

Abstract
Lentiviral vectors (LVs) are robust delivery vehicles for gene therapy as they can efficiently integrate transgenes into host cell genomes. However, LVs with lengthy or complex expression cassettes typically are produced at low titers and have reduced gene transfer capacity, creating barriers for clinical and commercial applications. Modifications of the packaging cell line and methods may be able to produce complex vectors at higher titer and infectivity and may improve production of many different LVs. In this study, we identified two host restriction factors in HEK293T packaging cells that impeded LV production, 2′-5′-oligoadenylate synthetase 1 (OAS1) and low-density lipoprotein receptor (LDLR). Knocking out these two genes separately led to ∼2-fold increases in viral titer. We created a monoclonal cell line, CRISPRed HEK293T to Disrupt Antiviral Response (CHEDAR), by successively knocking out OAS1, LDLR, and PKR, a previously identified factor impeding LV titers. Packaging in CHEDAR yielded ∼7-fold increases in physical particles, full-length vector RNA, and vector titers. In addition, overexpressing transcription elongation factors, SPT4 and SPT5, during packaging improved the production of full-length vector RNA, thereby increasing titers by ∼2-fold. Packaging in CHEDAR with over-expression of SPT4 and SPT5 led to ∼11-fold increases of titers. These approaches improved the production of a variety of LVs, especially vectors with low titers or with internal promoters in the reverse orientation, and may be beneficial for multiple gene therapy applications.
Keywords: gene and cell therapy, CAR-T, hemoglobinopathy, lentiviral vector, packaging, HEK293T cells
Graphical Abstract
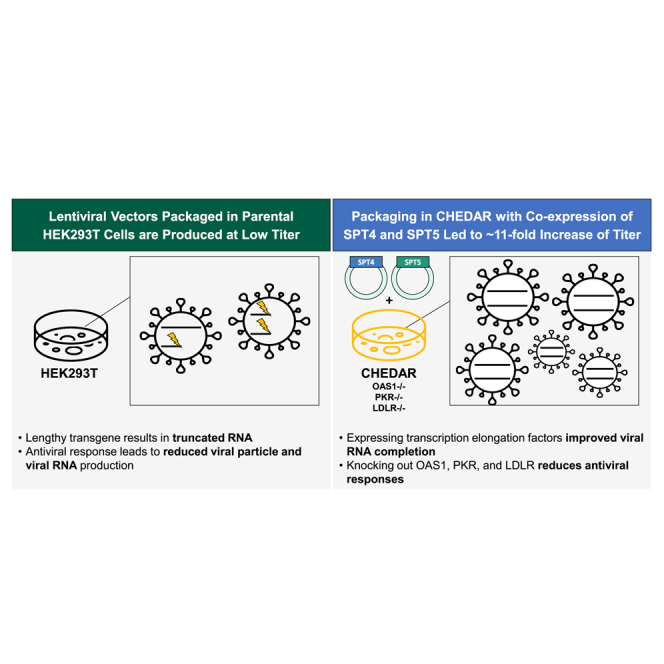
Lentiviral vectors with lengthy or complex expression cassettes are produced at low titer and infectivity, creating barriers to clinical gene therapy applications. We elucidated mechanisms behind low titer and infectivity and showed that packaging with transcription elongation factors in a genetically modified cell line, CHEDAR, increased titer of β-globin vector by ∼11-fold.
Introduction
Transplantation of autologous hematopoietic stem cells modified by lentiviral-based gene therapy has successfully treated multiple genetic blood cell diseases.1, 2, 3, 4 Lentiviral vectors (LVs) allow integration of transgenes into the host cell genomes of both dividing and nondividing cells, providing long-term stable expression of the gene of interest. However, we and others have observed that LV titers and infectivity decrease with increasing proviral length, resulting in less efficient transduction of patient cells as well as increased costs for clinical and commercial applications. Optimizing the expression cassette is one strategy to create LVs with optimal titer, infectivity, and expression5,6 but may be a prolonged process and have to be applied to each individual LV. Improvements of the vector packaging platform or the manufacturing protocol can provide a global solution to the production of many different LVs.
Recent research has shown that many cellular restriction factors (RFs) inhibit specific steps of the lentiviral life cycle.7, 8, 9 Some RFs are inducible by sensing viral components and activating the interferon (IFN) signaling cascade, while other RFs are ubiquitously expressed for direct antiviral functions. Ferreira et al. reported that LV production titer is not limited by induced intracellular innate immunity in HEK293T cells, because there was no detectable IFN cytokine release during the packaging process.10 This is likely due to the large T-antigen (TAg) and adenovirus E1A, which are expressed in HEK293T and inactivate the tumor suppressors p53, IRF3, and other IFN-dependent transcription downstream of RNA and DNA sensing.11, 12, 13, 14
On the other hand, constitutively expressed antiviral effectors appeared to regulate vector production in HEK293T cells. An example is protein kinase R (PKR), an IFN-stimulated gene that regulates protein synthesis. PKR is constitutively expressed in all tissues in an inactive form and is upregulated by type I and type III IFNs.15 PKR can be activated by TAR sequence or double-stranded RNA to inhibit general translation and hence viral protein production.16,17 Knocking out PKR in HEK293T cells increased titers of LVs, particularly for vectors with internal promoters in the reverse orientation, a common configuration for β-globin expressing LVs.18,19 Based on these previous studies, it is conceivable that the constitutively expressed antiviral effectors can still restrict LV production in HEK293T cells.
Moreover, RFs are not limited only to antiviral effectors but also include genes that regulate the lentiviral life cycle. To date, the most common envelope glycoprotein used to pseudotype LVs is the vesicular stomatitis virus spike protein G (VSVG), due to its robust and pantropic infectivity. The low-density lipoprotein receptor (LDLR) serves as the major entry port for VSVG-pseudotyped LVs, while other LDLR family members serve as alternative receptors.20,21 Otahal et al. showed that LDLR prematurely interacts with VSVG in an ER-Golgi intermediate compartment and reroutes the LDLR-VSVG complex to aggresome/autophagosome degradation prior to particle release.22 Therefore, LDLR is likely to be an RF that regulates the levels of VSVG that are available for vector pseudotyping. The effects on vector production from RFs that regulate antiviral responses or interfere in the lentiviral life cycle remain to be fully explored.
Another impediment to efficient vector production is the truncation of vRNA (vRNA). Previously we reported that vRNAs were truncated in a vector length-dependent manner in packaging cells and these truncated genomes were exported into the viral particles.19 These truncated vRNAs failed reverse transcription at the first strand transfer step due to the absence of the 3′ long terminal repeat (LTR) and could not form the double-stranded viral DNA to be integrated into the host cell genome. Expression during packaging of Tat, an HIV-1 accessory protein that functions to increase transcriptional processivity, modestly increased titers and the levels of complete vRNA, but the percentage of complete vRNA did not increase. New strategies are needed to improve genomic RNA completeness for more efficient complex LVs. Here, we conducted a targeted CRISPR-Cas9-mediated knockout (KO) screen in HEK293T cells to disrupt RFs that inhibit specific steps of the lentiviral life cycle. We showed that knocking out PKR, OAS1, and LDLR additively increased the titer of various LVs by ∼7-fold, particularly complex and lengthy LVs with low titers. We named the triple-KO cell line CRISPRed HEK293T to Disrupt Antiviral Response (CHEDAR). In addition, overexpressing transcription elongation factors SPT4 and SPT5 improved vRNA completeness, thereby increasing the titer of LVs by ∼2-fold. Packaging with the transcription elongation factors in CHEDAR cell line increased titer of the β-globin vector by ∼11-fold. These modifications of the packaging cell line and protocol should support the applications of different LVs in gene and cell therapy by improving the yield of vector production.
