

Abstract
Background
Glioblastoma (GBM) is the most common primary brain tumor in adults with a median survival of approximately 15 months; therefore, more effective treatment options for GBM are required. To identify new drugs targeting GBMs, we performed a high-throughput drug screen using patient-derived neurospheres cultured to preferentially retain their glioblastoma stem cell (GSC) phenotype.
Methods
High-throughput drug screening was performed on GSCs followed by a dose-response assay of the 5 identified original “hits.” A PI3K/mTOR dependency to a proteasome inhibitor (carfilzomib), was confirmed by genetic and pharmacologic experiments. Proteasome Inhibition Response Signatures were derived from proteomic and bioinformatic analysis. Molecular mechanism of action was determined using three-dimensional (3D) GBM-organoids and preclinical orthotopic models.
Results
We found that GSCs were highly sensitive to proteasome inhibition due to an underlying dependency on an increased protein synthesis rate, and loss of autophagy, associated with PTEN loss and activation of the PI3K/mTOR pathway. In contrast, combinatory inhibition of autophagy and the proteasome resulted in enhanced cytotoxicity specifically in GSCs that did express PTEN. Finally, proteasome inhibition specifically increased cell death markers in 3D GBM-organoids, suppressed tumor growth, and increased survival of mice orthotopically engrafted with GSCs. As perturbations of the PI3K/mTOR pathway occur in nearly 50% of GBMs, these findings suggest that a significant fraction of these tumors could be vulnerable to proteasome inhibition.
Conclusions
Proteasome inhibition is a potential synthetic lethal therapeutic strategy for GBM with proteasome addiction due to a high protein synthesis rate and autophagy deficiency.
Keywords: glioblastoma, neurospheres, organoids, proteasome, PTEN
Key Points.
Glioblastomas with hyper-activation of the PI3K/mTOR pathway are vulnerable to proteasome inhibition.
Autophagy inhibition enhances proteasome inhibition therapy in glioblastomas.
Dual inhibition of proteasome and autophagy is a new synthetic lethal therapeutic strategy for brain cancers.
Importance of the Study.
Alterations in RTK signaling and downstream PI3K/mTOR pathways are observed in a majority of GBMs, and here we show that these events result in a metabolic dependence on increased protein synthesis. Disruption of proteostasis, by pharmacological inhibition of the proteasome and autophagy, results in a synthetic lethality in GBM preclinical models, potentially identifying a new therapeutic avenue for treating GBM patients.
Glioblastoma (GBM) is the most lethal type of cancer that originates in the brain. The average survival for patients with GBM ranges from 12 to 16 months even after surgery followed by radiotherapy plus concomitant and adjuvant temozolomide treatment.1 Genome-wide profiling studies have identified important genetic events in human GBM including dysregulation of the phosphatidylinositol 3-kinase signaling (PI3K/AKT) and mammalian target of rapamycin (mTOR) pathways, which are also affected in other types of cancer.2 Indeed the primary negative regulator of the PI3K/AKT/mTOR pathway, the tumor suppressor gene PTEN (encoding the protein phosphatase and tensin homolog), is frequently deleted or inactivated in GBM.3,4
The PI3K/AKT and mTOR pathways control tumorigenesis in part by upregulating anabolic pathways, such as protein synthesis, and suppressing protein catabolism; while PTEN negatively regulates these features. Protein synthesis is regulated by mTOR through the phosphorylation of the p70S6 kinase 1 (S6K1) and the eIF4E-binding protein (4EBP) which are hyperactivated and inhibited, respectively, in the context of PTEN loss.2,5 Additionally, PI3K/mTOR activation contributes to malignancy by suppressing autophagy induction6 and by controlling proteasome activity2,7; in contrast, PTEN positively regulates catabolism in different cancer types.8 Numerous drugs have been developed targeting different nodes of the PI3K/AKT/mTOR pathway that regulate protein homeostasis and metabolism in cancer.9 Alternatively, cellular functions activated or inhibited by the PI3K/mTOR pathway, such as protein turnover, can be vulnerable targets in cancers.5 Indeed, proteasome inhibitors show great efficacy as single agents and in combination with other chemotherapeutic agents in hematological cancers.10 Additionally, proteasome inhibition has been proposed as a therapeutic opportunity for solid tumors, including brain tumors, where variable effects on proliferation, growth arrest, invasion, and apoptosis have been observed using cell lines.11,12 Herein, we uncover proteasome inhibition as a targeted therapy for GBMs by performing a drug screen in glioblastoma stem cells (GSCs). Proteasome inhibition specifically induced cell death in three-dimensional (3D) PTEN-deficient GBM-organoids and suppressed orthotopic tumor growth of PTEN-null GSCs in mice. This study reveals that proteasome inhibition is a potential synthetic lethal therapeutic strategy for brain cancers with proteasome addiction.
Materials and Methods
Cell Culture
GSCs were kindly provided by Dr. Cameron Brennan, MSKCC; Dr. Harley Kornblum, UCLA; Dr. Frederick Lang, MD Anderson; Dr. David James, Northwestern University; and Dr. Paul Mischel, Ludwig Institute. Detailed description of cell culture methods is presented in the Supplementary Methods.
Antibodies and Reagents
Description of antibodies, drugs, and kits is presented in the Supplementary Methods.
Drug Screening
GSCs were dissociated and plated in 384-well tissue culture plates at 2500 cells per well in 25 μL of medium and 3D neurospheres allowed to form for 72 hours. 25 nL of 1000× drug stock solution were then added using a Labcyte Echo acoustic dispensing device and the cells were incubated for 96 hours. Cell viability was assessed using CellTiterGlo (Promega) on an Envision plate reader. Detailed description of the drug screen is presented in the Supplementary Methods.
Proteomics Analysis
TS576 (PTEN-WT) and TS576-1A3 (PTEN-KO) GSCs were used for proteomic analysis. Sample preparation, LC-MS/MS analysis, and data analysis are described in the Supplementary Methods. Four comparisons were conducted, PTEN-WT vs PTEN-null before and after proteasome inhibition (placebo and drug), and Drug-treated vs Placebo for both phenotypes PTEN-WT and PTEN-null. Data have been deposited in ProteomeXchange PXD022934.
RNA-Sequencing
RNA was isolated from GSCs using RNeasy plus kit (Qiagen) and quantified with a Nanodrop (Thermo Fisher Scientific). Isolated RNA that passed quality control was prepared into libraries for sequencing on the Illumina HiSeq platform at 150 bp length paired-end. Raw reads with at least 20 million reads per sample were filtered based on sequencing quality and aligned with STAR13 to GRCh38 reference. Quantification was performed against Ensembl 88 transcriptome using RSEM.14 Data have been deposited in NCBI, GSE163906.
Bioinformatic Analysis
Correlative analyses between drug response and altered expression of molecular pathways can be found in the Supplementary Methods.
Cell Viability Assay
About 10 000 cells seeded per well in 96-well plates were incubated for 48 hours, treated with compounds, and assessed for cell viability 48- or 72-hour post-treatment by ATPlite assay (Perkin-Elmer). Detailed description of the assay is found in the Supplementary Methods.
Proteasome and Protein Activity Assays
20S proteasome activity and protein synthesis were determined according to the manufacturer’s instructions (Cayman Chemical).
Genetic Engineering of Cells
CRISPR/Cas9-edited clones were generated as described previously.15 Detailed description of the assay is found in the Supplementary Methods.
Intracranial Xenograft Tumor Model
Orthotopic models were conducted as previously described16 and detailed in the Supplementary Methods. Animal research experiments were conducted under the regulation of the UCSD Animal Care Program, #S00192M.
GBM-Organoids
GBM-organoids were established using a previously reported protocol17 and cerebral organoid kit (Stemcell Technologies). Detailed description of the assay is found in the Supplementary Methods.
Immunofluorescence Microscopy
GBM-organoids were fixed with 10% formalin (Sigma-Aldrich), embedded in OCT (Fisher Healthcare), and processed for staining and microscopy as detailed in the Supplementary Methods.
Statistical Analysis
Data sets were analyzed by unpaired t-test or multiple comparisons one-way ANOVA or two-way ANOVA according to the experiment using GraphPad Prism software. *P < .05, **P < .01, ***P < .001, and ****P < .0001. Kaplan-Meier curves and comparison of survival were analyzed using the log-rank (Mantel-Cox) test.
