

Abstract
Favipiravir has demonstrated efficacy against the SARS-CoV-2 virus in several preliminary studies. This study aimed to evaluate the efficacy and safety of favipiravir for treatment of mild to moderate COVID-19 in outpatients and hospitalized patients. We conducted an open-label, randomized, active-controlled trial of a generic form of favipiravir in patients with COVID-19 confirmed by PCR-test. Eligible patients (18-60 years) after stratification were randomly assigned (in a 2:1 ratio) to receive either favipiravir (1800 mg BID on day 1, followed by 800 mg BID for up to 9 days), or standard of care (SOC) treatment (umifenovir + intranasal interferon alpha-2b, or hydroxychloroquine) for up to 10 days. The co-primary outcomes were the time to clinical improvement and the time to viral clearance. Among 190 patients assessed for eligibility 168 were randomized to favipiravir (n=112) or to SOC (n=56) group. The median time to clinical improvement was 6.0 days (IQR 4.0; 9.3) in the favipiravir group and 10.0 (IQR 5.0; 21.0) days in the SOC group; the median difference was 4 days (HR 1.63; 95% CI 1.14-2.34; P=0.007). The statistically significant difference in the median time to viral clearance was observed only for hospitalized patients: 3.0 (IQR 3.0; 3.0) days in the favipiravir group vs. 5.0 (IQR 4.5; 5.5) days in the SOC group (HR 2.11; 95% CI 1.04-4.31; P=0.038). The rate of viral elimination on Day 5 in the favipiravir group was significantly higher than in SOC group: 81.2% vs. 67.9% (RR 1.22; 05% CI 1.00-1.48; P=0.022). The rate of clinical improvement on Day 7 in the favipiravir group was 1.5-fold higher than in SOC group: 52.7% vs. 35.8% (RR 1.50; 95% CI 1.02-2.22; P=0.020). Favipiravir was well-tolerated and the most common adverse reactions were asymptomatic hyperuricemia, transient elevation of ALT & AST, and mild gastrointestinal disorders. Favipiravir was superior to the SOC in shortening the time to clinical improvement in patients with mild to moderate COVID-19.
Keywords: COVID-19, SARS-CoV-2, coronavirus, favipiravir
Introduction
The global pandemics of coronavirus disease 2019 (COVID-19) introduced a significant challenge for the medical community for rapid identification of an effective and safe therapy against this new infection. The main strategy followed so far by majority of researchers was to evaluate the potential of existing drugs with suitable mechanism of action for treatment of SARS-CoV-2 infection [1]. One of such drugs is favipiravir [2]-a purine analogue, which active form favipiravir-ribofuranosyl-5’-triphosphate [3] selectively inhibits RNA dependent RNA polymerase (RdRP), an enzyme needed for RNA viral replication within infected cells [4]. The catalytic domain of RdRP is evolutionarily conserved in various RNA viruses, which results in the observed antiviral activity against a broad spectrum of RNA viruses, including arenaviruses, phleboviruses, hantaviruses, flaviviruses, enteroviruses, an alphavirus, a paramyxovirus, respiratory syncytial virus and noroviruses [5].
Favipiravir was initially developed for the treatment of influenza and approved in Japan in 2014, for use in an outbreak of novel or re-emerging influenza viral infections, where other antiviral drugs are not effective. Wang et al. demonstrated SARS-CoV-2 inhibition in vitro in the culture of model Vero E6 cells [6]. Since then, the results of several preliminary clinical trials have been reported, which have demonstrated the efficacy of favipiravir in COVID-19. Chen et al. have studied favipiravir in comparison to umifenovir in hospitalized patients with moderate to severe COVID-19 in a prospective, randomized, multicenter trial, organized in Wuhan, China. Favipiravir showed superior efficacy in the rate of clinical recovery at Day 7 compared to umifenovir in patients with moderate disease: 71.4% vs. 55.9% (P=0.019) [7]. Cai et al. demonstrated high efficacy of favipiravir for the treatment of COVID-19 in a small non-randomized clinical trial compared to the combination of lopinavir/ritonavir: in the favipiravir arm viral clearance was observed after 4 (2.5-9) days vs. 11 (8-13) days in the control arm [8]. Despite promising data, previous studies had relatively poor evidence due to a sub-optimal design (low patient numbers, retrospective enrolment, no PCR tests for SARS-CoV-2 confirmation, or combination with other antiviral agents).
The objective of our multicenter, open-label, randomized, active-controlled phase III trial was to evaluate the efficacy and safety of generic formulation of favipiravir in out- and inpatients with laboratory-confirmed mild to moderate COVID-19 disease.
Materials and methods
Patients
Eligible patients were 18-60 years of age; had a diagnosis of mild to moderate COVID-19 without respiratory failure; with symptom manifestation no more than 6 days before the randomization; with SARS-CoV-2 confirmed by PCR of oro- or nasopharyngeal swabs, and had received no previous antiviral therapy for COVID-19. Patients were excluded if they had respiratory failure (SpO2≤93%); the need for mechanical ventilation at screening; severe or extremely severe COVID-19; severe lung damage on computed tomography (CT) scans (subtotal diffuse ground-glass induration of pulmonary tissue, the involvement of ≥75% of the lung parenchyma, hydrothorax); unstable hemodynamics; or any of the following laboratory abnormalities at screening: AST or ALT level >2.5 × upper limit of normal (ULN), platelet count <50 × 109/L. All eligibility criteria are published on ClinicalTrials.gov, identifier: NCT04501783.
The trial protocol was approved by the Russian Ministry of Health (MoH) (permission #201 dated by May 20. 2020 [9]), including the Central Ethics Council, and by local ethics committees in 10 clinical centers. All patients provided written informed consent (IC) before the enrollment (Supplementary Materials).
