

Abstract
Since the beginning of the COVID‐19 pandemic, scientists across the globe are racing to find a cure for the highly contagious infectious disease caused by the SARS‐CoV‐2 virus. Despite many promising ongoing progress, there are currently no FDA approved drug to treat infected patients. Recently, the crowdsourcing of drug discovery for inhibiting the main protease (Mpro) of SARS‐CoV‐2 have yielded a plenty of drug fragments resolved inside the active site of Mpro via the crystallography method. Following the principle of fragment‐based drug design (FBDD), we are motivated to design a potent drug candidate (named B19) by merging three fragments JFM, U0P, and HWH. Through extensive all‐atom molecular dynamics simulation and molecular docking, we found that B19 among all designed ones is most stable inside the Mpro's active site and the binding free energy of B19 is comparable to or even a little better than that of a native protein ligand processed by Mpro. Our promising results suggest that B19 and its derivatives can potentially be efficacious drug candidates for COVID‐19.
Keywords: COVID‐19, fragment‐based drug design, main protease, SARS‐CoV‐2
1. INTRODUCTION
The ongoing coronavirus 2019 (COVID‐19) pandemic is caused by an enveloped, positive‐sense, single‐stranded RNA virus, that is known as the severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2). 1 SARS‐CoV‐2 was first identified in China and is the third pathogenic novel coronavirus to emerge over the last two decades. The first one was discovered in 2003 named SARS‐CoV‐1 which caused the severe acute respiratory syndrome (SARS), a serious and atypical pneumonia. 2 The second one emerged a decade later and was named MERS‐CoV which caused a similar respiratory ailment called Middle East respiratory syndrome (MERS). 3 These three coronaviruses are all zoonotic pathogens which begin in infected animals and are transmitted from animals to humans. Compared with the other two, SARS‐CoV‐2 has significant person‐to‐person transmission. Indeed, it took just a few months for COVID‐19 to spread to almost every corner of the world, resulting in detrimental effect on global healthcare systems with a ripple effect on every aspect of human life. To combat this disease, dozens of drugs have been tested in clinical trials as possible treatments for SARS‐CoV‐2 infections. These drugs fall into two general groups: repurposed antiviral drugs (e.g., remdesivir 4 ) to help stop the virus from reproducing, and immune‐based therapies (e.g., dexamethasone 5 ) to calm the “cytokine storm” from overreaction of the immune system which can lead to organ failure and death. However, at this time there are still no FDA‐approved drugs to cure or prevent COVID‐19 except treatments to ease the symptoms from a supportive care perspective.
Alternatively, new antiviral drugs have been explored simultaneously, targeting mainly at the main protease (Mpro) of SARS‐CoV‐2 that is highly conserved 6 when compared with Mpros of SARS‐CoV‐1 and MERs‐CoV. It is well known that Mpro processes many polyproteins translated from the viral RNA, such as the RNA‐dependent RNA polymerase (RdRp, Nsp12) and the helicase (Nsp13), playing an essential role in viral maturation. Targeting at the key residue cysteine 145 (C145) in the catalytic site of Mpro, many mechanism‐based ligands such as N3, 7 O6K, 8 and carmofur 9 were designed to irreversibly (or covalently) bind C145, which yields inhibited or nonfunctional Mpro. Additionally, dozens of fragment drugs were shown to be covalently bound to C145 inside Mpro, as listed on the website of the COVID Moonshot open initiative (https://postera.ai/covid), a project aiming to accelerate the development of antiviral drugs that can inhibit the SARS‐CoV‐2's Mpro with joined forces from scientists around the world. 10 Besides the covalently bound ligands, dozens of noncovalently bound drug fragments that span the entire active site of the SARS‐CoV‐2's Mpro have also been identified using large scale crystallographic and mass‐spectrometry screening. 11 These structures (listed on COVID Moonshot website and deposited in Protein Data Bank) reveal an exceptionally rich set of information, with extensive opportunities for fragment‐based drug discovery (FBDD) for inhibiting the SARS‐CoV‐2 Mpro.
Generally, FBDD provides an effective alternative to explore the chemical space for binding to a specific target protein. Since its inception two decades ago, FBDD has attracted a lot of interest from researchers and emerged as a mainstream approach in the pharmaceutical industry for reducing attrition and finding high‐quality lead candidates as a part of the drug discovery process. 12 FBDD is based on screening very small chemical compounds (fragments) with low molecular weights which could bind only weakly to the biological target as starting points. The binding affinity between the initial fragment hits to their target is usually in a μM–mM range. 13 This is due to the fact that there are fewer heavy atoms in the fragments to form multiple attractive interactions with the protein when compared with larger molecules. 14 Once the fragment hits are identified, their potency can be enhanced by applying fragment linking, merging, or growing strategies to form drug‐like leads optimized for potential clinical candidates.
Given a plethora of drug fragments resolved in the Mpro's crystal structure 11 and the established protocol for FBDD, in this work, we are motivated to design a potent drug molecule structurally merged from several of these drug fragments for inhibiting Mpro. Complimentary to experimental efforts, in silico approaches such as the all‐atom molecular dynamics (MD) 15 , 16 and docking 17 methods have been widely used to investigate the molecular mechanism of proteins as well as the ligand–protein interaction, and proven to produce results consistent with experimental ones. Here, we carried out MD simulations to explore a total of 19 designed ligands based on the FBDD approach and compare them with a native ligand (a protein peptide) previously resolved in the crystal structure of Mpro of SARS‐CoV‐1. 18 Out of these 19 designed ligands, we found that the binding affinity of a designed drug molecule (named B19) to Mpro is comparable or even slightly better than that of the native ligand, suggesting that B19 is highly promising for inhibiting Mpro of SARS‐CoV‐2.