Results
Identification of PTEN-Null Specific, Synthetic Lethal, Therapeutics
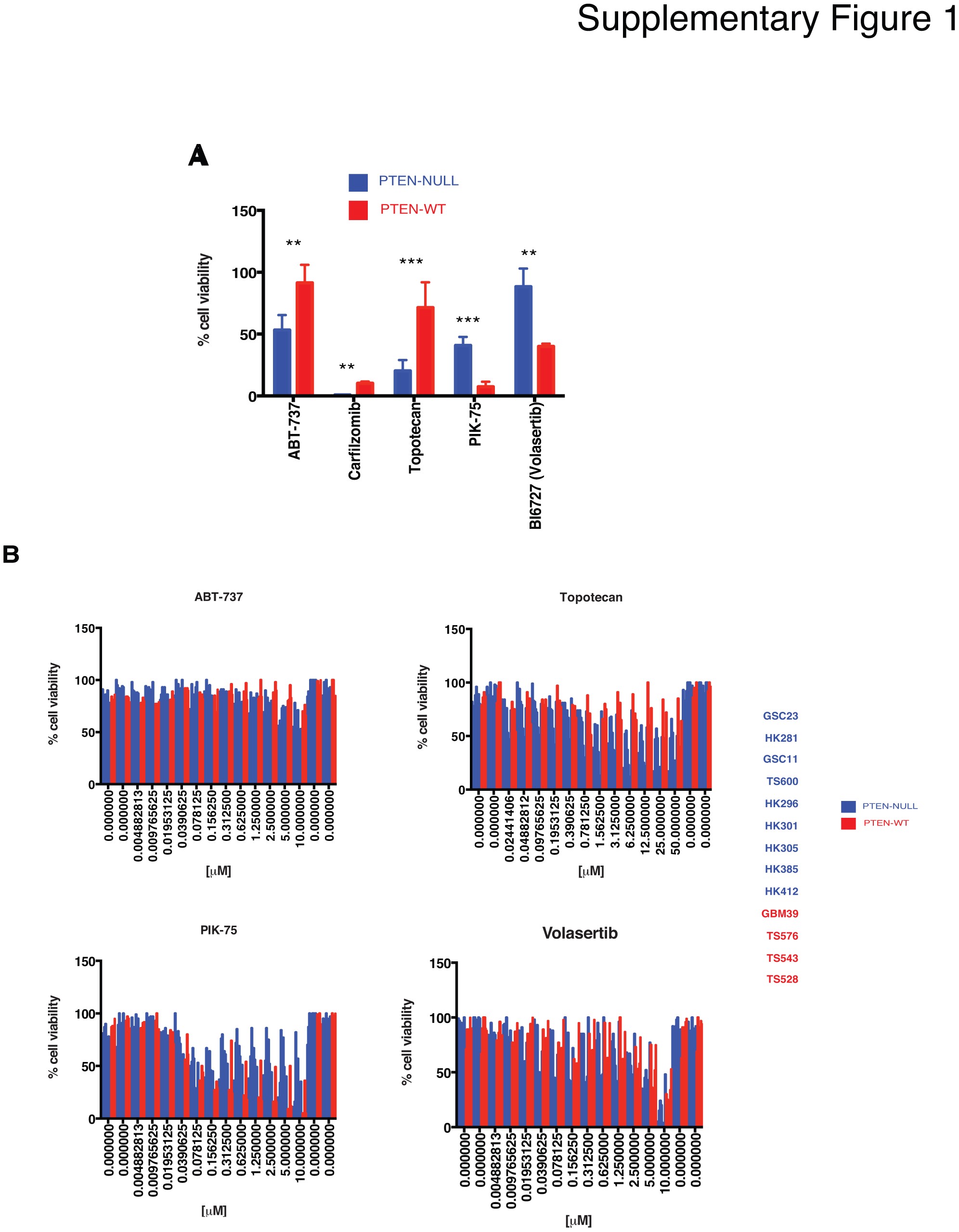
To identify novel drugs that specifically target glioblastoma stem cells (GSCs), high-throughput drug screening was performed using the 320 compound Informer Set collection developed as part of the NCI Cancer Target Discovery and Development2 Consortium (https://ocg.cancer.gov/programs/ctd2). The Informer Set, containing 80 FDA-approved oncology drugs and 240 tool compounds, was screened at two concentrations (10 µM and 1 µM) in 5 individual GSCs (3 PTEN-null and 2 PTEN-WT) as described in the Supplementary Methods. This was followed by a second, confirmatory dose-response assay (12 points), of 5 identified hits (ABT-737, carfilzomib [CFZ], topotecan, PIK-75, and volasertib) in a total of 15 distinct patient-derived GSCs (Fig. 1a, Supplementary Table 1). In follow-up drug dose-response curve (DRC) studies, only one drug, the proteasome inhibitor CFZ, showed specificity for PTEN-null cells as compared to PTEN-expressing GSCs (Fig. 1b and c, Supplementary Fig. 1). Proteasome inhibition sensitivity (IC50) ranged from 0.1 nM up to 1400 nM in PTEN-null cells. In contrast, drug sensitivity was correlated with total PTEN protein levels and AKT/mTOR activation in PTEN-WT cells (Fig. 1d and e). For example, TS543, with the lowest PTEN expression level, was the most sensitive cells (IC50 = 0.37 nM), while GBM39, with highest PTEN expression, was the most resistant patient sample to proteasome inhibition (IC50 = 32.48 μM, Fig. 1d, P = .0215). These data suggest that GBM sensitivity to proteasome inhibition inversely correlates with PTEN protein levels.
Fig. 1.

High-throughput drug screening of glioblastoma stem cells (GSCs). A, 320 compounds were tested against five GSCs (two GSCs expressing PTEN and three GSCs with PTEN deletion); five compounds (H2L, hits to lead) were selected based on their specificity to decrease cell viability in PTEN-null GSCs relative to PTEN-expressing samples and drug dose-response curves (DRC) were performed on 15 GSCs. B, Bar plot of proteasome inhibition dose-response. Cell viability was determined 48-hour post-treatment with carfilzomib at the indicated concentrations in PTEN-null (blue) and -wt GSCs (red) (n = 5 replicas per concentration, two-way ANOVA, multiple comparisons). IC50 values are indicated on the right side of the panel. C, “Box and whisker” dot-plot comparing PTEN status against carfilzomib IC50 (n = 4-9, *P < .05). D, Linear correlation between PTEN protein levels and IC50 values in GSCs PTEN-WT (r2 = 0.9575, P (two-tailed) = .0215). E, Immunoblots of different components of the PI3K/mTOR pathway in GSCs parental cells (PTEN-null, blue, and -wt, red; P: phosphorylated, T: total). The band observed with anti-PTEN in GBM HK301 is larger than predicted and therefore nonspecific.
We do note, however, an unexpected, yet reproducible, “biphasic” cell viability response to CFZ in PTEN-expressing samples (Fig. 1b, TS576 and TS543). We have also observed similar responses of patient-derived GBM and medulloblastoma samples to DNA replication interfering drugs (eg, topotecan, doxorubicin, idarubicin, etc.). We cannot at this time find any other examples of this phenomenon in the literature but intend to continue our investigations into possible mechanisms.
To discard nonspecific responses to proteasome inhibition due to other genomic alterations present in the GSCs, PTEN dependency was interrogated by performing gain-of-function and loss-of-function experiments. PTEN was overexpressed by lentiviral transduction in parental PTEN-null GSCs (GSC11 and GSC23) and drug response assays were conducted. GSC11 and GSC23 PTEN-overexpressing (PTEN-OE) cells showed resistance to proteasome inhibition as compared to the PTEN-null parental cells (GSC23-PTEN, IC50 = 3.167 μM, Fig. 2a, P < .0001). Conversely, PTEN-knockout (PTEN-KO) in PTEN-expressing GSCs (TS543 and TS576) and inducible neuronal progenitor cells (iNPCs), by CRISPR/Cas9 editing, conferred sensitivity to proteasome inhibition (TS543-PTEN-KO, IC50 = 1.89 × 10−5 μM; TS576-PTEN-KO-1A1, IC50 = 0.061 μM; TS576-PTEN-KO-1A3, IC50 = 0.0015 μM, Fig. 2b, P < .0001; Supplementary Fig. 2); demonstrating that proteasome inhibition specificity relies on PTEN loss.
Fig. 2.

