

Abstract
Malaysia has experienced three waves of coronavirus disease 2019 (COVID‐19) as of March 31, 2021. We studied the associated molecular epidemiology and SARS‐CoV‐2 seroprevalence during the third wave. We obtained 60 whole‐genome SARS‐CoV‐2 sequences between October 2020 and January 2021 in Kuala Lumpur/Selangor and analyzed 989 available Malaysian sequences. We tested 653 residual serum samples collected between December 2020 to April 2021 for anti‐SARS‐CoV‐2 total antibodies, as a proxy for population immunity. The first wave (January 2020) comprised sporadic imported cases from China of early Pango lineages A and B. The second wave (March–June 2020) was associated with lineage B.6. The ongoing third wave (from September 2020) was propagated by a state election in Sabah. It is due to lineage B.1.524 viruses containing spike mutations D614G and A701V. Lineages B.1.459, B.1.470, and B.1.466.2 were likely imported from the region and confined to Sarawak state. Direct age‐standardized seroprevalence in Kuala Lumpur/Selangor was 3.0%. The second and third waves were driven by super‐spreading events and different circulating lineages. Malaysia is highly susceptible to further waves, especially as alpha (B.1.1.7) and beta (B.1.351) variants of concern were first detected in December 2020/January 2021. Increased genomic surveillance is critical.
Keywords: COVID‐19, Malaysia, phylogenetic analysis, SARS‐CoV‐2, seroprevalence, whole genome sequencing
Highlights
As of March 2021, Malaysia has had 3 waves of COVID‐19
The 2nd wave was driven by lineage B.6 viruses
The 3rd wave was driven by lineage B.1.524 viruses
Seroprevalence in Kuala Lumpur/Selangor is only 3%
Malaysia is susceptible to further waves
1. INTRODUCTION
The coronavirus disease 2019 (COVID‐19) pandemic has now entered its second year, having caused over 4.5 million deaths worldwide as of September 2021. The cause, SARS‐CoV‐2, is a positive‐sense RNA betacoronavirus with a ~30 kb genome. Unprecedented efforts have been expended for global genomic surveillance. This utilizes whole genome sequencing to identify genetic lineages and variants of concern (VOC) carrying mutations that may increase transmission, enable immune escape, or impact vaccine responses or diagnostic tests. 1
Malaysia is a southeast Asian country comprising 13 states and 3 federal territories, with a population of about 32 million. The first wave of COVID‐19, caused by SARS‐CoV‐2, consisted of 22 mainly imported cases from China and lasted for 3 weeks from late January 2020. 2 A second, much larger wave occurred between March and June, mainly driven by a religious mass gathering linked to at least 3375 confirmed cases, a third of national cases at the time. 3 A nationwide movement control order and other public health measures led to a considerable reduction in numbers. 2 However, an ill‐timed election in the state of Sabah in September led to an even larger, nationwide third wave extending into 2021. 4 As of March 31, 2021, there have been 345 500 confirmed cases and 1272 deaths. 5
In this study, we performed whole‐genome sequencing from 60 SARS‐CoV‐2 cases from the recent third wave in Kuala Lumpur and Selangor. We analyzed them with other complete genome sequences from Malaysia available on the GISAID database (www.gisaid.org) from samples collected before March 31, 2021. Our objective was to relate the molecular epidemiology of circulating SARS‐CoV‐2 to the waves of reported cases in Malaysia. Additionally, having previously reported seroprevalence of 0.4% in Kuala Lumpur/Selangor after the second wave, 6 we carried out a follow‐up study to determine seroprevalence progression during the third wave.
2. MATERIALS AND METHODS
2.1. Samples for sequencing
This study was carried out in the Universiti Malaya Medical Centre (UMMC), a teaching hospital serving the populations of Kuala Lumpur federal territory and Selangor state, which accounted for 44.5% of national cases up to March 31, 2021. Patients admitted to UMMC were diagnosed with COVID‐19 by real‐time polymerase chain reaction (PCR) detection of SARS‐CoV‐2 in nasopharyngeal/oropharyngeal swabs, using the WHO‐recommended Berlin Charité protocol 7 and abTES COVID‐19 qPCR I Kit (AITbiotech, Singapore). Cases were diagnosed between October 2020 and January 2021. Demographic details and location of infection were collected for each case. This study was approved by the UMMC ethics committee (no. 2020730–8928). Our institution does not require informed consent for retrospective studies of archived and anonymized samples.
