

Abstract
While cancers evolve during disease progression and in response to therapy, temporal dynamics remain difficult to study in humans due to the lack of consistent barcodes marking individual clones in vivo. We employ mitochondrial single-cell assay for transposase-accessible chromatin with sequencing to profile 163,279 cells from 9 patients with chronic lymphocytic leukemia (CLL) collected across disease course and utilize mitochondrial DNA (mtDNA) mutations as natural genetic markers of cancer clones. We observe stable propagation of mtDNA mutations over years in the absence of strong selective pressure indicating clonal persistence, but dramatic changes following tight bottlenecks including disease transformation and relapse post-therapy, paralleled by acquisition of copy number variants, changes in chromatin accessibility and gene expression. Furthermore, we link CLL subclones to distinct chromatin states, providing insight into non-genetic sources of relapse. mtDNA mutations thus mirror disease history and provide naturally-occurring genetic barcodes to enable patient-specific study of cancer subclonal dynamics.
Keywords: Chronic lymphocytic leukemia, mitochondrial DNA mutations, clonal evolution, single cell ATAC sequencing, transcriptional state
Introduction
Cancer is a clonal disease originating from a founder cell population and is typically characterized by occurrence of divergent subclones with different fitness over time through the acquisition of somatic mutations, epigenetic reprogramming, and changes in transcriptional state (1–3). When exposed to evolutionary bottlenecks in the form of systemic therapy or disease transformation, fitter clones with distinct genomic properties may emerge or be selected for, which form the basis for acquired therapeutic resistance (4,5). Long-term studies of the clonal evolution of cancer cells alongside their phenotypic characterization on a molecular and genomic level are therefore pivotal for a better understanding of evolving biology and differential therapeutic susceptibilities within heterogeneous cancer cell populations.
Chronic lymphocytic leukemia (CLL) is an opportune setting for assessing clonal evolution due to ease of access to high purity tumor cells from blood or lymphoid tissue. This, combined with the long-term nature of the disease, often characterized by an initial period of watchful waiting and subsequent disease progression requiring intermittent therapies with eventual relapse, allows for sequential sampling. Indeed, the genetic heterogeneity and clonal dynamics of CLL have been well-described based on somatic nuclear mutations during disease evolution and in response to treatment (4,6). Although identification of mutations conferring resistance has the potential to provide insights into the mechanisms of relapse, one hurdle is the identification of non-genetic contributions to disease recurrence, of which there is growing evidence (7). Related to this shortcoming is the lack of bona fide markers or in vivo barcodes that allow individual subclones to be consistently tracked over time within a patient at the single-cell level. Although somatic nuclear mutations can be utilized, these are limited to known mutations, often requiring additional targeted amplification (8) and may be subject to selective pressure.
Mitochondrial DNA (mtDNA) mutations are located on a small 16.6kB circular genome that is replicated independently from nuclear DNA. Due to the high copy number of mtDNA per cell, variant allele frequencies of mtDNA mutations (heteroplasmy) may have a wide dynamic range from <1% to 100% (homoplasmy). While mtDNA mutations at low heteroplasmy are likely functionally silent, there is mounting evidence that truncating mtDNA mutations can play a role in cancer (9,10). Recent studies have demonstrated the ability to detect mtDNA mutations in single cells based on the Assay for Transposase-Accessible Chromatin using sequencing (mtscATAC-seq) (11,12). This technology leverages naturally occurring mtDNA mutations to mark subclonal populations, providing a complementary and unbiased approach to the temporal study of clonal evolution. Of note, mtscATAC-seq enables the pairing of clonal information based on mtDNA mutations and inferred chromosomal copy number variants (CNV) with an accessible chromatin readout, thus allowing the characterization of non-genetic mechanisms of clonal evolution (13).
Here, we evaluate the chromatin and CNV changes as well as mtDNA mutations in serially collected peripheral blood samples from 9 CLL patients across a span of up to 10 years and across different clinical scenarios. These include watchful waiting (W/W), chemoimmunotherapy with fludarabine, cyclophosphamide and rituximab (FCR), targeted inhibition of Bruton’s tyrosine kinase (BTK) through ibrutinib, and immunotherapy in the form of reduced-intensity conditioning allogeneic transplantation (RIC allo-HSCT) that reflect the evolving therapeutic strategies for CLL (14). We also track CLL subclones during their progression to Richter’s syndrome (RS), the transformation to an aggressive lymphoma which is associated with dismal prognosis (15). We observe mtDNA mutations follow natural disease evolution and their profiles are dramatically shaped by tight therapeutic bottlenecks, such as chemotherapy or RIC allo-HSCT, but remain largely unaffected by ibrutinib treatment. These changes in mtDNA mutations after chemoimmunotherapy are paralleled by acquisition of additional CNV and chromatin changes that are associated with profound shifts in transcriptional state, thus demonstrating the contribution of genetic and non-genetic features to disease evolution and therapeutic resistance.
Results
mtscATAC-seq on serially collected samples representative of the clinical spectrum of CLL
We performed mtscATAC-seq on 26 serial samples (24 peripheral blood [PB], 1 bone marrow [BM], 1 lymph node [LN]) collected from 9 CLL patients at time of active disease (median white blood cell [WBC] count 43.8x109/L, range 6.7 – 607x109/L; median %CD19+ CD5+ 99.5, range 42.4 – 100, where available) (Fig. 1A, Suppl. Table 1). 24 of 26 samples were also co-analyzed using single cell RNA-sequencing (scRNA-seq). The cohort included representative scenarios of CLL disease course such as watchful waiting (CLL1), chemotherapy with FCR (CLL1-3), FCR followed by RIC allo-HSCT (CLL4-6), treatment with ibrutinib (CLL6-8) and transformation to RS (CLL 9). In total, we obtained scATAC-seq profiles from 163,279 high-quality cells (Transcriptional Start Site (TSS) enrichment >4; unique nuclear fragments >1000; median cells per sample 5,425 [range 501-29,833]) (Suppl. Fig. 1A–C, Suppl. Table 1). While scATAC-seq profiles of non-CLL immune cells from CLL1-9 cells were highly similar, each CLL formed a distinct cluster distinguished by 41,384 marker peaks (Fig. 1B), illustrating patient-specific chromatin states. For mtDNA analysis, 97,690 cells were used (mean >10-20x coverage per base). To extend our analysis of mtDNA mutations in CLL, we also included two previously reported mtscATAC-seq CLL profiles (CLL A and B), for a total of 109,118 cells (11). CLL cells could be distinguished from physiologic B cells based on chromatin accessibility profiles. The estimated median CLL content of the CLL/B cell compartment was 99% (range 66% - 100%) across samples (Suppl. Fig. 1D–I).
Fig. 1. Longitudinal accessible chromatin and mtDNA mutation dynamics across a cohort of 9 CLL patients.
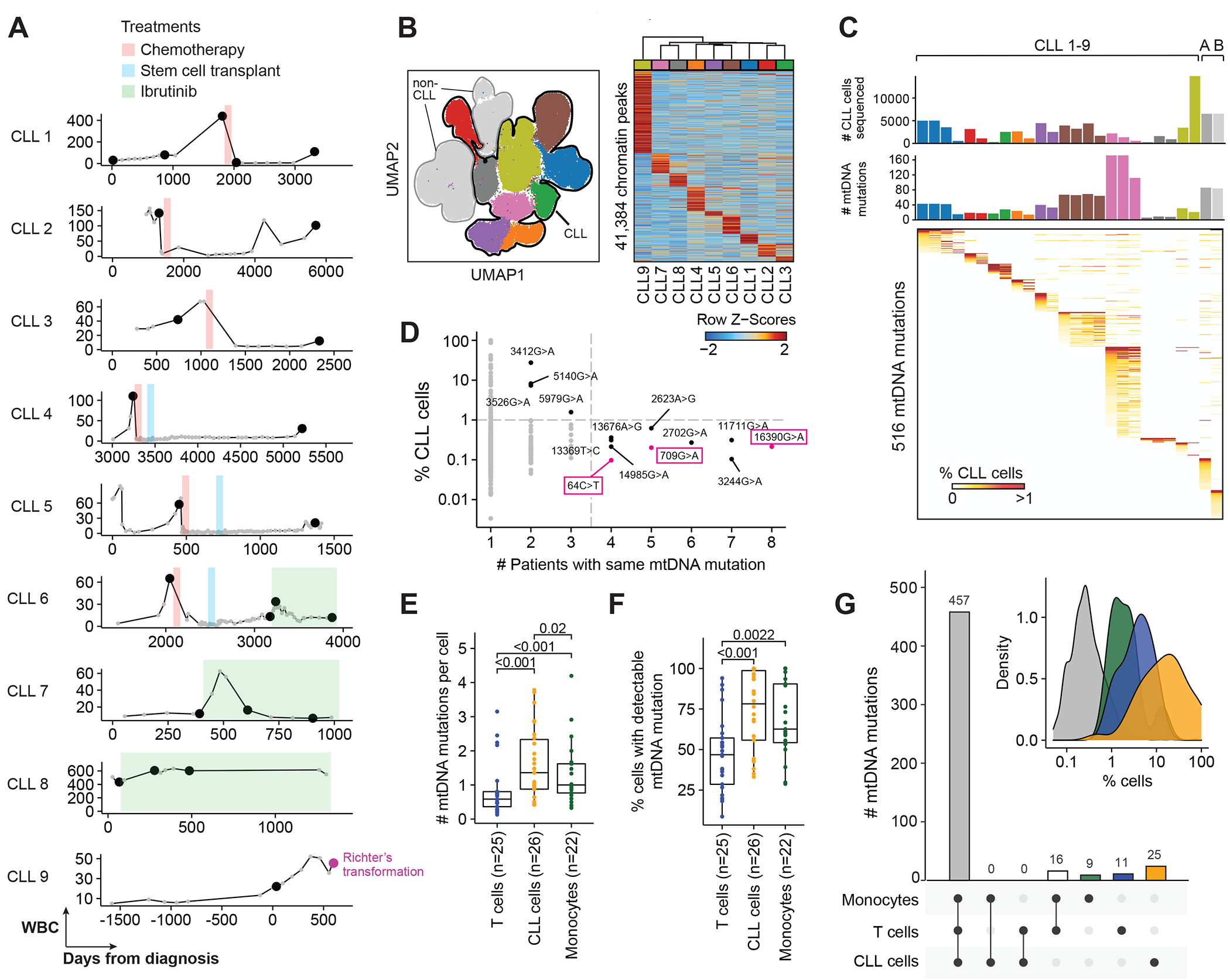
(A) Circulating white blood cell (WBC) counts of 9 CLL patients across disease course. Black dots - timepoints at which mtscATAC-seq samples were obtained. Day 0 - day of diagnosis. Treatments are indicated.