Proteasome inhibition sensitivity relies on PTEN. A and B, Drug dose responses in glioblastoma stem cell (GSC) parental cells and -derived clones. A, PTEN-null parental cells (GSC11 and GSC23, blue) and engineered to overexpress PTEN (GSC11-PTEN, GSC23-PTEN, red). B, PTEN-WT parental cells (TS543 and TS576, red) and PTEN-KO CRISPR clones (TS543-2C4, TS543-3B3, TS576-1A1, TS576-1A3, blue). Left panels, immunoblots with anti-PTEN and anti-ACTIN (as loading control). Right panels, drug dose-response curves (n = 5 replicas per concentration, two-way ANOVA, multiple comparisons against vehicle (0), *P < .05, ****P < .0001). C, Cell viability in GSC cells overexpressing a PTEN enzymatic dead construct (G129R). Left panels, immunoblots with anti-AKT, -mTOR, and -S6 (P: phosphorylated, T: total). Right panels, cell viability assay at 48 hours in GSCs overexpressing PTEN-WT or PTEN-G129R (n = 3 replicas per concentration, two-way ANOVA, multiple comparisons against vehicle, ****P < .0001). D, Bar plots showing changes in cell viability in PTEN-null cells (GSC23 upper panel and TS576-PTEN-KO bottom panel) treated with carfilzomib alone (CFZ, 1 μM and 0.1 μM) or in combination with a PI3K inhibitor (PIK-75, 100 nM) or mTOR inhibitor (Temsirolimus, 1 μM) for 48 hours (n = 3 replicas per concentration, two-way ANOVA, multiple comparisons against vehicle, ***P < .001, ****P < .0001). E, Cell viability assay with different chemically distinct proteasome inhibitors (0.1 μM) (n = 3 replicas per concentration, two-way ANOVA, multiple comparison against GSC23 (*) or TS576-1A3 (+), ****/++++P < .0001). Error bars represent the SEM from different independent experiments.
Loss of PTEN Enzymatic Activity and Activation of PI3K/mTOR Pathway Result in Proteasome Inhibition Sensitivity
PTEN’s tumor suppressor function mainly depends on its lipid- and protein phosphatase activity.18,19 To uncover if proteasome inhibition sensitivity was dependent on PTEN enzymatic activity, an enzymatically dead PTEN-construct (G129R) was ectopically overexpressed in PTEN-deficient GSCs and cell viability assays were conducted. Cells overexpressing the enzymatically dead mutant retained high sensitivity to proteasome inhibition comparable to PTEN-null cells (Fig. 2c, P < .0001), indicating that drug response depends on PTEN enzymatic activity. In support of this, a link between PI3K/mTOR activation and proteasome inhibition sensitivity was corroborated by blocking proteasome inhibition sensitivity with a PI3K (PIK-75) or mTOR inhibitor (Temsirolimus) in PTEN-null cells (GSC23 and TS576-PTEN-KO) (Fig. 2d, P < .0001; Supplementary Figure 3). Moreover, immunoblot analysis in GSC parental cells and derived clones confirmed activation of PI3K/AKT/mTOR in PTEN-deficient cells by showing increased phosphorylation of AKT-S473, GSK3-S9, PRAS40-T246, p70S6K1-S235, and 4EBP1-T37 (Fig. 1e and Supplementary Figure 4), correlating with previous reports.2 Taken together, these data demonstrate that loss of PTEN and activation of PI3K/mTOR signaling confers sensitivity to a proteasome inhibition in GBM.
Chemically Diverse Proteasome Inhibitors Demonstrate the Same Therapeutic Profile
Several proteasome inhibitors, with different potency and selectivity for proteasome subunits, have been tested in the clinic for several malignancies including GBM.10,12 To investigate if PTEN-targeted selectivity was shared with other proteasome inhibitors, cell viability response was tested using clinically approved proteasome inhibitors (CFZ, ixazomib, delanzomib, oprozomib, marizomib). In general, all pan-proteasome inhibitors showed specificity for PTEN-deficient cells with a range of potencies (Fig. 2e, P < .0001). These results indicate that PTEN-deficient GSCs are sensitive to a broad class of proteasome inhibitors.
Proteasome Inhibition Response Signature in GBM Relies on Protein Synthesis and Autophagy
As we have shown that PTEN deficiency and PI3K/mTOR activation result in sensitivity to pan-proteasome inhibitors in GSCs, we next investigated the molecular mechanism of action. First- and second-generation proteasome inhibitors prevent proper proteasome function by binding to the catalytic site of the proteasome, the chymotrypsin-like β5 subunit of the 20S proteolytic core.20 To interrogate if proteasome activity confers selectivity to proteasome inhibition in the context of presence or absence of PTEN in GSCs, the activity of the chymotrypsin-like proteasome was evaluated. No differences in proteasome activity were observed between the two genotypes (PTEN-WT and -null) or in the derived clones (PTEN-OE and PTEN-KO) (Fig. 3a), indicating that proteasome activity per se was not responsible for PTEN-targeted specificity.
Fig. 3.

Protein synthesis and autophagy regulate proteasome inhibition response. A, Proteasome activity quantification in glioblastoma stem cells (GSCs) (upper panel) and derived clones (bottom panel). Proteasome activity was determined by quantification of the fluorophore 7-amino-4-methylcoumarin coupled to substrate LLVY-AMC (chymotrypsin-like substrate) (n = 3, no significant differences). B, Relative protein synthesis quantification in GSCs (upper panel) and PTEN-derived clones (bottom panel). Protein synthesis rate was determined by quantification of O-propargyl-puromycin (OPP) incorporation into translating polypeptides after 2 hours (top panel, n = 3, two-way ANOVA, multiple comparisons, ***P < .001, ****P < .0001; bottom panel, n = 3, t-test ***P < .001, ****P < .0001). C, Immunoblot analysis of poly-ubiquitinated (Ub) proteins and apoptosis markers (PARP and caspase-3) in PTEN-WT (TS576) and PTEN-null (TS576-1A3) cells treated with 100 nM carfilzomib at indicated time points. D, Right panel, apoptosis quantification by Annexin V FACS staining in PTEN-null (GSC23 and TS576-PTEN-KO) and PTEN-WT (GSC23-PTEN and TS576) cells treated with 100 nM carfilzomib per 24 hours (n = 3, t-test, ***P < .001, ****P < .0001). Left panel, representative quadrant dot-plot of percentages of cells in each subpopulation. E, Immunoblot analysis of markers of autophagy (LC3-II and SQSTM1) in GSCs PTEN-null (GSC23 and TS576-PTEN-KO) and PTEN-WT (GSC23-PTEN and TS576) untreated or treated with 100 nM carfilzomib for 48 hours. Densitometric analysis of LC3-II normalized to untreated group for each GSC condition. PI, propidium iodide; Prot inhibit, proteasome inhibitor;. Error bars represent SEM from 3 different independent experiments.