2. RESULTS
Among the 50 known non‐covalently bound fragments inside the active site, 11 we selected three ligands JFM, U0P, and HWH (Figure 1A) that bind the active site at different locations (Figure 1B–D) and contain their own specific features as described in the following. Namely, from head to tail, JFM contains a methanesulfonamide and a benzene groups; U0P contains a cyclopropane and a carbonyl groups in the head part and an isoxazole group in the tail part; HWH contains an indole group with an attached fluorine atom (pink, Figure 1A) in the head part and an acetamide in the tail part. These chemical groups form either hydrogen bonds or hydrophobic interactions with Mpro's residues inside the active site, stabilizing these bound fragments.
FIGURE 1.
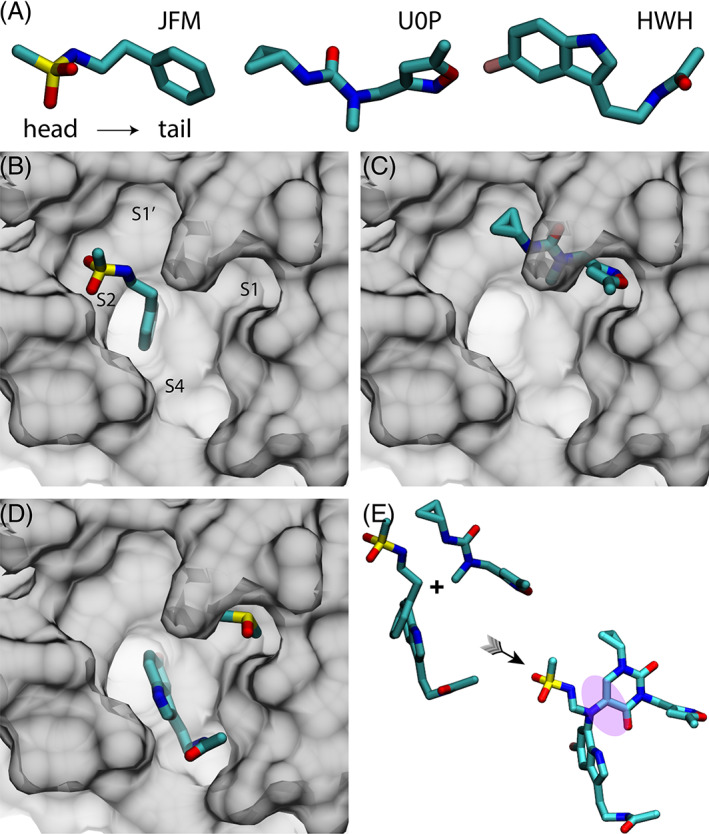
Scheme of fragment ‐based drug design of B19. (A) Three selected drug‐fragments: JFM, U0P, and HWH. For each of three fragment drugs, the head, and tail parts are on the left and right sides, respectively. (B) The crystal structure of JFM in the ligand‐binding pocket of Mpro (PDB ID: 5R7Y). (C) The crystal structure of U0P in the ligand‐binding pocket of Mpro (PDB ID: 5RGI). (D) The crystal structure of HWH in the ligand‐binding pocket of Mpro (PDB ID: 5R7Z). A DMSO molecule was co‐crystallized inside the subsite S1. (E) The assembly of JFM, U0P, and HWH into B19 with a linker (shaded)
Following previous studies, 19 the active site of Mpro is composed of four key subsites that are respectively labeled as S1′, S1, S2, and S4 (Figure 1B). As shown in Figure 1B–D, the fragment U0P binds the S1 and S1′ subsites, and the fragments JFM and HWH bind the S2 and S4 subsites, respectively. Interestingly, a co‐crystallization agent dimethyl sulfoxide (DMSO) is also present inside the active site (S1) along with HWH as shown in Figure 1D. Due to their proximity (Figure 1E), with an added linker molecule we combined JFM, U0P and HWH into one drug molecule (named B19 with the molecular formula C26H30N7O6SF), occupying all four subsites. B19 has a molecular weight of 587.6, which is comparable to O6K (595.7) and less than N3 (680.8). Totally, we designed 19 drug molecules using various linker molecules, with B19 shown in Figure 1E being the most stable one (evaluated from our MD simulations) and the rest (from B1 to B18) shown in Figure S1. In our design of B19, we first merged JFM and HWH together because their benzene rings overlap with each other. After that, we employed the linker molecule shown in Figure 1E to bridge the merged compound and U0P. In B19, the carbon atom attached to the benzene ring (originally in JFM) was replaced by a nitrogen atom, which can form a stable three‐way junction, maintaining the overall conformation of B19 inside the active site of Mpro (see below). Importantly, the catalytic dyad comprising H41 and C145 is completely covered by the designed B19 molecule.
Note that the Mpro's active site is formed by two protein domains, residues 1–100 (I) and residues 101–200 (II). The subsites S1, S2 are, respectively, in domains II and I, while the subsites S1′ and S4 are at the interface of domains I and II. Therefore, the active site of Mpro is not rigid and can deform slightly to harbor a ligand (such as B19) bridging two Mpro domains. Generally, the host's flexibility is amenable to the FBDD method, that is, capable to accommodate a merged complex containing slightly displaced fragments.
We performed MD simulations to verify the stability of each designed drug molecule (from B1 to B19) bound inside the active site. Figure 2A illustrates the simulation system: a Mpro dimer with B19 bound inside the active site of one monomer was solvated in a 0.15 M electrolyte. Details are provided in the Section 4. We carried out two independent MD simulations, each of which lasted about 190 ns. Figure 2B,C illustrate a representative conformation of B19 in Mpro shown in the molecular surface and cartoon representations, respectively.
FIGURE 2.

