

Abstract
The coronavirus disease 2019 (COVID‐19) has affected the lives of millions of people around the world. In an effort to develop therapeutic interventions and control the pandemic, scientists have isolated several neutralizing antibodies against SARS‐CoV‐2 from the vaccinated and convalescent individuals. These antibodies can be explored further to understand SARS‐CoV‐2 specific antigen–antibody interactions and biophysical parameters related to binding affinity, which can be utilized to engineer more potent antibodies for current and emerging SARS‐CoV‐2 variants. In the present study, we have analyzed the interface between spike protein of SARS‐CoV‐2 and neutralizing antibodies in terms of amino acid residue propensity, pair preference, and atomic interaction energy. We observed that Tyr residues containing contacts are highly preferred and energetically favorable at the interface of spike protein–antibody complexes. We have also developed a regression model to relate the experimental binding affinity for antibodies using structural features, which showed a correlation of 0.93. Moreover, several mutations at the spike protein–antibody interface were identified, which may lead to immune escape (epitope residues) and improved affinity (paratope residues) in current/emerging variants. Overall, the work provides insights into spike protein–antibody interactions, structural parameters related to binding affinity and mutational effects on binding affinity change, which can be helpful to develop better therapeutics against COVID‐19.
Keywords: binding affinity, COVID‐19, mutational analysis, neutralizing antibodies, regression analysis, SARS‐CoV‐2
1. INTRODUCTION
Coronavirus disease 2019 (COVID‐19) has caused severe global economic depression and the loss of millions of human lives. As of November 2021, there have been more than 246 million confirmed cases and over 5 million deaths due to SARS‐CoV‐2 infection (https://covid19.who.int/). The causative agent of the COVID‐19 pandemic, SARS‐CoV‐2, is a single‐stranded RNA virus that is around 30 000 base pairs in length. SARS‐CoV‐2 infection in humans is initiated by the interaction of the receptor binding domain (RBD), present in S1 subunit of the spike (S) protein, with the human ACE2 receptor leading entry to the host cells. 1 Researchers have identified several neutralizing antibodies induced against SARS‐CoV‐2, either by vaccines or natural infection. The major portion of these neutralizing antibodies controls viral infection by blocking the binding of SARS‐CoV‐2 to the human ACE2 receptor. 2 In other neutralization mechanisms, antibodies may bind the N‐terminal domain in the S1 subunit to potentially restrain the conformational changes of the S protein 3 , 4 or bind S2 to inhibit membrane fusion and subsequent viral entry. 5
An in depth analysis of virus‐host interaction is essential to understand the pathophysiology of infection and to develop therapeutic intervention. 6 The studies on SARS‐CoV‐2 protein interaction networks have revealed various drug targets for drug discovery. 7 , 8 The deep mutational scanning study by Greaney et al. 9 assessed all possible single amino acid variants in the spike protein and provided immune escape maps for mutations in the presence of antibodies. They applied these maps to five potent neutralizing antibodies (COV2‐2094, COV2‐2165, COV2‐2479, COV2‐2050, and COV2‐2499) isolated from SARS‐CoV‐2 convalescent patients and found K378E, K378N, E484K, G446D, and Q498R as immune escape mutations. Another study detected variants that escaped either neutralizing SARS‐CoV‐2 mAbs or convalescent plasma and observed that mutations S477N and E484K rank prominently among mAb escape mutations. 10 Although these mutational studies have significantly improved our knowledge on immune escape variants, they lack a large‐scale analysis to understand the biophysical factors leading to decreased/loss of binding. The analysis of SARS‐CoV‐2 neutralizing antibodies would provide better insights, allowing engineering of more potent antibodies for current/emerging strains. 11 , 12 , 13
In this work, we have analyzed the interface of SARS‐CoV‐2 spike protein and neutralizing antibody complexes with respect to ACE2 binding to understand the residue propensity, pair preference, and atomic interaction energy. Tyr residues show the highest propensity, and the YY pairs have highest pair preference at the interface of spike–antibody complexes. Further, we use structural features including atomic interactions, interface area, surrounding hydrophobicity, hydrophobic interactions, aromatic–aromatic interactions to develop a regression model to relate the binding affinity (K D ) for each antibody. The comprehensive mutational study of the residues at the spike–antibody interface revealed the important sites in spike protein, which lead to decreased binding affinity. The study is in agreement with experimental studies performed on selected neutralizing antibodies, C121, C144, and C135. 14 We extended the analysis to mutate the residues at the antibody interface to identify mutations that may improve the affinity of neutralizing antibodies. The analysis will be helpful to (a) understand the interactions between the spike protein and the neutralizing antibodies, (b) develop better therapeutics against COVID‐19 based on interaction preferences, (c) understand immune escape for emerging variants of SARS‐CoV‐2, and (d) improve stability and binding affinity of neutralizing antibodies.
2. MATERIALS AND METHODS
2.1. Dataset preparation
2.1.1. Construction of a dataset for spike–neutralizing antibody complex structures
The experimentally reported structures of 77 SARS‐CoV‐2 neutralizing antibodies in complex with spike protein were obtained from the literature. The dataset was further screened based on (a) structure resolution (highest resolution among available structures for particular antibody complexes), (b) single antibody per structure, and (c) sequence identity (<95% for whole sequence and <80% for CDRH3 region). The final antibody dataset contained 29 spike protein–antibody complexes (Table 1). The ACE2 bound spike protein structure (PDB id: 6M0J) was used as a reference for comparison.
TABLE 1.
K D values for neutralizing antibodies and RBD–ACE2 complex
| S. no. | PDB ID | Antibody | K D (nM) | ΔG (kcal/mol) |
|---|---|---|---|---|
| 1 | 6WPT | S309 | <0.001 | −16.3 |
| 2 | 6XC2 | CC12.1 | 17 | −10.55 |
| 3 | 6XC4 | CC12.3 | 14 | −10.67 |
| 4 | 6XCN | C105 | 14 | −10.67 |
| 5 | 6XE1 | CV30 | 3.63 | −11.47 |
| 6 | 6XEY | Fab2‐4 | N/A | N/A |
| 7 | 6XKP | CV07‐270 | N/A | N/A |
| 8 | 6XKQ | CV07‐250 | 0.04 | −14.14 |
| 9 | 7B3O | STE90‐C11 | 8.1 | −10.99 |
| 10 | 7BWJ | P2B‐2F6 | 5.14 | −11.26 |
| 11 | 7BYR | BD23 | N/A | N/A |
| 12 | 7BZ5 | B38 | 70.1 | −9.72 |
| 13 | 7C01 | CB6 | 2.49 | −11.69 |
| 14 | 7CAK | H014 | 0.1 | −13.6 |
| 15 | 7CHB | BD‐236 | 2.8 | −11.61 |
| 16 | 7CHH | BD‐368‐2 | 0.54 | −12.59 |
| 17 | 7CJF | P4A1 | 1.78 | −11.89 |
| 18 | 7CWO | P17 | N/A | N/A |
| 19 | 7JMW | COVA1‐16 | 0.2 | −13.18 |
| 20 | 7JV2 | S2H13 | N/A | N/A |
| 21 | 7K43 | S2‐M11 | 66 | −9.75 |
| 22 | 7K45 | S2‐E12 | 1.6 | −11.95 |
| 23 | 7K8U | C002 | 11 | −10.81 |
| 24 | 7K8V | C110 | 1.3 | −12.07 |
| 25 | 7K8W | C119 | 10 | −10.87 |
| 26 | 7K8Y | C121 | 0.5 | −12.63 |
| 27 | 7K8Z | C135 | 6 | −11.17 |
| 28 | 7K90 | C144 | 18 | −10.52 |
| 29 | 7KFV | C1A‐B3 | 76.3 | −9.67 |
| 30 | 6M0J | RBD‐ACE2 | 14.95 a | −10.65 |
Note: N/A, data are not available.
