

Abstract
Background: Necrotizing enterocolitis (NEC) is an acquired disease, which mainly occurs in premature infants or sick newborns. microRNA (miR), as a common non-coding RNA in recent years, is found in many diseases. In this research, miR usefulin NEC is analyzed by GEO. Method: The differentially expressed miRs in NEC were screened by analyzing GSE68054, and miR-200a-3p in IEC-6 cells induced by lipopolysaccharide (LPS) and serum of NEC children were detected by qRT-PCR. The role of miR-200a-3p in LPS-induced IEC-6 cells was tested using CCK-8, PI dyeing, and inflammatory cytokine detection. The direct downstream molecules of miR-200a-3p were identified using TargetScanHuman and verified by luciferase reporter gene assay. The mechanism of action was explored using western blot. Results: miR-200a-3p in IEC-6 treated with NEC and LPS was significantly decreased. In vitro experiments revealed that miR-200a-3p mimetic could inhibit IL-6 and TNF-α in IEC-6 cells induced by LPS and reduce the positive rate of PI. In addition, it was determined that receptor-interacting protein kinase 1 (RIPK1) was a downstream molecule of miR-200a-3p, and overexpression of RIPK1 could aggravate LPS-induced IEC-6 injury, while miR-200a-3p mimics could alleviate the overexpression of RIPK1. miR-200a-3p mimics inhibited the elevation of necrosis-related molecules and the interaction between RIPK1 and RIPK3 in LPS-induced IEC-6 cells. Conclusion: miR-200a-3p can protect intestinal epithelial cells from LPS injury by inhibiting inflammation and necrosis mediated by RIPK1, which provides a possible target for NEC.
Keywords: miR-200a-3p, necrotizing enterocolitis, RIPK1, GEO
Introduction
Necrotizing enterocolitis (NEC) is the most common cause of death in neonatology. Clinical statistics show that the mortality of NEC is about 20~30% [1,2]. There are many risk factors for the development of NEC, among which premature birth, bacterial colonization and the management of formula food are common [3,4]. NEC damages the digestive system of newborns greatly, which can cause shock, acidosis and jaundice in severe cases [5]. Providing adequate parenteral nutrition and maintaining the balance of water, electrolytes, acids, and bases are important strategies to improve NEC [6]. Although the disease has been effectively controlled at present, NEC is still a main cause of premature infant morbidity and death [7]. Therefore, it is particularly crucial to explore the mechanism of NEC and find latent diagnostic and therapeutic targets.
miR (microRNA) is a non-coding RNA with 18-24nt, which is an important part of the non-coding RNA [8,9]. miR can regulate the downstream mRNA by pairing with complementary sequences of downstream targeted mRNA [10]. Studies [11-13] have shown that miR is involved in a variety of acute and chronic diseases, inflammation, cancer, infection, and organ damage. An early study reported that the inhibition of miR-124 improved the development of NEC [14]. In this research, we tested the difference of GSE68054, and found that miR-200a-3p in NEC was inhibited. miR-200a-3p, as an early discovered miR, has been reported to be involved in inflammatory reactions. Therefore, we speculate that miR-200a-3p may be involved in NEC.
Cell death is a basic process that controls body development and homeostasis by regulating the number of cells and eliminating damaged or infected cells [15]. Receptor-interacting protein kinase 1 (RIPK1) is a multi-domain protein containing N-terminal kinase domain, intermediate domain and c-terminal death domain (DD) [16]. Recent studies have revealed that there is a relationship between RIPK1 and apoptosis. For example, RIPK1 can counteract ZBP1-mediated necrosis, thus inhibiting inflammation [17]. Other studies have shown that RIPK1 is a key regulator of inflammation and cell death [18]. In this study, we have found that miR-200a-3p has a targeted binding site with RIPK1, so we speculate that miR-200a-3p may participate in NEC by regulating RIPK1.
Lipopolysaccharide (LPS), as an effective component of the outer cell wall of the Gram-negative bacteria, has been proven to be an inducer of NEC [19]. The mechanism of miR-200a-3p in NEC was investigated by constructing NEC models in vivo and in vitro, aiming to provide reference strategies for NEC.
Methods and materials
GEO (Gene expression omnibus) analysis
We logged in to https://www.ncbi.nlm.nih.gov/gds, downloaded the Series Matrix File(s) in GSE68054, and obtained GPL16384 annotation file to annotate miR. A total of 12 samples were collected from the chip, including 4 NEC samples, 4 spontaneous intestinal perforation (SIP) cases and 4 intestinal tissues form the infant who received surgical treatment. Patients with NEC and infants who received surgical treatment were selected to collect intestinal tissue samples for analysis, and limma (threshold, Log fold change =1, P=0.05) was used to analyze the differentially expressed miRs and visualize volcano map, and then pheatmap was used to visualize the heat map.