MD simulation of B19 in Mpro. (A) Illustration of the MD system. The two monomers (gray and purple) in Mpro are in the cartoon representation. B19 is in the van der Waals sphere representation. Water is shown transparently. K+ and Cl− ions are colored in tan and cyan, respectively. (B, C) The equilibrated conformation of B19 in the active site of Mpro. Mpro is in the surface representation (B) and in the cartoon representation (C)
Overall, during the 190 ns simulation, B19 maintained its initial conformation in Mpro, with slight local adjustments. For example, compared with its initial pose (Figure 1B,E) the SO2 group turned itself away from the Mpro surface and faced the water environment, because of its hydrophilicity. Meanwhile, the hydrophobic methyl (CH3) group changed its initial unfavorable pose facing water to the one contacting the hydrophobic M49 and T25 on the Mpro surface. Additionally, the isoxazole group (present in the S1 subsite) could turn 180° from time to time. Thus, different from the one shown in Figure 2B,C, the N and O atoms in the five‐membered isoxazole ring could also be in contact with the Mpro surface (Figure 1C and Movie S1).
From the simulation trajectories, we calculated the root mean square deviations (RMSD) for backbone atoms in the monomer (of Mpro) that harbors the B19. Figure 3A shows that after about 75 ns RMSDs from two independent MD simulations saturate around 1.7 Å, suggesting that the Mpro's crystal structure was properly equilibrated in the physiology‐like (a 0.15 M electrolyte) environment. The saturated RMSDs for B19 inside the active site of Mpro are about 1.2 Å (Figure 3B), corroborating the stable conformation of B19 in the active site as shown in the Movie S1.
FIGURE 3.
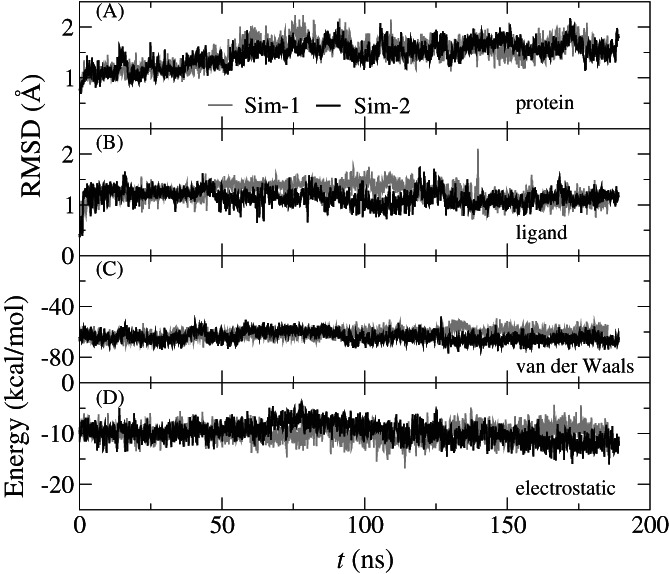
Dynamics and energetics of B19 inside the Mpro's pocket. (A) Time‐dependent RMSDs of the backbone atoms in the protein monomer that harbors B19. (B) Time‐dependent RMSDs of B19. (C) The van der Waals interaction energies (between B19 and Mpro) versus time. (D) The electrostatic interaction energies (between B19 and Mpro) versus time. The dielectric constant used in electrostatic interaction is 4
We further calculated the pairwise interaction energy between B19 and Mpro, including the van der Waals (vdW, Figure 3C) and electrostatic (Figure 3D) ones. Over the entire 190 ns, both the vdW and electrostatic potential energies are nearly constant for both simulations (Sim‐1 and Sim‐2), with the electrostatic ones (Figure 3D) showing larger fluctuations partly due to the omission of interfacial water molecules in these calculations. In comparison, results for Sim‐2 are slightly better (more negative) than those for Sim‐1, with the vdW energy about −68.3 kcal/mol and the electrostatic one about −12.3 kcal/mol. Besides demonstrating the binding stability of B19 in the active site, these results will be further used in the MM/GBSA calculations to obtain the binding free energy of B19 (see below).
In the other 18 designs (B1–B18) as shown in Figure S1, these drug molecules with unsuccessfully designed linker molecules moved more or less away from their initial positions during MD simulations, that is, at least one fragment in each designed drug molecule either rotated or shifted away from the one as seen in the crystal environment. For example, observed in two independent MD simulations, the U0P fragment in B15 was not stable and moved out of the S1 and S1′ subsites, however, the other two fragments of B15 remained inside the subsites S2 and S4, respectively, indicating the problematic design of the link molecule in B15 (Figure S1).
In Figure 4, we highlight key coordinations between B19 and Mpro to unveil the molecular mechanism of the binding. In Figure 4A, the carbonyl group in the original drug fragment U0P formed two hydrogen bonds with the backbones (NH groups) of residues G143 and C145, which pins the entire U0P fragment in the subsites S1 and S1′ (Figure 2B). Remarkably, this same local coordination can also be found in the complex of Mpro and O6K (PDB code: 6LU7). Furthermore, through hydrophobic interactions the cyclopropane group (originally in U0P) was stabilized by hydrophobic interactions with residues C145, L27, and T25 of Mpro.
FIGURE 4.