Average of 14.7 and 15.2 nM.
The residues at the interface of spike protein and antibody were identified using all heavy atom distance cutoff of 4 Å. For structural analysis, we removed heteroatom coordinates from the PDB files and retained one subunit of the spike protein RBD and antibody (heavy and light chain). A set of 983 heterodimer complexes were collected from the PDB database 15 and used as a reference to compare the propensities of interface residues. An overview of the study is represented in Figure 1.
FIGURE 1.
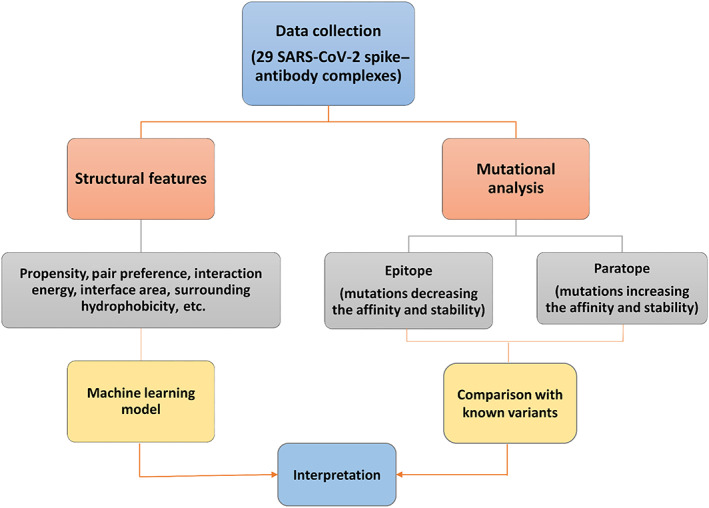
Overview of the workflow
2.1.2. Binding affinity of spike–neutralizing antibodies
The experimental binding affinities (K D ) were obtained from the literature for 24 spike–antibody complexes. The binding free energy (ΔG) was calculated from K D using the following equation:
| (1) |
where, R = 8.31 J/mol/K, is the gas constant and T is the temperature (298 K). The experimental ΔG values are presented in Table 1.
2.2. Computational procedures
2.2.1. Binding propensity
We have calculated the propensity of 20 amino acids to be present at the interface (P interface) using the following equation 16 :
| (2) |
where, n interface(i) and N(i) are the number of residues of type i at the interface and protein, respectively. n interface is the number of residues at the interface, and N is the total number of residues in the protein.
2.2.2. Residue pair preference and interaction energy
The pair preference (Pair[i,j]) for the spike–antibody residue pairs at the interface was computed using the following equation 17 :
| (3) |
where, i and j stand for the interface residues in spike and antibodies, respectively. N i,j is the number of interacting residues of type i in spike and j in antibodies. ∑N i and ∑N j are the total number of residues of type i in spike and j in antibodies, respectively.
The atomic interaction free energy (E inter) between interface residues of the spike and antibody was calculated using AMBER potential. 18 It is given as Equation (4) 17 :
| (4) |
where, 12 and 6; and 1/2; and are, respectively, the van der Waals radius and well depth, q i and q j are, respectively, the charges for the atoms i in spike and j in antibodies, and r ij is the distance between them.
2.2.3. Computation of interface area
Accessible surface areas of the RBD region in the spike protein (S E ), antibody (S H+L ) as well as the entire complex (S complex) were calculated by rolling a water molecule of radius 1.4 Å on the protein/complex surface as described in our previous study. 19 Further, the interface area (S interface) was calculated using Equation (5).
| (5) |
2.2.4. Computation of surrounding hydrophobicity
The surrounding hydrophobicity of residues in spike protein–antibody complexes was computed using the following equation. 20
| (6) |
where, H p (i) is surrounding hydrophobicity of ith residue of the protein. n ij is the total number of surrounding residues of type j around residue i within 8 Å distance (between Cα atoms). h j is hydrophobicity value for the residue type j (in kcal/mol) obtained from thermodynamic transfer experiments. 21 , 22 The average hydrophobicity indices (H p ) was calculated for the interface residues in each spike–antibody complex using PDBparam. 23
2.3. Development of regression model
2.3.1. Features used in the regression model
In addition, we computed the interaction energy and number of contacting residues using PRODIGY, 24 and various types of atomic interactions from PIC (Protein Interactions Calculator) server. 25 The details of all the features for the spike‐neutralizing complexes considered in the present study are presented in Table S1.
2.3.2. Development of multiple regression models
We have developed multiple regression equations to relate structure‐based features with binding affinity of spike–antibody complexes. It is defined as,
| (7) |
where, i is the number of observations, is a dependent variable (binding affinity), are structure‐based parameters, are regression coefficients, and is the error term of the model.
A systematic forward feature selection approach was utilized to select the optimum number of features with the best performance. The regression model was evaluated by the Pearson correlation coefficient to measure the strength of the relationship between two variables and the mean absolute error (MAE) to examine the absolute difference between predicted and experimental affinity values. The model was further validated using a jackknife test, where regression equations were developed to predict ΔG using (n − 1) data points to predict the performance on nth datapoint, recursively.
2.4. Mutational scanning based on change in affinity and stability
Change in affinity and stability upon mutation of the interface residues (paratope and epitope) for all antibody complexes were obtained using sequence and structure based methods. The ProAffiMuSeq server predicts protein–protein binding affinity change upon mutation using sequence‐based features and functional class. 26 mCSM relies on graph‐based signatures to study missense mutations and predicts change in stability and affinity. 27 The CUPSAT prediction model uses amino acid–atom potentials and torsion angle distribution to assess the amino acid environment of the mutation site and predicts the change in stability upon mutation. 28 FoldX software uses empirical function to evaluate the effect of mutations on the stability, interaction, folding, and dynamics of proteins. 29 For FoldX, “RepairPDB” command was used to rectify pdb files, the residues present at the interface of spike protein and antibody were mutated systematically using “BuildModel” command, and the interaction energy was calculated using “AnalyseComplex” command, and stability was calculated as “Stability” command. The change in interaction energy was further calculated by subtracting the values of mutant with wild‐type. 30
3. RESULTS AND DISCUSSION
3.1. Comparison of binding site residues in antibodies and ACE2 with spike protein
The SARS‐CoV‐2 binds with the human ACE2 at the receptor binding domain (RBD) of the spike protein. We compared the epitope regions in spike protein for each antibody and observed that four antibodies (COVA1‐16, H014, S309, and C135) have different epitopes than the ACE2 binding site. COVA1‐16 and H014 antibodies inhibit ACE‐2 binding through steric hindrance 31 , 32 whereas, S309 and C135 antibodies do not compete with ACE2 binding and recognize a proteoglycan epitope on the SARS‐CoV‐2 spike, distinct from the receptor‐binding motif. 4 , 33 Hence, for a fair comparison with similar binding regions, these four antibodies were not included in the interface analysis. Epitopes of the remaining 25 antibodies overlap with the ACE2 binding site.