Clinical data
Twelve children with NEC from January 2018 to May 2019 were selected as the research group (RG), and 12 healthy newborns during the same period were collected as the control group (CG). Inclusion criteria: The mother of the child was aged 23-30 years old and a primipara; The mother did not have diseases such as infection and malignant tumor; The delivery time was within 37-41 weeks. Exclusion criteria: The child was unable to cooperate with the experimental tests; The child had congenital defects after birth. This study conformed to the Medical Ethics Committee (ZJ1706841F), and the informed consent of all children was signed by their guardians. Peripheral blood of children was collected, placed at room temperature for 30 min, and centrifuged at 3000 rpm for 10 min. The supernatant was obtained, and then placed at laboratory for testing.
Cell cultivation
The intestinal epithelial cell line IEC-6 was obtained from ATCC (Manassas, VA), and cultured in modified DMEM (Gibco, Grand Island, NY) including 10% FBS (Gemini, West Sacramento, CA) at 37°C with 5% CO2. 100 μg/ml of LPS (Sigma, St. Louis, MO) was used to induce the cells and construct the NEC model in vitro.
Establishment of animal model
Twenty male C57BL/6 mice (1 week old) were obtained from Charles River (Wilmington, MA). The mice were grouped into the control group, NEC, miR-NC+NEC, and miR-200a-mimics + NEC. All mice were breast-fed, and the mice in the control group were given enema of normal saline. As Yu and Chen et al. [20,21] suggested, the mice in the NEC group were treated with artificial formula feeding combined with hypoxia and cold stimulus. Control group: Five neonatal rats were fed by the mother rats and clystered with normal saline (0.2 ml) (all rats were lightly anesthetized with 5% chloral hydrate before enema). NEC model group: Five mice were reared artificially and exposed to hypoxic cold stimulation. One hour before hypoxic cold stimulation, the normal saline (0.2 ml) was used for enema once a day. miR-NC+NEC: Five mice were raised artificially and exposed to hypoxic cold stimulation. One hour before hypoxic cold stimulation, they were fed with breast milk and miRNA NC (0.2 mL), once a day. miR-200a-mimics + NEC: Five mice were raised artificially and exposed to hypoxic cold stimulation. One hour before hypoxic cold stimulation, they were fed with breast milk and miR-200a-mimics (0.2 mL), once a day. In the case of abdominal distention, abdominal wall discoloration, hematochezia, or other clinical symptoms, the animals were executed 96 h later, and ileum tissue and blood samples were taken for further analysis. The death scheme was carbon dioxide inhalation, followed by cervical dislocation. The collected samples were frozen by liquid nitrogen for testing, and the remaining samples were stored in the refrigerator at -80°C.
Cell transfection
miR-200a-3p mimeics (5’-UAACACUGUCUGGUAACGAUGU-3’), anti-miR-200a-3p and miR-NC (5’-UUCUCCGAACGUGUCACUGUU-3’) were constructed. si-RIPK1 (5’-GAAUGAGGCUUACAACAGTT-3’) and si-NC (5’-AAUUCUCCGAACGUUCACGU-3’) were constructed to reduce the expression of RIPK1. In addition, pcDNA3.1-RIPK1 overexpression plasmid and control (pcDNA3.1) were constructed. All primer sequences were synthesized by GenePharma. Transfection was carried out using Lipofectamine 3000 (Invitrogen, USA).
qRT-PCR detection
Total RNA was obtained using TRIzol reagent (Ambion, USA). Reverse transcription was performed with PrimeScriptTM RT kit (Takara, Dalian, China), and cDNA was collected for amplification. PCR amplification was performed with PowerUpTM SYBRTM Green Master Mix and ABI 7500 (ABI, Foster, CA, USA). PCR reaction system was as follows: 5× PrimeScript® Buffer 4 μl , PrimeScript® RT Enzyme Mix I 1 μl , RT Primer Mix 1 μl , Genome Removal Solution 10 μl. RNase-free ddH2O was used to complete to 20 μL. PCR reaction conditions were as follows: pre-denaturation at 95°C for 5 min, denaturation at 95°C for 10 s, annealing at 55°C for 30 s and extension at 72°C for 34 s. U6 and GAPDH were internal references of miR and RIPK1, and 2-ΔΔCt was applied to test mRNA [22]. Primer sequences are shown in Table 1.
Table 1.