(B) Distinct accessible chromatin profiles in 163,279 CLL and non-CLL cells across 9 study subjects.
(C) mtDNA mutations detected in CLL cells from 26 samples collected at the time of detectable circulating disease. Available samples from CLL9 at time of transformation to Richter’s syndrome (lymph node, peripheral blood and bone marrow; n=3) are pooled. Colors of bars indicate individual patients. mtscATAC-seq profiles from a previously published dataset originating from two additional CLL patients (CLL A and B) are included (11).
(D) Recurrent mtDNA mutations (black) in the CLL cell compartment from 11 CLL patients. Grey dots - non-recurrent mtDNA mutations (presence in <4 patients or in <1% of cells). Magenta boxed mutations mark known frequent mtDNA variants.
(E, F) Number of mtDNA mutations detected per cell and %cells with detectable mtDNA mutations in T cells, CLL cells and monocytes across the samples. Statistical testing was performed using Wilcoxon signed rank test.
(G) mtDNA mutations enriched in T cells, CLL cells, or monocytes individually or as combinations. The majority of mutations (n=457) was equally distributed among all 3 compartments. Inset shows distribution of %monocytes (green), %T cells (blue) or %B cells (orange) marked by mtDNA mutations enriched in either cell compartment. Grey - Distribution of %cells not marked by enriched mtDNA mutations.
Within CLL cells we detected a total of 516 mtDNA mutations across the 28 samples (median 38 mutations per sample, range 6 – 171, Suppl. Table 2), with each patient displaying a largely distinct mutational profile (Fig. 1C). We confirmed a strong enrichment of T>C and C>T base substitutions among these mutations, verifying our experimental and computational workflow (Suppl. Fig. 2A) (11). We found no association between the number of mutations detected and the number of CLL cells sequenced or age at diagnosis (Suppl. Fig. 2B–C). In addition, samples with fewer mtDNA mutations did not show a more skewed distribution as measured by the normalized Shannon Index (Suppl. Fig. 2D). 10 mtDNA mutations were found in more than 3 subjects, all identifiable in <1% of cells at <1% mean (pseudo-bulk) heteroplasmy (Fig. 1D, Suppl. Fig. 2E). These recurring mutations at low frequency were not unexpected, and were in keeping with the relatively small mitochondrial genome and its 10-fold higher mutation rate compared to genomic DNA (16). In fact, three of these mutations (64C>T, 16390G>A, 709G>A) have been reported as frequently occurring variants and none are known to be pathogenic or have a functional role (17). However, 3244G>A has been associated with mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes (MELAS) syndrome (18). One mutation within MT-ND1 (3412G>A) was detectable in >10% of cells in two patients, but was not found in a previously reported cohort of 20 CLL patients (19). Overall, we did not observe substantial evidence of specific and recurrent mtDNA mutations that could be directly implicated in CLL pathogenesis within our cohort. These results support a model where the observed somatic mtDNA mutations occur randomly to mark clonal lineages within human cells, as previously suggested (12).
When comparing mtDNA mutations between the T cell, CLL, and monocyte compartments, we observed CLL cells to have the highest number of mtDNA mutations (median mutations per cell 0.59 vs. 1.36 vs. 1.0; p < 0.05 [pairwise comparisons; Wilcoxon signed-rank test]) and the highest percentage of cells with at least one detectable mutation (median percentage of cells with mutations 46.8% vs. 78.2% vs. 62.6%; p < 0.01 [pairwise comparisons; Wilcoxon signed-rank test]) (Fig. 1E–F). This difference is likely explained by a higher mtDNA coverage in CLL cells (median 38x, range 16-103) than in T cells (median 31x, range 15-60) or monocytes (median 34x, range 10-51). While 457 of 518 mutations (88.2%) were equally distributed among these three compartments, only 11 (2.1%), 25 (4.8%) and 9 (1.7%) were enriched in either T cells, CLL cells, or monocytes, respectively (adjusted p-value <0.05 [pairwise comparisons; Fisher’s exact test]) (Fig. 1G). Mutations that were equally distributed across these tissue compartments were typically detectable in <1% cells. Enrichment of mtDNA mutations in CLL cells was most pronounced with some mutations detectable in up to 99% of cells, while such mutations were least enriched in monocytes (Fig. 1G-inset), consistent with a lack of antigen-driven clonal expansion (Suppl. Fig. 2F–H).
Long-term tracking of mtDNA mutations and chromatin remodeling
To assess the stability of mtDNA mutations within CLL cells over a prolonged disease course and to associate their kinetics with chromatin states, we focused first on CLL1, from whom we obtained mtscATAC-seq profiles of circulating CLL samples over a period of 10 years. Two of five serially collected samples were procured across a 6-year treatment-free watchful waiting period [W/W-1 and −2], during which elevated WBC counts remained relatively stable before entering a phase of exponential growth requiring the initiation of FCR chemotherapy [Pre-FCR]. CLL1 experienced disease relapse after a 3-year remission, for which we profiled two further samples [remission and relapse] (Fig. 2A).
Fig. 2. Mitochondrial DNA mutations as long-term markers of disease evolution.
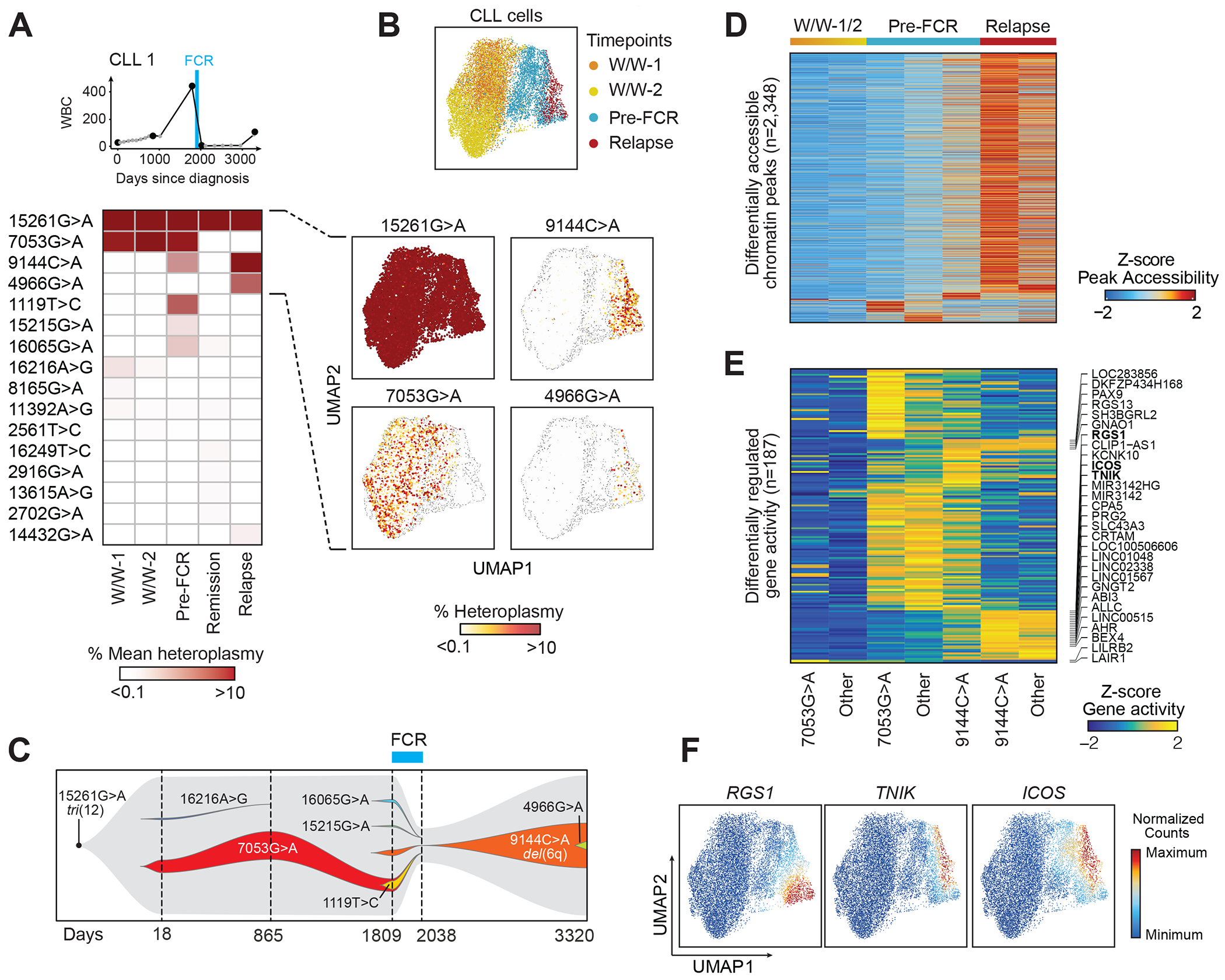
(A) Clinical information and mean heteroplasmy of mtDNA mutations at 5 timepoints (CLL1). FCR - fludarabine, cyclophosphamide and rituximab chemoimmunotherapy. 16 of 43 identified mtDNA mutations (mean heteroplasmy >0.01%) are displayed. For a complete list see Suppl. Table 2.
(B) UMAP plots of single CLL cells, clustered based on chromatin accessibility profiles, demonstrating distinct clusters based on timepoints (top). The heteroplasmy of 15261G>A, 7053G>A, 9144C>A and 4966G>A are shown in the individual panels, and segregate in distinct cell clusters.
(C) Trajectory of subclonal structure inferred from mean heteroplasmy of mtDNA mutations and copy number changes, derived from scATAC-seq data (see Suppl. Fig. 3).
(D, E) Chromatin accessibility of differentially expressed peaks and gene activity scores in CLL cells during watch and wait (W/W-1/2), before FCR chemotherapy (Pre-FCR) or at relapse marked by 7053G>A, 9144C>A or other mutations.
(F) Integrated scRNA-seq expression counts of RGS1, TNIK and ICOS projected onto the CLL1 scATAC-seq UMAP.