3.2. Amino acid propensity at the spike–antibody interface
To identify the residues that have a high frequency of occurrence at the spike–antibody interface, we calculated the propensity of interface residues (Equation (2). The propensity of Tyr was observed to be the highest at the interface of spike–antibody complexes (Figure 2). Interestingly, Tyr predominantly occurred at the interface of antigen–antibody 34 and other protein–protein complexes. 35 It has been reported that tyrosine residues are exceptionally versatile for mediating contacts at interfaces 36 as they can form stable aromatic interactions. Other residues with high propensity are Asn, Ile, Phe, and Gly from antibody interface and Gly, Phe. Gln and Arg from spike protein interface, respectively (Figure 2). On the other hand, Met, His, Trp, and Ile residues are absent at the spike interface, and Cys has the lowest propensity of 0.09 at the antibody interface. The residues which show a difference of greater than 1 in residue propensity (in spike and antibody) are Ile, Trp, Gln, and Lys (Figure 2 ).
FIGURE 2.
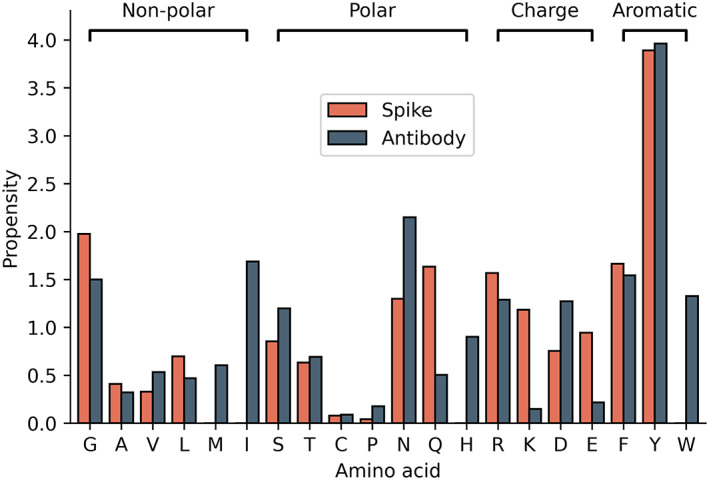
Propensity of interface residues for spike and antibody
Further comparison of the interface of antibodies with ACE2 showed that Arg has the highest propensity in ACE2 compared with Tyr in antibodies. Other residues in ACE2 with higher propensity include Asp, Tyr, Gln, and His (Figure 3). Similarly, a comparison of antibody and heterodimer protein interfaces showed that Tyr and Phe had high frequency in both interfaces (Figure 3). The Gly and Ser had a lower propensity in heterodimers and ACE2 but a higher propensity in antibodies. Cys and Ala had a lower propensity in all of the three datasets (Figure 3 ), which agrees well with previous studies reported in the literature for ACE2 and heterodimer interfaces. 37 , 38
FIGURE 3.
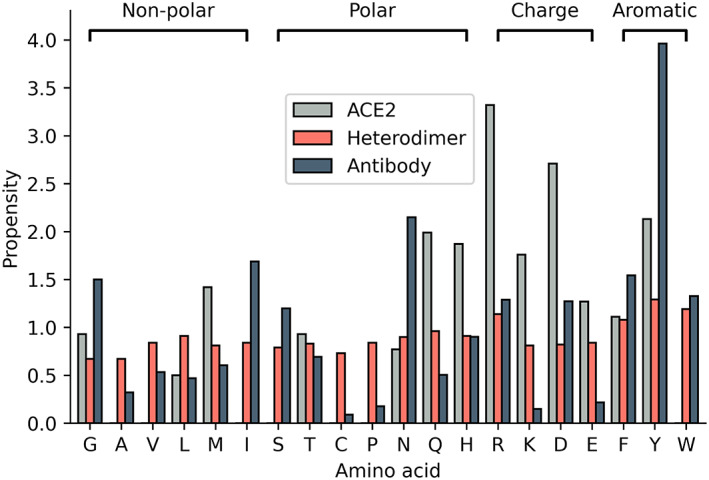
Binding propensity of amino acid residues at the interfaces of ACE2, heterodimers, and neutralizing antibodies
3.3. Residue pair preference and interaction energy
The analysis of residue pair preference at the interface of the spike–antibody complexes showed that Tyr containing residue pairs (YY, FY, GY, YG, YS) dominate the topmost preferred pairs. These residue pairs are present in 70% of the antibodies in the dataset. These highly preferred pairs are also more energetically favorable (Table 2) indicating that aromatic interactions are important for binding of antibodies to the SARS‐CoV‐2 RBD. Along with aromatic–aromatic interactions, nonpolar–π and polar–π interactions are also preferred, and the polar–π interactions are energetically significant to protein folding and function. 39 A previous study on 200 antibody–antigen complexes has also shown that Tyr residues are preferred at both the epitope and paratope regions. 40 In our previous mutational study, we also observed that Tyr residue at the ACE2 binding regions of spike protein are highly conserved along with Gly residues in all ACE2‐binding coronaviruses. 41 Hence, Tyr residue interactions are potentially important for developing highly specific antibodies or small drug molecules against SARS‐CoV‐2.
TABLE 2.
