

Abstract
Severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) has evolved into eight fundamental clades with four of these clades (G, GH, GR, and GV) globally prevalent in 2020. To explain plausible epistatic effects of the signature co‐occurring mutations of these circulating clades on viral replication and transmission fitness, we proposed a hypothetical model using in silico approach. Molecular docking and dynamics analyses showed the higher infectiousness of a spike mutant through more favorable binding of G614 with the elastase‐2. RdRp mutation p.P323L significantly increased genome‐wide mutations (p < 0.0001), allowing for more flexible RdRp (mutated)‐NSP8 interaction that may accelerate replication. Superior RNA stability and structural variation at NSP3:C241T might impact protein, RNA interactions, or both. Another silent 5′‐UTR:C241T mutation might affect translational efficiency and viral packaging. These four G‐clade‐featured co‐occurring mutations might increase viral replication. Sentinel GH‐clade ORF3a:p.Q57H variants constricted the ion‐channel through intertransmembrane–domain interaction of cysteine(C81)‐histidine(H57). The GR‐clade N:p.RG203‐204KR would stabilize RNA interaction by a more flexible and hypo‐phosphorylated SR‐rich region. GV‐clade viruses seemingly gained the evolutionary advantage of the confounding factors; nevertheless, N:p.A220V might modulate RNA binding with no phenotypic effect. Our hypothetical model needs further retrospective and prospective studies to understand detailed molecular events and their relationship to the fitness of SARS‐CoV‐2.
Keywords: clades, co‐occurring mutations, COVID‐19, fitness, infection paradox, SARS‐CoV‐2, virulence
Highlights
Most dominant spike mutation favors elastase‐2 binding.
The polymerase mutant (P323L) virus may speed up replication that corresponds to higher mutations.
ORF3a viroporin substitution (Q57H) decreases ion permeability.
N protein mutation (RG203‐204KR) can increase nucleocapsid stability and help evade immunity.
Co‐occurring mutations might modulate viral replication and transmission fitness through epistasis.
1. INTRODUCTION
Coronavirus disease (COVID‐19) has caused 239 642 888 infection cases with 4 882 436 deaths worldwide until October 15, 2021 (https://coronavirus.jhu.edu/map.html). Severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2), the etiological agent of COVID‐19 pandemic, has gained some extraordinary attributes that make it extremely infectious: High replication rate, large burst size, high stability in the environment, strong binding efficiency of spike glycoprotein (S) receptor‐binding domain (RBD) with human angiotensin‐converting enzyme 2 (ACE2) receptor, and additional furin cleavage site in S protein. 1 , 2 , 3 In addition to those, it has proof‐reading capability ensuring relatively high‐fidelity replication. 4 The virus contains four major structural proteins: spike glycoprotein (S), envelope (E), membrane (M), and nucleocapsid (N) protein, along with 16 nonstructural proteins (NSP1–NSP16) and seven accessory proteins (ORF3a, ORF6, ORF7a, ORF7b, ORF8a, ORF8b, and ORF10). 5 , 6 Mutational spectra within the SARS‐CoV‐2 genome, 7 , 8 spike protein, 9 RdRp, 10 ORF3a, 11 and N protein 12 were reported earlier.
SARS‐CoV‐2 was classified into eight major clades, such as G, GH, GR, GV, S, V, L, and O by Global Initiative on Sharing All Influenza Data (GISAID) consortium (https://www.gisaid.org/) based on the dominant core mutations in genomes. Four of those clades (G, GH, GR, and GV) are globally and geographically prevalent in 2020. 13 Yin 14 reported that the 5ʹ‐untranslated region (5ʹ‐UTR) mutation 241C>T is co‐occurring with three other mutations, 3037 C>T (NSP3: C318T), 14408C>T (RdRp: p.P323L), and 23403A>G (S: p.D614G). GISAID referred to these co‐occurring mutations containing viruses as clade G (named after the spike D614G mutation) or PANGO (https://cov-lineages.org/) lineage B.1. 15 , 16 The GR clade or lineage B.1.1.* is classified with additional trinucleotide mutations at 28 881–28 883 (GGG>AAC), creating two consecutive amino acid (aa) changes, R203K and G204R, in N protein. Another derivative of G clade is GH or lineage B.1.*, characterized by an additional ORF3a:p.Q57H mutation. The variant GV or lineage B.1.177 featured an A222V mutation in the S protein and other mutations of the clade G. 13 , 16 Also, N: A220V, ORF10: V30L and three other synonymous mutations T445C, C6286T, and C26801G are observed for this clade. 17
The most frequently observed mutation is D614G of the S protein, 18 which has direct roles in receptor binding and immunogenicity, thus viral immune‐escape, transmission, and replication fitness. 19 , 20 Mutations in proteins other than spike could also affect viral pathogenicity and transmissibility, but the role of those dominant clade‐featured mutations has mainly remained underestimated. Although the possible role of ORF3a:p.Q57H in the replication cycle 21 has recently been investigated, the molecular perspective was not fully explored. The effects of 5′‐UTR: C241T, Leader: T445C, NSP3: C318T, RdRp:p.P323L, N:p.RG203‐203KR, and N:p.A220V are still being overlooked.
Different mutation(s) of SARS‐CoV‐2 may work independently or through epistatic interactions. 22 , 23 It is difficult to precisely determine how these co‐occurring mutations, if not all, might have gained their relative evolutionary fitness through alterations of protein/RNA structure, function, and cross‐talk (protein‐protein or protein‐RNA interaction). 22 , 24 , 25 Overall, this in silico study aims to determine any plausible individual or epistatic impact of those mutants during replication in terms of viral entry and fusion, evasion of host cell lysis, replication rate, ribonucleoprotein stability, protein–protein interactions, translational capacity, and ultimately the probable combined effect on viral transmission and fitness.
2. MATERIALS AND METHODS
2.1. Retrieval of sequences and mutation analyses
This study analyzed 240 207 high‐coverage (<1% Ns and <0.05% unique aa mutations) and complete (>29 000 nucleotides) genome sequences from a total of 3 22 142 sequences submitted to GISAID (https://www.epicov.org/epi3/frontend#18d55a) from January 1, 2020, to January 3, 2021 (Supplementary Material 1) for calculating the yearly percentage of the clades in 2020. We removed the nonhuman host‐generated sequences during data set preparation.
For the genome‐wide mutation analysis, we initially selected 37 179 sequences from our whole data set before performing an alignment with MAFFT v7 (https://mafft.cbrc.jp/alignment/server/) against the Wuhan‐Hu‐1 (Accession ID‐ NC_045512.2) isolate as the reference genome. A python script (https://github.com/hridoy04/counting-mutations) was used to partition that data set into two subsets (RdRp wild‐type or “C” variant: 9,815; and mutant or “T” variant: 27 364) based on the presence of RdRp: C14408T mutation and then estimate the genome‐wide single nucleotide polymorphisms (SNPs) for each strain (Supplementary Material 2). The SNP frequency was tested for significance with the Wilcoxon rank‐sum test between the RdRp “C” and “T” variants implemented in IBM SPSS statistics 25. We chose this non‐parametric test because a Kolmogorov–Smirnov test of mutational frequency showed these data do not fit a normal distribution (p < 0.001).
2.2. Stability, secondary and three‐dimensional structure prediction analyses of S, RdRp, ORF3a, and N proteins
We used DynaMut 26 and FoldX 5.0 27 , 28 to determine the stability of both wild and mutated variants of N, RdRp, S, and ORF3a proteins. The following NCBI reference sequences were used as the wild and subsequently generated mutated aa sequence of N, RdRp, S, and ORF3a proteins, respectively, YP_009724397.2, YP_009725307.1, YP_009724390.1, and YP_009724391.1. We further used PredictProtein 29 for analyzing and predicting the possible secondary structure and solvent accessibility of both wild and mutant variants of those proteins. The SWISS‐MODEL homology modeling webtool 30 was utilized for generating the three‐dimensional (3D) structures of the RdRp, S, and ORF3a protein using 7c2k.1.A, 6xr8.1.A, and 6xdc.1.A PDB structure as the template, respectively. We also used Modeller v9.25 31 to generate the structures against the same templates to check the validity of SWISS‐MODEL‐derived structures. I‐TASSER 32 with default protein modeling mode was employed to construct the N protein 3D structure of wild and mutant type since there was no template structure available for the protein. The built‐in structural assessment tools (Ramachandran plot, MolProbity, and Quality estimate) of SWISS‐MODEL were used to check the quality of the generated structures.
2.3. Molecular docking and dynamics of RdRp–NSP8 and Spike–Elastase2 complexes
Determination of the active sites affected by binding is a prerequisite for docking analysis. We chose aa residue 323 along with the surrounding residues (315–324) of RdRp and the residues 110–122 of NSP8 monomer as the active sites based on the previously reported structure. 33 The passive residues were defined automatically, where all surface residues were selected within the 6.5°A radius around the active residues. The molecular docking of the wild and predicted mutated RdRp with the NSP8 monomer from the PDB structure 7C2K was performed using the HADDOCKv2.4 to evaluate the interaction. 34 The binding affinity of the docked RdRp–NSP8 complex was predicted using the PRODIGY. 35 The number and specific interfacial contacts (IC) for each of the complexes were identified.