Results
Knocking out IFNAR1, ATR, OAS1, and LDLR in 293T cells increased titers
To identify RFs that limit LV production, we conducted a targeted CRISPR-Cas9 KO screen in HEK293T cells with a focus on 16 genes, each of which regulates one of the following biological properties: the immune response, DNA damage response, receptor-mediated virus entry, and transcription. The Cas9 and single guide RNAs (sgRNAs) were delivered to HEK293T cells as ribonucleoprotein (RNP) via electroporation, and the edited cells were sorted for single-cell clones (Figure S1A). KO or knockdown (KD) clones were validated on the genomic level by Inference of CRISPR Edits (ICE) analysis and on the protein level by western blot or flow cytometry. The sequences of the sgRNAs used in this study and the genomic profile of the clones are listed in Table 1 and Table S1.
Table 1.
Genomic profile of KO cells assessed by ICE Synthego analysis
| Gene | sgRNA sequence | Indel % | KO score | Indel pattern |
|---|---|---|---|---|
| IFNAR1 | AAACACTTCTTCATGGTATG | 99 | 99 | 99% + 1 |
| OAS1 | CTGAAGGAAAGGTGCTTCCG | 96 | 96 | 49% + 2 47% + 1 |
| ATR | AAAGTGCTAGCTGGTTGTGC | 63 | 63 | 35% −2 33% 0 28% −13 |
| LDLR | GACAACGGCTCAGACGAGCA | 93 | 93 | 48% + 1 45% −11 |
Isogenic cells were harvested for genomic DNA extraction. The sgRNA targeted region was PCR amplified and sent for Sanger sequencing. The sequencing results were then analyzed using ICE Synthego.
These single-cell clones were expanded to test whether they increased the titer of the Lenti/βAS3-FB LV, which is known to have low titer and infectivity. Previously described by Romero et al. and Han et al.,19,23 Lenti/βAS3-FB is an 8.9-kb LV carrying a complex anti-sickling β-globin gene cassette in reverse orientation, as shown in Figure S2. PKR−/− cells were used as a positive control for the packaging process because we previously showed that knocking out PKR increased the titers of reverse-oriented LVs, such as Lenti/βAS3-FB.24
Because homozygous deletion of ATR leads to chromosome breaks and proliferative failure in culture,25, 26, 27 we selected an ATR KD clone with 63% indel and 63% KO score that showed a significant reduction in protein expression (Table 1 and Figure 1A). Interferon Alpha and beta receptor subunit 1 (IFNAR1), OAS1, and LDLR were successfully knocked out, as shown in Figures 1A and 1B. Unstimulated CD34+ hematopoietic stem and progenitor cells (HSPCs) are known to have no or low expression of LDLR28 and therefore were used as a negative control when measuring LDLR expression in the LDLR KO cells by flow cytometry (Figure 1B).
Figure 1.

Knocking out IFNAR1, ATR, OAS1, and LDLR in 293T cells increased titers
(A) IFNAR1, ATR, and OAS1 protein expression in parental and gene-edited cells measured by western blot. HEK293T cells were edited by CRISPR-Cas9 targeting each of the genes. The edited cells were sorted for single-cell clones, and an isogenic clone with no parental allele, except ATR, was expanded for protein expression analysis. Because ATR is an essential gene for cell survival, an ATR KD clone with 33% parental allele remaining was selected. β-Actin and Vinculin were used as the loading controls. (B) LDLR protein expression in parental, LDLR−/− 293T cells, and unstimulated CD34+ HSPCs measured by flow cytometry. Unstimulated CD34+ HSPCs are known to not express LDLR and were used as a negative control. A mouse IgG1 antibody was used as the isotype control. (C) Titers of Lenti/βAS3-FB packaged in parental and gene-edited cells (n = 9–29 dishes of identical cultures from three to 10 independent experiments; bars represent mean with SD; unpaired t test, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001). PKR−/− cells are known to increase LV titers and were used as a positive control to evaluate titer increase. LVs in unconcentrated viral supernatant were assayed for titer by transducing HT29 cells at 10-fold serial dilution and VCN measured by ddPCR.
The targeted CRISPR-Cas9 screen revealed that disruption individually of IFNAR1, OAS1, ATR, and LDLR genes each increased titers by ∼2-fold (Figure 1C; n = 9–29 dishes of identical cultures from three to 10 independent experiments; bars represent mean with standard deviation (SD); unpaired t test, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001). The PKR−/− cells increased titer by 4.1- ± 1.1-fold. The IFNAR1−/− cells increased titer by 2.4- ± 0.7 -fold. The OAS1−/− cells increased titer by 2.3- ± 0.8-fold. The ATR knockdown cells increased titer by 2.2- ± 0.8-fold. The LDLR−/− cells increased titer by 2.8- ± 0.9-fold. Knocking out RLR, RIG-I, MDA5, STING, TLR3, APOBEC, TRIM5a, TRIM56, ZAP, TASOR, and PCF11 in HEK293T cells did not increase the titer of Lenti/βAS3-FB more than 2-fold (Figure S1B; n = 3–22 dishes of identical cultures from one to six independent experiments; bars represent mean with SD). A potential explanation is that HEK293T cells do not have intact cellular pathways for the actions of these genes. Therefore, we decided to not prioritize these targets for this study.
Decreased titers are associated with rescued LDLR and OAS1 expression
To validate the RFs that increased titer, we restored the protein expression of the four RFs in their respective KO clones. The wild-type (WT) cDNAs of the longest open reading frames were cloned into a lentiviral backbone with an IRES-GFP reporter gene driven by the MND U3 promoter. The KO cells were transduced with a lentivirus encoding the WT cDNA linked to ires-GFP or a lentivirus encoding MND-GFP as a transduction control. The cells were subsequently sorted for the GFP+ population to exclude the non-transduced cells.
We first confirmed the restoration of the protein expressions of LDLR, OAS1, ATR, and IFNAR1 in the KO/KD cells by western blot or flow cytometry (Figures 2A, 2B, 2D, S3A, and S3C). The cells transduced with lentivirus encoding the WT cDNAs showed protein expression levels that were comparable with expression in the parental HEK293T cells, while the cells transduced with the control MND-GFP LV did not show restoration of the target proteins.
Figure 2.