Energetically favorable pairs (<−0.2 kcal/mol) within topmost 50 preferred pairs
| S. no. | Pair | Pair preference a | Energy (kcal/mol) | S. no. | Pair | Pair preference a | Energy (kcal/mol) |
|---|---|---|---|---|---|---|---|
| 1 | YY | 11.69 | −0.32 | 24 | KD | 2.30 | −0.23 |
| 2 | FY | 5.84 | −0.38 | 25 | RY | 2.28 | −0.43 |
| 3 | YI | 5.75 | −0.21 | 26 | FD | 2.20 | −0.26 |
| 4 | YN | 5.60 | −0.25 | 27 | NG | 2.19 | −0.43 |
| 5 | KY | 4.70 | −0.39 | 28 | EY | 1.91 | −0.38 |
| 6 | YR | 4.61 | −0.35 | 29 | KS | 1.81 | −0.38 |
| 7 | GY | 4.03 | −0.28 | 30 | YL | 1.80 | −0.39 |
| 8 | EH | 3.91 | −0.42 | 31 | FF | 1.78 | −0.25 |
| 9 | YG | 3.73 | −0.24 | 32 | NF | 1.77 | −0.34 |
| 10 | YS | 3.72 | −0.40 | 33 | LY | 1.65 | −0.42 |
| 11 | YF | 3.60 | −0.36 | 34 | YD | 1.65 | −0.37 |
| 12 | FR | 3.42 | −0.44 | 35 | EW | 1.60 | −0.23 |
| 13 | YW | 3.30 | −0.37 | 36 | QN | 1.54 | −0.56 |
| 14 | AN | 3.28 | −0.49 | 37 | AF | 1.49 | −0.22 |
| 15 | FM | 2.67 | −0.42 | 38 | FW | 1.47 | −0.22 |
| 16 | QW | 2.54 | −0.21 | 39 | EN | 1.46 | −0.30 |
| 17 | RN | 2.54 | −0.64 | 40 | GG | 1.42 | −0.31 |
| 18 | NR | 2.42 | −0.30 | 41 | YT | 1.38 | −0.35 |
| 19 | YM | 2.40 | −0.39 | 42 | LM | 1.35 | −0.30 |
| 20 | DY | 2.40 | −0.26 | 43 | GF | 1.33 | −0.32 |
| 21 | QY | 2.39 | −0.37 | 44 | QS | 1.32 | −0.40 |
| 22 | ER | 2.33 | −0.43 | 45 | TF | 1.26 | −0.27 |
| 23 | QM | 2.32 | −0.36 | 46 | RS | 1.25 | −0.28 |
The pair preference values are multiplied by 103.
Out of 20 topmost preferred pairs at the spike–antibody interface (Table S2) 17 contains hydrophobic residues (9 hydrophobic–hydrophobic interactions) and these hydrophobic residues play a vital role at protein–protein interfaces. 42 , 43 It has been shown that the dominance of hydrophobic contacts between SARS‐CoV‐2 and ACE2 enhances the binding affinity of SARS‐CoV‐2 for ACE2. 44 In the heterodimer dataset, CC is the most preferred pair, followed by KD, WI, RD, and VF, which does not contain Tyr and is different from the spike–antibody interface. It is important to note that Cys has the lowest propensity at the neutralizing antibody interfaces, and therefore, such pairing is not preferred at the spike–antibody interface.
3.4. Structural features influencing experimental binding affinity ( K D )
The binding free energy values for antibodies discussed in this work range from −9.7 to −16.3 kcal/mol (Table 1 ). The binding affinity values for spike–ACE2 complexes are reported as 4.7, 1 14.7, 45 and 15.2 nM 46 in the literature. In comparison to ACE2 binding affinity (~15 nM), most of the antibodies showed greater binding affinities leading to effective competitive inhibition of SARS‐CoV‐2 (Table 1).
3.4.1. Analysis of antibodies with experimental binding affinity
We also calculated the interface surface area and average surrounding hydrophobicity of the spike protein–ACE2 complex and spike–antibody complexes. The surface area of the interacting region for antibodies ranged between 370 and 1300 Å2 on the spike protein. SARS‐CoV‐2 spike–ACE2 complex had a relatively larger surface area of 1860 Å2 than antibodies. The contact surface area in the spike–antibody proteins shows a low positive correlation with enhanced binding affinity (r = .13), which is consistent with previous studies. 47 Similarly, the average surrounding hydrophobicity for the interacting region in antibodies ranged between 10.24 and 14.84 kcal/mol compared with 12.31 kcal/mol for ACE2. We have shown previously on coronaviruses that a higher hydrophobic environment at the interface can improve the binding of the complex. 41 The surrounding hydrophobicity also correlates positively with the binding affinity (r = .26).
3.4.2. Factors influencing binding free energy (ΔG )
The single property correlation of structural features to ΔG showed the highest negative correlation of −0.55 for charged–charged interfacial contacts and a highest positive correlation of 0.5 for aromatic–aromatic interactions, calculated within the distance of 4.5–7 Å. 48 A systematic forward feature selection approach was further utilized to select the optimum number of features with the best performance. We obtained a correlation of 0.93 and a mean absolute error (MAE) of 0.48 kcal/mol for the final regression model with three features (Figure 4). The features include total energy (E t , in kcal/mol), aromatic–aromatic interactions (I aro–aro), and number of nonpolar–polar atomic contacts (A np ). The multiple regression equation obtained from the model for binding free energy (ΔG, in kcal/mol) is given below:
| (8) |
We have also performed a jack‐knife test to validate the robustness of the model and observed the correlation of 0.90 and MAE of 0.58 between experimental and predicted binding energies (Figure 4). For comparison, we used PRODIGY (structure based method) 24 and PPA‐Pred (sequence based method) 49 for predicting the binding affinity of the same set of 24 protein–protein complexes and we obtained an MAE of 3.05 and 2.64 kcal/mol, respectively. Further, we tested our model for five typical antibodies [CT‐P59 (7CM4), P5A‐3C12 (7D0B), P5A‐1B9 (7CZX), Fab2‐43 (7L56), and P5A‐2F11 (7CZY)] that are not considered in our dataset, which showed an MAE of 1.15 kcal/mol between experimental and predicted ΔG. Development of a robust model is in progress.
FIGURE 4.
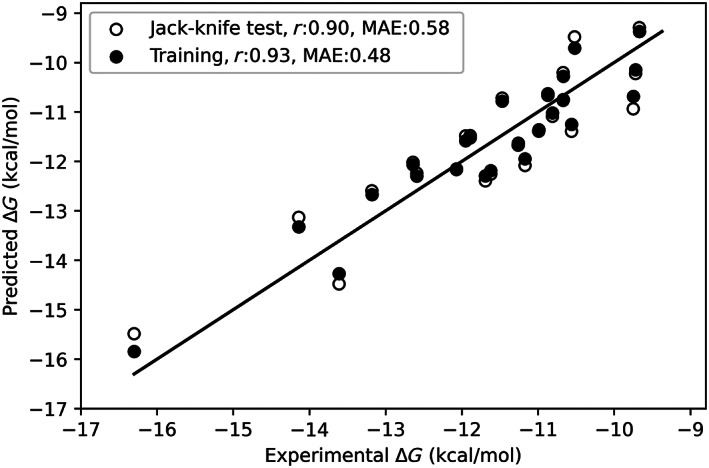
A scatter plot showing the relationship between experimental and predicted binding energies on training and jackknife test
3.5. Mutational analysis
We have performed a mutational scanning for the epitope and paratope residues for each neutralizing antibody to identify mutants that improve stability and binding affinity of the spike–antibody complex or vice versa. The analysis of changes in interaction energy upon point mutations showed that 85% of the residues in the epitope and paratope are important for binding as they reduce the binding affinity of the complex upon mutation. There are certain binding sites in antibodies, where mutation leads to increased binding affinity and stability of the paratope to the RBD. The mutation sites which improve affinity and stability for more than 10 amino acid mutations are given in Table 3. These paratope residues could be considered for engineering more potent antibodies against COVID‐19.
