviewed for patients with history of cancer treated with systemically administered chemotherapy, patients with history of cancer who did not receive chemotherapy, and patients with neither history of cancer nor treatment with chemotherapy. These 3 groups are hereafter referred to as the cancer with chemotherapy cohort, cancer without chemotherapy cohort, and cancer-negative cohort, respectively. We selected autopsy cases that included frontal cortex as part of the tissue sampling process. Patients with history of cranial radiation, intraparenchymal brain metastases, pathologically confirmed neurodegenerative disease, or macroinfarct/hemorrhage adjacent to the sampled frontal lobe were excluded from the study. To investigate time-related effects of chemotherapy on measures of oxidative stress and DNA damage, the cancer with chemotherapy cohort was divided into 3 subgroups based on interval between last dose of chemotherapy and death (<1 month, 1–6 months, and >6 months, hereafter referred to as short, intermediate, and long-term interval subgroups, respectively). Patients in the cancer with chemotherapy cohort were also placed into subgroups based on the number of total cycles of chemotherapy administered (≤4 cycles, between 5 and 9 cycles, and ≥10 cycles) to determine whether elevated neuronal oxidative stress and DNA damage were associated with increased exposure to chemotherapeutic agents; these subgroups were organized to ensure roughly equal number of patients in each. Patient clinical data were abstracted from the available electronic medical records. The study was conducted with Institutional Review Board approval.
Immunohistochemistry and Immunofluorescence
Immunohistochemistry was performed on 5-micron (μm) formalin-fixed paraffin-embedded (FFPE) sections of frontal lobe following routine heat antigen retrieval (10 mM sodium citrate buffer, pH 6.0). Sections were blocked in 2% non-fat dry milk in PBST and then incubated with one of the following primary antibodies: nitrotyrosine (Millipore AB5411, Burlington, MA; rabbit polyclonal, 1:200) or 4-hydroxynonenal (4-HNE) (Abcam 46545, Cambridge, MA; rabbit polyclonal, 1:200). Sections were then incubated with biotinylated goat anti-rabbit secondary antibody (Southern Biotech, Birmingham, AL; 1:200). The stains were then visualized with the avidin-biotin complex detection system (Vector Laboratories, Burlingame, CA) with 3,3′-diaminobenzidine. Nuclei were counterstained with hematoxylin. Coverslips were mounted using Permount (Thermo Fisher Scientific, Waltham, MA). Neurons were identified on the basis of their nuclear histologic features.
Immunofluorescence for the following antibodies was performed on 5-μm FFPE sections of frontal lobe following routine heat antigen retrieval and blocking in 2% non-fat dry milk in PBST: phospho-H2AX ser139 (pH2AX) (Millipore JBW301; mouse monoclonal, 1:800) and phospho-ataxia telangiectasia mutated kinase (pATM) (Rockland ser1981, Limerick, PA; mouse monoclonal, 1:200). Neurons were labeled with a microtubule-associated protein 2 (MAP2) antibody (Millipore ab5622; rabbit polyclonal, 1:200). Sections were then incubated with Alexa Fluor 555 goat anti-mouse (Invitrogen, Carlsbad, CA; 1:200) and Alexa Fluor 488 goat anti-rabbit (Invitrogen; 1:200) secondary antibodies. Coverslips were mounted with 4′,6-diamidino-2-phenylindole (DAPI)-containing Fluoromount medium (Southern Biotech).
A total of 200 cortical neurons were counted per stain per case. Neurons were analyzed from adjacent, non-overlapping high-power fields with a Nikon Eclipse E600 microscope with SPOT software. The high-power fields did not include sulcal areas. Immunohistochemistry photos were taken with an Olympus DP25 camera, and immunofluorescence photos were taken with a Zeiss LSM 800 confocal microscope.
Statistical Analysis
For nitrotyrosine and 4-HNE, H-scores for each case were calculated by multiplying the intensity of the stain (absent staining assigned value of 0, weak staining assigned value of 1, intermediate staining assigned value of 2, and strong staining assigned value of 3) by the percent of neurons exhibiting that staining pattern and then adding these values together. The average H-scores per stain per cohort were compared. For pH2AX and pATM, the percent of positive cortical neurons was calculated per stain per case, and then the average percent of positive cortical neurons per cohort was calculated. GraphPad Prism (v.8) was used to make comparisons between cohorts and to generate graphs. Comparisons were performed using one-way ANOVA with the Holm-Sidak’s multiple comparisons test. P-values <0.05 were considered statistically significant.