The human neutrophil elastase (hNE) or elastase‐2 (PDB id: 5A0C) was chosen for docking of the S protein, based on earlier reports. 36 Here we employed CPORT 37 to find out the active and passive protein‐protein interface residues of hNE. The S protein active sites were chosen based on the target region (594–638) interacting with the elastase‐2. The passive residues of S protein were automatically defined as mentioned for RdRp–NSP8 docking analysis. Afterward, we individually docked wild (614D) and mutated (614G) S protein with the hNE using HADDOCK 2.4. The binding affinity of the docked complexes and the number and specific interfacial contacts (IC) were predicted as performed after RdRP–NSP8 docking. We also employed HDOCK server 38 by specifying the active binding sites residues for predicting the molecular docking energy.
The structural stability of the protein complexes (RdRP–NSP8 and Spike–Elastase2) and their variants were assessed through the YASARA Dynamics software package. We used the AMBER14 force field for these four systems, and the cubic simulation cell was created with the TIP3P (at 0.997 g/L−1, 25°C, and 1 atm) water solvation model. The PME or particle mesh Ewald methods were applied to calculate the long‐range electrostatic interaction by a cut‐off radius of 8Å. 39 We used the Berendsen thermostat to maintain the temperature of the simulation cell. The time step of the simulation was set as 1.25fs, 40 and the simulation trajectories were saved after every 100 ps. Finally, we conducted the molecular dynamics simulation for 100 ns. 41 , 42 , 43 , 44
2.4. Mutational analysis of transmembrane domain 1 of ORF3a and serine‐rich (SR) domain of N protein
The complete genome of 12 pangolin‐derived coronavirus strains and 38 bats, civets, and human SARS‐CoVs were downloaded from GISAID and NCBI, respectively, for the mutational comparison between the SARS‐CoV and SARS‐CoV‐2 (Supplementary Material 2). We mainly targeted transmembrane domain 1 (TM1), which covers 41–63 residues, of ORF3a to find the identical mutation and scan overall variation in TM1. A generalized comparison between SARS‐CoV and SARS‐CoV‐2 reference sequences was performed to identify the mutations in the SR‐rich region that will help to postulate on N protein functions of novel coronavirus based on previous related research on SARS‐CoV.
2.5. Analyzing RNA folding prediction of 5ʹ‐UTR, leader protein, and NSP3
The Mfold web server 45 was used with default parameters to check the folding pattern of RNA secondary structure in the mutated 5ʹ‐UTR, synonymous leader (T445C), and NSP3 region (C3037T). The change C6286T is in between the nucleic acid‐binding (NAB) domain and betacoronavirus‐specific marker (βSM) domain of the NSP3 region. The change C26801G is at the transmembrane region 3 (TM3) of the virion membrane. Thus, changing in C6286T and C26801G will not affect the function significantly and was not predicted here. The structure of complete mutant 5ʹ‐UTR (variant “T”) was compared with the wild‐type (variant “C”) secondary pattern as mentioned in the Huston et al. study. 46 From the Mfold web server, we also estimated free energy change (∆G) for wild and mutant leader and NSP3 RNA fold to find any variation in stability.
3. RESULTS AND DISCUSSION
Our analysis showed possible individual effect of a total of nine mutations in S, RdRp, ORF3a, N, 5ʹ‐UTR, leader protein (NSP1), and NSP3 found in the dominant clades G (15.2%), GH (20.8%), GR (32.6%), and GV (22.6%) of 2020 on viral replication cycle and transmission. We uniquely approached to dock spike with elastase‐2 and RdRp with NSP8. Zeng et al. 23 showed the links of these mutations toward possible epistatic effects on fitness using statistical analysis that duly suits our purpose of presenting how the mutations might play the combined roles. The overall epistatic interactions of the mutated proteins and/or RNA were depicted in Figure 1 as a hypothetical model.
Figure 1.
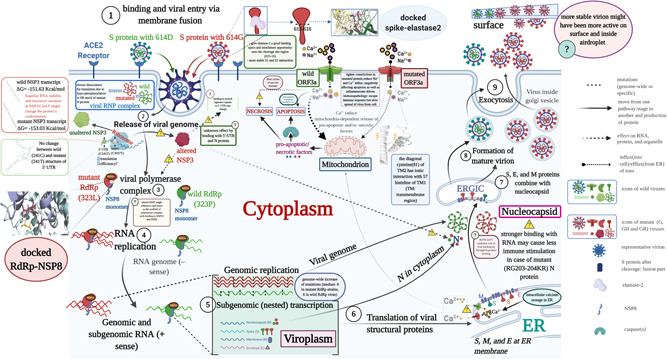
Schematic diagram of SARS‐CoV‐2 replication in the cell showcasing S, N, ORF3a, RdRp, NSP3, and 5′‐UTR based epistatic interactions. The replication cycle starts with the ACE2 receptor binding of the spike glycoprotein (S) as cornered at the top‐left and finishes with the exocytosis at the top‐right. The viruses which do not carry G‐, GH‐ and/or GR‐featured mutations in the S, N, ORF3a, RdRp, NSP3, and 5′‐UTR are denoted as the wild‐type where mutants contain those. The red and green color icons throughout the diagram, such as proteins, genome, and virion, represent the wild and mutant types, respectively. For a generalized virion, we used the blue color. Although this theme does not show the co‐infection of both types, which might occur on rare occasions, we showed the comparative epistatic effects side‐by‐side fashion during the whole replication cycle, making it easy to grasp. Related figure(s) for each protein are shown in the enclosed box. We used the “question” mark in a pathway and an explanatory box to mean the uncertainty or unknown effects of any mutant proteins/RNA structure. ACE2, angiotensin‐converting enzyme 2; ER, endoplasmic reticulum; ERGIC‐endoplasmic; RdRp, RNA dependent RNA polymerase; NSP14‐proof‐reading enzyme of SARS‐CoV‐2; SARS‐CoV‐2; severe acute respiratory syndrome coronavirus 2; 5′‐UTR, 5′‐untranslated region
3.1. Spike protein D614G mutation favors Elastase‐2 binding
This study found interesting structural features of the S protein while comparing and superimposing the wild protein (D614) over mutated protein (G614). The secondary structure prediction and surface accessibility analyses showed a slight mismatch at the S1‐S2 junction (681 PRRAR↓S686), where serine at 686 (S686) was found covered in G614 and exposed to the surface in D614. However, S686 in both G614 and D614 were exposed to an open‐loop region to have possible contact with the proteases (Figure S1). However, further investigation on the aligned 3D structures showed no conformational change at the S1–S2 cleavage site (Figure 2C). We also observed no structural variation in the surrounding residues of the protease‐targeting S1–S2 site (Figure 2C), which eliminated the assumption of Phan. 47 The predictive 3D models and structural assessment of D614 and G614 variants confirmed the cleavage site at 815–816 of the S2 subunit (812PSKR↓S816) or S2′ 1 , 48 had no structural and surface topological variation (Figure 2D,E). Instead, the superimposed 3D structures suggested a conformational change in the immediate downstream region (618TEVPVAIHADQLTPT632) of the 614th position of mutated protein (G614) that was not observed in D614 variants (Figure 2A,B).
Figure 2.