TABLE 3.
Mutation sites affecting binding affinity and stability in the epitope(s) and paratope(s) of spike–antibody interface
| Pdb id | Interface (chain) | Interface residues |
|---|---|---|
| 6WPT | Paratope (L) | D93 |
| Epitope (C) | N334 , L335 , P337 , G339 , E340 , N343 , A344, T345 , R346, N354, K356 , R357 , S359 , N360, C361 , L441 , R509 | |
| 6XC2 | Paratope | – |
| Epitope (A) | R403 , D405 , R408 , T415 , G416 , K417 , D420 , Y421 , Y453 , L455 , F456 , R457 , K458 , S459 , N460 , Y473 , Q474, A475 , G476 , F486 , N487 , Y489 , Q493 , S494, Y495 , G496 , Q498, T500 , N501 , G502, Y505 | |
| 6XC4 | Paratope | – |
| Epitope (A) | R403 , D405, T415 , G416 , K417 , D420 , Y421 , Y453, L455 , F456 , R457 , K458 , N460, Y473 , A475 , G476 , S477 , F486 , N487 , Y489 , Y495 , N501, Y505 | |
| 6XCN | Paratope (H,L) | G54, S56, G29, Y30, K31 |
| Epitope (C) | D405 , T415 , G416, K417 , Y421 , Y453 , F456 , R457 , K458 , N460, Y473 , A475 , G476, F486 , N487 , G502 , Y505 | |
| 6XE1 | Paratope (H,L) | D97, S27A |
| Epitope (E) | R403 , T415 , G416 , K417 , D420 , Y421 , Y453 , L455 , F456 , R457 , K458, N460, Y473 , A475 , G476 , F486 , N487 , Y489 , Q493 , N501 , G502, Y505 | |
| 6XEY | Paratope (J) | A99 |
| Epitope (C) | Y449 , L455 , F456 , V483 , E484 , G485 , F486 , Y489 , F490 , L492 , Q493 , S494 | |
| 6XKP | Paratope (H) | R96 |
| Epitope (A) | R346 , F347 , S349 , Y351 , K444 , V445 , G446 , G447 , N448 , Y449 , N450 , Y451 , L452 , T470 , E484 , F490 , L492 , Q493 , S494 , Q498 | |
| 6XKQ | Paratope (H,L) | M100E, D101, S27A, A29 |
| Epitope (A) | R403 , G446, Y449 , Y453, L455 , F456 , A475 , G476 , S477, T478 , G485, F486 , N487 , Y489 , Q493 , Y495 , Q498, N501 , Y505 | |
| 7B3O | Paratope (H,L) | N92, S31 , R97, A100 , D101 |
| Epitope (E) | R403 , D405 , E406, R408, Q409 , T415 , G416 , K417 , D420 , Y421 , Y453, L455 , F456 , R457 , K458, S459, N460, Y473 , Q474, A475 , G476, F486 , N487 , Y489 , Q493 , S494, Y495 , G496 , Q498, T500 , N501 , G502 , Y505 | |
| 7BWJ | Paratope (L) | G31 |
| Epitope (E) | R346 , K444 , G446, G447 , N448 , Y449 , N450 , L452 , V483 , E484 , G485, F490 , S494 | |
| 7BYR | Paratope (H) | S31, Y32, T53 , N54, D73, Q100, S103, W105 |
| Epitope (B) | G446, Y449, E484 , G485, F486 , Y489 , F490 , L492 , Q493 , G496 , Q498 , N501, Y505 | |
| 7BZ5 | Paratope | – |
| Epitope (A) | R403 , D405, E406, Q409 , T415 , G416 , K417 , D420 , Y421 , Y453 , L455 , F456 , R457 , K458, N460 , Y473 , Q474, A475 , G476 , E484 , F486 , N487 , Y489 , F490 , L492 , Q493 , Y495 , G496 , Q498, T500, N501 , G502 , Y505 | |
| 7C01 | Paratope (H,L) | D104, S30 |
| Epitope (A) | R403 , D405 , E406, R408 , Q409 , T415 , G416 , K417 , D420 , Y421 , L455 , F456 , R457 , K458, N460 , Y473 , Q474 , A475 , G476 , S477 , F486 , N487 , Y489 , Q493 , Y495 , G502, Y505 | |
| 7CAK | Paratope (D,E) | I2, S27, S30, N91, W93, D60, D102 , Y105 |
| Epitope (A) | Y365 , Y369, A372 , S373 , F374 , S375 , T376 , F377 , K378 , S383 , P384 , T385 , R408 , P412 , G413, Q414 , N437, V503 | |
| 7CHB | Paratope (H,L) | G26, S31, A102, D106, Q27, S30 |
| Epitope (R) | R403 , T415 , G416 , K417 , D420 , Y421 , Y453 , L455 , F456 , R457 , K458, S459 , N460 , Y473 , A475 , G476 , F486 , N487 , Y489 , Q493 , Y495 , G496, Q498, T500 , N501 , G502 , V503, G504 , Y505 | |
| 7CHH | Paratope (D) | S25, D106 |
| Epitope (A) | K444, Y449 , N450, L452 , N481 , G482 , V483 , E484 , G485, F490 | |
| 7CJF | Paratope (A,B) | S30, F58, D106, S93 |
| Epitope (C) | R403 , D405, Q409 , T415 , G416, K417 , D420 , Y421 , Y453 , L455 , F456 , R457 , K458 , N460, Y473 , Q474, A475 , G476 , S477 , F486 , N487 , Y489 , Q493 , Y495 , G496 , Q498, T500 , N501 , G502 , Y505 | |
| 7CWO | Paratope (H) | S31 |
| Epitope (A) | L455 , T470, N481 , G482 , V483 , E484 , G485 , F486 , Y489 , F490 , L492 , Q493 | |
| 7JMW | Paratope (H) | P96 |
| Epitope (A) | Y369 , S371 , F377 , K378 , C379 , Y380 , G381, V382 , S383 , P384 , T385 , R408 , P412 , G413, Q414 , T415 , G416 , D427 , F429 | |
| 7JV2 | Paratope (H,L) | Y102, R63 |
| Epitope (A) | G446, Y449, N481 , G482, V483 , E484, G485 , F486 , F490 | |
| 7K43 | Paratope (H) | G31 , T74 |
| Epitope (A) | G446, Y449, L452 , L455 , F456 , E484, G485 , F486 , Y489 , F490 , L492 , Q493 , S494 , G496 | |
| 7K45 | Paratope (H) | G54, G104, S105 |
| Epitope (B) | L455 , Y473 , A475 , G476 , S477 , G485 , F486 , N487 , C488 , Y489 | |
| 7K8U | Paratope (H,L) | G26, S30, A31, Y92, G93, T95 |
| Epitope (A) | K444 , V483 , F486 , F490 | |
| 7K8V | Paratope (H) | D99, V100D, P100G |
| Epitope (A) | T345 , R346, L441 , D442 , N448 , Y449 , N450 , L452 , F490 , Q498 , P499 , T500 , R509 | |
| 7K8W | Paratope (H,L) | S58, D100B, Y100D, Y100E , K30, S92 |
| Epitope (A) | K444, V445, G446 , Y449, N450, E484, Q493 , S494, Q498, Y505 | |
| 7K8Y | Paratope (G) | T58, T74, G110 |
| Epitope (B) | K444 , Y449 , F456 , E484, G485 , F486 , N487 , Y489, F490 , Q498 | |
| 7K8Z | Paratope | – |
| Epitope (A) | T345 , R346 , S438 , N439 , N440 , P499 | |
| 7K90 | Paratope (H) | G54, G55, S56, K73, N76 |
| Epitope (B) | Y449 , L455 , F456 , V483, G485, F486 , N487 , Y489 , F490 , Q493 , S494 | |
| 7KFV | Paratope (H,L) | S98, S67 |
| Epitope (A) | R403 , T415 , G416 , K417 , D420 , Y421 , Y453 , L455 , F456 , R457 , K458, S459 , N460 , Y473 , A475 , G476 , F486 , N487 , Y489 , Q493 , S494, Y495 , G496 , Q498, T500 , N501 , G502 , G504, Y505 |
Note: (i) The residues shown in italics for paratope increase the binding affinity, bold residues increase the stability, while residues shown for epitope decrease the binding affinity and stability. (ii) The residues are highlighted if two out of three methods satisfy the criteria that at least 50% of the mutations in each residue increases (for paratope) or decreases (for epitope) the affinity and stability. (iii) Notation for residues; Wild type residue followed by residue number.