2.2. Genome sequencing of SARS‐CoV‐2
Viral RNA was extracted from 60 positive clinical samples using a QIAamp Viral RNA Mini Kit and subjected to whole‐genome sequencing following the ARTIC network protocol (v3). 3 , 8 Briefly, extracted RNA was reverse transcribed using SuperScript IV First‐Strand Synthesis System (Invitrogen) with random hexamers. The cDNA was subsequently amplified with Q5 High‐Fidelity DNA polymerase (NEB) using two pools of nCoV‐2019/V3 primer sets. Amplicon libraries were prepared using the iTrue method and sequenced on the iSeq 100 System (Illumina), with an output of 1 × 300 bp reads.
2.3. Bioinformatic analysis
Generated reads were first imported into Geneious Prime 2020 (Biomatters) and trimmed using a BBDuk trimmer plugin (version 1.0) and mapped to reference genome Wuhan‐Hu‐1 (GenBank accession number: MN908947). The threshold to generate consensus sequences was set at “highest quality (adjusted),” with other settings at default, including calling an N if coverage depth is <2. We aimed to produce sequences that would fulfill GISAID inclusion criteria as complete sequences, that is >29 000 nucleotides with <50% Ns. The consensus sequences have been deposited in the GISAID database 9 with the accession numbers: EPI_ISL_2769381‐2769438 and EPI_ISL_2784328‐27843289. The raw sequence data are also available at BioProject (accession number: PRJNA776394, Sequence Read Archive numbers: SRR16641605‐SRR16641655). A total of 929 complete genome sequences from Malaysian samples collected on or before March 31, 2021, available from the GISAID database as of June 26, 2021, were aligned with the 60 sequences from this study using MAFFT with default parameters. 10 Maximum likelihood phylogenetic analysis was performed in IQ‐TREE v2.1.3 based on the best‐fit model chosen with Bayesian information criterion, with 1000 ultra‐fast bootstrap replicates. 11 Substitution rate and node dates were estimated using the least square dating method. Lineages were classified using Phylogenetic Assignment of Named Global Outbreak Lineages (PANGOLIN) software (v.3.1.5 2021‐06‐15). 12
2.4. Serological testing
To estimate the seroprevalence of the Kuala Lumpur/Selangor population, we tested residual serum samples from UMMC inpatients collected between December 2020 to April 2021 for diagnostic testing for nonrespiratory infections. Residual serum has been an adequate proxy for general population serosurveys for COVID‐19. 13 The sample size was calculated with an expected seroprevalence of 5% (95% CI, 3%–7%), and found to be 457. A total of 653 archived samples were tested, including 43–103 samples from every 10‐year age group (<10, 10–19, 20–29, 30–39, 40–49, 50–59, 60–69, and ≥70 years). The serum samples were screened using a SARS‐CoV‐2 total antibody enzyme‐linked immunosorbent assay (ELISA) (Beijing Wantai Biological Pharmacy Enterprise). This assay has received emergency use authorization by the US Food and Drug Administration and has high reported sensitivity (96.7%) and specificity (99.5%). 14 Samples testing indeterminate (optical density/cut‐off ratio: 0.9–1.1) were retested and considered positive if the retest was reactive. Crude seroprevalence rates are given with 95% exact binomial confidence intervals (CIs).
2.5. Epidemiological data and analysis
Daily SARS‐CoV‐2 case numbers in Malaysia from January 2020 to March 31, 2021, were retrieved from the Ministry of Health, Malaysia (https://kpkesihatan.com/). Age‐stratified population data for Kuala Lumpur and Selangor were obtained from the Department of Statistics, Malaysia (http://pqi.stats.gov.my/searchBI.php), and used to calculate incidence rates per 100 000 population and a direct age‐standardized seroprevalence rate. A map of incidence was constructed using R v4.1.0 software.