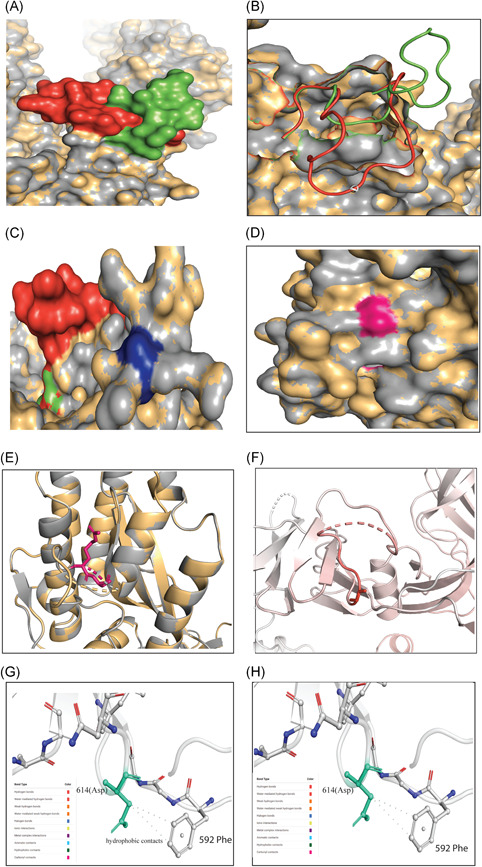
Different structural and stability comparisons of the wild and mutant spike protein. Structural superposition of wild and mutant spike proteins (A) and (B); conformation in S1–S2 (C) and S2ʹ sites (d) and (E); representation of vibrational entropy energy change on the mutant type structure (F); and interatomic interaction prediction of both wild (G) and mutant (H) types. For Figure (A)–(E), the gray and yellow colors represent the wild and mutant protein, respectively. (A) The downstream (617–636) of D614G in wild (green) and mutant (red) S protein was focused. Overlapping the wild (D614) and mutant (G614) S protein showed a conformational change in the 3D structures. (B) However, the conformational change is in the loop region (618–632) of the proteins thus may potentially play a role in interacting with other proteins or enzymes, such as elastase‐2, as we focused on in this study. (C) No change was found in the S1–S2 cleavage site (685–686), depicted in blue color, of the wild and mutant protein. (D) Surface and (E) cartoon (2°) structure of the superimposed wild and mutant proteins where the S2ʹ (pink) is situated in the surface region and does not show any change in accessibility in the residual loop region. (F) The mutant (G614) protein showed higher flexibility in the G614 (sticks) and its surroundings (red). The intramolecular interaction determined the overall stability of the (G) wild and (H) mutant structure where Cβ of D614 (aspartic acid at 614; green stick modeled) had two hydrophobic interactions with the benzene rings. These intramolecular contacts stabilize the S protein of wild‐type. The missing of this bond destabilizes the mutant (G614) protein. The mutant protein has glycine at 614, which has less chance of interacting with neighboring aa due to its shorter and nonpolar R‐group. The color code representing the bond type is presented in each (G) and (H)
Several experiments suggested that mutated (G614) protein contains a novel serine protease cleavage site at 615–616 that is cleaved by host elastase‐2, a potent neutrophil elastase. The level of this elastase at the site of infection during inflammation will facilitate the host cell entry for G614. 18 , 36 , 49 , 50 The elastase‐2 restrictedly cut valine at 615 due to its valine‐dependent constriction of catalytic groove. 51 The current sequence setting surrounding G614 (P6‐610VLYQGV↓NCTEV620‐P′5) showed higher enzymatic activity on the spike, 36 which cannot be entirely aligned with the previous works on the sequence‐based substrate specificity of elastase‐2. 52 However, the first non‐aligned residue of the superimposed G614, located at the P′4 position (T618), may also be essential for binding with the elastase‐2, and further down the threonine (T) at 618, the residues may affect the bonding with the respective aa of elastase‐2 (Figure 2A,B). This changed conformation at the downstream binding site of G614 may help overcome unfavorable adjacent sequence motifs around G614 residue. Therefore, the simultaneous or sequential processing of the mutated S protein by TMPRSS2/Furin/Cathepsin and/or elastase‐2 facilitates a more efficient SARS‐CoV‐2 entry into the host cells and cell–cell fusion. 36 , 49 , 50
This study further observed the possible association of the S protein with elastase‐2 and found an increased binding affinity in the case of G614 (Table 1). Hence, the active sites of the mutated protein interacted efficiently with more aa of elastase‐2 (Table 2), possibly providing a better catalytic activity, as shown by Hu et al. 36 The mutation may induce a better structural configuration of the elastase‐2 cleavage site of mutated spike protein for an easier and more accessible enzymatic cleavage (Figures 2A,B and 3). The efficient cleaving of this enzyme, although located in an upstream position of the S1–S2 junction, may assist in releasing S1 from S2 and change the conformation in a way that later helps in cleaving at the S2′ site by other protease(s) before fusion. 53 , 54 The complex of mutated spike protein and the elastase‐2 was more flexible than the wild spike‐elastase complex, and the interactions with enzyme were also different, as shown in root‐mean‐square deviation (RMSD) between the complexes (Figure 4).
Table 1.
The scores of HADDOCK, PRODIGY (ΔG and Kd (M) at 37.0℃) for RdRp/NSP8 and Spike–Elastase docked complex
| Variables | types | RdRp/NSP8 | Spike–Elastase |
|---|---|---|---|
| HADDOCK score | Wild | −82.2 ± 7.8 | −43.0 ± 8.9 |
| Mutant | −118.3 ± 2.5 | −61.9 ± 4.5 | |
| ΔG (kcal mol−1) | Wild | −10.6 | −13.3 |
| Mutant | −10.5 | −13.7 | |
| Kd (M) at 37.0℃ | Wild | 3.5E−08 | 4.5E−10 |
| Mutant | 3.9E−08 | 2.3E−10 | |
| Number of interfacial contacts (ICs) per property | Wild | Charged‐charged (5); charged‐polar (9); charged‐apolar (15); polar‐polar (2); polar‐apolar (16); and apolar‐apolar (21) | Charged‐charged (17); charged‐polar (22); charged‐apolar (32); polar‐polar (5); polar‐apolar (31); and apolar‐apolar (23) |
| Mutant | Charged‐charged (5); charged‐polar (16); charged‐apolar (19); polar‐polar (3); polar‐apolar(15); apolar‐apolar (23) | Charged‐charged (13); charged‐polar (18); charged‐apolar (27); polar‐polar (4); polar‐apolar (28), and apolar‐apolar (36) | |
| Associated amino acids of Elastase‐2 with possible docking interactions (for spike) or NSP8 (for RdRp) | Wild | P323: Asp (112), Cys (114), Val (115) and Pro (116) | 605 (Ser) and 607 (Gln): 36 (Arg); 618 (Thr): 199 (Phe); 619 (Glu): 199 (Phe), Cys (227); 620 (Val): 198 (Cys), 225:227 (Gly, Gly, Cys) |
| Mutant | P323: Asp (112), Cys (114), Val (115) and Pro (116) | 614 (Gly): 101 (Val); 618 (Thr): 181 (Arg), 223–226 (Val, Arg, Gly, Gly); 619 (Glu): 103 (Leu), 181 (Arg), 222–225 (Phe, Val, Arg, Gly), 236 (Ala); 620 (Val): 223–227 (Val, Arg, Gly, Gly, Cys) |
Table 2.
Assess the effect of mutations on structural dynamics of NSP‐12/RDRP, Spike, NS3, and N Protein of SARS‐CoV‐2 using DynaMut
| Protein name | Mutation with position | ΔΔG DynaMut kcal/mol | ΔΔG ENCoM kcal/mol | ΔΔG mCSM kcal/mol | ΔΔG SDM kcal/mol | ΔΔG DUET kcal/mol | ΔΔGFoldX (kcal/mol) | Results* | ΔΔSVib ENCoMkcal. mol−1.K−1 |
|---|---|---|---|---|---|---|---|---|---|
| RdRp | P323L | 1.054 | −0.441 | −0.264 | 0.700 | 0.118 | −0.733 | Stabilizing | −0.551 |
| Spike | D614G | −0.769 | +0.408 | −0.492 | 2.530 | 0.195 | +0.289 | Destabilizing | 0.510 |
| ORF3a | Q57H | 0.275 | −0.128 | 0.788 | 0.520 | −0.464 | −1.438 | Stabilizing | −0.160 |
| N Protein | RG203‐04KR | – | – | – | – | – | −3.42262 | Highly Destabilizing | – |
| N protein | A220V | 0.109 | 0.458 | −0.586 | −1.460 | −0.567 | +1.6 | Stabilizing | −0.572 |
Note: The value of ΔΔG < 0 indicates that the mutation causes destabilization and ΔΔG > 0 represents protein stabilization. For ΔΔSVibENCoM, positive and negative value denotes the increase and decrease of molecular flexibility, respectively.
Abbreviation: SARS‐CoV‐2, severe acute respiratory syndrome coronavirus 2.
The final result of the stability for each protein was determined based on the intramolecular interactome analysis.
Figure 3.
The molecular docking of wild and mutant with elastase‐2. Both the (upper figure) wild (D614) and (lower figure) mutant (G614) version of S protein was shown in golden color, whereas the elastase‐2 docked to D614 and G614 in blue and green color, respectively. The enlarged views of the docked site were shown in separate boxes. (A) The possible docked residues (stick model) on the wild S protein (warm pink) and elastase‐2 (green) are 618(Thr)−619(Glu)−620(Val) and 198(Cys)−199(Phe)−225:227 (Gly, Gly, Cys), respectively. The aspartic acid at 614 is 17.3°A far away from the valine (101), apparently the nearest aa of elastase‐2 to the cleavage site (615–616). (B) The possible interacting residues (stick model) on the mutant S protein (blue) and elastase‐2 (warm pink) are 614(Gly)−618(Thr)−619(Glu)−620(Val) and 101(Val)−103(Leu)−181(Arg)−222:227(Phe, Val, Arg, Gly, Gly, Cys), and 236 (Ala), respectively. In this case, the glycine at 614 is 5.4°Afar from the valine (101), the nearest aa of elastase‐2 to the cleavage site (615–616)
Figure 4.