The SARS‐CoV‐2 RBD is prone to many mutations that could escape the neutralization. We have identified several mutations in the RBD epitopes that decrease the binding affinity and stability of the antibodies (Table 3 ). The epitope residues which decrease the binding affinity and stability for more than 50% of mutations are F486, Y489, Q493, L455, and F456 (residues present in more than 50% of the antibodies). These positions have also been reported as having mutations emerging upon exposure (co‐incubation) to mAbs. 50 , 51 The mutation site F486 has shown immune escape from a mAb, COV2‐2832. 9 Experimental studies have identified the key immune escape mutant residues as E484, S477, N439 with mutations E484Q, E484K, E484P, S477G, S477N, S477R, and N439K 10 , 50 for a set of antibodies or convalescent sera, and we also observed a decrease in binding affinity and stability at these sites upon mutation. The delta variant (B.1.617.2) showed reduced sensitivity to neutralization by antibodies. 52 The amino acid changes in the RBD in this variant, K417N (present in delta plus, B.1.617.2.1), L452R, and T478K show a decrease in binding affinity to antibodies in our study. We noticed that the residues K417, L452, and T478 within the epitopes are mutated frequently. Further, mutations at these sites reduced the binding free energy (Table 3). Among the Gamma variant (P.1) mutations, 53 K417T and N501Y are observed frequently in the RBD of the spike protein and these mutations decrease the binding affinity (Table 3). Our results are consistent with the observations that the variants of concern and variants of interest highlighted by WHO (Table 4) also decreased the binding free energy and stability.
TABLE 4.
Spike protein mutations in variants of concern and variants of interest
| Variants of concern | ||||
|---|---|---|---|---|
| WHO label | Lineage + additional mutations | Country first detected (community) | Spike mutations of interest | Year and month first detected |
| Beta | B.1.351 | South Africa | K417N, E484K, N501Y, D614G, A701V | September 2020 |
| Gamma | P.1 | Brazil | K417T, E484K, N501Y, D614G, H655Y | December 2020 |
| Delta | B.1.617.2 | India | L452R, T478K, D614G, P681R | December 2020 |
| Variants of interest | ||||
| n/a | B.1.620 | Unclear | S477N, E484K, D614G, P681H | February 2021 |
| Mu | B.1.621 | Colombia | R346K, E484K, N501Y, D614G, P681H | January 2021 |
| Lambda | C.37 | Peru | L452Q, F490S, D614G | December 2020 |
Note: The data are taken from European Center for Disease Prevention and Control (https://www.ecdc.europa.eu/en/covid‐19/variants‐concern). n/a: no WHO label has been assigned to this variant (accessed on October 6, 2021).
We have further illustrated the results with two examples: (a) E484K and (b) K417N. E484 in the spike protein of the complex (PDB: 7BZ5) forms hydrogen bonds with Y100 of the antibody heavy chain (Figure 5A) and the mutation of E484K disrupt these interactions (Figure 5B) and reduced the binding affinity of 1.83 kcal/mol. On the other hand, K417 forms hydrogen bonds and cation–π interactions with Y33 and Y52 of the antibody heavy chain and hydrogen bonds with N92 of the antibody light chain (Figure 5C). The mutation K417N abolished the interactions with Y33 and N92 (Figure 5D) and decreased the binding affinity by 1.23 kcal/mol.
FIGURE 5.
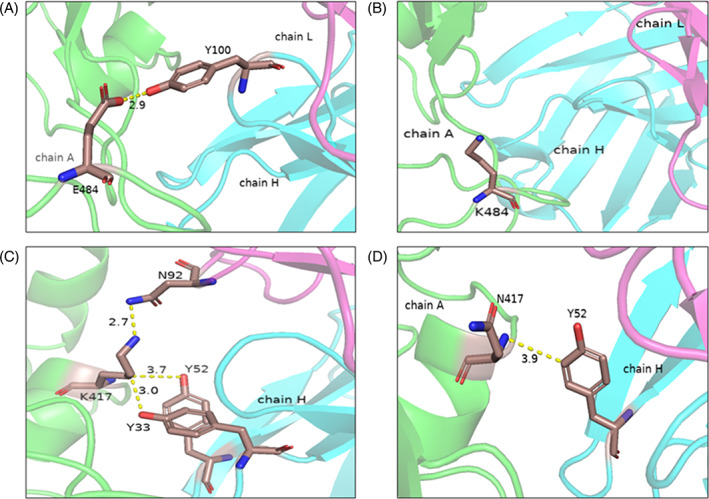
The interactions at the epitope positions and mutants (A) E484, (B) E484K, (C) K417, and (D) K417N in the spike–antibody complex (PDB: 7BZ5)
4. CONCLUSIONS
Our study revealed that Tyr is the key residue involved in the binding of the spike and antibodies, which contributes to aromatic and hydrophobic interactions at the interface. YY pairs are the most preferred and energetically favored pairs present at the spike–antibody interface. The regression analysis to predict ΔG shows that interaction energy and atomic interactions play an important role. The mutational analysis of interface residues revealed that mutations in epitope sites F486, Y489, Q493, L455, and F456 decrease the binding affinity of spike–antibody complexes and could be considered as potential immune escape sites. In conclusion, the study will be helpful in understanding the interactions between the spike protein and the neutralizing antibodies in order to reveal the mechanisms of neutralization, which could help curb the infection and in developing better therapeutics against COVID‐19. It could also provide an understanding of immune escape for emerging variants and on improving stability and binding affinity of neutralizing antibodies.
