

Abstract
Balanced immune regulation is crucial for recognizing an invading pathogen, its killing, and elimination. Toll‐like receptors (TLRs) are the key regulators of the innate immune system. It helps in identifying between self and nonself‐molecule and eventually eliminates the nonself. Endosomal TLR, mainly TLR3, TLR7, TLR8, and membrane‐bound TLR4, has a role in the induction of cytokine storms. TLR7/8 recognizes the ssRNA SARS‐COV‐2 and when it replicates to dsRNA, it is recognized by TLR3 and drives the TRIF‐mediated inflammatory signaling like NF‐κB, MAPK. Such signaling leads to significant transcription and translation of pro‐inflammatory genes, releasing inflammatory molecules into the systemic circulation, causing an imbalance in the system. So, whenever an imbalance occurs, a surge in the pro‐inflammatory mediators is observed in the blood, including cytokines like interleukin (IL)‐2, IL‐4, IL‐6, IL‐1β, IL‐8, interferon (IFN)‐γ, tumor necrosis factor (TNF)‐α. IL‐6 and IL‐1β are one of the driving factors for bringing the cytokine storm into the systemic circulation, which migrates into the other organs, causing multiple organ failures leading to the death of the individual with severe illness.
Keywords: COVID‐19, cytokines, multiple‐organ failure, SARS‐COV‐2, ssRNA, TLRs
Highlights
The imbalanced and hyper responsive immune system leads to a surge leading to death of the infected patients in COVID‐19.
It has been observed that cytokine surge is TLR induced, mainly through activation of TLR3, TLR4, TLR7, TLR8 receptors.
The cytokine storm migrates into the other organ through systemic circulation. The inflammation and the organ damage occur due to the TLR mediated NF‐κB, MAPK pathway. Hence blocking these specific TLRs may alleviate the chance of SARS‐COV‐2 infection.
1. INTRODUCTION
A pandemic and millions of deaths; though not as simple as it sounds. People are vexed by queries like, “Is our immune system not fighting enough with this guest organism? How is it transmitting in such a mass population? Most importantly, how is the virus so potent as to spread into other organs besides the lungs, causing tissue damage, and rendering multiorgan failure, driving the severely ill patient to death? Severe acute respiratory syndrome coronavirus 2 (SARS‐COV‐2) has been found as the causative agent for the COVID‐19 outbreak. 1 The virus differs from that of SARS‐COV and the Middle East respiratory syndrome (MERS). Influenced by its crown‐like appearance, the virus has been named “coronavirus.” 2 The virus has powerful transmissibility and the infection spreads from the cough or sneeze of the infected individual through respiratory droplets >5 µm in diameter within 1‐m distance; or by the nucleic acid of the virus <5 µm in diameter that remain in the air for a longer period of time. 3 The viral life cycle and its replication begin after its incorporation into angiotensin‐converting enzyme‐2 (ACE2) receptors present in the epithelium of oral mucosa, lung, heart, kidney, and so forth. ACE2 receptors are highly expressed in adults than that in children, as the expression of ACE2 increases with age. 4 ACE2 expression also varies between genders. 5 However, World Health Organization (WHO) has reported multiple inflammatory syndromes in children (MIS‐C) and adolescents (between 1 and 19 years of age), infected with SARS‐COV‐2. 6 The typical symptoms of a SARS‐COV‐2 infected individual includes fever, fatigue, dry cough, dysgeusia, which may proceed with gastric distress in some patients like vomiting and diarrhea. The defensive mechanism in SARS‐COV‐2 involves both adaptive and innate immunity. The serum analysis of symptomatic patients reveals an excessively high level of cytokines like interferon (IFN)‐γ, tumor necrosis factor (TNF)‐α, interleukin (IL)‐2, IL‐4, IL‐6, IL‐8, IL‐1β, and so forth. Characterized as “cytokine storm” that may have occurred due to an imbalanced immune system. 7 , 8 The cytokine storm is also considered to be one of the main reasons for extrapulmonary organ failure, which fully contributes to disease severity and mortality of the severely infected patients.
