

Abstract
Purpose
We investigated the change, if any, in prostaglandin E2 (PGE2) signaling in endometriotic lesions of different developmental stages in mouse. In addition, we evaluated the effect of treatment of mice with induced deep endometriosis (DE) with inhibitors of PGE2 receptor subtypes EP2 and EP4 and metformin.
Methods
Three mouse experimentations were conducted. In Experiment 1, female Balb/C mice were induced with endometriosis or DE and were serially sacrificed after induction. Experiments 2 and 3 evaluated the effect of treatment with EP2 and EP4 inhibitors and metformin, respectively, in mice with induced DE. Immunohistochemistry analysis of COX‐2, EP2, and EP4, along with the extent of lesional fibrosis, was evaluated.
Results
The immunostaining of COX‐2, EP2, and EP4 turned from activation to a stall as lesions progressed. Treatment with EP2/EP4 inhibitors in DE mice exacerbated endometriosis‐associated hyperalgesia and promoted fibrogenesis in lesions even though it suppressed the PGE2 signaling dose‐dependently. In contrast, treatment with metformin resulted in increased PGE2 signaling, concomitant with improved hyperalgesia, and retarded lesional fibrogenesis.
Conclusions
The PGE2 signaling diminishes as endometriotic lesions progress. Treatment with EP2/EP4 inhibitors in DE mice exacerbates endometriosis, but metformin appears to be promising seemingly through the induction of the PGE2 signaling.
Keywords: cyclooxygenase 2, E series prostanoid receptor, endometriosis, fibrosis, mouse
1. INTRODUCTION
Endometriosis is an estrogen‐dependent and inflammatory gynecologic disease affecting 6%–10% of women of reproductive age. 1 Despite advances in surgery and expansion of our knowledge on its molecular aberrations, the clinical management of endometriosis remains a challenge, 1 and the development of more efficacious and safer medical treatment is still an unfulfilled need for endometriosis. Unfortunately, the development of non‐hormonal drugs for endometriosis has been painfully stagnant. 2 , 3 Recognizing the chronic proinflammatory nature of the disease, many non‐hormonal anti‐inflammation drugs have been tested but all apparently failed. 3
Complexity aside, this stagnancy is likely attributable also to the fibrotic content in lesions, 3 probably a result of the well‐documented diagnostic delay in endometriosis. 4 By the time when the patient is finally diagnosed unequivocally with endometriosis, her lesions may already become highly fibrotic, and, as such, become very difficult to treat by medication. 5
In endometriosis, prostaglandin E2 (PGE2), a bioactive prostanoid, sits right at the nexus of estrogen biosynthesis and inflammation. 6 Abundant data indicate that COX‐2 is overexpressed in endometriotic lesions. 7 , 8 , 9 , 10 , 11 , 12 , 13 In addition, the lesional expression of microsomal PGE2 synthases (mPGESs)—genes coding for the terminal enzymes that specifically convert PGH2 to PGE2—is reported also to be elevated in endometriosis, 12 suggesting that the PGE2 signaling pathway is activated in endometriosis.
Preclinical studies have shown that several selective COX‐2 inhibitors, called COXIBS, are effective in suppressing endometriosis. 14 , 15 , 16 One clinical study went even further to demonstrate that treatment with rofecoxib postoperatively for 6 months is effective in the management of pelvic pain associated with endometriosis. 17 However, due to the elevated risk of cardiovascular events, several commercial COXIBS were pulled out from the market in 2004–2005, quelling the interest in COXIBS as a therapeutics for endometriosis. In addition, the possible disruption of ovulation also is of concern. 18
The biological actions of PGE2 are mediated via its receptors EP1–4 by integrating multiple signaling pathways. 19 In endometriosis, it has been reported that EP2 and/or EP4 are overexpressed. 13 , 20 It also has been shown that an EP2/EP4 inhibition induces apoptosis 21 and suppresses invasion. 22 One study also demonstrates that selective antagonism against EP2/EP4 attenuated DNA synthesis, cAMP accumulation, and IL‐1β‐induced secretion of IL‐6 and IL‐8 and aromatase expression. 23 EP2/EP4 antagonism also resulted in reduced expression of vascular endothelial growth factor (VEGF), and chemokine ligand 2 (CXCL2) and CXCL3. 23 Mouse studies indicated that inhibiting EP2/EP4 decreased the growth and survival of endometriotic tissues, 24 and treatment with an EP2 antagonist reduced hyperalgesia. 25
However, we recently found that increased fibrotic content in endometriotic lesions is accompanied by significantly reduced PGE2 signaling, manifesting as reduced expression of COX‐2, EP2, and EP4, 26 consistent with the reported attenuation of PGE2 signaling in several fibrotic conditions. Indeed, as the extent of fibrosis—and thus tissue stiffness—increases, it interferes with multiple steps of PGE2 biosynthesis, including the suppression of PGESs, 27 and the PGE2 signaling becomes subsided. 28 , 29
A close scrutiny of the published studies would reveal that while both EP2 and EP4 have been shown to be overexpressed in endometriotic lesions, 13 the supporting data were based on 16 tissue samples, of which 12 (75%) were from ovarian endometrioma (OE) and the rest 4 (25%) were from deep endometriosis (DE) lesions. 13 Yet DE lesions are known to be more fibrotic than OE lesions. 5 Another study failed to show EP2 overexpression. 20 More curiously, a recent study demonstrating the therapeutic potentials of EP2/EP4 inhibitors actually showed that the EP2/EP4 staining in ectopic endometrium appears to be lower than that in normal endometrium. 23
As for the study showing the efficacy of EP2/EP4 inhibitor treatment, it can be seen that the results were based on the treatment that was started merely 2 weeks after induction of endometriosis on immunodeficient mice. 24 In another study using immune‐competent mouse, 24 , 30 the treatment with EP2/EP4 inhibitors started 3 weeks after the induction of endometriosis. Typically, 2 or 3 weeks are not long enough to permit lesional fibrogenesis to consummate. 3 , 31 This raises the question as whether PGE2 signaling would change as endometriotic lesions progress, and, if so, whether EP2/EP4 inhibition would be truly efficacious when lesions induced in mouse can recapitulate some salient features of their human counterpart.
In this study, we first carried out a serial experimentation to evaluate the immunohistochemistry staining of COX‐2, EP2, and EP4 in lesions in mice with induced endometriosis and DE. Based on the finding that the PGE2 signaling is attenuated when lesions are fibrotic enough, we further evaluated the efficacy of EP2/EP4 inhibitors in mice with induced DE. Finally, we evaluated the effect of metformin treatment on mice with induced DE and on PGE2 signaling in lesions, since endometriosis is increasingly recognized as a fibrotic condition 32 , 33 and metformin is recently shown to reverse lung fibrosis. 34 , 35
2. MATERIALS AND METHODS
2.1. Animals
All mice used for this study were female Balb/C mice, 6‐week old, and ~18–21 g in bodyweight and were purchased from the SLAC Experimental Animal Company. They were maintained under controlled conditions with a light/dark cycle of 12/12 h and had access to chows and water ad libitum. All mice underwent experimental procedures after two weeks of acclimatization. All experiments were performed under the National Research Council's Guide for the Care and Use of Laboratory Animals 36 and were approved by the institutional review board on experimental animals of Shanghai OB/GYN Hospital, Fudan University.
2.2. Experiment protocols
Three mouse experiments were conducted. The purpose and protocol of these three experiments are detailed below.
2.2.1. Experiment 1. To evaluate the changes, if any, in the PGE2 signaling pathway in the mouse model of (regular) endometriosis (EM) and DE
Ninety mice were used for this experiment, and 30 of them were randomly selected as donors to provide uterine tissue fragments while the remaining 60 mice were designated as recipients that received the fragments. We used the established mouse model of endometriosis by Somigliana et al. 37 and of the mouse DE model. 38
The recipient mice were randomly divided into three equal‐sized groups: Control or CT group, in which mice received intraperitoneal (i.p.) injection of fat tissues, instead of uterine fragments, for sham modeling, EM group, in which mice were induced with endometriosis by i.p. injection of uterine fragments harvested from donor mice, 37 and DE group, in which mice also had i.p. injection of uterine fragments but also received infusion of substance P. 38 We designated Day 0 as the induction day when mice received i.p. injection of either uterine or fat tissue fragments. At day −1, mice in the DE group were inserted with Alzet osmotic pumps (model 1004, DURECT Corp) containing substance P (0.1 mg/kg/day; Abcam), 39 while those in the EM and CT groups were inserted with identical pumps containing the same volume of sterile saline. On Day 0, all donor mice were sacrificed and their uteri were harvested. To induce endometriosis, uterine tissues were harvested from one donor mouse were processed and then injected i.p. to two recipient mice, one each from EM and DE groups, and an equal volume (half of that uterine tissues because of only one recipient) of fat tissues from the parametrium of the same donor mouse was harvested, processed similarly as the uterine tissues, and then injected i.p. to one CT mouse. Two and four weeks after induction, 10 mice each from the three groups were selected at random and sacrificed. Eutopic (for CT mice) and ectopic (for EM and DE mice) endometrial tissue samples were carefully excised, collected, weighed, and then processed for further analyses. Bodyweight and hotplate latency were evaluated before induction and sacrifice.
2.2.2. Experiment 2. To evaluate the treatment effect of different doses of EP2/4 inhibitors in mice with induced DE
This experiment used 48 mice, and 16 of them were randomly selected as donors while remaining 32 mice were recipients. The recipient mice were induced with DE as described previously 39 and above. Four weeks after induction, the recipient mice were randomly divided into four groups in equal sizes: Control, Low (low‐dose), Medium (medium‐dose), and High (high‐dose) groups, and were treated, through daily oral gavage, with different doses of PF‐04418948 (PZ0213, Sigma), an EP2 inhibitor (EP2I), and ONO‐AE3‐208 (SML2076, Sigma), an EP4 inhibitor (EP4I). We chose these two inhibitors since they both have excellent specificity. 40 In addition, they both have superior oral bioavailability. 41 , 42 PF‐04418948 was also used by. 25 In this experiment and in Experiment 3, the duration of treatment was set to be 3 weeks, was based on the principle that treatment period should be long enough to realize its therapeutic potential yet not overly long, especially compared with the length of the induction period. Here, we used the rough conversion of average human lifespan, which is about 70 years old, to mouse, of which the average life span is 2 years old. Thus, 3 weeks in mouse would be equivalent to 3 × 35 = 105 weeks or about 2 years in humans. The 2 years of medication appears to be long enough to exert treatment effect but not too long.