In total, we identified 43 mtDNA mutations across all cell types with mean pseudo-bulk heteroplasmy ranging from 0.0001% to 98.7%. These mutations each followed distinct patterns over time, suggesting their ability to mark clonal substructure. The mutation 15261G>A, not known to be pathogenic, was detectable at high mean heteroplasmy in all samples (>95% in CLL cells and >30% in T cells and monocytes), indicating it to broadly mark hematopoietic cells. In contrast, all other detected mutations followed dynamic patterns: 7053G>A had a stably high mean heteroplasmy amongst CLL cells during watchful waiting and at the time of accelerated growth (>8%) but disappeared after chemotherapy. Other mutations appeared only during exponential growth before therapy at low frequency (i.e. 15215G>A [1.3% mean heteroplasmy], 16065G>A [2.3%], 9144C>A [4.3%], 1119T>C [6.3%]) (Fig. 2B–C). Amongst the mtDNA mutations that appeared before FCR, CLL cells shared 7053G>A and 1119T>C, but presence of 7053G>A rarely co-occurred with 16065G>A and 9144C>A (Suppl. Fig. 3A–E). Of these emerging mutations, only cells carrying the 9144C>A mutation survived FCR therapy and reemerged at relapse (32.5% mean heteroplasmy in CLL cells) along with partial loss of chromosome 6q which was also detected through clinical karyotyping (Table 1), and an additional novel mutation (4966G>A). This latter mutation was present virtually exclusively within cells that also had the relapse-dominating 9144C>A mutation (Suppl. Fig. 3F–H), suggesting this to be a marker of a diverging subclone that emerged within the relapse population.
Table 1.
Molecular and clinical karyotypic characterization of CLL1-9.
| CLL | Age | Sex | IGHV | Cytogenetics before FCR | Cytogenetics at relapse post-FCR | Karyotype before FCR | Karyotype at relapse post-FCR |
|---|---|---|---|---|---|---|---|
| 1 | 37 | M | unmutated | tri(12) | tri(12), del(6) | N/A | 47,XY,t(2;14)(p15;q32),+12,t(14;19)(q32;q13.3)[11]/ 47,sl,del(6)(q13q27)[2]/ 46,XY,del(20)(q11.2q13.3)[5]/ 46,XY,t(7;8)(p14;q11.2)[1]/46,XY[1] |
| 2 | 37 | M | unmutated | del(13q), del(p53) | del(13q), del(p53) | 46,XY nuc ish 13q14.3 (D13S319x1),17p13.1(p53x1) |
45,XY,der(17;18)(q10;q10)[cp6]/46,XY[cp14] nuc ish(ATMx2,P53x1)[48/200], (D12Z3x2,D13S319x1,LSI13q34x2)[55/200]/ (D12Z3x2,D13S319x0,LSI13q34x2)[138/200] |
| 3 | 58 | M | unmutated | normal | normal | 46,XY | 46,XY |
| CLL | Age | Sex | IGHV | Cytogenetics before FCR | Cytogenetics at relapse post allo-HSCT | Karyotype before FCR | Karyotype at relapse post allo-HSCT |
| 4 | 54 | F | unmutated | del(11q) | del(17p) | nuc ish(CCND1,IGH)x2[100], (ATMx1,P53x2)[93/100] |
45,X,-X[7]/45,X,-X,inv(3)(p21p25)[7]/ 45,X,-X,inv(3),t(1;6)(p34;p23)[cp4]/ 45,X,-X,t(1;5)(p36;p13)[cp2] nuc ish(CCND1,IGH)x2[200], (ATMx2,TP53x1)[155/200], (D12Z3x2,D13S319x2,LSI13q34x2)[200], (DXZ1x2)[2/200]/(DXZ1x1)[131/200]// (DXZ1,DYZ1)x1[67/200] |
| 5 | 59 | F | unmutated | der(6)t(6;17), del(13q), del(17p) |
del(13q) | 45,XX,der(6)t(6;17)(q2?1;q21),-17[6]/46,XX[cp14] nuc ish (D13S319x1)[10/100], (P53x1)[15/100], (ATM,D12Z3,LSI13q34,)x2[100] |
nuc ish(DXZ1x2)[12/200]/(DXZ1,DYZ1)x1[92/200] nuc ish(DXZ1x1)[96/200] nuc ish(DYZ1x0), (D13S319x1),(LSI13q34x2)[13/100] nuc ish(DYZ1x0),(P53x2)[100] |
| 6 | 40 | M | mutated | del(14q) | del(14q), der(13;17) | nuc ish(P53x1)[7/100], (CCND1,ATM,D12Z3,D13S319, LSI13q34,IGH)x2[100] |
45,XY,der(13;17)(q10;q10),del(14)(q22q32)[cp5] nuc ish(DXZ1,DYZ1)x1[45/200]// (DXZ1x2)[155/200] |
| CLL | Age | Sex | IGHV | Cytogenetics before Ibrutinib | |||
| 7 | 52 | F | unmutated | del(13q) | |||
| 8 | 65 | M | n/a | del(13q) | |||
| CLL | Age | Sex | IGHV | Cytogenetics CLL | Cytogenetics Richter’s syndrome | Karyotype CLL | Karyotype Richter’s syndrome |
| 9 | 56 | M | mutated | del(17p) | del(17p), t(8;22) | nuc ish(TP53x1)[107/200], (ATM,D12Z3,DLEU1, DLEU2,TFDP1)x2[200] nuc ish(CCND1, IGH)x2[200] |
44,XY,t(3;4)(q28;q25), der(8)t(8;17)(p11.2;q11,2)t(8;22)(q24;q11), -9,add(14)(q24),-17,add(20)(q13.3), der(22)t(8;22)[cp7]/78-88<4n>, idemx2,ider(3)(q10)add(3)(q27)[cp9]/46,XY[4] |
| CLL | Age | Sex | IGHV | Cytogenetics | Karyotype | ||
| A | 84 | M | n/a | tri(12), del(13q14.3) | nuc ish(MYBx2)[200] nuc ish(ATMx2)[200] nuc ish(D12Z3x3)[82/200] nuc ish(D13S319,D13S25x1,LAMP 1x2)[24/200] nuc ish(CCND1, IGH)x2[200] nuc ish(TP53,D17Z1)[200] |
||
| B | 45 | F | mutated | del(13q) | n/a | ||
FCR: fludarabine, cyclophosphamide, rituximab; allo-HSCT: allogeneic hematopoietic stem cell transplantation; F: female; M: male; n/a: not available
We wondered whether the two dynamic mutations 7053G>A (contraction at relapse) and 9144C>A (expansion at relapse) could mark CLL populations with different chromatin states. Indeed, cells bearing 7053G>A or 9144C>A did not colocalize on the UMAP plot derived from scATAC-seq profiles and marked distinct populations (Fig. 2B). Differential analysis of chromatin accessibility showed higher accessibility of transcription factor motifs associated with B cell development (SPI1, REL, RELA, SPIC) in cells marked by 7053G>A than those marked by 9144C>A (Suppl. Fig. 3I). Notably, CLL cells marked by 9144C>A before FCR had chromatin peaks that were already more similar with cells at relapse than those marked by 7053G>A (Fig. 2D). These differences in chromatin peaks led to higher gene activity scores of genes previously described to play a role in B cell malignancies such as RGS1(20), TNIK (21) and ICOS (22) in CLL cells marked by 9144C>A (Fig. 2E). Indeed, we observed higher expression of these genes at relapse through the integration of matched scRNA-seq profiles of CLL1 cells into the scATAC-seq space (Fig. 2F). Finally, we wondered whether clonal dynamics inferred with mtDNA mutations would be consistent with analyses based on somatic nuclear mutations from matched whole-exome sequencing (WES) of the same tumors. Reanalysis of WES data (23) similarly showed that a number of therapy-sensitive clones were eradicated by FCR, while therapy-resistant clones expanded at relapse (Suppl. Fig. 3J). WES also confirmed the acquisition of del(6q) at relapse (Suppl. Fig. 3K–L).
Together, our analysis revealed that the dynamics of mtDNA mutations mirrored disease evolution in CLL1 over a period of 10 years and marked putative subclones with distinct chromatin profiles and differential therapeutic sensitivity. Further, single cell chromatin and mtDNA profiles demonstrated continued evolution and selection of a therapy-resistant phenotype.
Dynamics of mtDNA mutations following chemotherapy and stem cell transplantation
Having observed evidence of selective pressure exercised by natural disease history or therapeutic intervention in CLL1, we sought to determine whether consistent patterns of change in mtDNA mutations and chromatin profiles during clinical inflection points could be also detected in other CLL patients. To this end, we evaluated the mtDNA mutation profiles of 2 additional patients undergoing FCR chemotherapy (CLL2-3), and 3 patients treated with FCR followed by RIC allo-HSCT (CLL4-6, see Table 1). We first focused on CLL4 who relapsed 6 years after allo-HSCT and asked whether this longer disease course was associated with even more extreme changes in mtDNA mutations than in CLL1. Indeed, while we observed that the 16247G>A mutation (mean heteroplasmy of 30.5% pre-FCR) was almost undetectable at relapse (mean heteroplasmy <0.2%), two other mutations (6426G>A, 16290C>T) that were initially almost undetectable reached high levels of mean heteroplasmy at relapse (50.7%, 43.3%). At the same time, 3538G>A continued to be detectable in practically all CLL cells with a mean heteroplasmy of almost 100% but was absent from T cells or monocytes, indicating this to be a CLL cell-lineage specific mutation (Fig. 3A). We observed a uniformly detectable chromosome 4p deletion in all CLL cells at both timepoints, which, along with the occurrence of the 3538G>A mutation, seems to have been an early genetic change in the disease history of this patient. Consistent with the profound shift in the detection of 6426G>A and 16290C>T mutations at relapse, and the corresponding loss of 16247G>A, a chromosome 11q deletion was only detectable before FCR and a 17p deletion was present in most CLL cells at relapse but undetectable before FCR (see arrows), consistent with the known cytogenetics (Fig. 3A–B, Table 1).
Fig. 3. Selection pressure during intensive chemoimmunotherapy revealed by mtDNA and copy number alterations.