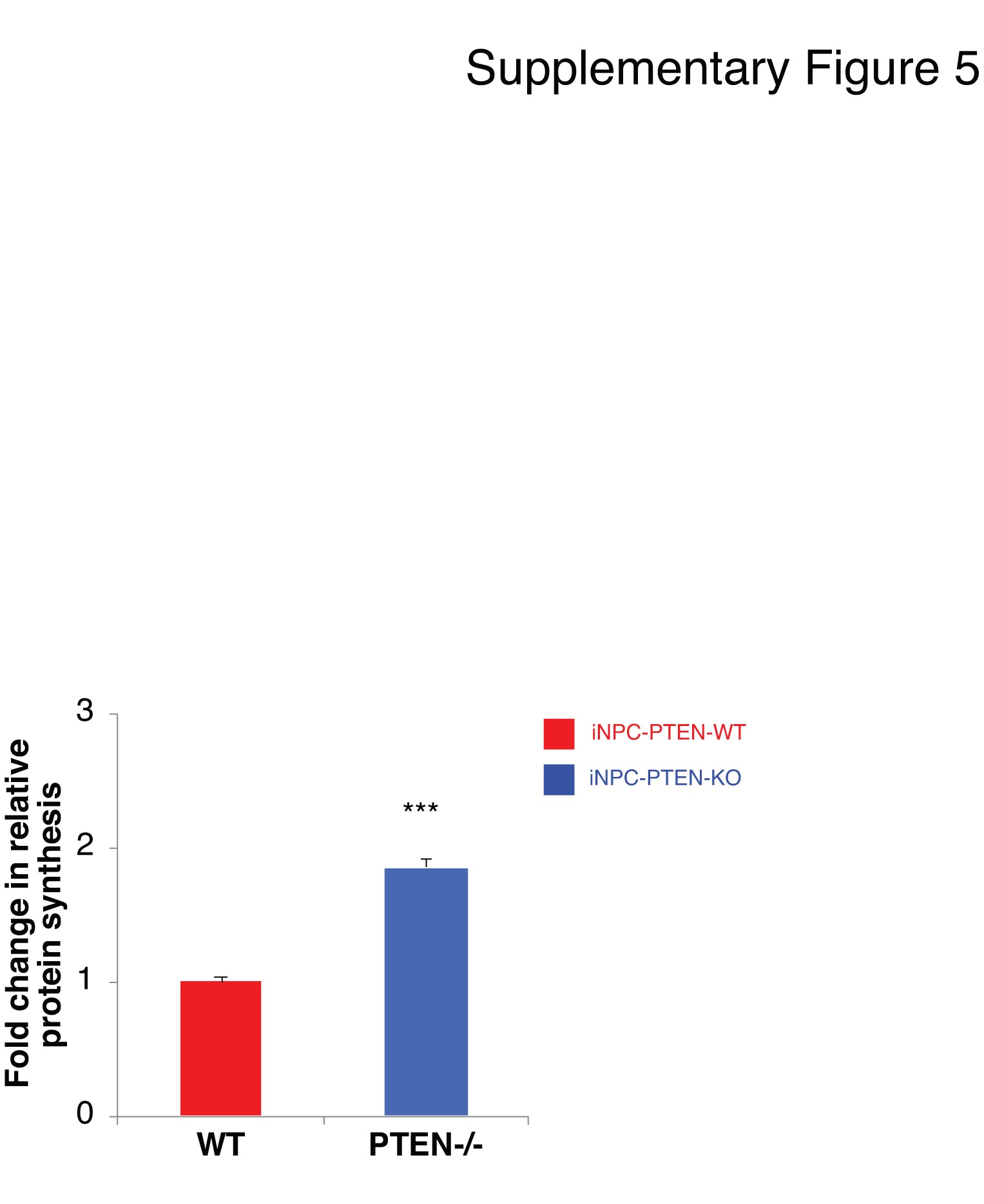
Proteasome inhibitors induce antitumor activity, in part, by impairing the balance between proteasome load and capacity10 that results in protein accumulation, and therefore disruption of multiple signaling pathways resulting in activation of apoptosis. One of the mechanisms that enhance proteasome loading is protein synthesis.21 Given that PTEN negatively regulates protein synthesis through the AKT/mTOR pathway which controls translation5, that PTEN-deficient hematopoietic stem cells (HSC) show high protein synthesis,22 and mTORC1 increases protein synthesis and protein degradation capacity,23 we investigated differences in protein synthesis rate in both genotypes. PTEN-null GBM cells exhibited higher protein synthesis compared with PTEN-WT cells (Fig. 3b top panel, P < .0001), which was abolished by PTEN-OE in PTEN-null clones and augmented by PTEN-KO in PTEN-expressing cells (Fig. 3b, bottom panel, P < .001). Similar results were observed in iNPCs with PTEN-deleted cells compared with parental iNPCs (Supplementary Figure 5). These findings suggest that the high protein synthesis rate observed in PTEN-deficient cells can confer sensitivity to proteasome inhibition.
To determine if a high protein synthesis rate correlated with accumulation of poly-ubiquitinated proteins after proteasome inhibition, anti-ubiquitin immunoblotting was performed. Poly-ubiquitinated proteins were specifically accumulated in PTEN-null GSCs at 6-hour post-treatment and reached a maximal accumulation at 12 hours (Fig. 3c, lanes 6-10). After 24 hours, markers of apoptosis, such as cleavage PARP and caspase-3, were specifically present in PTEN-deleted cells that retained poly-ubiquitinated proteins (Fig. 3c, lane 12) yet completely absent in the treated parental wild-type counterparts. Apoptosis induction after proteasome inhibition was confirmed by Annexin V staining in PTEN-null GSCs (Fig. 3d, P < .0001).
We next decided to monitor autophagy in GSCs before and after proteasome inhibition since pharmacological inhibition of the proteasome leads to activation of the autophagic-lysosomal pathway (ALP) by inducing accumulation of LC3-II24—a marker of autophagy induction25—and upregulating SQSTM1 (P62)26—an autophagy flux regulator. Moreover, mTOR directly regulates autophagy and controls protein accumulation.27 When we compared LC3-II and SQSTM1 protein levels we found that PTEN-null cells showed less LC3-II accumulation (Fig. 3e) and a consistent upregulation of SQSTM1 before and after proteasome inhibition, compared with PTEN-expressing cells (Fig. 3e; Supplementary Figure 6), indicating a potential autophagy dysregulation in the context of PTEN loss that can result in proteotoxic stress.28 Collectively, these data indicate that PTEN-deficient GSC cells are sensitive to proteasome inhibition because of a high protein synthesis rate and autophagy deficiency, which result in accumulation of poly-ubiquitinated proteins and ultimately cell death through apoptosis.
Proteomic and Bioinformatic Analyses Identify a Proteasome Inhibition Response Signature That Predicts Sensitivity in GBM With High PI3K/mTOR Expression
To resolve the proteins accumulated and pathways activated after proteasome inhibition (100 nM CFZ, 12 hours), a mass-spectrometry proteomic analysis was conducted in PTEN-WT and -KO GSCs before and after drug treatment (see Supplementary Methods). From this analysis, we identified that approximately 900 proteins were differentially enriched (DE) in the PTEN-WT group compared with the PTEN-KO placebo group (Supplementary Figure 7, Supplementary Table 2), while 58 proteins were DE in the PTEN-WT group (Fig. 4a, left panel, adj. P value < .001, and log 2-fold change) and 137 proteins in the PTEN-KO group (Fig. 4a, right panel, adj. P value < .001, and log 2-fold change) after CFZ treatment compared with their respective placebo control (Supplementary Table 2). We next performed gene set enrichment analysis (GSEA) using the complete Log2(FC) ranked gene lists from differential proteomic analysis of CFZ-treated and untreated PTEN-KO and -WT GSCs (see Materials and Methods; Supplementary Table 2). From this analysis we found a slight enrichment of the PI3K/mTOR signaling gene sets in the PTEN-WT CFZ-treated group (Fig. 4b) that was significantly higher in the PTEN-KO CFZ-treated group (mTORC1 signaling, WT-FDR = 0.0603 vs KO-FDR = 0.0020; PI3K/AKT/mTOR signaling, WT-FDR = 0.1316 vs KO-FDR = 0.0230, Fig. 4b and c; Supplementary Table 2), corroborating the role of PI3K/mTOR signaling in response to proteasome inhibition. Furthermore, we identified 102 proteins that were specifically and significantly accumulated after proteasome inhibition in the PTEN-KO group compared with placebo-treated group and were not statistically present in the PTEN-WT drug vs placebo groups (Supplementary Table 3). We defined this gene set as our Proteasome Inhibition Response Signature (PIRS), which did not exhibit substantial overlap with any of the gene sets previously deposited in the molecular signatures database (MSigDB).29
Fig. 4.

Proteomic and bioinformatic analysis in proteasome inhibitor-treated GSCs. A, Volcano plot of DE proteins, highlighted in dark green and dark blue, in PTEN-WT (left panel) and -KO (right panel) GSCs after carfilzomib treatment (CFZ 100 nM, 12 hours) compared with placebo group (adj. P-value < .001 and log 2-fold change). B, Table showing representative gene sets enriched in PTEN-WT (red) and PTEN-KO (blue) after CFZ treatment. C, GSEA plot of MTORC1 signaling (NES = 1.9040, FDR = 0.0020) and PI3K/AKT/mTOR signaling (NES = 1.9879, FDR = 0.0) in PTEN-KO GSCs treated with CFZ. D, pIC50 (−log10(IC50[M])) sorted heatmap of gene expression in GSCs for the PIRS. Expression values represented as colors (red = high, pink = moderate, light blue = low, dark blue = lowest). Representative genes upregulated in GBM indicated at the right side (n = 28 GSC samples). E, GSEA enrichment plot of PIRS (NES = 1.27, FDR = 0.191) and FOXO (NES = 1.89, FDR = 0.1012). F, AU-ROC analysis of the PIRLE signature in TCGA GBM samples subset by MTOR, AKT1, PIK3CA, and PTEN gene expression level (high vs low). True-positive rate = high expression; False-positive rate = low expression. G, Boxplot of ssGSEA enrichment scores of the PIRLE signature for AKT1, MTOR, PIK3CA, and PTEN high vs low TCGA GBM samples (Wilcox test, **P < .01, ***P < .001). CFZ: carfilzomib 100 nM for 12 hours. Abbreviations: AU-ROC, area under the receiver operating characteristic curve; DE, differentially enriched; FDR, false discovery rate; GSCs, glioblastoma stem cells; NES, normalized enrichment score; PIRLE, Proteasome Inhibition Response Leading Edge; PIRS, Proteasome Inhibition Response Signature; Placebo, DMSO; Prot Inhibit, proteasome inhibitor.