Restoring protein expression of LDLR and OAS1 decreased titers
(A) LDLR protein expression in parental 293T, LDLR−/− 293T, LDLR−/− 293T transduced with Lenti/GFP, and LDLR−/− 293T transduced with Lenti/LDLR measured by flow cytometry. LDLR−/− cells were transduced with an LV encoding LDLR to restore its expression, or an LV encoding GFP as a transduction control. (B) Percentage of LDLR+ cells in parental, LDLR−/−, and LDLR restored cells measured by flow cytometry. (C) Titers of Lenti/βAS3-FB packaged in parental 293T, LDLR−/− 293T, LDLR−/− 293T transduced with Lenti/GFP, and LDLR−/− 293T transduced with Lenti/LDLR. Because Lenti/LDLR carries an ires-GFP cassette, all transduced cells were sorted for the GFP+ population. The sorted GFP+ cells were expanded for VCN analysis by ddPCR and packaging (n = 9 dishes of identical cultures from three independent experiments; bars represent mean with SD; unpaired t test, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001). (D) OAS1 protein expression in parental 293T, OAS1−/− 293T, OAS1−/− 293T transduced with Lenti/GFP, and OAS1−/− 293T transduced with Lenti/OAS1 measured by western blot. (E) Titers of Lenti/βAS3-FB packaged in parental 293T, OAS1−/− 293T, OAS1−/− 293T transduced with Lenti/GFP, and OAS1−/− 293T transduced with Lenti/OAS1 (n = 12 dishes of identical cultures from four independent experiments; bars represent mean with SD; unpaired t test, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001).
Next, we packaged Lenti/βAS3-FB in parental HEK293T, KO cells, KO cells transduced with an LV encoding the corresponding cDNA, or KO cells transduced with control MND-GFP LV. Restoring LDLR expression decreased the titer to the WT level, while the LDLR−/− cells transduced only with MND-GFP showed persistence of the increased titer (n = 9 dishes of identical cultures from three independent experiments; bars represent mean with SD; unpaired t test, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001). Restoring OAS1 expression decreased titer compared with the OAS1−/− cells and OAS1−/− cells transduced with MND-GFP (n = 12 dishes of identical cultures from four independent experiments; bars represent mean with SD; unpaired t test, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001). The decrease in titer in the cells in which LDLR and OAS1 were restored suggested that the titer changes were associated with these genes. Although we observed titer increase in multiple IFNAR1−/− clones (data not shown), restoring IFNAR1 and ATR did not decrease titers (Figures S3B and S3D; n = 8 or 9 dishes of identical cultures from three independent experiments; bars represent mean with SD; unpaired t test; ns, not significant). The failure to restore decreased titers could be due to the introduction of an isoform of the protein that was not responsible for the antiviral activity observed upon gene disruption.
Combining OAS1, LDLR, and PKR gene knockouts led to increased physical particle formation, RNA production, and titers
Because OAS1, LDLR, and PKR are likely to have nonredundant functions, it is conceivable that knocking out multiple RFs may additively improve the vector production process. Next, we consecutively knocked out these RFs to study the effects on titer. Cas9 and sgRNA targeting OAS1 were electroporated into the PKR−/− isogenic clone. After confirming the cutting efficiency at OAS1, some of the edited cells were sorted for single-cell clones; other cells were again electroporated with Cas9 and sgRNA targeting LDLR. These cells were used again to further knock out IFNAR1 and then ATR. Electroporation of two sgRNAs simultaneously was avoided to minimize chromosome translocation. The sgRNAs were introduced in the following order to generate single, double, triple, quadruple, and quintuple KO/KD cells: PKR, OAS1, LDLR, IFNAR1, and ATR.
Lenti/βAS3-FB was packaged in the single-, double-, and triple-KO cells in parallel and the unconcentrated viruses titered on HT-29 cells. As shown in Figure 3A (n = 9 dishes of identical cultures from three independent experiments; bars represent mean with SD; unpaired t test, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001), the OAS1 PKR double-KO cells increased titer by 5.9- ± 1.8-fold over the parental 293T cells. The OAS1 PKR double-KO cells slightly increased titer compared with the PKR−/− cells (4.5- ± 1.5-fold over the parental 293T cells). Furthermore, the OAS1 PKR LDLR triple-KO cells showed a significant increase in titer compared with the single KO cells, resulting in a 6.7- ± 0.7-fold increase over parental 293T cells. OAS1 PKR LDLR IFNAR1 quadruple-KO cells as well as OAS1 PKR LDLR IFNAR1 ATR quintuple-KO/KD cells did not further increase titer (Figure S4; n = 3 dishes of identical culture from one independent experiment; base represent mean with SD). These data suggest that OAS1, PKR, and LDLR triple-KO led to the highest titer, and the data were reproducible in more than one clone (Figure S4). In addition, these edited cells shared a similar morphology and growth rate to the parental 293T cells (Figure S5). We named the OAS PKR LDLR triple-KO cells CHEDAR.
Figure 3.

Knocking out PKR, OAS1, and LDLR in 293T cells additively increased titer, RNA, and physical particles
(A) Fold difference of titers of Lenti/βAS3-FB packaged in single, double, and triple-KO cells (n = 12 dishes of identical cultures from four independent experiments; bars represent mean with SD; unpaired t test, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001). The double-KO isogenic clones were created by electroporating RNP targeting the OAS1 gene in the PKR−/− isogenic clones and selecting an isogenic clone with OAS1 and PKR knocked out. The triple-KO isogenic clones were created by electroporating RNP targeting LDLR gene in the PKR and OAS1 double-KO clone and selecting an isogenic clone with OAS1, PKR, and LDLR knocked out. (B and C) The absolute quantification and percentage of complete vRNA in Lenti/βAS3-FB viral particles measured by ddPCR (n = 9 dishes of identical cultures from three independent experiments; bars represent mean with SD; unpaired t test, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001). (D) Concentrations of physical particles in Lenti/βAS3-FB unconcentrated virus quantified by p24 ELISA (n = 9 dishes of identical cultures from three independent experiments; bars represent mean with SD; unpaired t test, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001). (E) Titers of Lenti/βAS3-FB, PYC-CAR, EFS-ADA, and Mini-G packaged in parental 293T and OAS1, PKR, and LDLR KO cells (n = 9 dishes of identical cultures from three independent experiments; bars represent mean with SD; unpaired t test, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001).
Next, we investigated the vRNA completeness and physical particle formation of the LVs produced from these genome-engineered cells. Using the method previously described in Han et al.24 to quantify complete vRNA, we observed that CHEDAR led to the highest absolute level of complete vRNA, which was significantly more than from the double-KO cells and the parental cells (Figure 3B; n = 9 dishes of identical cultures from three independent experiments; bars represent mean with SD; unpaired t test, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001). None of the different genome modifications affected the percentage of RNA completeness (Figure 3C; n = 9 dishes of identical cultures from three independent experiments; bars represent mean with SD; unpaired t test, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001). CHEDAR led to a significant increase in the concentration of physical particles compared with the parental HEK293T cells but did not further increase physical particles release compared with the PKR KO alone (Figure 3D; n = 9 dishes of identical cultures from three independent experiments; bars represent mean with SD; unpaired t test, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001). These data suggested that CHEDAR produced more physical particles and LVs with higher absolute complete vRNA yet a similar percentage of complete vRNA, resulting in higher titers.
