

Summary
Molecular mechanisms mediating cardiac hypertrophy by glucose metabolism are incompletely understood. Hexosamine biosynthesis pathway (HBP), an accessory pathway of glycolysis, is known to be involved in the attachment of O-linked N-acetylglucosamine motif (O-GlcNAcylation) to proteins, a post-translational modification. We here demonstrate that glutamine-fructose-6-phosphate amidotransferase 2 (GFAT2), a critical HBP enzyme, is a major isoform of GFAT in the heart and is increased in response to several hypertrophic stimuli, including isoproterenol (ISO). Knockdown of GFAT2 suppresses ISO-induced cardiomyocyte hypertrophy, accompanied by suppression of Akt O-GlcNAcylation and activation. Knockdown of GFAT2 does not affect anti-hypertrophic effect by Akt inhibition. Administration of glucosamine, a substrate of HBP, induces protein O-GlcNAcylation, Akt activation, and cardiomyocyte hypertrophy. In mice, 6-diazo-5-oxo-L-norleucine, an inhibitor of GFAT, attenuates ISO-induced protein O-GlcNAcylation, Akt activation, and cardiac hypertrophy. Our results demonstrate that GFAT2 mediates cardiomyocyte hypertrophy by HBP-O-GlcNAcylation-Akt pathway and could be a critical therapeutic target of cardiac hypertrophy.
Subject areas: Classification Description: Cellular physiology, Molecular physiology, Cell biology
Graphical abstract
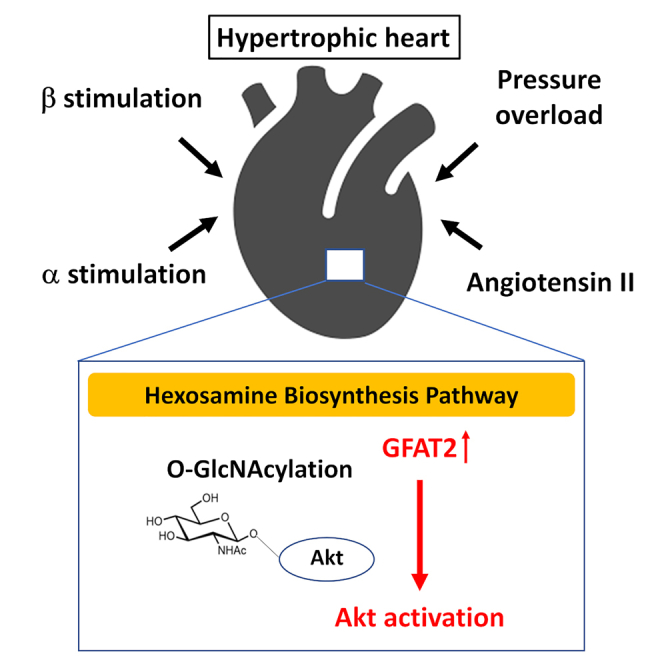
Highlights
-
•
GFAT2 is universally upregulated in the heart in response to hypertrophic stimuli
-
•
GFAT2 mediates cardiomyocyte hypertrophy by enhancing Akt O-GlcNAcylation and activation
-
•
HIF-1α, CaMKIIγ, and CaMKIIδ are major regulators of GFAT2 in cardiomyocyte hypertrophy
-
•
GFAT inhibitor attenuates cardiac hypertrophy by suppressing Akt activation
Classification Description: Cellular physiology; Molecular physiology; Cell biology
Introduction
Cardiac hypertrophy is an adaptive mechanism triggered in the heart in response to increased mechanical stress or neurohormonal stimuli. However, the prolonged presence of hypertrophy eventually leads to development of cardiac dysfunction (Okin et al., 2006). Cardiac hypertrophy is a risk factor for heart failure (HF) and cardiac events (Haider et al., 1998; Levy et al., 1990; Schillaci et al., 2000). Fatty acid oxidation (FAO) is a predominant metabolic pathway to produce adenosine triphosphate (ATP) in normal hearts (Stanley et al., 2005), whereas glucose becomes the preferential substrate for metabolism in hypertrophied hearts, which is the so-called metabolic shift from FAO to glucose metabolism (Nascimben et al., 2004). Although energy metabolism is known to be intimately involved in cardiac hypertrophy, its regulatory mechanism by the metabolic shift is not fully understood.
Hexosamine biosynthesis pathway (HBP) is a branch of glycolytic metabolism. It generates uridine diphosphate N-acetyl-glucosamine (UDP-GlcNAc), a substrate used for the attachment of O-linked N-acetylglucosamine motif (O-GlcNAcylation) to serine and threonine residues of proteins, using less than 0.01% of total cellular glucose in the heart (Olson et al., 2020). O-GlcNAc transferase (OGT) and O-GlcNAcase (OGA) catalyze the addition and removal of O-GlcNAc, respectively. O-GlcNAcylation is a post-translational modification, which is highly sensitive to various cellular stress such as heat shock, hypoxia, and nutrient deprivation (Hart et al., 2011). Abnormality of O-GlcNAcylation is implicated in the pathogenesis of several diseases, including cancer, diabetes mellitus, neurodegeneration, and cardiovascular diseases (Yang et al., 2008; Yi et al., 2012; Yuzwa et al., 2012). Cardiac-specific OGT knockout mice showed exacerbation of cardiac dysfunction after myocardial infarction, indicating that O-GlcNAcylation is indispensable for preserving cardiac function (Watson et al., 2010). However, several studies have demonstrated pro-hypertrophic actions of O-GlcNAcylation in hearts. Pathological cardiac hypertrophy was associated with increased total protein O-GlcNAcylation (Lunde et al., 2012; Sansbury et al., 2014; Young et al., 2007). Overexpression of OGT induced cardiomyocyte hypertrophy by increasing total protein O-GlcNAcylation (Facundo et al., 2012). c-Myc knockout mice showed attenuation of protein O-GlcNAcylation and cardiac hypertrophy (Ledee et al., 2015). Recently, adenosine monophosphate (AMP)-activated protein kinase (AMPK) has been shown to inhibit cardiac hypertrophy by reducing O-GlcNAcylation (Gelinas et al., 2018). Therefore, O-GlcNAcylation could be one of potential target for cardiac hypertrophy. However, the involvement of protein O-GlcNAcylation in hypertrophic signaling remains unclear.
Glutamine fructose-6-phosphate amidotransferase (GFAT) is a rate-limiting enzyme of HBP (Marshall et al., 1991). Mammalian cells express two GFAT isoforms, GFAT1 and GFAT2, which show 75% amino acid sequence identity in mice and humans (Oki et al., 1999). Although GFAT1 mRNA is ubiquitously expressed in several tissues including pancreas, placenta, testis skeletal muscle, and heart, and GFAT2 mRNA is mostly expressed in heart and the central nervous system (Oki et al., 1999), their protein levels remain unknown. Recently, GFAT1 has been reported to mediate cardiac hypertrophy in response to pressure overload (Tran et al., 2020). However, to date, the impact of GFAT2 in cardiac O-GlcNAcylation and cardiomyocyte hypertrophy have not been determined. If GFAT2 could be involved in the development of cardiomyocyte hypertrophy by HBP, elucidating its downstream signaling, including O-GlcNAcylation, is important to establish a novel therapeutic strategy for cardiac hypertrophy.
The aims of this study were to determine the impact and functional roles of GFAT2 in cardiomyocyte hypertrophy, to elucidate its downstream signaling, and to establish a strategy of anti-hypertrophy by targeting GFAT2.
Results
GFAT2 is a major GFAT isoform in hearts and cardiomyocytes
First, we investigated mRNA and protein levels of GFAT1 and GFAT2 in various tissues. GFAT1 mRNA was most abundantly expressed in brain (Figure 1A). The GFAT1 protein is predominantly expressed in liver, testis, and kidney (Figure 1B). On the other hand, GFAT2 mRNA levels in brain and heart were higher than those in other organs (Figure 1A). GFAT2 protein was abundant in lung, testis, and heart (Figure 1B). Next, we evaluated GFAT1, GFAT2, and protein O-GlcNAcylation levels in cardiac hypertrophy model by isoproterenol (ISO, β-adrenergic receptor agonist) infusion (15 mg/kg body weight/day, 7 days). ISO infusion increases in left ventricular wall thickness evaluated by echocardiography and heart weight (Table S2). ISO increased mRNA levels of GFAT2, but not GFAT1, in the heart (Figure 1C). At protein levels, GFAT2 and protein O-GlcNAcylation, but not GFAT1, were increased in ISO-infused hearts (Figure 1D). ISO did not affect protein levels of OGT and OGA, other regulators of protein O-GlcNAcylation, in hearts (Figure 1D). To assess the impact of GFAT2 in pathological cardiac hypertrophy in vivo, we evaluated GFAT2 and protein O-GlcNAcylation levels in several cardiac hypertrophy models, including the PE-infusion model (100 mg/kg body weight/day, 7 days), Ang II-infusion model (1.44 mg/kg body weight/day, 7 days), and transverse aortic constriction model (7 days). All of these hypertrophic stimuli induced increases in left ventricular wall thickness evaluated by echocardiography and heart weight (Tables S3–S5), which were accompanied by increases in GFAT2 and protein O-GlcNAcylation in the heart (Figures S1A–S1C). These data indicate that GFAT2 is universally upregulated and a major GFAT isoform involved in protein O-GlcNAcylation during pathological cardiac hypertrophy.
Figure 1.

GFAT2 is a major GFAT isoform in the heart and cardiomyocytes
(A) mRNA expression levels of GFAT1 and GFAT2 adjusted by18S mRNA in lung, liver, brain, heart, testis, and kidney (n = 3).
(B) Representative immunoblots of GFAT1, GFAT2, and β-actin and Coomassie brilliant blue (CBB) staining in lung, liver, brain, heart, testis, and kidney. Quantitative analysis of the absolute value of intensity of GFAT1 and GFAT2 in lung, liver, brain, heart, testis, and kidney (n = 3).