2. ROLE OF TLRS IN COVID‐19
2.1. Toll‐like receptors (TLRs)
TLRs are the central regulators of the immune system and belong to a class of pathogen‐recognition receptors (PRR). The invading pathogens are recognized initially by the innate immune system by sensing their pathogen‐associated molecular pattern (PAMP) with PRR. Macrophage, monocyte, and dendritic cells (DCs) express PRR either on their surface or inside the cell depending on the type of invading pathogens. 9 Viral PAMP are recognized by their genomic materials' nucleic acid by the cell surface or cytosolic PRR. 10 TLRs are a class of membrane PRR that detect the microbes on the cell surface as well as in the cytoplasm. These receptors detect both the DNA as well as RNA of the invading pathogen. Each TLR is composed of leucine‐rich repeats (LRR) ectodomain, a transmembrane domain, and a cytoplasmic domain, through which it mediates the downstream signaling. 11 A total of 11 types of TLRs have been found in human beings, among those some are membrane‐bound and some are intracellular receptors. TLR3, TLR7, TLR8, TLR9 are endosomal in nature. 12 , 13 These are expressed in alveolar and bronchial epithelial cells, whereas TLR2, TLR4, TLR6 are restricted to the cellular membrane. The TLRs can also be classified based on detecting PAMP. TLR4 detects glycoprotein, TLR7 and TLR8 detects viral single‐stranded ribonucleic acid (ssRNA), TLR3 detects viral double‐stranded RNA (dsRNA), while TLR9 detects viral deoxyribonucleic acid (DNA). 14 The known recognition of genetic material of SARS‐COV‐2 (i.e., ssRNA) occurs through TLR4, TLR7, or TLR8. Interaction of TLRs with PAMP stimulates NF‐κB and IFN regulatory factor (IRF) leading to the production of Type I IFN, which activates the adaptive immune responses considered as the main antiviral response. 15
2.2. TLR4 receptor
TLR4 is a membrane receptor that regulates the inflammatory response by recognizing PAMP as well as DAMP. TLR4 remains attached to alveolar cells and bronchial epithelial cells in the lungs. TLR4 is widely known for its function in recognizing bacterial LPS but their association in SARS‐COV‐2 pathogenesis has been found as well. SARS‐COV‐2 bind with TLR4 and activate transcription factors like AP‐1, NF‐kB, and IRF. TLR4 was also found to regulate IL‐6 via NF‐kB. Some researchers have found oxidized phospholipid (OxPL) mediated TLR4 response in SARS‐COV‐1 infection resulting in cytokine production and lung injury. 15 A few other research studies have found the involvement of TLR4 in SARS‐COV‐2 through molecular docking study revealing the interaction between spike protein and cell surface TLRs. The binding interaction of spike protein and TLR1, TLR4, TLR6 is found significant with the respective binding energy of −57.3, −120.3, and −68.4. 13 , 16 The interaction of spike protein of SARS‐COV‐2 with TLR4 was found to be the highest than other TLRs. However, the reason behind such strong recognition of TLR4 by SARS‐COV‐2 having ssRNA was not found. SARS‐COV‐2 should be recognized by endosomal TLR7 and TLR8 because it has ssRNA; however, due to the concept that immune response always happens on the basis of host‐viral interaction, it interacts with cell surface stimulating the downstream pro‐inflammatory signaling. 9 TLR mediate their function by two pathways MyD88 and toll/IL‐1 receptor/resistance protein domain‐containing adapter‐inducing interferon‐β (TRIF)‐dependent and MyD88 and TRIF‐independent pathways (Figure 1). TLR4 mediates through both the MyD88 and TRIF pathways, TLR3 signals through TRIF, and all the other TLRs mediate through the MyD88 pathway. Activation of MyD88 leads to activation of type‐1 IFN responses and increases the expression of pro‐inflammatory cytokine. Both the pathways contribute to the augmented cytokine in body, directly or indirectly. 13 , 17 , 18 , 19
Figure 1.
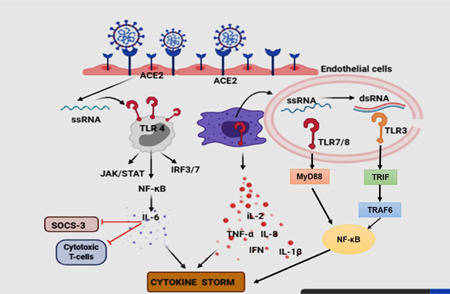
Endothelial cells are loaded with ACE2 receptors and their density depends on the tissue/organ in which they are present. The interaction of ACE2 with SARS‐COV‐2 may mediate the viral entry. Toll‐like receptor 4 (TLR4) is a cell‐surface receptor, whereas TLR3, TLR7, TLR8 are endosomal. TLR4 will induce JAK/STAT, NF‐κB inflammatory signaling, whereas endosomal TLRs also induce similar inflammatory signaling via different adapter molecules. For example, TLR7/8 via MyD88 and TLR3 via TRIF adapter molecule (TRAM)
2.3. TLR7/8 receptor
The virus may get recognition by endosomal‐located TLR7 and activate the MyD88‐dependent MAPK ‐ NF‐κB pathway leading to the production of TNF‐α, ILs, especially IL‐6. The level of IL‐6 is found to be significantly elevated in the serum of SARS‐COV‐2 patients during cytokine storms. 15 , 20 IL‐6 inactivates cell‐mediated antiviral response by inhibiting the expression of cytotoxic T‐cells, SOCS‐3 signaling, and increase in expression of PD‐1 cells. 21 However, the recognition of the ssRNA fragments in SARS‐COV‐2 by TLR7/8 has also been reported. 13 TLR7 and TLR8 are considered phylogenetically and structurally similar, but different TLR7 and TLR8 agonists produce different types of cytokines. The elevated level of pro‐inflammatory cytokines is also found to be mediated by TLR8, proved by bioinformatic analysis. 13 , 22 On the other hand, a few genetic studies have shown a decrease in the level of Type I and Type II IFN and IRF‐7, 22 via missense variant of TLR7 particularly in male patients. 23
2.4. TLR 9 receptor
TLR9 receptors are abundantly expressed on epithelial cells of lungs and nasal mucosa, brain cells, and immune cells. It senses RNA and DNA rich in unmethylated cytosine‐phosphate‐guanine (CpG) sequence. Some viruses as well as human mitochondrial DNA are rich with such motifs, which trigger inflammatory responses via TLR9 receptor‐mediated infection. 24 The expression of TLR9 may increase due to predisposition of genetic factors, gender, and carboxy‐alkyl‐pyrrole protein adducts (CAPs)—mainly released by damaged host cells in response to oxidative stress and known to induce TLR9/Myd88 pathway. 16 , 24 CAP is responsible for platelet activation and its promotion, granule secretion, and thrombosis in‐vivo. TLR9 drives the release of several pro‐inflammatory cytokines such as IL‐6, IL‐1B, IL‐10, IL‐17, TNF‐α, type‐I IFN and may lead to cytokine storm and thrombotic complications. 24