To test whether the engineered packaging cell can be used to improve the titer of other LVs, we packaged four different LVs in CHEDAR cells. The vector maps are shown in Figure S2. PYC-CAR is an LV encoding a chimeric antigen receptor (CAR) that targets B cell maturation antigen (BCMA), an antigen present on multiple myeloma cells.29 The CAR was constructed with four anti-BCMA single-chain variable fragments, fused to the CD137 (4-1BB) co-stimulatory and CD3ζ signaling domains under the control of an MND promoter.29 Mini-G is a reverse-oriented β-globin vector with redefined enhancer element boundaries of the β-globin locus control region.6 The elongation factor-α gene short (EFS)-adenosine deaminase (ADA) vector consists of the human EFS promoter driving the expression of a codon-optimized human ADA gene cassette followed by the wPRE, all in the sense (forward) orientation.30
Our engineered cell line was able to improve the titer of all four LVs (Figure 3E). We were able to improve the titer of Lenti/βAS3-FB by 5.9- ± 1.7-fold, PYC-CAR by 4.2- ± 0.9-fold, Mini-G by 3- ± 0.5-fold, and EFS-ADA by 1.8- ± 0.3-fold (n = 9 dishes of identical cultures from three independent experiments; bars represent mean with SD; unpaired t test, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001). These data suggest that the OAS1 PKR LDLR triple-KO cells can be used to improve the packaging process for different LVs, especially LVs with low titer and LVs in reverse orientation.
Packaging with transcription elongation factors improved RNA completeness and titer
In our previous study, we reported that Lenti/βAS3-FB vRNA is truncated during vRNA production, and the truncated RNA cannot be reverse transcribed and integrated into the host cell genome.19 We hypothesized that packaging cells overexpressing transcription elongation factors SPT4 and SPT5 may result in more complete vRNA. SPT4 and SPT5 are components of the 5,6-Dichloro-1-β-D-ribofuranosylbenzimidazole sensitivity-inducing factor complex (DSIF complex), which regulates transcription elongation by RNA polymerase II (RNA Pol II). SPT4-SPT5 complex promotes Pol II processivity by traveling with Pol II throughout transcription elongation.31 Recent evidence suggests that the SPT4-SPT5 complex binds to the DNA exit region on Pol II, assisting the rewinding of DNA and preventing aberrant backtracking of Pol II.32,33
To test our hypothesis, we packaged Lenti/βAS3-FB with co-transfection of SPT4/5 expression plasmids. SPT4 and SPT5 were cloned into expression plasmids under the control of an MND promoter and transiently transfected into the 293T cells with the rest of the packaging plasmids 24 h after plating the cells. Four conditions were tested: 5 μg of GFP plasmids alone, 2.5 μg of SPT4 plasmids with 2.5 μg of GFP plasmids, 2.5 μg of SPT5 plasmids with 2.5 μg of GFP plasmids, and 2.5 μg of SPT4 plasmids with 2.5 μg of SPT5 plasmid. GFP plasmids were used as fillers to ensure an equal mass of plasmids were transfected in each condition.
We first assessed the effect of SPT4 and SPT5 on vRNA production (Figure 4A; n = 9 dishes of identical cultures from three independent experiments; bars represent mean with SD; unpaired t test, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001). Compared with the transfection control, adding SPT5 alone increased the level of initial vRNA by 1.7- ± 0.4-fold, the level of intermediate vRNA by 1.9- ± 0.5-fold, and the level of complete vRNA by 2.4- ± 1.5-fold; adding SPT4 alone increased the level of initial vRNA by 1.5- ± 0.4-fold, the level of intermediate vRNA by 1.6- ± 0.5-fold, and the complete vRNA by 2- ± 1.2-fold. These data suggest that there was a gradual enrichment in vRNA level toward the 3′ end, when the transcription elongation factors were added. Moreover, adding both SPT4 and SPT5 expression plasmids during packaging increased the level of initial vRNA by 2.2- ± 0.6-fold, the level of intermediate vRNA by 2.4- ± 0.7-fold, and the level of complete vRNA by 4.4- ± 2-fold compared with the transfection control. We did not observe an increase in the percentage of intermediate RNA, probably because the primers and probes to detect the initial vRNA (in RU5) and intermediate vRNA (in the primer binding site sequence) are very close to each other (Figure 4B; n = 9 dishes of identical cultures from three independent experiments; bars represent mean with SD; unpaired t test, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001). Nevertheless, we indeed observed a significant increase in the percentage of complete vRNA when both SPT4 and SPT5 were added. These data suggest that SPT4 and SPT5 additively facilitates transcription elongation.
Figure 4.

Packaging with transcription elongation factors SPT4/5 increased vRNA completeness and vector titer
(A) The fold difference of initial, intermediate, and complete vRNA compared with the transfection control and (B) a percentage of intermediate and complete vRNA in parental HEK293T cells with SPT4/5 plasmids or a filler plasmid (n = 9 dishes identical cultures from three independent experiments; bars represent mean with SD; unpaired t test, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001). The percentage of intermediate vRNA was calculated as the copies of intermediate vRNA divided by the copies of initial RNA. The percentage of complete vRNA was calculated as the copies of complete vRNA divided by the copies of initial vRNA. (C) Titers of Lenti/βAS3-FB packaged in parental HEK293T cells with SPT4/5 plasmids or a filler plasmid as the transfection control (an unpackageable GFP plasmid without lentiviral sequences) (n = 9 dishes identical cultures from three independent experiments; bars represent mean with SD; unpaired t test, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001). (D) Concentration of physical particles measured by p24 ELISA (n = 9–12 dishes with identical cultures from three to four independent experiments; bars represent mean with SD; unpaired t test, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001).
Next, we examined whether the increase in vRNA correlated with titer (Figure 4C; n = 9 dishes of identical cultures from three independent experiments; bars represent mean with SD; unpaired t test, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001). Compared with the transfection control, adding SPT5 alone increased titer by 1.4- ± 0.2-fold, adding SPT4 alone increased titer by 1.3- ± 0.1-fold, and adding both SPT4 and SPT5 increased titer by 2.2- ± 0.4-fold.
We also observed increased levels of physical particles measured by p24 ELISA when SPT4 and SPT5 were added (Figure 4D; n = 9–12 dishes of identical cultures from three to four independent experiments; bars represent mean with SD; unpaired t test, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001). This observation suggested that SPT4 and SPT5 potentially enhanced transcription elongation not only for the transfer plasmid but also the packaging and envelope plasmids, resulting in increased physical particle release.