(C) mRNA expression levels of GFAT1 and GFAT2 adjusted by18S mRNA in ISO (15 mg/kg body weight/day, 7 days)- or control vehicle (saline)-treated C57B/6J mouse hearts (n = 5).
(D) Representative immunoblots of GFAT1, GFAT2, OGT, OGA, protein O-GlcNAcylation, and GAPDH in ISO (15 mg/kg body weight/day, 7 days)- or control vehicle (saline)-treated C57B/6J mouse hearts. Quantitative analysis of GFAT1, GFAT2, OGT, OGA, and protein O-GlcNAcylation in ISO- or control vehicle-treated C57B/6J mouse hearts (n = 5).
(E) Representative immunoblots of GFAT1, GFAT2, and β-actin and CBB staining in neonatal rat ventricular cardiomyocytes (NRVMs), fibroblasts (FBs), endothelial cells (ECs), C2C12 cells, and HEK293 cells. Quantitative analysis of the absolute value of intensity of GFAT1, GFAT2 in NRVMs, FBs, ECs, C2C12 cells, and HEK293 cells (n = 3).
(F) Representative immunoblots of GFAT1, GFAT2, OGT, OGA, protein O-GlcNAcylation, and GAPDH in NRVMs treated with ISO (1 μM) at the indicated time point (n = 6). Quantitative analysis of GFAT1, GFAT2, OGT, OGA, and protein O-GlcNAcylation in NRVMs treated with ISO (1 μM) at the indicated time point (n = 6). ∗p < 0.05, ∗∗p < 0.01: post hoc Tukey's comparison test.
Next, we evaluated protein levels of GFAT1 and GFAT2 in several types of cells. GFAT1 was detected in cardiomyocytes (neonatal rat ventricular myocytes, NRVMs), fibroblasts (FBs), endothelial cells (ECs), C2C12 cells, and HEK293 cells, whereas GFAT2 predominantly was expressed in cardiomyocytes, fibroblasts, and HEK293 cells (Figure 1E). We also evaluated protein levels of GFAT1, GFAT2, OGT, OGA, and protein O-GlcNAcylation in cardiomyocytes with hypertrophic stimuli. GFAT1, OGT, and OGA were not altered after ISO stimulation, whereas GFAT2 was increased in a time-dependent manner (Figure 1F). Consistent with GFAT2, protein O-GlcNAcylation was increased after ISO stimulation (Figure 1F). These data indicate that GFAT2 is universally upregulated and a major GFAT isoform involving in protein O-GlcNAcylation during pathological cardiac hypertrophy.
GFAT2 mediates cardiomyocyte hypertrophy by protein O-GlcNAcylation
To elucidate the specific role of GFAT2 in cardiomyocyte hypertrophy, we conducted knockdown of GFAT2 in NRVMs by small interfering RNA (siRNA). We confirmed that knockdown of GFAT2 equally decreased its protein levels in each cardiomyocyte (Figure S2). Knockdown of GFAT2 significantly suppressed ISO-induced increases in GFAT2 in cardiomyocytes without affecting GFAT1, OGT, and OGA protein levels (Figures 2A–2E). It also attenuated ISO-induced increases in protein O-GlcNAcylation (Figures 2A and 2F). ISO-induced cardiomyocyte hypertrophy evaluated by cell surface area (CSA) and knockdown of GFAT2 significantly ameliorated it (Figure 2G). Furthermore, we examined whether GFAT2 medicates cardiomyocyte hypertrophy in response to other hypertrophic stimulation. PE increased GFAT2, protein O-GlcNAcylation, atrial natriuretic factor (ANF) mRNA levels, and CSA in cardiomyocytes, and knockdown of GFAT2 attenuated them (Figures S3A–S3C). Taken together, GFAT2 mediates cardiomyocyte hypertrophy in response to hypertrophic stimuli through protein O-GlcNAcylation.
Figure 2.

GFAT2 mediates cardiomyocyte hypertrophy by protein O-GlcNAcylation
(A) Representative immunoblots of GFAT2, GFAT1, OGT, OGA, protein O-GlcNAcylation, and GAPDH in NRVMs treated with indicated small interfering RNA (siRNA) in the presence or absence of ISO (1 μM, 12 h).
(B–F) Quantitative analysis of GFAT2, GFAT1, OGT, OGA, and protein O-GlcNAcylation in NRVMs treated with indicated small interfering RNA (siRNA) in the presence or absence of ISO (1 μM, 12 h) (n = 6).
(G) Cell surface area of NRVMs treated with indicated siRNA in the presence or absence of ISO (1 μM, 24 h) (n = 150 cells in each group). Scale bar, 50 μm. ∗p < 0.05, ∗∗p < 0.01: post hoc Tukey's comparison test.
We identified factors regulating basal level of O-GlcNAcylation in cardiomyocytes. Interestingly, neither GFAT1 nor OGT knockdown changed the basal level of O-GlcNAcylation (Figure S4). Furthermore, double knockdown of GFAT1 and GFAT2 did not affect it (Figure S4). On the other hand, knockdown of OGA increased it (Figure S4). These data indicate that OGA critically regulates the basal level of O-GlcNAcylation in cardiomyocytes.
GFAT2-Akt axis positively regulates ISO-induced cardiomyocyte hypertrophy
To elucidate GFAT2 downstream signaling, we evaluated the activation of proteins related to cardiac hypertrophy. ISO increased phosphorylation of Akt and c-jun N-terminal kinase (JNK), not extracellular signal-regulated kinase (ERK) and p38, in cardiomyocytes (Figures 3A–3E). Among them, knockdown of GFAT2 significantly suppressed only phosphorylation at S473 of Akt, its active form (Figures 3A and 3B). Paralleled with S473, phosphorylation at T308 was changed (Figure S5). Diazo-5-oxo-L-norleucine (DON: 50 μM, 12 h), an inhibitor of GFAT, also suppressed protein O-GlcNAcylation and Akt phosphorylation in cardiomyocytes treated with ISO or PE (Figures S6A and S6B). Transient activation of hypertrophic signaling is important. We evaluated phosphorylation and total protein levels of ERK 1, 3, and 6 h after ISO stimulation. GFAT2 knockdown did not change phosphorylation of ERK (Figure S7). These data suggest that Akt is a critical downstream target of GFAT2 in cardiomyocyte hypertrophy. Protein phosphorylation is known to be involved in O-GlcNAcylation. Thus, we evaluated the involvement of O-GlcNAcylation in Akt. Immunoprecipitated O-GlcNAcylated protein levels were similar in si-scramble and si-GFAT2-treated cardiomyocytes after ISO stimulation (middle panel in Figure 3F). ISO-induced O-GlcNAcylation of Akt and knockdown of GFAT2 suppressed it (Figure 3F). To elucidate whether the antihypertrophic effects of GFAT2 are mediated by protein O-GlcNAcylation, we investigated the effect of OGA knockdown in GFAT2-knocked-down cardiomyocytes after ISO stimulation. OGA knockdown reversed the anti-hypertrophic effects of GFAT2 knockdown, accompanied by an increase in protein O-GlcNAcylation and Akt phosphorylation (Figures S8A and S8B). These data indicate that the anti-hypertrophic effects of GFAT2 knockdown are mediated by protein O-GlcNAcylation.
Figure 3.

GFAT2-Akt axis positively regulates ISO-induced cardiomyocyte hypertrophy
(A) Representative immunoblots of Akt, phospho-Akt(S473), ERK, phospho-ERK, p38, phospho-p38, JNK, phospho-JNK, and GAPDH in NRVMs treated with indicated small interfering RNA (siRNA) in the presence or absence of ISO (1 μM, 12 h).
(B–E) Quantitative analysis of phospho-Akt(S473), phospho-ERK, phospho-p38, phospho-JNK in NRVMs treated with indicated siRNA in the presence or absence of ISO (1 μM, 12 h) (n = 6).
(F) Immunoprecipitation assays using lysates of NRVMs treated with indicated siRNA in the presence or absence of ISO (1 μM, 12 h) (n = 6). After immunoprecipitation with control IgG or an O-GlcNAcylation antibody, immunoblotting for Akt was performed. Immunoblots of input controls (10% lysates) are also shown.
(G) Representative immunoblots of Akt, phospho-Akt(S473), and GAPDH in NRVMs treated with adenovirus harboring wild-type Akt or mutant Akt in T479A in the presence of ISO (1 μM, 24 h). Quantitative analysis of phospho-Akt(S473) in NRVMs treated with adenovirus harboring wild-type Akt or mutant Akt in T479A in the presence of ISO (1 μM, 24 h).
(H) Cell surface area of NRVMs treated with adenovirus harboring wild-type Akt or mutant Akt in T479A in the presence of ISO (1 μM, 24 h) (n = 150 cells in each group). Scale bar, 50 μm.
(I) Cell surface area of NRVMs treated with GFAT2 siRNA and/or MK-2206(1 nM, 24 h) in the presence of ISO (1 μM, 24 h) (n = 150 cells in each group). Scale bar, 50 μm. IP, immunoprecipitation. ∗p < 0.05, ∗∗p < 0.01: post hoc Tukey's comparison test.
To further elucidate the impact of O-GlcNAcylation of Akt on cardiac hypertrophy, we conducted an experiment using adenovirus harboring wild-type Akt or mutant Akt in T479A, which is known as its O-GlcNAcylation-resistant mutation. Of importance, overexpression of mutant Akt exhibited decreases in ISO-induced Akt phosphorylation compared with wild-type Akt in cardiomyocytes (Figure 3G). Consistently, the mutant Akt exhibited ISO-induced increases in CSA of cardiomyocytes compared with wild-type Akt (Figure 3H). These data indicate that Akt O-GlcNAcylation is critically involved in its phosphorylation and cardiomyocyte hypertrophy. MK-2206 (a specific Akt inhibitor; 10 nM, 24 h) and/or knockdown of GFAT2 did not change CSA of cardiomyocytes at baseline (Figure S9). MK-2206 suppressed ISO-induced cardiomyocyte hypertrophy (Figure 3I). Important, knockdown of GFAT2 did not affect the suppressive effects of MK-2206 on cardiomyocyte hypertrophy (Figure 3I). These data indicate that the GFAT2-Akt axis positively regulates ISO-induced cardiomyocyte hypertrophy.