Randomization and masking
Eligible patients were randomly assigned at a 2:1 ratio to receive either favipiravir or standard of care (SOC) therapy. Randomization was performed using a web-response system after stratification by COVID-19 severity (mild or moderate), age (<45 or ≥45 years), and CT severity (CT-0-1 or CT-2-3) at enrollment.
Study procedures
Favipiravir was administered orally with a loading dose of 1800 mg BID on Day 1, followed by 800 mg BID on Days 2-10. Patients in the control group received, depending on the severity of the condition of the patient, either umifenovir (200 mg 4 QID orally) + intranasal interferon alpha-2b (10000 IU/ml-3 drops in each nasal channel 5 times a day), or hydroxychloroquine (400 mg BID on Day 1 followed by 200 mg BID or 200 mg BID on Day 1 followed by 100 mg BID) during the period up to 10 days. These therapies represented a SOC treatment of COVID-19 in Russian MoH guidelines at the time of clinical study. All patients received supportive care, including antipyretics, antibiotics, anticoagulants, and vasoconstrictor drugs.
Patients had follow-up for a total period of 28 days. Outpatients had to be quarantined at home and monitored using telemedicine technology. They visited the trial sites using specialized transport with self-isolation measures. All patients were provided all necessary equipment for self-monitoring of their condition (pulse oximeter, tonometer, thermometer) and the diaries for symptom assessment.
Efficacy evaluation included clinical status (using WHO 8-Category Ordinal Scale [10]) and COVID-19 symptoms assessment, oxygen saturation levels and the body temperature that were performed daily during the first ten days of the study and on Days 14, 21, and 28. Oropharyngeal swabs for PCR were taken on Days 3, 5, 7, 10, 14, 21 and 28. CT scan of the chest was scheduled at screening and on Days 5, 14, and 28.
Adverse events (AE) were reported throughout the study up to Day 28 and were graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE), version 5.0. Clinical laboratory tests and electrocardiographic monitoring were performed at screening and on Days 5, 14, and 28.
Outcomes
The two primary endpoints were: (1) time to clinical improvement (a reduction of patient clinical status on at least 1 score according to WHO 8-Category Ordinal Scale compared to screening) and; (2) time to viral clearance (the absence of SARS-CoV-2 virus according to PCR in two consecutive swabs with an interval of at least 24 hours). The categories of WHO 8-Category Ordinal Scale were as follows: 0-no clinical or virological evidence of infection; 1-ambulatory, no limitation of activities; 2-ambulatory, limitation of activities; 3-hospitalized, no oxygen therapy; 4-hospitalized, oxygen by mask or nasal prongs; 5-hospitalized, severe disease, non-invasive ventilation or high-flow oxygen; 6-hospitalized, severe disease, intubation, and mechanical ventilation; 7-hospitalized, severe disease, intubation + additional organ support-pressors, RRT, ECMO; and 8-death [10].
The secondary and exploratory endpoints were the rate of clinical improvement at Day 7 and 14 and the rate of viral clearance at separate time points (Days 3, 5, 7, 10, 14, 21 and 28), the time to body temperature normalization (<37°C without antipyretics for at least 48 hours), the rate of resolution of lung changes on CT at Day 14, average score according to WHO 8-Category Ordinal Scale at Days 7 and 14, the time to resolution of the main disease symptoms, the rate of hospitalization for outpatients, the rate of use of artificial lung ventilation (ALV), the rate of transfer to intensive care unit (ICU) and the mortality rate during the 28 days. The standard safety endpoints were the rate and severity of AEs and serious AEs (SAE), the rate of severe AEs, and the rate of study discontinuation due to AE/SAE.
Statistical analysis
Efficacy was assessed in the intent-to-treat (ITT) population, which included all the patients who underwent randomization. Safety was assessed in the as-treated population, which included all patients who received at least one dose of the investigational therapy.
The Kaplan-Meier method was applied to estimate primary endpoints. The log-rank test was used to assess the differences between groups in these endpoints. Hazard ratios (HR) and relative risks (RR) with 95% confidence intervals were calculated using the Cox proportional-hazards model. Pre-specified subgroups in these analyses were defined according to patient cohort (outpatients or hospitalized), disease severity (mild or moderate), time from the first symptom onset to randomization (≤3 or 4-6 days), patient age (<45 or ≥45 years) and CT severity grade at baseline (CT-0-1 or CT-2-3).
In the full statistical analysis plan it was assumed to perform an interim (60 patients) and a final analysis.
We estimated that a sample of 168 patients, would have 90% power for the trial to show a difference in the time to clinical improvement in the whole ITT population at a one-sided alpha level of 0.025 (as calculated based on 129 events of clinical improvement). The type I error rate for the primary outcomes in this trial was strictly controlled at a one-sided alpha level of 0.025. For the other efficacy and safety outcomes, a two-sided alpha level of 0.05 was applied.
Results
Between May 23, 2020 and June 30, 2020, 190 patients were assessed for eligibility, of whom 168 were randomly assigned to receive either favipiravir (n=112), or SOC (n=56). Most patients (94.6%), in both groups completed the study according to the protocol. Six (5.4%) and 3 (5.4%) patients withdrew the informed consent and discontinued the study in the favipiravir and SOC groups, respectively (Figure 1). 127 subjects (75.5%) of the study population were outpatients and 41 (24.5%) were hospitalized (3:1 ratio).
Figure 1.