Packaging with transcription elongation factors in the CHEDAR cell line increased physical particle formation, RNA production, and titers
Finally, we explored the combinatorial effects of packaging with the transcription elongation factors in the CHEDAR cell line. As shown in Figure 5A, packaging in the parental HEK293T cells with the addition of SPT4 and SPT5 increased titer by 2- ± 0.4-fold compared with the parental HEK293T transfection control. Packaging in the CHEDAR cell line with the transfection control increased titer by 7- ± 1-fold over the parental HEK293T transfection control. Packaging in the CHEDAR cell line with the addition of SPT4 and SPT5 increased titer by 10.7- ± 3.2-fold over the parental HEK293T transfection control (n = 9 dishes of identical cultures from three independent experiments; bars represent mean with SD; unpaired t test, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001). We also observed a significant increase in the level of complete vRNA, the percentage of complete and the level of physical particles when SPT4 and SPT5 were added to the CHEDAR cell line compared with the parental transfection control (Figures 5B–5D; n = 6–9 dishes of identical cultures from two to three independent experiments). Packaging with the addition of SPT4 and SPT5 in the CHEDAR cell line further improved titer and RNA production compared with the CHEDAR transfection control, suggesting that these two strategies work additively to increase titer.
Figure 5.

Packaging with SPT4 and SPT5 in the CHEDAR cell line increased titer, vRNA, and physical particles
(A) Titers of Lenti/βAS3-FB packaged in parental HEK293T cells and CHEDAR cells with SPT4/5 plasmids or a filler plasmid as the transfection control (an unpackageable GFP plasmid without lentiviral sequences) (n = 9 dishes identical cultures from three independent experiments; bars represent mean with SD; unpaired t test, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001). (B) The absolute quantification of complete vRNA and (C) the percentage of complete vRNA of Lenti/βAS3-FB packaged in parental HEK293T cells and CHEDAR cells with SPT4/5 plasmids or a filler plasmid as the transfection control. (D) The concentration of physical particles measured by p24 ELISA (n = 6–9 dishes identical cultures from two to three independent experiments; bars represent mean with SD; unpaired t test, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001).
Discussion
Low titer and infectivity of complex lentiviral vectors, like β-globin LVs and CAR-T LVs, create barriers for clinical and commercial applications of gene and cell therapy. We elucidated the mechanisms leading to low titer and infectivity: (1) RFs impeding various steps of the lentiviral life cycle; (2) vRNA truncations. RFs are not only IFN-dependent genes in innate immunity but also cellular proteins inhibiting lentiviral life cycle. We first conducted a targeted CRISPR-Cas9 mediated KO screen focusing on genes in the immune response, receptor-mediated virus entry, transcription factors, and DNA damage response pathway. We showed that knocking out OAS1, LDLR, and PKR in the HEK293T cells increased the titer of LVs, and knocking out all three genes in a monoclonal cell line, CHEDAR, further increased titer of Lenti/βAS3FB 4–8-fold higher than the titer of the vectors produced from the parental HEK293T cells.
LDLR serves as the major cellular entry port of VSVG-pseudotyped LVs, and other LDLR family proteins serve as the alternative, yet less effective, binding target.34 One initial hypothesis of the LDLR-dependent decrease of vector titer is that LVs interact with HEK293T LDLR at the plasma membrane and re-enter the packaging cells, resulting in the loss of LVs. However, Otahal er al. showed that VSVG granules colocalized with LDLR in the ER-Golgi intermediate compartment (ERGIC) and aggresome/autophagosome.35 They proposed a model that LDLR and VSVG interact prematurely in ERGIC, and these VSVG-LDLR complexes are subsequently rerouted to be degraded in aggresome/autophagosome.35 We also observed that the LDLR−/− cells and the parental HEK293T cells were equally infected (data not shown), suggesting that knocking out LDLR did not prevent LVs from reentering the cells. The latter hypothesis of aggresome/autophagosome degradation of the LDLR-VSVG complex is more likely to explain the increase in titer in the LDLR−/− cells.
The 2′-5′ oligoadenylate synthetases (OASs) are a family of antiviral proteins consisting of OAS1, OAS2, OAS3, and OASL. The OAS proteins are expressed at low levels and are augmented upon IFN induction.36 The OAS1, OAS2, and OAS3 proteins can be activated upon detecting double-stranded RNA (dsRNA) to produce 2′-5′ oligoadenylates (2-5As),37 which subsequently activates RNase L.38 RNase L degrades both cellular and vRNA, thereby inhibiting viral replication.39 While OAS1-3 proteins are well known for their RNase L-dependent activity, recent evidence suggests that OAS1 can directly inhibit viral replication independently of RNase L. Kristiansen et al. reported that OAS can be released to the extracellular space and acts as a paracrine antiviral agent without activation of RNase L. In our study, we did not observe an increase in titer packaging in RNASEL KD 293T cells, suggesting that the RNase L-independent antiviral activity may explain the increase in titer with the OAS1−/− 293T cells. Future studies to explore the mechanisms leading to the titer increase in OAS1−/− cells are needed. Although the IFN signaling is uncoupled from cytokine secretion in 293T cells, we showed that some of the antiviral effectors, such as OAS1 and PKR, are still active in 293T cells, and knocking them out improved packaging efficiency.
In addition to exploring the lentiviral RFs, we overexpressed transcription elongation factors SPT4 and SPT5 to enhance the production of complete vRNA. Expressing either SPT4 or SPT5 individually led to a ∼2-fold increase in complete vRNA. SPT4 and SPT5 worked synergistically, because expressing both transcription elongation factors together led to a ∼4-fold increase in complete vRNA and a 2-fold increase in the percentage of complete vRNA. Moreover, the transcription elongation factors showed an additive effect with the CHEDAR KO cell line to further increase complete vRNA by 1.5-fold, resulting in a 1.2-fold increase in titer. These results suggest that overexpressing transcription elongation factors facilitates the vRNA production in both parental HEK293T and the triple-KO cells. Future studies are needed to explore the addition of other transcription elongation factors.
In summary, we present a new packaging cell line, CHEDAR, with OAS1, LDLR, and PKR knocked out to prevent the antiviral responses and inhibition of the lentiviral life cycle. We also showed that including transcription elongation factors SPT4 and SPT5 improved vRNA completion. Combining the cell line and packaging with the transcription elongation factors improved titer by 10.7- ± 3.2-fold.
Methods
LV production and titration
The LVs used in this study include EFS-ADA,30 Lenti/βAS3-FB,23 Mini-G,6 and PYC-CAR. The LV packaging and titration protocols were previously described in Han et al.24 Briefly, genetically modified and WT 293T cells were plated in six-well plates and transiently transfected with fixed amount of HIV Gag/Pol, Rev, VSV-G expression plasmids, and equimolar amounts of transfer plasmids with TransIT-293 (Mirus Bio, Madison, WI). For certain experiments, SPT4 and SPT5 expression plasmids or a non-packageable GFP plasmid were added to the transfection mix. About 20 h after transfection, transfected cells were incubated in D10 containing 10 mM sodium butyrate and 20 mM HEPES for 6–8 h. Cells were then washed with PBS and cultured in fresh D10 for approximately 40 h. Viral supernatants were collected and filtered through a 0.45-μm filter. If needed, viral supernatants were concentrated by ultracentrifugation at 26,000 rpm for 90 min. Both raw and concentrated viruses were kept at −80°C for long-term storage.