We also investigated phosphorylation of mTOR, S6K, and p70S6K, downstream targets of Akt. Consistent with a decrease in phosphorylation of Akt, GFAT2 knockdown decreased ISO-induced phosphorylation of mTOR, S6K, and p70S6K (Figure S5). These data indicate that O-GlcNAcylation of Akt mediates GFAT2-related cardiomyocyte hypertrophy by regulating the mTOR-S6K-p70S6K axis and that the O-GlcNAcylation of Akt plays a pro-hypertrophic role through its activation.
GFAT1 is known to be involved in cardiac hypertrophy. Thus, we investigated the role of GFAT1 in O-GlcNAcylation and cardiomyocyte hypertrophy. GFAT1 knockdown did not affect ISO-induced protein O-GlcNAcylation in cardiomyocytes, accompanied with an increase in GFAT2 (Figure S10A). This finding suggests that GFAT2 could redundantly regulate O-GlcNAcylation in GFAT1-knocked-out cardiomyocytes in response to ISO stimulation. Nonetheless, interestingly, GFAT1 knockdown attenuated ISO-induced cardiomyocyte hypertrophy (Figure S10B), suggesting that GFAT1 is involved in ISO-induced cardiac hypertrophy independently of O-GlcNAcylation.
Glucosamine induces cardiomyocyte hypertrophy by protein O-GlcNAcylation and Akt activation
Glucosamine enters the HBP downstream of GFAT and bypasses this limiting step (Figure 4A). Next, we assessed the relationship between HBP and cardiomyocyte hypertrophy. Administration of glucosamine (5 mM, 12 h) induced both protein O-GlcNAcylation and phosphorylation of Akt (Figure 4B). It also increased cardiomyocyte hypertrophy (Figure 4C). Glucosamine canceled the suppressive effects of knockdown of GFAT2 on protein O-GlcNAcylation and phosphorylation of Akt (Figure 4D). Glucosamine also abolished the anti-hypertrophic effect of knockdown of GFAT2 (Figure 4E). Furthermore, Akt inhibitor suppressed glucosamine-induced cardiomyocyte hypertrophy (Figure S11). These data suggest that the HBP-O-GlcNAcylation-Akt pathway is a downstream signaling of GFAT2.
Figure 4.

Glucosamine induces cardiomyocyte hypertrophy by protein O-GlcNAcylation and Akt activation
(A) Schematic representation of the HBP/O-GlcNAcylation pathway. P, phosphate. UDP-GlcNAc, uridine diphosphate N-acetylglucosamine.
(B) Representative immunoblots of protein O-GlcNAcylation, phospho-Akt(S473), Akt, and GAPDH in NRVMs treated with or without glucosamine (5 mM, 12 h) (n = 6). Quantitative analysis of protein O-GlcNAcylation and phospho-Akt in NRVMs treated with or without glucosamine (5 mM, 12 h) (n = 6).
(C) Cell surface area of NRVMs treated with or without glucosamine (5 mM, 24 h) (n = 150 cells in each group). Scale bar, 50 μm.
(D) Representative immunoblots of protein GFAT2, protein O-GlcNAcylation, phospho-Akt(S473), Akt, and GAPDH in NRVMs treated with or without glucosamine (5 mM, 12 h) in the presence or absence of ISO (1 μM, 12 h) in addition to treatment of GFAT2 siRNA (n = 12). Quantitative analysis of protein GFAT2, protein O-GlcNAcylation, and phospho-Akt in NRVMs treated with or without glucosamine (5 mM, 12 h) in the presence or absence of ISO (1 μM, 12 h) in addition to treatment of GFAT2 siRNA (n = 6).
(E) Cell surface area of NRVMs treated with or without glucosamine (5 mM, 24 h) in the presence or absence of ISO (1 μM, 24 h) in addition to treatment of GFAT2 siRNA (n = 150 cells in each group). Scale bar, 50 μm. ∗p < 0.05, ∗∗p < 0.01: post hoc Tukey's comparison test.
HIF-1α positively regulates GFAT2 in cardiomyocyte hypertrophy
HBP is known to be involved in not only glucose metabolism but also calcium handling. We investigated whether glucose metabolism was related to the regulation of GFAT2 expression in cardiomyocyte hypertrophy. ISO administration increased the protein levels of HIF-1α, a master regulator of glucose metabolism, and its knockdown by siRNA significantly attenuated GFAT2 mRNA and protein levels (Figures 5A–5C, and 5E). In addition, knockdown of HIF-1α attenuated ISO-induced phosphorylation of Akt (Figures 5A and 5D). Consistent with these changes in GFAT2 and phosphorylation of Akt, it also suppressed ISO-induced cardiomyocyte hypertrophy (Figure 5F). We next examined the role of CaMKII, a major regulator of calcium handling in cardiomyocytes, in regulation of GFAT2. Knockdown of CaMKIIγ by siRNA suppressed ISO-induced increased GFAT2 at protein levels but not mRNA levels (Figures S12A and S12B). In addition, knockdown of CaMKIIδ also ameliorated ISO-induced increased GFAT2 at protein levels (Figures S13A and S13B). These data suggest that HIF-1α, CaMKIIγ, and CaMKIIδ are critical positive regulators of GFAT2 in cardiomyocyte hypertrophy.
Figure 5.

HIF-1α positively regulates GFAT2 in cardiomyocyte hypertrophy
(A) Representative immunoblots of HIF-1α, GFAT2, phospho-Akt(S473), Akt, and GAPDH in NRVMs treated with indicated siRNA in the presence or absence of ISO (1 μM, 12 h) (n = 5).
(B–D) Quantitative analysis of protein HIF-1α, GFAT2, and phospho-Akt in NRVMs treated with indicated siRNA in the presence or absence of ISO (1 μM, 12 h) (n = 5).
(E) Expression of GFAT2 and 18S mRNA in NRVMs treated with indicated siRNA in the presence or absence of ISO (1 μM, 12 h) (n = 5).
(F) Cell surface area of NRVMs treated with indicated siRNA in the presence or absence of ISO (1 μM, 12 h) (n = 150 cells in each group). Scale bar, 50 μm. ∗p < 0.05, ∗∗p < 0.01: post hoc Tukey's comparison test.
We evaluated the direct effect of hyperglycemia on GFAT2 protein levels. Interestingly, treatment of high glucose concentration (200 and 450 mg/dL) did not affect GFAT2 protein levels in cardiomyocytes and ISO increased them independently of glucose levels (Figure S14A). Furthermore, we investigated the effects of glucose concentration on protein O-GlcNAcylation in cardiomyocytes. Protein O-GlcNAcylation did not depend on glucose concentrations (100, 200, or 450 mg/dL) (Figure S14B).
DON attenuates ISO-induced cardiac hypertrophy in vivo
We examined the pharmacological therapeutic effect of DON (0.05 mg/kg body weight/day) on ISO-induced cardiac hypertrophy. DON effectively suppressed protein O-GlcNAcylation in ISO-infused hearts (Figure 6A). However, it did not change GFAT1, GFAT2, OGT, and OGA protein levels (Figure S15). DON significantly suppressed ISO-induced increases in wall thickness without affecting blood pressure and heart rate (Figures 6B, 6C, and S16). It did not alter left ventricular (LV) diameter and LV ejection fraction (LVEF), an index of cardiac function (Figures 6B and 6D–6F). DON attenuated ISO-induced increases in heart weight and LV weight (Figure 6G). ISO-induced cardiomyocyte hypertrophy evaluated by CSA and DON significantly suppressed it (Figure 6H). Consistent with these anti-hypertrophic effects, DON attenuated ISO-induced increases in ANF mRNA levels in the hearts (Figure 6I). In contrast, DON did not affect ISO-induced interstitial fibrosis and mRNA levels of collagen I and III (Figure S17). Taken together, DON effectively suppressed ISO-induced cardiac hypertrophy.
Figure 6.

DON attenuates ISO-induced cardiac hypertrophy in vivo
(A) Representative immunoblots of protein O-GlcNAcylation and GAPDH in ISO (15 mg/kg body weight/day, 7 days)- or control vehicle (saline) treated-C57B/6J mouse hearts subjected to either 6-Diazo-5-oxo-L-norleucine (DON, 0.05 mg/kg body weight/day) or saline for 7 days. Quantitative analysis of protein O-GlcNAcylation in ISO- or control vehicle-treated C57B/6J mouse hearts subjected to either DON (0.05 mg/kg body weight/day) or saline for 7 days (n = 9).
(B) The representative echocardiographic images of ISO- or control vehicle-treated C57B/6J mouse hearts subjected to either DON (0.05 mg/kg body weight/day) or saline for 7 days. Long two-way arrows and short two-way arrows indicate left ventricular end-diastolic diameter (LVDd) and left ventricular end-systolic diameter (LVDs), respectively.
(C–F) Wall thickness, LVDd, LVDs, and LV ejection fraction (LVEF) measured by echocardiography at day 7 in each group (n = 12).
(G) Heart weight to tibial length (TL) ratio and LV weight to TL ratio in each group (n = 12).
(H) Masson-trichrome-stained heart section in each group. Scale bar, 50 μm. Cardiomyocyte hypertrophy as assessed by cross-sectional area in each group (n = 6).
(I) Expression of atrial natriuretic factor (ANF) and 18S mRNA in the indicated mouse hearts (n = 6). ∗p < 0.05, ∗∗p < 0.01: post hoc Tukey's comparison test.