RESULTS
Patient Clinical Characteristics
The cancer with chemotherapy cohort consisted of 15 patients (8 female, 7 male; mean age 64.6 ± 11.1 years). A total of 21 different chemotherapeutic agents were administered to patients in the cohort. The chemotherapeutic regimen for one patient was not specified in the available medical records, and the number of cycles of chemotherapeutic regimens administered to 3 patients could not be ascertained. For patients with known chemotherapeutic regimens, the most frequently administered drugs were cisplatin (42.9%), carboplatin (28.6%), cyclophosphamide (28.6%), doxorubicin (28.6%), pemetrexed (28.6%), paclitaxel (28.6%), cytarabine (21.4%), etoposide (21.4%), vincristine (21.4%), docetaxel (14.3%), gemcitabine (14.3%), ifosfamide (14.3%), azacitidine (7.1%), decitabine (7.1%), fluorouracil (7.1%), irinotecan (7.1%), eribulin (7.1%), busulfan (7.1%), fludarabine (7.1%), daunorubicin (7.1%), and oxaliplatin (7.1%). Patients received a median of 3 chemotherapeutic agents (range 2–8) and were treated with a median of 5.5 cycles of chemotherapeutic regimens (range 1–17); of the 12 patients whose number of administered chemotherapy cycles could be ascertained, 4 patients received 4 or fewer cycles, 4 patients received between 5 and 9 cycles, and 4 patients received 10 or greater cycles. Additional anticancer therapies given to patients in this cohort included rituximab (21.4%), checkpoint inhibitor therapy (14.3%), CDK4/6 inhibitor therapy (7.1%), brentuximab vedotin (7.1%), selective inhibitor of nuclear export (SINE) compound (7.1%), venetoclax (7.1%), cabozantinib (7.1%), and erlotinib (7.1%). The median time from last chemotherapy dose to death was 2 months (range: 1 day to 18 years); there were 5 patients in the short (<1 month) interval subgroup, 6 patients in the intermediate (1–6 months) interval subgroup, and 4 patients in the long-term (>6 months) interval subgroup. Sixty percent of patients underwent cytoreductive surgery, and 33.3% had documented local radiation therapy. In order of decreasing frequency, cancer diagnoses were of pulmonary/pleural (40.0%), hematologic (33.3%), breast (13.3%), genitourinary (13.3%), gynecologic (6.7%), colonic (6.7%), dermatologic (6.7%), and pancreatic (6.7%) origin. Twenty percent of patients had multiple primary malignancies. The reported causes of death in this cohort were (metastatic) cancer (40.0%), respiratory failure (26.7%), pneumonia/sepsis (13.3%), myocardial infarction (6.7%), multifactorial lung disease (6.7%), and multiorgan failure (6.7%).
The cancer without chemotherapy cohort consisted of 10 patients (4 female, 6 male; mean age 71.7 ± 9.2 years). Within the cancer without chemotherapy cohort, 80.0% of patients underwent cytoreductive/curative surgery, and 30.0% underwent local radiation therapy. In order of decreasing frequency, cancer diagnoses were of genitourinary (30.0%), pulmonary (20.0%), breast (20.0%), gynecologic (10.0%), hepatic (10.0%), adrenal (10.0%), oropharyngeal (10.0%), and dermatologic (10.0%) origin. Twenty percent of patients had multiple primary malignancies. The reported causes of death in this cohort were respiratory failure (20.0%), cardiogenic or septic shock (20.0%), multisystem organ failure (20.0%), metastatic cancer (10.0%), heart failure (10.0%), cardiac arrhythmia (10.0%), and cirrhosis (10.0%).
The cancer-negative cohort consisted of 10 patients (7 females, 3 males; mean age 68.9 ± 11.2 years). Within the cancer-negative cohort, the reported causes of death were respiratory failure (20.0%), myocardial infarction (20.0%), cardiac arrest (10.0%), cardiomyopathy (10.0%), ruptured thoracic aneurysm (10.0%), hemothorax (10.0%), pulmonary embolism (10.0%), and pneumonia (10.0%).
The mean interval between time of death and time of autopsy for all patients in the study was 20.4 ± 12.6 hours. The death-to-autopsy interval was not statistically different between cohorts. Importantly, immunohistochemical staining performed on human autopsy brain tissue remains consistent for many proteins across a range of postmortem intervals, including those that exceed 50 hours (42).
Brain MRIs were performed on 12 patients based on the available radiology reports: 10 patients in the cancer with chemotherapy cohort, 1 patient in the cancer without chemotherapy cohort, and 1 patient in the cancer-negative cohort. Within the cancer with chemotherapy cohort, 2 patients had imaging performed prior to initiation of chemotherapy only. Ultimately, the low number of control patients with brain MRIs and the significant variation within the cancer with chemotherapy cohort in terms of timing of MRI in relation to (1) chemotherapy treatment and (2) proximity to death (ranging from 1 month to 5 years) preclude meaningful radiologic comparisons.
Immunohistochemical/Immunofluorescent Staining for Oxidative Stress and DNA Damage
There were elevated markers of oxidative stress associated with history of chemotherapy. Nitrotyrosine and 4-HNE show cytoplasmic positivity and are well-validated markers of oxidative stress, reflecting protein oxidation and lipid oxidation, respectively (43). Patients in the cancer with chemotherapy cohort had a higher neuron nitrotyrosine H-score (48.8) compared to controls (cancer without chemotherapy cohort, 19.3, p < 0.05; cancer-negative cohort, 26.3, p < 0.05) (Fig. 1). In addition, patients in the cancer with chemotherapy cohort had a higher neuron 4-HNE H-score (33.3) compared to patients in the cancer-negative cohort (19.0, p < 0.05) and patients in the cancer without chemotherapy cohort (20.4), though the latter comparison did not reach statistical significance (p = 0.056) (Fig. 2).