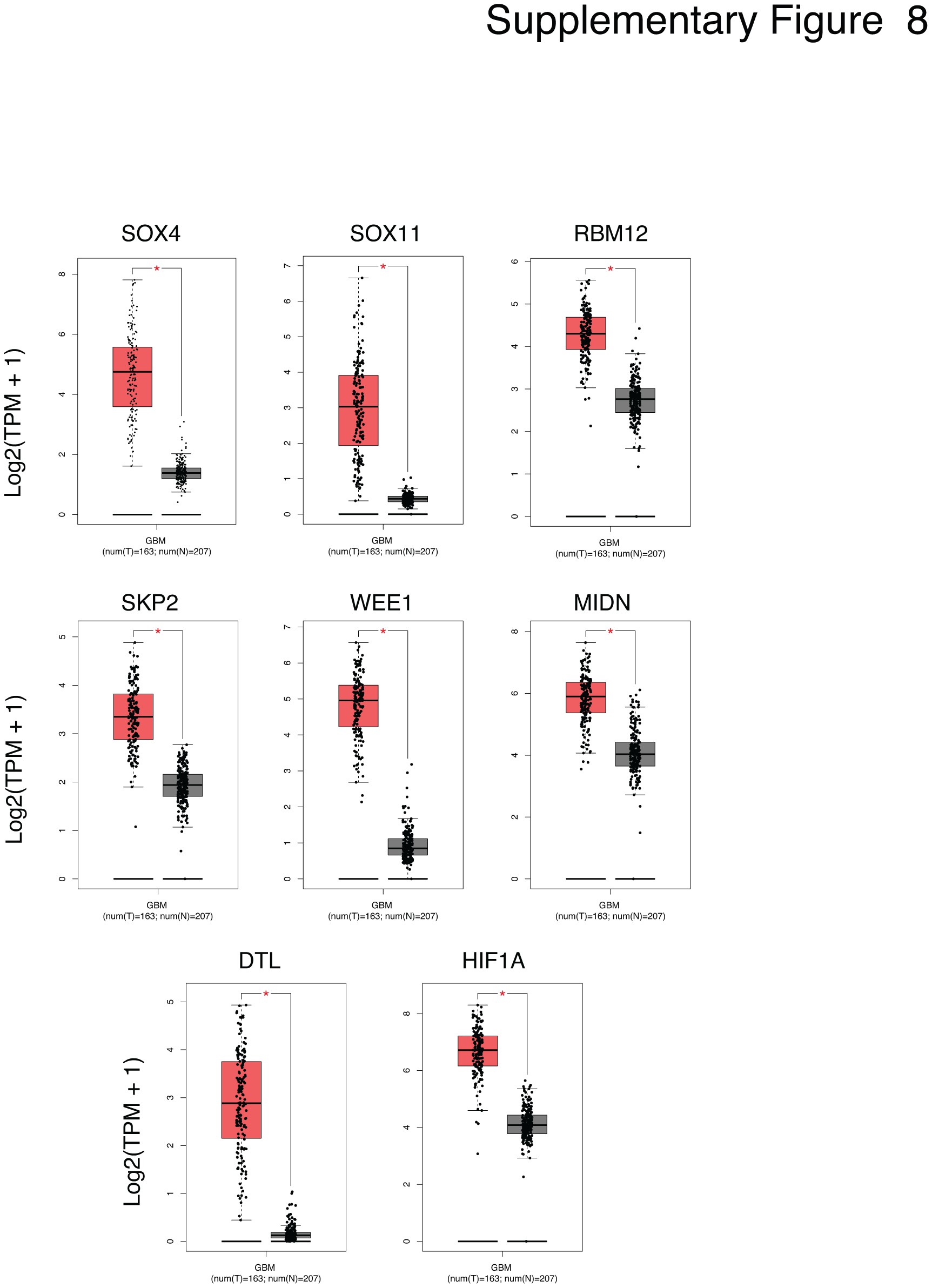
Next, to correlate the PIRS with CFZ response in GSCs (28 samples) we conducted standard GSEA by correlating drug response, as defined by pIC50 (−log10(IC50[M])), with gene expression quantification from RNA-seq analysis (see Materials and Methods). Enrichment analysis identified positive correlations between drug response and several genes upregulated in GBMs (Fig. 4d; Supplementary Figure 8), and gene sets involved in mRNA and rRNA regulation, cell cycle control, sumoylation, and DNA damage (Supplementary Figure 9; Supplementary Table 3). Importantly, FOXO and AKT gene sets, as well as the PIRS derived from the proteomic analysis correlated with CFZ response in our panel of GSCs (Fig. 4e; Supplementary Table 3).
Finally, we implemented ROC (Receiver Operating Characteristic) analysis to predict how much the PIRS can discriminate between expression levels of genes involved in the PI3K/mTOR pathway (PIK3CA, AKT1, MTOR, and PTEN) in GBM.2 For that, the original proteomic signature was filtered to the subset of genes contained in the leading edge of the GSEA results (48 genes) as expression of these genes was the strongest signal for drug response. This filtered Proteasome Inhibition Response Leading Edge (PIRLE) signature was used as input for single-sample GSEA on TPM normalized GBM data from TCGA (see Materials and Methods, Supplementary Table 3). ssGSEA enrichment of the PIRLE signature was found to be highly discriminative between MTOR, AKT1, and PIK3CA high and low GBM samples by calculating the area under the curve (AUC) (0.9386, 0.8289, and 0.7222, respectively) (Fig. 4f and g; Supplementary Table 3), however, the signature did not discriminate between high and low PTEN expression (AUC = 0.4230). In general, these data show that combining proteomic and transcriptomic bioinformatic analyses generates a PIRS predictive of drug response in GBM with high PI3K/AKT/mTOR expression.
Sensitization of GBM to Proteasome Inhibition via Autophagy Modulation
Informed by these data, we hypothesized that proteasome inhibition sensitivity can be observed in PTEN-WT cells with PI3K/mTOR activation and disruption of protein homeostasis and autophagy (Fig. 5a). This hypothesis was supported by our in vitro and in silico findings showing that PTEN-null cells had high protein synthesis and autophagy deficiencies in part driven by the PI3K/mTOR pathway. To test this premise, we genetically and pharmacologically mimicked a PTEN-null phenotype in PTEN-expressing cells by constitutively activating AKT (Myr-AKT) and inhibiting autophagy (bafilomycin A1 or chloroquine).25 We first overexpressed Myr-AKT30 in PTEN-WT cells, resulting in an increase in protein synthesis rate (Fig.e 5b, P < .001). We next treated the Myr-AKT expressing cells alone or in combination with proteasome inhibitor (CFZ) and autophagy inhibitors (bafilomycin A1 or chloroquine). Single inhibition of the proteasome or autophagy slightly reduced cell viability in cells that overexpress Myr-AKT (Fig. 5c). In contrast, combinatory inhibition of autophagy with proteasome inhibition resulted in a pronounced reduction in proliferation of PTEN-WT Myr-AKT cells (Fig. 5c, P < .001). Similar results were observed in PTEN-null cells (GSC23 and TS576-PTEN-KO), where cotreatment with an autophagy inhibitor enhanced proteasome inhibition sensitivity compared with single treatment (P < .0001, Fig. 5d), proposing a dual therapeutic strategy for GBM.
Fig. 5.

Autophagy modulation sensitizes glioblastoma stem cells (GSCs) to proteasome inhibition. A, Schematic diagram of the PI3K/mTOR pathway indicating key components involved in the proteasome inhibition response (PTEN, AKT, mTORC2, mTORC1, and SQSTM1). B, Upper panel, immunoblot analysis of AKT activation. Bottom panel, relative protein synthesis rate in parental cells and overexpressing constitutively active AKT (n = 3, two-way ANOVA, multiple comparisons, *P < .05, **P < .01, ***P < .001). C and D, Cell viability assays on cells treated with carfilzomib alone (CFZ 100 nM, 48 hours) and in combination with autophagy inhibitors bafilomycin A1 (BafA1) or chloroquine (CHQ) in PTEN-expressing (C) and PTEN-KO GSCs (D) (n = 3, two-way ANOVA, multiple comparisons against CFZ; parental (*) or AKT (+) or KO (+), **/++P < .01, ***/+++P < .001, ****/++++P < .0001). E, Left panel, immunoblots of cells transduced with lentivirus shRNA anti-RICTOR and anti-SQSTM1. Right panel, relative protein synthesis rate quantification in GSCs sh-Control (shLuc), shRNA anti-RICTOR and anti-SQSTM1 (n = 3, one-way ANOVA, multiple comparisons against shLuc, ****P < .0001). F, Cell viability assay in shControl, shRICTOR and shSQSTM1 GSCs untreated or treated with 100 nM carfilzomib per 48 hours (n = 3, two-way ANOVA, multiple comparisons against shLuc, *P < .05, ***P < .001, ****P < .0001). Error bars represent the SEM from 3 different independent experiments.