3. RESULTS
3.1. Incidence rates
There were 345 500 cases reported in Malaysia as of March 31, 2021. The cumulative incidence was highest in the more densely populated states/federal territories of western and southern Peninsular Malaysia and in East Malaysia (Figure 1A). Plotting the state‐stratified 7‐day incidence per 100 000 population over time (Figure 1B) shows the second wave occurring between March and June 2020. This is followed by the much larger third wave, which began and peaked in Sabah (and the small neighboring federal territory of Labuan) in September before waves appear in all other states several weeks later. Peak 7‐day incidence rates in Sabah and Labuan reached 122 and 400 per 100 000, respectively. In Kuala Lumpur and Selangor, where our hospital is based, peak 7‐day incidence rates were 232 and 200 per 100 000, respectively. As of March 31, case numbers had begun to fall in some states.
Figure 1.
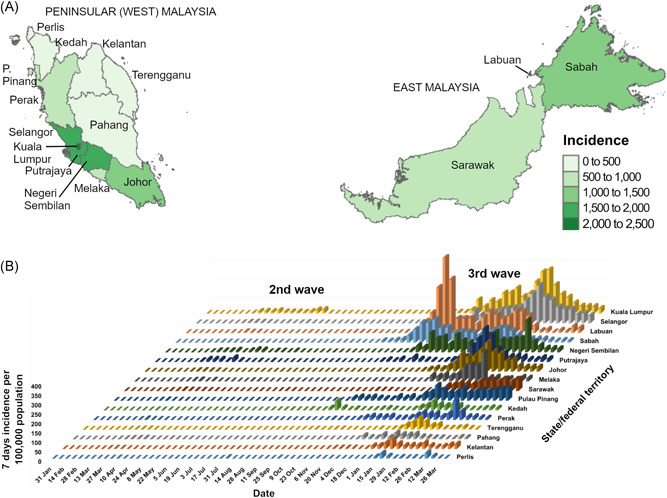
Incidence rates of COVID‐19 in the states and federal territories of Malaysia, as of March 31, 2021. (A) Cumulative incidence per 100 000 population and (B) 7‐day incidence per 100 000 population over time are shown. The map was obtained from the Database of Global Administrative Areas (https://gadm.org/)
3.2. Virus phylogeny and lineages
The samples sequenced in this study came from 60 patients, comprising 31 females and 29 males, with a median age of 35 years (range: 3–81). Sequencing of the 60 samples generated an average of >99.7% breadth of coverage of the genome and average coverage depth of 1721 (range: 313–2802, Table S1). All were successfully accepted in GISAID, and all were considered as “high coverage” with <1% Ns except for 1 sequence (hCoV‐19/Malaysia/8827/2021) with 9.52% Ns. All cases were acquired locally. The Pango lineages were identified, and 56 (93.3%) were from B.1.524, while the remaining two were from B.1 and two were from B.1.428.3. Nonsynonymous mutations seen in the B.1.524 lineage sequences were nsp3‐T1198I, nsp4‐T28I, nsp5‐T24A, nsp12‐P323L, nsp13‐L428F, spike‐D614G, spike‐A701V, and N‐S194L.
There were 989 Malaysian sequences (including the 60 from this study) collected on or before March 31, 2021, in GISAID, of which 427 (43%) were from the East Malaysian state of Sarawak. The Pango lineages are shown in Figure 2. The most frequently identified were B.1.524 (354, 35.8%); B.1466.2 and sublineages B.1466.2.1 (AU.1), B.1466.2.2 (AU.2), and B.1466.2.3 (AU.3) (224, 22.6%), B.6 and sublineages B.6.1, B.6.2, and B.6.6 (109, 11.0%), and B.1.36.16 (53, 5.4%).
Figure 2.