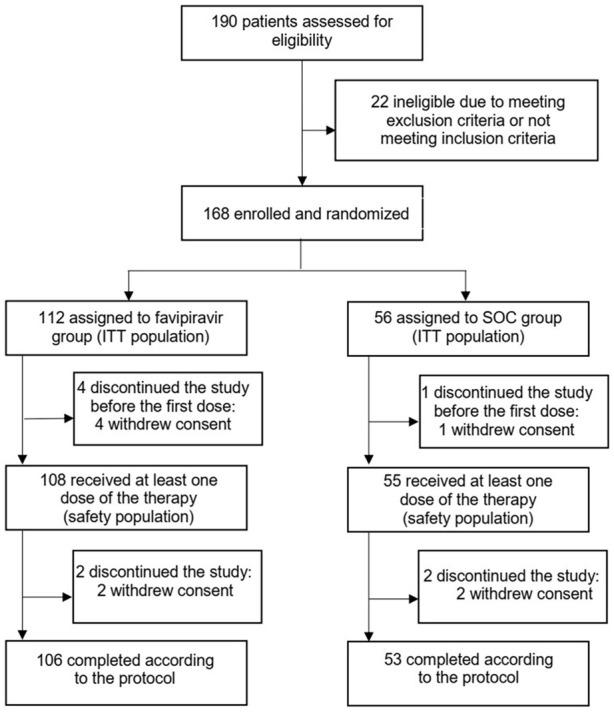
Trial profile.
Baseline demographics and disease characteristics were balanced between the treatment groups (Table 1). The mean times from the onset of symptoms to randomization were 3.5 (1.4) and 3.6 (1.4) days in favipiravir and SOC groups, respectively. A slightly larger proportion of the patients in the favipiravir group had extended lung damage on CT scan (CT-3 corresponded to damage of 50%-75% of lungs): 5.4% vs. 1.8% of the patients, respectively.
Table 1.
Baseline characteristics of the intent-to-treat population
| Favipiravir (N=112) | SOC (N=56) | |
|---|---|---|
| Age, years | ||
| Mean (SD) | 41.7 (10.6) | 42.0 (10.4) |
| Sex | ||
| Female | 63 (56.2%) | 26 (46.4%) |
| Male | 49 (43.8%) | 30 (53.6%) |
| Race* | ||
| Caucasian | 111 (99.1%) | 55 (98.2%) |
| Asian | 1 (0.9%) | 1 (1.8%) |
| Patient cohort | ||
| Outpatients-no. (%) | 83 (74.1%) | 44 (78.6%) |
| Hospitalized patients-no. (%) | 29 (25.9%) | 12 (21.4%) |
| Time from the first symptom onset to randomization | ||
| Mean (SD), days | 3.5 (1.4) | 3.6 (1.4) |
| ≤3 days | 53 (47.3%) | 24 (42.9%) |
| 4-6 days | 59 (52.7%) | 32 (57.1%) |
| Disease severity at baseline | ||
| Mild | 28 (25.0%) | 15 (26.8%) |
| Moderate | 84 (75.0%) | 41 (73.2%) |
| Score on Ordinal scale at baseline | ||
| 1-ambulatory. no limitation of activities | 14 (12.5%) | 13 (23.2%) |
| 2-ambulatory. limitation of activities | 69 (61.6%) | 31 (55.4%) |
| 3-hospitalized. no oxygen therapy | 28 (25.0%) | 10 (17.9%) |
| 4-hospitalized. oxygen by mask or nasal prongs | 1 (0.9%) | 2 (3.6%) |
| Severity according to CT | ||
| CT-0 | 28 (25.0%) | 13 (23.2%) |
| CT-1 | 66 (58.9%) | 33 (58.9%) |
| CT-2 | 12 (10.7%) | 8 (14.3%) |
| CT-3 | 6 (5.4%) | 1 (1.8%) |
| Hyperthermia at baseline | 79 (70.5%) | 43 (76.8%) |
Data are n (%) unless otherwise stated.
Race was reported by the patients.
Primary outcome
The analysis of time to clinical improvement (the first co-primary endpoint) showed that patients in the ITT population in the favipiravir group had a shorter median time to clinical improvement compared to the SOC group: 6.0 (IQR 4.00; 9.25) days vs. 10.0 (IQR 5.0; 21.0) days respectively (HR 1.63; 95% CI 1.14-2.34; P=0.007) (Figure 2 and Table 2). The median difference was 4 days. Clinical improvement occurred faster in the outpatient cohort (6.0 (IQR: 4.0; 12.0) vs. 14 (IQR: 5.0; 28.0) days) compared to the cohort of the hospitalized patients (7.0 (IQR: 5.0; 9.0) vs. 8.5 (IQR: 6.0; 10.0) days), as well as in the cohorts of moderate disease, patients ≥45 years, patients with CT-0-1 at baseline, and patients with therapy onset within 4-6 days (Figure 2).
Figure 2.

Kaplan-Meier estimates and subgroup analysis of time to clinical improvement.
Table 2.