The low‐dose group were administrated with EP2I 10 mg/kg/day and EP4I 5 mg/kg/day, and the medium‐ and high‐dose groups were EP2I 10 mg/kg/day + EP4I 10 mg/kg/day and EP2I 30 mg/kg/day + EP4I 10 mg/kg/day, respectively. The choice of these three dosages was based on published studies on mouse. 43 , 44 The mice in the Control group received an equal volume of saline with the same administration route. Three weeks of the start of the treatment, all groups of mice were sacrificed, and all their lesions were carefully excised, evaluated, and processed for further analyses. Bodyweight and hotplate latency were evaluated on Day 0 before induction, 4 weeks after induction but before treatment, and 5, 6, and 7 weeks after induction.
2.2.3. Experiment 3. To evaluate the treatment effect of metformin in mice with induced DE
Thirty mice were used for this experiment, and 10 of them were randomly selected as donors and the remaining 20 were designated as recipients. The recipient mice were induced with DE as described above. Four weeks after the induction, the 20 mice were randomly divided into two equal‐sized groups: Control group and Metformin (Met) group. Mice in Met group were injected i.p. with metformin (PHR1084, Sigma) 200 mg/kg/day dissolved in 300 μl sterile saline, while Control mice were injected i.p. with an equal volume of saline. The choice of this dosage was based on the conversion of human dosage, which ranges from 1000 to 2000 mg per day, to mouse, which ranges from 150 to 300 mg/kg, as well as on published studies on mouse. 45 , 46 The choice of the i.p. administration route was based on a previous report. 47
After 3 weeks of treatment, all mice were sacrificed, and the endometriotic lesions, bodyweight, and hotplate latency were evaluated as described in Experiment 2.
2.3. The hotplate test and lesion measurement
We used the hotplate test to evaluate the endometriosis‐associated hyperalgesia in mice as reported previously. 48 Hot Plate Analgesia Meter (Model BME‐480, Institute of Biomedical Engineering, Chinese Academy of Medical Sciences) was used to administrate hotplate test to all mice. Before each test, the mouse was brought into the testing room to acclimatize for 10 min. During the test, the temperature of the metal plate was kept to 55.0°C. The mouse was put inside a plastic cylinder with a height of 20 cm and a diameter of 18 cm on the metal plate of 25 cm × 25 cm in size. The responding latency was defined as the time interval (in seconds) from the time when the mouse was put into the cylinder to the time when it licked or flicked its hind paws, or jolted or jumped off the hot plate. The extent of endometriosis was measured by measuring the dry weight of all lesions excised from mice as described previously. 37 All endometriotic lesions were excised and carefully weighed and then fixed with 10% formalin (w/v) for further analyses.
2.4. Immunohistochemistry (IHC) staining
All mice were sacrificed by cervical dislocation, and their lesions were harvested, weighed, and fixed with 10% formalin (w/v) and paraffin‐embedded for further evaluation or immunohistochemistry. Serial 4‐μm sections were obtained from each block, with the subsequent slides for immunohistochemistry (IHC) analysis for cyclooxygenase‐2 (COX‐2, 1:300; #ab15191; Abcam), EP2 (1:300; #ab167171; Abcam), EP4 (1:300; #bs‐8538r; Bioss), and αsmooth muscle actin (α‐SMA, 1:100; #ab5694. Abcam). The tissue slides were incubated at 60℃ for 1 h and then were deparaffinized in xylene and rehydrated in a series of graded ethanol concentrations. For antigen retrieval, the slides were soaked in a citrate buffer (pH 6.0) and put into pressure cooker to boil for 3 min. Cool down naturally to room temperature. After three‐time rinsing in phosphate‐buffered saline (PBS), the slides were incubated with 0.5% goat blocking serum for 30 min at room temperature for blocking nonspecific binding sites. Then incubate with the primary antibodies overnight at 4°C. After three‐time rinsing in PBS, the slides were incubated with biotinylated secondary antibody, Supervision TM Universal (Anti‐Mouse/Rabbit) Detection Reagent (HRP; Shanghai GeneTech Company) for 30 min at room temperature. The bound antibody complexes were stained for 3–5 min or until appropriate for microscopic examination with diaminobenzidine and then counterstained with hematoxylin (1 min) and mounted. Images were obtained with an Olympus BX51 microscope (Olympus) fitted with a digital camera (Olympus DP70, Olympus) at 400× magnification and export as a TIFF‐format digital file. Images taken from the same experiment were obtained under identical microscopy conditions and were evaluated on the same day. When replacing the sample, the microscope settings were kept constant except the necessary adjustment of focal length and field of view, with the automatic white balance function turned off and to keeping all images with the identical exposure time and aperture. Three to five images of each sample were randomly selected to obtain a mean optional density value by Image Pro‐Plus 6.0 (Media Cybernetics, Inc.). For negative controls, we used IgG from rabbit or mouse serum instead of the primary antibodies. For positive controls, human colorectal tumor tissues were used for COX‐2, human colon tissues for EP2, lung cancer tissues for EP4, and mouse intestine tissues for α‐SMA (Figure S1).
2.5. Masson trichrome staining
Masson trichrome staining was employed to evaluate the extent of lesional and tissue fibrosis. The tissue slides were incubated at 60°C for 1 h and then were deparaffinized in xylene and rehydrated in a series of graded ethanol concentrations. Then, slides were immersed at 37°C for 2 h in Bouin's solution, which consisted of a mixture of saturated 75 ml of picric acid, 25 ml of 10% (w/v) formalin solution, and 5 ml of acetic acid. Sections were stained using the Masson's Trichrome Staining kit (Servicebio) following the manufacturer's instructions. Image Pro‐Plus 6.0 was used to calculate the blue‐stained areas of the collagen fiber layer in proportion to the entire field of the ectopic endometrium.
2.6. Statistical analysis
The lesion weight data and the immunostaining data from Experiments 2 and 3 were presented in boxplots. In a boxplot, the line in the box represents the median, and the upper and lower sides represent the 3rd and the 1st quantile, respectively, while the upper and lower whiskers represent respectively the maximum and the minimum values in the data. The comparison of distributions of continuous variables between or among two or more groups was made using the Wilcoxon and Kruskal tests, respectively, and the paired Wilcoxon test was used when the before‐after comparison was made for the same group of subjects. A Pearson rank correlation coefficient was used when evaluating correlations between two variables. Multiple linear regression analysis was used to identify which factor(s) were associated with the lesion weight or IHC measure (square‐root‐ or log‐transformed to improve normality, where appropriate).
P‐values of <0.05 were considered statistically significant. All computations were made with R 4.1.0 49 (www.r‐project.org).
3. RESULTS
3.1. The diminishing PGE2 signaling in lesions as lesions progressing in mice with (regular) endometriosis (EM) and deep endometriosis (DE)
All 60 mice survived the Experiment 1. As expected, there was no difference in bodyweight at the baseline (P = 0.94; Figure 1A) and 2 weeks after the induction of endometriosis (P = 0.64; Figure 1A) among the 3 groups of mice. However, 4 weeks after the induction, there was a significant difference in bodyweight (P = 7.4 × 10−5; Figure 1A), with mice in the EM and DE groups having significantly reduced bodyweight as compared with the CT mice (P = 0.0046 and P = 0.0002, respectively; Figure 1A). In particular, the bodyweight of the DE mice was significantly lower than that of the EM mice (P = 0.013; Figure 1A). Multiple linear regression on bodyweight using time of measurement, induction of endometriosis or not, and infusion with substance P (SP) or not indicates that the time of measurement was positively associated with the bodyweight (P < 2.2 × 10−16), and both endometriosis induction and SP infusion interacted negatively with the time of measurement (P = 0.0013 and P = 0.036, R 2 = 0.62), suggesting that while there is a general trend for increasing bodyweight, both endometriosis and SP infusion attenuated the increase, due possibly to the endometriosis‐associated pain‐suppressive food intake. This is further evidenced by the correlation between the hotplate latency and bodyweight at the end of the 4th week (r = 0.67, P = 4.6 × 10−5) and between the change in latency and bodyweight since the start of the experiment (r = 0.65, P = 0.00011).
FIGURE 1.
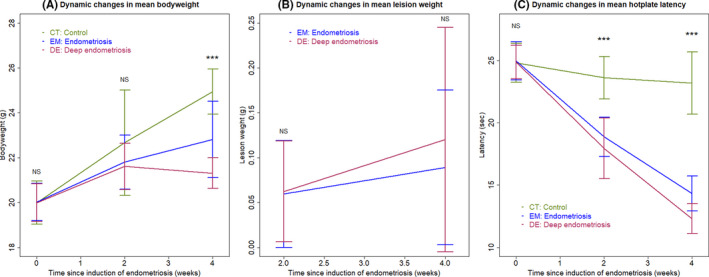
(A) Dynamic changes in the mean bodyweight in three different groups of mice. (B) Dynamic changes in mean lesion weight between mice with induced (regular) endometriosis and deep endometriosis. (C) Dynamic changes in the mean hotplate latency in three different groups of mice. In all panels, the data are represented by the means ± SDs. Symbols of statistical significance levels: ***P < 0.001; NS: not statistically significant (P > 0.05). In panels (A) and (C), Kruskal's rank test was used, while Wilcoxon's test was used in panel (B)
No difference in lesion weight between EM and DE groups was found at 2 and 4 weeks after the induction of endometriosis (both P's ≥ 0.80; Figure 1B), despite the apparent trend for the increase in lesion weight in both the EM and DE groups, especially in the DE mice (Figure 1B).