FIGURE 1.
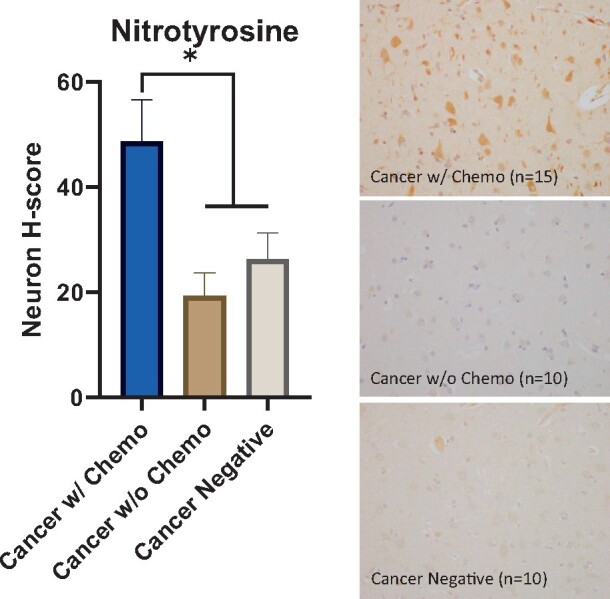
Elevated oxidative stress in cortical neurons after chemotherapy. (Left) Cancer patients treated with chemotherapy have a higher nitrotyrosine H-score in frontal lobe cortical neurons (48.8) compared to cancer patients not treated with chemotherapy (19.3, p < 0.05) and patients without history of cancer (26.3, p < 0.05). Error bars represent the standard error of the mean. (Right) Representative nitrotyrosine immunostain images from all 3 cohorts counterstained with hematoxylin (400×), *p < 0.05.
FIGURE 2.
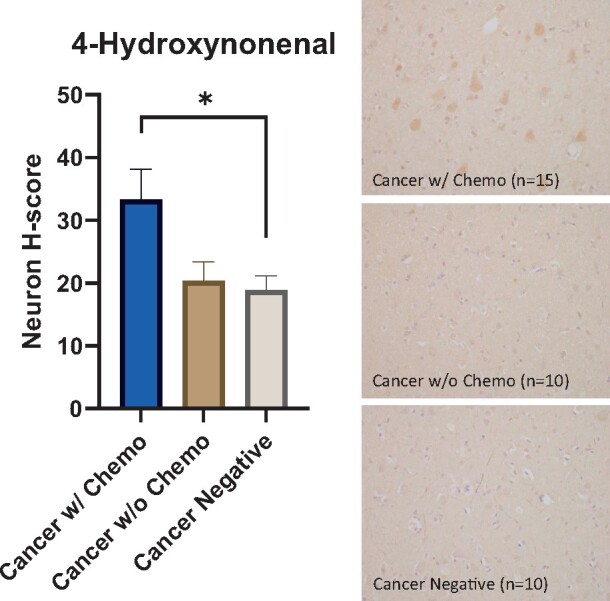
Elevated oxidative stress in cortical neurons after chemotherapy. (Left) Cancer patients treated with chemotherapy have a higher 4-hydroxynonenal (4-HNE) H-score in frontal lobe cortical neurons (33.3) compared to patients without history of cancer (19.0, p < 0.05) and cancer patients not treated with chemotherapy (20.4), though the latter comparison did not reach statistical significance (p = 0.056). Error bars represent the standard error of the mean. (Right) Representative 4-HNE immunostain images from all 3 cohorts counterstained with hematoxylin (400×), *p < 0.05.
There was evidence for increased DNA damage in patients treated with chemotherapy. H2AX is a histone H2A variant that becomes phosphorylated at serine residue 139 in response to double-stranded DNA breaks (44). ATM is a component of the cellular DNA damage response and autophosphorylates in response to double-stranded DNA breaks (45). pH2AX shows a nuclear punctate staining pattern, and pATM shows a more diffuse nuclear staining pattern. Patients in the cancer with chemotherapy cohort had a higher percentage of cortical neurons with pH2AX-positive foci (28.8%) compared to patients in the cancer without chemotherapy cohort (12.8%, p < 0.01) and cancer-negative cohort (16.2%, p < 0.05) (Fig. 3). Furthermore, patients in the cancer with chemotherapy cohort had a higher percentage of pATM-positive cortical neurons (40.0%) compared to patients in the cancer without chemotherapy cohort (19.2%, p < 0.05) and patients in the cancer-negative cohort (19.0%, p < 0.05) (Fig. 4).
FIGURE 3.