DON suppresses protein O-GlcNAcylation and Akt activation in ISO-induced cardiac hypertrophy
To further investigate the mechanism mediating anti-hypertrophic effects of DON in ISO-induced cardiac hypertrophy, we evaluated the activation of proteins related to cardiac hypertrophy. ISO increased the phosphorylation of Akt and JNK, not ERK and p38 (Figures 7A–7E). Among them, DON significantly suppressed only phosphorylation of Akt (Figures 7A and 7B). Consistent with this change, DON attenuated ISO-induced phosphorylation of S6 and p70S6K in the heart (Figure S18). These data suggest that suppression of the GFAT2-protein O-GlcNAcylation-Akt pathway mediates the anti-hypertrophic effect of DON.
Figure 7.

DON suppresses Akt activation in ISO-induced cardiac hypertrophy
(A) Representative immunoblots of Akt, phospho-Akt(S473), ERK, phosphor-ERK, p38, phosphor-p38, JNK, phospho-JNK, and GAPDH in ISO (15 mg/kg body weight/day)- or control vehicle (saline)- treated C57B/6J mouse hearts subjected to either DON (0.05 mg/kg body weight/day) or saline for 7 days.
(B–E) Quantitative analysis of phospho-Akt, phospho-ERK, phospho-p38, phospho-JNK, and GAPDH in ISO- or control vehicle-treated C57B/6J mouse hearts subjected to either DON (0.05 mg/kg body weight/day) or saline for 7 days (n = 9). ∗p < 0.05: post hoc Tukey's comparison test.
Discussion
This study demonstrated that GFAT2 was universally upregulated in the heart in response to hypertrophic stimuli and GFAT2 critically mediated cardiomyocyte hypertrophy by enhancing protein O-GlcNAcylation and activation of Akt. In addition, HIF-1α, CaMKIIγ, and CaMKIIδ were the major regulators of GFAT2 in cardiomyocyte hypertrophy. Furthermore, DON, an inhibitor of GFAT, exerted anti-hypertrophic effects by suppressing GFAT2 downstream signaling in vivo. This study is the first report demonstrating the critical role of GFAT2 and its downstream signaling in cardiomyocyte hypertrophy (Figure S19).
HBP flux is regulated by nutrient intake of glucose and glutamine, which is elevated under several stress conditions (Brownlee, 2001). In cardiac hypertrophy, the metabolic shift from FAO to glucose metabolism has been identified (Dorn, 2007). HBP is thought to be intimately involved in cardiac hypertrophy. Of note, a recent study demonstrated that GFAT1 mediated pathological cardiac hypertrophy in response to pressure overload (Tran et al., 2020). In the present study, we revealed that GFAT2 was upregulated by not only pressure overload but also other hypertrophic stimuli such as α1-adrenergic receptor, β-adrenergic receptor, and renin-angiotensin system stimulation (Figures 1 and S1). Furthermore, GFAT2, but not GFAT1, was increased by ISO administration and GFAT2 knockdown attenuated cardiomyocyte hypertrophy (Figure 2). GFAT2 knockdown did not affect GFAT1 protein levels (Figure 2C), whereas GFAT1 knockdown increased GFAT2 protein levels in cardiomyocytes with or without ISO stimulation (Figure S10A). Furthermore, GFAT1 knockdown did not affect ISO-induced protein O-GlcNAcylation in spite of its anti-hypertrophic effect (Figures S10A and S10B). These data raise a possibility that GFAT2 is upregulated in compensation for GFAT1 downregulation and provide evidence that GFAT2 is a critical regulator of protein O-GlcNAcylation and cardiomyocyte hypertrophy by ISO stimulation. In addition, GFAT1 is ubiquitously expressed in several organs, whereas GFAT2 is predominantly expressed in hearts (Figure 1). Therefore, GFAT2 could be a selective target of cardiac hypertrophy without affecting the HBP in other tissues except for hearts.
HBP positively regulates protein O-GlcNAcylation, which is involved in pathophysiology in the heart (Cannon et al., 2015). Acute increases in O-GlcNAcylation levels were protective against cardiac ischemia/reperfusion injury (Chatham and Marchase, 2010; Laczy et al., 2010). Deletion of O-GlcNAcylation accelerated the progression to HF following coronary artery ligation (Watson et al., 2010). OGT promoted compensated cardiac function in response to pressure overload (Zhu et al., 2019). On the other hand, OGA suppressed myofilament O-GlcNAcylation and restores Ca2+ sensitivity in diabetic cardiac muscle in diabetes (Ramirez-Correa et al., 2015). The role of O-GlcNAcylation in cardiovascular diseases depends on the pathological state and disease phase (Wright et al., 2017). In the context of cardiac hypertrophy, protein O-GlcNAcylation has pro-hypertrophic effects. In this study, knockdown of GFAT2 attenuated ISO-induced protein O-GlcNAcylation, especially Akt O-GlcNAcylation. Among several hypertrophic signaling including JNK, ERK, p38, and Akt, only Akt activation was suppressed by knockdown of GFAT2 (Figure 3A). In addition, the combination of GFAT2 knockdown and MK-2206 administration did not show additive anti-hypertrophic effect compared with MK-2206 alone (Figure 3I). Akt is a critical mediator of physiological and pathological cardiac hypertrophy (Aoyagi and Matsui, 2011). O-GlcNAcylation is known to positively or negatively regulate protein function. Indeed, several previous studies demonstrated that Akt was activated by its O-GlcNAcylation (Heath et al., 2014; Liu et al., 2018). In this study, the mutant Akt in T479A was resistant to ISO-induced phosphorylation of Akt and cardiomyocyte hypertrophy (Figures 3G and 3H). Therefore, O-GlcNAcylation of Akt by GFAT2 is thought to mediate cardiomyocyte hypertrophy. However, other pro-hypertrophic factors such as NFAT and Troponin T have been shown to be O-GlcNAcylated (Mailleux et al., 2016). We could not completely exclude the effect of O-GlcNAcylation of NFAT and Troponin T on cardiac hypertrophy. Further investigations are needed to elucidate all targets of O-GlcNAcylation during cardiac hypertrophy.
Implication of protein O-GlcNAcylation in physiological and pathological hypertrophy is an important issue. Swim-exercised mice showed a decreased level of protein O-GlcNAcylation in heart (Belke, 2011). Therefore, there might be a different regulatory mechanism of protein O-GlcNAcylation between pathological and physiological hypertrophy. The findings from this previous study and our results raise a possibility that Akt activation during pathological hypertrophy depends on O-GlcNAcylation, whereas its activation during physiological hypertrophy does not. Akt O-GlcNAcylation is thought to be a potential therapeutic target for pathological hypertrophy without affecting physiological hypertrophy.
Mammalian target of rapamycin (mTOR) is reported to be a downstream target of HBP in cardiac hypertrophy (Tran et al., 2020). Akt positively regulates activation of mTORC1 (Dan et al., 2014). Based on these findings, it is speculated that activation of Akt by O-GlcNAcylation could induce activation of mTOR and its downstream signaling such as S6 and p70S6K in cardiac hypertrophy. Our results showed that GFAT2 knockdown and DON attenuated ISO-induced phosphorylation of S6 and p70S6K in cardiomyocytes and hearts, respectively (Figures S5 and S18).
In this study, glucosamine, a substrate of hexokinase in HBP, increased protein O-GlcNAcylation and Akt activation, leading to cardiomyocyte hypertrophy. Interestingly, knockdown of GFAT2 did not inhibit glucosamine-induced O-GlcNAcylation-Akt signaling and cardiomyocyte hypertrophy (Figure 4). These findings indicate that HBP-O-GlcNAcylation-Akt axis is a downstream of GFAT2. The role of glucosamine in cardiac hypertrophy is controversial. Tran et al. demonstrated that glucosamine (5 mM, 48 h) was sufficient to increase cell size in cardiomyocytes (Tran et al., 2020). On the other hand, Gelinas et al. demonstrated that glucosamine (5 mM, 24 h) did not increased cell size in cardiomyocytes (Gelinas et al., 2018). In this study, glucosamine (5 mM, 12 h) increased cell size in cardiomyocytes. All these studies used the same concentration of glucosamine at 5 mM. In addition, the result of the shortest incubation time (12 h) was the same as that of the longest one (24 h). Therefore, the concentration and duration do not seem to be a cause of the discrepancy. Based on these findings, we speculate several potential reasons for this discrepancy. First, differences in components of culture medium might affect the hypertrophic effect of glucosamine. Second, related to above, unknown factors might be needed for O-GlcNAcylation of specific proteins, such as Akt, by glucosamine administration. However, to clarify these points, further investigations are needed.
To date, the regulatory mechanism of GFAT2 has not been elucidated. In this study, high glucose did not increase GFAT2 protein levels but ISO stimulation increased them independently of glucose levels (Figure S14A). Consistently, protein O-GlcNAcylation did not depend on glucose concentrations (Figure S14B). Thus, hypertrophic stimuli might be main drivers of protein GFAT2 and O-GlcNAcylation rather than glucose levels during cardiac hypertrophy. In addition, we here found that HIF-1α, a master regulator of glycolysis, positively regulated GFAT2 mRNA and protein levels (Figure 5). CaMKIIγ and CaMKIIδ, major factors of calcium handling, positively regulated GFAT2 protein levels (Figures S12 and S13). Based on these findings, glucose metabolism and calcium handling by hypertrophic stimuli might be implicated with cardiac hypertrophy at least partially through GFAT2. However, CaMKIIγ and CaMKIIδ did not change GFAT2 mRNA levels (Figures S3 and S4). Thus, it is thought that HIF-1α transcriptionally regulates GFAT2 but CaMKIIγ and CaMKIIδ regulate it post-translationally.