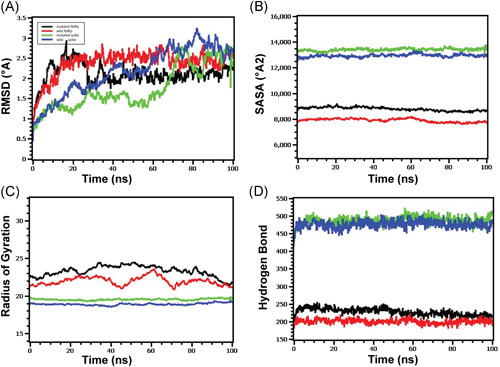
Molecular dynamics of spike‐elastase2 and RdRp‐NSP8 complex (A) Both the wild and mutated spike protein had lower RMSD profile till 60 ns, then it rose and maintained a steady state. Although the spike protein had a higher degree of deviation in the RMSD profile than RdRp, they did not exceed 3.0 Å. The RMSD demonstrated that mutant and wild RdRp protein complex has an initial rise in RMSD profile due to flexibility. Therefore, both RdRp complexes stabilized after 30 ns and maintained a steady peak. The wild‐type RdRp complex had a slightly higher RMSD peak than mutant RdRp, indicating the more flexible nature of the wild‐type. (B) The spike protein complex had a similar SASA profile, did not change its surface volume, and maintained a similar trend during the whole simulation time. The higher deviation of SASA indicates that mutant and wild‐type RdRp had a straight line. Still, the mutant structure had a higher SASA profile, indicating the protein complex had enlarged its surface area. Therefore, the mutation in RdRp protein leads to more surface area expansion than wild types as their average SASA value had a significant difference. (C) Mutated spike exhibits a little more Rg profile than the wild‐type, which correlates with the comparative labile nature of the mutant. The higher level of Rg value defines the loose packaging system and mobile nature of the protein systems. The mutant RdRp had a lower level of fluctuations and maintained its integrity during the whole simulation time. The wild‐type RdRp complexes had higher deviations and more mobility than the mutant complex. (D) Any aberration in hydrogen bond numbers can lead to higher flexibility. Therefore, the mutant and wild spike proteins exhibit the same flexibility in terms of H‐bonding. The mutant RdRp protein had more hydrogen bonding than the wild types, but they did not differentiate too much, and a relatively straight line was observed for the protein. RMSD, root‐mean‐square deviation
This G614 aa replacement may destabilize the overall protein structure (Table 2 and Figure 2A,B), and the deformed flexible region at or near G614 proves this destabilizing change (Figures 2F and S3). The S1 will release from S2 more effectively in G614 protein by introducing glycine that will break the hydrogen bond between the D614 (wild) and T859 (threonine) of the neighboring protomer. 55 , 56 Increasing the chance of 1‐RBD‐up conformation due to breakage of both intra‐and interprotomer interactions of the spike trimer and symmetric conformation will increase binding potential with an ACE2 receptor and increase antibody‐mediated neutralization. 55 Our analyses provided the in silico proof of this fact by showing that the mutated protein was more flexible than the wild‐type protein by missing a hydrophobic interaction between G614 and Phe592 (Figure 2G,H). The overall structural flexibility may assist the mutated S protein by providing elastase‐2 a better binding space and attachment opportunity onto the cleavage site (Figure 3A,B), thus providing a more stable interaction that increases the credibility of an efficient infection (Figure 1).
3.2. Increased flexibility of RdRp–NSP8 complex: Compromise proof‐reading efficiency with replication speed
The binding free energy (ΔG) of the RdRp–NSP8 complexes have been predicted to be −10.6 and −10.5 kcal mol−1, respectively, in wild (P323) and mutated (L323) type, which suggests a more flexible interaction for the mutated protein (Table 2). The increased number of contacts found in the L323–NSP8 complex (Table 1) was possibly due to slightly more hydrogen bonds, which had no considerable impact on protein flexibility (Figure 4D). Our analyses identified that proline (P323) or leucine (L323) of RdRp can interact with the aspartic acid (D112), cysteine (C114), valine (V115), and proline (P116) of NSP8 (Table 1 and Figure 5). RdRp binds with NSP8 in its interface domain (from residues alanine: A250 to arginine: R365), forming positively charged or comparatively neutral “sliding poles” for RNA exit, and enhance the replication speed probably by extending the RNA‐binding surface in that domain area. 57 , 58 Molecular dynamics of the mutated RdRp–NSP8 complex supported this by showing a more expanded surface area in the interacting site (Figure 4B) and maintained integrity throughout the simulation (Figure 4C). Besides, we did not find any interaction of NSP8 with the zinc‐binding residues (H295, C301, C306, and C310) of the RdRp protein (Table 1 and Figure 5). 59 , 60 Therefore, the P323L mutation within the RdRp interface domain's conserved site may only affect the RdRp–NSP8 interaction without changing metal binding affinity.
Figure 5.
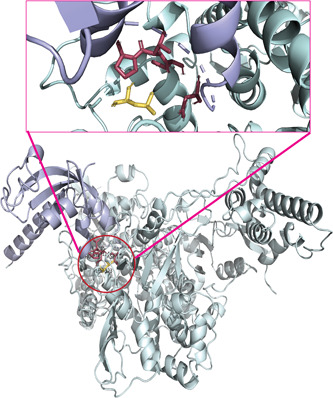
The molecular interaction of mutant RdRp with NSP8. The mutant (L323) RdRp (pale green) and NSP8 (light blue) are interacting as shown in the center of the lower figure, and an enlarged view of the docked site is presented above within a box. The leucine at 323 interacted with the Asp (112), Cys (114), Val (115), and Pro (116). The wild (P323) RdRp has identical docking interactions with NSP8 (Table 1), thus it is not presented as a separate figure
The results from six state‐of‐the‐art tools of protein stability suggested that mutated (L323) protein cannot be concluded as “stable,” only because of ambiguous ΔΔG estimates (Table 2); instead, the interaction with the adjacent aa mainly defined the stability. 61 The superimposed 3D structures and secondary structure analyses showed no deviation in the loop/turn structure of the mutated protein, even though hydrophobic leucine (L323) was embedded (Figures S1 and S4). The mutation stabilized the L323 structure to some extent making the protein more rigid and bound less firmly with the NSP8 by expanding the interacting region.
Together, these structural variations may increase the replication speed by helping exit the processed RNA genome from the RdRp groove structure more swiftly (Figure 1). The increasing replication speed might be due to the perturbation of interaction between RdRp and NSP8, 57 , 61 or less possibly, the complex tripartite interactions (RdRp, NSP8, and NSP14) responsible for the speculated decrease of proof‐reading efficiency. 4 Thus, RdRp mutants might increase the mutation rate by a trade‐off between high replication speed and low fidelity of the mutated polymerase. 62 The Wilcoxon rank‐sum test revealed that the frequency of mutation (median = 8) in L323 mutants (n = 27 364) is significantly higher (p < 0.0001) than the frequency (median = 6) of wild‐type (P323)strains (n = 9815). This increased mutation rate in L323 mutants can surpass the constant proof‐reading fidelity, 63 which might help adapt more quickly in adverse climatic conditions, evade the immune response, and survive within different selective pressure. 64 , 65
3.3. Q57H substitution in ORF3a viroporin: The roles of decreased ion permeability
Our study found that the replacement of glutamine (Q57) with positively charged histidine (H57) at aa position 57 of ORF3a transmembrane region 1 (TM1) does not change secondary transmembrane helical configuration (Figure S1). Aligned 3D structures have also shown no variation of TM1 in the monomeric state (Figure 6A). The mutant (H57) protein has a nonsignificant increase in structural stabilization and a minimal decrease in molecular flexibility (Table 2 and Figure S5). This higher stability is because of the weak ionic interaction of H57‐Cαwith the sulfur atom of cysteine (C81) that is present in TM2 and the hydrogen bond of terminal Nζ of lysine (K61) with one of the endocyclic nitrogens of H57 (Figure 6B). The Q57 in wild‐type protein forms the major hydrophilic constriction within the ORF3a channel pore. 66 Thus, further favorably increasing constrictions within the H57 protein channel pore due to diagonal H57 (TM1)‐C81 (TM2) ionic interaction (Figure 6B) and the replacement of charge‐neutral glutamine with a positively charged histidine in the selectivity filter may reduce the passing of positive ions, such as Ca2+, Na+, and K+, by either electrostatic repulsion or blocking. 67 , 68 , 69 , 70 This speculation for ORF3a mutated protein was supported by another study showing the reduction of ion permeability of Na2+ and Ca2+ through the H57; however, that decrease was not found statistically significant (p > 0.05). 66
Figure 6.
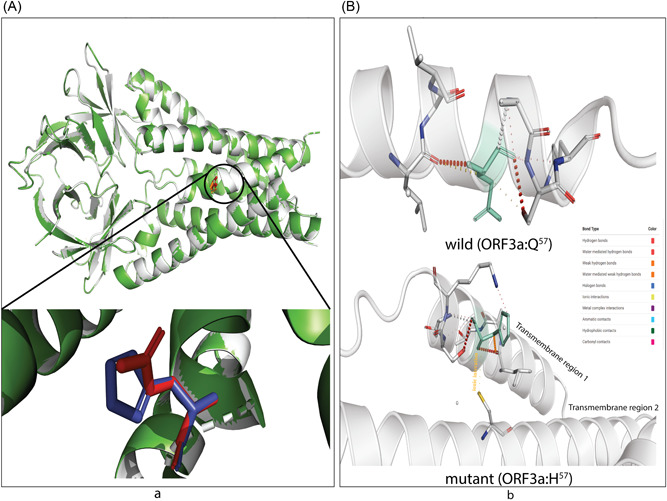
The effect on transmembrane channel pore of ORF3a viroporin due to p.Q57H mutation. (A) The wild (Q57) and mutant (H57) ORF3a proteins are presented in light gray and green colors. The structural superposition displays no overall conformation change; however, the histidine at the position 57 of mutant ORF3a (deep blue) is slightly rotated from glutamine at the exact position of the wild protein (bright red). This change in rotamer state at the residue 57 may influence (B) the overall stability of H57 (upper part) overQ57 (lower part)because of ionic interaction of histidine (green; stick model) of transmembrane domain 1 (TM1) with cysteine at 81 (yellow stick) of TM2. The color code defined different bond types is shown in the inlet
The decreased intracellular concentration of cytoplasmic Ca2+ ions potentially reduces caspase‐dependent apoptosis of the host cell, 71 mainly supporting viral spread without affecting replication, 21 as shown in Figure 1. Moreover, the ORF3a can drive necrotic cell death 72 wherein the permeated ions into cytoplasm 73 and the insertion of ORF3a as viroporin into lysosome 74 play vital roles. The H57 mutant may thus decrease pathogenicity and symptoms during the early stages of the infection, that is, reducing “cytokine storm” in the host. 75 Besides, ORF3a was shown to affect inflammasome activation, virus release, and cell death, as detailed by Castaño‐Rodriguez et al. 76 that the deletion of ORF3a reduced viral load and morbidity in animal models.