We also evaluated the severity of hyperalgesia resulting from endometriosis using hotplate test. As expected, there was no difference in hotplate latency before the induction of endometriosis among the three groups (P = 1.0; Figure 1C). However, 2 and 4 weeks after the induction, there was a significant difference among the 3 groups (P = 0.0001 and P = 1.1 × 10−5; Figure 1C). Of note, while there was no significant reduction in latency at 2 weeks after induction in the CT mice (P = 0.47; Figure 1C), there was a marginally significant reduction at 4 weeks (P = 0.058; Figure 1C), but still no difference in latency between 2 and 4 weeks after induction (P = 0.70). In contrast, at both 2 and 4 weeks after the induction, both EM and DE groups had significantly reduced latency either compared with their baseline levels or the levels at 2 weeks (all P's < 0.006; Figure 1C). At both 2 and 4 weeks after induction, both EM and DE groups had significantly shorter latency than that of the CT mice (all P's < 0.0005; Figure 1C). Multiple linear regression on latency using time of measurement, induction of endometriosis or not, and infusion with SP or not indicates that the time of measurement was negatively associated with the latency (P = 0.019), and both endometriosis induction and SP infusion interacted negatively with the time of measurement (P < 2.2 × 10−16 and P = 0.0013, R 2 = 0.90).
Next, we evaluated the extent of tissue fibrosis and markers of the PGE2 pathway by immunohistochemistry, including COX‐2, EP2, and EP4, in ectopic endometrium of mice from EM and DE groups as well as in endometrium from CT mice, matched by time of tissue harvest. As shown in Figure 2, the extent of lesional fibrosis increased as lesions progressed, especially in DE lesions. In addition, the immunostaining of COX‐2, EP2, and EP4 was seen both in epithelial and stromal cells, and COX‐2 staining was localized in the cell cytoplasm, while staining of EP2 and EP4 was in the cell membrane (Figure 2).
FIGURE 2.
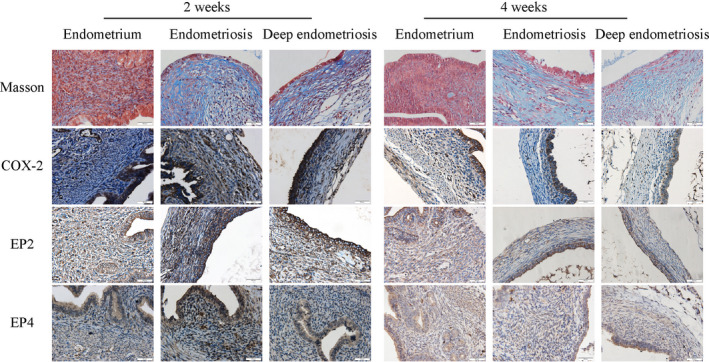
Representative photomicrographs of immunostaining and histochemistry analysis of endometrial samples from control mice and endometriotic lesions from mice with (regular) endometriosis and deep endometriosis at 2 and 4 weeks after induction. Different rows show different markers as indicated. Different columns represent different tissue samples from normal endometrium from control mice, endometriotic lesions from mice with induced endometriosis and deep endometriosis, respectively, grouped under time point at which the tissue samples were harvested. In Masson trichrome staining, the collagen fibers in lesions were stained in blue. In all figures, magnification: ×400. Scale bar = 50 μm
Further analysis revealed two general trends (Figure 3). First, with the only exception of EP2, the extent of tissue fibrosis and the staining levels of COX‐2 and EP4 in endometrium from CT mice were largely unchanged during the 2‐week time period (all P‐values > 0.27; Figure 3A–C,F,G). The EP2 staining levels, however, were significantly elevated in both stromal and epithelial components during this time period (both P‐values ≤ 0.0020, Figure 3D,E). Second, starting from 2 weeks after induction, the extent of lesional fibrosis in both EM and DE mice was significantly higher than that of endometrium from CT mice (Table 1; Figure 3A). Third, there was a general trend that the PGE2 signaling was activated during the early stage of lesional progression (at 2 weeks after induction) but became largely abrogated during the later stage (Table 1; Figure 3B–G) despite increasing EP2 staining in normal endometrium. Lastly, this reverse in trend appears to be more prominent in DE lesions (Table 1; Figure 3B–G), which had higher fibrotic content than in EM lesions (P = 0.0003 at week 4; Figure 3A).
FIGURE 3.
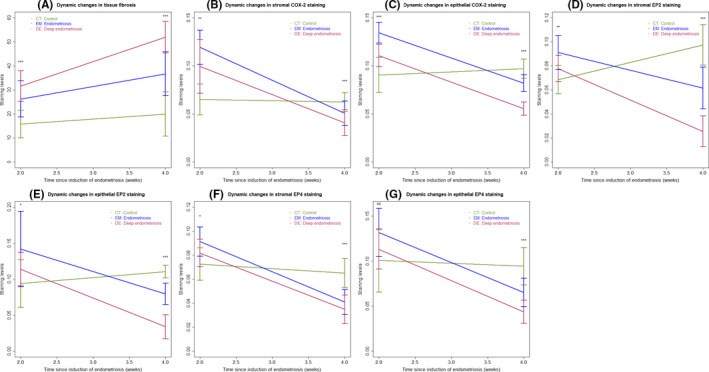
Summary result of immunohistochemistry and Masson trichrome staining in human samples showing the dynamic changes as endometriotic lesions progress over time. Dynamic changes in the extent of fibrosis in tissues/lesions in different groups of mice (A). Dynamic changes in the immunostaining of COX‐2 the stromal (B) and epithelial (C) components in tissues/lesions in different groups of mice. Dynamic changes in the immunostaining of EP2 the stromal (D) and epithelial (E) components in tissues/lesions in different groups of mice. Dynamic changes in the immunostaining of EP4 the stromal (F) and epithelial (G) components in tissues/lesions in different groups of mice. In all panels, the data are represented by the means ± SDs, and Kruskal's rank test was used. Symbols of statistical significance levels: *P < 0.05; **P < 0.01; ***P < 0.001; NS: not statistically significant (P > 0.05)
TABLE 1.
P‐values for the difference between the lesions of interest and the normal endometrium based on linear regression analysis
| Marker name | Type of lesions | 2 weeks | 4 weeks |
|---|---|---|---|
| Extent of tissue fibrosis | Endometriosis | ↑0.00095 | ↑0.00013 |
| Deep endometriosis | ↑1.1 × 10−5, R 2 = 0.53 |
↑4.0 × 10−9, R 2 = 0.73 |
|
| COX−2 | Endometriosis |
↑3.9 × 10−6 (↑1.5 × 10−7) |
↓0.039 (↓0.0005) |
| Deep endometriosis |
↑0.00091, R 2 = 0.56 (↑0.003, R 2 = 0.65) |
↓0.00043, R 2 = 0.37 (↓2.6 × 10−11, R 2 = 0.82) |
|
| EP2 | Endometriosis |
↑0.00032 (↑0.009) |
↓0.00023 (↓3.2 × 10−5) |
| Deep endometriosis |
0.11, R 2 = 0.39 (0.25, R 2 = 0.23) |
↓9.4 × 10−11, R 2 = 0.80 (↓1.3 × 10−12, R 2 = 0.85) |
|
| EP4 | Endometriosis |
↑0.0023 (↑0.022) |
↓7.6 × 10−5 (↓0.0011) |
| Deep endometriosis |
0.082, R 2 = 0.30 (0.34, R 2 = 0.18) |
↓3.0 × 10−6, R 2 = 0.59 (↓1.9 × 10−7, R 2 = 0.64) |
The red and blue arrows indicate the significant increase and decrease, respectively. The numbers shown in parenthesis are for the epithelial component.
The arrows ↑ and ↓ indicate, respectively, an increasing or decreasing trend by the sign of the regression coefficient. If an arrow is absent, then the regression coefficient is not statistically significant, indicating no association.
While the hotplate latency was not correlated well with the lesion weight (r = −0.20, P = 0.20), it was correlated negatively with the extent of lesional fibrosis (r = −0.78, P = 3.7 × 10−9; Figure S2A). The extent of lesional fibrosis correlated negatively with lesional staining levels of COX‐2, EP2, and PE4 in both stromal and epithelial components (all 6 r's≤‐0.63, all 6 P‐values ≤1.5 × 10−5; Figure S2B–G).
3.2. Treatment with EP2 and EP4 inhibitors exacerbates endometriosis in mice with deep endometriosis
Given the above finding that the PGE2 signaling is progressively diminished in endometriotic lesions as they progressed, one would infer that the further suppression of EP2 and EP4 by pharmacological means may not yield desired therapeutic effect, as shown previously. 22 , 24 , 50 , 51 To find out the efficacy of EP2 and EP4 inhibitors on endometriosis, we conducted Experiment 2 using the DE mouse model and then treated the mice with vehicle or PF‐04418948 (an EP2 inhibitor) and ONO‐AE3‐208 (an EP4 inhibitor) at low, medium, or high doses for 3 weeks. And we evaluated bodyweight, lesion weight, hotplate latency, and immunostaining of COX‐2, EP2, EP4, and α‐SMA, as well as the extent of lesional fibrosis.
Similar to Experiment 1 as presented above, there was no significant difference in bodyweight before and after the induction of DE but before the treatment (both P's > 0.83; Figure 4A). In addition, the bodyweight in all 4 groups of mice was increased steadily and significantly during the induction period (P = 8.3 × 10−7; Figure 4A). While there was no significant difference in bodyweight among the four groups of mice 1 and 2 weeks of treatment (both P's ≥ 0.18; Figure 4A), the difference was marginally significant at the end of the experiment (P = 0.060; Figure 4A). Multiple linear regression on the weight gain/loss in reference to the bodyweight at the start of the treatment using the duration of treatment and the dosages of EP2 and EP4 inhibitors as covariables, we found that both the duration of treatment and the dosage of the EP4 inhibitor are both negatively associated with the bodyweight (P = 0.038, P = 7.0 × 10−5; R 2 = 0.19). The correlation coefficient between the hotplate latency and bodyweight at the end of the experiment was 0.27 but not statistically significant (P = 0.13), so was the correlation coefficient between the change in latency and bodyweight since the start of the treatment (r = 0.30, P = 0.099).
FIGURE 4.