Development of a pharmacological approach targeting GFAT2-HBP- O-GlcNAcylation axis is an important strategy for establishing anti-hypertrophic therapy. We investigated anti-hypertrophic effects of DON, an inhibitor of GFAT, in this study. DON effectively suppressed ISO-induced protein O-GlcNAcylation and cardiac hypertrophy without changes in hemodynamics such as heart rate and blood pressure (Figure 6 and S16). Since DON inhibits GFAT activity as a glutamine analog, its anti-hypertrophic effect might be due to inhibition of both GFAT1 and GFAT2. However, DON attenuated Akt activation in addition to protein O-GlcNAcylation (Figure 7). These data indicate that DON exerts anti-hypertrophic effect by inhibition of GFAT2-O-GlcNAcylation-Akt axis. We speculate that GFAT2 is a major regulator of HBP in the heart and inhibition of not only GFAT1 but also GFAT2 is indispensable for treatment of cardiac hypertrophy and that glutamine analogues such as DON could be a novel potential anti-hypertrophic drug.
GFAT2 was expressed in not only cardiomyocytes but also fibroblasts. This finding raises a possibility that GFAT2 could be involved in the function of fibroblast such as fibrosis. We confirmed that ISO increased GFAT2 protein levels in cardiac fibroblasts (shown in Figure S20A but not in results section). However, GFAT2 knockdown did not change the mRNA levels of collagen I and III in cardiac fibroblasts treated with ISO (Figure S20B). These data suggest that GFAT2 is not involved in fibrosis in cardiac fibroblasts and are consistent with no anti-fibrotic effect of DON in vivo (Figure S17).
We did not evaluate diastolic function such as mitral inflow and e’. However, the echocardiographic analysis demonstrated that DON attenuated ISO-indued cardiac hypertrophy in mice. Furthermore, DON decreased heart weight, an index of cardiac hypertrophy. These hypertrophic phenotypes are intimately related to diastolic dysfunction (Schnelle et al., 2018). DON might have a beneficial effect on diastolic dysfunction.
DON has been studied for more than 50 years as a potential anticancer therapeutic drug (Lemberg et al., 2018). DON binds competitively to the glutamine active site of enzymes and forms a covalent adduct with them, leading to their inhibition (Thomas et al., 2013). DON has been reported to inhibit GFAT at low micromolar levels (Walker et al., 2000). Although clinical studies regarding a low dose of DON demonstrated antitumor activity, phase I and II trials of a high dose of DON were hampered by dose-limiting gastrointestinal symptoms such as nausea and vomiting (Lemberg et al., 2018). However, recently, prodrug strategies for DON have been developed as cancer therapies (Rais et al., 2016). A pre-clinical study demonstrated that an oral DON prodrug suppressed MYC-driven medulloblastoma (Hanaford et al., 2019). Based on these studies and our finding, DON prodrugs could be clinically applicable for anti-hypertrophic therapy without toxicity.
Although several studies regarding GFAT, HBP, and cardiac hypertrophy has been already reported (Gelinas et al., 2018; Tran et al., 2020), our study revealed the role of GFAT2 in Akt O-GlcNAcylation and cardiac hypertrophy. This study provides proof-of-principal evidence that GFAT2 inhibition can ameliorate pathological cardiac hypertrophy. However, the optimal approach for targeting this pathway still needs to be developed.
In summary, we demonstrated that GFAT2 critically mediates cardiomyocyte hypertrophy through the HBP-O-GlcNAcylation-Akt pathway. GFAT2 might be a potential novel therapeutic target for cardiac hypertrophy.
Limitation of the study
We acknowledge several limitations of our study. First, we could not measure GFAT activity because of technical difficulty. To further elucidate the role of GFAT isoforms, GFAT activity needs to be evaluated. However, knockdown of GFAT2 efficiently suppressed protein O-GlcNAcylation, suggesting that GFAT2 could regulate HBP. Second, we conducted loss of GFAT2 function experiments with a chemical inhibitor rather than cardiomyocyte-specific genetically altered animals. We did not use specific inhibition of GFAT2 in in vivo experiment. However, we found that GFAT2 critically mediated cardiomyocyte hypertrophy in in vitro study. In addition, DON suppressed the O-GlcNAcylation-Akt axis, which is a downstream signaling of GFAT2. We provide novel evidence that GFAT2 is involved in cardiomyocyte hypertrophy. Finally, we could not detect O-GlcNAcylation of Akt in the heart. However, we confirmed it in cardiomyocytes in in vitro experiment, suggesting a direct modification of Akt by O-GlcNAcylation. Despite several limitations described above, this study shed light on a glutamine analogue as an anti-hypertrophic drug. The therapeutic strategy to interfere with the GFAT2-HBP-O-GlcNAcylation pathway has a potential for development of treatment of HF because cardiac hypertrophy is a major cause of HF. This study provides a better understanding of the mechanism regarding GFAT and HBP in cardiac hypertrophy.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-GFAT1 | Abcam | Cat#ab125069; RRID: AB_10975709 |
| Rabbit monoclonal anti-GFAT2 | Abcam | Cat#ab190966; RRID: AB_2868470 |
| Rabbit monoclonal anti-OGT | Cell Signaling Technology | Cat#24083; RRID: AB_2716710 |
| Rabbit polyclonal anti-OGA | Novus Biologicals | Cat#NBP1-81244; RRID:AB_11053819 |
| Mouse monoclonal anti-O-GlcNAc(CTD110.6) | Cell Signaling Technology | Cat#9875; RRID: AB_10950973 |
| Mouse monoclonal anti-O-GlcNAc(RL2) | Novus Biologicals | Cat#NBP300-524; RRID: AB_10001871 |
| Rabbit monoclonal anti-Akt | Cell Signaling Technology | Cat#4691; RRID: AB_915783 |
| Rabbit monoclonal anti-phospho-Akt(S473) | Cell Signaling Technology | Cat#4060; RRID: AB_2315049 |
| Rabbit monoclonal anti-phospho-Akt(T308) | Cell Signaling Technology | Cat#13038; RRID: AB_2629447 |
| Rabbit monoclonal anti-mTOR | Cell Signaling Technology | Cat#2972; RRID: AB_330978 |
| Rabbit monoclonal anti-phospho-mTOR | Cell Signaling Technology | Cat#5536; RRID: AB_10691552 |
| Rabbit monoclonal anti-S6 | Cell Signaling Technology | Cat#2217; RRID: AB_331355 |
| Rabbit monoclonal anti-phospho-S6(Ser235/236) | Cell Signaling Technology | Cat#4858; RRID: AB_916156 |
| Rabbit monoclonal anti-p70 S6 Kinase | Cell Signaling Technology | Cat#2708; RRID: AB_390722 |
| Rabbit monoclonal anti-phospho-p70 S6 Kinase | Cell Signaling Technology | Cat#9234; RRID: AB_2269803 |
| Rabbit polyclonal anti-ERK | Cell Signaling Technology | Cat#9102; RRID: AB_330744 |
| Rabbit polyclonal anti-phospho-ERK | Cell Signaling Technology | Cat#9101 |
| Rabbit polyclonal anti-p38 | Cell Signaling Technology | Cat#9212; RRID: AB_330713 |
| Rabbit monoclonal anti-phospho-p38 | Cell Signaling Technology | Cat#4511; RRID: AB_2139682 |
| Rabbit polyclonal anti-JNK | Cell Signaling Technology | Cat#9252; RRID: AB_2250373 |
| Rabbit monoclonal anti-phospho-JNK | Cell Signaling Technology | Cat#4668; RRID: AB_823588 |
| Rabbit monoclonal anti-HIF-1α | Cell Signaling Technology | Cat#36169; RRID: AB_2799095 |
| Rabbit monoclonal anti-CaMKII | Cell Signaling Technology | Cat#4436; RRID: AB_10545451 |
| Mouse monoclonal anti-β-Actin | Santa Cruz Biotechnology | Cat#sc47778; RRID:AB_2714189 |
| Mouse monoclonal anti-GAPDH | Santa Cruz Biotechnology | Cat#sc32233; RRID:AB_627679 |
| Rabbit monoclonal anti-Troponin I | Cell Signaling Technology | Cat#13083; RRID: AB_2798114 |
| Fluorescein goat anti-rabbit IgG antibody | Vector Laboratories | Cat#FI-1000; RRID:AB_2336197 |
| Bacterial and virus strains | ||
| pAV[Exp]-EGFP-CMV>6xHis/rAkt1(T479A) | VectorBuilder | N/A |
| pAV[Exp]-EGFP-CMV>6xHis/rAkt1 | VectorBuilder | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Angiotensin II | Sigma Aldrich | Cat#A9525; CAS:4474-91-3 |
| 6-Diazo-5-oxo-L-norleucine crystalline (DON) | Sigma Aldrich | Cat#D2141; CAS:157-03-9 |
| D-glucosamine hydrochloride | Sigma Aldrich | Cat#G1514; CAS:66-84-2 |
| MK-2206 | Selleck Chemicals | Cat#S1078; CAS:1032350-13-2 |
| Isoproterenol hydrochloride | Tokyo Chemical Industry | Cat#I0260; CAS:51-30-9 |
| Phenylephrine hydrochloride | Sigma Aldrich | Cat#P6126; CAS:61-76-7 |
| Experimental models: Cell lines | ||
| C2C12 | Masataka Ikeada | NA |
| HEK293 | Masataka Ikeada | NA |
| Experimental models: Organisms/strains | ||
| Mouse C57BL/6, male | CREA Japan | https://www.clea-japan.com/en/products/inbred/item_a0420 |
| Oligonucleotides | ||
| qPCR primers | See Table S1 | NA |
| siRNA targeting sequence: GFAT2 | Thermo Fisher Scientific | Cat#s165752 |
| siRNA targeting sequence: GFAT1 | Thermo Fisher Scientific | Cat#s150682 |
| siRNA targeting sequence: OGT | Thermo Fisher Scientific | Cat#s130676 |
| siRNA targeting sequence: OGA | Thermo Fisher Scientific | Cat#s139145 |
| siRNA targeting sequence: HIF-1α | Takara Bio | Cat#RS0035615 |
| siRNA targeting sequence: CaMKIIγ | Thermo Fisher Scientific | Cat#s139829 |
| siRNA targeting sequence: CaMKIIδ | Thermo Fisher Scientific | Cat#s127546 |
| Software and algorithms | ||
| Fusion Capt Advance | Vilber Lourmat | NA |
| BZ-X800 Analyzer (1.1.1.8) | Keyence corporation | NA |
| JMP pro 15 | SAS Institute | NA |
| Other | ||
| BZ-X800:fluorescent microscope | Keyence corporation | NA |
| NIBP Monitor for Rats & Mice | Muromachi Kikai | Cat#Model MK-1030 |
| Vevo 2100 ultrasonography system | Visual Sonics | NA |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Shouji Matsushima (shouji-m@cardiol.med.kyushu-u.ac.jp).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Cell cultures
Primary cultures of neonatal rat ventricular cardiomyocytes (NRVMs) were prepared from the ventricles of neonatal Sprague Dawley rats as described previously, with some modifications (Tadokoro et al., 2020). Neonatal rats were euthanized with an overdose of isoflurane, after which the hearts were rapidly excised and digested. After digestion of the myocardial tissues with trypsin (Thermo Fisher Scientific, 25300-062) and collagenase type 2 (Worthington Biochemical Corporation, LS004176), the cells were suspended in DMEM (Sigma-Aldrich, D5796) containing 10% FBS (HyClone Laboratories, SH30910.03) and 1% penicillin/streptomycin (P/S; Nacalai, 26253-84). Cells were plated twice in 100-mm culture dishes for 70 min each to reduce the number of non-myocytes. Non-adherent cells were plated in culture dishes (Primaria) at an appropriate density for each experiment as NRVMs. Adherent cells were passaged 2 times, and passaged cells (P2) were used as fibroblasts. They were maintained at 37°C in humidified air with 5% CO2. C2C12 cells and HEK cells were kindly gifted from Dr.Ikeda. C2C12 cells and HEK cells were maintained in DMEM containing 10% FBS and P/S at 37°C in humidified air with 5% CO2. In in vitro experiments, NRVMs and fibroblasts were treated with ISO (1 μM, 12-24 h), glucosamine (5 mM, 12-24 h), phenylephrine (100 μM, 12 h), DON (50 μM, 12 h), and MK-2206 (10 nM, 24 h). We used phosphate-buffered saline (PBS) for ISO, glucosamine, and phenylephrine as vehicle controls with a final volume concentration of 0.1%.