Primer sequences
| Gene | Upstream sequence (5’-3’) | Downstream sequence (5’-3’) |
|---|---|---|
| miR-200a-3p | TAACACTGTCTGGTAACGATGT | CATCTTACCGGACAGTGCTGGA |
| RIPK1 | CCAGCCTCAGCATAGCACCT | ACCCCAAGGGGAGCCATAAC |
| U6 | CTCGCTTCGGCAGCACA | AACGCTTCACGAATTTGCGT |
| GAPDH | GCTTCGGCAGCACATATACTAAAAT | CGCTTCACGAATTTGCGTGTCAT |
Hematoxylin and eosin (H&E) dyeing
The collected intestinal tissues were fixed with 10% neutral formaldehyde, embedded in paraffin, and cut into 3 μm sections. H&E staining was performed to observe the changes of intestinal tissue in mice. Ten most representative visual fields were randomly selected and observed under a high-power microscope (×400) to select representative pictures. The degree of intestinal injury was determined using the score of intestinal histopathology in mice, and a score more than 2 points was considered NEC. The classification was as follows: 0: normal, no damage; 1: slight submucosal and/or lamina propria separation; 2: moderate separation of submucosa and/or lamina propria and/or edema in submucosa and muscular layer; 3: severe separation of submucosa and/or lamina propria and/or severe edema of submucosa and muscular layer, and shedding of regional villi; 4: loss and necrosis of villi.
Evaluation of inflammatory cytokines
IL-6, IL-1β and TNF-α (Mlbio, China) in cells and intestinal tissues were tested using ELISA. The method was as follows: The tissues were added into appropriate amount of normal saline, mashed, and centrifuged at 1000×g for 10 minutes, and then the supernatant was obtained. After dilution, 50 μl of the standard was added to the reaction well, and 50 μl of the sample to be tested was added to the reaction well. The biotin-labeled antibody (50 μl) was added. The plates were sealed, and then they were mixed and incubated at 37°C for 1 h. The plates were washed, and 80 ul of streptavidin-HRP was added to each well, mixed, and incubated at 37°C for 30 min. The plates were washed, and 50 μl of substrate A and B were added to each well, shaken and mixed gently, and incubated at 37°C for 10 min out of light. The enzyme-labeling plate was taken out, and 50 μl of the termination solution was added immediately. Then, the results were determined immediately. The OD value of each well was measured with a microplate reader at 450 nm.
Evaluation of cell activity
MTT (3-(4,5)-Dimethylthiahiazo (-z-y1)-3, 5-di- phenytetrazoliumromide) (Abcam, USA) was applied to test the cell activity. The transfected cells were collected, adjusted to 6*104/cell, inoculated into 96-well plate, and cultivated for 24 h. Then 20 μL MTT solution (5 mg/mL) was added and set for 4 h. 150 μl of DMSO was added to accelerate the dissolution. Micro-plate reader (BioTek, Winooski, VT, USA) was applied to test the absorbance at 570 nm.
Evaluation of apoptosis
The apoptosis was tested using flow cytometry (BD Biosciences, Heidelberg, Germany) and TUNEL dyeing (Shanghai Yeasen BioTechnologies Co., Ltd., China, Alexa Fluor 640). Annexin V-PI kit (Invitrogen, Life Technologies, USA) was applied to test the apoptosis of IEC-6 cells. The cells were washed with PBS and re-suspended in binding buffer. A 5 μL of Annexin V-FITC and 5 μL of PI were added to cell suspension (400 μL) and cultivated for 30 min at 4°C in the dark. The apoptosis was monitored by FACSCalibur flow cytometry (BD Biosciences) and detected by FlooJo7.6. The apoptosis of NEC tissues was evaluated using DeadEnd TM fluorescent TUNEL kit. The operation steps were carried out according to the research of Chen et al. [21].
WB detection
Transfected cells and mouse tissues were collected, the protein concentration was tested using BCA protein kit, and the separated protein samples were loaded into SDS-PAGE 12% gel with the same protein concentration, and then electrophoresed on Bio-Rad Mini-PROTEAN 3. Subsequently, the samples were transferred with nitrocellulose membrane and sealed at room temperature (PBS containing 0.05% Tween and 5% skimmed milk powder) for 1 hour. Primary antibodies RIPK1 (1:500, Abcam, USA) and GAPDH (1:1000, Abcam, USA) were added and cultivated at 4°C overnight, washed with PBS-tween, and added with secondary antibodies for 1 h. ECL (Amersham Biosciences Corp., Piscataway, NJ) was applied to test the protein bands, and Quantity One was applied for quantification.