Efficacy outcomes in ITT population and the results of subgroup analysis
| Measure | Favipiravir | SOC | Hazard ratio/Risk ratio (95% CI) | P-value |
|---|---|---|---|---|
| Primary outcomes and subgroup analysis | ||||
| Time to clinical improvement | ||||
| All patients (N=168) | ||||
| N | 112 | 56 | - | - |
| Median (IQR). days | 6.0 (4.00; 9.25) | 10.0 (5.00; 21.0) | 1.63 (1.14; 2.34) | 0.007§,* |
| Outpatients (N=127) | ||||
| N | 83 | 44 | - | - |
| Median (IQR). days | 6.0 (4.0; 12.0) | 14.0 (5.0; 28.0) | 1.65 (1.08; 2.52) | 0.019§,* |
| Hospitalized patients (N=41) | ||||
| N | 29 | 12 | - | - |
| Median (IQR). days | 7.0 (5.0; 9.0) | 8.5 (6.0; 10.0) | 1.42 (0.72; 2.81) | 0.271§ |
| Time to viral clearance | ||||
| All patients (N=168) | ||||
| N | 112 | 56 | - | - |
| Median (IQR). days | 3.0 (3.0; 3.5) | 3.0 (3.0; 7.0) | 1.28 (0.92; 1.79) | 0.161§ |
| Outpatients (N=127) | ||||
| N | 83 | 44 | - | - |
| Median (IQR). days | 3.0 (3.0; 3.5) | 3.0 (3.0; 7.0) | 1.11 (0.76; 1.61) | 0.459§ |
| Hospitalized patients (N=41) | ||||
| N | 29 | 12 | - | - |
| Median (IQR). days | 3.0 (3.0; 3.0) | 5.0 (4.5; 5.5) | 2.11 (1.04; 4.31) | 0.038§,* |
| Key secondary and exploratory outcomes | ||||
| Rate of clinical improvement | ||||
| N | 112 | 56 | - | - |
| Day 7 | 59 (52.7%) | 20 (35.7%) | 1.50 (1.02; 2.22) | 0.020* |
| Day 14 | 93 (83.0%) | 37 (66.1%) | 1.28 (1.05; 1.56) | 0.005* |
| Day 28 | 96 (85.7%) | 44 (78.6%) | 1.11 (0.96; 1.29) | 0.098 |
| Rate of viral clearance | ||||
| N | 112 | 56 | - | - |
| Day 3 | 80 (71.4%) | 32 (57.1%) | 1.27 (0.99; 1.64) | 0.030* |
| Day 5 | 91 (81.2%) | 38 (67.9%) | 1.22 (1.00; 1.48) | 0.022* |
| Day 7 | 95 (84.8%) | 46 (82.1%) | 1.05 (0.92; 1.21) | 0.296 |
| Day 10 | 101 (90.2%) | 49 (87.5%) | 1.05 (0.95; 1.17) | 0.244 |
| Day 14 | 103 (92.0%) | 53 (94.6%) | 0.99 (0.93; 1.06) | 0.750 |
| Day 21 | 105 (93.8%) | 53 (94.6%) | 1.01 (0.95; 1.07) | 0.549 |
| Day 28 | 105 (93.8%) | 53 (94.6%) | 1.01 (0.95; 1.07) | 0.549 |
| Rate of pneumonia resolution on CT scans | ||||
| All patients (N=168) | ||||
| N | 112 | 56 | - | - |
| Day 5 | 3 (2.7%) | 0 (0.0%) | n.a. | 0.553 |
| Day 14 | 14 (12.5%) | 5 (8.9%) | n.a. | 0.613 |
| Day 28& | 27 (24.1%) | 12 (21.4%) | n.a. | 0.847 |
| Hospitalized patients (N=41) | ||||
| N | 29 | 12 | - | - |
| Day 5 | 0 (0.0%) | 0 (0.0%) | n.a. | - |
| Day 14 | 3 (10.3%) | 1 (8.3%) | n.a. | 1.000 |
| Day 28& | 9 (31.0%) | 1 (8.3%) | n.a. | 0.231 |
| Rate of other disease outcomes | ||||
| N | 112 | 56 | - | - |
| Hospitalization | 3 (3.6%) | 2 (4.5%) | 0.94 (0.78; 1.14) | 0.494 |
| Transfer to ICU | 3 (2.7%) | 1 (1.8%) | 1.51 (0.16; 4.21) | 1.000 |
| Invasive ventilation | 1 (0.9%) | 0 (0.0%) | - | 1.000 |
| Hospitalization | 3 (3.6%) | 2 (4.5%) | 0.94 (0.78; 1.14) | 0.494 |
Data are n (%) unless otherwise stated. Abbreviations: ITT-intent-to-treat population; CI-confidential intervals; SOC-standard of care; n.a.-not applicable; IQR-interquartile range; CT-computed tomography; ICU-intensive care unit.
The difference is statistically significant.
P-values were calculated using a log rank test.
On Day 28, the best response for the study was taken into account, since for a number of patients had resolution on CT at previous visits. CT on the Day 28 was not performed according to the protocol and by the decision of the investigator.
In the ITT population, there was no difference in the time to viral clearance between the groups (medians were 3 days in both groups); however, in the cohort of hospitalized patients viral clearance occurred faster in the favipiravir group (Figure 3): 3.0 (IQR: 3.0; 3.0) vs. 5.0 (IQR: 4.5; 5.5) days (HR 2.11; 95% CI 1.04-4.31; P=0.038). A difference in favor of favipiravir was also observed in the cohorts of patients with therapy onset within 4-6 days.
Figure 3.

Kaplan-Meier estimates and subgroup analysis of viral clearance.
Secondary and exploratory outcomes
The analysis of secondary and exploratory endpoints also revealed statistically significant differences between the groups. The rates of clinical improvement on Days 7 and 14 were 1.5-fold and 1.25-fold higher in the favipiravir group compared to SOC group: on Day 7-52.7% (59 of 112) of the patients vs. 35.5% (20 of 56) (RR 1.50; 95% CI 1.02-2.22; P=0.020) and on Day 14-83.0% (93 of 112) of the patients vs. 66.1% (37 of 56) (RR 1.28; 95% CI 1.05-1.56; P=0.005).
The rates of viral clearance were also significantly higher on Days 3 and 5 in favipiravir group compared to SOC group: on Day 3-71.4% (80 of 112) vs. 57.1% (32 of 56) (RR 1.27; 95% CI 0.99-1.64; P=0.030) and on Day 5-81.2% (91 of 112) vs. 67.9% (38 of 56) (RR 1.22; 95% CI 1.00-1.48; P=0.022). On the following days of follow-up (7, 10, 14, 21 and 28), no significant difference was noted.