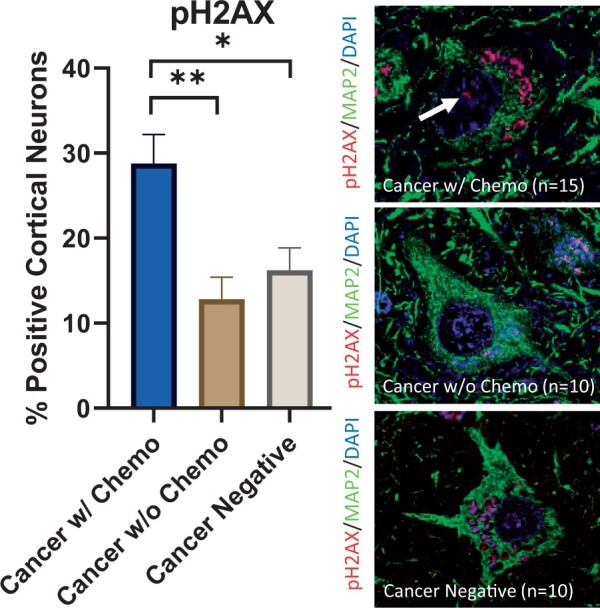
Elevated DNA damage in cortical neurons after chemotherapy. (Left) An increased percentage of frontal lobe cortical neurons in cancer patients treated with chemotherapy contain nuclear pH2AX-positive foci (28.8%) compared to frontal lobe cortical neurons in cancer patients not treated with chemotherapy (12.8%, p < 0.01) and patients without history of cancer (16.2%, p < 0.05). Error bars represent the standard error of the mean. (Right) Representative pH2AX and MAP2 double-labeled immunofluorescent images from all 3 cohorts counterstained with DAPI; the top panel shows a neuron with a pH2AX-positive focus (arrow) in a cancer patient treated with chemotherapy; the middle and bottom panels show neurons without pH2AX-positive foci in control patients. Nonspecific pH2AX immunopositivity is seen in cytoplasmic lipofuscin granules. DAPI, 4',6-diamidino-2-phenylindole; MAP2, microtubule-associated protein 2; pH2AX, phospho-H2AX; *p < 0.05, **p < 0.01.
FIGURE 4.
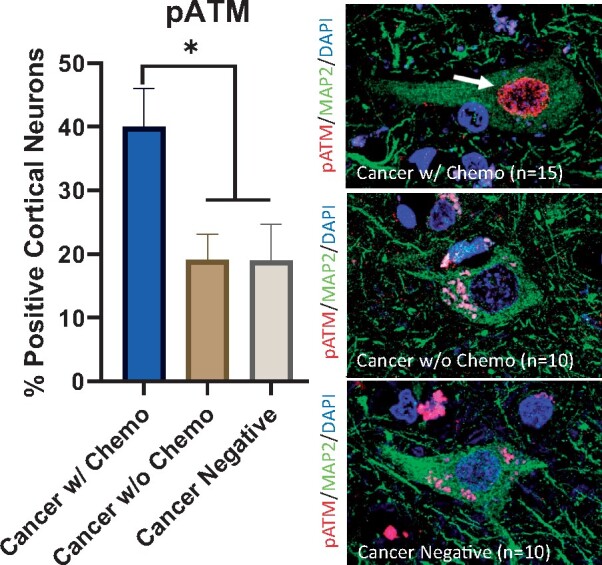
Elevated DNA damage in cortical neurons after chemotherapy. (Left) An increased percentage of frontal lobe cortical neurons in cancer patients treated with chemotherapy show nuclear pATM positivity (40.0%) compared to frontal lobe cortical neurons in cancer patients not treated with chemotherapy (19.2%, p < 0.05) and patients without history of cancer (19.0%, p < 0.05). Error bars represent the standard error of the mean. (Right) Representative pATM and MAP2 double-labeled immunofluorescent images from all 3 cohorts counterstained with DAPI; the top panel shows a neuron with positive nuclear pATM staining (arrow) in a cancer patient treated with chemotherapy; the middle and bottom panels show neurons without nuclear pATM staining in control patients. Nonspecific pATM immunopositivity is seen in cytoplasmic lipofuscin granules. DAPI, 4',6-diamidino-2-phenylindole; MAP2, microtubule-associated protein 2; pATM, phospho-ataxia telangiectasia mutated kinase; *p < 0.05.
No statistical differences in markers of oxidative stress or DNA damage were detected among the chemotherapy patient subgroups organized by time interval between last chemotherapy dose and death. The neuron nitrotyrosine H-scores for patients in the short-term interval subgroup (<1 month), intermediate-term interval subgroup (1–6 months), and long-term interval subgroup (>6 months) were 55.5, 42.8, and 49.3, respectively (p > 0.05 for all comparisons) (Fig. 5A). The neuron 4-HNE H-scores for patients in the short-term interval subgroup, intermediate-term interval subgroup, and long-term interval subgroup were 30.1, 33.0, and 37.8, respectively (p > 0.05 for all comparisons) (Fig. 5B). The percentage of cortical neurons with pH2AX-positive foci in the short-term interval subgroup, intermediate-term interval subgroup, and long-term interval subgroup were 32.1%, 26.7%, and 27.8%, respectively (p > 0.05 for all comparisons) (Fig. 5C). The percentage of pATM-positive cortical neurons in the short-term interval subgroup, intermediate-term interval subgroup, and long-term interval subgroup were 41.3%, 34.3%, and 46.9%, respectively (p > 0.05 for all comparisons) (Fig. 5D).
FIGURE 5.