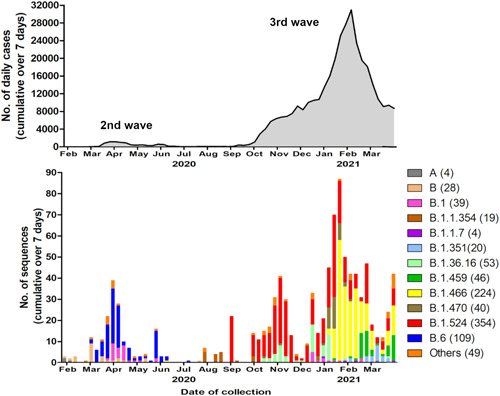
Total reported daily cases (above) and circulating lineages of SARS‐CoV‐2 (below) in Malaysia, from sequences available on GISAID as of March 31, 2021. Lineages are named using the Pango system
When the lineages were analyzed over time (Figures 2 and 3), the earliest identified sequences during the first wave in January/February 2020 were from lineages A and B. The second wave in March to June 2020 was predominantly associated with lineage B.6. In July/August, there were reported sequences from lineage B.1.1.354. The large third wave starting in Sabah in September 2020 was mainly associated with B.1.524. The B.1.524 sequences are diverse, from different states around the country. There were also limited sequences from lineages B.1.36.16. However, 91.5% of the Malaysian sequences from lineages B.1.466.2 (mainly AU.2), and 100% of B.1.459 and B.1.470 were reported from Sarawak starting January 2021, and these form relatively tight clusters consistent with geographic restriction. Of note, small numbers of the VOCs B.1.1.7 (alpha; n = 4) and B.1.351 (beta; n = 20) were reported in December 2020 and January 2021, respectively.
Figure 3.
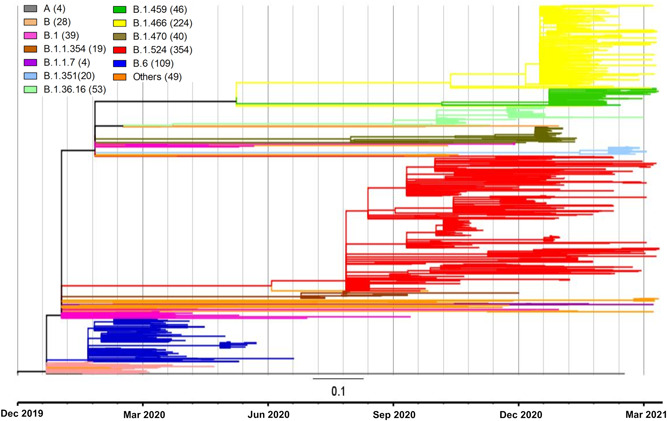
Phylogenetic tree of 989 SARS‐CoV‐2 whole‐genome sequences collected on or before March 31, 2021, from Malaysia, available on GISAID as of June 26, 2021. The key shows the color‐coded Pango lineages with the number of available sequences in brackets
3.3. Seroprevalence rate
Serosurveys provide better tracking of the extent of population infection and immunity, as previously undiagnosed cases can also be identified. A total of 653 serum samples collected between December 2020 to April 2021 from UMMC inpatients were tested for total anti‐SARS‐CoV‐2 antibodies (Table 1), and the crude seropositive rate was 4.1% (95% CI: 2.7–6.0). The highest rates were seen in those aged 30–49 years and >60 years. No seropositives were seen in the 123 samples from patients aged 0–19 years. Using age‐stratified population data for Kuala Lumpur and Selangor, a direct age‐standardized seroprevalence rate was calculated as 3.0%.
Table 1.
SARS‐CoV‐2 total antibodies seropositivity rate from Kuala Lumpur/Selangor samples collected between December 2020 to April 2021
| Age group (years) | Number of samples | Seropositive rate % (95% CI) |
|---|---|---|
| 0–9 | 80 | 0 (0–4.5) |
| 10–19 | 43 | 0 (0–8.2) |
| 20–29 | 99 | 1.0 (0.03–5.5) |
| 30–39 | 102 | 5.9 (2.2–12.4) |
| 40–49 | 84 | 4.8 (1.3–11.8) |
| 50–59 | 81 | 1.2 (0.03–6.7) |
| 60–69 | 78 | 10.3 (4.5–19.2) |
| >70 | 86 | 8.1 (3.3–16.1) |
| Total | 653 | 4.1 (2.7–6.0) |
4. DISCUSSION
There were clear changes in circulating lineages over the course of the pandemic. The first wave cases were from early lineages A and B, as most were imported from China. 12 We previously showed that the second wave from March to June was driven mainly by lineage B.6 spread at a religious mass gathering of thousands of participants from Malaysia, other countries in the Southeast Asian region, India and Australia. 3 Although B.6 viruses were reported from India in GISAID as recently as May 2021, B.6 and its sublineages seem to have disappeared from Malaysia, with none reported since July 2020.