Viral titers were determined by transducing HT-29 human colon carcinoma cells with different dilutions of the LVs and pelleted the cells ∼60 h after transduction. Genomic DNA was extracted using either the PureLink Genomic DNA Mini Kit (Invitrogen, Waltham, MA) or QuickExtract DNA Extraction Solution (Lucigen, Middleton, WI). Viral titers were calculated as vector copy number (VCN) times cell number at the time of transduction times dilution factor. VCN was defined as the ratio of the copies of the HIV-1 PBS region to the copies of the SDC4 endogenous reference gene and measured by Droplet Digital PCR (ddPCR). The droplet generation process was described in Hindson et al.,40 and the ddPCR cycling conditions and protocols were described previously in Han et al.24
sgRNA construction
sgRNAs were designed using the Benchling CRISPR online tool, and oligonucleotides were synthesized by Integrated DNA Technologies (San Diego, CA). sgRNAs were in vitro transcribed following the protocol as previously described.41 Briefly, sgRNA was assembled as DNA and PCR amplified to generate enough DNA templates. The DNA template was in vitro transcribed into sgRNAs using the HiScribe T7 Quick High Yield RNA Synthesis Kit (New England Biolabs; Ipswich, MA). RNA was purified using the RNeast MinElute Cleanup Kit (Qiagen; Valencia, CA), following the manufacturer's protocol.
Electroporation
At room temperature, 2 × 105 cells per condition were pelleted at 100 × g for 10 min and resuspended in 10 μL of SF solution with supplements (Lonza; Basel, Switzerland). Then 100 pmol of Cas9 was added to 240 pmol of sgRNA to form the RNP complex, and the RNP was incubated on ice for 10–20 min. The cells and RNP were combined and electroporated with the Lonza 4D Nucleofector system (program CM-130). After electroporation, cells were recovered at room temperature for 10 min and then transferred to a 24-well plate. To analyze the cutting efficiency, cells were pelleted for DNA extraction 24–48 h after electroporation using the PureLink Genomic DNA Mini Kit (Invitrogen/Thermo Fisher Scientific; Carlsbad, CA). DNA regions around the cut site were PCR amplified, and the PCR products were sent for Sanger sequencing. The cutting efficiencies were analyzed using the Synthego ICE Analysis tool (2019. v3.0). After confirming the cutting efficiency, electroporated cells were single-cell sorted at the University of California, Los Angeles (UCLA) Broad Stem Cell Research Center Flow Cytometry Core. About 2 weeks after sorting, isogenic cell clones were collected for genomic DNA extraction, PCR amplification, Sanger sequencing, and ICE analysis to identify KO or knockdown clones.
Flow cytometry
All flow cytometry analyses were performed on BD LSRFortessa and BD FACSAria II. The cells were harvested and washed with PBS and stained for 30 min at 4°C or 15 min at room temperature with the anti-LDLR antibody (R&D Systems, Minneapolis, MN) and DAPI. For fluorescence-activated cell sorting (FACS) of single cells, cells were stained with DAPI, and live cells sorted into one cell per well in 96-well plates.
Western blot
The western blot protocol was previously described by Crowell et al.43 All samples except OAS1 did not require prior treatments. To induce OAS1 expression, cells were incubated with 10 ng/mL recombinant human IFN-beta protein (R&D Systems, Minneapolis, MN) at 37°C for 24 h prior to harvesting the cells. Then 2×106 cells were collected and lysed in RIPA buffer (50 mM Tris-HCL pH8.0, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS) with HALT protease inhibitor cocktail (Thermo Fisher, Waltham, MA). To determine the protein concentration, the BCA assay was conducted using the Pierce BCA Protein assay kit (Thermo Fisher, Waltham, MA) following the manufacturer's protocol. An equal amount of proteins was run on NuPAGE 4%–12% Bis-Tris Gel (Novex), transferred onto polyvinylidene fluoride (PVDF) membranes (Millipore Sigma), and incubated with the primary antibodies at 4°C overnight. The primary antibodies used in this study include anti-SPT4 (Cell Signaling Technology, product #648285, Danvers, MA), anti-SPT5 (Santa Cruz Biotechnology, sc-390961, Dallas, TX), anti-ATR (Cell Signaling Technology, Product #13934, Danvers, MA), anti-IFNAR1 (Abcam, ab124764, Cambridge, MA), anti-β-Actin (Cell Signaling Technology, product #3700, Danvers, MA), anti-OAS1 (Cell Signaling Technology, product #14498, Danvers, MA), and anti-tubulin (abcam, ab56676). The primary antibodies were then detected with horseradish peroxidase (HRP)-conjugated secondary antibodies followed by HRP chemiluminescence (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6710009/).
vRNA analysis by ddPCR
RNA was extracted from 140 μL of unconcentrated viral supernatant using the QIAmp Viral RNA Mini Kit (Qiagen), treated with DNAse I (Invitrogen, Waltham, MA), reverse transcribed with Moloney Murine Leukemia Virus (M-MLV) reverse transcriptase (Invitrogen, Waltham, MA), as previously described in Han et al.24
The abundance of initial, intermediate, and complete RNA was quantified by ddPCR using different primer and probe sets. Initial RNA was quantified by the amplification with the R/U5 primers and probe: forward primer 5′-GCTAACTAGGGAACCCACTGCT-3′, reverse primer 5′-GGGTCTGAGGGATCTCTAGTTACCA-3′, and probe 5′-FAM- CTTCAAGTAGTGTGTGCCCGTCTGT-31ABFQ-3′ (Integrated DNA Technologies). Intermediate RNA was quantified by the amplification with the PBS primers and probe: forward primer 5′-AAGTAGTGTGTGCCCGTCTG-3′, reverse primer 5′-CCTCTGGTTTCCCTTTCGCT-3′, and probe 5′-FAM-CCCTCAGACCCTTTTAGTCAGTGTGGAAAATCTCTAG-31ABFQ-3′. Complete RNA was quantified by the amplification with the U3/R primers and probe: forward primer 5′-AGCAGTGGGTTCCCTAGTTAG-3′, reverse primer 5′-GGGACTGGAAGGGCTAATTC-3′, and probe 5′-FAM-AGAGACCCAGTACAAGCAAAAAGCAG-31ABFQ-3′. The cycling conditions were 95°C for 10 min for one cycle (94°C for 30 s and 60°C for 1 min) for 40 cycles, 10 min at 98°C for one cycle, and a 12°C hold.
P24 assay
UCLA/Center for AIDS Research (CFAR) Virology Core kindly conducted all the p24 ELISA to quantify p24 antigen concentration in unconcentrated viral supernatants using the Alliance HIV-1 p24 Antigen ELISA Kit (catalog # NEK050, PerkinElmer, Waltham, MA), following the manufacturer's manual.
Statistical analyses
Descriptive statistics, such as a number of observations, mean, and SD were reported and presented graphically for quantitative measurements. Unpaired t tests were used to compare columns for outcome measures, such as titers, the concentration of physical particles, copies, and percentage of RNA. In the case of normality assumption violation, nonparametric Wilcoxon rank-sum tests were used. For all statistical investigations, tests for significance were two-tailed. A p value of less than 0.05 significance level was considered to be statistically significant. All statistical analyses were conducted using GraphPad Prism version 8.0 (GraphPad Software, San Diego, CA).