The analysis of data of radiological endpoints revealed a trend towards a greater proportion of patients with resolution of ground-glass changes on CT in the favipiravir group (with or without residual signs), but with no statistical significance (Table 2). The resolution of signs of pneumonia at Day 5 was observed only in 2.7% (3 of 112) of the patients in the favipiravir group. At Day 14 resolution was observed in 12.5% (14 of 112) of the patients in the favipiravir group and 8.9% (5 of 56) in the SOC group. On Day 28 in the hospitalized cohort, the signs of pneumonia were resolved in 31.0% (9 of 29) of the patients in the favipiravir group vs. 8.3% (1 of 12) in the SOC group. This result was also observed in patients with severe lung damage at baseline (CT-3) (Figure 4).
Figure 4.
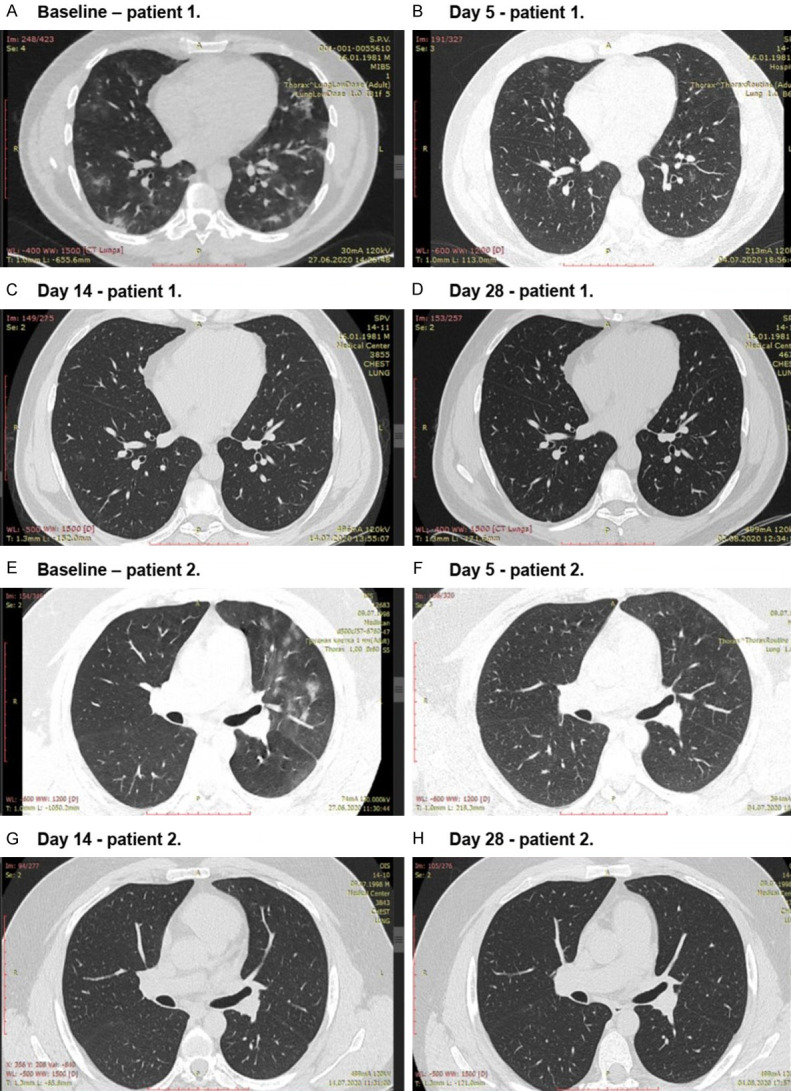
Dynamics of disease-specific changes on CT in 2 patients with CT-3 at baseline from the favipiravir group during the study.
The resolution of main clinical symptoms tended to occur on average 0.5-1 day faster in the favipiravir group in comparison to the SOC group (Table 3), but the difference was not significant.
Table 3.
Duration of main clinical symptoms for therapy groups
| Symptom | Measure | Favipiravir (N=112) | SOC (N=56) | P-value1 |
|---|---|---|---|---|
| Cough | N | 77 | 38 | 0.202 |
| Average | 3.64 | 4.92 | ||
| 95% CI | 3.00; 4.28 | 3.54; 6.30 | ||
| Headache | N | 91 | 40 | 0.794 |
| Average | 8.14 | 8.50 | ||
| 95% CI | 6.96; 9.33 | 6.67; 10.33 | ||
| Dyspnoea | N | 50 | 29 | 0.281 |
| Average | 3.04 | 3.69 | ||
| 95% CI | 2.65; 3.43 | 3.01; 4.37 | ||
| Myalgia | N | 55 | 17 | 0.592 |
| Average | 6.36 | 7.24 | ||
| 95% CI | 5.13; 7.60 | 5.90; 8.58 | ||
| Weakness | N | 92 | 47 | 0.421 |
| Average | 7.54 | 8.49 | ||
| 95% CI | 6.39; 8.69 | 6.86; 10.11 | ||
| Fever | N | 85 | 46 | 0.244 |
| Average | 3.49 | 4.57 | ||
| 95% CI | 3.01; 3.98 | 3.03; 6.10 | ||
| Decrease of mental activity | N | 46 | 24 | 0.719 |
| Average | 5.00 | 5.50 | ||
| 95% CI | 4.12; 5.88 | 4.08; 6.92 | ||
| Decrease of physical activity | N | 88 | 43 | 0.464 |
| Average | 8.59 | 9.60 | ||
| 95% CI | 7.30; 9.88 | 7.76; 11.45 |
P-value was calculated by Student’s t-test. The normality of distribution was assessed by Kolmogorov-Smirnov test (the normality was confirmed).