AUTHOR CONTRIBUTIONS
Conceptualization: M. Michael Gromiha; Methodology: M. Michael Gromiha, Puneet Rawat; Software/code: Divya Sharma, Puneet Rawat; Investigation: Divya Sharma, Puneet Rawat; Discussion: Divya Sharma, Puneet Rawat, Vani Janakiraman, M. Michael Gromiha; Writing Original Draft: Divya Sharma; Review & Editing: M. Michael Gromiha, Puneet Rawat, Vani Janakiraman; Supervision: M. Michael Gromiha. All authors read and approved the manuscript.
Supporting information
Figure S1 Workflow illustrating the steps followed to prepare the dataset for SARS‐CoV‐2 neutralizing antibodies.
Table S1. Details of features used in this study
Table S2. Pair preference for top 20 pairs at the spike–antibody interface.
Table S3. Interaction energy in kcal/mol for top 20 pairs at the spike–antibody interface.
Table S4. Interface residues affecting binding affinity and stability in the paratope and epitope of spike–antibody interface.
ACKNOWLEDGMENTS
We thank Bioinformatics Infrastructure facility, Department of Biotechnology and Indian Institute of Technology Madras for computational facilities and Ministry of human resource and development (MHRD) for HTRA scholarship to DS. This work is partially supported by the Robert Bosch Center for Data Science and Artificial Intelligence (RBCDSAI), Indian Institute of Technology Madras to MMG and VJ, and Department of Science and Technology, Government of India to MMG (MSC/2020/000319).
Sharma D, Rawat P, Janakiraman V, Gromiha MM. Elucidating important structural features for the binding affinity of spike ‐ SARS‐CoV‐2 neutralizing antibody complexes. Proteins. 2022;90(3):824-834. doi: 10.1002/prot.26277
Funding information Robert Bosch Center for Data Science and Artificial Intelligence (RBCDSAI), Indian Institute of Technology Madras; Department of Science and Technology, Government of India, Grant/Award Number: MSC/2020/000319
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Lan J, Ge J, Yu J, et al. Structure of the SARS‐CoV‐2 spike receptor‐binding domain bound to the ACE2 receptor. Nature. 2020;581(7807):215‐220. [DOI] [PubMed] [Google Scholar]
- 2. Wang C, Li W, Drabek D, et al. A human monoclonal antibody blocking SARS‐CoV‐2 infection. Nat Commun. 2020;11(1):2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chi X, Yan R, Zhang J, et al. A neutralizing human antibody binds to the N‐terminal domain of the spike protein of SARS‐CoV‐2. Science. 2020;369(6504):650‐655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Barnes CO, Jette CA, Abernathy ME, et al. SARS‐CoV‐2 neutralizing antibody structures inform therapeutic strategies. Nature. 2020;588(7839):682‐687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zheng Z, Monteil VM, Maurer‐Stroh S, et al. Monoclonal antibodies for the S2 subunit of spike of SARS‐CoV‐1 cross‐react with the newly‐emerged SARS‐CoV‐2. Euro Surveill. 2020;25(28):2000291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Perrin‐Cocon L, Diaz O, Jacquemin C, et al. The current landscape of coronavirus‐host protein–protein interactions. J Transl Med. 2020;18(1):1‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gordon DE, Jang GM, Bouhaddou M, et al. A SARS‐CoV‐2‐human protein‐protein interaction map reveals drug targets and potential drug‐repurposing. bioRxiv. 2020. doi: 10.1101/2020.03.22.002386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhou Y, Hou Y, Shen J, Huang Y, Martin W, Cheng F. Network‐based drug repurposing for novel coronavirus 2019‐nCoV/SARS‐CoV‐2. Cell Discov. 2020;6(1):14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Greaney AJ, Starr TN, Gilchuk P, et al. Complete mapping of mutations to the SARS‐CoV‐2 spike receptor‐binding domain that escape antibody recognition. Cell Host Microbe. 2021;29(1):44‐57.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Liu Z, VanBlargan LA, Bloyet L‐M, et al. Identification of SARS‐CoV‐2 spike mutations that attenuate monoclonal and serum antibody neutralization. Cell Host Microbe. 2021;29(3):477‐488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dispinseri S, Secchi M, Pirillo MF, et al. Neutralizing antibody responses to SARS‐CoV‐2 in symptomatic COVID‐19 is persistent and critical for survival. Nat Commun. 2021;12(1):2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tortorici MA, Czudnochowski N, Starr TN, et al. Structural basis for broad sarbecovirus neutralization by a human monoclonal antibody. bioRxiv. 2021. doi: 10.1101/2021.04.07.438818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chen RE, Zhang X, Case JB, et al. Resistance of SARS‐CoV‐2 variants to neutralization by monoclonal and serum‐derived polyclonal antibodies. Nat Med. 2021;27(4):717‐726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Weisblum Y, Schmidt F, Zhang F, et al. Escape from neutralizing antibodies by SARS‐CoV‐2 spike protein variants. Elife. 2020;9:e61312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Burley SK, Bhikadiya C, Bi C, et al. RCSB protein data bank: powerful new tools for exploring 3D structures of biological macromolecules for basic and applied research and education in fundamental biology, biomedicine, biotechnology, bioengineering and energy sciences. Nucleic Acids Res. 2021;49(D1):D437‐D451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gromiha MM, Saranya N, Selvaraj S, Jayaram B, Fukui K. Sequence and structural features of binding site residues in protein‐protein complexes: comparison with protein‐nucleic acid complexes. Proteome Sci. 2011;9:S13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gromiha MM, Yokota K, Fukui K. Energy based approach for understanding the recognition mechanism in protein–protein complexes. Mol Biosyst. 2009;5(12):1779‐1786. [DOI] [PubMed] [Google Scholar]
- 18. Cornell WD, Cieplak P, Bayly CI, et al. A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J Am Chem Soc. 1995;117(19):5179‐5197. [Google Scholar]
- 19. Rawat P, Sharma D, Srivastava A, Janakiraman V, Gromiha MM. Exploring antibody repurposing for COVID‐19: beyond presumed roles of therapeutic antibodies. Sci Rep. 2021;11(1):10220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Manavalan P, Ponnuswamy PK. Hydrophobic character of amino acid residues in globular proteins. Nature. 1978;275(5681):673‐674. [DOI] [PubMed] [Google Scholar]
- 21. Nozaki Y, Tanford C. The solubility of amino acids and two glycine peptides in aqueous ethanol and dioxane solutions. Establishment of a hydrophobicity scale. J Biol Chem. 1971;246(7):2211‐2217. [PubMed] [Google Scholar]
- 22. Jones DD. Amino acid properties and side‐chain orientation in proteins: a cross correlation approach. J Theor Biol. 1975;50(1):167‐183. [DOI] [PubMed] [Google Scholar]