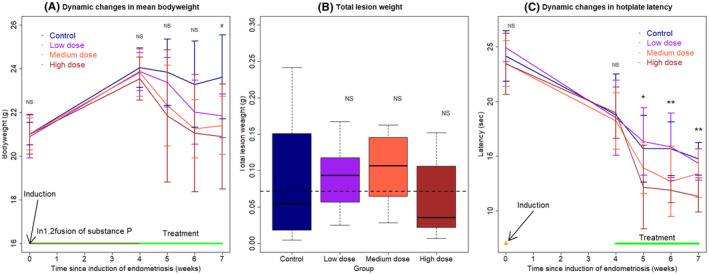
(A) Dynamic changes in the mean bodyweight in 4 different treatment groups of mice. (B) Boxplot of the lesion weight among different groups. The dashed line represents the median value of all mice, and the upper and lower sides represent the 3rd and the 1st quantile, respectively. (C) Dynamic changes in the mean hotplate latency in four treatment different groups of mice. In panels (A) and (C), the data are represented by the means ± SDs, and Kruskal's rank test was used. In addition, the time point at which endometriosis was induced is indicated, and the treatment period also is indicated. In panel (B), Wilcoxon's test was used, and the comparison was made in reference to the control group. Symbols of statistical significance levels: *P < 0.05; **P < 0.01; NS: not statistically significant (P > 0.05)
There was no significant difference in lesion weight among the 4 groups of mice (P = 0.38; Figure 4B). Pairwise comparison in reference to the control group also yielded no significant result (all P's ≥ 0.38; Figure 4B). In fact, the mean lesion weight of the low‐, medium‐, and high‐dose groups was 5.0%, 19.0%, and 29.1% higher than that of the control group. Multiple linear regression using the dosages of EP2 and EP4 inhibitors as covariables did not yield any meaningful results.
As expected, there was no difference in hotplate latency among the 4 groups of mice before the induction of endometriosis and before the start of treatment (both P's ≥ 0.19; Figure 4C). Again, 4 weeks after the induction, there was a significant reduction in latency (P = 8.1 × 10−7; Figure 4C). Starting from 1 week after the treatment, there was a statistically significant difference in hotplate latency among the 4 groups of mice (P = 0.049, P = 0.0069, and P = 0.0059, respectively; Figure 4C). After 3 weeks of treatment, mice treated with high dose, but not low and medium dose (P = 0.65 and P = 0.27, respectively), of EP2 and EP4 inhibitors had significantly shorter latency than that of the control mice (P = 0.0006; Figure 4C).
We further performed immunohistochemistry analysis of COX‐2, EP2, EP4, and α‐SMA (a marker of FMT) in endometriotic lesions and evaluated the extent of lesional fibrosis by Masson trichrome staining (Figure 5). The staining levels of COX‐2, EP2, and EP4 in the epithelial and stromal components were highly correlated (all r's≥0.57, and all P‐values ≤ 0.0006). We found that lesions from mice treated with all doses of EP2 and EP4 inhibitors had significantly reduced stromal COX‐2 staining as compared with that of control mice (all P‐values ≤ 0.012; Figure 5). For the epithelial component, all doses reduced the COX‐2 staining, but only those treated with medium and high doses yielded significant reduction (both P‐values ≤ 0.010). For EP2 and EP4, with the only exception for the low‐dose group in the epithelial component, lesions from all treated mice had significantly reduced staining levels (all P's ≤ 0.038; Figure 5). Multiple linear regression using the EP2 and EP4 inhibitor doses as covariables indicated that EP2 and EP4 inhibitors dose‐dependently reduced stromal EP2 and EP4 staining levels (all P‐values ≤ 0.016; both R 2 ≥ 0.84). However, the joint use of EP2 and EP4 inhibitors did not yield synergistic effect in suppressing EP2 and EP4 expression. In fact, for EP4 staining, the joint use slightly but significantly abrogated the suppression (P = 0.003).
FIGURE 5.
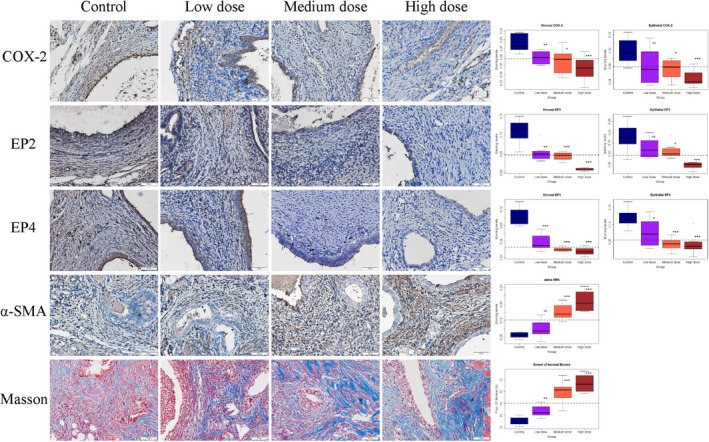
Representative photomicrographs of immunostaining and histochemistry analysis (left panel), along with data summary (right panel). On the left panel, different rows show different markers as indicated. Different columns represent different tissue samples from endometriotic lesions taken from control mice, and mice treated with low‐, medium‐, and high dose of EP2 and EP4 inhibitors (see text for more details), respectively. All mice were induced with deep endometriosis. In Masson trichrome staining, the collagen fibers in lesions were stained in blue. In all figures, magnification: ×400. Scale bar = 50 μm. On the right panel, the results are summarized by the boxplot, separated by the stromal or epithelial component, when applicable. The dashed line represents the median value of all mice. All comparison was made in reference to the control group, and Wilcoxon's test was used. Symbols of statistical significance levels: *P < 0.05; **P < 0.01; ***P < 0.001; NS: not statistically significant (P > 0.05)
The staining levels of ɑ‐SMA in mice treated with medium and high dose, but not low dose (P = 0.13; Figure 5), of EP2/EP4 inhibitor were significantly higher than that of the control mice (both P's = 0.00016; Figure 5). Remarkably, all treatment groups had significantly higher extent of lesional fibrosis than the control mice (all three P's < 0.0011; Figure 5). Multiple linear regression using dosages as covariables indicated that the EP4 inhibitor and the joint use of EP2 and EP4 inhibitors significantly and positively associated with the both ɑ‐SMA staining levels and the extent of lesional fibrosis (all P's < 0.04; R 2 ≧ 0.79).
The hotplate latency at the end of experiment correlated negatively with the extent of lesional fibrosis (r = −0.71, P = 6.3 × 10−7; Figure S3A), but did not correlate with the lesion weight at all (r = −0.10, P = 0.60). The staining levels of COX‐2, EP2, and EP4 in the stromal as well as the epithelial components all correlated negatively with the extent of lesional fibrosis (r = −0.59, P = 0.0004, r = −0.75, P = 5.9 × 10−7, r = −0.80, P = 2.9 × 10−8, r = −0.50, P = 0.0010, r = −0.66, P = 4.6 × 10−5, and r = −0.77, P = 3.4 × 10−7, respectively; Figure S3B–G), but positively (except epithelial COX‐2 (r = 0.26, P = 0.1) with the hotplate latency (r = 0.50, P = 0.0036, r = 0.66, P = 3.6 × 10−5, r = 0.48, P = 0.0057, r = 0.57, P = 0.0007, and r = 0.50, P = 0.003, respectively). Taken together, these results indicate that the treatment with EP2 and EP4 inhibitors in mice did not impact much on the lesion weight, but accelerated the lesional progression and fibrogenesis, exacerbating the hyperalgesia.
3.3. Metformin attenuates the extent of lesional fibrosis concomitant with elevated PGE2 signaling in mice with induced deep endometriosis
We also carried out a mouse experimentation to see whether metformin can reduce the fibrosis of endometriotic lesions and increase the expression of PGE2 pathway. As expected, before the induction of endometriosis and the start of the treatment, there was no significant difference in bodyweight between the two groups of mice (both P's ≧ 0.57; Figure 6A), even though the bodyweight was increased during the 4‐week induction period (P = 1.9 × 10−6).
FIGURE 6.
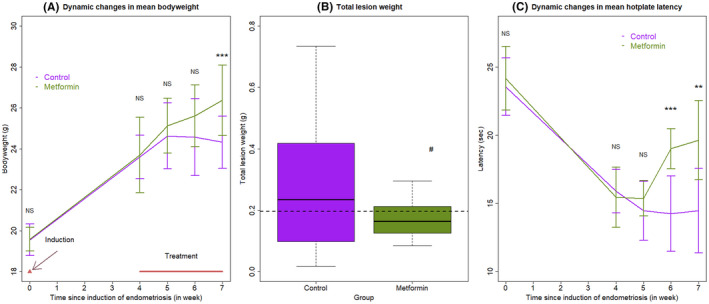
(A) Dynamic changes in the mean bodyweight between the two different treatment groups. (B) Boxplot of the lesion weight between the two groups. The dashed line represents the median value of all mice. (C) Dynamic changes in the mean hotplate latency between the two treatment groups. In panels (A) and (C), the data are represented by the means ± SDs. In addition, the time point at which endometriosis was induced is indicated, and the treatment period also is indicated. In all panels, Wilcoxon's test was used, and the comparison was made in reference to the control group. Symbols of statistical significance levels: *P < 0.05; **P < 0.01; *** P < 0.001; NS: not statistically significant (P > 0.05)
Starting from 1 week after the treatment, the bodyweight between the two groups of mice appeared to diverge, with the metformin‐treated mice gaining weight continuously but the untreated mice losing weight, even though the difference did not reach the statistical significance level 1 and 2 weeks after treatment (both P's ≧ 0.47; Figure 6A). By the end of the treatment, there was a significant difference in bodyweight between the two groups (P = 0.011; Figure 6A).
The mice treated with metformin also had marginally significantly reduced lesion weight (P = 0.084; Figure 6B). There was no statistically significant difference in hotplate latency between the two groups before the induction of endometriosis and the start of the treatment (both P's = 0.42; Figure 6C), even though the latency was significantly reduced before the start of the treatment (P = 9.5 × 10−5; Figure 6C). Consistent with the change in bodyweight, the latency in the two groups of mice began to diverge and the difference became marginally and significantly significant at 1, 2, and 3 weeks after treatment (P = 0.082, P = 0.0010, and P = 0.0028, respectively; Figure 6C), with those treated metformin having increasingly longer latency. In fact, compared with the before‐treatment levels, the metformin‐treated, but not untreated (P = 0.22), mice had significantly longer latency (P = 0.014). There was a positive correlation between the change in latency and bodyweight since the start of the treatment (r = 0.51, P = 0.020), but the correlation coefficient between the hotplate latency and bodyweight at the end of the treatment did not reach statistical significance (r = 0.43, P = 0.060).