Even though similar proteins of ORF3a have been identified in the sarbecovirus lineage infecting bats, pangolins, and humans, 77 only one pangolin derived strain from 2017 in Guangxi, China contains H57 residue as shown by mutation analyses (Figure S5), and also reported by Kern et al. 66 A possible explanation behind that presence could be the more adaptive nature of the virus toward reverse transmission by being less virulent, that is, from human to other animals, as observed in recent reports. 78 , 79
3.4. N protein mutation: Augmenting nucleocapsid stability and exerting miscellaneous effects
Our study has observed that the combined mutation (N: p.RG203‐204KR) causes no conformational change in secondary and 3D structures (Figures S1 and Figure 7, respectively) of the conserved SR‐rich site (184 → 204) in the linker region (LKR: 183 → 254) of the N protein (Figure S7). But there is a minor alteration among buried or exposed residues (Figures S7C and 8B). The superimposed 3D structures showed structural deviation, rather at 231ESKMSGKGQQQQGQTVT247 of the LKR (Figure 7), corresponding to the high destabilization of the mutated (KR203‐204) protein (Table 2).
Figure 7.
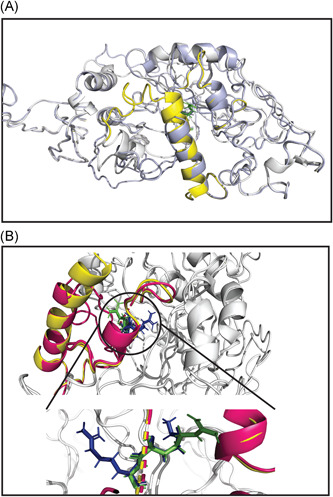
Structural superposition of wild and mutant N protein. The light gray color represents both wild (RG203–204) and mutant (KR203–204) N protein. The linker region (LKR: 183–247 aa) of wild (RG203–204) and mutant (KR203–204) are in pale yellow and warm pink color, respectively. (A) The aligned structures showed a highly destabilizing (Table 2) conformational change from 231 to 247 aa within LKR. Other regions of the N protein, especially the SR‐rich region (184–204 aa) where the mutations occur, do not change. (B) A more emphasized look into the SR‐rich and mutated sites (RG203–204KR) of wild and mutant N protein represent a slight deviation in the Ser (197) and Thr (198) where only glycine (green) to arginine (blue) substitution at position 204 shows changing at rotamer state. The enlarged view is shown in the bottom part
Impedance to form particular SR‐motif due to RG→KR mutation might disrupt the phosphorylation catalyzed by host glycogen synthase kinase‐3. 80 After the virus enters the cell, this synchronized hypo‐phosphorylation of KR203‐204 protein should make the viral ribonucleoprotein (RNP) unwind in a slower but more organized fashion that might impact translation and immune‐modulation. 81 , 82 , 83 In KR203‐204, replacing glycine with arginine may increase the nucleocapsid (N protein–RNA complex) stability by forming stronger electrostatic and ionic interactions due to increased positive charge. 84 , 85 Besides, the more disordered orientation of the downstream LKR site 81 and the highly destabilizing property of KR203‐204 may assist in packaging a stable RNP. 86 , 87 N protein also utilizes the intrinsically disordered dynamic linker region (LKR) that controls its affinity toward M protein, self‐monomer, 5′‐UTR, and cellular proteins. 88 , 89 , 90 The phosphorylation at the LKR site may play an essential role in regulating these interactions. 84 These plausible interactions and impacts upon mutations are depicted in Figure 1.
3.5. Silent mutations may not be silent
The C241T of 5′‐UTR, a single nucleotide “silent” mutation, is located at the UUCGU pentaloop part of the stem‐loop region (SLR5B). This pentaloop of 5′‐UTR remains unchanged and maintains a particular structure with a potential role in viral packaging. 89 , 91 The RNA secondary structures predicted no change in the 241T structure (Figure S8A). However, C241T is present just upstream to the ORF1a start codon (266–268 position) and may be involved in differential RNA binding affinity to the ribosome and translational factors. 92
In the case of multi‐domain NSP3 (papain‐like protease), we have observed superior stability of the RNA after gaining the synonymous mutation 3037C<T (C318T) where wild and mutant RNA structure has −151.63 and −153.03 Kcal/mol, respectively (Figure S7B,C). A more stable secondary structure of (+)‐ssRNA as observed in the mutated NSP3 protein‐coding sequence corresponds to the slower translational elongation that generally contributes to a range of abnormalities resulting in low translation efficiency affecting posttranslational modifications as a part of protein regulation. 93 This silent mutation is located within the flexible loop of the NSP3 ubiquitin‐like domain 1 (Ubl1). In SARS‐CoV, Ubl1 was reported to bind with single‐stranded RNA containing AUA patterns and interact with the N protein. 94 , 95 Besides, Ubl1 was likely to bind with several signature repeats in 5′‐UTR in the SARS‐CoV‐2 genome. 96 Finally, a change in T445C in leader protein may not cause any change in expression or others since the structure (data not shown) and energy is the same −172.34 kcal/mol. Figure 1 represents the overall possible scenario due to these silent mutations.
3.6. Epistatic effects of the co‐occurring mutations on viral replication and transmission: A plausible hypothesis
The co‐occurring mutations, simultaneous multisite variations in the same or different proteins or genes, have provided new insights into the dynamic epistatic network by employing differential molecular interactions. The epistatic effects of the mutations were reported to control viral fitness and virulence through modulating the replication cycle and virus‐host interactions, as observed before for Influenza and Ebola virus. 97 , 98 , 99 , 100 , 101 , 102 The co‐occurring mutations of the major SARS‐CoV‐2 clades discussed in this study contained epistatic links 22 , 23 and positive selection pressure except for the synonymous mutations. 22 , 24 , 25 Observable (https://observablehq.com/) also presents those mutated sites as positively selected.
We speculated no interlinked functional relationship between p:D614G of the S protein and p:P323L of the RdRp, two important G clade‐featured co‐occurring mutations (Figure 1). The sequence‐based prediction showed no potentially significant epistatic link as well. 23 These seemingly unrelated mutations can cumulatively escalate the infectiousness of the virus because of a higher viral load and shorter burst time. The S:p.D614G might assist in rapid entry into the host cells followed by quick dissemination, and the RdRp:p:P323L may instead boost the replication by a faster RNA processing (exiting).
NSP3 is a scaffolding protein for the replication–transcription complex, and the possible change in its structure may affect the overall dynamics of viral replication. 94 , 95 P323L mutation of RdRp may change binding affinity to the Ubl1 region of NSP3 103 (Figure 1). Significant epistatic links of NSP3:C3037T with spike and RdRp mutations were also reported. 23 In contrast, we could not predict any possible association of the 5ʹ‐UTR:C241T mutation with the S, RdRp, and NSP3 mutated proteins, as shown by sequence analysis in Zeng et al. 23 The rapid within‐host replication and modified replication dynamics might be correlated with the fitness of G clade strains. 104
The mutant N protein may impact viral replication and transcription, like other coronaviruses, 90 through binding with the NSP3 protein, which linked the RdRp‐centered replication complex. The N protein can also affect the membrane stability through an uncharacterized interaction with the M protein, ultimately producing more stable virion particles. 105 , 106 , 107 A more robust N protein–RNA complex provokes a slower intracellular immune response. 82 At the same time, the mutated virus can remain highly contagious and aggressive because of the simultaneous presence of G clade‐featured S protein and RdRp mutations (Figure 1). The GR strains could hence attain a plausible advantage over G and GH by a more orchestrated, delicately balanced synergistic effects on replication and transmission fitness. These epistatic effects might increase the fitness by hiding the virus from host cellular immunity of the host and increasing stability in the environment.