Acknowledgments
The Flow Cytometry Core of the UCLA Eli and Edythe Broad Center of Regenerative Medicine and Stem Cell Research and the Virology Core of the UCLA Center for AIDS Research (5P30 AI028697) supported these studies. The PYC-CAR vector was created by Dr. Yvonne Chen and kindly provided by Dr. Satiro De Oliveira.
Author contributions
J.H., R.P.H., and D.B.K. conceived and designed all experiments. J.H., K.T., and C.T. helped execute portions of the experiments. D.B.K. provided financial and administrative support. J.H. wrote the manuscript. J.H. and D.B.K. approved the final manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omto.2021.11.012.
Supplemental information
and Table S1
References
- 1.Kohn D.B., Booth C., Shaw K.L., Xu-Bayford J., Garabedian E., Trevisan V., et al. Autologous ex vivo lentiviral gene therapy for adenosine deaminase deficiency. N. Engl. J. Med. 2021;384:2002–2013. doi: 10.1056/NEJMoa2027675. NEJMoa2027675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aiuti A., Cattaneo F., Galimberti S., Benninghoff U., Cassani B., Callegaro L., Scaramuzza S., Andolfi G., Mirolo M., Brigida I., et al. Gene therapy for immunodeficiency due to adenosine deaminase deficiency. N. Engl. J. Med. 2009;360:447–458. doi: 10.1056/NEJMoa0805817. [DOI] [PubMed] [Google Scholar]
- 3.Cartier N., Hacein-Bey-Abina S., von Kalle C., Bougnères P., Fischer A., Cavazzana-Calvo M., Aubourg P. [Gene therapy of x-linked adrenoleukodystrophy using hematopoietic stem cells and a lentiviral vector] Bull. Acad. Natl. Med. 2010;194:255–264. discussion 264-8. [PubMed] [Google Scholar]
- 4.de Ravin S.S., Wu X., Moir S., Anaya-O’Brien S., Kwatemaa N., Littel P., Theobald N., Choi U., Su L., Marquesen M., et al. Lentiviral hematopoietic stem cell gene therapy for X-linked severe combined immunodeficiency. Sci. Transl. Med. 2016;8:335ra57. doi: 10.1126/scitranslmed.aad8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Morgan R.A., Ma F., Unti M.J., Brown D., Ayoub P.G., Tam C., Lathrop L., Aleshe B., Kurita R., Nakamura Y., et al. Creating new β-globin-expressing lentiviral vectors by high-resolution mapping of locus control region enhancer sequences. Mol. Ther. Methods Clin. Dev. 2020;17:999–1013. doi: 10.1016/j.omtm.2020.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morgan R.A., Unti M.J., Aleshe B., Brown D., Osborne K.S., Koziol C., et al. Improved titer and gene transfer by lentiviral vectors using novel, small β-globin locus control region elements. Mol. Ther. 2020;28:328–340. doi: 10.1016/j.ymthe.2019.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Strebel K., Luban J., Jeang K.T. Human cellular restriction factors that target HIV-1 replication. BMC Med. 2009;7:48. doi: 10.1186/1741-7015-7-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kajaste-Rudnitski A., Pultrone C., Marzetta F., Ghezzi S., Coradin T., Vicenzi E. Restriction factors of retroviral replication: the example of Tripartite Motif (TRIM) protein 5α and 22. Amino Acids. 2010;39:1–9. doi: 10.1007/s00726-009-0393-x. [DOI] [PubMed] [Google Scholar]
- 9.Kajaste-Rudnitski A., Naldini L. Cellular innate immunity and restriction of viral infection: implications for lentiviral gene therapy in human hematopoietic cells. Hum. Gene Ther. 2015;26:201–209. doi: 10.1089/hum.2015.036. [DOI] [PubMed] [Google Scholar]
- 10.Ferreira C.B., Sumner R.P., Rodriguez-Plata M.T., Rasaiyaah J., Milne R.S., Thrasher A.J., Qasim W., Towers G.J. Lentiviral vector production titer is not limited in HEK293T by induced intracellular innate immunity. Mol. Ther. Methods Clin. Dev. 2020;17:209–219. doi: 10.1016/j.omtm.2019.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Juang Y.T., Lowther W., Kellum M., Au W.C., Lin R., Hiscott J., Pitha P.M. Primary activation of interferon A and interferon B gene transcription by interferon regulatory factor 3. Proc. Natl. Acad. Sci. U S A. 1998;95:9837–9842. doi: 10.1073/pnas.95.17.9837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fonseca G.J., Thillainadesan G., Yousef A.F., Ablack J.N., Mossman K.L., Torchia J., Mymryk J.S. Adenovirus evasion of interferon-mediated innate immunity by direct antagonism of a cellular histone posttranslational modification. Cell Host Microbe. 2012;11:597–606. doi: 10.1016/j.chom.2012.05.005. [DOI] [PubMed] [Google Scholar]
- 13.Ronco L.v., Karpova A.Y., Vidal M., Howley P.M. Human papillomavirus 16 E6 oncoprotein binds to interferon regulatory factor-3 and inhibits its transcriptional activity. Genes Dev. 1998;12:2061–2072. doi: 10.1101/gad.12.13.2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Levine A.J. The common mechanisms of transformation by the small DNA tumor viruses: the inactivation of tumor suppressor gene products: p53. Virology. 2009;384:285–293. doi: 10.1016/j.virol.2008.09.034. [DOI] [PubMed] [Google Scholar]
- 15.Ank N., West H., Bartholdy C., Eriksson K., Thomsen A.R., Paludan S.R. Lambda interferon (IFN-λ), a type III IFN, is induced by viruses and IFNs and displays potent antiviral activity against select virus infections in vivo. J. Virol. 2006;80:4501–4509. doi: 10.1128/JVI.80.9.4501-4509.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Williams B.R.G. PKR; a sentinel kinase for cellular stress. Oncogene. 1999;18:6112–6120. doi: 10.1038/sj.onc.1203127. [DOI] [PubMed] [Google Scholar]
- 17.Garcia M.A., Gil J., Ventoso I., Guerra S., Domingo E., Rivas C., Esteban M. Impact of protein kinase PKR in cell biology: from antiviral to antiproliferative action. Microbiol. Mol. Biol. Rev. 2006;70:1032–1060. doi: 10.1128/MMBR.00027-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hu P., Bi Y., Ma H., Suwanmanee T., Zeithaml B., Fry N.J., Kohn D.B., Kafri T. Superior lentiviral vectors designed for BSL-0 environment abolish vector mobilization. Gene Ther. 2018;25:454–472. doi: 10.1038/s41434-018-0039-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Han J., Tam K., Ma F., Tam C., Aleshe B., Wang X., Quintos J.P., Morselli M., Pellegrini M., Hollis R.P., et al. β-Globin lentiviral vectors have reduced titers due to incomplete vector RNA genomes and lowered virion production. Stem Cell Rep. 2021;16:198–211. doi: 10.1016/j.stemcr.2020.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Finkelshtein D., Werman A., Novick D., Barak S., Rubinstein M. LDL receptor and its family members serve as the cellular receptors for vesicular stomatitis virus. Proc. Natl. Acad. Sci. U S A. 2013;110:7306–7311. doi: 10.1073/pnas.1214441110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Amirache F., Lévy C., Costa C., Mangeot P.E., Torbett B.E., Wang C.X., Nègre D., Cosset F.L., Verhoeyen E. Mystery solved: VSV-G-LVs do not allow efficient gene transfer into unstimulated T cells, B cells, and HSCs because they lack the LDL receptor. Blood. 2014;123:1422–1424. doi: 10.1182/blood-2013-11-540641. [DOI] [PubMed] [Google Scholar]