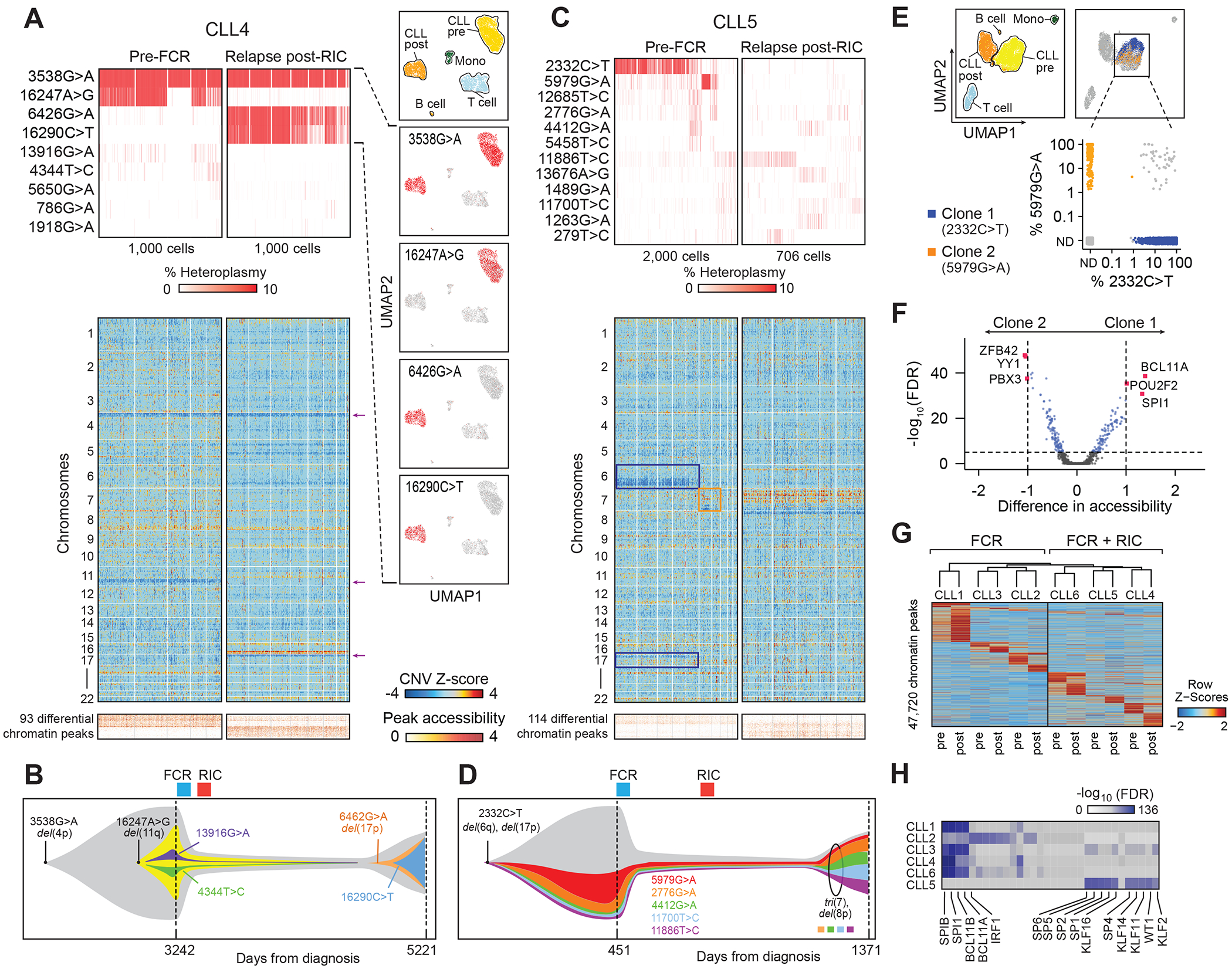
(A) mtDNA mutations, inferred copy number changes from scATAC-seq data and differential chromatin peaks in CLL cells (CLL4) before chemoimmunotherapy with fludarabine, cyclophosphamide, rituximab (FCR) and reduced intensity conditioning allogeneic hematopoietic stem cell transplantation (RIC allo-HSCT) as well as at relapse. For the heat maps, only CLL cells with at least one detectable mtDNA mutation are shown. UMAP plots show cell populations and localization of cells marked by the 3538G>A, 16247A>G, 6426G>A and 16290C>T mutation. Analogous analyses on CLL2 and 3 in Suppl. Fig. 4.
(B) Schematic representation of the trajectory of subclonal structure inferred from mean heteroplasmy of mtDNA mutations and copy number changes (CLL4).
(C) mtDNA mutations, inferred copy number changes and differential chromatin peaks of CLL cells (CLL5) before FCR chemotherapy and RIC allo-HSCT (Pre-FCR) and at relapse (Relapse post-RIC). For the heat maps, only CLL cells with at least one detectable mtDNA mutation are shown.
(D) Schematic representation of the trajectory of subclonal structure inferred from mean heteroplasmy of mtDNA mutations and copy number changes (CLL5).
(E, F) Identification of CLL subclones 1 (2332C>T; blue) and 2 (5979G>A; yellow) in CLL5 with distinct chromatin states.
(G) Changes in chromatin peaks at relapse after FCR (CLL1-3) or FCR and RIC allo-HSCT (CLL4-6).
(H) Transcription factor motifs depleted from marker peaks shown in G at relapse. Colors indicate statistical significance of enrichment before treatment as adjusted p-value (FDR) ranging from 10−136 (blue) to 0 (white).
The analysis of the mtscATAC-seq profiles of CLL4 illustrated how samples separated by years of disease history may contain distinct mtDNA mutational profiles, suggesting a model of clonal succession. We repeatedly observed similar changes in mtDNA mutations across 4 more patients with mtscATAC-seq profiles available before treatment and at relapse after FCR chemotherapy and RIC allo-HSCT, although to different extents and with distinct dynamics (Suppl. Fig. 4A–C). For example, in CLL5 we observed how two mutations with high mean heteroplasmy 2332C>T (15.8%) and 5979G>A (6.6%) disappeared at relapse, while an additional copy of chromosome 7 was uniformly present across all CLL5 cells at relapse, illustrating the utility of combining the detection of distinct types of somatic mutation events to trace clonal structure over time (Fig. 3C–D). In CLL5, we were able to identify 2 subpopulations within CLL cells before treatment marked by two different mtDNA mutations, 2332C>T (clone 1) and 5979G>A (clone 2). These two subpopulations had detectable differences in accessibility of transcription factor motifs implicated in B cell malignancies such as YY1, BCL11A, POU2F2, and SPI1 (24–27), and clone 1 harbored evidence of a 17p deletion that was absent in clone 2 (Fig. 3C–F). Chromatin accessibility of CD38 and ITGA4 (encoding for CD49d), two unfavorable prognostic markers in CLL, differed between these clones (Suppl. Fig. 4D–F).
Fig. 4. Stability of mtDNA mutations during ibrutinib treatment.
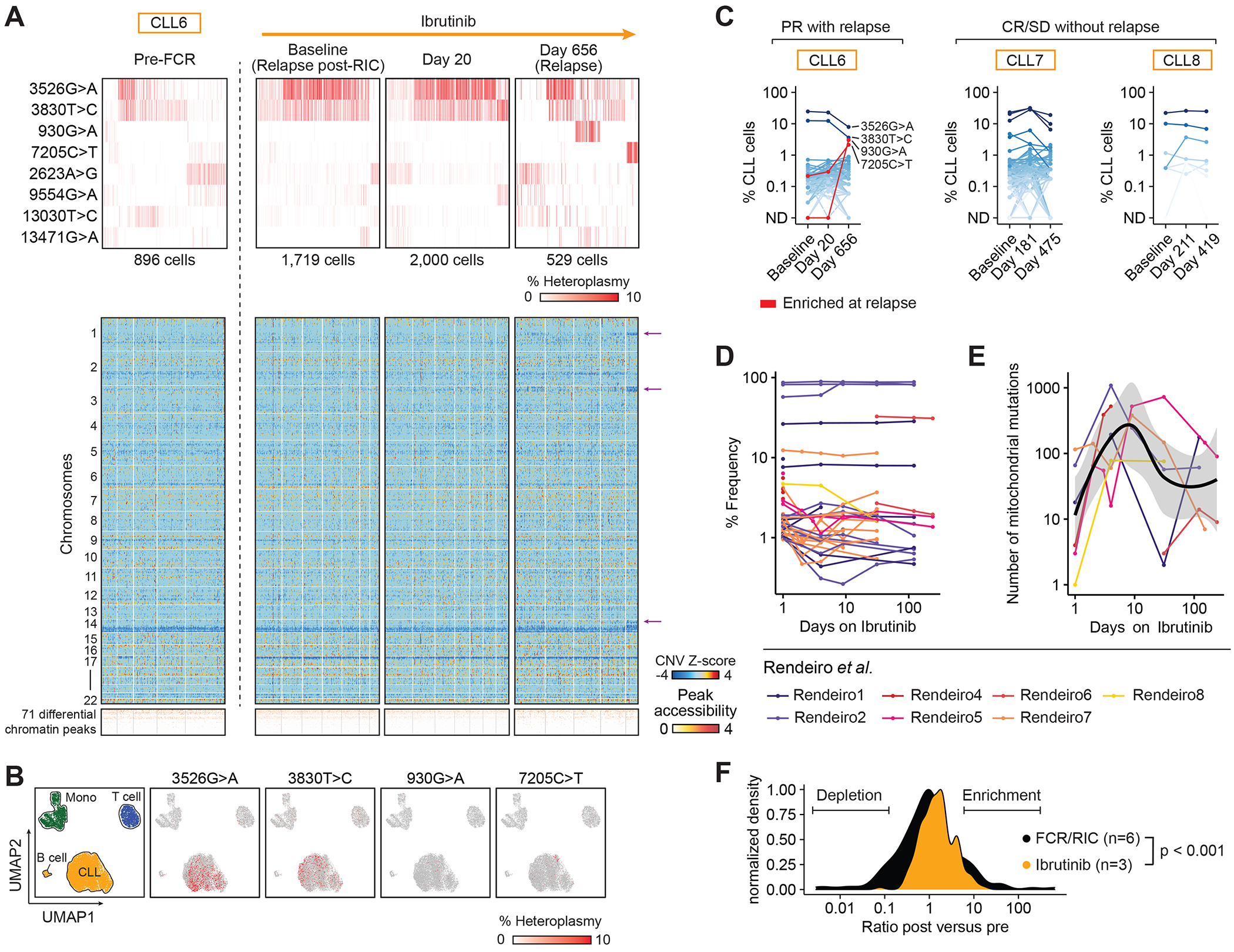
(A, B) mtDNA mutations, inferred copy number changes from scATAC-seq data and differential chromatin peaks in CLL cells (CLL6) (1) before fludarabine, cyclophosphamide, rituximab (Pre-FCR), (2) at relapse after reduced-intensity conditioning allogeneic stem cell transplantation (Relapse post-RIC), (3) after 20 days of ibrutinib treatment as well as (4) at relapse on ibrutinib. Arrows indicate copy number changes unique to cells marked by 7205C>T at relapse on ibrutinib. UMAP plots show cell populations and localization of cells marked by 3526G>A, 3830T>C, 930G>A and 7205C>T (B). In the heatmaps, only CLL cells with at least one detectable mtDNA mutation are shown.
(C) %CLL cells marked by mtDNA mutations during ibrutinib treatment in CLL cells in CLL6 (partial remission - PR with relapse), CLL7 (complete remission - CR) and CLL8 (stable disease - SD).
(D, E) Frequency of individual mtDNA mutations (D) and total number of mtDNA mutations (E) detected per sample in reanalysis of public bulk ATAC-seq profiles of CD19+ CD5+ CLL cells from 7 patients during ibrutinib treatment (29).