We next evaluated immunoreactivity against α‐SMA, COX‐2, EP2, and EP4 and estimated the extent of fibrosis by Masson trichrome staining in endometriotic lesions (Figure 7). After 3 weeks of metformin treatment, we found that the ɑ‐SMA staining levels and the extent of lesional fibrosis in the stromal component were both reduced significantly (both P's < 1.1 × 10−5; Figure 7). In addition, the PGE2 signaling pathway in endometriotic lesions appeared to be activated, as manifested by the elevated immunostaining levels of COX‐2, EP2, and EP4 in both stromal and epithelial components (all P‐values ≤ 0.043; Figure 7).
FIGURE 7.
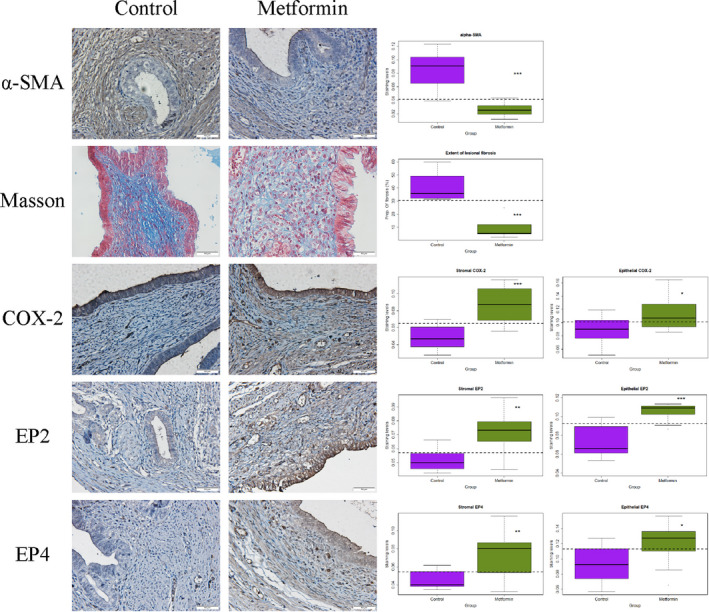
Representative photomicrographs of immunostaining and histochemistry analysis (left panel), along with data summary (right panel). On the left panel, different rows show different markers as indicated. Different columns represent different tissue samples from endometriotic lesions taken from control or metformin‐treated mice, respectively. All mice were induced with deep endometriosis. In Masson trichrome staining, the collagen fibers in lesions were stained in blue. In all figures, magnification: ×400. Scale bar = 50 μm. On the right panel, the results are summarized by the boxplot, separated by the stromal or epithelial component, when applicable. The dashed line represents the median value of all mice. All comparison was made in reference to the control group, and Wilcoxon's test was used. Symbols of statistical significance levels: *P < 0.05; **P < 0.01; ***P < 0.001; NS: not statistically significant (P > 0.05)
The hotplate latency correlated negatively with the extent of lesional fibrosis (r = −0.64, P = 0.0025; Figure S4A), but not with the lesion weight (r = 0.04, P = 0.86). The extent of lesional fibrosis, however, correlated negatively with the staining levels of COX‐2 in both stromal and epithelial components (r = −0.80, P = 1.9 × 10−5, and r = −0.60, P = 0.0054, respectively; Figure S4B,C), EP2 (r = −0.81, P = 1.5 × 10−5, and r = −0.69, P = 0.0007, respectively; Figure S4D,E) and EP4 (r = −0.74, P = 0.0002, and r = −0.63, P = 0.0027, respectively; Figure S4F,G).
4. DISCUSSION
In this study, we have shown that the PGE2 signaling, manifested as the immunostaining activity against COX‐2, EP2, and EP4, turned from a state of overdrive to a stall as endometriotic lesions progress, especially in mice with induced DE in which fibrotic content is higher. In addition, treatment with EP2/EP4 inhibitors in mice with induced DE had no effect on lesion weight, but exacerbated endometriosis‐associated hyperalgesia and promoted myofibroblast activation and fibrogenesis in lesions even though they appeared to suppress the PGE2 signaling in a dose‐dependent manner. Moreover, treatment with metformin in mice with induced DE resulted in elevated PGE2 signaling, concomitant with somewhat reduced lesion weight, improved pain behavior, and attenuated myofibroblast activation and fibrogenesis in lesions.
Our results are consistent with the finding that the PGE2 signaling is attenuated progressively in human endometriosis as lesions become more fibrotic. 26 In particular, our results are consistent with the finding that OE lesions in adolescents had higher PGE2 signaling than those in adults. 26 They are also consistent with the reports that the percentage of COX‐2 staining positivity is significantly lower in DE lesions as compared with that in OE lesions. 52 , 53
Endometriotic lesions are fundamentally wounds undergoing repeated tissue injury and repair (ReTIAR) due to cyclic bleeding. 33 , 54 , 55 Increased lesional fibrosis begets increased lesional stiffness, 56 which, in turn, facilitates the actions of TGF‐β1 and promotes myofibroblast activation. 57 , 58 , 59 In fact, stiff extracellular matrix (ECM) alone can facilitate, propagate, and even start fibrosis. 60 Elevated ECM stiffness alone can activate a profibrotic positive feedback loop. 28 , 61 , 62 In contrast, soft ECM has been shown to reverse myofibroblast activation. 29 In this sense, our results, in retrospect, are not entirely surprising.
Metformin is a commonly prescribed anti‐diabetic medication that improves insulin sensitivity and has an excellent safety profile. 63 It is also used to treat polycystic ovarian syndrome (PCOS) 64 and exhibits pleiotropic effects on cellular biology such as anti‐inflammation 65 and anti‐fibrosis. 34 , 35 In endometriosis, metformin has been shown to suppress PGE2‐induced induction of steroidogenic acute regulatory protein (StAR) 66 and aromatase 67 through induction of AMP‐activated protein kinase (AMPK) in endometriotic stromal cells, the two pivotal proteins involved in estrogen biosynthesis. Metformin can suppress interleukin (IL)‐1β‐induced IL‐8 production, aromatase activation, and proliferation of endometriotic stromal cells 68 and disrupt the stroma‐epithelium communication via Wnt2‐mediated signaling. 69 Two preclinical studies have also shown that metformin is promising in treating endometriosis in rats. 70 , 71 One clinical study reported very promising effect of metformin treatment in women with endometriosis. 72 Thus, metformin is considered to have a unique therapeutic potential. 73
Our data demonstrate that treatment with metformin in mice with induced DE resulted in increased PGE2 signaling in lesions concomitant with reduced fibrosis. This may be consistent with the reported anti‐fibrotic role of PGE2, 74 , 75 , 76 , 77 , 78 mediated mostly by EP2 and EP4. 74
However, given its multitude of mode of actions, 79 it is unlikely that metformin suppresses endometriosis through the PGE2 signaling pathway exclusively and alone. For example, since metformin is known to induce AMPK, 80 which is known to be a key regulatory enzyme in cellular energy homeostasis. 81 Through induction of AMPK, metformin improves lipid metabolism and reverses the Warburg effect/glycolysis, 82 suppresses collagen formation, activates the PPARγ signaling, 35 and/or facilitates deactivation and apoptosis of myofibroblasts. 34 Endometriotic lesions are known to experience Warburg effect manifesting as increased glycolysis 83 , 84 , 85 and, as such, PDK1 inhibitors such as dichloroacetate have been proposed as a therapeutics for endometriosis. 86 Along the same line, metformin, which is known to have an excellent safety profile, can also serve as a possible treatment for the purpose of reversing glycolysis. Regardless, future studies are warranted to further elucidate the underlying mechanism of action.
Our results contradict directly with the report that EP2/EP4 inhibition resulted in significant reduction in lesion growth in a dose‐dependent manner. 24 There are several reasons for this discrepancy. First, we used two inhibitors that are known to have a high specificity 40 and excellent oral bioavailability. 41 , 42 In contrast, both AH6809 (an EP2 inhibitor) and AH23848 (an EP4 inhibitor) as used in 24 are of mediocre specificity. 40 In fact, AH6809 is an inhibitor for both EP1 and EP2, and it has the highest affinity with DP (fact sheet by Sigma; Catalog #: A1221).
Second, there is an issue of which mouse model most likely resembles the human condition. The xenograft of mixed populations of immortalized endometriotic epithelial and stromal cells, both derived from humans, in ovariectomized and estrogen‐treated nude mice was used in, 24 and the mouse model induced with artificially induced menses was used in. 25 In contrast, we used a mouse DE model. The nude mouse is known to be immune compromised, while the mice used in this study are, in contrast, immune‐competent. The lesions established in the menses model in 25 have the tendency of disappearing 5–6 weeks of induction (Dr. Erin Greaves, personal communication). Given the role in immune system in the development of endometriosis, 87 , 88 and given the observation that endometriotic lesions tend to progress if undisturbed, 89 it can be argued that the mouse model we used is most likely to recapitulate its human condition.
Lastly, the two reports showing the beneficial effect of EP2/EP4 inhibition in mouse models started the treatment just 2 24 or 3 25 weeks after induction, not long enough for lesions to become fibrotic. 31 However, fibrosis is a prominent feature of endometriosis, 5 , 90 especially given the well documented diagnostic delay. 4 So much so that a redefinition of endometriosis to incorporate this feature has been proposed recently. 32 , 33 In contrast, lesions in our mouse DE model displayed extensive fibrosis and adhesion, 38 closely recapitulating its human counterpart. With increased fibrotic content and ensuing elevated lesional stiffness, the mechanical microenvironment within and surrounding lesions would change accordingly, attenuating PGE2 signaling and EP2/EP4 expression. 27 , 28 This difference alone would account for this discrepancy.
Once we understand these subtle yet biologically plausible and important differences between early and more advanced lesions, we can envision several important implications. First, endometriotic lesions are not monolithic, static, and immutable. Rather, they are dynamic and progressive, with vast differences between early and more advanced lesions—the PGE2 signaling levels being one of these differences. Second, with this in mind, we can now understand as why there are often conflicting reports in endometriosis literature. For example, the critical role of PGE2 in inducing aromatase expression is widely accepted, 6 but most, if not all, of the data in support for this notion were based on OE samples. 67 If a sizeable portion of lesion samples came from more fibrotic lesions such as DE lesions, the evidence for aromatase positivity could easily vanish. 91 , 92 , 93 , 94 Thus, we need to pay more attention to the source of endometriotic tissue samples and also to the quantification of lesional fibrosis when conducting investigation and reporting. Lastly, care needs to be exercised when treating endometriosis, since “older” and more fibrotic lesions are unlikely to respond well to NSAIDs but “early” lesions likely would.