There followed a relatively quiet period between June to early September when ≤43 daily cases were reported. In July/August 2020, clusters were reported in Kedah state in northern Peninsular Malaysia. In early September, outbreaks in an immigration detention center and prison in Sabah state, in East Malaysia, spread to the community. These Kedah and Sabah clusters were not genetically linked. 15 Amidst rising community cases in Sabah, a state election was held in late September, which involved in‐person campaigning, rallies, and voting. The inter‐ and intrastate movement of political party workers and 1.1 million voters (some flying to/from Peninsular Malaysia) led to extensive spread throughout Malaysia, particularly in Kuala Lumpur, Selangor, and Johor, where most Sabah migrants live. 16 This led to the third pandemic wave, due mainly to viruses from lineage B.1.524. The third wave also involved large outbreaks among foreign migrants incarcerated in detention centers and prisons, or working in cramped environments in factories and construction sites. 17 As of March 31, 2021, over 22% of COVID‐19 cases affected non‐Malaysians. The non‐Malaysian population (including undocumented migrants) is estimated at 3‐5.5 million, 17 or 10%–18% of the total population.
Sequences of the B.1.524 lineage were first reported in Germany in March 2020 and Switzerland in July 2020, but of the 441 reported sequences, 68% are from Malaysia, and almost all the rest are from neighboring Southeast Asian countries (Singapore, Thailand, Philippines, and Indonesia). B.1.524 viruses carry the spike mutations D614G (present in most SARS‐CoV‐2 viruses) and A701V, which is also found in the VOC B.1.351 and the previously designated variant of interest B.1.526 (iota). 1 The mutation D614G, which has become predominant worldwide, is associated with increased cell entry, replication, and transmissibility, possibly by stabilizing the spike receptor‐binding domain and enhancing binding to the host cell receptor angiotensin‐converting enzyme 2. 1 A701V is close to the S2ʹ cleavage site which when cleaved leads to spike‐mediated membrane fusion, but its significance remains unknown. The nsp12 (RNA‐dependent RNA polymerase) P323L mutation present in lineage B.1.524 is almost always associated with D614G; the significance of P323L is also unclear, but it may impact the interaction between nsP12 and nsP8 and affect RNA polymerase activity. 18 To date, B.1.524 does not contain other spike mutations associated with increased pathogenicity or immune escape, such as K417N, L452R, E484K, and N501Y. The main factor associating it with Malaysia's most severe wave to date is likely the Sabah election as a super‐spreading event.
Apart from the predominant B.1.524, other lineages made contributions, indicating introduction from other countries in the region despite the strict border controls. The Kedah clusters in July/August 2020 originated from a traveler to India 19 and were associated with lineage B.1.1.354, which was first reported in GISAID from India in May 2020. A third of reported B.1.1.354 sequences are from India, with a further quarter from Malaysia and Singapore, indicating regional spread. There were also Malaysian sequences from lineages B.1.36.16 reported in October, November 2020, and January 2021; preceding this, sequences of this lineage were reported from Bangladesh, England, and Thailand. B.1.459, B.1.466.2 (and sublineages), and B.1.470 sequences were reported from January 2021, almost exclusively from Sarawak state. 20 The majority of reported sequences from these lineages originate from neighboring Indonesia. Border controls with strictly enforced quarantines continue to be important for pandemic control, but Malaysia's extensive porous land and maritime international borders pose a major challenge. The probable explanation for why B.1.459, B.1.466.2, and B.1.470 viruses are confined mainly in Sarawak state, for now, is that following introduction, the rigid restrictions on interstate travel imposed after the onset of the third wave have prevented spread elsewhere in Malaysia.