Double luciferase reporter
The three major untranslated regions (3’-UTR) of RIPK1-wt and RIPK1-Mut of RIPK1 were cloned and inserted into the downstream of luc2 firefly luciferase of pmiRGLO vector, and then co-transfected with miR-200a-3p-mimics into IEC-6. After 48-hour transfection, the cells were lysed and luciferase activity was tested using Dual Luciferase Assay Kit (Stratagene, La Jolla, CA, USA). Luciferase activities of fireflies and sea renilla were tested using micro-plate reader, which was standardized by sea renilla luciferase.
Statistical analysis
All data were represented as Mean ± SD. Prism package was applied for statistical analysis and figure visualizing. The differences between the two groups were compared by Student t-test, and the multiple groups comparison was conducted by ANOVA, and then Tukey back testing was conducted. If P<0.05, the difference was considered significant.
Result
miR-200a-3p decreased in NEC
The differentially expressed miRs of NEC in GSE68054 were first tested, and miR-200a-3p was found to be downregulated (Figure 1A, 1B). Then, in order to verify miR-200a-3p in NEC, we found that the expression of miR-200a-3p in NEC children’s serum was significantly lower than that in the control group by qRT-PCR (Figure 1C, P<0.001). In addition, miR-200a-3p in IEC-6 cells and NEC animal models induced by LPS also decreased (Figure 1D, 1E, P<0.01), suggesting that miR-200a-3p may be involved in NEC.
Figure 1.

Relative expression of miR-200a-3p in NEC. A. Limma analysis of differential miR volcano map in GSE68054 (n=6). B. Pheatmap analysis of differential miR volcano map in GSE68054 (n=6). C. qRT-PCR was applied to test miR-200a-3p in serum of NEC children (n=24). D. qRT-PCR was applied to test miR-200a-3p after IEC-6 was induced by LPS (n=6). E. qRT-PCR was applied to test miR-200a-3p in intestinal tissue of NEC animal model (n=12). **indicates P<0.01; ***indicates P<0.001.
Increase of miR-200a-3p promoted the activity of IEC-6 cells and inhibited apoptosis and inflammation
To investigate the role of miR-200a-3p in NEC, we constructed miR-200a-3p-mimics plasmid and transferred it into LPS-induced IEC-6 (Figure 2A, 2B, P<0.01). MTT assay revealed that the activity of IEC-6 cells transfected with miR-200a-3p-mimics was significantly higher than that of miR-NC (Figure 2C, P<0.01), and the apoptosis was inhibited (Figure 2D, P<0.01). In addition, ELISA revealed that the concentration levels of IL-6, IL-1β and TNF-α in IEC-6 cells after transfection of miR-200a-3p-mimics were significantly lower than those in miR-NC (Figure 2E-G, P<0.01), indicating that miR-200a-3p could effectively inhibit LPS-induced IEC-6 apoptosis and inflammatory reaction.
Figure 2.
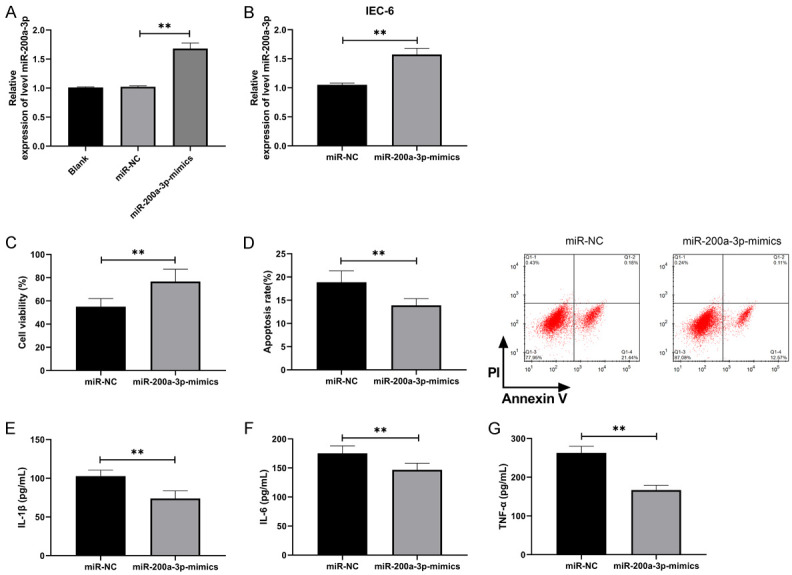
miR-200a-3p inhibited LPS-induced apoptosis and inflammation of IEC-6 and promoted cell growth. A. qRT-PCR was applied to test miR-200a-3p in the plasmid after constructing miR-200a-3p-mimics (n=9). B. qRT-PCR was applied to test miR-200a-3p in IEC-6 cells after transfection of miR-200a-3p-mimics (n=6). C. MTT assay was applied to test the activity of IEC-6 cells induced by LPS after transfection of miR-200a-3p-mimics (n=6). D. Flow cytometry was applied to test the change of apoptosis rate of IEC-6 induced by LPS after transfection of miR-200a-3p-mimics (n=6). E. ELISA was applied to test the concentration level of IL-1β in IEC-6 induced by LPS after transfection of miR-200a-3p-mimics (n=6). F. ELISA was applied to test IL-6 concentration in IEC-6 induced by LPS after transfection of miR-200a-3p-mimics (n=6). G. ELISA was applied to test TNF-α concentration in IEC-6 induced by LPS after transfection of miR-200a-3p-mimics (n=6). **indicates P<0.01.