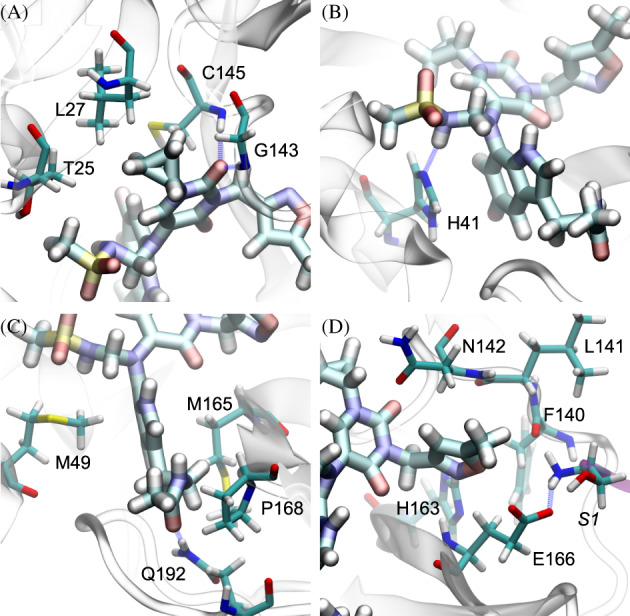
Illustrations of local interactions between B19 and Mpro. (A) The head part of the U0P fragment. (B) The head part of the JFM fragment. (C) The HWH fragment. (D) The tail part of the U0P fragment
Figure 4A also shows the hydrophobic interaction between T25 and the methyl group in the methanesulfonamide group (originally in JFM). Notably, in Figure 4B the NH group in the methanesulfonamide group (in the subsite S2) formed a hydrogen bond with the residue H41 at the bottom of the active site. Furthermore, as shown in Figure 4C, the methyl group in the methanesulfonamide group also interacts hydrophobically with the residue M49. Overall, the residues T25 (Figure 4A), H41 (Figure 4B), and M49 (Figure 4C) stabilize the methanesulfonamide group in the subsite S2.
Illustrated in Figure 4C, the indole group (originally in HWH) insert itself deeply into the gap between the two protein domains I and II. Namely, the indole group interacts hydrophobically with the residue M49 in the protein domain I (of Mpro) on one side and interacts also hydrophobically with the residue M165 in the protein doamin II (of Mpro) on the other side. In addition, the acetamide group (originally in the tail part of HWH) can form a hydrogen bond with the residue Q192 and also have the hydrophobic interaction with the residue P168 (Figure 4C).
The isoxazole group (originally in U0P) occupies the subsite S1, a pocket formed by residues H163, N142, L141, F140, E166 as well as the residue S1 in the N‐terminal of the other protein monomer (purple, Figure 4D). There is no hydrogen bond formed in this local interaction, which is consistent with the observation that the isoxazole group can rotate from time to time (see Movie S1). Taking all together, the isoxazole group and the methanesulfonamide group are, respectively, hosted by the domains II and I, while the cyclopropane group and the indole group lie between the domains I and II (see Figure 2B,C). Thus, fitting nicely inside the active site, B19 appears to act like a glue bonding the two protein domains together.
One restriction of the above MD simulation is that we cannot determine whether the discovered binding pose of B19 inside the active site is correspondent with the lowest energy state (or the best pose), due to the limited simulation time. Here, we resort to the docking method to explore the ligand–receptor conformation space for all plausible poses of B19 as well as their binding affinities. Among many docking tools, Autodock Vina 20 has been used as a powerful one for in silico drug design as it can help understand the structural determinants for protein–ligand complex at the atomic level which is crucial for designing ligands with high specificity and affinity to a target protein. The detailed docking protocol can be found in the Section 4.
Figure 5 shows the docking results for the top 200 poses, starting from a random pose of B19. Remarkably, from a total of more than 700 docked poses of B19 inside the active site, the best one (Figure 5, inset) indeed agrees with the one that was discovered in MD simulations (Figure 2B,C). The binding affinity for the best pose of B19 is −10.1 kcal/mol, which is significantly better than many repurposed drug molecules (typically < −7.0 kcal/mol), N3 (−7.1 kcal/mol), and O6K (−7.4 kcal/mol) without their covalent bonds with C145, 21 compared when using the same docking protocols. In insets of Figure 5, we also show several other binding poses ranked as the 20th (−8.8 kcal/mol), 40th (−8.4 kcal/mol), and 60th (−8.2 kcal/mol) ones. Therefore, the docking study reasonably confirmed that the B19's pose from MD simulation is the best one, residing in the global minimum on the binding free energy surface.
FIGURE 5.
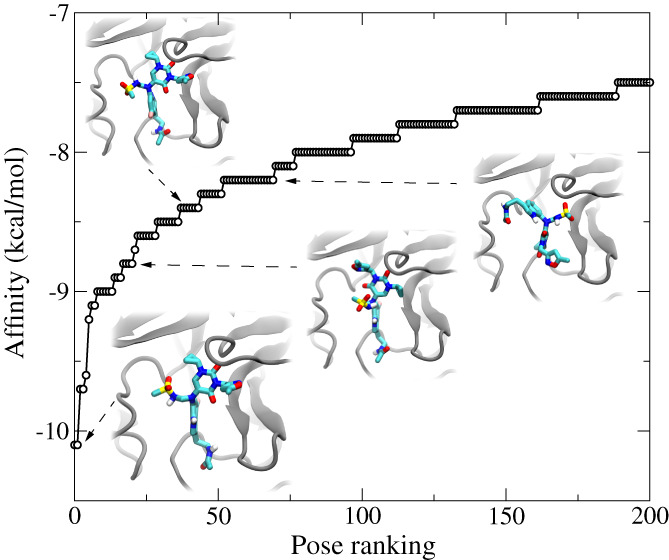
The docking study of top 200 poses of B19 inside the pocket of Mpro. The insets illustrate four different docking poses (1st, 20th, 40th, and 60th) of B19
Next, we compare the binding affinities between B19 and a native ligand. So far, there is no crystal structure for Mpro of SARS‐CoV‐2 bound with a native ligand. However, due to the highly conserved Mpro structures among all coronavirus, we refer to the crystal structure (PDB code: 2Q6G) of SARS‐CoV‐1's Mpro bound with a native ligand (peptide sequence: TSAVLQSGFRK). Note that residues in the active site are identical for both Mpros in SARS‐CoV‐1 and SARS‐CoV‐2. Thus, we carried out two independent MD simulations (Sim‐3 and Sim‐4) of Mpro with the bound native ligand whose binding pose is same as that in the crystal structure (PDB code: 2Q6G), as shown in Figure S2a. After about 190 ns, the equilibrated structure of the native ligand is shown in Figure 6A,B. The RMSD of protein backbones saturated around 2.2 and 1.7 Å for Sim‐3 and Sim‐4, respectively (Figure S2b). The difference in RMSDs resulted from the different thermal fluctuations of some disordered cords in the secondary structure of Mpro. The saturated RMSDs for the native ligand are about 1.5 Å (Figure S2c), which is slightly larger than the one for B19 due to a few flexible residues outside the active site. Overall, the native ligand is stable inside the active site, which is also demonstrated in Movie S2.