Our studies have several strengths. First, we used the two inhibitors with documented high specificity and excellent bioavailability. This should ensure that the inhibition is precisely on target, and our results are not an off‐target, and thus spurious, finding. Second, by capitalizing on our findings of differences in PGE2 signaling between OE and DE lesions and between adolescent and adult patients with OE, we complemented that finding with our first experiment, providing experimental evidence that indeed there is a change from overexpression to suppression in PGE2 signaling as endometriotic lesions progress. Third, based on the results of the first experiment, we tested the efficacy of EP2/EP4 inhibitor treatment in a mouse DE model. The two experiments are thus complementary and consistent. Our results may thus help researchers to guard against unrealistic expectation and minimize the risk of failure in further clinical studies. Lastly, by evaluating the PGE2 signaling after metformin treatment in mice with induced DE, our study provides a biologically plausible clue as how endometriosis could be managed through induction of the signaling pathway.
Our study also has some limitations. First, this study is limited by the use of histologic and IHC analyses only and the lack of molecular data. Second, we did not measure lesional PGE2 concentrations in all three experiments, hence we do not know exactly whether it is the reduced PGE2 production, reduced EP2/EP4 expression or both that are responsible for the exacerbation of endometriosis in mouse with induced DE receiving treatment. Nor do we know whether it is the induction of COX‐2, or EP2/EP4, or both that are responsible for the treatment effect of metformin. Third, this study only evaluated the immunostaining of COX‐2, EP2, and EP4 that are related with the PGE2 production and possible action (through receptors EP2 and EP4). While the supposedly downregulation of EP2/EP4 is likely to impact on their downstream genes/proteins, we actually did not evaluate any genes/proteins downstream of EP2/EP4, such as ERK, cAMP/PKA, PI3K/AKT, and NF‐κB, 95 , 96 , 97 hence caution should be exercised when the word, PGE2 signaling, is used. Future studies are needed to illuminate this issue. Fourth, we used i.p. injection for metformin administration in this study due to the ease to control the dosage of metformin delivered to the mice. While different routes of delivery seem to be able to achieve similar blood concentrations, 98 , 99 it is possible that i.p. injection may have the added advantage of exposing endometriotic lesions more easily to the drug, thus achieving greater efficacy. Whether or not this is true would await further investigation. Fifth, painful states in laboratory animals is reliably associated with decreases in feeding and bodyweight. 100 , 101 While there were signs of retarded growth due to endometriosis‐induced pain‐suppressed intake in all three experiments (especially in Experiment 3 where mice taking metformin had increased bodyweight as compared with those untreated, given that metformin use leads to a reduction in food intake and bodyweight 102 ), these results are merely correlative but certainly not conclusive. Lastly, while we have shown reduced staining (and thus presumably expression) of COX‐2, the protein responsible for the rate‐limiting enzyme in PGE2 production, and of EP2 and EP4, we did not demonstrate the reduction of molecules downstream of the PGE2 signaling. Future studies are warranted to illuminate these issues.
To conclude, the PGE2 signaling in endometriotic lesions goes from a state of overdrive to a stall as lesions become more fibrotic. For mouse with induced DE, treatment with EP2/EP4 inhibitors results in worsening hyperalgesia and increased fibrosis. On the other hand, treatment with metformin in mice with induced DE resulted in increased PGE2 signaling, concomitant with somewhat reduced lesion weight, improved hyperalgesia, and attenuated myofibroblast activation and fibrogenesis in lesions. Our findings highlight the dynamic nature of endometriotic lesions, which has important implications for research, drug development, and clinical management of endometriosis.
CONFLICT OF INTEREST
Qingqing Huang, Xishi Liu, and Sun‐Wei Guo declare that they have no conflict of interest.
ETHICS APPROVAL
The protocol for this study has been approved by Institutional Ethics Board, Shanghai Obstetrics and Gynecology, Fudan University.
ANIMAL STUDIES
All institutional and national guidelines for the care and use of laboratory animals were followed.
Supporting information
Fig S1‐S4
ACKNOWLEDGEMENTS
This research was funded in part by grants 81771553 (SWG), 82071623 (SWG), and 81871144 (XSL) from the National Natural Science Foundation of China, an Excellence in Centers of Clinical Medicine grant (2017ZZ01016) from the Science and Technology Commission of Shanghai Municipality, and Clinical Research Plan grant SHDC2020CR2062B from Shanghai Shenkang Center for Hospital Development.
Huang Q, Liu X, Guo S‐W. Changing prostaglandin E2 (PGE2) signaling during lesional progression and exacerbation of endometriosis by inhibition of PGE2 receptor EP2 and EP4. Reprod Med Biol. 2021;21:e12426. doi: 10.1002/rmb2.12426
REFERENCES
- 1. Giudice LC, Kao LC. Endometriosis. Lancet. 2004;364(9447):1789‐1799. [DOI] [PubMed] [Google Scholar]
- 2. Vercellini P, Crosignani P, Somigliana E, Vigano P, Frattaruolo MP, Fedele L. ‘Waiting for Godot’: a commonsense approach to the medical treatment of endometriosis. Hum Reprod. 2011;26(1):3‐13. [DOI] [PubMed] [Google Scholar]
- 3. Guo SW, Groothuis PG. Is it time for a paradigm shift in drug research and development in endometriosis/adenomyosis? Hum Reprod Update. 2018;24(5):577‐598. [DOI] [PubMed] [Google Scholar]
- 4. Hadfield R, Mardon H, Barlow D, Kennedy S. Delay in the diagnosis of endometriosis: a survey of women from the USA and the UK. Hum Reprod. 1996;11(4):878‐880. [DOI] [PubMed] [Google Scholar]
- 5. Liu X, Zhang Q, Guo SW. Histological and immunohistochemical characterization of the similarity and difference between ovarian endometriomas and deep infiltrating endometriosis. Reprod Sci. 2018;25(3):329‐340. [DOI] [PubMed] [Google Scholar]
- 6. Bulun SE, Lin Z, Imir G, et al. Regulation of aromatase expression in estrogen‐responsive breast and uterine disease: from bench to treatment. Pharmacol Rev. 2005;57(3):359‐383. [DOI] [PubMed] [Google Scholar]
- 7. Ota H, Igarashi S, Sasaki M, Tanaka T. Distribution of cyclooxygenase‐2 in eutopic and ectopic endometrium in endometriosis and adenomyosis. Hum Reprod. 2001;16(3):561‐566. [DOI] [PubMed] [Google Scholar]
- 8. Chishima F, Hayakawa S, Sugita K, et al. Increased expression of cyclooxygenase‐2 in local lesions of endometriosis patients. Am J Reprod Immunol. 2002;48(1):50‐56. [DOI] [PubMed] [Google Scholar]
- 9. Matsuzaki S, Canis M, Pouly JL, Wattiez A, Okamura K, Mage G. Cyclooxygenase‐2 expression in deep endometriosis and matched eutopic endometrium. Fertil Steril. 2004;82(5):1309‐1315. [DOI] [PubMed] [Google Scholar]
- 10. Buchweitz O, Staebler A, Wulfing P, Hauzman E, Greb R, Kiesel L. COX‐2 overexpression in peritoneal lesions is correlated with nonmenstrual chronic pelvic pain. Eur J Obstet Gynecol Reprod Biol. 2006;124(2):216‐221. [DOI] [PubMed] [Google Scholar]
- 11. Carli C, Metz CN, Al‐Abed Y, Naccache PH, Akoum A. Up‐regulation of cyclooxygenase‐2 expression and prostaglandin E2 production in human endometriotic cells by macrophage migration inhibitory factor: involvement of novel kinase signaling pathways. Endocrinology. 2009;150(7):3128‐3137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rakhila H, Carli C, Daris M, Lemyre M, Leboeuf M, Akoum A. Identification of multiple and distinct defects in prostaglandin biosynthetic pathways in eutopic and ectopic endometrium of women with endometriosis. Fertil Steril. 2013;100(6):pp. 1650‐1659.e1–e2. [DOI] [PubMed] [Google Scholar]
- 13. Santulli P, Borghese B, Noel JC, et al. Hormonal therapy deregulates prostaglandin‐endoperoxidase synthase 2 (PTGS2) expression in endometriotic tissues. J Clin Endocrinol Metab. 2014;99(3):881‐890. [DOI] [PubMed] [Google Scholar]
- 14. Efstathiou JA, Sampson DA, Levine Z, et al. Nonsteroidal antiinflammatory drugs differentially suppress endometriosis in a murine model. Fertil Steril. 2005;83(1):171‐181. [DOI] [PubMed] [Google Scholar]
- 15. Ozawa Y, Murakami T, Tamura M, Terada Y, Yaegashi N, Okamura K. A selective cyclooxygenase‐2 inhibitor suppresses the growth of endometriosis xenografts via antiangiogenic activity in severe combined immunodeficiency mice. Fertil Steril. 2006;86(4 Suppl):1146‐1151. [DOI] [PubMed] [Google Scholar]
- 16. Machado DE, Berardo PT, Landgraf RG, et al. A selective cyclooxygenase‐2 inhibitor suppresses the growth of endometriosis with an antiangiogenic effect in a rat model. Fertil Steril. 2010;93(8):2674‐2679. [DOI] [PubMed] [Google Scholar]
- 17. Cobellis L, Razzi S, De Simone S, et al. The treatment with a COX‐2 specific inhibitor is effective in the management of pain related to endometriosis. Eur J Obstet Gynecol Reprod Biol. 2004;116(1):100‐102. [DOI] [PubMed] [Google Scholar]
- 18. Norman RJ, Wu R. The potential danger of COX‐2 inhibitors. Fertil Steril. 2004;81(3):493‐494. [DOI] [PubMed] [Google Scholar]
- 19. Narumiya S, Sugimoto Y, Ushikubi F. Prostanoid receptors: structures, properties, and functions. Physiol Rev. 1999;79(4):1193‐1226. [DOI] [PubMed] [Google Scholar]