(F) Changes in abundance of mtDNA mutations detected in CLL cells relapsing after FCR/RIC allo-HSCT (CLL1-6) compared to CLL cells during ibrutinib treatment (CLL6-8). Statistical testing performed using Kolmogorov-Smirnov test.
Finally, we assessed the ability to identify common changes at the chromatin level at relapse following fludarabine-based treatment in CLL1-6. The CLL cells from all six patients demonstrated chromatin remodeling. With the exception of CLL5, a number of transcription factor motifs (SPIB, SPI1, BCL11B, BCL11A, and IRF1) were depleted from differentially accessible chromatin peaks at relapse, consistent with a less differentiated B cell phenotype than prior to treatment (26) (Fig. 3G–H, Suppl. Fig. 5A). Similarly, although corresponding scRNA-seq profiles were essentially unique for each CLL, they showed concordant expression changes of B cell-associated genes such as down-regulation of MEF2C or CD24 and upregulation of CXCR4 and RGS1/2 at relapse (Suppl. Fig. 5B–E), reenforcing the idea of defined selective pressure through therapeutic interventions. Altogether, these observations demonstrate how somatic events (CNV and mtDNA mutations) in CLL cells demarcate subclones with distinct genomic properties within the pool of tumor cells and their evolution under profound selective pressure.
Dynamics of mtDNA mutations under ibrutinib treatment
The targeted BTK inhibitor ibrutinib is known to deepen clinical responses over months to years (28), and we therefore compared changes of clonal cell populations as measured by mtDNA mutations in this setting with those seen in chemotherapy or allo-HSCT. We first focused on CLL6 with the longest follow-up on ibrutinib and from whom we obtained mtscATAC-seq profiles of peripheral blood at four timepoints: (1) before FCR and RIC allo-HSCT, (2) at relapse after RIC allo-HSCT shortly before ibrutinib treatment, (3) at the peak of ibrutinib-induced lymphocytosis 20 days after initiation and (4) at relapse after 656 days with mixed nodal and blood manifestation (Fig. 4A).
At initiation of ibrutinib (after relapse from FCR and RIC allo-HSCT), CLL6 was dominated by cells with a 17p deletion and two mtDNA mutations (3526G>A, 3830T>C) which had expanded from a minor clone (13.2% of CLL cells) before FCR. During ibrutinib treatment, mtDNA mutations remained stable: only negligible change was observed after 20 days of ibrutinib and both predominant mutations marked large fractions of cells (36.2% and 37.3% [3526G>A], 25.1% and 25.5% [3830T>C] in cells with ≥1 detectable mtDNA mutation). At relapse, CLL cells marked by these two mutations diminished to 16.2% (3526G>A) and 12.4% (3830T>C), and distinct groups marked by two independent mutations became apparent in 6.8% (930G>A) and 5.8% (7205C>T) of cells. Although differences in chromatin accessibility were undetectable in CLL cells harboring either the decreased (3526G>A, 3830T>C) or increased mutations (930G>A, 7205C>T), we observed cells marked by 7205C>T to demonstrate additional copy number changes in chromosomes 1, 3 and 13, indicating they represent a newly emerging relapse associated subclone (arrows, Fig. 4A, B).
We also observed relatively stable mtDNA genotypes over the course of ibrutinib treatment in CLL7 and 8, whose best response was complete remission and stable disease respectively (Fig. 4C). Ibrutinib did not appear to affect the T cells and monocytes in CLL6 and 7, as we did not observe any major shifts of mtDNA mutations in these populations (Suppl. Fig. 6A–C). Despite the different clinical responses in CLL6-8, a reduction in NFKB1 motif accessibility under ibrutinib treatment was observed in all 3 subjects (Suppl. Fig. 7A–B), consistent with recently reported data (29). Similarly, scRNA-seq profiles showed a consistent pattern of ibrutinib-induced changes in 20 differentially expressed genes, some of which have critical roles in CLL such as MS4A1 (30) or MIR155HG (31) (Suppl. Fig. 7C–D). These ibrutinib-induced gene expression changes largely reversed in CLL6 at relapse, suggesting acquired treatment-resistance to ibrutinib (Suppl. Fig. 7E–F).
To determine if the observed clonal stability as captured via mtDNA mutations in CLL6-8 was generalizable, we took advantage of public bulk ATAC-seq profiles obtained from FACS-sorted CD19+CD5+ cells from 7 independent CLL patients, serially sampled before and during ibrutinib treatment (29). We similarly observed stability in the frequency of individual mtDNA mutations during 6 months of ibrutinib treatment (Fig. 4D). Some mtDNA mutations with low abundance showed small changes at ~day 10 of ibrutinib treatment, which coincided with a transiently increased number of detectable mtDNA mutations (Fig. 4E) and mirrored the kinetics of ibrutinib-induced lymphocytosis. The stability of mtDNA mutations in this dataset was thus highly concordant with the dynamics observed in CLL6-8 and contrasted with the dramatic changes observed in CLL1-6 at relapse after chemoimmunotherapy (Fig. 4F). Taken together, ibrutinib and chemotherapy appear to induce distinct selective pressures, with ibrutinib inducing more uniform chromatin remodeling across CLL subclones.
Clonal dynamics during CLL transformation to Richter’s syndrome
Although CLL is typically characterized as an indolent B cell malignancy, a subset of patients can transform into an aggressive lymphoma, a process known as Richter’s Syndrome (RS). Since RS cells expand from a subclone of antecedent CLL in the majority of cases, we reasoned that they are enriched for distinct mtDNA mutations. CLL9 progressed to RS, with evidence of circulating disease, 20 months after the initial CLL diagnosis and in the absence of any intervening therapy. PB samples were procured early in this patient’s CLL course and at the time of RS, as well as BM and LN specimens at the time of RS (Fig. 5A–B). Comparison of mtDNA mutations in CLL and RS demonstrated clonal outgrowth: within the CLL peripheral blood cells (CLL PB1 and PB2), 24% of cells contained the 3412G>A or 9553G>A mutations at a high heteroplasmy (>5%) and 4.7% of cells had the 2343G>A mutation also at high heteroplasmy, marking two distinct subclones (Fig. 5C, Suppl. Fig. 8A–D). Upon transformation to RS, cells with 2343G>A became undetectable while those with 3412G>A or 9553G>A were observed in a higher proportion of Richter’s cells at comparable percentages in PB, BM and LN (57.4-64.2%) (Fig. 5C). Consistent CNV changes were detected across these compartments, such as deletion of chromosomes 8p, 9q and 14q in addition to the 17p deletion already present at time of CLL (purple arrows), which were confirmed by WES. WES also revealed enrichment of subclones at RS compared to CLL and was consistent with our observations made using mtDNA mutations (Fig. 5C, Suppl. Fig. 8E–G). Within the lymph node, we also identified a subclone with additional copy number changes of chromosome 4 and 13 that were marked by high heteroplasmy of 3412G>A (92.7%) and 9553G>A (71.1%) (green arrows).
Fig. 5. Accessible chromatin and mtDNA mutational dynamics during transformation to Richter’s syndrome.

(A) Circulating white blood cell count (CLL9). Analyzed samples are peripheral blood during CLL phase (CLL; black) and peripheral blood, bone marrow and lymph node at time of Richter’s transformation (Richter; magenta).
(B) UMAP plot of CLL, Richter’s and immune cells based on chromatin profiles (n=30,395) with identification of CD4+ T cells, CD8+ T cells, physiologic B cells, CLL cells from peripheral blood (PB), Richter’s cells from lymph node (LN), peripheral blood and bone marrow (BM).
(C) Heteroplasmy of mtDNA mutations and inferred copy number changes from scATAC-seq data of cells in clusters identified in (B). Only cells with at least one detectable mtDNA mutation are shown.
(D) Heteroplasmy of 3412G>A and 9553G>A in Richter’s cells. Coloring of dots indicates sequencing coverage (20-350x). Inset table shows number of cells with both, either one or none of the mutations detectable.
(E) Distribution of heteroplasmy of 3412G>A (blue) or 9553G>A (red) in Richter’s cells.
(F) Differential chromatin accessibility of transcription factor motifs between CLL (CLL PB1, CLL PB2) and Richter’s cells (Richter LN1, Richter LN2, Richter PB, Richter BM). Precision limit for adjusted p-value 2.6e-297.
(G) Chromatin accessibility of POU2F2 motif across clusters.
(H) Differential gene expression of scRNA-seq profiles of CLL cells from peripheral blood (CLL PB; black) and Richter’s cells from peripheral blood (Richter PB; magenta) or bone marrow (Richter BM; magenta).
We noted that the heteroplasmy of both 3412G>A and 9553G>A mutations in RS cells was highly correlated (R = 0.89) (Fig. 5D), suggesting co-occurrence within the same clone. Notably, both mutations showed a wide range of heteroplasmy, which we cannot attribute to variation in coverage (Suppl. Fig. 8H). The distribution of heteroplasmy of both mutations was such that the maximum value was ~10% and fewer than 1% of cells had a heteroplasmy <1% (Fig. 5E). This analysis confirms that mtDNA mutations in monoclonal populations can have a wide range of heteroplasmy (12); mtDNA mutations thus behave more dynamically than somatic nuclear mutations due to processes such as asymmetrical segregation of mitochondria, relaxed replication, or mitophagy which should be considered when interpreting longitudinal mtDNA kinetics.
Given the clear changes in mtDNA mutations and CNV associated with RS, we asked if similarly profound chromatin and transcriptional changes could be detected. Analysis of chromatin accessibility revealed RS cells to have strong reduction of accessibility in motifs related to the AP-1 transcription factor complex and higher accessibility of POU2F transcription factor motifs (Fig. 5F–G), which are known to play an important role in B cell lymphomagenesis (25). Likewise, scRNA-seq profiles of RS cells were quite distinct from those of antecedent CLL cells, with down-regulation of genes such as JUNB, JUN, FOS and FOSB and upregulation of B cell-related genes such as LTB, CD24 and CD70 (Fig. 5H, Suppl. Fig. 8I). Of note, the RS cells in bone marrow and peripheral blood were transcriptionally indistinguishable (Suppl. Fig. 8J) and matched our finding that the same mtDNA mutation profiles could be found between these two compartments. Our analyses thus revealed the dramatic changes in accessible chromatin and transcriptional profiles along with the acquisition of numerous CNVs in the transformation from CLL to RS.