- 23. Nagarajan R, Archana A, Thangakani AM, Jemimah S, Velmurugan D, Gromiha MM. PDBparam: online resource for computing structural parameters of proteins. Bioinform Biol Insights. 2016;10:BBI‐S38423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Xue LC, Rodrigues JP, Kastritis PL, Bonvin AM, Vangone A. PRODIGY: a web server for predicting the binding affinity of protein–protein complexes. Bioinformatics. 2016;32(23):3676‐3678. [DOI] [PubMed] [Google Scholar]
- 25. Tina KG, Bhadra R, Srinivasan N. PIC: protein interactions calculator. Nucleic Acids Res. 2007;35:W473‐W476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jemimah S, Sekijima M, Gromiha MM. ProAffiMuSeq: sequence‐based method to predict the binding free energy change of protein–protein complexes upon mutation using functional classification. Bioinformatics. 2019;36(6):1725‐1730. [DOI] [PubMed] [Google Scholar]
- 27. Pires DEV, Ascher DB, Blundell TL. mCSM: predicting the effects of mutations in proteins using graph‐based signatures. Bioinformatics. 2014;30(3):335‐342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Parthiban V, Gromiha MM, Schomburg D. CUPSAT: prediction of protein stability upon point mutations. Nucleic Acids Res. 2006;34:W239‐W242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Delgado J, Radusky LG, Cianferoni D, Serrano L. FoldX 5.0: working with RNA, small molecules and a new graphical interface. Bioinformatics. 2019;35(20):4168‐4169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schymkowitz J, Borg J, Stricher F, Nys R, Rousseau F, Serrano L. The FoldX web server: an online force field. Nucleic Acids Res. 2005;33:W382‐W388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liu H, Wu NC, Yuan M, et al. Cross‐neutralization of a SARS‐CoV‐2 antibody to a functionally conserved site is mediated by avidity. Immunity. 2020;53(6):1272‐1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lv Z, Deng Y‐Q, Ye Q, et al. Structural basis for neutralization of SARS‐CoV‐2 and SARS‐CoV by a potent therapeutic antibody. Science. 2020;369(6510):1505‐1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pinto D, Park Y‐J, Beltramello M, et al. Cross‐neutralization of SARS‐CoV‐2 by a human monoclonal SARS‐CoV antibody. Nature. 2020;583(7815):290‐295. [DOI] [PubMed] [Google Scholar]
- 34. Wang M, Zhu D, Zhu J, Nussinov R, Ma B. Local and global anatomy of antibody‐protein antigen recognition. J Mol Recognit. 2018;31(5):e2693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ma B, Wolfson HJ, Nussinov R. Protein functional epitopes: hot spots, dynamics and combinatorial libraries. Curr Opin Struct Biol. 2001;11(3):364‐369. [DOI] [PubMed] [Google Scholar]
- 36. Koide S, Sidhu SS. The importance of being tyrosine: lessons in molecular recognition from minimalist synthetic binding proteins. ACS Chem Biol. 2009;4(5):325‐334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shang J, Ye G, Shi K, et al. Structural basis of receptor recognition by SARS‐CoV‐2. Nature. 2020;581(7807):221‐224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhanhua C, Gan JG‐K, Lei L, Sakharkar MK, Kangueane P. Protein subunit interfaces: heterodimers versus homodimers. Bioinformation. 2005;1(2):28‐39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pace CJ, Gao J. Exploring and exploiting polar‐π interactions with fluorinated aromatic amino acids. Acc Chem Res. 2013;46(4):907‐915. [DOI] [PubMed] [Google Scholar]
- 40. Soga S, Kuroda D, Shirai H, Kobori M, Hirayama N. Use of amino acid composition to predict epitope residues of individual antibodies. Protein Eng Des Sel. 2010;23(6):441‐448. [DOI] [PubMed] [Google Scholar]
- 41. Rawat P, Jemimah S, Ponnuswamy PK, Michael Gromiha M. Why are ACE2 binding coronavirus strains SARS‐CoV / SARS‐CoV ‐2 wild and NL63 mild? Proteins: Struct Funct Genet. 2021;89(4):389‐398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Janin J, Miller S, Chothia C. Surface, subunit interfaces and interior of oligomeric proteins. J Mol Biol. 1988;204(1):155‐164. [DOI] [PubMed] [Google Scholar]
- 43. Tsai CJ, Nussinov R. Hydrophobic folding units at protein‐protein interfaces: implications to protein folding and to protein‐protein association. Protein Sci. 1997;6(7):1426‐1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ali A, Vijayan R. Dynamics of the ACE2‐SARS‐CoV/SARS‐CoV‐2 spike protein interface reveal unique mechanisms. Sci Rep. 2020;10(1):1‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wrapp D, Wang N, Corbett KS, et al. Cryo‐EM structure of the 2019‐nCoV spike in the prefusion conformation. bioRxiv. 2020. doi: 10.1101/2020.02.11.944462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tian X, Li C, Huang A, et al. Potent binding of 2019 novel coronavirus spike protein by a SARS coronavirus‐specific human monoclonal antibody. Emerg Microbes Infect. 2020;9(1):382‐385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chen J, Sawyer N, Regan L. Protein‐protein interactions: general trends in the relationship between binding affinity and interfacial buried surface area. Protein Sci. 2013;22(4):510‐515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Burley SK, Petsko GA. Aromatic‐aromatic interaction: a mechanism of protein structure stabilization. Science. 1985;229(4708):23‐28. [DOI] [PubMed] [Google Scholar]
- 49. Yugandhar K, Gromiha MM. Protein–protein binding affinity prediction from amino acid sequence. Bioinformatics. 2014;30(24):3583‐3589. [DOI] [PubMed] [Google Scholar]
- 50. Harvey WT, Carabelli AM, Jackson B, et al. SARS‐CoV‐2 variants, spike mutations and immune escape. Nat Rev Microbiol. 2021;19(7):409‐424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Muecksch F, Weisblum Y, Barnes CO, et al. Affinity maturation of SARS‐CoV‐2 neutralizing antibodies confers potency, breadth, and resilience to viral escape mutations. Immunity. 2021;54(8):1853‐1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Planas D, Veyer D, Baidaliuk A, et al. Reduced sensitivity of SARS‐CoV‐2 variant Delta to antibody neutralization. Nature. 2021;596(7871):276‐280. [DOI] [PubMed] [Google Scholar]
- 53. Faria NR, Mellan TA, Whittaker C, et al. Genomics and epidemiology of the P.1 SARS‐CoV‐2 lineage in Manaus, Brazil. Science. 2021;372(6544):815‐821. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Workflow illustrating the steps followed to prepare the dataset for SARS‐CoV‐2 neutralizing antibodies.
Table S1. Details of features used in this study
Table S2. Pair preference for top 20 pairs at the spike–antibody interface.
Table S3. Interaction energy in kcal/mol for top 20 pairs at the spike–antibody interface.
Table S4. Interface residues affecting binding affinity and stability in the paratope and epitope of spike–antibody interface.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.