Animals
All mice are on the C57BL/6 background. Littermates of male mice were randomly assigned to experimental groups. Mice were maintained on a 12-hour light/dark cycle with free access to standard chow and water. All procedures involving animals and animal care protocols were approved by the Committee on Ethics of Animal Experiments of the Kyushu University Graduate School of Medicine and Pharmaceutical Sciences (A29-390) and were performed in accordance with the Guideline for Animal Experiments of Kyushu University and the Guideline for the Care and Use of Laboratory Animals published by the US National Institutes of Health (revised in 2011). Isoproterenol (ISO, 15 mg/kg body weight/day) or vehicle (saline) was continuously infused into 8 to 9-week-old mice using a miniosmotic pump (model 1007D, Alzet) for 1 week. In addition to ISO, 6-diazo-5-oxo-L-norleucine crystalline (DON, 0.05 μg/kg body weight/day) was also continuously infused into mice using another miniosmotic pump for 1 week. Phenylephrine (PE, 100 mg/kg body weight/day) or vehicle (saline) was continuously infused into 9 to 10-week-old mice using a miniosmotic pump (model 1007D, Alzet) for 1 week. Angiotensin II (Ang II, 1.44 mg/kg body weight/day) or vehicle (saline) was continuously infused into 8 to 9-week-old mice using a miniosmotic pump (model 1007D, Alzet) for 1 week. The methods used to impose PO in mice have been described (Furihata et al., 2016). 8 to 9-week-old mice were anesthetized with a mixture of ketamine (0.065 mg/g), xylazine (0.013 mg/g), and acepromazine (0.002 mg/g) and mechanically ventilated. The left side of the chest was opened at the second intercostal space. Transverse aortic constriction (TAC) was performed by ligation of the transverse thoracic aorta between the innominate artery and left common carotid artery with a 28-gauge needle using a 7-0 nylon suture. Sham mice (Sham) had their aorta mobilized, but no suture tightened. We performed our experiments after 1 week of TAC.
Method details
Quantitation of cell surface area of NRVMs
NRVMs grown glass plate dish were washed 3 times with PBS. The cells were fixed with 4% paraformaldehyde for 15 min and washed 3 times with PBS. The cells were then permeabilized with 0.1% Triton X-100 for 15 min and washed 3 times with PBS. The cells were incubated with blocking buffer (1% BSA in PBS) for 1 hour and then in primary anti-Troponin I antibody (1:400, #13083, Cell Signaling Technology) for another hour. The cells were washed with PBS 3 times and incubated with Fluorescein goat anti-rabbit IgG antibody (1:400, Fl-1000, Vector Laboratories) for 1 hour. Excess antibody was removed by 3 times washes of PBS. The cells then mounted with VECTASHIELD mounting medium with DAPI (H-1200, Vector Laboratories). Pictures of cardiomyocytes were obtained with a fluorescent microscope (BZ-X800, Keyence). Cell surface area of 150 cardiomyocytes randomly selected from five dishes in each group was evaluated with the BZ-X800 Analyzer (1.1.1.8) software.
Immunoblot analyses
Cardiomyocyte lysates and heart homogenates were prepared in RIPA lysis buffer containing 50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholae, and 0.1% SDS. Lysates were centrifuged at 4°C, 13,200 rpm for 10 minutes to remove cell debris and protein concentration of the resulting supernatant was measured by BCA assay. The samples were separated by SDS-PAGE gels and proteins were transferred to nitrocellulose membranes at 100 Volts for 1 hour. Membranes were blocked in 5% skim milk diluted in TBST for 1 hr at room temperature, incubated with primary antibodies overnight at 4°C and secondary antibodies for 1 hr at room temperature. Membranes were exposed by enhanced chemiluminescence method and signal intensities were quantified by Fusion Capt software(Vilber Lourmat). For immunoblot analyses, we used monoclonal antibodies against GFAT1 (1:1000, ab125069, Abcam), GFAT2 (1:5000, ab190966, Abcam), OGT (1:5000, #24083, Cell Signaling Technology), O-GlcNAc (CTD110.6) (1:1000, #9875, Cell Signaling Technology), Akt (1:10000, #4691, Cell Signaling Technology), phospho-Akt (S473) (1:5000, #4060, Cell Signaling Technology), phospho-Akt (T308) (1:5000, #13038, Cell Signaling Technology), mTOR (1:5000, #2972, Cell Signaling Technology), phospho-mTOR (1:5000, #5536, Cell Signaling Technology), S6 (1:5000, #2217, Cell Signaling Technology), phospho-S6 (S235/236) (1:5000, #4858, Cell Signaling Technology), p70S6K (1:5000, #2708, Cell Signaling Technology), phospho-p70S6K (1:5000, #9234, Cell Signaling Technology), phospho-p38 (1:1000, #4511, Cell Signaling Technology), phospho-JNK (1:1000, #4668, Cell Signaling Technology), HIF-1α (1:5000, #36169, Cell Signaling Technology), CaMKII (1:5000, #4436, Cell Signaling Technology), β-Actin (1:10000, sc47778, Santacruz), and GAPDH (1:10000, sc32233, Santacruz), and polyclonal antibodies against OGA (1:5000, NBP1-81244, Novus), ERK (1:5000, #9102, Cell Signaling Technology), phospho-ERK (1:5000, #9101, Cell Signaling Technology), p38 (1:1000, #9212 Cell Signaling Technology), and JNK (1:1000, #9252, Cell Signaling Technology).
Immunoprecipitation
Cardiomyocytes were homogenized in RIPA lysis buffer containing 50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholae, and 0.1% SDS on ice. Lysates were centrifuged at 13,200 rpm for 10 minutes, and the supernatants were collected at 4°C. Lysates were made to a protein concentration of 1 μg/μl with RIPA buffer and incubated with anti-O-GlcNAc antibody (RL-2, NB300-524, Novus) or control IgG for 2 h with slow rotation at 4°C. Lysates were added protein G beads, and incubated for 1 h with slow rotation at 4°C. Lysates were centrifuged at 13,200 rpm for 5 minutes, pellets washed with RIPA buffer and centrifuged again. This process was repeated 3 times. Pellets eluted by boiling for 5 min in Laemmli buffer, and subjected to Western blotting. Lysate diluted to the immunoprecipitation (IP) concentration was used for input.
Quantitative real-time PCR reaction
Methods of quantitative RT-PCR have been described previously (Zhai et al., 2005). In brief, total RNA was extracted using an RNeasy Mini Kit (Qiagen), RNA was converted to cDNA using ReverTra Ace qPCR RT Kit (TOYOBO), and the reactions were run in an Applied Biosystems QuantStudio3 (Thermo Fisher Scientific) for the THUNDERBIRD SYBR qPCR Mix(TOYOBO). The specific oligonucleotide primers for 18S, ANF, collagen I, collagen III, GFAT1, and GFAT2 were selected using Perfect Real Time Primer (TAKARA). The following oligonucleotide primers were used in this study: 18S (rat), sense 5′-AAGTTTCAGCACATCCTGCGAGTA-3′ and antisense 5′-TTGGTGAGGTCAATGTCTGCTTTC-3′; ANF (rat), sense 5-TGACAGGATTGGAGCCCAGAG-3′ and antisense 5′-TCGATCGTGATAGATGAAGACAGGA-3′; GFAT1 (rat), sense 5′-GGCGACAGAGCTTTACCACCA-3′ and antisense 5′-CACCAGCAAGGATGCCTTCA-3′; GFAT2 (rat), sense 5′-CATCCGTGGCCTCAGATCTTTA-3′ and antisense 5′-CTTCCAGGCATGTGGCATAGTTA-3′; 18S (mus), sense 5′-TTCTGGCCAACGGTCTAGACAAC-3′ and antisense 5′-CCAGTGGTCTTGGTGTGCTGA-3′; ANF (mus), sense 5′-TGACAGGATTGGAGCCCAGA-3′ and antisense 5′-GACACACCACAAGGGCTTAGGA-3′; GFAT1 (mus), sense 5′-GCAGAGCTAGACCCTCAGAGCAA-3′ and antisense 5′-CCCAGCATTTAATACCTCAGCACAG-3′; GFAT2 (mus), sense 5′-TCCTGAGCGTGATTCCACTCC-3′ and antisense 5′-GCAGGTGACGACAGTCTTGTGATAG-3′; CollagenI (mus), sense 5′-GACATGTTCAGCTTTGTGGACCTC-3′ and antisense 5′-GGGACCCTTAGGCCATTGTGTA-3′; CollagenIII (mus), sense 5′-CAGGAGCCAGTGGCCATAA-3′ and antisense 5′-TCTCGACCTGGCTGACCATC-3′.