Characteristic chromatin profiles and mtDNA mutations in T cell populations
Since T cells undergo antigen-driven clonal expansion, we also evaluated whether mtDNA mutations from the 33,573 available scATAC-seq profiles of T cells could mark distinct T cell subpopulations co-existing with CLL. The scATAC-seq profiles of T cells from PB and BM were highly similar across all 9 subjects but clearly differed from T cells isolated from LN, which were collected from RS (Fig. 6A–B). Based on gene activity scores, we identified CD4+ and CD8+ T cell populations (Suppl. Fig. 9A). For CLL4, CLL6 and CLLA, we could identify 6 mutations that marked subsets of clonally expanded CD8+ T cells. In CLL4, 1918G>A marked donor-derived CD8+ T cells and was undetectable in CLL cells or host CD8+ T cells (Suppl. Fig. 9B). Similarly, in CLL6, 2647G>A and 10408T>C were only detectable in donor-derived CD8+ T cells (Fig. 6C, Suppl. Fig. 9C) and marked two distinct CD8+ T cell clones with shared chromatin state which expanded and contracted at different timepoints (Fig. 6D). Finally, in CLLA, 3 mtDNA mutations marked two CD8+ T cell clones with distinct chromatin states (Fig. 6E–G, Suppl. Fig. 9D–E). 2464G>A and 4573T>C marked CD8+ T cells with higher chromatin accessibility of GNLY and GZMH, indicative of an effector memory phenotype, while 7762G>A marked cells with higher chromatin accessibility of IL7R, TCF7, NFATC1 and CD28, consistent with a naïve T cell phenotype (32). Taken together, these 6 mtDNA mutations likely mark individual expanded T cell clones with distinct specificities, thus demonstrating the potential of mtscATAC-seq to track phenotypically diverse cell populations.
Fig. 6. T cell phenotypes and clonal dynamics revealed through mtDNA mutations and accessible chromatin profiles.

(A, B) UMAP plots showing clustering of 33,573 scATAC-seq profiles of T cells from CLL1-9 isolated from peripheral blood (PB), bone marrow (BM) or lymph node (LN).
(C, D) Identification of two CD8+ T cell clones marked by 2647G>A (clone 1; yellow) and 10408T>C (clone 2; brown) in CLL6 that expand at relapse after reduced intensity conditioning allogeneic hematopoietic stem cell transplantation (RIC allo-HSCT) (clone 1) or after 21 months of ibrutinib treatment (clone 2).
(E-G) Identification of two CD8+ T cell clones marked by 4573T>C and 2464G>A (clone 1; yellow) or 7762G>A (clone 2; magenta) in CLLA. Clone 1 and 2 show different chromatin accessibility of T cell-related genes associated with an effector memory (GZMH, GNLY) and naïve (TCF7, NFATC1, IL7R and CD28) T cell phenotype.
(H) Differential analysis of gene activity scores between T cells isolated from PB/BM (CLL1-9) and LN (CLL9 at time of Richter’s syndrome).
(I) Browser tracks showing chromatin accessibility of TOX gene across T cell clusters (Suppl. Fig. 9A)
For CLL9, we had the opportunity to compare T cells from the CLL phase with the RS phase of disease. LN-associated T cells (CLL9 at the time of RS) showed higher gene activity of TOX and immune checkpoint molecules such as PDCD1, TIGIT, CTLA4 and ICOS than T cells from PB (CLL1-9), consistent with the immunophenotype of follicular T helper cells (33) (Fig. 6H–I). In particular, LN-associated CD8+ T cells (CLL9 at the time of RS) had higher gene scores of EOMES and GZMK while CD8+ T cells from PB (CLL1-9) expressed TBX21 and GZMB, which may point to different effector memory populations (34) (Suppl. Fig. 9F). LN-associated CD8+ T cells also highly expressed HAVCR2 and PDCD1 (Suppl. Fig. 9G). These observations are especially intriguing given that PD-1 inhibition has shown clinical activity in Richter’s syndrome.
Discussion
Longitudinal tracking of distinct clones within heterogenous cell populations and linking clonal lineages to distinct functional states at the single cell level has been subject of numerous efforts aimed at understanding disease evolution and mechanisms leading to therapeutic resistance in a wide range of cancers. While in vitro and in vivo model systems have the potential to exploit technologies that introduce synthetic genetic markers into cells (35) and can provide high-resolution clonal tracking capabilities, in vivo human studies primarily rely on the presence and detection of naturally occurring barcodes. The most common approach to monitoring cancer subclones has been through the tracking of somatic nuclear mutations, although for many diseases such as CLL, these are often sparse events and frequently subject to selective pressures confounding the inferences that can be made from such mutations. For non-cancer populations, such as B cells and T cells, B cell receptor (BCR) (36) and T cell receptor (TCR) sequences (37,38), respectively have been commonly exploited for clone tracking. While such sequences cannot be extracted from scATAC-seq data, we confirm herein that mtDNA mutations lend themselves as natural markers of clonal lineage, and as a single entity, enable the tracking of multi-lineage populations from both cancer and non-cancer fractions of tissues. We further demonstrate their promise in linking subclonal structures to distinct functional states combined with co-evolving CNV changes.
Our study of mtscATAC-seq applied to 9 CLL patients to longitudinally assess their clonal and accessible chromatin dynamics in the context of distinct treatment modalities revealed several notable new biologic insights. First, the extent to which cells carrying distinct mtDNA mutation profiles evolved with therapy appeared to reflect the degree of therapeutic bottleneck. We observed mutational stability over years in the absence of treatment but also saw dramatic changes in mtDNA mutations with disease acceleration, transformation or relapse after strong therapeutic pressure such as chemoimmunotherapy and allo-HSCT. In contrast, ibrutinib therapy was associated with less dramatic changes of mtDNA genotypes. In specific instances, we also noted that mtDNA mutations behaved more dynamically than somatic nuclear mutations. For example, in CLL9, two mutations showed a wide range of heteroplasmy within RS cells. In CLL1 and CLL4, cells carrying mtDNA mutations in a high percentage of cells at baseline were absent at relapse, suggesting vulnerability to prior treatment. Our data highlight the potential utility of mtDNA mutations for direct ex vivo tracking of clonal populations of human cells, but we also caution that the unique nature of mitochondrial genetics warrants further high-resolution exploration of mtDNA mutational dynamics to understand these processes in greater detail. The large mtDNA copy number, the diverse levels of heteroplasmy present among these mutations, the relaxed replication of mtDNA, mitophagy, and largely stochastic allocation to daughter cells during cell division are all processes that require further consideration when making inferences about clonal dynamics (39).
Second, we observed that mtDNA mutations can identify CLL subpopulations with distinct functional states, demonstrating how mtscATAC-seq can capture information on cell state, clonal lineage, and copy number alterations at single cell resolution. In the case of CLL1, we identified a specific mtDNA subclone at the time of disease acceleration that not only expanded following FCR chemotherapy but had a distinct chromatin state more similar to relapse even prior to therapy. In keeping with parallel genomic and non-genomic evolution, we identified changes in CNVs that co-occurred with changes in mtDNA mutations and chromatin accessibility such as in CLL5, CLL6 and CLL9, thus strengthening the link between disease evolution and composition of mtDNA mutations within the CLL compartment. Given that mtDNA mutations are enriched in CLL cells, key open questions remain, such as at what stage during lymphoid development these occur; the impact of IGHV mutational status on mtDNA mutations; whether they are already detectable at the stage of lymphoid progenitor or even hematopoietic stem cell cells; or whether they are result of later events in the disease pathogenesis (40) and the relationship with prognosis.
We noted that chromatin profiles were dynamic in relation to disease history: In CLL9, Richter’s cells, compared to CLL cells, showed increased chromatin accessibility for POU transcription factor motifs, which was most pronounced in lymph node-associated cells and potentially reflects that POU2F2 (encoding Oct-2) is known to play a role in diffuse large B cell lymphoma (DLBCL) and has even been proposed as a diagnostic marker alic (25,41). Although underpowered to generalize, our study identified common changes at the level of chromatin accessibility and gene expression across 6 CLLs who had undergone fludarabine-based treatment. Transcription factor motifs involved in B cell development (SPIB, SPI1) were depleted at relapse which speaks to the selection of a less differentiated CLL phenotype, in line with a recent study reporting enrichment of stem cell gene sets in CLL late relapse specimens (42). We also detected higher expression of CXCR4 at relapse, which is consistent with studies that have defined high CXCR4 expression as an adverse prognostic factor in DLBCL and CLL (43) and as a mechanism of acquired resistance to fludarabine/mafosfamide in an in vitro model (35). Altogether, these observations support the idea that despite marked heterogeneity across patients, defined therapeutic interventions lead to common principles of response and acquired resistance, thereby confirming other studies documenting convergent epigenomic evolution with acquired drug resistance (44). This justifies further research into epigenetic mechanisms of acquired resistance given that genomic somatic mutations only identify mechanisms of resistance in a proportion of patients (45) and that it is still unclear how many known recurrent somatic mutations drive complex changes in transcriptional state and resistance (46).
Third, although the majority of cells were CLL cells, analysis of chromatin profiles of single T cells was feasible and showed stark phenotypic differences between lymph node and peripheral blood. Furthermore, we could identify mtDNA mutations and associated chromatin profiles to define subclones of non-CLL immune cells. These analyses thus demonstrate their ability to mark physiological cell subpopulations with diverging chromatin states.
Clonal tracking with mtscATAC-seq is an alternative to other DNA and RNA-based single cell approaches that utilize somatic nuclear mutations as natural barcodes. Compared to somatic nuclear mutations, mtDNA mutations have the advantage that a priori knowledge is not required for their detection. However, the exact extent to which mtDNA mutations contribute to tumorigenesis is not yet clearly established (9,10,16). Additionally, they may be shared between cell compartments at different frequencies, which needs to be taken into account when inferring clonal lineage. The high copy number and small size of mitochondrial genome allows mtscATAC-seq to read out mtDNA mutations in single cells at high coverage and avoids the necessity of additional targeted amplification steps, while simultaneously providing information on cell states. Although scRNA-seq can provide higher resolution of cell states, genotyping using transcriptomic data has a far lower detection rate than mtscATAC-seq (8). Emerging multi-omic technologies for the combined capture of accessible chromatin, transcriptome, and protein expression along with sufficient mtDNA profiling are being developed to overcome current limitations and will surely enable the future improved detection of subtle but important differences in functional states between subclones identified by mtDNA mutations or other clonal markers (47,48). Combined capture of mtDNA mutations and different types of nuclear mutations would further enhance the elucidation of both genomic and non-genomic evolution of malignant cells. Higher throughput and increased coverage of single cells in the future will increase the level of detection of mtDNA mutations, thereby improving the tracking of clones using mtDNA mutations in settings such as premalignant states or minimal residual disease. Our observations using mtscATAC-seq support the use of mtDNA mutations as natural barcodes to track changes in the subclonal structure of disease populations over time and the contribution of non-genetic changes in disease progression, transformation, and therapeutic resistance.