Conversely, we have not found any literature for even other coronaviruses that showed ORF3a: p.Q57H correlating with the rest of the co‐occurring mutations. The H57 mutant, possibly linked to the mild or asymptomatic cases, may allow the silent transmission and increase the chance of viral spread by lowering the activation of the inflammatory response (Figure 1), such as reduced viral particle release and cytokine storm. 21 , 108 , 109 According to our hypothesis and Wolf et al. (2021), the social interventions on movement could also play a role in disseminating these G, GH, and GR clades at the early and later pandemic phases well. 110
The GV strains featuring an A222V mutation in the S protein have probably no effect on the viral transmission, severity, and antibody escape due to its structural position; rather, super‐spreading founder events might be the reason behind its faster spreading. 17 , 111 The A220V mutation stabilized the mutated N protein's linker region (Table 2) might affect RNA binding affinity; 86 however, different mutations at positions 220 in N found in other major lineages showed no phenotypic consequence. 17 There was also no epistatically linked pairing between GV clade co‐occurring mutations. 22 , 23 Altogether, the co‐occurring mutations of GV strains might not affect transmission fitness.
Vaccine inequity, immunocompromised patients, and a tremendous number of hosts are now frequently introducing variants with mutations in the RBD of the spike protein. The introduced mutations in a lineage (a variant of concern/interest) on top of the original clade‐featured ones in the genome might play the most crucial role in increasing transmission fitness and a slightly reduced neutralization to antibodies by showing epistatic effects. 112 , 113 Future studies are necessary to investigate the roles of the “mutation package” in each of these variants of concern/interest.
4. CONCLUSION
In 2020, the course of the COVID‐19 pandemic was dominated by the G, GH, GR, and GV clades. The G clade‐featured co‐occurring mutations might increase the viral load, alter host immune responses, and modulate intrahost virus genome plasticity that raises the speculation of their potential role in continuous transmission. The GR and GH clade mutant with the signature mutation, respectively, in nucleocapsid and ORF3a protein, might contribute to the host's immune response and viral transmission. The GV strains, however, could have spread quickly by superspreading events with no apparent epistatic effect. Therefore, the fitness of SARS‐CoV‐2 may increase in terms of replication and transmission where viral strains are always giving their spread capacity within a population the top priority by calibrating the infection cycle. However, further in vivo and ex vivo studies and more investigations are required to prove and bolster this hypothesis.
CONFLICT OF INTERESTS
The authors declare that there are no conflict of interests.
AUTHOR CONTRIBUTIONS
Iqbal Kabir Jahid, Ovinu Kibria Islam, and A. S. M. Rubayet Ul Alam hypothesized about the work. A. S. M. Rubayet Ul Alam performed the sequence analysis part after Ovinu Kibria Islam and Md. Shazid Hasan compiled the data set. The Python coding, structural (both RNA and protein), and protein docking were done by A. S. M. Rubayet Ul Alam. MSH predicted protein structure in I‐TASSER and performed stability analysis in DynaMut. Shafi Mahmud performed the molecular dynamics study. Hassan M. Al‐Emran and Mir Raihanul Islam performed the statistical analysis. Hassan M. Al‐Emran then reviewed and organized the manuscript expertly. Iqbal Kabir Jahid, Keith A. Crandall, and M. Anwar Hossain supervised, suggested, and revised the write‐up to produce the final draft.
Supporting information
Supporting information.
Supporting information.
Supporting information.
ACKNOWLEDGMENTS
We want to acknowledge the team at GISAID and all contributing authors, who generated and submitted sequence data, for creating the SARS‐CoV‐2 global database. Jashore University of Science and Technology provided the funding for the research. We want to thank the Microbial Genetics and Bioinformatics Laboratory of the University of Dhaka for supporting high‐performance computer access. We especially appreciate the efforts of M. Shaminur Rahman and Spencer Mark Mondol in protein structure prediction and stability analyses.
Alam ASM Rubayet Ul, Islam OK, Hasan MS, et al. Dominant clade‐featured SARS‐CoV‐2 co‐occurring mutations reveal plausible epistasis: An in silico based hypothetical model. J Med Virol. 2022;94:1035‐1049. 10.1002/jmv.27416
A. S. M. Rubayet Ul Alam and Ovinu Kibria Islam contributed equally to the study.
DATA AVAILABILITY STATEMENT
All the sequence and structural data were taken from the GISAID (https://www.gisaid.org/) and RCSB PDB (https://www.rcsb.org/), as mentioned in the methodology section. We provide all the necessary information, such as accession numbers and the date‐based data source for helping readers and reviewers to check the authenticity of the work. The data that support the findings of this study are available in GISAID at https://www.gisaid.org/. These data were derived from the resources available in the public domain: GISAID, https://www.gisaid.org/.
REFERENCES
- 1. Hoffmann M, Kleine‐Weber H, Pöhlmann S. A multibasic cleavage site in the spike protein of SARS‐CoV‐2 is essential for infection of human lung cells. Mol Cell. 2020;78:779‐784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hoque MN, Chaudhury A, Akanda MAM, Hossain MA, Islam MT. Genomic diversity and evolution, diagnosis, prevention, and therapeutics of the pandemic COVID‐19 disease. PeerJ. 2020;8:e9689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Petersen E, Koopmans M, Go U, et al. Comparing SARS‐CoV‐2 with SARS‐CoV and influenza pandemics. Lancet Infect Dis. 2020;20:238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Romano M, Ruggiero A, Squeglia F, Maga G, Berisio R. A Structural view of SARS‐CoV‐2 RNA replication machinery: RNA synthesis, proofreading and final capping. Cells. 2020;9(5):1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rahman MS, Hoque MN, Islam MR, et al. Epitope‐based chimeric peptide vaccine design against S, M and E proteins of SARS‐CoV‐2 etiologic agent of global pandemic COVID‐19: an in silico approach. Peer J. 2020;8:e9572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yoshimoto FK. The proteins of severe acute respiratory syndrome coronavirus‐2 (SARS CoV‐2 or n‐COV19), the cause of COVID‐19. Protein J. 2020;39(3):198‐216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Callaway E. The coronavirus is mutating‐does it matter? Nature. 2020;585(7824):174‐177. [DOI] [PubMed] [Google Scholar]
- 8. Islam MR, Hoque MN, Rahman MS, et al. Genome‐wide analysis of SARS‐CoV‐2 virus strains circulating worldwide implicates heterogeneity. Sci Rep. 2020;10(1):1‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rahman MS, Islam MR, Hoque MN, et al. Comprehensive annotations of the mutational spectra of SARS‐CoV‐2 spike protein: a fast and accurate pipeline. Transbound Emerg Dis. 2020;68:1625‐1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Eskier D, Karakülah G, Suner A, Oktay Y. RdRp mutations are associated with SARS‐CoV‐2 genome evolution. PeerJ. 2020;8(e):9587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hassan SS, Choudhury PP, Basu P, Jana SS. Molecular conservation and Differential mutation on ORF3a gene in Indian SARS‐CoV2 genomes. Genomics. 2020;112:3226‐3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shaminur Rahman M, Rafiul Islam M, Rubayet Ul Alam A, et al. Evolutionary dynamics of SARS‐CoV‐2 nucleocapsid (N) protein and its consequences. J Med Virol. 2021;93(4):2177‐2195. [DOI] [PubMed] [Google Scholar]
- 13. Hamed SM, Elkhatib WF, Khairalla AS, Noreddin AM. Global dynamics of SARS‐CoV‐2 clades and their relation to COVID‐19 epidemiology. Sci Rep. 2021;11(1):1‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yin C. Genotyping coronavirus SARS‐CoV‐2: methods and implications. Genomics. 2020;112(5):3588‐3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mercatelli D, Giorgi FM. Geographic and genomic distribution of SARS‐CoV‐2 mutations. Front Microbiol. 2020;11:1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rambaut A, Holmes EC, O'toole Á, et al. A dynamic nomenclature proposal for SARS‐CoV‐2 lineages to assist genomic epidemiology. Nat Microbiol. 2020;5(11):1403‐1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hodcroft EB, Zuber M, Nadeau S, et al. Emergence and spread of a SARS‐CoV‐2 variant through Europe in the summer of 2020. medRxiv. Published online March 24, 2021. [DOI] [PubMed]
- 18. Grubaugh ND, Hanage WP, Rasmussen AL. Making sense of mutation: what D614G means for the COVID‐19 pandemic remains unclear. Cell. 2020;182:794‐795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Plante JA, Liu Y, Liu J, et al. Spike mutation D614G alters SARS‐CoV‐2 fitness. Nature. 2020;592:1‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hou YJ, Chiba S, Halfmann P, et al. SARS‐CoV‐2 D614G variant exhibits efficient replication ex vivo and transmission in vivo. Science. 2020;370:1464‐1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Issa E, Merhi G, Panossian B, Salloum T, Tokajian S. SARS‐CoV‐2 and ORF3a: nonsynonymous mutations, functional domains, and viral pathogenesis. mSystems. 2020;5(3):e00266‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rochman ND, Wolf YI, Faure G, Mutz P, Zhang F, Koonin EV. Ongoing global and regional adaptive evolution of SARS‐CoV‐2. Proc Natl Acad Sci USA. 2021;118(29):e2104241118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zeng H‐L, Dichio V, Horta ER, Thorell K, Aurell E. Global analysis of more than 50,000 SARS‐CoV‐2 genomes reveals epistasis between eight viral genes. Proc Natl Acad Sci USA. 2020;117(49):31519‐31526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Presti AL, Rezza G, Stefanelli P. Selective pressure on SARS‐CoV‐2 protein coding genes and glycosylation site prediction. Heliyon. 2020;6(9):e05001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Berrio A, Gartner V, Wray GA. Positive selection within the genomes of SARS‐CoV‐2 and other coronaviruses independent of impact on protein function. PeerJ. 2020;8:e10234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rodrigues CH, Pires DE, Ascher DB. DynaMut: predicting the impact of mutations on protein conformation, flexibility and stability. Nucleic Acids Res. 2018;46(W1):W350‐W355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schymkowitz J, Borg J, Stricher F, Nys R, Rousseau F, Serrano L. The FoldX web server: an online force field. Nucleic Acids Res. 2005;33(suppl_2):W382‐W388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Delgado J, Radusky LG, Cianferoni D, Serrano L. FoldX 5.0: working with RNA, small molecules and a new graphical interface. Bioinformatics. 2019;35(20):4168‐4169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rost B, Yachdav G, Liu J. The predictprotein server. Nucleic Acids Res. 2004;32(suppl_2):W321‐W326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Waterhouse A, Bertoni M, Bienert S, et al. SWISS‐MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018;46(W1):W296‐W303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Webb B, Sali A. Comparative protein structure modeling using MODELLER. Curr Protoc Bioinformatics. 2016;54(1):1‐37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Roy A, Kucukural A, Zhang Y. I‐TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc. 2010;5(4):725‐738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang Q, Wu J, Wang H, et al. Structural basis for RNA replication by the SARS‐CoV‐2 polymerase. Cell. 2020;182(2):417‐428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Van Zundert G, Rodrigues J, Trellet M, et al. The HADDOCK2. 2 web server: user‐friendly integrative modeling of biomolecular complexes. J Mol Biol. 2016;428(4):720‐725. [DOI] [PubMed] [Google Scholar]
- 35. Xue LC, Rodrigues JP, Kastritis PL, Bonvin AM, Vangone A. PRODIGY: a web server for predicting the binding affinity of protein–protein complexes. Bioinformatics. 2016;32(23):3676‐3678. [DOI] [PubMed] [Google Scholar]
- 36. Hu Y, Ma C, Szeto T, Hurst B, Tarbet B, Wang J. The D614G mutation of SARS‐CoV‐2 spike protein enhances viral infectivity. bioRxiv. Published online July 22, 2020.