We further validated our model by performing a reciprocal experiment in PTEN-null cells, by either reactivating autophagy31 or blocking protein synthesis,22 by decreasing SQSTM1 (P62) and RICTOR expression, respectively. SQSTM1 or RICTOR knockdown in PTEN-null cells reactivated autophagy, as determined by LC3-II accumulation (Fig. 5e, left panel), and decreased protein synthesis (Fig. 5e, right panel, P < .00001) compared with a non-silencing shRNA control (shLuc). Moreover, knockdown of these autophagy and protein synthesis modulators induced resistance to proteasome inhibition in PTEN-null GSCs, compared with shRNA control (Fig. 5f, right panel, P < .001). Taken together, our results suggest that GBM cells are highly sensitive to proteasome inhibition when protein synthesis and autophagy are deregulated, particularly in the context of aberrant PI3K/mTOR activity, which is frequent in this disease.
Proteasome Inhibition Is Therapeutically Effective In Vivo
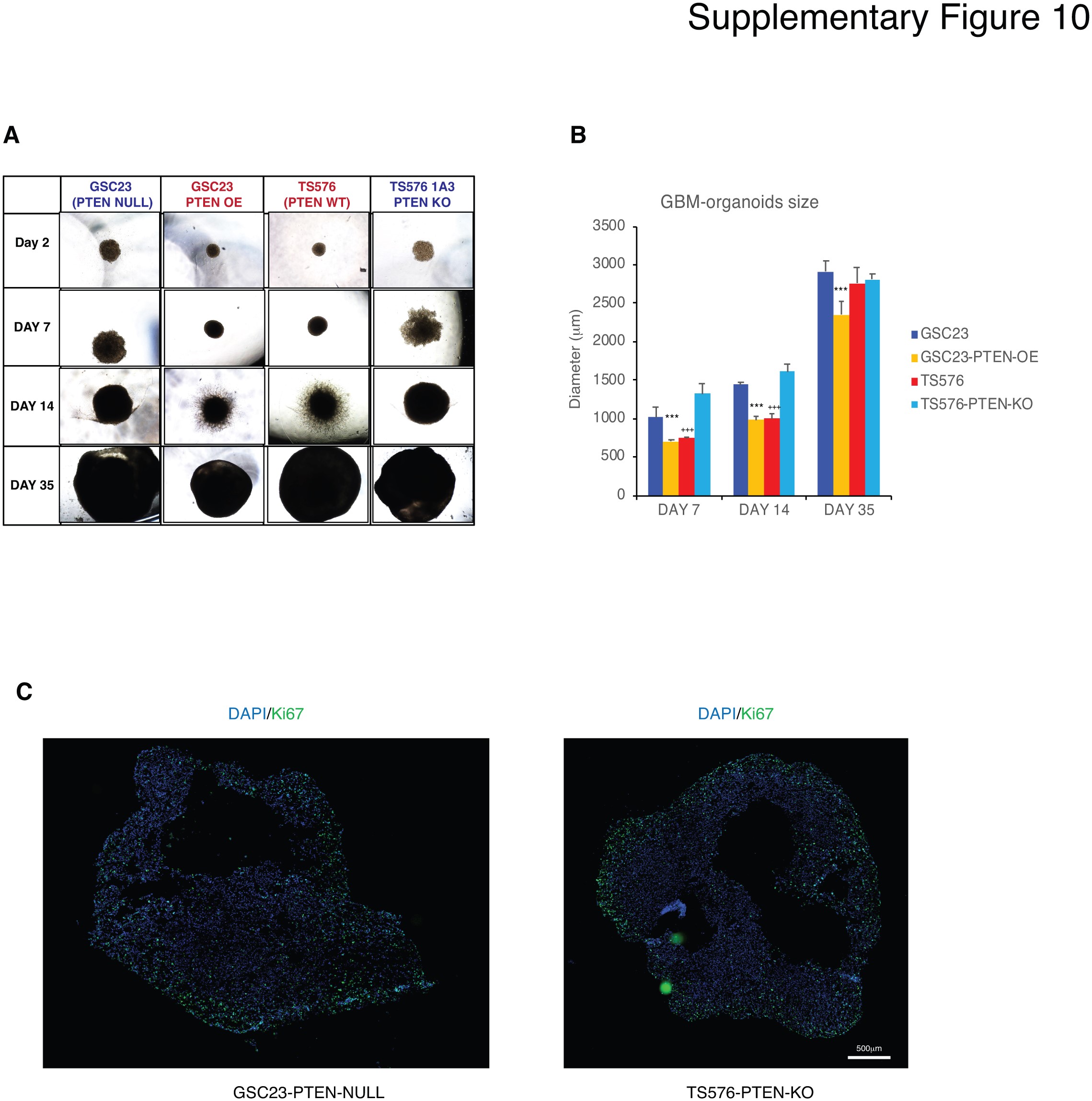
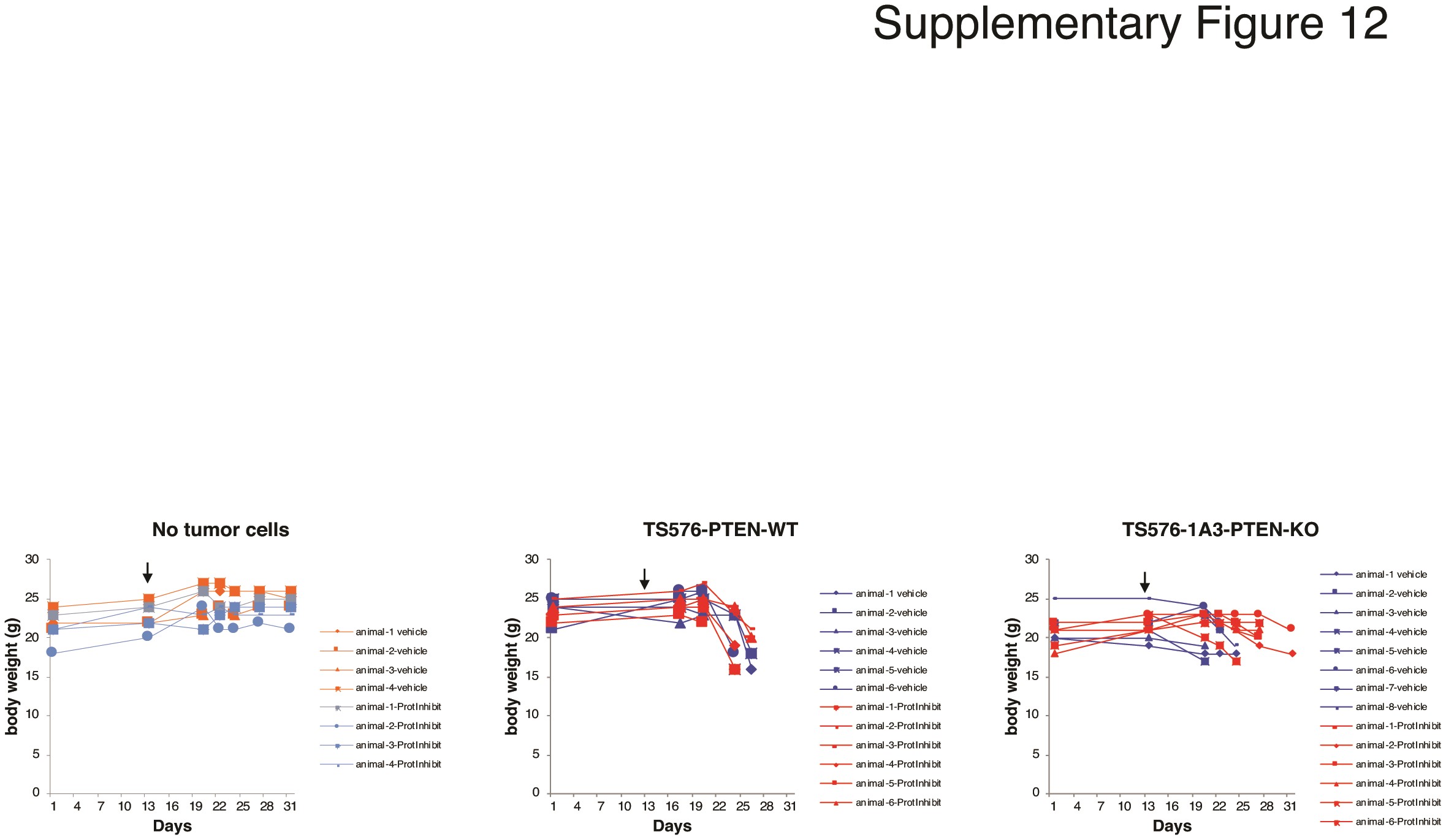
Lastly, proteasome inhibition response was investigated using more clinically relevant models that better recapitulate the heterogeneity of the tumor such as 3D GBM-organoids and in in vivo preclinical models. To distinguish differences in organoid maturation and drug response by PTEN expression, 3D GBM-organoids were established in parental PTEN-wild type (TS576) and PTEN-null (GSC23) GSCs and their derived clones (TS576-PTEN-KO and GSC23-PTEN-OE), respectively. Consistent with previous reports,17 PTEN-deficient organoids showed differences in growth rate during formation (weeks 2-4) compared with PTEN-expressing organoids (Supplementary Figure 10a and b). At 8 weeks, both PTEN-WT and -null mature GBM-organoids showed high cellular heterogeneity of proliferation as revealed by Ki-67 staining (Supplementary Figure 10c). Furthermore, proteasome inhibition showed a significant increase (8-fold) in the apoptosis marker cleaved caspase-3, specifically in PTEN-null GBM-organoids compared with the PTEN-expressing group (Fig. 6a, P < .005 and Supplementary Figure 11a). In contrast, drug treatment significantly reduced the cancer stem cell marker CD133 in PTEN-null organoids (Fig. 6b, P < .005 and Supplementary Figure 11b), corroborating PTEN target specificity by proteasome inhibition in 3D GBM-organoid models. Similarly, a significant decrease in tumor burden16 was observed in mice intracranially engrafted with PTEN-KO GSCs and treated with CFZ (5 mg/kg daily for 2 weeks) as compared with mice implanted with PTEN-WT GSC (Fig. 6c, P < .001). These results were further correlated with an increase in survival only in PTEN-KO-treated mice (Fig. 6d, P = .0014) but not in the PTEN-WT-treated group (Fig. 6c, P = .1637). No changes in body weight were observed between vehicle and drug-treated groups (Supplementary Figure 12), indicating this therapeutic approach is well tolerated in mice. In summary, these data demonstrate that proteasome inhibition specifically reduces tumor growth and increases survival in PTEN-deficient GBM preclinical models.
Fig. 6.

Proteasome inhibition induces cell death in GBM-organoids and inhibits intracranial tumor growth. A and B, Left panels, representative immunofluorescence staining of active caspase-3 (A) or, stem-cell marker, CD133 (B) in PTEN-deficient and -expressing GBM-organoids before and after treatment with 100 nM carfilzomib (CFZ) per 48 hours. DAPI, nuclear staining. Right panels, bar plot quantification of fluorescent signal intensities (n = 2-3 organoids and 5-10 fields, two-way ANOVA, multiple comparisons, *P < .05, **P < .01). Scale bar length 500 μm. C and D, Upper panels, representative FMT images of mice engrafted with TS576 PTEN-WT (C) or TS576-1A3 PTEN-null (D) GSCs and untreated (vehicle) or treated with proteasome inhibitor (5 mg/kg daily for 2 weeks). Bottom panels, tumor burden (relative fluorescence quantification) before (left) and after drug treatment (middle) (n = 5-7 animals per group, t-test comparison, **P < .01). Error bars represent the SEM from different independent experiments. Kaplan-Meier survival plots of mice treated with either a proteasome inhibitor (red) or vehicle only control (blue) (right). Abbreviations: DAPI, 4′,6-diamidino-2-phenylindole; GNM, glioblastoma; GSCs, glioblastoma stem cells; Prot Inhibit, proteasome inhibitor.
Discussion
Most GBM patients do not respond to standard of care therapy and very few show durable therapeutic response.1 Therefore, more effective treatment options for GBM are needed. There are approximately 1000 active clinical trials for GBM in the United States, mostly divided into chemotherapy approaches, immunotherapy strategies, and targeted therapies (clinicaltrials.gov).