Materials and Methods
Sample acquisition
Peripheral blood, bone marrow and lymph node samples were collected from patients treated at Dana-Farber Cancer Institute or in collaboration with CLL Research Consortium (CRC) under Institutional Review Board-approved protocols from patients who provided written informed consent. Peripheral blood and bone marrow mononuclear cells were isolated by Ficoll-Hypaque density gradient centrifugation. Lymph node mononuclear cells were isolated through (Miltenyi GentleMACS, RRID:SCR_020280) dissociation. All samples were stored in vapor-phase liquid nitrogen after cryopreservation with 10% dimethyl sulfoxide until analysis.
mtscATAC-seq cell preparation
The mtscATAC-seq cell preparation was performed as previously described (11). Briefly, cells were fixated with 1% formaldehyde for 10 minutes at room temperature and quenched with 0.125M glycine final. After cell lysis in 100 μl with a modified lysis buffer (10 mM Tris-HCl (pH 7.4), 10 mM NaCl, 3 mM MgCl2, 0.1% Nonidet P40 Substitute, 1% bovine serum albumin, nuclease-free H2O) for 3 minutes, cells were washed with a modified wash buffer (10 mM Tris-HCl (pH 7.4), 10 mM NaCl, 3 mM MgCl2, 1% bovine serum albumin, nuclease-free H2O) and resuspended in nuclei buffer (10x Genomics) at a concentration of 5,000 cells/μl.
scATAC-seq sequencing
After loading (targeted recovery of 7,000 cells) onto a Chromium Chip E (10x Genomics cat. 1000082), library preparation was performed using the Chromium Single Cell ATAC Library & Gel Bead Kit (cat. 1000110) according to manufacturer’s instructions. Following quality control using a (BioAnalyzer 2100, RRID:SCR_019715) High Sensitivity DNA Kit (Agilent), pooled libraries were sequenced on a (Illumina NovaSeq 6000 Sequencing System, RRID:SCR_016387) S1 or S2 platform with 50 bp paired-end reads, 8 bp for index 1 and 16 bp for index 2.
scRNA-seq sequencing
17,000 cells per sample were loaded onto a Chromium Chip A (10x Genomics cat. 120236). Single cell gene expression was obtained using the Chromium Single Cell 3’ or 5′ Library & Gel Bead Kit (cat. 120237 or 1000006). Library preparations were performed according to manufacturer’s instructions. Following quality control with a Bioanalyzer High Sensitivity DNA Kit (Agilent), pooled libraries were sequenced on a (Illumina NovaSeq 6000 Sequencing System, RRID:SCR_016387) with 150 bp paired-end reads, and 8 bp for index 1.
scRNA-seq data analysis
Raw sequencing reads were demultiplexed and aligned using (Cell Ranger, RRID:SCR_017344) version 3.1.0 with the GRCh38-3.0.0 reference genome. Downstream analyses of scRNA-seq data were performed in (RStudio, RRID:SCR_000432) using the (Seurat, RRID:SCR_007322) package version 3.2.0.(49). Low quality cells were excluded from downstream analyses based on %mitochondrial reads <20, features per cell >200 and <2,500, and number of reads per cell <10,000. Differential gene expression analysis was performed on pseudobulk transcriptome profiles using Seurat::FindMarkers() and visualized using (ggplot2, RRID:SCR_014601).
mtscATAC-seq data analysis
Demultiplexing and alignment of raw sequencing reads was performed using (Cell Ranger ATAC, RRID:SCR_021160) version 1.2.0 with a custom GRCh38 reference genome. Mitochondrial DNA mutations were called with (mgatk, RRID:SCR_021159) 0.5.6 using default parameters (11) and call_mutations_mgatk() downloaded from https://github.com/caleblareau/mtscATACpaper_reproducibility or the (Signac, RRID:SCR_021158) functionality for mtscATAC-seq data. Mitochondrial DNA mutations were selected for further analysis based on variant mean ratio >0.01 and strand concordance >0.6. Downstream analyses of mtscATAC-seq data were performed using the (ArchR, RRID:SCR_020982) package (50). The ArchR manual was followed for exclusion of low-quality cells (transcription start site (TSS) enrichment <4 and fragments per cell <1000) and doublets as well as analyses of chromatin marker peaks, calculation of imputed gene activity scores and transcription factor deviation scores.
Mitochondrial mutations and copy number alterations were analyzed as previously described (https://github.com/caleblareau/mtscATACpaper_reproducibility) in cells with coverage of mitochondrial DNA >10-20x. Specifically, copy number variants were calculated using patient-specific mtscATAC-seq data from non-CLL immune cells as healthy controls (11).
Statistical testing
Statistical significance of differences between median numbers of mtDNA mutations per cell of CLL, T cells and monocytes was calculated using a Wilcoxon signed-rank test by pairwise comparison of CLL with T cells and monocytes, and between T cells and monocytes. Mitochondrial DNA mutations with significant enrichment in CLL, T cells and monocytes were detected with a Fisher’s exact test using the number of cells with a heteroplasmy >0.05% for each mutation between CLL and T cells as well as CLL cells and monocytes. Adjustment for multiple hypothesis testing was performed using a Benjamini-Hochberg correction (FDR).
Data and materials availability:
Single cell ATAC and RNA sequencing data for CLL1-9 can be downloaded from Gene Expression Omnibus (GEO) (GSE163579 and GSE165087). Single cell ATAC sequencing data for CLL A-B were previously published (11) and can be accessed from GEO (GSE142745) and at https://osf.io/bupge. Whole-exome sequencing data from CLL1 were accessed from dbGap (phs001431.v1.p). Whole-exome sequencing data from CLL9 is available on dbGap (Phs002458.v1).
Supplementary Material
Statement of Significance:
Single cell multi-omic profiling of chronic lymphocytic leukemia reveals the utility of somatic mitochondrial DNA mutations as in vivo barcodes, which mark subclones that can evolve over time along with changes in accessible chromatin and gene expression profiles to capture dynamics of disease evolution.
Acknowledgments:
We thank Doreen Hearsey and the members of the DFCI Ted and Eileen Pasquarello Tissue Bank in Hematologic Malignancies for provision of samples and the patients who generously consented for the research use of these samples; Stacey Fernandes for expert technical assistance. We are grateful to Hayley Lyons, Sam Pollock, Oriol Olive and Brian Danysh for project management support. We thank the members of the Wu Lab for constructive discussions of the data.
Financial support:
This work was supported by grants from the National Cancer Institute and from the National Institute of Diabetes and Digestive and Kidney Diseases of the National Institutes of Health (R01DK103794 to V.G.S.; UG1 CA233338, 1U24CA224331-01, P01CA206978 and P01CA229092 to C.J.W.). L.Pe. is supported by a research fellowship from the German Research Foundation (DFG, PE 3127/1-1). S.H.G. is supported by a Kay Kendall Leukaemia Fund Fellowship. S.L. is supported by the NCI Research Specialist Award (R50CA251956). C.A.L. is supported by a Stanford Science Fellowship. L.S.L. is supported by an Emmy Noether fellowship by the German Research Foundation (DFG, LU 2336/2-1). V.G.S. is a New York Stem Cell Foundation-Robertson Investigator. E.M.P. is supported by a Doris Duke Charitable Foundation (DDCF) Physician-Scientist Fellowship, a Dana-Farber FLAIR Fellowship in Lymphoma/Leukemia Research and an American Society of Clinical Oncology Young Investigator Award (ASCO YIA). J.R.B. is supported by NIH R01 CA 213442 (PI: Brown, Jennifer). This work was partially supported by the Broad/IBM Cancer Resistance Research Project.
Footnotes
Conflicts of interest:
C.J.W. holds equity in BioNTech,Inc; and receives research support from Pharmacyclics. M.S.D. received an institutional research grant from Bristol-Myers Squibb during the conduct of this study, as well as personal fees from AbbVie, Adaptive Biotechnologies, Ascentage Pharma, AstraZeneca, Beigene, Celgene, Genentech, Eli Lilly, Janssen, Merck, Novartis, Pharmacyclics, Research to Practice, TG Therapeutics, and Verastem, and institutional research funding from Ascentage Pharma, Astra-Zeneca, Genentech, MEI Pharma, Novartis, Pharmacyclics, Surface Oncology, TG Therapeutics and Verastem, outside the submitted work. C.A.L., L.S.L. and V.G.S. are listed as co-inventors on a patent related to mtscATAC-seq (US provisional patent application 62/683,502). J.R.B. has served as a consultant for Abbvie, Acerta, Astra-Zeneca, Beigene, Catapult, Dynamo Therapeutics, Eli Lilly, Genentech/Roche, Gilead, Juno/Celgene/Bristol Myers Squibb, Kite, Loxo, MEI Pharma, Nextcea, Novartis, Octapharma, Pfizer, Pharmacyclics, Rigel, Sunesis, TG Therapeutics, Verastem; received research funding from Gilead, Loxo, Sun, TG Therapeutics and Verastem; and served on data safety monitoring committees for Invectys. G.G. receives research funds from IBM and Pharmacyclics, and is an inventor on patent applications related to MuTect, ABSOLUTE, MutSig, MSMuTect, MSMutSig, MSIdetect, POLYSOLVER and TensorQTL. G.G. is a founder, consultant and holds privately held equity in Scorpion Therapeutics. D.N. and T.J.K. receive research funding from Pharmacyclics.
The remaining authors declare no competing financial interests.