FIGURE 6.

MD simulation of the native ligand inside the active site of Mpro. (A) Illustration of the complex in the cartoon representation. The native ligand is colored in cyan. (B) Illustration of the complex, with residues 2–8 of the native ligand in the stick representation and Mpro in the molecular surface representation. (C) Illustration of essential interactions between the native ligand and Mpro. The Mpro residues in contact with the native ligand are in the van der Waals sphere representation
As shown in Figure 6A, the native ligand is stabilized by forming two antiparallel beta sheets with residues (e.g., T26) in the protein domain I and with residues (e.g., E166) in the protein domain II. Inside the active site, Q6 (of the native ligand) occupies the S1 subsite; S7 and G8 are in the subsite S1′; A3 and S2 are in the subsite S4; and L5 occupies the subsite S2. The side chain of V4 is oriented toward outside the active site (Figure 6B). The other residues T1, F9, R10, and K11 are all outside of the active site.
Figure 6C highlights detailed interactions between the native ligand and Mpro. We found that the C=O group in the backbone of Q6, similar to the same group of B19 as shown in Figure 4A, also formed two hydrogen bonds with G143 and C145. It is worth noting that Q6 is very specific to the subsite S1, as shown in many other native protein strands for Mpro of SARS‐CoV‐1. 22 However, the side chain of Q6 (also the isoxazole group in B19) neither forms any hydrogen bond nor has any hydrophobic interactions with the nearby Mpro residues. Presumably, the orientation of the Q6's side chain and the isoxazole group in B19 inside the subsite S1 enables the neighboring C=O group to form strong hydrogen bonds with G143 and C145. Notably, S7 forms a hydrogen bond with H41. The latter also forms a hydrogen bond with the NH group in the JFM fragment in B19 (Figure 4B). Additionally, G8 forms a hydrogen bond with T26 in Mpro via their backbones, forming antiparallel β‐sheets. All these hydrogen bonds tightly positioned the native ligand in the front of the catalytic center C145, illustrating the molecular mechanism of catalytic processing of the bound peptide.
The binding of L5 inside the S2 subsite is also highly conserved among all native protein strands in Mpro of SARS‐CoV‐1. 22 As shown in Figure 6C, L5 is sandwiched between two hydrophobic residues M49 and M165 in Mpro. In contrast, the benzene ring in the HWH fragment in B19 resides between M49 and M165 (Figure 4C). Although the side chain of V4 is outside the active site, its backbone forms two hydrogen bonds with backbone atoms in E166 of Mpro, forming antiparallel β‐sheets locally. For comparison, B19 does not directly coordinate E166. The side chain of A3 of the native ligand interacts hydrophobically with L167 of Mpro, and the oxygen atom in the A3's backbone forms a hydrogen bond with the NH2 group in Q189. Overall, S2, A3, V4, and Q6 mainly bind the protein domain II, while L5, S7, and G8 mainly bind the protein domain I (of Mpro), demonstrating a sophisticated binding mechanism of the native ligand with Mpro.
To compare the binding affinities between B19 and the native ligand, we utilized the MM/GBSA method that is generally applied to estimate the ligand–protein binding free energy and can yield results highly correlated with experimental ones. 23 The detailed description of the calculation procedures are summarized in the Section 4. Briefly, the MM/GBSA binding free energy contains three parts: (a) the molecular mechanics (MM); (b) the Generalized Born (GB) for solvation energy, and (c) the nonpolar energy proportional to the solvent accessible surface area (SA). Using the NAMD package, the MM part was calculated from the trajectory analysis on a pairwise interaction between a ligand and Mpro. Here, the MM part for B19 is the addition of vdW and electrostatic interactions as shown in Figure 3C,D, which is −80.6 kcal/mol (Table 1). Similarly, we obtained the MM energy for the native ligand in the active site of Mpro, which is −83.5 kcal/mol (Table 1). Here, for the MM part only, the binding is stronger for the native ligand, mainly due to more hydrogen bonds (i.e., stronger electrostatic interaction) formed between the native ligand and Mpro.
TABLE 1.
Comparisons of MM/GPSA binding free energies for the native ligand and B19 inside the active site of Mpro
| Ligand | MM (kcal/mol) | GBSA (kcal/mol) | Total (kcal/mol) |
|---|---|---|---|
| Native | −83.5 | −44.7 | −128.2 |
| B19 | −80.6 | −54.6 | −135.2 |
With the NAMD package, the GBSA part can be calculated together during the trajectory analysis 24 (see Section 4). The GBSA binding free energies for B19 and the native ligand are −54.6 and −44.7 kcal/mol (Table 1), respectively. For the GBSA part, because B19 is less hydrophilic than the native ligand, the desolvation penalty for B19 after its entry into the active site of Mpro is less than that for the native ligand. Thus, it is energetically more beneficial for B19 to move from the solution into the Mpro's active site. Adding together the MM and GBSA parts, the total binding free energies for B19 and the native ligand are −135.2 and −128.2 kcal/mol, respectively, suggesting that B19 is slightly (∼5%) better than the native ligand when competing for Mpro. Here, the entropy contribution that typically yields noisy data is ignored. Qualitatively, the native protein ligand is more flexible in the solution (due to peptide bonds) than B19, causing more entropy loss (i.e., unfavorable) for the native ligand when entering the active site and thus an even larger difference in binding free energies between B19 and the native ligand. Note that typically the MM/GBSA binding free energy is about 5–10 times larger than the experimental value. Nevertheless, the MM/GBSA method provides good ranking results that are generally consistent with experimental ones.