- 37. de Vries SJ, Bonvin AM. CPORT: a consensus interface predictor and its performance in prediction‐driven docking with HADDOCK. PLoS One. 2011;6(3):e17695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yan Y, Tao H, He J, Huang S‐Y. The HDOCK server for integrated protein–protein docking. Nat Protoc. 2020;15(5):1829‐1852. [DOI] [PubMed] [Google Scholar]
- 39. Krieger E, Nielsen JE, Spronk CA, Vriend G. Fast empirical pKa prediction by Ewald summation. J Mol Gr Model. 2006;25(4):481‐486. [DOI] [PubMed] [Google Scholar]
- 40. Krieger E, Vriend G. New ways to boost molecular dynamics simulations. J Comput Chem. 2015;36(13):996‐1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chowdhury KH, Chowdhury MR, Mahmud S, et al. Drug repurposing approach against novel coronavirus disease (COVID‐19) through virtual screening targeting SARS‐CoV‐2 main protease. Biology. 2021;10(1):2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pramanik SK, Mahmud S, Paul GK, et al. Fermentation optimization of cellulase production from sugarcane bagasse by Bacillus pseudomycoides and molecular modeling study of cellulase. Curr Res Microb Sci. 2021;2:100013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Swargiary A, Mahmud S, Saleh MA. Screening of phytochemicals as potent inhibitor of 3‐chymotrypsin and papain‐like proteases of SARS‐CoV2: an in silico approach to combat COVID‐19. J Biomol Struct Dyn. 2020:1‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yan H, Yu C. Repair of full‐thickness cartilage defects with cells of different origin in a rabbit model. Arthroscopy. 2007;23(2):178‐187. [DOI] [PubMed] [Google Scholar]
- 45. Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31(13):3406‐3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Huston NC, Wan H, Tavares RdCA, Wilen C, Pyle AM. Comprehensive in‐vivo secondary structure of the SARS‐CoV‐2 genome reveals novel regulatory motifs and mechanisms. BioRxiv. Published online July 10, 2020. [DOI] [PMC free article] [PubMed]
- 47. Phan T. Genetic diversity and evolution of SARS‐CoV‐2. Infect Genet Evol. 2020;81:104260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Belouzard S, Chu VC, Whittaker GR. Activation of the SARS coronavirus spike protein via sequential proteolytic cleavage at two distinct sites. Proc Natl Acad Sci USA. 2009;106(14):5871‐5876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Korber B, Fischer WM, Gnanakaran S, et al. Tracking changes in SARS‐CoV‐2 Spike: evidence that D614G increases infectivity of the COVID‐19 virus. Cell. 2020;182:812‐827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bhattacharyya C, Das C, Ghosh A, et al. SARS‐CoV‐2 mutation 614G creates an elastase cleavage site enhancing its spread in high AAT‐deficient regions. Infect Genet Evol. 2021;90:104760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Perona JJ, Craik CS. Structural basis of substrate specificity in the serine proteases. Prot Sci. 1995;4(3):337‐360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Fu Z, Thorpe M, Akula S, Chahal G, Hellman LT. Extended cleavage specificity of human neutrophil elastase, human proteinase 3, and their distant ortholog clawed frog PR3—three elastases with similar primary but different extended specificities and stability. Front Immunol. 2018;9:2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Li F. Structure, function, and evolution of coronavirus spike proteins. Ann Rev Virol. 2016;3:237‐261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Walls AC, Tortorici MA, Snijder J, et al. Tectonic conformational changes of a coronavirus spike glycoprotein promote membrane fusion. Proc Natl Acad Sci USA. 2017;114(42):11157‐11162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Weissman D, Alameh M‐G, de Silva T, et al. D614G spike mutation increases SARS CoV‐2 susceptibility to neutralization. Cell Host Microbe. 2021;29(1):23‐31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wrapp D, Wang N, Corbett KS, et al. Cryo‐EM structure of the 2019‐nCoV spike in the prefusion conformation. Science. 2020;367(6483):1260‐1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hillen HS, Kokic G, Farnung L, Dienemann C, Tegunov D, Cramer P. Structure of replicating SARS‐CoV‐2 polymerase. Nature. 2020;584:1‐6. [DOI] [PubMed] [Google Scholar]
- 58. Yin W, Mao C, Luan X, et al. Structural basis for inhibition of the RNA‐dependent RNA polymerase from SARS‐CoV‐2 by remdesivir. Science. 2020;368:1499‐1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kirchdoerfer RN, Ward AB. Structure of the SARS‐CoV nsp12 polymerase bound to nsp7 and nsp8 co‐factors. Nat Commun. 2019;10(1):1‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Gao Y, Yan L, Huang Y, et al. Structure of the RNA‐dependent RNA polymerase from COVID‐19 virus. Science. 2020;368(6492):779‐782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Chand GB, Banerjee A, Azad GK. Identification of novel mutations in RNA‐dependent RNA polymerases of SARS‐CoV‐2 and their implications on its protein structure. bioRxiv. 2020;8:e9492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Eskier D, Karakülah G, Suner A, Oktay Y. RdRp mutations are associated with SARS‐CoV‐2 genome evolution. bioRxiv. Published online May 20 2020. [DOI] [PMC free article] [PubMed]
- 63. Smith EC, Sexton NR, Denison MR. Thinking outside the triangle: replication fidelity of the largest RNA viruses. Ann Rev Virol. 2014;1:111‐132. [DOI] [PubMed] [Google Scholar]
- 64. Pfeiffer JK, Kirkegaard K. Increased fidelity reduces poliovirus fitness and virulence under selective pressure in mice. PLoS Pathog. 2005;1(2):e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Vignuzzi M, Stone JK, Arnold JJ, Cameron CE, Andino R. Quasispecies diversity determines pathogenesis through cooperative interactions in a viral population. Nature. 2006;439(7074):344‐348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Reid MS, Kern DM, Brohawn SG. Cryo‐EM structure of the SARS‐CoV‐2 3a ion channel in lipid nanodiscs. bioRxiv. Published online January 26, 2020.