- 20. Rakhila H, Bourcier N, Akoum A, Pouliot M. Abnormal expression of prostaglandins E2 and F2alpha receptors and transporters in patients with endometriosis. Biomed Res Int. 2015;2015:808146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Banu SK, Lee J, Speights VO Jr, Starzinski‐Powitz A, Arosh JA. Selective inhibition of prostaglandin E2 receptors EP2 and EP4 induces apoptosis of human endometriotic cells through suppression of ERK1/2, AKT, NFkappaB, and beta‐catenin pathways and activation of intrinsic apoptotic mechanisms. Mol Endocrinol. 2009;23(8):1291‐1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lee J, Banu SK, Subbarao T, Starzinski‐Powitz A, Arosh JA. Selective inhibition of prostaglandin E2 receptors EP2 and EP4 inhibits invasion of human immortalized endometriotic epithelial and stromal cells through suppression of metalloproteinases. Mol Cell Endocrinol. 2011;332(1–2):306‐313. [DOI] [PubMed] [Google Scholar]
- 23. Makabe T, Koga K, Nagabukuro H, et al. Use of selective PGE2 receptor antagonists on human endometriotic stromal cells and peritoneal macrophages. Mol Hum Reprod. 2021;27(1):gaaa077. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Arosh JA, Lee J, Balasubbramanian D, et al. Molecular and preclinical basis to inhibit PGE2 receptors EP2 and EP4 as a novel nonsteroidal therapy for endometriosis. Proc Natl Acad Sci USA. 2015;112(31):9716‐9721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Greaves E, Horne AW, Jerina H, et al. EP2 receptor antagonism reduces peripheral and central hyperalgesia in a preclinical mouse model of endometriosis. Sci Rep. 2017;7:44169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Huang Q, Liu X, Guo SW. Higher fibrotic content of endometriotic lesions is associated with diminished prostaglandin E2 (PGE2) signaling. Reprod Med Biol. 2021;In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Berhan A, Harris T, Jaffar J, et al. Cellular microenvironment stiffness regulates eicosanoid production and signaling pathways. Am J Respir Cell Mol Biol. 2020;63(6):819‐830. [DOI] [PubMed] [Google Scholar]
- 28. Liu F, Mih JD, Shea BS, et al. Feedback amplification of fibrosis through matrix stiffening and COX‐2 suppression. J Cell Biol. 2010;190(4):693‐706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Marinkovic A, Liu F, Tschumperlin DJ. Matrices of physiologic stiffness potently inactivate idiopathic pulmonary fibrosis fibroblasts. Am J Respir Cell Mol Biol. 2013;48(4):422‐430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Arosh JA, Lee J, Starzinski‐Powitz A, Banu SK. Selective inhibition of prostaglandin E2 receptors EP2 and EP4 modulates DNA methylation and histone modification machinery proteins in human endometriotic cells. Mol Cell Endocrinol. 2015;409:51‐58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhang Q, Liu X, Guo SW. Progressive development of endometriosis and its hindrance by anti‐platelet treatment in mice with induced endometriosis. Reprod Biomed Online. 2017;34(2):124‐136. [DOI] [PubMed] [Google Scholar]
- 32. Vigano P, Candiani M, Monno A, Giacomini E, Vercellini P, Somigliana E. Time to redefine endometriosis including its pro‐fibrotic nature. Hum Reprod. 2018;33(3):347‐352. [DOI] [PubMed] [Google Scholar]
- 33. Guo SW. Fibrogenesis resulting from cyclic bleeding: the Holy Grail of the natural history of ectopic endometrium. Hum Reprod. 2018;33(3):353‐356. [DOI] [PubMed] [Google Scholar]
- 34. Rangarajan S, Bone NB, Zmijewska AA, et al. Metformin reverses established lung fibrosis in a bleomycin model. Nat Med. 2018;24(8):1121‐1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kheirollahi V, Wasnick RM, Biasin V, et al. Metformin induces lipogenic differentiation in myofibroblasts to reverse lung fibrosis. Nat Commun. 2019;10(1):2987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. National Research Council Institute for Laboratory Animal R . Guide for the Care and Use of Laboratory Animals. Academies Press (US). Copyright 2011 by the National Academy of Sciences. All rights reserved; 2011. [Google Scholar]
- 37. Somigliana E, Vigano P, Filardo P, Candiani M, Vignali M, Panina‐Bordignon P. Use of knockout transgenic mice in the study of endometriosis: insights from mice lacking beta(2)‐microglobulin and interleukin‐12p40. Fertil Steril. 2001;75(1):203‐206. [DOI] [PubMed] [Google Scholar]
- 38. Yan D, Liu X, Guo SW. The establishment of a mouse model of deep endometriosis. Hum Reprod. 2019;34(2):235‐247. [DOI] [PubMed] [Google Scholar]
- 39. Yan D, Liu X, Guo S‐W. The establishment of a mouse model of deep endometriosis. Hum Reprod (Oxford, England). 2019;34(2):235‐247. [DOI] [PubMed] [Google Scholar]
- 40. Markovic T, Jakopin Z, Dolenc MS, Mlinaric‐Rascan I. Structural features of subtype‐selective EP receptor modulators. Drug Discov Today. 2017;22(1):57‐71. [DOI] [PubMed] [Google Scholar]
- 41. af Forselles KJ, Root J, Clarke T, et al. In vitro and in vivo characterization of PF‐04418948, a novel, potent and selective prostaglandin EP(2) receptor antagonist. Br J Pharmacol. 2011;164(7):1847‐1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Thieme K, Majumder S, Brijmohan AS, et al. EP4 inhibition attenuates the development of diabetic and non‐diabetic experimental kidney disease. Sci Rep. 2017;7(1):3442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yang L, Huang Y, Porta R, et al. Host and direct antitumor effects and profound reduction in tumor metastasis with selective EP4 receptor antagonism. Cancer Res. 2006;66(19):9665‐9672. [DOI] [PubMed] [Google Scholar]
- 44. Aoki T, Frosen J, Fukuda M, et al. Prostaglandin E2‐EP2‐NF‐kappaB signaling in macrophages as a potential therapeutic target for intracranial aneurysms. Sci Signal. 2017;10(465):eaah6037. [DOI] [PubMed] [Google Scholar]
- 45. Al‐Hashem F, Al‐Humayed S, Amin SN, et al. Metformin inhibits mTOR‐HIF‐1alpha axis and profibrogenic and inflammatory biomarkers in thioacetamide‐induced hepatic tissue alterations. J Cell Physiol. 2019;234(6):9328‐9337. [DOI] [PubMed] [Google Scholar]
- 46. Tang X, Li J, Xiang W, et al. Metformin increases hepatic leptin receptor and decreases steatosis in mice. J Endocrinol. 2016;230(2):227‐237. [DOI] [PubMed] [Google Scholar]
- 47. Niu C, Chen Z, Kim KT, et al. Metformin alleviates hyperglycemia‐induced endothelial impairment by downregulating autophagy via the Hedgehog pathway. Autophagy. 2019;15(5):843‐870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ding D, Liu X, Duan J, Guo SW. Platelets are an unindicted culprit in the development of endometriosis: clinical and experimental evidence. Hum Reprod. 2015;30(4):812‐832. [DOI] [PubMed] [Google Scholar]
- 49. Team RDC . R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing; 2016. [Google Scholar]
- 50. Lee J, Banu SK, Burghardt RC, Starzinski‐Powitz A, Arosh JA. Selective inhibition of prostaglandin E2 receptors EP2 and EP4 inhibits adhesion of human endometriotic epithelial and stromal cells through suppression of integrin‐mediated mechanisms. Biol Reprod. 2013;88(3):77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lee J, Banu SK, Rodriguez R, Starzinski‐Powitz A, Arosh JA. Selective blockade of prostaglandin E2 receptors EP2 and EP4 signaling inhibits proliferation of human endometriotic epithelial cells and stromal cells through distinct cell cycle arrest. Fertil Steril. 2010;93(8):2498‐2506. [DOI] [PubMed] [Google Scholar]
- 52. Fagotti A, Ferrandina G, Fanfani F, et al. Analysis of cyclooxygenase‐2 (COX‐2) expression in different sites of endometriosis and correlation with clinico‐pathological parameters. Hum Reprod. 2004;19(2):393‐397. [DOI] [PubMed] [Google Scholar]
- 53. Fanfani F, Fagotti A, Ferrandina G, et al. Increased cyclooxygenase‐2 expression is associated with better clinical outcome in patients submitted to complete ablation for severe endometriosis. Hum Reprod. 2005;20(10):2964‐2968. [DOI] [PubMed] [Google Scholar]
- 54. Zhang Q, Duan J, Liu X, Guo SW. Platelets drive smooth muscle metaplasia and fibrogenesis in endometriosis through epithelial‐mesenchymal transition and fibroblast‐to‐myofibroblast transdifferentiation. Mol Cell Endocrinol. 2016;428:1‐16. [DOI] [PubMed] [Google Scholar]
- 55. Zhang Q, Duan J, Olson M, Fazleabas A, Guo SW. Cellular changes consistent with epithelial‐mesenchymal transition and fibroblast‐to‐myofibroblast transdifferentiation in the progression of experimental endometriosis in baboons. Reprod Sci. 2016;23(10):1409‐1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ding D, Chen Y, Liu X, Jiang Z, Cai X, Guo SW. Diagnosing deep endometriosis using transvaginal elastosonography. Reprod Sci. 2020;27(7):1411‐1422. [DOI] [PubMed] [Google Scholar]
- 57. Huang X, Yang N, Fiore VF, et al. Matrix stiffness‐induced myofibroblast differentiation is mediated by intrinsic mechanotransduction. Am J Respir Cell Mol Biol. 2012;47(3):340‐348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Marinkovic A, Mih JD, Park JA, Liu F, Tschumperlin DJ. Improved throughput traction microscopy reveals pivotal role for matrix stiffness in fibroblast contractility and TGF‐beta responsiveness. Am J Physiol Lung Cell Mol Physiol. 2012;303(3):L169‐L180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Gimenez A, Duch P, Puig M, Gabasa M, Xaubet A, Alcaraz J. Dysregulated collagen homeostasis by matrix stiffening and TGF‐beta1 in fibroblasts from idiopathic pulmonary fibrosis patients: role of FAK/Akt. Int J Mol Sci. 2017;18(11):2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Herrera J, Henke CA, Bitterman PB. Extracellular matrix as a driver of progressive fibrosis. J Clin Invest. 2018;128(1):45‐53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Parker MW, Rossi D, Peterson M, et al. Fibrotic extracellular matrix activates a profibrotic positive feedback loop. J Clin Invest. 2014;124(4):1622‐1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Shimbori C, Gauldie J, Kolb M. Extracellular matrix microenvironment contributes actively to pulmonary fibrosis. Curr Opin Pulm Med. 2013;19(5):446‐452. [DOI] [PubMed] [Google Scholar]