Increase of miR-200a-3p improved intestinal tissue damage and inflammatory response in NEC mice
miR-200a-3p expression in NEC in vitro model was tested and NEC mouse model was constructed. Through H&E staining, we found that we successfully constructed the NEC model. Compared with the control group, the pathologic changes of mice in the NEC and miR-NC+NEC groups showed severely necrotic intestinal tissue with increased number of inflammatory cells, but the intestinal tissue damage in miR-200a-3p-mimics+NEC group improved and the number of inflammatory cells declined compared with the NEC group and miR-NC+NEC group (Figure 3A). In addition, the NEC scores of miR-200a-3p-mimics+NEC group were significantly lower than those of NEC group and miR-NC+NEC group, indicating that miR-200a-3p-mimics could improve intestinal tissue injury (Figure 3B, P<0.05). TUNEL detection also suggested that most of the nuclei in the NEC and miR-NC+NEC groups were red and strongly positive, but were faint in the miR-200a-3p-mimics+NEC group (Figure 3C). In addition, the changes of inflammatory factors concentration in the intestinal tissue of NEC mice were tested. The levels of IL-6, IL-1β, and TNF-α inflammatory factors in other groups were increased significantly compared with the control group, but those in miR-200a-3p-mimics+NEC group were lower compared with NEC group and miR-NC+NEC group (Figure 3D-F, P<0.05), indicating that miR-200a-3p could improve intestinal tissue damage and inflammatory reaction in NEC mice.
Figure 3.

miR-200a-3p improved intestinal tissue damage and inflammation in NEC mice. A. H&E stain confirmed damage of miR-200a-3p-mimics to intestinal tissue in NEC mice (n=5). B. Evaluation of animal model after intervention with miR-200a-3p-mimics by NEC score (n=20). C. TUNEL was applied to test intestinal apoptosis of NEC mice after miR-200a-3p-mimic intervention (n=5). D. ELISA was applied to test the concentration level of TNF-α in intestinal tissue of NEC mice after intervention with miR-200a-3p-mimics (n=5). E. ELISA was applied to test IL-1β concentration in intestinal tissue of NEC mice after intervention with miR-200a-3p-mimics (n=5). F. ELISA was applied to test IL-6 concentration in intestinal tissue of NEC mice after miR-200a-3p-mimic intervention (n=5). *indicates P<0.05; **indicates P<0.01; ***indicates P<0.001.
miR-200a-3p regulated RIPK1 in a targeted manner
In order to further determine the potential mechanism of miR-200a-3p, we predicted the miR downstream of miR-200a-3p. Through analysis, we found that there was a targeted binding site between RIPK1 and miR-200a-3p. To verify the binding between them, we found that the fluorescence activity of RIPK1-WT was inhibited by miR-200a-3p-mimics through double luciferase reporter detection (Figure 4A, 4B, P<0.05). Furthermore, qRT-PCR detection revealed that RIPK1 in NEC children’s serum was higher than that in the control group (Figure 4C, P<0.001), and correlation analysis suggested that there was a negative correlation of miR-200a-3p with RIPK1 (Figure 4D). In addition, WB detection also revealed that RIPK1 protein in IEC-6 cells induced by LPS and NEC animal model was significantly elevated, but RIPK1 protein in tumor tissues of NEC mice was inhibited after miR-200a-3p-mimic intervention (Figure 4E, P<0.05), suggesting that miR-200a-3p could target RIPK1.
Figure 4.

miR-200a-3p regulated RIPK1 in a targeted manner. A. Target binding site of miR-200a-3p and RIPK1 (n=12). B. Double luciferase reporter analysis of miR-200a-3p and RIPK1 target binding (n=24). C. qRT-PCR was applied to test RIPK1 in serum of children with NEC (n=24). D. Pearson test was applied to test the correlation between RIPK1 and miR-200a-3p (n=6). E, F. RIPK1 protein in NEC models in vivo and in vitro was tested using WB (n=12). *indicates P<0.05; ***indicates P<0.001.