- 67. Malasics A, Gillespie D, Nonner W, Henderson D, Eisenberg B, Boda D. Protein structure and ionic selectivity in calcium channels: selectivity filter size, not shape, matters. Biochim Biophys Acta (BBA)‐Biomembr. 2009;1788(12):2471‐2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Naranjo D, Moldenhauer H, Pincuntureo M, Díaz‐Franulic I. Pore size matters for potassium channel conductance. J Gen Physiol. 2016;148(4):277‐291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Stephens RF, Guan W, Zhorov BS, Spafford JD. Selectivity filters and cysteine‐rich extracellular loops in voltage‐gated sodium, calcium, and NALCN channels. Front Physiol. 2015;6:153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Suárez‐Delgado E, Islas LD. Ion Channels: a novel origin for calcium selectivity. eLife. 2020;9:e55216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kondratskyi A, Kondratska K, Skryma R, Prevarskaya N. Ion channels in the regulation of apoptosis. Biochim Biophys Acta (BBA)‐Biomembr. 2015;1848(10):2532‐2546. [DOI] [PubMed] [Google Scholar]
- 72. Yue Y, Nabar NR, Shi C‐S, et al. SARS‐coronavirus open reading frame‐3a drives multimodal necrotic cell death. Cell Death Dis. 2018;9(9):1‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Pinton P, Giorgi C, Siviero R, Zecchini E, Rizzuto R. Bcl‐2 and Ca2+ homeostasis in the endoplasmic reticulum. Cell Death Differ. 2006;2:1409‐1418. [DOI] [PubMed] [Google Scholar]
- 74. Nieva JL, Madan V, Carrasco L. Viroporins: structure and biological functions. Nat Rev Microbiol. 2012;10(8):563‐574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Ren Y, Shu T, Wu D, et al. The ORF3a protein of SARS‐CoV‐2 induces apoptosis in cells. Cell Mol Immunol. 2020;17:1‐3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Castaño‐Rodriguez C, Honrubia JM, Gutiérrez‐Álvarez J, et al. Role of severe acute respiratory syndrome coronavirus viroporins E, 3a, and 8a in replication and pathogenesis. mBio. 2018;9(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Boni MF, Lemey P. Evolutionary origins of the SARS‐CoV‐2 Sarbecovirus lineage responsible for the COVID‐19 pandemic. Nat Microbiol. 2020;5:1408‐1417. [DOI] [PubMed] [Google Scholar]
- 78. Halfmann PJ, Hatta M, Chiba S, et al. Transmission of SARS‐CoV‐2 in domestic cats. N Engl J Med. 2020;383:592‐594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Shi J, Wen Z, Zhong G, et al. Susceptibility of ferrets, cats, dogs, and other domesticated animals to SARS–coronavirus 2. Science. 2020;368(6494):1016‐1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Tylor S, Andonov A, Cutts T, et al. The SR‐rich motif in SARS‐CoV nucleocapsid protein is important for virus replication. Can J Microbiol. 2009;55(3):254‐260. [DOI] [PubMed] [Google Scholar]
- 81. Järvelin AI, Noerenberg M, Davis I, Castello A. The new (dis) order in RNA regulation. Cell Commun Signal. 2016;14(1):9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Kikkert M. Innate immune evasion by human respiratory RNA viruses. J Innate Immun. 2020;12(1):4‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Kopecky‐Bromberg SA, Martínez‐Sobrido L, Frieman M, Baric RA, Palese P. Severe acute respiratory syndrome coronavirus open reading frame (ORF) 3b, ORF 6, and nucleocapsid proteins function as interferon antagonists. J Virol. 2007;81(2):548‐557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. McBride R, Van Zyl M, Fielding BC. The coronavirus nucleocapsid is a multifunctional protein. Viruses. 2014;6(8):2991‐3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Sokalingam S, Raghunathan G, Soundrarajan N, Lee S‐G. A study on the effect of surface lysine to arginine mutagenesis on protein stability and structure using green fluorescent protein. PLoS One. 2012;7(7):e40410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Chang C‐k, Hou C‐D, Huang T‐h, et al. The SARS coronavirus nucleocapsid protein–forms and functions. Antiviral Res. 2014;103:39‐50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Haynes C, Iakoucheva LM. Serine/arginine‐rich splicing factors belong to a class of intrinsically disordered proteins. Nucleic Acids Res. 2006;34(1):305‐312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Carlson CR, Asfaha JB, Ghent CM, et al. Phosphoregulation of Phase Separation by the SARS‐CoV‐2 N Protein Suggests a Biophysical Basis for its Dual Functions. Mol Cell. 2020;11:025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Schuster NA. Using the nucleocapsid protein to investigate the relationship between SARS‐CoV‐2 and closely related bat and pangolin coronaviruses. BioRxiv. Published online June 25, 2020.
- 90. de Haan CA, Rottier PJ. Molecular interactions in the assembly of coronaviruses. Adv Virus Res. 2005;64:165‐230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Huston NC, Wan H, Strine MS, Tavares RdCA, Wilen CB, Pyle AM. Comprehensive in vivo secondary structure of the SARS‐CoV‐2 genome reveals novel regulatory motifs and mechanisms. Mol Cell. 2021;81(3):584‐598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Kristofich J, Morgenthaler AB, Kinney WR, et al. Synonymous mutations make dramatic contributions to fitness when growth is limited by a weak‐link enzyme. PLoS Genet. 2018;14(8):e1007615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Mitra S, Ray SK, Banerjee R. Synonymous codons influencing gene expression in organisms. Res Rep Biochem. 2016;6:57‐65. [Google Scholar]
- 94. Hurst KR, Ye R, Goebel SJ, Jayaraman P, Masters PS. An interaction between the nucleocapsid protein and a component of the replicase‐transcriptase complex is crucial for the infectivity of coronavirus genomic RNA. J Virol. 2010;84(19):10276‐10288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Hurst KR, Koetzner CA, Masters PS. Characterization of a critical interaction between the coronavirus nucleocapsid protein and nonstructural protein 3 of the viral replicase‐transcriptase complex. J Virol. 2013;87(16):9159‐9172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Serrano P, Johnson MA, Almeida MS, et al. Nuclear magnetic resonance structure of the N‐terminal domain of nonstructural protein 3 from the severe acute respiratory syndrome coronavirus. J Virol. 2007;81(21):12049‐12060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Du X, Wang Z, Wu A, et al. Networks of genomic co‐occurrence capture characteristics of human influenza A (H3N2) evolution. Genome Res. 2008;18(1):178‐187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Rimmelzwaan G, Berkhoff E, Nieuwkoop N, Smith DJ, Fouchier R, Osterhaus A. Full restoration of viral fitness by multiple compensatory co‐mutations in the nucleoprotein of influenza A virus cytotoxic T‐lymphocyte escape mutants. J Gen Virol. 2005;86(6):1801‐1805. [DOI] [PubMed] [Google Scholar]
- 99. Deng L, Liu M, Hua S, et al. Network of co‐mutations in Ebola virus genome predicts the disease lethality. Cell Res. 2015;25(6):753‐756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Chen H, Zhou X, Zheng J, Kwoh C‐K. Rules of co‐occurring mutations characterize the antigenic evolution of human influenza A/H3N2, A/H1N1 and B viruses. BMC Med Genomics. 2016;9(3):69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Rimmelzwaan GF, Kreijtz JH, Bodewes R, Fouchier RA, Osterhaus AD. Influenza virus CTL epitopes, remarkably conserved and remarkably variable. Vaccine. 2009;27(45):6363‐6365. [DOI] [PubMed] [Google Scholar]
- 102. Lyons DM, Lauring AS. Mutation and epistasis in influenza virus evolution. Viruses. 2018;10(8):407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Lei J, Kusov Y, Hilgenfeld R. Nsp3 of coronaviruses: Structures and functions of a large multi‐domain protein. Antiviral Res. 2018;149:58‐74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Skjesol A, Skjæveland I, Elnæs M, et al. IPNV with high and low virulence: host immune responses and viral mutations during infection. Virol J. 2011;8(1):1‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Schoeman D, Fielding BC. Coronavirus envelope protein: current knowledge. Virol J. 2019;16(1):1‐22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Escors D, Ortego J, Laude H, Enjuanes L. The membrane M protein carboxy terminus binds to transmissible gastroenteritis coronavirus core and contributes to core stability. J Virol. 2001;75(3):1312‐1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Alsaadi J, Jones EA. IM. Membrane binding proteins of coronaviruses. Future Virol. 2019;14(4):275‐286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Bai Y, Yao L, Wei T, et al. Presumed asymptomatic carrier transmission of COVID‐19. JAMA. 2020;323(14):1406‐1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Quan‐Xin L, Xiao‐Jun T, Qiu‐Lin S, et al. Clinical and immunological assessment of asymptomatic SARS‐CoV‐2 infections. Nature Med. 2020. [DOI] [PubMed] [Google Scholar]
- 110. Wolf J M, Streck A F, Fonseca A, Ikuta N, Simon D, Lunge V R. Dissemination and evolution of SARS‐CoV‐2 in the early pandemic phase in South America. Journal of Medical Virology. 2021;93(7):4496–4507. 10.1002/jmv.26967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. McCallum M, De Marco A & Lempp F et al. N‐terminal domain antigenic mapping reveals a site of vulnerability for SARS‐CoV‐2. bioRxiv. Published online January 14, 2021. [DOI] [PMC free article] [PubMed]
- 112. McCarthy KR, Rennick LJ, Nambulli S, et al. Recurrent deletions in the SARS‐CoV‐2 spike glycoprotein drive antibody escape. Science. 2021;371:1139‐1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Kemp S, Collier D, Datir R, et al. Neutralising antibodies drive Spike mediated SARS‐CoV‐2 evasion (medRxiv). bioRxiv. Published online December 19, 2020.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information.
Supporting information.
Supporting information.
Data Availability Statement
All the sequence and structural data were taken from the GISAID (https://www.gisaid.org/) and RCSB PDB (https://www.rcsb.org/), as mentioned in the methodology section. We provide all the necessary information, such as accession numbers and the date‐based data source for helping readers and reviewers to check the authenticity of the work. The data that support the findings of this study are available in GISAID at https://www.gisaid.org/. These data were derived from the resources available in the public domain: GISAID, https://www.gisaid.org/.