Quantification of oxidative stress and DNA damage in cortical neurons by time interval between last chemotherapy dose and patient death: (A) nitrotyrosine, (B) 4-hydroxynonenal, (C) pH2AX and (D) pATM staining did not differ significantly by time interval (p > 0.05 for all comparisons; n = 5 for <1-month-interval subgroup, n = 6 for 1–6-month-interval subgroup, n = 4 for >6-month-interval subgroup). Error bars represent the standard error of the mean. pATM, phospho-ataxia telangiectasia mutated kinase; pH2AX, phospho-H2AX.
No statistical differences in markers of oxidative stress or DNA damage were detected among the chemotherapy patient subgroups organized by total number of cycles of chemotherapy received. In addition, since all chemotherapy patients had been placed on regimens consisting of at least 2 agents, no clear relationship could be ascertained between a single chemotherapeutic drug and markedly higher staining with nitrotyrosine, 4-HNE, pH2AX, or pATM compared to other chemotherapies (data not shown).
DISCUSSION
Mechanisms underlying chemotherapy-induced neurotoxicity are understudied despite the high prevalence and morbidity of CRCI. While valuable insights have been gathered from preclinical and human biomarker studies, identifying contributory molecular pathways supported by human data is limited by a dearth of neuropathologic studies examining patient brain tissue. We demonstrate, for the first time to our knowledge, that cancer patients treated with chemotherapy have elevated markers of oxidative stress and DNA damage in cortical neurons compared to cancer patients not treated with chemotherapy and to patients without history of cancer. Importantly, our observations highlight the relevance of human neuronal oxidative stress and DNA damage in the study of mechanisms that may contribute to CRCI.
The finding of elevated oxidative stress and DNA damage in frontal lobe cortical neurons of chemotherapy patients complements the existent CRCI literature. Frontal lobe dysfunction is supported by patient cognitive (37–39) and radiologic studies (6, 40, 41). One study reported that pediatric patients diagnosed with acute lymphoblastic leukemia had elevated oxidized phospholipids in their cerebrospinal fluid after methotrexate administration, supporting the presence of chemotherapy-induced oxidative stress in the CNS (17). Furthermore, there was an association between the level of oxidative stress and worse measures of executive function (17). In a mouse model of CRCI (22), administration of doxorubicin resulted in increased oxidative stress in the brain and worse performance on the novel object recognition test, a task that interrogates the function of the frontal cortex and hippocampus.
Elevated oxidative stress and DNA damage were seen in our chemotherapy patient cohort despite the heterogeneity of chemotherapeutic regimens and the variability in how readily they permeate the blood–brain barrier (BBB), suggesting that some mechanisms underlying chemotherapy-induced neurotoxicity and CRCI may converge on general downstream pathways. For example, chemotherapeutic agents such as doxorubicin that do not readily cross the BBB (46) may induce oxidative stress and DNA damage in the brain via peripheral circulating cytokines including TNF-α. These proinflammatory cytokines can across the BBB (47) to mediate oxidative damage and mitochondrial dysfunction (21), as well as microglial activation (48). Chemotherapeutic agents that can cross the BBB such as cisplatin and paclitaxel (49) may cause oxidative stress and DNA damage in CNS cell populations both indirectly through peripheral circulating cytokines and directly by inducing mitochondrial dysfunction and generation of reactive oxygen species (50, 51) in the brain. Although some general downstream pathways may be shared by different chemotherapies, we cannot exclude the possibility that specific agents or chemotherapy class types (i.e. anthracyclines, alkylating agents, taxols, etc.) are more neurotoxic than others.
Because the mean age of patients in our study was over 65-years-old, some degree of age-related oxidative damage and DNA damage is expected in cortical neurons (52, 53). It should be noted, however, that the cancer patients treated with chemotherapy in this study were younger on average (64.6-years-old) than the cancer patients not treated with chemotherapy (71.7-years-old) and patients without history of cancer (68.9-years-old). In addition, cancer-associated cachexia and other cancer-induced metabolic derangements are associated with oxidative stress (54). Although cancer itself and/or its associated metabolic derangements may have contributed to the elevated oxidative stress and DNA damage in cortical neurons of cancer patients treated with chemotherapy in our study, we attempted to address these potential confounders by including a cohort of cancer patients not treated with chemotherapy in our analysis.
Given that the cognitive deficits of CRCI (9, 55) and observed radiologic brain alterations (55–57) can abate over time, we looked at neuronal oxidative stress and DNA damage at different time intervals between last administration of chemotherapy and death. Our expectation was that these markers would be maximally elevated in the short-term interval (<1 month) and lowest in the long-term interval (>6 months). However, we found no statistically significant difference in staining for any of the markers of oxidative stress and DNA damage by interval from last administration of chemotherapy to death. One possible explanation is that a source of oxidative stress and DNA damage persists in the CNS of chemotherapy patients after treatment has ended. Ultimately, larger cohorts are needed to improve the sensitivity to detect smaller differences in markers of oxidative stress and DNA damage when chemotherapy patients are grouped by interval from last administration of chemotherapy to death and by number of cycles of chemotherapy received.