To evaluate the synthetic accessibility of the drug‐like molecule B19, we employed the recently debuted synthetic analysis provided by the RXN for Chemistry (https://rxn.res.ibm.com) that predicts chemical reactions and the development of molecules. The newly added retrosynthesis module is based on an extension of the artificial intelligence (AI) model called the Molecular Transformer combined with a hyper‐graph exploration strategy to automate the planning of the retrosynthesis route without human supervision. 25 , 26 When compared with other works, statistical analysis indicates that the retrosynthetic architecture of RXN performs very well for a broad class of disconnections. 26 Here, due to the complexity of B19, we applied the software to delineate the steps for synthesizing B19 without the methanesulfonamide group, that is, the complex including fragments U0P, HWH, and the linker. The result shows that the complex can be synthesized in merely eight steps (see Appendix S1). The synthesized complex can be further linked with the methanesulfonamide group (terminated with a bromine atom) through the Bromo N‐alkylation reaction in the NaOH solution.
3. DISCUSSION AND CONCLUSIONS
In summary, by merging drug fragments occupying different subsites of the Mpro's active site, we designed the drug‐like molecule B19 that was confirmed to bind the active site stably from two independent MD simulations. Similar to the previously designed mechanism‐based N3 and O6K ligands, B19 occupies four subsites (S1, S1′, S2, and S4), through various hydrogen bonds and hydrophobic interactions. Using the docking method with a large exhaustiveness number (i.e., improved searching for the global minimum), we were able to screen over 700 different conformations of B19 inside the active site of Mpro and verified that the equilibrated B19 structure in MD simulation is the most stable one (with the highest binding affinity). Additionally, we compared the binding free energies between B19 and a native protein ligand processed by Mpro, which indicates that B19 can bind Mpro more strongly than the native ligand. Taking all these encouraging results together, B19 is a promising candidate for inhibiting Mpro of SARS‐CoV‐2 and deserves further investigation in vitro/in vivo. With the unveiled molecular mechanism of B19's binding in the active site of Mpro, it is possible to further optimize B19 through the in silico alchemical free‐energy perturbation (FEP) method 27 , 28 to improve its stability and druglikeness. Last but not least, the in silico approach described in this work might be applied to yield other efficacious drug molecules with the ongoing crowdsourcing efforts to fight collectively and efficiently against COVID‐19.
4. METHODS
4.1. MD simulations
We performed all‐atom MD simulations for designed ligand (drug) molecules inside Mpro, using the NAMD2.13 package 29 running on the Power Cluster. To model the ligand–Mpro complex, we obtained the previously resolved crystal structure for Mpro (PDB code: 5R7Y) 11 from the Protein Data Bank (PDB) and assembled together two to three fragments (PDB codes: 5R7Y, 5RGI, and 5R7Z) with a short linker molecule into a ligand in Mpro. Overall, we designed 19 ligands with various linker molecules. The position of each fragment in a ligand was kept as close to the crystallized one in Mpro as possible. The complex was solvated in a cubic water box measuring about 97.2 × 97.2 × 97.2 Å3. The 88 K+ and 80 Cl− were added in order to neutralize the entire simulation system and set the ion concentration to be 0.15 M (Figure 2A). Because the active sites of Mpro among all coronavirus Mpros are highly conserved, we also modeled a native ligand (with the sequence TSAVLQSGFRK) in Mpro of SARS‐CoV‐2 (Figure 6A), with the ligand's position adopted from the crystal structure (PDB code: 2Q6G) for Mpro in SARS‐CoV‐1. The final system containing about 94 K atoms was first minimized for 10 ps and further equilibrated for 500 ps in the NPT ensemble (P ∼ 1 bar and T ∼ 300 K), with atoms in the Mpro's backbone and nonhydrogen atoms in the ligand harmonically restrained (spring constant k = 1 kcal/mol/Å2). After removing all restraints, the production run was carried out in the NVT ensemble. This same protocol was successfully applied to study the molecular mechanism of SARS‐CoV‐2's variants: Alpha 30 and Beta. 31
The CHARMM36 force field 32 was chosen for proteins (Mpro and the native ligand); the TIP3P model 33 , 34 was chosen for water; the standard force field 35 was used for ions. The force field for the designed ligands were obtained from SwissParam. 36 We provided the .psf and .pdb files for B19 in the Appendices S2 and S3. The periodic boundary conditions (PBC) were applied in all three dimensions. Long‐range Coulomb interactions were calculated using particle‐mesh Ewald (PME) full electrostatics with the grid size about 1 Å in each dimension. The van der Waals (vdW) energies between a pair of atoms were calculated using a smooth (10–12 Å) cutoff. The temperature T was maintained at 300 K by applying the Langevin thermostat, 37 while the pressure was kept constant at 1 bar using the Nosé–Hoover method. 38 With the SETTLE algorithm 39 applied to keep all bonds rigid, the simulation time‐step was set to be 2 fs for bonded and non‐bonded (e.g., vdW, angle, and dihedral) interactions, and electric interactions were calculated every 4 fs, with the multiple time‐step algorithm. 40
4.2. MM/GBSA free energy calculations
We used the GBIS module in NAMD to run postprocessing of MD simulation trajectories, for obtaining the GBSA binding free energy that comprises the polar solvation free energy estimated from the Generalized Born (GB) method 41 and the nonpolar one obtained as a function of the solvent accessible surface area (SASA), −γ ·SASA where γ (=0.00542 kcal/molÅ2) is the surface tension. The solvent dielectric constant was set to be 78.5 and the cutoff distance was set to be 16 Å. Overall, the GBSA free energy change ΔG = ΔG complex − ΔG protein − ΔG ligand. For the MM part, we used the NAMD‐energy module for calculating the pair‐wise energy between a ligand and Mpro, with the solute dielectric constant set to be 4.