- 63. Bailey CJ. Metformin: historical overview. Diabetologia. 2017;60(9):1566‐1576. [DOI] [PubMed] [Google Scholar]
- 64. Lord J, Wilkin T. Metformin in polycystic ovary syndrome. Curr Opin Obstet Gynecol. 2004;16(6):481‐486. [DOI] [PubMed] [Google Scholar]
- 65. Kita Y, Takamura T, Misu H, et al. Metformin prevents and reverses inflammation in a non‐diabetic mouse model of nonalcoholic steatohepatitis. PLoS One. 2012;7(9):e43056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Xu JN, Zeng C, Zhou Y, Peng C, Zhou YF, Xue Q. Metformin inhibits StAR expression in human endometriotic stromal cells via AMPK‐mediated disruption of CREB‐CRTC2 complex formation. J Clin Endocrinol Metab. 2014;99(8):2795‐2803. [DOI] [PubMed] [Google Scholar]
- 67. Zhou Y, Xu JN, Zeng C, et al. Metformin suppresses prostaglandin E2‐induced cytochrome P450 aromatase gene expression and activity via stimulation of AMP‐activated protein kinase in human endometriotic stromal cells. Reprod Sci. 2015;22(9):1162‐1170. [DOI] [PubMed] [Google Scholar]
- 68. Takemura Y, Osuga Y, Yoshino O, et al. Metformin suppresses interleukin (IL)‐1beta‐induced IL‐8 production, aromatase activation, and proliferation of endometriotic stromal cells. J Clin Endocrinol Metab. 2007;92(8):3213‐3218. [DOI] [PubMed] [Google Scholar]
- 69. Zhang H, Xue J, Li M, Zhao X, Wei D, Li C. Metformin regulates stromal‐epithelial cells communication via Wnt2/beta‐catenin signaling in endometriosis. Mol Cell Endocrinol. 2015;413:61‐65. [DOI] [PubMed] [Google Scholar]
- 70. Oner G, Ozcelik B, Ozgun MT, Serin IS, Ozturk F, Basbug M. The effects of metformin and letrozole on endometriosis and comparison of the two treatment agents in a rat model. Hum Reprod. 2010;25(4):932‐937. [DOI] [PubMed] [Google Scholar]
- 71. Yilmaz B, Sucak A, Kilic S, et al. Metformin regresses endometriotic implants in rats by improving implant levels of superoxide dismutase, vascular endothelial growth factor, tissue inhibitor of metalloproteinase‐2, and matrix metalloproteinase‐9. Am J Obstet Gynecol. 2010;202(4):368.e1‐e8. [DOI] [PubMed] [Google Scholar]
- 72. Foda AA, Abdel Aal IA. Metformin as a new therapy for endometriosis, its effects on both clinical picture and cytokines profile. Middle East Fertil Soc J. 2012;17:262‐267. [Google Scholar]
- 73. Stochino‐Loi E, Major AL, Gillon TER, Ayoubi JM, Feki A, Bouquet de Joliniere J. Metformin, the rise of a new medical therapy for endometriosis? A systematic review of the literature. Front Med (Lausanne). 2021;8:581311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Huang S, Wettlaufer SH, Hogaboam C, Aronoff DM, Peters‐Golden M. Prostaglandin E(2) inhibits collagen expression and proliferation in patient‐derived normal lung fibroblasts via E prostanoid 2 receptor and cAMP signaling. Am J Physiol Lung Cell Mol Physiol. 2007;292(2):L405‐L413. [DOI] [PubMed] [Google Scholar]
- 75. Huang SK, White ES, Wettlaufer SH, et al. Prostaglandin E(2) induces fibroblast apoptosis by modulating multiple survival pathways. FASEB J. 2009;23(12):4317‐4326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Penke LR, Huang SK, White ES, Peters‐Golden M. Prostaglandin E2 inhibits alpha‐smooth muscle actin transcription during myofibroblast differentiation via distinct mechanisms of modulation of serum response factor and myocardin‐related transcription factor‐A. J Biol Chem. 2014;289(24):17151‐17162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Wettlaufer SH, Scott JP, McEachin RC, Peters‐Golden M, Huang SK. Reversal of the transcriptome by prostaglandin e2 during myofibroblast dedifferentiation. Am J Respir Cell Mol Biol. 2016;54(1):114‐127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Mukherjee S, Sheng W, Michkov A, et al. Prostaglandin E2 inhibits profibrotic function of human pulmonary fibroblasts by disrupting Ca(2+) signaling. Am J Physiol Lung Cell Mol Physiol. 2019;316(5):L810‐L821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Kaneto H, Kimura T, Obata A, Shimoda M, Kaku K. Multifaceted mechanisms of action of metformin which have been unraveled one after another in the long history. Int J Mol Sci. 2021;22(5):2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Zhou G, Myers R, Li Y, et al. Role of AMP‐activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108(8):1167‐1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Kahn BB, Alquier T, Carling D, Hardie DG. AMP‐activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab. 2005;1(1):15‐25. [DOI] [PubMed] [Google Scholar]
- 82. Liu Y, Bai F, Liu N, et al. Metformin improves lipid metabolism and reverses the Warburg effect in a canine model of chronic atrial fibrillation. BMC Cardiovasc Disord. 2020;20(1):50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Young VJ, Brown JK, Maybin J, Saunders PT, Duncan WC, Horne AW. Transforming growth factor‐beta induced Warburg‐like metabolic reprogramming may underpin the development of peritoneal endometriosis. J Clin Endocrinol Metab. 2014;99(9):3450‐3459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Young VJ, Ahmad SF, Brown JK, Duncan WC, Horne AW. ID2 mediates the transforming growth factor‐beta1‐induced Warburg‐like effect seen in the peritoneum of women with endometriosis. Mol Hum Reprod. 2016;22(9):648‐654. [DOI] [PubMed] [Google Scholar]
- 85. Lee HC, Lin SC, Wu MH, Tsai SJ. Induction of pyruvate dehydrogenase kinase 1 by hypoxia alters cellular metabolism and inhibits apoptosis in endometriotic stromal cells. Reprod Sci. 2019;26(6):734‐744. [DOI] [PubMed] [Google Scholar]
- 86. Horne AW, Ahmad SF, Carter R, et al. Repurposing dichloroacetate for the treatment of women with endometriosis. Proc Natl Acad Sci USA. 2019;116(51):25389‐25391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Duan J, Liu X, Wang H, Guo SW. The M2a macrophage subset may be critically involved in the fibrogenesis of endometriosis in mice. Reprod Biomed Online. 2018;37(3):254‐268. [DOI] [PubMed] [Google Scholar]
- 88. Xiao F, Liu X, Guo SW. Platelets and regulatory T cells may induce a type 2 immunity that is conducive to the progression and fibrogenesis of endometriosis. Front Immunol. 2020;11:610963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Ding D, Wang X, Chen Y, Benagiano G, Liu X, Guo S‐W. Evidence in support for the progressive nature of ovarian endometriomas. J Clin Endocrinol Metab. 2020;105(7):2189‐2202. [DOI] [PubMed] [Google Scholar]
- 90. Cornillie FJ, Oosterlynck D, Lauweryns JM, Koninckx PR. Deeply infiltrating pelvic endometriosis: histology and clinical significance. Fertil Steril. 1990;53(6):978‐983. [DOI] [PubMed] [Google Scholar]
- 91. Delvoux B, Groothuis P, D'Hooghe T, Kyama C, Dunselman G, Romano A. Increased production of 17beta‐estradiol in endometriosis lesions is the result of impaired metabolism. J Clin Endocrinol Metab. 2009;94(3):876‐883. [DOI] [PubMed] [Google Scholar]
- 92. Colette S, Lousse JC, Defrere S, et al. Absence of aromatase protein and mRNA expression in endometriosis. Hum Reprod. 2009;24(9):2133‐2141. [DOI] [PubMed] [Google Scholar]
- 93. Goncalves HF, Zendron C, Cavalcante FS, et al. Leptin, its receptor and aromatase expression in deep infiltrating endometriosis. J Ovarian Res. 2015;8:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Szaflik T, Smolarz B, Mroczkowska B, et al. An analysis of ESR2 and CYP19A1 gene expression levels in women with endometriosis. In Vivo. 2020;34(4):1765‐1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Xu S, Zhou W, Ge J, Zhang Z. Prostaglandin E2 receptor EP4 is involved in the cell growth and invasion of prostate cancer via the cAMPPKA/PI3KAkt signaling pathway. Mol Med Rep. 2018;17(3):4702‐4712. [DOI] [PubMed] [Google Scholar]
- 96. Ching MM, Reader J, Fulton AM. Eicosanoids in cancer: prostaglandin E2 receptor 4 in cancer therapeutics and immunotherapy. Front Pharmacol. 2020;11:819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Liu J, Zhang YD, Chen XL, et al. The protective effect of the EP2 receptor on TGF‐beta1 induced podocyte injury via the PI3K/Akt signaling pathway. PLoS One. 2018;13(5):e0197158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Thompson MD, Grubbs CJ, Bode AM, et al. Lack of effect of metformin on mammary carcinogenesis in nondiabetic rat and mouse models. Cancer Prev Res (Phila). 2015;8(3):231‐239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Dowling RJ, Lam S, Bassi C, et al. Metformin pharmacokinetics in mouse tumors: implications for human therapy. Cell Metab. 2016;23(4):567‐568. [DOI] [PubMed] [Google Scholar]
- 100. Flecknell PA, Roughan JV, Stewart R. Use of oral buprenorphine (‘buprenorphine jello’) for postoperative analgesia in rats–a clinical trial. Lab Anim. 1999;33(2):169‐174. [DOI] [PubMed] [Google Scholar]
- 101. Stasiak KL, Maul D, French E, Hellyer PW, VandeWoude S. Species‐specific assessment of pain in laboratory animals. Contemp Top Lab Anim Sci. 2003;42(4):13‐20. [PubMed] [Google Scholar]
- 102. Gerstein HC, Pare G, Hess S, et al. Growth differentiation factor 15 as a novel biomarker for metformin. Diabetes Care. 2017;40(2):280‐283. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1‐S4