However, other TLRs like TLR3 have been reported to impart protective function in SARS‐COV‐2 infection. Mice having TLR3 gene regulates TRIF‐mediated downstream signaling of the interferon‐stimulated gene (ISG) as a defensive mechanism against SARS‐COV infection, whereas mice having TLR3‐/‐ gene are much prone to the SARS‐COV infection and suffered weight loss and display increased mortality rate. Thus, it was concluded that lacking a TLR3 gene does not render the mice to SARS‐COV infection, but the absence of TRIF‐mediated adaptor molecules (TRAM) signaling renders the mice susceptible to SARS‐COV infection. 25
3. ROLE OF TLR IN CYTOKINE STORM INDUCTION
The innate immune system identifies the antigen‐presenting cells (APC) by recognizing their PAMP. The cells derived from myeloid plays an important role in the immune system including macrophage and monocyte. Macrophages express TLRs, which recognize the invading virus by their nucleic acid pattern (ssRNA). These TLRs include TLR4, TLR7, TLR8, TLR9; however, no specific TLRs have been identified in SARS‐COV‐2 pathogenesis yet. 10 , 26 The binding of TLRs occurs with the virus and may lead to the activation of NF‐κB and MAPK signaling pathways to facilitate viral clearance and produce antiviral responses. To enhance the pathogen elimination, the TLRs also initiate the process of phagocytosis, cytokine release, and various other mediators in order to amplify the local anti‐inflammatory processes as a part of immune regulation. 12 , 16 , 26 The adaptive response shows a fine recognition of virus through T‐cell receptors, which are of two types, cytotoxic T‐cells and T‐helper cells. The cytotoxic T‐cells kill the APC directly and stimulate the local inflammatory processes by inducing the differentiation of T‐helper cells recruiting IFN‐γ and pro‐inflammatory cytokines. 27 The significant enormous release of pro‐inflammatory cytokines during such immune dysregulation is considered to be one of the main reasons for the COVID‐19 pathogenesis leading to cytokine storm and organ failure. 23 The ssRNA of SARS‐COV‐2 also acts as PAMP for the TLR7 receptors, expressed in the endosomes of monocyte‐macrophages and DCs. When the viral ssRNA replicates to form dsRNA, it is recognized by endosomal TLR3. The TLR7‐mediated recognition proceeds through the activation of the JAK/STAT signaling pathway. This may further lead to activation of NF‐κβ, activator protein‐1 (AP‐1), IRF3, IRF7, which ultimately results in amplification of pro‐inflammatory cytokines like IL‐1, IL‐6, IL‐8, TNF‐α, Type‐1 IFN responses. The damage‐associated molecular pattern (DAMP) is sensed by Nod‐like receptors (NLR), which are present in the cytosol of immune cells. Ligation of DAMP with NLRs triggers the release of the inflammasome. 28 These inflammasome are responsible for the conversion of procaspase‐I to active caspase‐I, which in turn activate pro‐IL‐1β into IL‐1β (Figure 2). This activates a process of cell pyroptosis. IL‐1β is the main activator of IL‐6 and contributes to the aggravation of cytokine storms. 15 , 29 , 30 , 31
Figure 2.
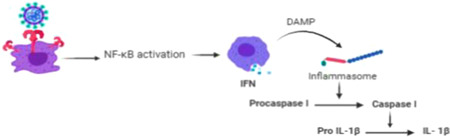
NF‐κB activation occurs due to toll‐like receptor (TLR)‐mediated inflammation results in DAMP production. The ligation of the DAMP with nod‐like receptor (NLR) produces inflammasomes, which activate the pro‐caspase I into active caspase I, ultimately activating interleukin (IL)‐1β
TLRs are solely responsible for mediating innate immune signaling. It is no wonder that blocking of TLR could suppress IFN‐related signaling, which could possibly increase the viral load, but in cases of preexisting chronic autoimmune diseases, where the immunity system is hyperactive like cancer, SLE, rheumatoid arthritis, and so forth, 4 TLR7, 8, 9 antagonists may prove to show their benefits. As SARS‐COV‐2 targets TLR3, 4, 7, 8, 9 receptors to enter the host system, hence TLR blockers may prove to be beneficial in this deadly infection. 8 There are some drugs like hydroxychloroquine under Phase III clinical trial (NCT04448756) that has shown the endosomal TLR signaling (TLR3, 7, 8, 9) inhibition, which possibly could delimit the viral entry into the host; whereas M5049 developed by Merck, Germany, is under Phase II trial have also shown the inhibition of TLR7 and TLR8 by recognizing the genomic RNA of invading virus. 32 Hence, by scrutinizing both the limitations as well as the benefits of TLR blockers, it can be said that it may not be a fruitful approach to completely block all TLRs nonspecifically. Specific targeting of TLR receptors, the treatment period, and the dose of TLR antagonists need to be optimized so that they must be efficient enough in reducing the release of inflammatory mediators thus mediating the cytokine storm effect in COVID‐19 patients. However, the basal activity of TLRs should also be maintained for the regulation of the immune homeostasis and prevention of opportunistic infections. 33 , 34
4. MULTIORGAN FAILURE
The population severely infected with SARS‐COV‐2 also suffered through extrapulmonary organ damage and organ failure, ultimately leading to death. Some comorbid risk factors such as coronary heart disease, hypertension, diabetes, kidney infection, and so forth. highly contributed to the death of the patients during COVID‐19. 4 , 8 SARS‐COV‐2 damages the pulmonary tissue and induce high levels of cytokines and chemokines that are released into the systemic circulation and migrate to the other vital organs like the kidney, pancreas, liver, heart and even brain, leading to damage to multiple organs (Figure 3).