We previously estimated an age‐standardized seroprevalence of 0.4% for Kuala Lumpur/Selangor during and after the second pandemic wave ending June 2020. 6 A national, population‐wide seroprevalence study carried out between August and October 2020 showed a compatible rate of 0.5%. 21 In this study, we estimated that the rate in Kuala Lumpur/Selangor had increased to 3.0% during the third wave, exceeding the cumulative incidence of reported cases of 1.9% as of March 31, 2021. Seropositive rates were lowest among children, consistent with the relatively low rate of pediatric cases reported here. 22 The overall very low seropositive rate points to population susceptibility to further disease, which is concerning amidst reports of extreme pressures on the healthcare system. 23 A recent review of population‐based studies up to March 2021 estimated a global seroprevalence of 9.5%, with the lowest region‐specific seroprevalence of 1.6% in the east and southeast Asia. 24 Differences in sample population, testing method and study timing make direct comparisons between countries difficult. However, in keeping with the overall lower regional seroprevalence rate and the perception that east and southeast Asian countries have been relatively less impacted by the pandemic, our seroprevalence finding was lower than the global average. Continued serosurveys are needed, as the risk of emerging VOCs continues and vaccine rollout remains slower in developing countries.
The main limitation of our study is that it is based on 989 cases, which is just 0.3% of total reported Malaysian cases, with unknown criteria for the selection of samples for sequencing. Of these sequences, 427 (43%) are from Sarawak and 122 (12%) are from our center, covering Kuala Lumpur/Selangor. The cases from our center were from inpatients, which may bias toward more severe cases. However, until January 2021, all confirmed cases in Malaysia were admitted to hospitals regardless of severity. The states of origin for many Malaysian sequences are not stated, and other minor lineages may have been missed. Sampling was also uneven over time, with low numbers in December 2020, for example. All these reflect the differing capacities and schedules of the limited number of centers performing sequencing at the time. The Malaysian government has recently announced a national genomic surveillance program involving institutions from the Ministry of Health and academia, which will optimize this currently fragmented sequencing capacity and improve the representativeness of sequenced cases. This program is critical to the need to monitor emerging variants and the commencement of the national immunization program in March 2021. In resource‐limited settings, whole‐genome sequencing can also be supplemented by partial sequencing or PCRs to detect key mutations found in VOCs. 25
5. CONCLUSIONS
The major second and third pandemic waves in Malaysia were driven by super‐spreading events of different predominant virus lineages B.6 and B.1.524, respectively. There was also more localized circulation of other minor lineages, notably in Sarawak. There is still a very low level of population immunity even in Kuala Lumpur/Selangor, the most heavily affected areas. With the recent first detections of the VOCs B.1.351 and B.1.1.7 in Malaysia, there is a continuing high risk of further waves of COVID‐19.
CONFLICT OF INTERESTS
The authors declare no conflict of interests.
AUTHOR CONTRIBUTIONS
I‐Ching Sam: Conceptualization, methodology, formal analysis, writing – original draft, project administration, funding acquisition. Yoong Min Chong: Methodology, investigation, formal analysis, writing – original draft, visualization. Azwani Abdullah: Investigation. Jolene Yin Ling Fu: Methodology, investigation, formal analysis. M. Shahnaz Hasan: Resources, investigation. Fadhil Hadi Jamaluddin: Resources, investigation. Adeeba Kamarulzaman: Resources, investigation. Koo Koon Lim: investigation. Mohd Afiq Mohd Nor: Investigation. Yong Kek Pang: Resources, investigation. Sasheela Ponnampalavanar: Resources, investigation. Muhammad Fadzil Shahib: Investigation. Sharifah Faridah Syed Omar: Resources, investigation. Jonathan Chia Jui Chan: Resources, investigation. David Perera: Resources, investigation. Yoke Fun Chan: Conceptualization, methodology, formal analysis, writing – review and editing, project administration, funding acquisition. All authors were involved in reviewing and editing the manuscript.