Proteasome inhibition, as a targeted therapy, has shown great efficacy in hematological malignancies, such as multiple myeloma, mantle-cell lymphoma, and acute myeloid leukemia,10,32 in part because plasma cells have high rate of protein production and hence are more vulnerable to deregulation of the protein degradation machinery (proteome homeostasis).10,21 Currently, proteasome inhibitors are being tested as a therapeutic strategy for solid tumors, including GBMs.11,12,33 Here, we report that a drug screen against GSCs identified proteasome inhibition as a targeted therapy for PTEN-null GBMs. Furthermore, we demonstrate that proteasome inhibition sensitivity relies on protein synthesis rates and autophagy deregulation, and combining a proteomic and bioinformatic analysis a PIRS was derived predicting response in GBM with high PI3K/mTOR activity. Our current model correlates with previous publications and data indicating that proteasome inhibition disrupts the protein homeostasis and induces proteotoxic stress by disrupting the balance between protein production and protein depletion, predominantly observed in cells with defective compensatory mechanism and/or increased translation.
Several oncogenes and tumor suppressors directly regulate ribosome biogenesis and protein synthesis, and MYC and PTEN are master regulators of these signatures.34 In this study, we demonstrate that PTEN deletion directly affects protein synthesis in GBM, in part, through PI3K/mTOR activation. However, additional investigation will be needed to determine the contribution of other alterations that affect protein homeostasis and ultimately proteasome inhibition response in GBM, such as receptor tyrosine kinase (RTKs) activation, MYC, TP53, and RB mutations. For example, Myc and p53 drive endoplasmic reticulum stress and autophagy in a SMARCB1-deficient rhabdoid murine tumor model and combinatory inhibition of autophagy with proteasome inhibition results in tumor regression.35 In our study, we did not see a correlation of TP53 mutations with deregulation of protein synthesis and autophagy; nevertheless, alterations in ubiquitin pathways (AREL1 and MARCH9) have been correlated with TP53 mutations.36
None of the current treatment strategies, either alone or in combination, have been effective for GBM patients. However, identifying new molecular signatures—a “cancer fingerprint”—could help to identify patient subpopulations that may respond to these new therapeutic opportunities. Our findings show that protein synthesis and autophagy are “metabolic signatures” that can identify GBMs vulnerable to proteasome inhibition. The ubiquitin-proteasome system and autophagy act as a cellular quality control network that maintains cellular fitness. For example, low protein synthesis rates are required in HSCs to maintain protein quality control and stemness function.22,37 Changes in protein synthesis rate due to genetic alterations in genes that control homeostasis results in proteome imbalance.22,23,27,34 As we reported in this manuscript, targeting proteome imbalance in GBM is a new therapeutic opportunity for cancers and has also been reported for diffuse intrinsic pontine gliomas (DIPGs)33 and in rhabdoid tumor models.35
Autophagy also plays an important role in protein homeostasis by controlling protein degradation and proteasome load.10,27 Autophagy activation has been observed as a response mechanism after proteasome inhibition to eliminate protein aggregation in cancer cells (proteotoxic stress).24 However, in the absence of autophagy induction and upregulation of SQSTM1 (P62),26 associated with mTOR activation,31 the balance between proteasome load and degradation capacity can be disrupted.10 Here we show that PTEN-deficient GSCs have high protein synthesis rate and are sensitive to proteasome inhibition in part associated with low autophagy activation and constant levels of SQSTM1 expression that result in protein homeostasis imbalance and ultimately cell death. Moreover, we show that pharmacological inhibition of autophagy enhances proteasome inhibitor sensitivity in GBM. This is similar to other studies showing that combination of autophagy inhibition with MEK (mitogen-activated protein kinase) or ERK (extracellular signal-regulated kinase) inhibition can mediate antitumor activity in RAS-driven cancers.38
Recently, a study of the rare cancer cholangiocarcinoma,39 supports our notion that PTEN deletion results in increased sensitivity to a proteasome inhibitor. In that model, however, the suggested mechanism of action is related to high protein synthesis rate and activation of FOXO1 target genes. Indeed, we provide a PIRS that accurately predicts (AUC = 0.9386 for MTOR) sensitivity in GBM with high PI3K/mTOR expression.