Figure 3.
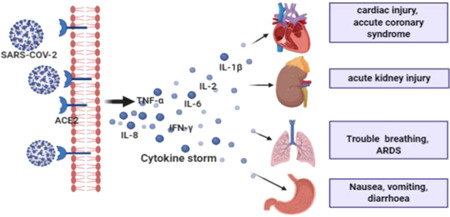
The cytokine storm originates in the lungs and is released into the bloodstream. After it reaches the systemic circulation, it produces hyperinflammation and imparts damage to the vital organs. In critically ill patients such inflammation spread into various other organs as well, may cause multiple organ failure
4.1. ACE2 polymorphism in multiorgan failure
One of the main mechanisms of such multiorgan damage is postulated as ACE2 polymorphism. 4 , 5 , 35 ACE2 is normally present in both Type 1 and Type 2 alveolar cells in lungs, epithelial cells of the oral mucosa, peripheral myocytes in cardiac cells, pancreas, enterocytes of the small intestine and are highly expressed on hemopoietic cells, also in monocytes and macrophages. 36 , 37 , 38 ACE2 acts as the port of entry of ssRNA virus into the human and plays a protective mechanism under free concentration. There a certain kind of pattern can be seen in SARS‐COV‐2 infection, be its high transmissibility, or its higher risk of infection in a certain population and rendering to multiorgan damage. The high transmissibility of SARS‐COV‐2 may be due to two reasons 36 , 38 , 39 : first, due to the high binding of SARS‐COV‐2 with ACE2 receptor in host cells, which was not observed earlier; and second, the presence of four amino acid residues between two subunits of spike protein (S): S1 and S2, which leads to the introduction of a novel “furin cleavage site.” The exact function of this cleavage site is yet to be known, but due to the expression of the furin proteases, it facilitates the processing of the spike protein subunit, which activates the binding of SARS‐COV‐2 with the ACE2 receptor. 4 , 35 These two reasons have a major impact on the expansion of SARS‐COV‐2 in other tissue or organ contributing to organ damage. Also, multiple organ failure has been observed in male patients more widely than that of females, particularly in aged individuals. This is because ACE2 is encoded by a gene expression on X‐chromosomes. Females possess a pair of X‐chromosomes; hence a much stronger immune mechanism is found in them, even if one of the X‐chromosomes stays inactivated. The expression of TLR7 is also higher in females, providing them antiviral response by producing a large amount of antibodies by TLR7, as a result of which they possess a much stronger immunity than males, and can fight against single‐stranded SARS‐COV‐2. 5 However, due to a single X‐chromosomes male are more prone to the ssRNA SARS‐COV‐2. In a recent study through single‐cell analysis, it has been found that Asian men have higher expression of ACE2 receptors in the lungs than women, hence making them more prone to SARS‐COV‐2 infection and disease severity. The expression of ACE2 increases with age, which is one of the possibilities why old people are more affected than young people. 5 However, a case report presented a frequent visitor young man of China seafood market, who was found to be severely affected by SARS‐COV‐2 infection and suffered multiple organ damage. 40 The patient was a nonsmoker and had no history of any chronic disease.
4.2. Acute respiratory distress syndrome (ARDS)
ARDS is one of the most common and serious complications of SARS‐COV‐2 infections. The severity of patients with pulmonary infection increased with increased TLR4 signaling on the endothelial membrane. 15 , 39 Upon entry of the virus, it is recognized by TLR, which initiates the intracellular inflammation signaling either by MyD88 or TRIF depending upon the specific type of TLR recognition, resulting in activation of transcription factors like NF‐κB and IRFs. All these results in release of the pro‐inflammatory cytokines in the lungs such as IL‐1, IL‐8, IL‐6, IL‐12, IFN‐γ, and chemokines like CXCL‐9, CXCL‐10, CCL‐2, CCL‐5. These pro‐inflammatory mediators have been found in a detectable quantity in the serum of the severely infected patient. When these inflammatory molecules are released into the systemic circulation via alveolar wall injury, it migrates to other tissues and causes systemic inflammation. 41 , 42 , 43 Apart from the hyperimmune response, there are some other molecular mechanisms that fully contribute to the inflammation and lung injury in a SARS‐COV‐2 infected patient such as endothelial dysfunction, intravascular coagulopathy, thrombosis ultimately hypoxemia. 44
The renin‐angiotensin‐system (RAS) and its receptor, ACE2 are highly correlated with invasion and initiation of inflammation associated with COVID‐19 in the lungs. 41 Angiotensin I (Ang I) is an inactive peptide that gets activated to Ang II by ACE activity. Ang II a potent vasoconstrictor, has an important role in maintaining blood pressure and in cell proliferation by mediating the release of pro‐inflammatory cytokines. Ang II gets converted to Ang (1–7) by ACE2. 4 Under normal circumstances, Ang II directly binds to the NF‐κB and expresses inflammatory cytokines; whereas Ang (1–7) maintains the anti‐inflammatory response by upregulation of P13K/Akt and ERK signaling. But due to the binding of SARS‐COV‐2 with ACE2 receptors, the free concentration of ACE2 decreases, and as a result, Ang II will not get converted into Ang (1–7) leading to the accumulation of Ang II. This may further augment inflammation and pulmonary injury. 43 , 45
Endothelial dysfunction is a result of cytokine storm, which activates the complement cascade, increasing vascular permeability and vasculitis. Due to the wide expression of ACE2 receptors on the endothelial cell surface, the virus entry into the host becomes easier. The complement activation is a defensive mechanism and will take place via the lectin pathway and followed by the classical pathway, resulting in the accumulation of complement 3b (C3b), which triggers the formation of C5 and its product C5a to C5b‐9. 46 Apart from inducing vascular inflammation, C5a and C5b‐9 promote thrombomodulin loss, P‐selectin exocytosis and von‐Willebrand factor triggering coagulation cascade. This may further result in vascular injury and platelet aggregation. 46 In a postmortem autopsy studies of COVID‐19 patient (n = 7), a higher deposition of platelet in the lung and heart vessels have been demonstrated. Several biomarkers have been identified for the ARDS exclusively, including pro‐inflammatory cytokines (IL‐6, IL‐8), TNF‐α, surface protein‐D, von‐Willebrand factor antigen, plasminogen‐activator inhibitor‐1, which were found significantly elevated in the critically ill COVID‐patients. 3 , 47 , 48 , 49
The induction of hypoxemia is a secondary response to ARDS but equally contributes to pulmonary inflammation, it causes the upregulation of hypoxia‐inducible factor‐1α (HIF‐1α), vascular adhesion molecule‐1 (VCAM), and other adhesion molecules, increasing the adhesiveness of leukocytes and endothelial cells. 23 , 43 , 44 This phenomenon not only causes the infiltration of inflammatory molecules and amplifies their response, but also may lead to upregulation of TLR4 signaling pathway.