Supporting information
Supplementary Table 1.
ACKNOWLEDGMENTS
We gratefully acknowledge the authors from originating and submitting laboratories of GISAID sequence data on which the analysis is based. This study was partly supported by the Defense Threat Reduction Agency, USA (grant number: HDTRA1‐17‐1‐0027) and the Ministry of Education, Malaysia (grant number: FRGS/1/2020/SKK0/UM/02/5). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The serology kits were kindly donated by Xiamen University, China.
Sam I‐C, Chong YM, Abdullah A, et al. Changing predominant SARS‐CoV‐2 lineages drives successive COVID‐19 waves in Malaysia, February 2020 to March 2021. J Med Virol. 2022;94:1146‐1153. 10.1002/jmv.27441
Contributor Information
I‐Ching Sam, Email: jicsam@ummc.edu.my.
Yoke Fun Chan, Email: chanyf@um.edu.my.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are openly available in GISAID at https://www.gisaid.org/, reference number EPI_ISL_2769381‐2769438, 2784328‐27843289, and at BioProject at https://www.ncbi.nlm.nih.gov/bioproject/ (accession number PRJNA776394, Sequence Read Archive numbers SRR16641605‐SRR16641655).
REFERENCES
- 1. Peacock TP, Penrice‐Randal R, Hiscox JA, Barclay WS. SARS‐CoV‐2 one year on: evidence for ongoing viral adaptation. J Gen Virol. 2021;102:001584. 10.1099/jgv.0.001584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ng CFS, Seposo XT, Moi ML, Tajudin MABA, Madaniyazi L, Sahani M. Characteristics of COVID‐19 epidemic and control measures to curb transmission in Malaysia. Int J Infect Dis. 2020;101:409‐411. 10.1016/j.ijid.2020.10.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chong YM, Sam IC, Chong J, et al. SARS‐CoV‐2 lineage B.6 was the major contributor to early pandemic transmission in Malaysia. PLOS Negl Trop Dis. 2020;14:e0008744. 10.1101/2020.08.27.269738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rampal L, Liew BS. Malaysia's third COVID‐19 wave–a paradigm shift required. Med J Malaysia. 2021;76:1‐4. [PubMed] [Google Scholar]
- 5. Ministry of Health Malaysia . Press statement from the Director‐General of Health, 31 March 2021: updates on the coronavirus disease 2019 (COVID‐19) situation in Malaysia. Accessed November 2, 2021. https://kpkesihatan.com/2021/03/31/kenyataan-akhbar-kpk-31-mac-2021-situasi-semasa-jangkitan-penyakit-coronavirus-2019-covid-19-di-malaysia/
- 6. Sam IC, Chong YM, Tan CW, Chan YF. Low postpandemic wave SARS‐CoV‐2 seroprevalence in Kuala Lumpur and Selangor, Malaysia. J Med Virol. 2021;93:647‐648. 10.1002/jmv.26426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Corman VM, Landt O, Kaiser M, et al. Detection of 2019 novel coronavirus (2019‐nCoV) by real‐time RT‐PCR. Euro Surveill. 2020;25:2000045. 10.2807/1560-7917.ES.2020.25.3.2000045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. ARTICnetwork, 2020 . nCoV‐2019 sequencing protocol. Accessed November 2, 2021. https://www.protocols.io/view/ncov-2019-sequencing-protocol-bbmuik6w
- 9. Shu Y, McCauley J. GISAID: Global initiative on sharing all influenza data–from vision to reality. Euro Surveill. 2017;22:30494. 10.2807/1560-7917.ES.2017.22.13.30494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772‐780. 10.1093/molbev/mst010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. To TH, Jung M, Lycett S, Gascuel O. Fast dating using least‐squares criteria and algorithms. Syst Biol. 2016;65:82‐97. 10.1093/sysbio/syv068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rambaut A, Holmes EC, O'Toole Á, et al. A dynamic nomenclature proposal for SARS‐CoV‐2 lineages to assist genomic epidemiology. Nat Microbiol. 2020;5:1403‐1407. 