Safety outcomes
Safety outcome analysis showed that favipiravir was well tolerated. AEs occurred in 74.1% (80 of 108) of the patients in the favipiravir group and 60.0% (33 of 55) in the SOC group without statistical significance (P=0.074) (Table 4). Most of the reported AEs were mild. The rates of severe AEs (which only included grade 3) were similar in both groups: 7.4% (8 of 108) in the favipiravir group and 7.3% (4 of 55) in the SOC group. SAEs occurred in 2 patients (1.9%) in the favipiravir group which included a bone fracture and secondly decreased oxygen saturation due to COVID-19 progression; both of thesewere assessed as not related to the investigational drug. Two patients (1.8%) in the favipiravir group and 1 (1.8%) in the SOC group discontinued treatment because of AEs: due to ALT & AST elevation of grade 3 in one patient and grade 2 in another patient in the favipiravir group; and due to ALT elevation of grade 3 in one patient receiving hydroxychloroquine in the SOC group. After discontinuation of therapy, AEs in all patients were completely resolved.
Table 4.
Adverse events of any cause that occurred in 5% or more patients in the as-treated population
| Event | Favipiravir (N=108) | SOC (N=55) | ||
|---|---|---|---|---|
| Grade (NCI CTCEA 5.0) | Any grade | Grade 3 | Any grade | Grade 3 |
| Any adverse event during treatment | 80 (74.1%) | 8 (7.4%) | 33 (60.0%) | 4 (7.3%) |
| Serious adverse event during treatment | 2 (1.9%)* | 2 (1.9%)* | 0 (0.0%) | 0 (0.0%) |
| Adverse event leading to discontinuation of treatment | 2 (1.9%) | 1 (0.9%) | 1 (1.8%) | 1 (1.8%) |
| Metabolism and nutrition disorders | ||||
| Hyperuricemia§ | 45 (41.7%) | - | 2 (3.6%) | - |
| Hyperglycemia | 14 (13.0%) | - | 7 (12.7%) | - |
| Hepatobiliary disorders | ||||
| ALT elevation | 38 (31.5%) | 3 (2.8%) | 12 (20.0%) | 2 (3.6%) |
| AST elevation | 25 (21.3%) | 2 (1.9%) | 6 (10.9%) | 1 (1.8%) |
| LDH elevation | 6 (5.6%) | - | 1 (1.8%) | - |
| Hyperbilirubinemia | 5 (4.6%) | - | 5 (9.1%) | - |
| Gastrointestinal disorders | ||||
| Diarrhea | 16 (14.8%) | - | 7 (12.7%) | - |
| Nausea | 9 (8.3%) | - | 5 (9.1%) | - |
| Epigastric pain | 7 (6.5%) | - | 1 (1.8%) | - |
| Abdominal pain | 8 (7.4%) | - | 4 (7.3%) | - |
| Investigations | ||||
| Creatine kinase elevation | 15 (13.9%) | 0 (0.0%) | 10 (18.2%) | 1 (1.8%) |
| Cardiac disorders | ||||
| Sinus bradycardia | 10 (9.3%) | - | 2 (3.6%) | - |
| Skin and subcutaneous tissue disorders | ||||
| Rash | 6 (5.6%) | 1 (0.9%) | 1 (1.8%) | 0 (0.0%) |
Data are n (%) unless otherwise stated.
Both cases of SAE were not associated with the investigational drug and included: 1 case of bone fracture (gr. 3) and decreased oxygen saturation due to COVID-19 progression (gr. 3).
The difference was statistically significant (P<0.0001).
The frequency of hyperuricemia was significantly higher in favipiravir group, and is known to be related to favipiravir treatment: 41.7% (45 of 108) vs. 3.6% (2 of 55), respectively (P<0.0001). The incidence of any other AEs was not found to be significantly different between the groups. The other most common AEs in both groups were transaminases (ALT & AST) elevation, creatine kinase elevation, gastrointestinal disorders (diarrhea, nausea, epigastric and abdominal pain) and hyperglycemia. ALT elevation was reported in 31.5% (38 of 108) of the patients in the favipiravir group vs. 20.0% (12 of 56) in the SOC group (P=0.106). AST elevation-was in 21.3% (25 of 108) vs. 10.9% (6 of 56), respectively (P=0.089). An increase in transaminase level during therapy with favipiravir was expected and transient.
Discussion
The results of this trial suggest that a 10-day course of favipiravir was superior to SOC in the treatment of hospitalized and outpatients with mild to moderate COVID-19. The difference in median time to clinical improvement was 4 days in the ITT population and 8 days in the outpatient cohort. This is a clinically significant period, given the general severity of the patient’s condition even with a mild or moderate disease course. There was also a significant difference in the rate of clinical improvement in favor of favipiravir (1.5-fold higher) on Day 7. In the subgroup analysis, favipiravir benefits were most apparent in the cohorts of moderate disease inpatients 45 years of age or older, patients with CT-0-1 at baseline, and patients with therapy onset within 4-6 days.