4.3. TLR‐induced cardiac disease
Severe cases of COVID‐19 may cause cardiovascular disorders like myocardial injury, acute coronary syndrome, thromboembolism, cardiac failure. 33 The cardiac symptoms could be a clinical symptom for the patients who are not showing the symptoms of SARS‐COV‐2 infection like dry cough and fever. Cardiac pericytes have a higher expression of ACE2 than cardiomyocytes. 35 Cardiac pericytes are responsible for endothelial stability, binding of SARS‐COV‐2 causes endothelial dysfunction and results in microcirculatory disorders. Hence, despite low expression of ACE2, the patient suffers cardiac injury. Patients with a history of cardiac failure have highly expressed ACE2, hence are more susceptible to cardiac injury. 33 , 48 , 50
TLR4 is expressed on cardiomyocytes, fibroblasts, and macrophages. Among all the TLRs, TLR4 is the most widely expressed on cardiomyocytes. 35 An association between TLR4 and Ang II is known to exist. Ang II is found to mediate vascular dysfunction through TLR4. SARS‐COV‐2 downregulates the ACE2 receptor, which decreases the conversion of ACE2 into Ang (1–7). Ang (1–7) has efficacy in opposing pro‐inflammatory, profibrotic, vasoconstriction activity, and its decreased level results in exaggeration of the cytokine storm. 51 TLR4 is also associated with high blood pressure in several models depicting hypertension. The pro‐inflammatory response to Ang II has been mediated by TLR7/8/9 in response to the exogenous ligand in spontaneously hypertensive rats. TLR9 is considered as a negative regulator for cardiac vagal tone as well as for baroreflex function, activation of TLR9 results in the increment in blood pressure and vascular dysfunction in a normotensive rat. 18 Hence, TLR9 activation through SARS‐COV‐2 may result in vascular dysfunction. TLR activation was also found to mediate calcium homeostasis in cells. 24 Activation of TLR4, TLR7, TLR9 in macrophages increases the calcium efflux and activates the calcium calmodulin protein kinase. The calcium calmodulin protein kinase releases a large amount of pro‐inflammatory cytokines and IFNs by both MyD88 and TRIF‐mediated pathways. TLR4 is shown to have a synergistic effect in the augmented efflux of calcium resulting in a decreased level of sarcoplasmic storage of calcium level, eventually decreasing the cardiac contractility. This effect has a direct relation with hypertension and vascular dysfunction, of which both factors contribute to organ damage. 12 , 16
4.4. TLR‐mediated kidney injury
ACE2 are highly expressed on proximal convoluted tubule (PCT), and absent in glomerular endothelial cells of the kidney. The direct effect of viral entry causing acute kidney injury (AKI) has not been evidenced yet. AKI has been seen widely in patients with ARDS suggesting lung–kidney crosstalk postinfection. 20 , 52 The possible mechanism was described as hemodynamic effect, hypoxemia, and hyperinflammation associated with ARDS. AKI may increase the levels of inflammatory cytokines production, especially IL‐6, and decrease their clearance. IL‐6 causes infiltration of neutrophils and macrophages, which increases lung permeability. 37 , 53 The viral entry showed the activation of the complement system in the kidney. The complement C5a is responsible for neutrophil attraction to the sites, platelet aggregation, and endothelial injury. Altogether these damaging effects may contribute to the worsening of AKI. In the kidney biopsy studies, platelets adhesion along with fibrin were seen in peritubular capillaries and venules. TLR4 mRNA expression has been found on endothelial cells as well as tubular cells of the kidney, activation of which by SARS‐COV‐2 could contribute to tubular necrosis, ischemia, inflammation, and injury. 20 , 53 However, a direct effect of cytokine storm has shown its effect on renal epithelial cells along with coagulopathy and hemodynamic changes. 12 , 46
Not only these vital organs are susceptible to SARS‐COV‐2, but also it has an impact on the metabolism of the host, it may also affect the gastric system causing diarrhea and vomiting postinfection. It also affects patients with diabetes mellitus, as it messes with the metabolism of the host, causing glucotoxicity and lipotoxicity.