Increase in RIPK1 reversed the growth function and inflammatory response of miR-200a-3p to IEC-6
At the end of the study, we constructed RIPK1 overexpression vector to determine the relationship between miR-200a-3p and RIPK1, and then transfected pcDNA3.1-RIPK1 into IEC-6 cells to further observe cell function (Figure 5A, 5B, P<0.01). Compared with pcDNA-3.1-NC, IEC-6 cells transfected with pcDNA3.1-RIPK1 were significantly inhibited in viability and had more apoptosis (Figure 5C, 5D, P<0.01). Furthermore, it was found that the concentration levels of IL-6, IL-1β, and TNF-α inflammatory factors in IEC-6 cells transfected with pcDNA3.1-RIPK1 were significantly elevated compared with pcDNA-3.1-NC. However, the above results were reversed when miR-200a-3p-mimics and pcDNA3.1-RIPK1 were co-transfected into IEC-6 cells (Figure 5E-G, P<0.01). Our results suggested that miR-200a-3p participated in NEC development by regulating RIPK1, which can be used as a therapeutic target.
Figure 5.

miR-200a-3p regulated RIPK1 to promote NEC cell activity and inhibit inflammatory reaction. A. qRT-PCR was applied to test RIPK1 after constructing pcDNA3.1-RIPK1 vector (n=12). B. qRT-PCR was applied to test RIPK1 after pcDNA3.1-RIPK1 was transfected into IEC-6 (n=6). C. MTT cells were applied to test the activity changes in IEC-6 after co-transfection (n=12). D. Flow cytometry was applied to test the change in apoptosis of IEC-6 after co-transfection (n=12). E. ELISA was applied to test the concentration level of IL-1β in IEC-6 after co-transfection (n=12). F. ELISA was applied to test the concentration level of IL-6 in IEC-6 after co-transfection (n=12). G. Detection of TNF-α concentration level in IEC-6 after co-transfection by ELISA. **indicates P<0.01 (n=12).
Discussion
NEC, a common neonatal acute necrotizing enteritis, has no cure in the clinic at present. Moreover, the mechanism of NEC is still under exploration. In this research, we found that miR-200a-3p in NEC was significantlylower by differential analysis of chips and qRT-PCR detection. After up-regulating miR-200a-3p, RIPK1 was inhibited, cell activity was improved, and the inflammatory reaction was inhibited, so it is a possible target for NEC.
Studies have shown that [23,24] miR regulates innate immunity, tumor formation, proliferation, and differentiation, and inflammation in humans. It is reported that miR participates in intestinal physiologic and pathologic processes by regulating the intestinal immune or inflammatory response [25,26]. In this study, we first screened out the differentially expressed miRs of NEC through the GEO database. As an early discovered miR, miR-200a-3p plays a regulatory role in the development of many diseases. For example, miR-200a and miR-200b inhibit inflammation by targeting ORMDL3 to regulate ERK/MMP-9 in asthma [27]. In addition, Xue et al. [28] revealed that miR-200-3p inhibits the proliferation of diabetic retinopathy cells and reduced apoptosis by blocking TGF-β2/Smad. In this study, miR-200a-3p in serum of children with NEC and in vitro cell models of NEC was significantly decreased, and miR-200a-3p overexpression could inhibit the occurrence of an inflammatory reaction in NEC cells and mice and increase cell activity.
RIPK1 is a member of serine/threonine protein kinase receptor interacting protein (RIP) family [29]. The protein encoded by this gene acts in inflammation and cell death in response to tissue injury and pathogen recognition, and it is a part of developmental regulation [30]. RIPK1 antagonizes necrotic sagging mediated by ZBP1 and inhibits inflammation [31]. RIPK1 and RIPK3 kinases promote death-independent inflammation through Toll-like receptor 4. However, recent studies have shown that RIPK1 is regulated by miR-141-3p to improve NEC [32,33]. In our research, online prediction analysis revealed that there was a targeted aggregation site between miR-200a-3p and RIPK1, and RIPK1 was highly expressed in serum of children with NEC . This was negatively correlated with miR-200a-3p, suggesting that there may be a targeted relationship between RIPK1 and miR-200a-3p, which was further verified by double luciferase reporter analysis. Further, we found that miR-200a-3p-mimics effectively inhibited RIPK1 protein in IEC-6 cells and NEC animal models induced by LPS. Therefore, miR-200a-3p could regulate RIPK1. At the end of the study, in order to verify the role of miR-200a-3p on NEC by adjusting RIPK1, a rescue experiment was carried out in an in vitro cell model. It was found that miR-200a-3p-mimics could reverse the inhibitory effect of pcDNA3.1-RIPK1 on IEC-6 cells, reduce apoptosis, and alleviate inflammatory reaction, making it a possible target for NEC.