4.3. Docking method
We employed Autodock Vina, 20 one of the most popular and highly cited open source docking software applications in the research community, to identify the conformation of the studied drug molecules and the corresponding binding affinity with Mpro. Autodock Vina was shown to have the highest scoring power in a study that performed a comprehensive evaluation of 10 famous currently available docking programs, including five commercial and five academic ones. 17 To prepare the input for the docking simulations we used the set of commands provided by AutoDockTools (ADT) specifically developed to support the AutoDock users downloaded from The Center for Computational Structural Biology (CCSB), and followed the protocol in our previous study. 21
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
PEER REVIEW
The peer review history for this article is available at https://publons.com/publon/10.1002/prot.26260.
Supporting information
Figure S1 Th unsuccessful designs of other 18 fragment‐based drug ligands, from B1 to B18. The ligands from B14 to B17 were merged from all three fragments as listed in the main text. The rest were merged from U0P and HWH only. Various linker molecules were tested for ligand stability. For each drug, at least two MD simulations were carried out. These ligands are not as stable as B19 inside the active site of Mpro.
Figure S2 MD simulation of Mpro bound with the native ligand. (A) Simulation system. The native ligand is colored in cyan and is in the cartoon representation. Others are illustrated same as those in Figure 2 in the main text. (B) RMSDs of backbone atoms in the host protein, that is, the momoner in gray in (A). (C) RMSDs of heavy atoms in the native ligand.
Movie S1 Supplementary Movie
Movie S2 Supplementary Movie
Appendix S1 Synthesis steps for B19 without the methanesulfonamide group.
Appendix S2 Supporting Information
Appendix S3 Supporting Information
ACKNOWLEDGMENTS
T.H. and B.L. gratefully acknowledge the help from the HCLS division for the computing resource in the Cognitive Computing Clusters (IBM).
Luan B, Huynh T. Crystal‐structures‐guided design of fragment‐based drugs for inhibiting the main protease of SARS‐CoV‐2. Proteins. 2022;90(5):1081‐1089. doi: 10.1002/prot.26260
Funding information IBM cognitive computating program
DATA AVAILABILITY STATEMENT
The data that supports the findings of this study are available in the supplementary material of this article.
REFERENCES
- 1. Wu F, Zhao S, Yu B, et al. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579(7798):265‐269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kuiken T, Fouchier RA, Schutten M, et al. Newly discovered coronavirus as the primary cause of severe acute respiratory syndrome. Lancet. 2003;362(9380):263‐270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bermingham A, Chand M, Brown C, et al. Severe respiratory illness caused by a novel coronavirus, in a patient transferred to the United Kingdom from the Middle East, September 2012. Eurosurveillance. 2012;17(40):20290. [PubMed] [Google Scholar]
- 4. Beigel JH, Tomashek KM, Dodd LE, et al. Remdesivir for the treatment of Covid‐19—preliminary report. N Engl J Med. 2020;383:1813‐1826. [DOI] [PubMed] [Google Scholar]
- 5. Villar J, Ferrando C, Martínez D, et al. Dexamethasone treatment for the acute respiratory distress syndrome: a multicentre, randomised controlled trial. Lancet Respir Med. 2020;8(3):267‐276. [DOI] [PubMed] [Google Scholar]
- 6. Ullrich S, Nitsche C. The SARS‐CoV‐2 main protease as drug target. Bioorg Med Chem Lett. 2020;30(17):127377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jin Z, Du X, Xu Y, et al. Structure of Mpro from COVID‐19 virus and discovery of its inhibitors. Nature. 2020;582:289‐293. 10.1038/s41586-020-2223-y [DOI] [PubMed] [Google Scholar]
- 8. Zhang L, Lin D, Sun X, et al. Crystal structure of SARS‐CoV‐2 main protease provides a basis for design of improved α‐ketoamide inhibitors. Science. 2020;368(6489):409‐412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jin Z, Zhao Y, Sun Y, et al. Structural basis for the inhibition of SARS‐CoV‐2 main protease by antineoplastic drug Carmofur. Nat Struct Mol Biol. 2020;27(6):529‐532. [DOI] [PubMed] [Google Scholar]
- 10. Chodera J, Lee AA, London N, Delft F. Crowdsourcing drug discovery for pandemics. Nat Chem. 2020;12(7):581‐581. [DOI] [PubMed] [Google Scholar]
- 11. Douangamath A, Fearon D, Gehrtz P, et al. Crystallographic and electrophilic fragment screening of the SARS‐CoV‐2 main protease. Nat Commun. 2020;11:5047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Erlanson DA. Introduction to Fragment‐Based Drug Discovery. Springer; 2011:1‐32. [DOI] [PubMed] [Google Scholar]
- 13. Murray CW, Rees DC. The rise of fragment‐based drug discovery. Nat Chem. 2009;1(3):187‐192. [DOI] [PubMed] [Google Scholar]
- 14. Jhoti H, Williams G, Rees DC, Murray CW. The 'rule of three' for fragment‐based drug discovery: where are we now? Nat Rev Drug Discov. 2013;12(8):644‐644, 645. [DOI] [PubMed] [Google Scholar]
- 15. Karplus M, McCammon J. Molecular dynamics simulations of biomolecules. Nat Struct Biol. 2002;265:654‐652. [DOI] [PubMed] [Google Scholar]
- 16. Freddolino PL, Harrison CB, Liu Y, Schulten K. Challenges in protein‐folding simulations. Nat Phys. 2010;6(10):751‐758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang Z, Sun H, Yao X, et al. Comprehensive evaluation of ten docking programs on a diverse set of protein–ligand complexes: the prediction accuracy of sampling power and scoring power. Phys Chem Chem Phys. 2016;18(18):12964‐12975. [DOI] [PubMed] [Google Scholar]