10.1038/s41564-020-0770-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bobrovitz N, Arora RK, Cao C, et al. Global seroprevalence of SARS‐CoV‐2 antibodies: a systematic review and meta‐analysis. PLoS One. 2021;16:e0252617. 10.1371/journal.pone.0252617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Harritshøj LH, Gybel‐Brask M, Afzal S, et al. Comparison of 16 serological SARS‐CoV‐2 immunoassays in 16 clinical laboratories. J Clin Microbiol. 2021;59:e02596‐20. 10.1128/JCM.02596-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. CodeBlue. Virus From Sabah's Benteng LD cluster not linked to Kedah clusters: IMR . September 25, 2020. Accessed November 2, 2021. https://codeblue.galencentre.org/2020/09/25/virus-from-sabahs-benteng-ld-cluster-not-linked-to-kedah-clusters-imr/
- 16. Lim JT, Maung K, Tan ST, et al. Estimating direct and spill‐over impacts of political elections on COVID‐19 transmission using synthetic control methods. PLoS Comput Biol. 2021;17:e1008959. 10.1371/journal.pcbi.1008959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wahab A. The outbreak of Covid‐19 in Malaysia: pushing migrant workers at the margin. Soc Sci Humanit Open. 2020;2:100073. 10.1016/j.ssaho.2020.100073 [DOI] [Google Scholar]
- 18. Ilmjärv S, Abdul F, Acosta‐Gutiérrez S, et al. Concurrent mutations in RNA‐dependent RNA polymerase and spike protein emerged as the epidemiologically most successful SARS‐CoV‐2 variant. Sci Rep. 2021;11:13705. 10.1038/s41598-021-91662-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ministry of Health Malaysia. Press statement from the Director‐General of Health, 27 August 2020: Updates on the coronavirus disease 2019 (COVID‐19) situation in Malaysia. Accessed November 2, 2021. https://kpkesihatan.com/2020/08/27/kenyataan-akhbar-kpk-27-ogos-2020-situasi-semasa-jangkitan-penyakit-coronavirus-2019-covid-19-di-malaysia/
- 20. Perera D. Sarawak COVID‐19 genomic surveillance statement. 2021. Accessed November 2, 2021. https://www.pustaka-sarawak.com/document_uploaded/1620051337.pdf [Google Scholar]
- 21. Chong ZL, Rodzlan Hasani WS, Mat Rini H, et al. The true burden of COVID‐19 in Malaysia: seroprevalence of SARS‐CoV‐2 infection. Institute for Public Health, National Health and Morbidity Survey (NHMS) 2020: Communicable Diseases, Cognitive, Attitude and Behaviour. Vol 2. Shah Alam: Ministry of Health Malaysia; 2021:18‐22. [Google Scholar]
- 22. Ng DC, Tan KK, Chin L, et al. Clinical and epidemiological characteristics of children with COVID‐19 in Negeri Sembilan, Malaysia. Int J Infect Dis. 2021;108:347‐352. 10.1016/j.ijid.2021.05.073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Idris AN PM: nation's healthcare system is at breaking point. January 11, 2021. Accessed November 2, 2021. https://www.theedgemarkets.com/article/pm-says-malaysias-healthcare-system-breaking-point
- 24. Rostami A, Sepidarkish M, Fazlzadeh A, et al. Update on SARS‐CoV‐2 seroprevalence ‐ regional and worldwide. Clin Microbiol Infect. 2021. 10.1016/j.cmi.2021.09.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kami W, Kinjo T, Arakaki W, et al. Rapid and simultaneous identification of three mutations by the Novaplex SARS‐CoV‐2 variants I assay kit. J Clin Virol. 2021;141:104877. 10.1016/j.jcv.2021.104877 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1.
Data Availability Statement
The data that support the findings of this study are openly available in GISAID at https://www.gisaid.org/, reference number EPI_ISL_2769381‐2769438, 2784328‐27843289, and at BioProject at https://www.ncbi.nlm.nih.gov/bioproject/ (accession number PRJNA776394, Sequence Read Archive numbers SRR16641605‐SRR16641655).