Transfection of siRNA and adenoviruses
Silencing of GFAT2, GFAT1, OGT, OGA, Hif-1α, CaMKIIγ, and CaMKIIδ gene expressions in primary neonatal rat cardiomyocytes were achieved by the small interfering RNA (siRNA) technique. Transfection of cultured cardiomyocytes was carried out by Lipofectamine RNAiMAX (Themo Fisher Scientific) according to proposed protocol. Briefly, 24 h after seeding, the cardiomyocytes were transfected with siRNA and incubated for 48 h. siRNAs against GFAT2 (s165752, used at 5 nmol/L, Thermo Fisher Scientific), GFAT1 (s150682, used at 5 nmol/L, Thermo Fisher Scientific), OGT (s130676, used at 5 nmol/L, Thermo Fisher Scientific), OGA (s139145, used at 5 nmol/L, Thermo Fisher Scientific), HIF-1α (RS0035615, used at 1 nmol/L, Takara Bio Inc), CaMKIIγ (s139829, used at 5 nmol/L, Thermo Fisher Scientific), and CaMKIIδ (s127546, used at 5 nmol/L, Thermo Fisher Scientific) were commercially purchased. Overexpression of wild-type Akt and mutant Akt in NRVMs were achieved by infection of adenoviruses (VectorBuilder Inc.) harboring wild-type Akt or mutant Akt in T479A 48 h after seeding.
Measurement of blood pressure and heart rate
The mice to be measured were placed into their restrainers, allowed the mice to acclimate to the restrainer for 5 min prior to initiating the blood pressure measurement. Following the 5-min acclimation period, blood pressure and heart rate were measured using a noninvasive tail cuff system (Model MK-1030 NIBP Monitor for Rats & Mice). The mice were warmed by heating pads under the restrainer platform during the acclimation and measurement periods to ensure sufficient blood flow to the tail.
Echocardiography
Under light anesthesia with 1-2% isoflurane, two-dimensional targeted M-mode images were obtained from the short axis view at the papillary muscle level using a Vevo 2100 ultrasonography system (Visual Sonics, Toronto, Canada). The LVEF was calculated by the following formula: LVEF = [(diastolic LV volume—systolic LV volume)/diastolic LV volume) × 100], where LV volume = [(7.0/(2.4 + LV diameter)] × LV diameter. LV wall thickness was calculated as average of interventricular septum thickness and posterior wall thickness.
Histological analyses
The LV accompanied by the septum was cut into base, midportion, and apex, fixed with 10% formalin, and submitted for Masson's trichrome staining. The slides imaged with an optical microscope (BZ-X800, Keyence) using a 40× objective. Myocyte cross-sectional area and collagen volume fraction were determined by quantitative morphometry of tissue sections from the mid-LV using the BZ-X800 Analyzer (1.1.1.8) software (Keyence).
Quantification and statistical analysis
All values are expressed as mean ± SEM. Comparison between two groups was analyzed by unpaired two-tailed Student's t-test and comparison among three or more groups was analyzed by one-way ANOVA test. JMP software was used for analyzing data. Difference was considered to be significant when p value < 0.05 (∗), p value < 0.01 (∗∗).
Acknowledgments
We are very grateful to A. Hanada and M. Sato for technical support. We also appreciate the technical assistance from the Research Support Center, Faculty of Medical Sciences, Kyushu University. This work was supported by Japan Society for the Promotion of Science (JSPS) KAKENHI (Grant Nos. 17K09581, 19K22622, and 19H03655) and Vascular Biology Innovation Conference (VBIC).
Author contributions
A.I. and S.M. designed the research. A.I., S.I., K.O., N.E., T.Y., M.S., and Y.T. performed in vivo experiments. A.I., S.I., R.N., T.T., and R.M. performed in vitro experiments. A.I., S.M., M.I., T.I., and H.T. analyzed data. M.I., T.I., S.K., and H.T. provided guidance on experimental designs and data analyses. A.I. and S.M. wrote the manuscript. All authors provided feedback on the manuscript.
Declaration of interests
The authors declare that they have no competing interests.
Published: December 17, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.103517.
Supplemental information
Data and code availability
Data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Aoyagi T., Matsui T. Phosphoinositide-3 kinase signaling in cardiac hypertrophy and heart failure. Curr. Pharm. Des. 2011;17:1818–1824. doi: 10.2174/138161211796390976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belke D.D. Swim-exercised mice show a decreased level of protein O-GlcNAcylation and expression of O-GlcNAc transferase in heart. J. Appl. Physiol. 2011;111:157–162. doi: 10.1152/japplphysiol.00147.2011. [DOI] [PubMed] [Google Scholar]
- Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813–820. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- Cannon M.V., Sillje H.H., Sijbesma J.W., Vreeswijk-Baudoin I., Ciapaite J., van der Sluis B., van Deursen J., Silva G.J., de Windt L.J., Gustafsson J.A., et al. Cardiac LXRalpha protects against pathological cardiac hypertrophy and dysfunction by enhancing glucose uptake and utilization. EMBO Mol. Med. 2015;7:1229–1243. doi: 10.15252/emmm.201404669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatham J.C., Marchase R.B. The role of protein O-linked beta-N-acetylglucosamine in mediating cardiac stress responses. Biochim. Biophys. Acta. 2010;1800:57–66. doi: 10.1016/j.bbagen.2009.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dan H.C., Ebbs A., Pasparakis M., Van Dyke T., Basseres D.S., Baldwin A.S. Akt-dependent activation of mTORC1 complex involves phosphorylation of mTOR (mammalian target of rapamycin) by IkappaB kinase alpha (IKKalpha) J. Biol. Chem. 2014;289:25227–25240. doi: 10.1074/jbc.M114.554881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorn G.W., 2nd The fuzzy logic of physiological cardiac hypertrophy. Hypertension. 2007;49:962–970. doi: 10.1161/HYPERTENSIONAHA.106.079426. [DOI] [PubMed] [Google Scholar]
- Facundo H.T., Brainard R.E., Watson L.J., Ngoh G.A., Hamid T., Prabhu S.D., Jones S.P. O-GlcNAc signaling is essential for NFAT-mediated transcriptional reprogramming during cardiomyocyte hypertrophy. Am. J. Physiol. Heart Circulatory Physiol. 2012;302:H2122–H2130. doi: 10.1152/ajpheart.00775.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furihata T., Kinugawa S., Takada S., Fukushima A., Takahashi M., Homma T., Masaki Y., Tsuda M., Matsumoto J., Mizushima W., et al. The experimental model of transition from compensated cardiac hypertrophy to failure created by transverse aortic constriction in mice. Int. J. Cardiol. Heart Vasc. 2016;11:24–28. doi: 10.1016/j.ijcha.2016.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelinas R., Mailleux F., Dontaine J., Bultot L., Demeulder B., Ginion A., Daskalopoulos E.P., Esfahani H., Dubois-Deruy E., Lauzier B., et al. AMPK activation counteracts cardiac hypertrophy by reducing O-GlcNAcylation. Nat. Commun. 2018;9:374. doi: 10.1038/s41467-017-02795-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haider A.W., Larson M.G., Benjamin E.J., Levy D. Increased left ventricular mass and hypertrophy are associated with increased risk for sudden death. J. Am. Coll. Cardiol. 1998;32:1454–1459. doi: 10.1016/s0735-1097(98)00407-0. [DOI] [PubMed] [Google Scholar]
- Hanaford A.R., Alt J., Rais R., Wang S.Z., Kaur H., Thorek D.L.J., Eberhart C.G., Slusher B.S., Martin A.M., Raabe E.H. Orally bioavailable glutamine antagonist prodrug JHU-083 penetrates mouse brain and suppresses the growth of MYC-driven medulloblastoma. Transl Oncol. 2019;12:1314–1322. doi: 10.1016/j.tranon.2019.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart G.W., Slawson C., Ramirez-Correa G., Lagerlof O. Cross talk between O-GlcNAcylation and phosphorylation: roles in signaling, transcription, and chronic disease. Annu. Rev. Biochem. 2011;80:825–858. doi: 10.1146/annurev-biochem-060608-102511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heath J.M., Sun Y., Yuan K., Bradley W.E., Litovsky S., Dell'Italia L.J., Chatham J.C., Wu H., Chen Y. Activation of AKT by O-linked N-acetylglucosamine induces vascular calcification in diabetes mellitus. Circ. Res. 2014;114:1094–1102. doi: 10.1161/CIRCRESAHA.114.302968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laczy B., Marsh S.A., Brocks C.A., Wittmann I., Chatham J.C. Inhibition of O-GlcNAcase in perfused rat hearts by NAG-thiazolines at the time of reperfusion is cardioprotective in an O-GlcNAc-dependent manner. Am. J. Physiol. Heart Circulatory Physiol. 2010;299:H1715–H1727. doi: 10.1152/ajpheart.00337.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledee D., Smith L., Bruce M., Kajimoto M., Isern N., Portman M.A., Olson A.K. c-Myc alters substrate utilization and O-GlcNAc protein posttranslational modifications without altering cardiac function during early aortic constriction. PLoS ONE. 2015;10:e0135262. doi: 10.1371/journal.pone.0135262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemberg K.M., Vornov J.J., Rais R., Slusher B.S. We're not “DON” yet: optimal dosing and prodrug delivery of 6-diazo-5-oxo-L-norleucine. Mol. Cancer Ther. 2018;17:1824–1832. doi: 10.1158/1535-7163.MCT-17-1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy D., Garrison R.J., Savage D.D., Kannel W.B., Castelli W.P. Prognostic implications of echocardiographically determined left ventricular mass in the Framingham Heart Study. New Engl. J. Med. 1990;322:1561–1566. doi: 10.1056/NEJM199005313222203. [DOI] [PubMed] [Google Scholar]