In conclusion, our findings propose that PTEN-null GBMs are vulnerable to proteasome inhibition, whilst PTEN-expressing GBMs can be sensitized to proteasome inhibitors with concomitant autophagy inhibition. This synthetic lethality is due to a high protein synthesis rate and autophagy deficiency, and thus opens potential new therapeutic strategies to treat brain tumors.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We would like to thank SBP’s Tumor Analysis, Chemical Screening, and Proteomics Shared Resources for assistance.
Funding
The Sanford Burnham Prebys (SBP) Shared Resources are supported by SBP’s the National Cancer Institute’s Cancer Center Support Grant (P30 CA030199). The Informer Set drug collection was developed as part of the National Cancer Institute CTD2 Consortium (https://ocg.cancer.gov/programs/ctd2) and was partly funded through the National Institutes of Health (U01 CA168397). Research was also supported by a Hannah’s Heroes St. Baldrick’s Scholar Award to L.C.; and National Institutes of Health grants (R21 NS116455) to L.C., (U01 CA184898, U24 CA220341) to J.P.M; (R01 NS080939) to F.F., and (T32GM008666) to A.D.P. The Defeat GBM Research Collaborative, a subsidiary of the National Brain Tumor Society, and Ludwig Cancer Research provided support to F.F.
Conflict of interest statement. J.A.B. is currently an employee and shareholder of BMS. BMS had no input into this study.
Author contribution. J.A.B., D.F., J.M., T.K., R.V., and A.I. performed and analyzed experiments. J.A.B., A.D.P., and C.L. performed organoids assays; J.A.B., C.L., and G.S. performed in vivo experiments. J.A.B., D.F., and A.C. performed the proteomic assay. J.A.B., A.C., T.L., and L.C. performed GSEA analysis; J.P.M., K.V., and F.F. contributed intellectually to the project and edited the manuscript. J.A.B. and D.F. designed the experiments and wrote the manuscript.
References
- 1. Stupp R, Mason WP, van den Bent MJ, et al. ; European Organisation for Research and Treatment of Cancer Brain Tumor and Radiotherapy Groups; National Cancer Institute of Canada Clinical Trials Group . Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987–996. [DOI] [PubMed] [Google Scholar]
- 2. Zhang Y, Kwok-Shing Ng P, Kucherlapati M, et al. A pan-cancer proteogenomic atlas of PI3K/AKT/mTOR pathway alterations. Cancer Cell. 2017;31(6):820–832.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Brennan CW, Verhaak RG, McKenna A, et al. ; TCGA Research Network . The somatic genomic landscape of glioblastoma. Cell. 2013;155(2):462–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Verhaak RG, Hoadley KA, Purdom E, et al. ; Cancer Genome Atlas Research Network . Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17(1):98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hsieh AC, Liu Y, Edlind MP, et al. The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature. 2012;485(7396):55–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13(2):132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhao D, Lu X, Wang G, et al. Synthetic essentiality of chromatin remodelling factor CHD1 in PTEN-deficient cancer. Nature. 2017;542(7642):484–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Arico S, Petiot A, Bauvy C, et al. The tumor suppressor PTEN positively regulates macroautophagy by inhibiting the phosphatidylinositol 3-kinase/protein kinase B pathway. J Biol Chem. 2001;276(38):35243–35246. [DOI] [PubMed] [Google Scholar]
- 9. Mayer IA, Arteaga CL. The PI3K/AKT pathway as a target for cancer treatment. Annu Rev Med. 2016;67:11–28. [DOI] [PubMed] [Google Scholar]
- 10. Manasanch EE, Orlowski RZ. Proteasome inhibitors in cancer therapy. Nat Rev Clin Oncol. 2017;14(7):417–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yin D, Zhou H, Kumagai T, et al. Proteasome inhibitor PS-341 causes cell growth arrest and apoptosis in human glioblastoma multiforme (GBM). Oncogene. 2005;24(3):344–354. [DOI] [PubMed] [Google Scholar]
- 12. Di K, Lloyd GK, Abraham V, et al. Marizomib activity as a single agent in malignant gliomas: ability to cross the blood-brain barrier. Neuro Oncol. 2016;18(6):840–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Koga T, Chaim IA, Benitez JA, et al. Longitudinal assessment of tumor development using cancer avatars derived from genetically engineered pluripotent stem cells. Nat Commun. 2020;11(1):550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Benitez JA, Zanca C, Ma J, Cavenee WK, Furnari FB. Fluorescence molecular tomography for in vivo imaging of glioblastoma xenografts. J Vis Exp. 2018(134):57448, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li Y, Muffat J, Omer A, et al. Induction of expansion and folding in human cerebral organoids. Cell Stem Cell. 2017;20(3):385–396.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Myers MP, Pass I, Batty IH, et al. The lipid phosphatase activity of PTEN is critical for its tumor suppressor function. Proc Natl Acad Sci U S A. 1998;95(23):13513–13518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Furnari FB, Lin H, Huang HS, Cavenee WK. Growth suppression of glioma cells by PTEN requires a functional phosphatase catalytic domain. Proc Natl Acad Sci U S A. 1997;94(23):12479–12484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Schwartz AL, Ciechanover A. Targeting proteins for destruction by the ubiquitin system: implications for human pathobiology. Annu Rev Pharmacol Toxicol. 2009;49:73–96. [DOI] [PubMed] [Google Scholar]
- 21. Cenci S, Oliva L, Cerruti F, et al. Pivotal advance: protein synthesis modulates responsiveness of differentiating and malignant plasma cells to proteasome inhibitors. J Leukoc Biol. 2012;92(5):921–931. [DOI] [PubMed] [Google Scholar]
- 22. Signer RA, Magee JA, Salic A, Morrison SJ. Haematopoietic stem cells require a highly regulated protein synthesis rate. Nature. 2014;509(7498):49–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhang Y, Nicholatos J, Dreier JR, et al. Coordinated regulation of protein synthesis and degradation by mTORC1. Nature. 2014;513(7518):440–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ding WX, Ni HM, Gao W, et al. Linking of autophagy to ubiquitin-proteasome system is important for the regulation of endoplasmic reticulum stress and cell viability. Am J Pathol. 2007;171(2):513–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140(3):313–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sha Z, Schnell HM, Ruoff K, Goldberg A. Rapid induction of p62 and GABARAPL1 upon proteasome inhibition promotes survival before autophagy activation. J Cell Biol. 2018;217(5):1757–1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell. 2017;168(6):960–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Deshaies RJ. Proteotoxic crisis, the ubiquitin-proteasome system, and cancer therapy. BMC Biol. 2014;12:94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kohn AD, Summers SA, Birnbaum MJ, Roth RA. Expression of a constitutively active Akt Ser/Thr kinase in 3T3-L1 adipocytes stimulates glucose uptake and glucose transporter 4 translocation. J Biol Chem. 1996;271(49):31372–31378. [DOI] [PubMed] [Google Scholar]
- 31. Duran A, Amanchy R, Linares JF, et al. p62 is a key regulator of nutrient sensing in the mTORC1 pathway. Mol Cell. 2011;44(1):134–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Richardson PG, Barlogie B, Berenson J, et al. A phase 2 study of bortezomib in relapsed, refractory myeloma. N Engl J Med. 2003;348(26):2609–2617. [DOI] [PubMed] [Google Scholar]
- 33. Lin GL, Wilson KM, Ceribelli M, et al. Therapeutic strategies for diffuse midline glioma from high-throughput combination drug screening. Sci Transl Med. 2019;11(519):1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pelletier J, Thomas G, Volarević S. Ribosome biogenesis in cancer: new players and therapeutic avenues. Nat Rev Cancer. 2018;18(1):51–63. [DOI] [PubMed] [Google Scholar]
- 35. Carugo A, Minelli R, Sapio L, et al. p53 is a master regulator of proteostasis in SMARCB1-deficient malignant rhabdoid tumors. Cancer Cell. 2019;35(2):204–220.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ge Z, Leighton JS, Wang Y, et al. Integrated genomic analysis of the ubiquitin pathway across cancer types. Cell Rep. 2018;23(1):213–226.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hidalgo San Jose L, Sunshine MJ, Dillingham CH, et al. Modest declines in proteome quality impair hematopoietic stem cell self-renewal. Cell Rep. 2020;30(1):69–80.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bryant KL, Stalnecker CA, Zeitouni D, et al. Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat Med. 2019;25(4):628–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jiang TY, Pan YF, Wan ZH, et al. PTEN status determines chemosensitivity to proteasome inhibition in cholangiocarcinoma. Sci Transl Med. 2020;12(562):1–18. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.