5. CYTOKINE STORM IN COVID‐19
The cytokine storm refers to the abnormal increment in the level of pro‐inflammatory cytokines following the viral invasion as a result of the hyperactive immune response. An abnormal high level of IL‐6, IL‐1, TNF‐α has been observed in intensive care unit (ICU) patients in comparison to non‐ICU patients. 1 , 36 The main cytokines involved are ILs, IFNs, TNF‐α, colony‐stimulating factor (CSF), chemokine family, growth factor (GF), and so forth. These cytokines are classified as pro‐inflammatory factors (such as IL‐1β, IL‐6, IL‐12, TNF‐α, IFN‐γ) and anti‐inflammatory factors (such as IL‐4, IL‐10, IL‐13, TGF‐β) based on their functions and their level of production to a stimulus. 39 , 54 , 55 , 56
A viral infection proceeds with three phases: (1) initiation phase, (2) resolution phase, (3) restoration phase. In the initiation phase, the SARS‐COV‐2 infects the host by the incorporation of ACE2 receptors present on the epithelial cells of multiple organs like lung, kidney, pancreas, and so forth. It enters the host and induces acute inflammation. Under normal conditions, the natural killer (NK) cells of innate immunity and T‐cell of adaptive immune response have the tendency to cause the apoptosis of APC and prevent unnecessary activation. 29 Hence a balanced immune regulation is maintained, but any alterations in the lymphocyte catalytic activity due to the acquired infection in SARS‐COV‐2 infected patients dysregulate the immune balance by inactivating the NK cells as well cytotoxic T‐cells to kill the APC, leading to the dysregulated interactions of innate and adaptive immunity. 14 This results in the release of several cytokines at the site of infection (primarily lungs). The abnormal secretion of cytokines is termed as “cytokine storm.” In the restoration phase, the barrier integrity of the lung is restored in other viral infections, unlike SARS‐COV‐2, where lung‐barrier integrity, as well as vascular permeability, is lost due to the cytokine storm, releasing the loads to cytokine into the systemic circulation. Hence wherever it reaches from the lung to other organs such as the kidney, heart, pancreas, and so forth, it may stimulate an exaggerated inflammation and cause damage to that organ. 29 , 57 Hence, it can be said that the cytokine storms are a crucial cause of ARDS, systemic inflammatory response, and multiple organ failure in SARS‐COV‐2. Based on these consequences, the cytokine storm has been categorized into two stages: the temporary immune‐deficient condition is described as the first stage, whereas the hyperactive immune state resulting due to the compensatory mechanism to eliminate the virus, failure of which spike up the cytokine levels, is considered as the second stage.
The cytokine storm can also be mediated either by ACE2‐mediated inflammatory response, by cell pyroptosis or by the delayed release of Type I IFNs. Cell pyroptosis is caused due to programmed cell deaths in SARS‐COV‐2, which occurs by activation of NLRP3 inflammasomes by viroporin 3, a protein from coronavirus having a role in IL‐1β production. 10 , 54 , 58 Delayed release of type‐I IFN is due to the activation of the JAK/STAT pathway initiating the transcription of the ISG and driving the production of cytokines like IL‐1β, IL‐6, and so forth, which are mainly pro‐inflammatory in nature. 59 This IL‐1β is highly responsible for the induction of bronchiolar and alveolar inflammation, whereas both the IL‐1B and IL‐6 activate the complement cascade that increases the vascular permeability. 56
5.1. IL‐6: The driving factors for cytokine storm
IL‐6 is a pleiotropic molecule that plays an important role in immune response as a pro‐inflammatory molecule. They are produced by almost all the immune cells like macrophages, monocytes, T‐cells, B‐cells as well as nonimmune cells like fibroblasts and epithelial cells. TLR‐7 mediated virus evasion into the host induces IL‐6 leading to hyperactive immune response in infected patients. These effects may have a protective role in other viral infections as evident in some murine model. However, 15 it has been found that IL‐6 is the major cause of hyper‐inflammation and death in SARS‐COV‐1 mice model. IL‐6 shows its anti‐inflammatory effects by classical (Cis) signaling cascades, whereas pro‐inflammatory effect is shown by the trans‐signaling pathway, prominent in case of viral infection (Figure 4). Cis signaling involves gp130 and membrane‐bound mIL‐6Rs, mainly restricted to immune cells; whereas trans‐signaling involves sIL‐6R (soluble form of IL‐6R). sIL‐6R remain soluble in blood and urine and form a complex with gp130, expressed in both hemopoietic and nonhemopoietic types of cells. The activation of gp130 by mIL‐6R activates STAT3; nuclear translocation of which results in the translation of pro‐inflammatory genes. Hence activation of cell other than the cells that express mIL‐6R like endothelial cells would result in an elevated level of IL‐6, which increase the vascular permeability and hence induces the release of pro‐inflammatory cytokine to the systemic circulation contributing to the cytokine storm. 15 , 18 A recent meta‐analysis of nine studies reported an increment in the level of IL‐6 in patients with COVID‐19 severity (p = 0.05). Some clinical reports suggested that critically ill patients with SARS‐COV‐2 have an abnormal increment in the level of pro‐inflammatory cytokines, which is possibly due to IL‐6‐driven immune dysregulation or macrophage‐activation‐syndrome. 15
Figure 4.