Although this study has revealed the mechanism of miR-200a-3p in NEC, there are still some shortcomings. First, we only used one kind of cell line for in vitro study, and it is unclear whether this is specific. Secondly, the number of children with NEC was small, and the correlation analysis results may be biased. Finally, it is not clear whether low miR-200a-3p was caused by other factors. Therefore, we hope to collect more samples, supplement the inclusion and exclusion criteria, increase more experimental samples, and improve our research conclusions in the follow-up exploration.
In sum, miR-200a-3p has low expression in NEC and can reduce the development of NEC by regulating RIPK1, which may be a target in NEC.
Disclosure of conflict of interest
None.
References
- 1.Rich BS, Dolgin SE. Necrotizing enterocolitis. Pediatr Rev. 2017;38:552–559. doi: 10.1542/pir.2017-0002. [DOI] [PubMed] [Google Scholar]
- 2.Wu YZ, Chan KYY, Leung KT, Lam HS, Tam YH, Lee KH, Li K, Ng PC. Dysregulation of miR-431 and target gene FOXA1 in intestinal tissues of infants with necrotizing enterocolitis. FASEB J. 2019;33:5143–5152. doi: 10.1096/fj.201801470R. [DOI] [PubMed] [Google Scholar]
- 3.Cotten CM. Modifiable risk factors in necrotizing enterocolitis. Clin Perinatol. 2019;46:129–143. doi: 10.1016/j.clp.2018.10.007. [DOI] [PubMed] [Google Scholar]
- 4.Samuels N, van de Graaf RA, de Jonge RCJ, Reiss IKM, Vermeulen MJ. Risk factors for necrotizing enterocolitis in neonates: a systematic review of prognostic studies. BMC Pediatr. 2017;17:105. doi: 10.1186/s12887-017-0847-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bazacliu C, Neu J. Necrotizing enterocolitis: long term complications. Curr Pediatr Rev. 2019;15:115–124. doi: 10.2174/1573396315666190312093119. [DOI] [PubMed] [Google Scholar]
- 6.Hackam DJ, Sodhi CP, Good M. New insights into necrotizing enterocolitis: from laboratory observation to personalized prevention and treatment. J Pediatr Surg. 2019;54:398–404. doi: 10.1016/j.jpedsurg.2018.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Qian T, Zhang R, Zhu L, Shi P, Yang J, Liu Y, Yu JL, Zhou XG, Yang Y, Qiu YP, Liu L, Wei QF, Xu FL, Li YF, Chen C. Analysis of clinical characteristics of necrotizing enterocolitis in term infants. Zhonghua Yi Xue Za Zhi. 2016;96:1766–1772. doi: 10.3760/cma.j.issn.0376-2491.2016.22.012. [DOI] [PubMed] [Google Scholar]
- 8.Li X, Chang Y, Ding Z, Guo Z, Mehta JL, Wang X. Functions of MicroRNAs in angiogenesis. Biochemical basis and therapeutic implications of angiogenesis. Springer; 2017. pp. 133–155. [Google Scholar]
- 9.Ritchie W. microRNA target prediction. Methods Mol Biol. 2017;1513:193–200. doi: 10.1007/978-1-4939-6539-7_13. [DOI] [PubMed] [Google Scholar]
- 10.Lu TX, Rothenberg ME. MicroRNA. J Allergy Clin Immunol. 2018;141:1202–1207. doi: 10.1016/j.jaci.2017.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cao RY, Li Q, Miao Y, Zhang Y, Yuan W, Fan L, Liu G, Mi Q, Yang J. The emerging role of MicroRNA-155 in cardiovascular diseases. Biomed Res Int. 2016;2016:9869208. doi: 10.1155/2016/9869208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mohr AM, Mott JL. Overview of microRNA biology. Semin Liver Dis. 2015;35:3–11. doi: 10.1055/s-0034-1397344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Krol J, Loedige I, Filipowicz W. The widespread regulation of microRNA biogenesis, function and decay. Nat Rev Genet. 2010;11:597–610. doi: 10.1038/nrg2843. [DOI] [PubMed] [Google Scholar]
- 14.Yin Y, Qin Z, Xu X, Liu X, Zou H, Wu X, Cao J. Inhibition of miR-124 improves neonatal necrotizing enterocolitis via an MYPT1 and TLR9 signal regulation mechanism. J Cell Physiol. 2019;234:10218–10224. doi: 10.1002/jcp.27691. [DOI] [PubMed] [Google Scholar]
- 15.Degterev A, Boyce M, Yuan J. A decade of caspases. Oncogene. 2003;22:8543–8567. doi: 10.1038/sj.onc.1207107. [DOI] [PubMed] [Google Scholar]