- 18. Xue X, Yu H, Yang H, et al. Structures of two coronavirus main proteases: implications for substrate binding and antiviral drug design. J Virol. 2008;82(5):2515‐2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dai W, Zhang B, Su H, et al. Structure‐based design of antiviral drug candidates targeting the SARS‐CoV‐2 main protease. Science. 2020;368(6497):1331‐1335. 10.1126/science.abb4489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31(2):455‐461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Huynh T, Wang H, Luan B. In silico exploration of molecular mechanism of clinically oriented drugs for inhibiting SARS‐CoV‐2's main protease. J Phys Chem Letts. 2020;11(11):4413‐4420. [DOI] [PubMed] [Google Scholar]
- 22. Muramatsu T, Takemoto C, Kim YT, et al. SARS‐CoV 3CL protease cleaves its C‐terminal autoprocessing site by novel subsite cooperativity. Proc Natl Acad Sci U S A. 2016;113(46):12997‐13002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Greenidge PA, Kramer C, Mozziconacci JC, Wolf RM. MM/GBSA binding energy prediction on the PDBbind data set: successes, failures, and directions for further improvement. J Chem Inf Model. 2013;53(1):201‐209. [DOI] [PubMed] [Google Scholar]
- 24. Vergara‐Jaque A, Comer J, Monsalve L, González‐Nilo FD, Sandoval C. Computationally efficient methodology for atomic‐level characterization of dendrimer–drug complexes: a comparison of amine‐and acetyl‐terminated PAMAM. J Phys Chem B. 2013;117(22):6801‐6813. [DOI] [PubMed] [Google Scholar]
- 25. Vaucher AC, Zipoli F, Geluykens J, Nair VH, Schwaller P, Laino T. Automated extraction of chemical synthesis actions from experimental procedures. Nat Commun. 2020;11:3601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schwaller P, Petraglia R, Zullo V, et al. Predicting retrosynthetic pathways using transformer‐based models and a hyper‐graph exploration strategy. Chem Sci. 2020;11(12):3316‐3325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chipot C, Pohorille A. Free Energy Calculations. Springer; 2007. [Google Scholar]
- 28. Luan B, Xu G, Feng M, Cong L, Zhou R. Combined computational–experimental approach to explore the molecular mechanism of SaCas9 with a broadened DNA targeting range. J Am Chem Soc. 2019;141(16):6545‐6552. [DOI] [PubMed] [Google Scholar]
- 29. Phillips JC, Braun R, Wang W, et al. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781‐1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Luan B, Wang H, Huynh T. Enhanced binding of the N501Y‐mutated SARS‐CoV‐2 spike protein to the human ACE2 receptor: insights from molecular dynamics simulations. FEBS Lett. 2021;595(10):1454‐1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Luan B, Huynh T. Insights into SARS‐CoV‐2's mutations for evading human antibodies: sacrifice and survival. J Med Chem. 2021. 10.1021/acs.jmedchem.1c00311 [DOI] [PubMed] [Google Scholar]
- 32. MacKerell A, Bashford D, Bellott M, et al. All‐atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B. 1998;102:3586‐3616. [DOI] [PubMed] [Google Scholar]
- 33. Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926‐935. [Google Scholar]
- 34. Neria E, Fischer S, Karplus M. Simulation of activation free energies in molecular systems. J Chem Phys. 1996;105(5):1902‐1921. [Google Scholar]
- 35. Beglov D, Roux B. Finite representation of an infinite bulk system: solvent boundary potential for computer simulations. J Chem Phys. 1994;100:9050‐9063. [Google Scholar]
- 36. Zoete V, Cuendet MA, Grosdidier A, Michielin O. SwissParam: a fast force field generation tool for small organic molecules. J Comput Chem. 2011;32(11):2359‐2368. [DOI] [PubMed] [Google Scholar]
- 37. Allen MP, Tildesley DJ. Computer Simulation of Liquids. Oxford University Press; 1987. [Google Scholar]
- 38. Martinetz T, Schulten K. Topology representing networks. Neural Netw. 1994;7(3):507‐522. [Google Scholar]
- 39. Miyamoto S, Kollman PA. SETTLE: an analytical version of the SHAKE and RATTLE algorithm for rigid water molecules. J Comput Chem. 1992;13(8):952‐962. [Google Scholar]
- 40. Tuckerman M, Berne BJ, Martyna GJ. Reversible multiple time scale molecular dynamics. J Chem Phys. 1992;97(3):1990‐2001. [Google Scholar]
- 41. Tsui V, Case DA. Theory and applications of the generalized born solvation model in macromolecular simulations. Biopolymers. 2000;56(4):275‐291. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Th unsuccessful designs of other 18 fragment‐based drug ligands, from B1 to B18. The ligands from B14 to B17 were merged from all three fragments as listed in the main text. The rest were merged from U0P and HWH only. Various linker molecules were tested for ligand stability. For each drug, at least two MD simulations were carried out. These ligands are not as stable as B19 inside the active site of Mpro.
Figure S2 MD simulation of Mpro bound with the native ligand. (A) Simulation system. The native ligand is colored in cyan and is in the cartoon representation. Others are illustrated same as those in Figure 2 in the main text. (B) RMSDs of backbone atoms in the host protein, that is, the momoner in gray in (A). (C) RMSDs of heavy atoms in the native ligand.
Movie S1 Supplementary Movie
Movie S2 Supplementary Movie
Appendix S1 Synthesis steps for B19 without the methanesulfonamide group.
Appendix S2 Supporting Information
Appendix S3 Supporting Information
Data Availability Statement
The data that supports the findings of this study are available in the supplementary material of this article.