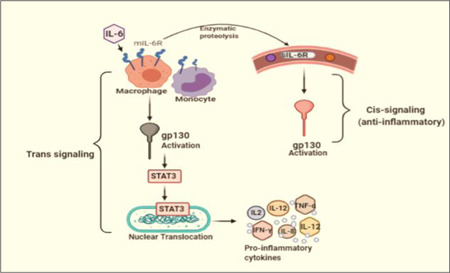
The cis‐ and trans‐signaling pathway of interleukin (IL)‐6: The cis signaling is mediated by soluble IL‐6R, whereas trans‐signaling is mediated by membrane‐bound IL‐6. Trans‐signaling proceeds with STAT3 activation, transcription of which would result in the synthesis and release of cytokines. However, both the signaling involves gp130 activation
IL‐6 may predict the disease severity in the patient who does not show any typical symptoms of COVID‐19 or are a poor prognosis of the disease. Hence it may be used as a biomarker in COVID‐19. Some other biomarkers of this disease are serum amyloid A (SAA), C‐reactive protein (CRP), and procalcitonin (Table 1). Some reports have shown a correlation (R = 0.902) between serum viral load (RNAaemia) and the occurrence of cytokine storm, which is directly related to the level of IL‐6. Apart from highly elevated IL‐6, a low count of lymphocytes and thrombocytes may also predict disease severity in COVID‐19. 18 , 20
Table 1.
Biomarkers associated with cytokine storm in COVID‐19
| Biomarker | Biology | Level in infected patients | References | |
|---|---|---|---|---|
| Complete blood count biomarkers | Neutrophil to lymphocyte ratio | Hemopoietic marker | Increase | Ye et al. 60 |
| Platelets count (cells/µl) | Hemopoietic marker | Decrease | Malik et al. 49 | |
| Metabolic biomarker | Aspartate transaminase (AST) | Hepatic injury | Increase | Ponti et al. 61 |
| Alanine transaminase (ALT) | Hepatic injury | Increase | Ponti et al. 61 | |
| Creatinine | Kidney infection | Increase | Kermali et al. 37 | |
| Cardiac troponin | Cardiac injury | Increase | Kermali et al. 37 | |
| Inflammatory biomarker | C‐reactive protein (CRP) (mg/L) | Plasma protein | Increase | Samprathi & Jayashree 3 |
| Procalcitonin (PCT) | Proinflammatory peptides | Increase | Henderson et al. 54 | |
| Interleukin‐2R (U/ml) | Cytokines | Increase | Kaur et al. 47 | |
| IL‐6 (pg/ml) | Cytokines | Increase | Chaudhary et al. 48 | |
| Serum amyloid A (SAA) (mg/L) | Acute‐phase protein | Increase | Kaur et al. 47 | |
| Lactate dehydrogenase (LDH) | Tissue injury | Increase | Samprathi & Jayashree 3 | |
| Others | d‐dimer (mg/dl) | Fibrin degradation product | Increase | Keddie et al. 36 |
6. CONCLUSION
Due to the imbalanced and hyper‐responsive immune system, a surge in the cytokines has been seen in patients, leading to the death of the infected in COVID‐19. It has been observed that cytokine surge is TLR induced, mainly through activation of TLR3, TLR4, TLR7, TLR8 receptors. However, the exact role of TLRs in the induction of cytokine storms has not been confirmed yet. The cytokine storm migrates into the other organ through the systemic circulation. The inflammation and the organ damage occur due to the TLR mediated NF‐κB, MAPK pathway. Hence blocking these specific TLRs may alleviate the chance of SARS‐COV‐2 infection, there are two TLR blockers hydroxyquione sulfate (NCT04448756) and M5049 32 under Phase III and Phase II of clinical trial, respectively. However, as these are the key players of innate immunity, these receptors should not be completely blocked. Targeting TLRs subtypes with the optimized dose and optimized time period of treatment might be one of the beneficial therapeutic strategies.
The TLR induced organ damage has also been related to ACE2 polymorphism and its expression across patient gender and age group. Recently WHO has reported MIS‐C in children, hence the reason behind this exception also needs to be explored.
CONFLICT OF INTERESTS
The authors declare that there are no conflict of interests.
AUTHOR CONTRIBUTIONS
Moumita Manik has prepared the first draft. Rakesh K. Singh has conceptualized, read, and modified the final draft of the manuscript for submission. Both the authors have read and finalized the submitted version of the manuscript.