- 16.Degterev A, Ofengeim D, Yuan J. Targeting RIPK1 for the treatment of human diseases. Proc Natl Acad Sci U S A. 2019;116:9714–9722. doi: 10.1073/pnas.1901179116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lin J, Kumari S, Kim C, Van TM, Wachsmuth L, Polykratis A, Pasparakis M. RIPK1 counteracts ZBP1-mediated necroptosis to inhibit inflammation. Nature. 2016;540:124–128. doi: 10.1038/nature20558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Newton K. RIPK1 and RIPK3: critical regulators of inflammation and cell death. Trends Cell Biol. 2015;25:347–353. doi: 10.1016/j.tcb.2015.01.001. [DOI] [PubMed] [Google Scholar]
- 19.Ling X, Linglong P, Weixia D, Hong W. Protective Effects of bifidobacterium on intestinal barrier function in LPS-induced enterocyte barrier injury of Caco-2 monolayers and in a rat NEC model. PLoS One. 2016;11:e0161635. doi: 10.1371/journal.pone.0161635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yu RQ, Wang M, Jiang SY, Zhang YH, Zhou XY, Zhou Q. Small RNA sequencing reveals differentially expressed miRNAs in necrotizing enterocolitis in rats. Biomed Res Int. 2020;2020:5150869. doi: 10.1155/2020/5150869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen H, Zeng L, Zheng W, Li X, Lin B. Increased expression of microRNA-141-3p improves necrotizing enterocolitis of neonates through targeting MNX1. Front Pediatr. 2020;8:385. doi: 10.3389/fped.2020.00385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 23.Lin YH. MicroRNA networks modulate oxidative stress in cancer. Int J Mol Sci. 2019;20:4497. doi: 10.3390/ijms20184497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yuan Y, Weidhaas JB. Functional microRNA binding site variants. Mol Oncol. 2019;13:4–8. doi: 10.1002/1878-0261.12421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Qian WH, Liu YY, Li X, Pan Y. MicroRNA-141 ameliorates alcoholic hepatitisinduced intestinal injury and intestinal endotoxemia partially via a TLR4-dependent mechanism. Int J Mol Med. 2019;44:569–581. doi: 10.3892/ijmm.2019.4221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lakhan R, Subramanian VS, Said HM. Role of MicroRNA-423-5p in posttranscriptional regulation of the intestinal riboflavin transporter-3. Am J Physiol Gastrointest Liver Physiol. 2017;313:G589–G598. doi: 10.1152/ajpgi.00238.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Duan XJ, Zhang X, Li LR, Zhang JY, Chen YP. MiR-200a and miR-200b restrain inflammation by targeting ORMDL3 to regulate the ERK/MMP-9 pathway in asthma. Exp Lung Res. 2020;46:321–331. doi: 10.1080/01902148.2020.1778816. [DOI] [PubMed] [Google Scholar]
- 28.Xue L, Xiong C, Li J, Ren Y, Zhang L, Jiao K, Chen C, Ding P. miR-200-3p suppresses cell proliferation and reduces apoptosis in diabetic retinopathy via blocking the TGF-beta2/Smad pathway. Biosci Rep. 2020;40:BSR20201545. doi: 10.1042/BSR20201545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Degterev A, Ofengeim D, Yuan J. Targeting RIPK1 for the treatment of human diseases. Proc Natl Acad Sci U S A. 2019;116:9714–9722. doi: 10.1073/pnas.1901179116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yuan J, Amin P, Ofengeim D. Necroptosis and RIPK1-mediated neuroinflammation in CNS diseases. Nat Rev Neurosci. 2019;20:19–33. doi: 10.1038/s41583-018-0093-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lin J, Kumari S, Kim C, Van TM, Wachsmuth L, Polykratis A, Pasparakis M. RIPK1 counteracts ZBP1-mediated necroptosis to inhibit inflammation. Nature. 2016;540:124–128. doi: 10.1038/nature20558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Najjar M, Saleh D, Zelic M, Nogusa S, Shah S, Tai A, Finger JN, Polykratis A, Gough PJ, Bertin J, Whalen M, Pasparakis M, Balachandran S, Kelliher M, Poltorak A, Degterev A. RIPK1 and RIPK3 kinases promote cell-death-independent inflammation by toll-like receptor 4. Immunity. 2016;45:46–59. doi: 10.1016/j.immuni.2016.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li X, Wang Y, Wang Y, He X. MiR-141-3p ameliorates RIPK1-mediated necroptosis of intestinal epithelial cells in necrotizing enterocolitis. Aging (Albany NY) 2020;12:18073–18083. doi: 10.18632/aging.103608. [DOI] [PMC free article] [PubMed] [Google Scholar]