- 22.Otahal A., Fuchs R., Al-Allaf F.A., Blaas D. Release of vesicular stomatitis virus spike protein G-pseudotyped lentivirus from the host cell is impaired upon low-density lipoprotein receptor overexpression. J. Virol. 2015;89:11723–11726. doi: 10.1128/JVI.01869-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Romero Z., Urbinati F., Geiger S., Cooper A.R., Wherley J., Kaufman M.L., Hollis R.P., de Assin R.R., Senadheera S., Sahagian A., et al. β-Globin gene transfer to human bone marrow for sickle cell disease. J. Clin. Invest. 2013;123:3317–3330. doi: 10.1172/JCI67930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Han J., Tam K., Ma F., Tam C., Aleshe B., Wang X., et al. β-Globin lentiviral vectors have reduced titers due to incomplete vector RNA genomes and lowered virion production. Stem Cell Rep. 2021;16:198–211. doi: 10.1016/j.stemcr.2020.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ruzankina Y., Pinzon-Guzman C., Asare A., Ong T., Pontano L., Cotsarelis G., Zediak V.P., Velez M., Bhandoola A., Brown E.J. Deletion of the developmentally essential gene ATR in adult mice leads to age-related phenotypes and stem cell loss. Cell Stem Cell. 2007;1:113–126. doi: 10.1016/j.stem.2007.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brown E.J., Baltimore D. Essential and dispensable roles of ATR in cell cycle arrest and genome maintenance. Genes Dev. 2003;17:615–628. doi: 10.1101/gad.1067403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cortez D., Guntuku S., Qin J., Elledge S.J. ATR and ATRIP: partners in checkpoint signaling. Science. 2001;294:1713–1716. doi: 10.1126/science.1065521. [DOI] [PubMed] [Google Scholar]
- 28.Baldwin K., Urbinati F., Romero Z., Campo-Fernandez B., Kaufman M.L., Cooper A.R., Masiuk K., Hollis R.P., Kohn D.B. Enrichment of human hematopoietic stem/progenitor cells facilitates transduction for stem cell gene therapy. Stem Cells. 2015;33:1532–1542. doi: 10.1002/stem.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Friedman K.M., Garrett T.E., Evans J.W., Horton H.M., Latimer H.J., Seidel S.L., Horvath C.J., Morgan R.A. Effective targeting of multiple B-cell maturation antigen-expressing hematological malignances by anti-B-cell maturation antigen chimeric antigen receptor T cells. Hum. Gene Ther. 2018;29:585–601. doi: 10.1089/hum.2018.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carbonaro D.A., Zhang L., Jin X., Montiel-Equihua C., Geiger S., Carmo M., Cooper A., Fairbanks L., Kaufman M.L., Sebire N.J., et al. Preclinical demonstration of lentiviral vector-mediated correction of immunological and metabolic abnormalities in models of adenosine deaminase deficiency. Mol. Ther. 2014;22:607. doi: 10.1038/mt.2013.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fitz J., Neumann T., Pavri R. Regulation of RNA polymerase II processivity by Spt5 is restricted to a narrow window during elongation. EMBO J. 2018;37:e97965. doi: 10.15252/embj.201797965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bernecky C., Plitzko J.M., Cramer P. Structure of a transcribing RNA polymerase II-DSIF complex reveals a multidentate DNA-RNA clamp. Nat. Struct. Mol. Biol. 2017;24:809–815. doi: 10.1038/nsmb.3465. [DOI] [PubMed] [Google Scholar]
- 33.Ehara H., Yokoyama T., Shigematsu H., Yokoyama S., Shirouzu M., Sekine S.I. Structure of the complete elongation complex of RNA polymerase II with basal factors. Science. 2017;357:921–924. doi: 10.1126/science.aan8552. [DOI] [PubMed] [Google Scholar]
- 34.Finkelshtein D., Werman A., Novick D., Barak S., Rubinstein M. LDL receptor and its family members serve as the cellular receptors for vesicular stomatitis virus. Proc. Natl. Acad. Sci. U S A. 2013;110:7306–7311. doi: 10.1073/pnas.1214441110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Otahal A., Fuchs R., Al-Allaf F.A., Blaas D. Release of vesicular stomatitis virus spike protein G-pseudotyped lentivirus from the host cell is impaired upon low-density lipoprotein receptor overexpression. J. Virol. 2015;89:11723–11726. doi: 10.1128/JVI.01869-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Baglioni C., Minks M.A., Clercq E. Structural requirements of polynucleotides for the activation of (2′-5′)An polymerase and protein kinase. Nucleic Acids Res. 1981;9:4939–4950. doi: 10.1093/nar/9.19.4939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dong B., Silverman R.H. A bipartite model of 2-5A-dependent RNase L. J. Biol. Chem. 1997;272:22236–22242. doi: 10.1074/jbc.272.35.22236. [DOI] [PubMed] [Google Scholar]
- 38.Clemens M.J., Williams B.R.G. Inhibition of cell-free protein synthesis by pppA2′ p5′ A2′ p5′ A: a novel oligonucleotide synthesized by interferon-treated L cell extracts. Cell. 1978;13:565–572. doi: 10.1016/0092-8674(78)90329-x. [DOI] [PubMed] [Google Scholar]
- 39.Kristiansen H., Scherer C.A., McVean M., Iadonato S.P., Vends S., Thavachelvam K., Steffensen T.B., Horan K.A., Kuri T., Weber F., et al. Extracellular 2′-5′ oligoadenylate synthetase stimulates RNase L-independent antiviral activity: a novel mechanism of virus-induced innate immunity. J. Virol. 2010;84:11898–11904. doi: 10.1128/JVI.01003-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hindson B.J., Ness K.D., Masquelier D.A., Belgrader P., Heredia N.J., Makarewicz A.J., Bright I.J., Lucero M.Y., Hiddessen A.L., Legler T.C., et al. High-Throughput Droplet Digital PCR System for Absolute Quantitation of DNA Copy Number. Anal. Chem. 2011;83:8604–8610. doi: 10.1021/ac202028g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Crowell P.D., Fox J.J., Hashimoto T., Diaz J.A., Navarro H.I., Henry G.H., Feldmar B.A., Lowe M.G., Garcia A.J., Wu Y.E., et al. Expansion of luminal progenitor cells in the aging mouse and human prostate. Cell Rep. 2019;28:1499–1510.e6. doi: 10.1016/j.celrep.2019.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
and Table S1