- Liu Y., Cao Y., Pan X., Shi M., Wu Q., Huang T., Jiang H., Li W., Zhang J. O-GlcNAc elevation through activation of the hexosamine biosynthetic pathway enhances cancer cell chemoresistance. Cell Death Dis. 2018;9:485. doi: 10.1038/s41419-018-0522-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunde I.G., Aronsen J.M., Kvaloy H., Qvigstad E., Sjaastad I., Tonnessen T., Christensen G., Gronning-Wang L.M., Carlson C.R. Cardiac O-GlcNAc signaling is increased in hypertrophy and heart failure. Physiol. Genomics. 2012;44:162–172. doi: 10.1152/physiolgenomics.00016.2011. [DOI] [PubMed] [Google Scholar]
- Mailleux F., Gelinas R., Beauloye C., Horman S., Bertrand L. O-GlcNAcylation, enemy or ally during cardiac hypertrophy development? Biochim. Biophys. Acta. 2016;1862:2232–2243. doi: 10.1016/j.bbadis.2016.08.012. [DOI] [PubMed] [Google Scholar]
- Marshall S., Bacote V., Traxinger R.R. Discovery of a metabolic pathway mediating glucose-induced desensitization of the glucose transport system. Role of hexosamine biosynthesis in the induction of insulin resistance. J. Biol. Chem. 1991;266:4706–4712. [PubMed] [Google Scholar]
- Nascimben L., Ingwall J.S., Lorell B.H., Pinz I., Schultz V., Tornheim K., Tian R. Mechanisms for increased glycolysis in the hypertrophied rat heart. Hypertension. 2004;44:662–667. doi: 10.1161/01.HYP.0000144292.69599.0c. [DOI] [PubMed] [Google Scholar]
- Oki T., Yamazaki K., Kuromitsu J., Okada M., Tanaka I. cDNA cloning and mapping of a novel subtype of glutamine:fructose-6-phosphate amidotransferase (GFAT2) in human and mouse. Genomics. 1999;57:227–234. doi: 10.1006/geno.1999.5785. [DOI] [PubMed] [Google Scholar]
- Okin P.M., Devereux R.B., Nieminen M.S., Jern S., Oikarinen L., Viitasalo M., Toivonen L., Kjeldsen S.E., Dahlof B., Investigators L.S. Electrocardiographic strain pattern and prediction of new-onset congestive heart failure in hypertensive patients: the Losartan Intervention for Endpoint Reduction in Hypertension (LIFE) study. Circulation. 2006;113:67–73. doi: 10.1161/CIRCULATIONAHA.105.569491. [DOI] [PubMed] [Google Scholar]
- Olson A.K., Bouchard B., Zhu W.Z., Chatham J.C., Des Rosiers C. First characterization of glucose flux through the hexosamine biosynthesis pathway (HBP) in ex vivo mouse heart. J. Biol. Chem. 2020;295:2018–2033. doi: 10.1074/jbc.RA119.010565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rais R., Jancarik A., Tenora L., Nedelcovych M., Alt J., Englert J., Rojas C., Le A., Elgogary A., Tan J., et al. Discovery of 6-Diazo-5-oxo-l-norleucine (DON) prodrugs with enhanced CSF delivery in monkeys: a potential treatment for glioblastoma. J. Med. Chem. 2016;59:8621–8633. doi: 10.1021/acs.jmedchem.6b01069. [DOI] [PubMed] [Google Scholar]
- Ramirez-Correa G.A., Ma J., Slawson C., Zeidan Q., Lugo-Fagundo N.S., Xu M., Shen X., Gao W.D., Caceres V., Chakir K., et al. Removal of abnormal myofilament O-GlcNAcylation restores Ca2+ sensitivity in diabetic cardiac muscle. Diabetes. 2015;64:3573–3587. doi: 10.2337/db14-1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansbury B.E., DeMartino A.M., Xie Z., Brooks A.C., Brainard R.E., Watson L.J., DeFilippis A.P., Cummins T.D., Harbeson M.A., Brittian K.R., et al. Metabolomic analysis of pressure-overloaded and infarcted mouse hearts. Circ. Heart Fail. 2014;7:634–642. doi: 10.1161/CIRCHEARTFAILURE.114.001151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schillaci G., Verdecchia P., Porcellati C., Cuccurullo O., Cosco C., Perticone F. Continuous relation between left ventricular mass and cardiovascular risk in essential hypertension. Hypertension. 2000;35:580–586. doi: 10.1161/01.hyp.35.2.580. [DOI] [PubMed] [Google Scholar]
- Schnelle M., Catibog N., Zhang M., Nabeebaccus A.A., Anderson G., Richards D.A., Sawyer G., Zhang X., Toischer K., Hasenfuss G., et al. Echocardiographic evaluation of diastolic function in mouse models of heart disease. J. Mol. Cell Cardiol. 2018;114:20–28. doi: 10.1016/j.yjmcc.2017.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley W.C., Recchia F.A., Lopaschuk G.D. Myocardial substrate metabolism in the normal and failing heart. Physiol. Rev. 2005;85:1093–1129. doi: 10.1152/physrev.00006.2004. [DOI] [PubMed] [Google Scholar]
- Tadokoro T., Ikeda M., Ide T., Deguchi H., Ikeda S., Okabe K., Ishikita A., Matsushima S., Koumura T., Yamada K.I., et al. Mitochondria-dependent ferroptosis plays a pivotal role in doxorubicin cardiotoxicity. JCI Insight. 2020;5:e132747. doi: 10.1172/jci.insight.132747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas A.G., Rojas C., Tanega C., Shen M., Simeonov A., Boxer M.B., Auld D.S., Ferraris D.V., Tsukamoto T., Slusher B.S. Kinetic characterization of ebselen, chelerythrine and apomorphine as glutaminase inhibitors. Biochem. Biophys. Res. Commun. 2013;438:243–248. doi: 10.1016/j.bbrc.2013.06.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran D.H., May H.I., Li Q., Luo X., Huang J., Zhang G., Niewold E., Wang X., Gillette T.G., Deng Y., et al. Chronic activation of hexosamine biosynthesis in the heart triggers pathological cardiac remodeling. Nat. Commun. 2020;11:1771. doi: 10.1038/s41467-020-15640-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker B., Brown M.F., Lynas J.F., Martin S.L., McDowell A., Badet B., Hill A.J. Inhibition of Escherichia coli glucosamine synthetase by novel electrophilic analogues of glutamine--comparison with 6-diazo-5-oxo-norleucine. Bioorg. Med. Chem. Lett. 2000;10:2795–2798. doi: 10.1016/s0960-894x(00)00565-5. [DOI] [PubMed] [Google Scholar]
- Watson L.J., Facundo H.T., Ngoh G.A., Ameen M., Brainard R.E., Lemma K.M., Long B.W., Prabhu S.D., Xuan Y.T., Jones S.P. O-linked beta-N-acetylglucosamine transferase is indispensable in the failing heart. Proc. Natl. Acad. Sci. United States America. 2010;107:17797–17802. doi: 10.1073/pnas.1001907107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright J.N., Collins H.E., Wende A.R., Chatham J.C. O-GlcNAcylation and cardiovascular disease. Biochem. Soc. Trans. 2017;45:545–553. doi: 10.1042/BST20160164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X., Ongusaha P.P., Miles P.D., Havstad J.C., Zhang F., So W.V., Kudlow J.E., Michell R.H., Olefsky J.M., Field S.J., et al. Phosphoinositide signalling links O-GlcNAc transferase to insulin resistance. Nature. 2008;451:964–969. doi: 10.1038/nature06668. [DOI] [PubMed] [Google Scholar]
- Yi W., Clark P.M., Mason D.E., Keenan M.C., Hill C., Goddard W.A., 3rd, Peters E.C., Driggers E.M., Hsieh-Wilson L.C. Phosphofructokinase 1 glycosylation regulates cell growth and metabolism. Science. 2012;337:975–980. doi: 10.1126/science.1222278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young M.E., Yan J., Razeghi P., Cooksey R.C., Guthrie P.H., Stepkowski S.M., McClain D.A., Tian R., Taegtmeyer H. Proposed regulation of gene expression by glucose in rodent heart. Gene Regul. Syst. Biol. 2007;1:251–262. doi: 10.4137/grsb.s222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuzwa S.A., Shan X., Macauley M.S., Clark T., Skorobogatko Y., Vosseller K., Vocadlo D.J. Increasing O-GlcNAc slows neurodegeneration and stabilizes tau against aggregation. Nat. Chem. Biol. 2012;8:393–399. doi: 10.1038/nchembio.797. [DOI] [PubMed] [Google Scholar]
- Zhai P., Yamamoto M., Galeotti J., Liu J., Masurekar M., Thaisz J., Irie K., Holle E., Yu X., Kupershmidt S., et al. Cardiac-specific overexpression of AT1 receptor mutant lacking G alpha q/G alpha i coupling causes hypertrophy and bradycardia in transgenic mice. J. Clin. Invest. 2005;115:3045–3056. doi: 10.1172/JCI25330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W.Z., El-Nachef D., Yang X., Ledee D., Olson A.K. O-GlcNAc transferase promotes compensated cardiac function and protein kinase A O-GlcNAcylation during early and established pathological hypertrophy from pressure overload. J. Am. Heart Assoc. 2019;8:e011260. doi: 10.1161/JAHA.118.011260. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.