A limitation of our study is the absence of patient cognitive data. Since the patients in our cohort did not undergo formal neurocognitive testing, we cannot correlate the degree of neuronal staining for markers of oxidative stress and DNA damage with the presence or extent of cognitive dysfunction. The lack of cognitive data highlights the need for prospective chemotherapy patient cohorts to be followed with formal neurocognitive testing and for clinicians to have discussions with these patients about the scientific value of autopsies.
Additional studies will need to be performed to identify the source of oxidative stress and DNA damage in the brain, characterize the downstream molecular alterations of chemotherapy-associated oxidative stress and DNA damage in cortical neurons, correlate CNS pathology with single nucleotide polymorphisms associated with cognitive impairment in chemotherapy patients (58, 59), and investigate the effects of chemotherapy in different brain regions (e.g. temporal lobe, hippocampus, cerebellum, white matter) and in non-neuronal cell populations in the CNS of patients. Animal studies are needed to investigate the roles of specific chemotherapeutic agents and combinations of chemotherapeutic agents in the generation of oxidative stress, DNA damage, and other molecular alterations in CNS cell populations. The potential contributions of non-chemotherapy anticancer agents on neuronal oxidative stress and DNA damage also need to be determined (60).
In summary, we provide evidence, for the first time to our knowledge, that there is elevated oxidative stress and DNA damage in cortical neurons of cancer patients treated with chemotherapy compared to control patients. Modulating oxidative stress and DNA damage pathways may therefore be promising targets to ameliorate chemotherapy-induced neurotoxicity and CRCI.
ACKNOWLEDGMENTS
We would like to acknowledge the staff members of the Histology Laboratory at Brigham and Women’s Hospital (Boston, MA) for their contributions and support.
MT is supported by the National Cancer Institute of the National Institutes of Health under award number F32 CA257210. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The authors have no conflicts of interest to declare.
REFERENCES
- 1. Miller KD, Nogueira L, Mariotto AB, et al. Cancer treatment and survivorship statistics, 2019. CA Cancer J Clin 2019;69:363–85 [DOI] [PubMed] [Google Scholar]
- 2. Giglio P, Gilbert MR. Neurologic complications of cancer and its treatment. Curr Oncol Rep 2010;12:50–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ahles TA, Root JC, Ryan EL. Cancer- and cancer treatment-associated cognitive change: An update on the state of the science. J Clin Oncol 2012;30:3675–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wefel JS, Schagen SB. Chemotherapy-related cognitive dysfunction. Curr Neurol Neurosci Rep 2012;12:267–75 [DOI] [PubMed] [Google Scholar]
- 5. Koppelmans V, de Ruiter MB, van der Lijn F, et al. Global and focal brain volume in long-term breast cancer survivors exposed to adjuvant chemotherapy. Breast Cancer Res Treat 2012;132:1099–106 [DOI] [PubMed] [Google Scholar]
- 6. McDonald BC, Conroy SK, Smith DJ, et al. Frontal gray matter reduction after breast cancer chemotherapy and association with executive symptoms: A replication and extension study. Brain Behav Immun 2013;30 Suppl:S117–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Deprez S, Amant F, Smeets A, et al. Longitudinal assessment of chemotherapy-induced structural changes in cerebral white matter and its correlation with impaired cognitive functioning. JCO 2012;30: 274–81 [DOI] [PubMed] [Google Scholar]
- 8. Kesler SR, Bennett FC, Mahaffey ML, et al. Regional brain activation during verbal declarative memory in metastatic breast cancer. Clin Cancer Res 2009;15:6665–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Schagen SB, Muller MJ, Boogerd W, et al. Late effects of adjuvant chemotherapy on cognitive function: A follow-up study in breast cancer patients. Ann Oncol 2002;13:1387–97 [DOI] [PubMed] [Google Scholar]
- 10. Wefel JS, Lenzi R, Theriault RL, et al. The cognitive sequelae of standard-dose adjuvant chemotherapy in women with breast carcinoma: Results of a prospective, randomized, longitudinal trial. Cancer 2004;100:2292–9 [DOI] [PubMed] [Google Scholar]
- 11. Wefel JS, Saleeba AK, Buzdar AU, et al. Acute and late onset cognitive dysfunction associated with chemotherapy in women with breast cancer. Cancer 2010;116:3348–56 [DOI] [PubMed] [Google Scholar]
- 12. Koppelmans V, Breteler MM, Boogerd W, et al. Neuropsychological performance in survivors of breast cancer more than 20 years after adjuvant chemotherapy. JCO 2012;30:1080–6 [DOI] [PubMed] [Google Scholar]
- 13. Falleti MG, Sanfilippo A, Maruff P, et al. The nature and severity of cognitive impairment associated with adjuvant chemotherapy in women with breast cancer: A meta-analysis of the current literature. Brain Cogn 2005;59:60–70 [DOI] [PubMed] [Google Scholar]