The time to viral clearance did not differ between groups in the whole ITT population but showed a trend in favor of favipiravir. There was a significant difference in time to viral clearance for the cohort of hospitalized patients (2 days). This observation was similar to those reported by Cai et al. (2020) for hospitalized patients [8]. Also, the rates of viral clearance were significantly higher in the favipiravir group at Days 3 (1.3-fold) and 5 (1.2-fold). It should be noted that viral clearance is not considered a predictive measure due to a lack of established methodology and a clear correlation with the clinical condition of the patient [11]. WHO recommends as a primary endpoint a measure of patients’ clinical status at a particular time point after enrollment, while all other biomarkers of illness, including viral clearance, could be considered only as a secondary endpoints [9]. The FDA has a similar position, considering that virological outcomes may be acceptable as a primary endpoint only in Phase 2 studies [12]. Finally, at the second Global regulatory workshop on COVID-19 therapeutics, it was agreed that virological endpoints are useful only for proof-of-concept studies [13]. In our study, we have shown a clear clinical improvement in the patient group treated with favipiravir. Virological outcomes were taken into account; however, following current recommendations of regulatory agencies, we do not consider them as a key measure of benefit. Thus, based on results we consider favipiravir is superior to SOC therapy.
Safety evaluation did not reveal any new unreported previously AEs for favipiravir. The incidence of hyperuricemia in the favipiravir group (41.7%) exceeded those reported previously: 9.9% in healthy adult Japanese volunteers [14] and 13.76% in patients with COVID-19 according to Chen et al. [7]. The observed discrepancies are likely to be explained by the specificity of AE reporting in the study (all observed laboratory deviations of any grade were reported as AEs). Alternatively, the increase AEs can be explained by the specificity of the studied patient population. The median concentration of uric acid did not increase significantly beyond the limits of normal, while there was a trend of an increase its level in the middle of the treatment course with further resolution (Figure 5).
Figure 5.
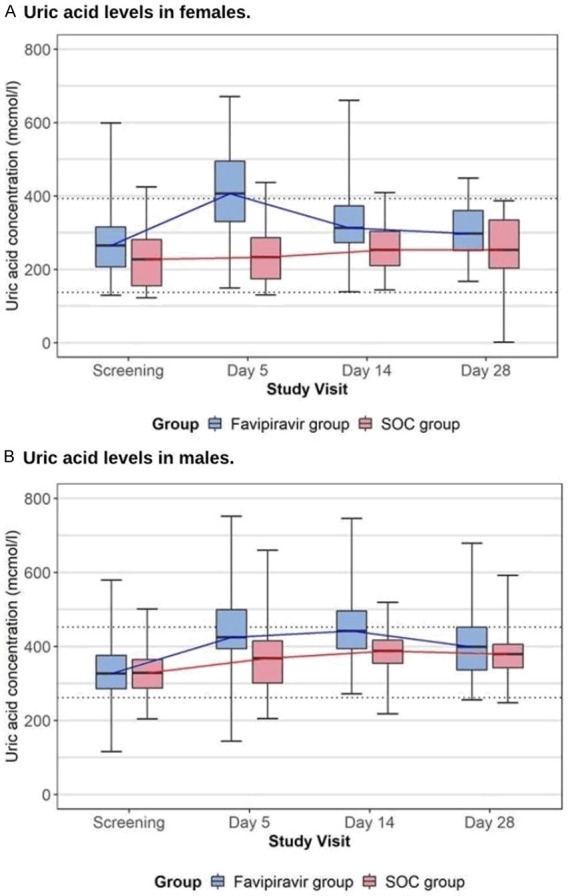
Dynamics of uric acid level in both treatment groups during the study (presented medians. IQR. Minimum and maximum; limits of normal by dotted line), µmol/ml.
Although placebo-controlled blinded trials provide the highest degree of evidence of efficacy, ethical considerations did not allow us to leave the control group in this trial without any treatment. For this reason, the study design was open-label, randomized against the active control with SOC treatment included ethiotropic therapies recommended by the Russian MoH guideline, which did not allow blinding. Nevertheless, the current design provided strong evidence of the efficacy of favipiravir among other currently published trials. Our results are also aligned with recently reported (September 2020) by FUJIFILM Toyama Chemical Co. Ltd. preliminary results of a placebo-controlled study of favipiravir (Avigan) in 156 patients with COVID-19. The median value of primary endpoints (included time to negative conversion of detectable SARS-CoV-2 viral RNA in the PCR assays and alleviation of symptoms - body temperature. oxygen saturation, and chest images) - was 11.9 days for the favipiravir group and 14.7 days for the placebo group (P=0.0136) [15].
In conclusion, the results of this study confirm that early treatment initiation with favipiravir brings clinical benefits to patients with mild to moderate COVID-19. It is also important to note that favipiravir can be used successfully in outpatient settings, providing an advantage to a wider coverage of the population suffering from COVID-19 and reducing expenditures of the healthcare system associated with the hospital setting. Due to the risks of the teratogenic effect of favipiravir observed in preclinical studies [15], the outpatient use of favipiravir has to include robust measures in the area of pregnancy prevention, such as information brochures on the risk of teratogenic and informed consent for patients with childbearing potential.
Acknowledgements
This study was funded by R-Pharm Group of Companies. This study was done with assistance from a large number of people in several different organizations. We thank the staff of the Clinical Research Department and Clinical Laboratory of “Central Research Institute for Epidemiology” of The Federal Service on Customers’ Rights Protection and Human Well-being Surveillance (P.V. Chukhliaev. MD; D.A. Khavkina. MD; A.A. Garbuzov. MD); the Medical Center “Eco-safety”. The Medical Center “Group of Companies “MEDSI” JSC (Prof. I.V. Shestakova. Ph.D.; A.Yu. Vafin. MD); the Clinical Pharmacology Department of the Clinical Hospital of Zhukovsky; the Medical center “Neuroprofi” LLC (T.N. Domostroeva. MD); the L.A. Vorokhobov City Clinical Hospital No. 67 of the Moscow City Healthcare Department (M.V. Otpushchennikova. MD); the City Clinical Hospital No. 52 of the Moscow City Healthcare Department (E.A. Kaplun. MD; D.V. Petina. MD); the Infectious Clinical Hospital No. 1 of the Moscow City Healthcare Department (D.A. Bistritskiy. MD); the Voronezh Regional Clinical Hospital No. 1 (V.S. Lesina. MD); the City Hospital No. 40 of the Kurortny District (Prof. S.G. Scherbak. Ph.D); N.I. Pirogov National Medical and Surgical Center of the Ministry of Health of the Russian Federation. Moscow. Russia (Y.F. Brook. MD; O.Y. Bronov. Ph.D.); the N.N. Burdenko National Medical Research Centr of Neurosurgery” of the Ministry of Health of the Russian Federation. Moscow. Russia (E.I. Shults. Ph.D.); and the employees of R-Pharm Group of Companies participating in organization and performance of the study (M.Y. Samsonov. Ph.D.; E.N. Krasavina. Ph.D.; A.V. Zintchenko. Ph.D.; M.V. Nikolskaya. MD; V.A. Razzhivina. Ph.D). We also thank all the patients for participating in this trial.