References and Notes:
- 1.McGranahan N, Swanton C. Clonal Heterogeneity and Tumor Evolution: Past, Present, and the Future. Cell. 2017;168:613–28. [DOI] [PubMed] [Google Scholar]
- 2.Suvà ML, Riggi N, Bernstein BE. Epigenetic Reprogramming in Cancer. Science. 2013;339:1567–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ashworth A, Lord CJ, Reis-Filho JS. Genetic Interactions in Cancer Progression and Treatment. Cell. 2011;145:30–8. [DOI] [PubMed] [Google Scholar]
- 4.Landau DA, Tausch E, Taylor-Weiner AN, Stewart C, Reiter JG, Bahlo J, et al. Mutations driving CLL and their evolution in progression and relapse. Nature. 2015;526:525–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Landau DA, Carter SL, Stojanov P, McKenna A, Stevenson K, Lawrence MS, et al. Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell. 2013;152:714–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Landau DA, Sun C, Rosebrock D, Herman SEM, Fein J, Sivina M, et al. The evolutionary landscape of chronic lymphocytic leukemia treated with ibrutinib targeted therapy. Nature Communications. 2017;8:2185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Salgia R, Kulkarni P. The Genetic/Non-genetic Duality of Drug ‘Resistance’ in Cancer. Trends in Cancer. 2018;4:110–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nam AS, Kim K-T, Chaligne R, Izzo F, Ang C, Taylor J, et al. Somatic mutations and cell identity linked by Genotyping of Transcriptomes. Nature. 2019;571:355–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yuan Y, Ju YS, Kim Y, Li J, Wang Y, Yoon CJ, et al. Comprehensive molecular characterization of mitochondrial genomes in human cancers. Nature Genetics. 2020;52:342–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gorelick AN, Kim M, Chatila WK, La K, Hakimi AA, Berger MF, et al. Respiratory complex and tissue lineage drive recurrent mutations in tumour mtDNA. Nature Metabolism. 2021;1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lareau CA, Ludwig LS, Muus C, Gohil SH, Zhao T, Chiang Z, et al. Massively parallel single-cell mitochondrial DNA genotyping and chromatin profiling. Nat Biotechnol. 2021;39:451–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ludwig LS, Lareau CA, Ulirsch JC, Christian E, Muus C, Li LH, et al. Lineage Tracing in Humans Enabled by Mitochondrial Mutations and Single-Cell Genomics. Cell. 2019;176:1325–1339.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marine J-C, Dawson S-J, Dawson MA. Non-genetic mechanisms of therapeutic resistance in cancer. Nature Reviews Cancer. 2020;20:743–56. [DOI] [PubMed] [Google Scholar]
- 14.Burger JA. Treatment of Chronic Lymphocytic Leukemia. New England Journal of Medicine. 2020;383:460–73. [DOI] [PubMed] [Google Scholar]
- 15.Rossi D, Spina V, Gaidano G. Biology and treatment of Richter syndrome. Blood. 2018;131:2761–72. [DOI] [PubMed] [Google Scholar]
- 16.Stewart JB, Chinnery PF. The dynamics of mitochondrial DNA heteroplasmy: implications for human health and disease. Nat Rev Genet. 2015;16:530–42. [DOI] [PubMed] [Google Scholar]
- 17.Lott MT, Leipzig JN, Derbeneva O, Xie HM, Chalkia D, Sarmady M, et al. mtDNA Variation and Analysis Using Mitomap and Mitomaster. Curr Protoc Bioinformatics. 2013;44:1.23.1–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kirino Y, Goto Y-I, Campos Y, Arenas J, Suzuki T. Specific correlation between the wobble modification deficiency in mutant tRNAs and the clinical features of a human mitochondrial disease. Proc Natl Acad Sci U S A. 2005;102:7127–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carew JS, Zhou Y, Albitar M, Carew JD, Keating MJ, Huang P. Mitochondrial DNA mutations in primary leukemia cells after chemotherapy: clinical significance and therapeutic implications. Leukemia. 2003;17:1437–47. [DOI] [PubMed] [Google Scholar]
- 20.Grünebach F, Erndt S, Häntschel M, Heine A, Brossart P. Generation of antigen-specific CTL responses using RGS1 mRNA transfected dendritic cells. Cancer Immunol Immunother. 2008;57:1483–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shkoda A, Town JA, Griese J, Romio M, Sarioglu H, Knöfel T, et al. The Germinal Center Kinase TNIK Is Required for Canonical NF-κB and JNK Signaling in B-Cells by the EBV Oncoprotein LMP1 and the CD40 Receptor. PLOS Biology. 2012;10:e1001376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fukuda T, Neuberg DS, Huynh L, Rassenti LZ, Toy TL, Rai KR, et al. Expression of T Cell Co-Stimulator (ICOS) and Its Ligand and Disease Progression in B-Cell Chronic Lymphocytic Leukemia. Blood. 2005;106:2943–2943. [Google Scholar]
- 23.Gruber M, Bozic I, Leshchiner I, Livitz D, Stevenson K, Rassenti L, et al. Growth dynamics in naturally progressing chronic lymphocytic leukaemia. Nature. 2019;570:474–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Castellano G, Torrisi E, Ligresti G, Nicoletti F, Malaponte G, Traval S, et al. Yin Yang 1 overexpression in diffuse large B-cell lymphoma is associated with B-cell transformation and tumor progression. Cell Cycle. 2010;9:557–63. [DOI] [PubMed] [Google Scholar]
- 25.Hodson DJ, Shaffer AL, Xiao W, Wright GW, Schmitz R, Phelan JD, et al. Regulation of normal B-cell differentiation and malignant B-cell survival by OCT2. Proc Natl Acad Sci U S A. 2016;113:E2039–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Batista CR, Lim M, Laramée A-S, Abu-Sardanah F, Xu LS, Hossain R, et al. Driver mutations in Janus kinases in a mouse model of B-cell leukemia induced by deletion of PU.1 and Spi-B. Blood Adv. 2018;2:2798–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Satterwhite E, Sonoki T, Willis TG, Harder L, Nowak R, Arriola EL, et al. The BCL11 gene family: involvement of BCL11A in lymphoid malignancies. Blood. 2001;98:3413–20. [DOI] [PubMed] [Google Scholar]
- 28.Ahn IE, Farooqui MZH, Tian X, Valdez J, Sun C, Soto S, et al. Depth and durability of response to ibrutinib in CLL: 5-year follow-up of a phase 2 study. Blood. 2018;131:2357–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rendeiro AF, Krausgruber T, Fortelny N, Zhao F, Penz T, Farlik M, et al. Chromatin mapping and single-cell immune profiling define the temporal dynamics of ibrutinib response in CLL. Nature Communications. 2020;11:577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kozlova V, Ledererova A, Ladungova A, Peschelova H, Janovska P, Slusarczyk A, et al. CD20 is dispensable for B-cell receptor signaling but is required for proper actin polymerization, adhesion and migration of malignant B cells. PLoS One. 2020;15:e0229170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cui B, Chen L, Zhang S, Mraz M, Fecteau J-F, Yu J, et al. MicroRNA-155 influences B-cell receptor signaling and associates with aggressive disease in chronic lymphocytic leukemia. Blood. 2014;124:546–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cossarizza A, Chang H-D, Radbruch A, Acs A, Adam D, Adam-Klages S, et al. Guidelines for the use of flow cytometry and cell sorting in immunological studies (second edition). Eur J Immunol. 2019;49:1457–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shi J, Hou S, Fang Q, Liu X, Liu X, Qi H. PD-1 Controls Follicular T Helper Cell Positioning and Function. Immunity. 2018;49:264–274.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Harari A, Bellutti Enders F, Cellerai C, Bart P-A, Pantaleo G. Distinct profiles of cytotoxic granules in memory CD8 T cells correlate with function, differentiation stage, and antigen exposure. J Virol. 2009;83:2862–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Al’Khafaji A, Gutierrez C, Brenner E, Durrett R, Johnson KE, Zhang W, et al. Expressed barcodes enable clonal characterization of chemotherapeutic responses in chronic lymphocytic leukemia. bioRxiv. 2019;761981. [Google Scholar]
- 36.Hansmann L, Han A, Penter L, Liedtke M, Davis MM. Clonal Expansion and Interrelatedness of Distinct B-Lineage Compartments in Multiple Myeloma Bone Marrow. Cancer Immunol Res. 2017;5:744–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Penter L, Dietze K, Ritter J, Cobo MFL, Garmshausen J, Aigner F, et al. Localization-associated immune phenotypes of clonally expanded tumor-infiltrating T cells and distribution of their target antigens in rectal cancer. OncoImmunology. 2019;8:e1586409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Penter L, Dietze K, Bullinger L, Westermann J, Rahn H-P, Hansmann L. FACS single cell index sorting is highly reliable and determines immune phenotypes of clonally expanded T cells. European Journal of Immunology. 2018;48:1248–50. [DOI] [PubMed] [Google Scholar]
- 39.Stewart JB, Chinnery PF. Extreme heterogeneity of human mitochondrial DNA from organelles to populations. Nat Rev Genet. 2021;22:106–18. [DOI] [PubMed] [Google Scholar]
- 40.Kikushige Y, Ishikawa F, Miyamoto T, Shima T, Urata S, Yoshimoto G, et al. Self-renewing hematopoietic stem cell is the primary target in pathogenesis of human chronic lymphocytic leukemia. Cancer Cell. 2011;20:246–59. [DOI] [PubMed] [Google Scholar]
- 41.Browne P, Petrosyan K, Hernandez A, Chan JA. The B-cell transcription factors BSAP, Oct-2, and BOB.1 and the pan-B-cell markers CD20, CD22, and CD79a are useful in the differential diagnosis of classic Hodgkin lymphoma. Am J Clin Pathol. 2003;120:767–77. [DOI] [PubMed] [Google Scholar]
- 42.Bachireddy P, Ennis C, Nguyen VN, Gohil SH, Clement K, Shukla SA, et al. Distinct evolutionary paths in chronic lymphocytic leukemia during resistance to the graft-versus-leukemia effect. Science Translational Medicine. 2020;12:eabb7661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen J, Xu-Monette ZY, Deng L, Shen Q, Manyam GC, Martinez-Lopez A, et al. Dysregulated CXCR4 expression promotes lymphoma cell survival and independently predicts disease progression in germinal center B-cell-like diffuse large B-cell lymphoma. Oncotarget. 2015;6:5597–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fleur-Lominy SS, Evensen NA, Bhatla T, Sethia G, Narang S, Choi JH, et al. Evolution of the Epigenetic Landscape in Childhood B Acute Lymphoblastic Leukemia and Its Role in Drug Resistance. Cancer Res. 2020;80:5189–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Quinquenel A, Fornecker L-M, Letestu R, Ysebaert L, Fleury C, Lazarian G, et al. Prevalence of BTK and PLCG2 mutations in a real-life CLL cohort still on ibrutinib after 3 years: a FILO group study. Blood. 2019;134:641–4. [DOI] [PubMed] [Google Scholar]
- 46.Byrd JC, Hillmen P, O’Brien S, Barrientos JC, Reddy NM, Coutre S, et al. Long-term follow-up of the RESONATE phase 3 trial of ibrutinib vs ofatumumab. Blood. 2019;133:2031–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhu C, Preissl S, Ren B. Single-cell multimodal omics: the power of many. Nature Methods. 2020;17:11–4. [DOI] [PubMed] [Google Scholar]
- 48.Mimitou EP, Lareau CA, Chen KY, Zorzetto-Fernandes AL, Takeshima Y, Luo W, et al. Scalable, multimodal profiling of chromatin accessibility and protein levels in single cells. bioRxiv. 2020;2020.09.08.286914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM, et al. Comprehensive Integration of Single-Cell Data. Cell. 2019;177:1888–1902.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Granja JM, Corces MR, Pierce SE, Bagdatli ST, Choudhry H, Chang HY, et al. ArchR is a scalable software package for integrative single-cell chromatin accessibility analysis. Nature Genetics. 2021;53:403–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.