- 14. Bijker R, Duijts SFA, Smith SN, et al. Functional impairments and work-related outcomes in breast cancer survivors: A systematic review. J Occup Rehabil 2018;28:429–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Von Ah D, Habermann B, Carpenter JS, et al. Impact of perceived cognitive impairment in breast cancer survivors. Eur J Oncol Nurs 2013;17:236–41 [DOI] [PubMed] [Google Scholar]
- 16. Hutchinson AD, Hosking JR, Kichenadasse G, et al. Objective and subjective cognitive impairment following chemotherapy for cancer: A systematic review. Cancer Treat Rev 2012;38:926–34 [DOI] [PubMed] [Google Scholar]
- 17. Caron JE, Krull KR, Hockenberry M, et al. Oxidative stress and executive function in children receiving chemotherapy for acute lymphoblastic leukemia. Pediatr Blood Cancer 2009;53:551–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lomeli N, Di K, Czerniawski J, et al. Cisplatin-induced mitochondrial dysfunction is associated with impaired cognitive function in rats. Free Radic Biol Med 2017;102:274–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Joshi G, Sultana R, Tangpong J, et al. Free radical mediated oxidative stress and toxic side effects in brain induced by the anti cancer drug adriamycin: Insight into chemobrain. Free Radic Res 2005;39:1147–54 [DOI] [PubMed] [Google Scholar]
- 20. Joshi G, Aluise CD, Cole MP, et al. Alterations in brain antioxidant enzymes and redox proteomic identification of oxidized brain proteins induced by the anti-cancer drug adriamycin: Implications for oxidative stress-mediated chemobrain. Neuroscience 2010;166:796–807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ren X, Keeney JTR, Miriyala S, et al. The triangle of death of neurons: Oxidative damage, mitochondrial dysfunction, and loss of choline-containing biomolecules in brains of mice treated with doxorubicin. Advanced insights into mechanisms of chemotherapy induced cognitive impairment (“chemobrain”) involving TNF-alpha. Free Radic Biol Med 2019;134:1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Keeney JTR, Ren X, Warrier G, et al. Doxorubicin-induced elevated oxidative stress and neurochemical alterations in brain and cognitive decline: Protection by MESNA and insights into mechanisms of chemotherapy-induced cognitive impairment (“chemobrain”). Oncotarget 2018;9:30324–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bagnall-Moreau C, Chaudhry S, Salas-Ramirez K, et al. Chemotherapy-induced cognitive impairment is associated with increased inflammation and oxidative damage in the hippocampus. Mol Neurobiol 2019;56:7159–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Krynetskiy E, Krynetskaia N, Rihawi D, et al. Establishing a model for assessing DNA damage in murine brain cells as a molecular marker of chemotherapy-associated cognitive impairment. Life Sci 2013;93:605–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Conroy SK, McDonald BC, Smith DJ, et al. Alterations in brain structure and function in breast cancer survivors: Effect of post-chemotherapy interval and relation to oxidative DNA damage. Breast Cancer Res Treat 2013;137:493–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Manchon JF, Dabaghian Y, Uzor NE, et al. Levetiracetam mitigates doxorubicin-induced DNA and synaptic damage in neurons. Sci Rep 2016;6:25705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gibson EM, Nagaraja S, Ocampo A, et al. Methotrexate chemotherapy induces persistent tri-glial dysregulation that underlies chemotherapy-related cognitive impairment. Cell 2019;176:43–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Acharya MM, Martirosian V, Chmielewski NN, et al. Stem cell transplantation reverses chemotherapy-induced cognitive dysfunction. Cancer Res 2015;75:676–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Geraghty AC, Gibson EM, Ghanem RA, et al. Loss of adaptive myelination contributes to methotrexate chemotherapy-related cognitive impairment. Neuron 2019;103:250–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kesler S, Janelsins M, Koovakkattu D, et al. Reduced hippocampal volume and verbal memory performance associated with interleukin-6 and tumor necrosis factor-alpha levels in chemotherapy-treated breast cancer survivors. Brain Behav Immun 2013;30:S109–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dietrich J, Han R, Yang Y, et al. CNS progenitor cells and oligodendrocytes are targets of chemotherapeutic agents in vitro and in vivo. J Biol 2006;5:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Christie LA, Acharya MM, Parihar VK, et al. Impaired cognitive function and hippocampal neurogenesis following cancer chemotherapy. Clin Cancer Res 2012;18:1954–65 [DOI] [PubMed] [Google Scholar]
- 33. Nokia MS, Anderson ML, Shors TJ. Chemotherapy disrupts learning, neurogenesis and theta activity in the adult brain. Eur J Neurosci 2012;36:3521–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Winocur G, Berman H, Nguyen M, et al. Neurobiological mechanisms of chemotherapy-induced cognitive impairment in a transgenic model of breast cancer. Neuroscience 2018;369:51–65 [DOI] [PubMed] [Google Scholar]