Disclosure of conflict of interest
None.
Supplementary Materials
References
- 1.Tu YF, Chien CS, Yarmishyn AA, Lin YY, Luo YH, Lin YT, Lai WY, Yang DM, Chou SJ, Yang YP, Wang ML, Chiou SH. A review of SARS-CoV-2 and the ongoing clinical trials. Int J Mol Sci. 2020;21:2657. doi: 10.3390/ijms21072657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Delang L, Abdelnabi R, Neyts J. Favipiravir as a potential countermeasure against neglected and emerging RNA viruses. Antiviral Res. 2018;153:85–94. doi: 10.1016/j.antiviral.2018.03.003. [DOI] [PubMed] [Google Scholar]
- 3.Furuta Y, Takahashi K, Shiraki K, Sakamoto K, Smee DF, Barnard DL, Gowen BB, Julander JG, Morrey JD. T-705 (favipiravir) and related compounds: novel broad-spectrum inhibitors of RNA viral infections. Antiviral Res. 2009;82:95–102. doi: 10.1016/j.antiviral.2009.02.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Furuta Y, Gowen BB, Takahashi K, Shiraki K, Smee DF, Barnard DL. Favipiravir (T-705), a novel viral RNA polymerase inhibitor. Antiviral Res. 2013;100:446–54. doi: 10.1016/j.antiviral.2013.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Furuta Y, Komeno T, Nakamura T. Favipiravir (T-705), a broad spectrum inhibitor of viral RNA polymerase. Proc Jpn Acad Ser B Phys Biol Sci. 2017;93:449–463. doi: 10.2183/pjab.93.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang M, Cao R, Zhang L, Yang X, Liu J, Xu M, Shi Z, Hu Z, Zhong W, Xiao G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020;30:269–271. doi: 10.1038/s41422-020-0282-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen C, Zhang Y, Huang J, Yin P, Cheng Z, Wu J, Chen S, Zhang Y, Chen B, Lu M, Luo Y, Ju L, Zhang J, Wang X. A randomized clinical trial. MedRxiv. 2020 [Google Scholar]
- 8.Cai Q, Yang M, Liu D, Chen J, Shu D, Xia J, Liao X, Gu Y, Cai Q, Yang Y, Shen C, Li X, Peng L, Huang D, Zhang J, Zhang S, Wang F, Liu J, Chen L, Chen S, Wang Z, Zhang Z, Cao R, Zhong W, Liu Y, Liu L. Experimental treatment with favipiravir for COVID-19: an open-label control study. Engineering (Beijing) 2020;6:1192–1198. doi: 10.1016/j.eng.2020.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.The portal of the Russian Ministry of Health. (https://grls.rosminzdrav.ru/CIPermissionMini.aspx?CIStatementGUID=1bf6ea37-2035-45cf-aeb0-08d087c57cdf&CIPermGUID=89427E53-84C2-483E-8B04-1E9496E2D8B3; date of reference: May 21. 2020)
- 10.Geneva, Switzerland: World Health Organization; 2020. WHO R&D Blueprint novel Coronavirus COVID-19 Therapeutic Trial Synopsis. (https://www.who.int/blueprint/priority-diseases/key-action/COVID-19_Treatment_Trial_Design_Master_Protocol_synopsis_Final_18022020.pdf; date of reference: August 30. 2020) [Google Scholar]
- 11.Liu WD, Chang SY, Wang JT, Tsai MJ, Hung CC, Hsu CL, Chang SC. Prolonged virus shedding even after seroconversion in a patient with COVID-19. J Infect. 2020;81:318–356. doi: 10.1016/j.jinf.2020.03.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.FDA Guidance for Industry. COVID-19: developing drugs and biological products for treatment or prevention guidance for industry. 2020. (https://www.fda.gov/regulatory-infor-mation/search-fda-guidance-documents/covid-19-developing-drugs-and-biological-products-treatment-or-prevention; date of reference: August 30. 2020)
- 13.International Coalition of Medicines Regulatory Authorities. ICMRA COVID-19 treatments and clinical trials workshop #2. 2020. (http://icmra.info/drupal/news/20july2020/summary; date of reference: August 30. 2020)
- 14.Anti-influenza drug Avigan® Tablet meets primary endpoint in phase III Clinical trial in Japan for COVID-19 patients. 2020. (https://www.fujifilm.com/jp/en/news/hq/5451; date of reference: September 27. 2020)
- 15.PMDA. Avigan tablet 200 mg. Report on the deliberation results. March 4. 2014 evaluation and licensing division. Pharmaceutical and food safety bureau ministry of health. Labour and Welfare. 2020. (https://www.pmda.go.jp/files/000210319.pdf; date of reference: June 30. 2020)
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.