- 35. Gibson EM, Monje M. Emerging mechanistic underpinnings and therapeutic targets for chemotherapy-related cognitive impairment. Curr Opin Oncol 2019;31:531–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nguyen LD, Ehrlich BE. Cellular mechanisms and treatments for chemobrain: Insight from aging and neurodegenerative diseases. EMBO Mol Me 2020;12:e12075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yao C, Bernstein LJ, Rich JB. Executive functioning impairment in women treated with chemotherapy for breast cancer: A systematic review. Breast Cancer Res Treat 2017;166:15–28 [DOI] [PubMed] [Google Scholar]
- 38. Hodgson KD, Hutchinson AD, Wilson CJ, et al. A meta-analysis of the effects of chemotherapy on cognition in patients with cancer. Cancer Treat Rev 2013;39:297–304 [DOI] [PubMed] [Google Scholar]
- 39. Jansen CE, Miaskowski C, Dodd M, et al. A metaanalysis of studies of the effects of cancer chemotherapy on various domains of cognitive function. Cancer 2005;104:2222–33 [DOI] [PubMed] [Google Scholar]
- 40. Li M, Caeyenberghs K. Longitudinal assessment of chemotherapy-induced changes in brain and cognitive functioning: A systematic review. Neurosci Biobehav Rev 2018;92:304–17 [DOI] [PubMed] [Google Scholar]
- 41. Kesler SR, J.S. Kent JS, O'Hara R. Prefrontal cortex and executive function impairments in primary breast cancer. Arch Neurol 2011;68:1447–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Blair JA, Wang C, Hernandez D, et al. Individual case analysis of postmortem interval time on brain tissue preservation. PLoS One 2016;11:e0151615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Liou GY, Storz P. Detecting reactive oxygen species by immunohistochemistry. Methods Mol Biol 2015;1292:97–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Burma S, Chen BP, Murphy M, et al. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J Biol Chem 2001;276:42462–7 [DOI] [PubMed] [Google Scholar]
- 45. Lee JH, Paull TT. Activation and regulation of ATM kinase activity in response to DNA double-strand breaks. Oncogene 2007;26:7741–8 [DOI] [PubMed] [Google Scholar]
- 46. Tangpong J, Cole MP, Sultana R, et al. Adriamycin-induced, TNF-alpha-mediated central nervous system toxicity. Neurobiol Dis 2006;23:127–39 [DOI] [PubMed] [Google Scholar]
- 47. Banks WA, Kastin AJ, Broadwell RD. Passage of cytokines across the blood-brain barrier. Neuroimmunomodulation 1995;2:241–8 [DOI] [PubMed] [Google Scholar]
- 48. Riazi K, Galic MA, Kuzmiski JB, et al. Microglial activation and TNFalpha production mediate altered CNS excitability following peripheral inflammation. Proc Natl Acad Sci USA 2008;105:17151–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Torre M, Feany MB. Iatrogenic neuropathology of systemic therapies. Surg Pathol Clin 2020;13:331–42 [DOI] [PubMed] [Google Scholar]
- 50. Chen Y, Jungsuwadee P, Vore M, et al. Collateral damage in cancer chemotherapy: Oxidative stress in nontargeted tissues. Mol Interv 2007;7:147–56 [DOI] [PubMed] [Google Scholar]
- 51. Conklin KA. Chemotherapy-associated oxidative stress: Impact on chemotherapeutic effectiveness. Integr Cancer Ther 2004;3:294–300 [DOI] [PubMed] [Google Scholar]
- 52. Lu T, Pan Y, Kao SY, et al. Gene regulation and DNA damage in the ageing human brain. Nature 2004;429:883–91 [DOI] [PubMed] [Google Scholar]
- 53. Castelli V, Benedetti E, Antonosante A, et al. Neuronal cells rearrangement during aging and neurodegenerative disease: Metabolism, oxidative stress and organelles dynamic. Front Mol Neurosci 2019;12:132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bjorklund G, Dadar M, Aaseth J, et al. Cancer-associated cachexia, reactive oxygen species and nutrition therapy. Curr Med Chem 2019;26:5728–44 [DOI] [PubMed] [Google Scholar]
- 55. Collins B, Mackenzie J, Tasca GA, et al. Persistent cognitive changes in breast cancer patients 1 year following completion of chemotherapy. J Int Neuropsychol Soc 2014;20:370–9 [DOI] [PubMed] [Google Scholar]
- 56. Inagaki M, Yoshikawa E, Matsuoka Y, et al. Smaller regional volumes of brain gray and white matter demonstrated in breast cancer survivors exposed to adjuvant chemotherapy. Cancer 2007;109:146–56 [DOI] [PubMed] [Google Scholar]
- 57. Dumas JA, Makarewicz J, Schaubhut GJ, et al. Chemotherapy altered brain functional connectivity in women with breast cancer: A pilot study. Brain Imaging Behav 2013;7:524–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lengacher CA, Reich RR, Kip KE, et al. Moderating effects of genetic polymorphisms on improvements in cognitive impairment in breast cancer survivors participating in a 6-week mindfulness-based stress reduction program. Biol Res Nurs 2015;17:393–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sharafeldin N, Richman J, Bosworth A, et al. Clinical and genetic risk prediction of cognitive impairment after blood or marrow transplantation for hematologic malignancy. J Clin Oncol 2020;38:1312–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Teppo HR, Soini Y, Karihtala P. Reactive oxygen species-mediated mechanisms of action of targeted cancer therapy. Oxid Med Cell Longev 2017;2017:1485283. [DOI] [PMC free article] [PubMed] [Google Scholar]