Manik M, Singh RK. Role of toll‐like receptors in modulation of cytokine storm signaling in SARS‐CoV‐2‐induced COVID‐19. J Med Virol. 2022;94:869‐877. 10.1002/jmv.27405
DATA AVAILABILITY STATEMENT
Data are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Wang HY, Li XL, Yan ZR, Sun XP, Han J, Zhang BW. Potential neurological symptoms of COVID‐19. Ther Adv Neurol Disord. 2020;13:1756286420917830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Naji HS. Cytokine storm of SARS‐CoV‐2, the virus that causes COVID‐19. European J Med Health Sci. 2020;2(3):245. [Google Scholar]
- 3. Samprathi M, Jayashree M. Biomarkers in COVID‐19: an up‐to‐date review. Front Pediatr. 2020;8:607647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Devaux CA, Rolain JM, Raoult D. ACE2 receptor polymorphism: susceptibility to SARS‐CoV‐2, hypertension, multi‐organ failure, and COVID‐19 disease outcome. J Microbiol Immunol Infect. 2020;53:425‐435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhao Y, Zhao Z, Wang Y, Zhou Y, Ma Y, Zuo W. Single‐cell RNA expression profiling of ACE2, thereceptor of SARS‐CoV‐2. Am J Respir Crit Care Med. 2020.202(5):756‐759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ziaee V, Assari R, Mamishi S, Zeinaloo A, Mohammadpour M, Malekzadeh I. An algorithmic approach to multisystem inflammatory syndrome in children with COVID‐19: Tehran Children's Medical Center Protocol. Iran J Ped. 2020;30:1‐9. [Google Scholar]
- 7. Sau S, Kumar A. COVID‐19 altered immune signalling pathways. Science. 2021;12:64‐75. [Google Scholar]
- 8. Yazdanpanah F, Hamblin MR, Rezaei N. The immune system and COVID‐19: friend or foe? Life Sci. 2020;256:117900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Choudhury A, Mukherjee S. In silico studies on the comparative characterization of the interactions of SARS‐CoV‐2 spike glycoprotein with ACE‐2 receptor homologs and human TLRs. J Med Virol. 2020;92:2105‐2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Livia O, Caraglia M, Facchini G, Margherita V, De Placido S, Buonerba C. Toll‐like receptors and COVID‐19: a two‐faced story with an exciting ending. Future Sci OA. 2020;6(8):FSO605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. El‐Zayat SR, Sibaii H, Mannaa FA. Toll‐like receptors activation, signaling, and targeting: an overview. Bull Nat Res Centre. 2019;43:187. [Google Scholar]
- 12. Goulopoulou S, McCarthy CG, Webb RC. Toll‐like receptors in the vascular system: sensing the dangers within. Pharmacol Rev. 2016;68:142‐167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Moreno‐Eutimio MA, Lopez‐Macias C, Pastelin‐Palacios R. Bioinformatic analysis and identification of single‐stranded RNA sequences recognized by TLR7/8 in the SARS‐CoV‐2, SARS‐CoV, and MERS‐CoV genomes. Microb Infect. 2020;22:226‐229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Choi Y, Bowman JW, Jung JU. Autophagy during viral infection–a double‐edged sword. Nat Rev Microbiol. 2018;16:341‐354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Magro G. SARS‐CoV‐2 and COVID‐19: is interleukin‐6 (IL‐6) the'culprit lesion'of ARDS onset? What is there besides Tocilizumab? SGP130Fc. Cytokine: X. 2020;2:100029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hennessy EJ, Parker AE, O'Neill LA. Targeting toll‐like receptors: emerging therapeutics? Nat Rev Drug Discov. 2010;9:293‐307. [DOI] [PubMed] [Google Scholar]
- 17. Cuevas AM, Clark JM, Potter JJ. Increased TLR/MyD88 signaling in patients with obesity: is there a link to COVID‐19 disease severity? Int J Obes (Lond). 2021;45:1152‐1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Roshanravan N, Seif F, Ostadrahimi A, Pouraghaei M, Ghaffari S. Targeting Cytokine Storm to Manage Patients with COVID‐19: A Mini‐Review. Arch Med Res. 2020;51:608‐612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sohn KM, Lee S‐G, Kim HJ, et al. COVID‐19 patients upregulate toll‐like receptor 4‐mediated inflammatory signaling that mimics bacterial sepsis. J Korean Med Sci. 2020. 35(38):e343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Su H, Lei CT, Zhang C. Interleukin‐6 Signaling Pathway and Its Role in Kidney Disease: An Update. Front Immunol. 2017;8:405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ahmadpoor P, Rostaing L. Why the immune system fails to mount an adaptive immune response to a COVID‐19 infection. Transpl Int. 2020;33:824‐825. [DOI] [PubMed] [Google Scholar]
- 22. Fallerini C, Daga S, Mantovani S. et al. Association of Toll‐like receptor 7 variants with life‐threatening COVID‐19 disease in males: findings from a nested case‐control study Elife. 2021;10:e67569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mustafa MI, Abdelmoneim AH, Mahmoud EM, Makhawi AM. Cytokine storm in COVID‐19 patients, its impact on organs and potential treatment by QTY code‐designed detergent‐free chemokine receptors. Mediators Inflamm. 2020;2020:8198963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bezemer GFG, Garssen J. TLR9 and COVID‐19: A multidisciplinary theory of a multifaceted therapeutic target. Front Pharmacol. 2020;11:601685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Totura AL, Whitmore A, Agnihothram S, et al. Toll‐like receptor 3 signaling via TRIF contributes to a protective innate immune response to severe acute respiratory syndrome coronavirus infection. mBio. 2015;6:e00638‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Catanzaro M, Fagiani F, Racchi M, Corsini E, Govoni S, Lanni C. Immune response in COVID‐19: addressing a pharmacological challenge by targeting pathways triggered by SARS‐CoV‐2. Signal Transduct Target Ther. 2020;5:84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hasan MZ, Islam S, Matsumoto K, Kawai T. SARS‐CoV‐2 infection initiates interleukin‐17‐enriched transcriptional response in different cells from multiple organs. Sci Rep. 2020;11:16814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Libby P. Inflammatory mechanisms: the molecular basis of inflammation and disease. Nutr Res. 2007;65:S140‐S146. [DOI] [PubMed] [Google Scholar]
- 29. McGonagle D, Sharif K, O'Regan A, Bridgewood C. The role of cytokines including interleukin‐6 in COVID‐19 induced pneumonia and macrophage activation syndrome‐like disease. Autoimmun Rev. 2020;19:102537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mishra CB, Pandey P, Sharma RD, et al. Identifying the natural polyphenol catechin as a multi‐targeted agent against SARS‐CoV‐2 for the plausible therapy of COVID‐19: an integrated computational approach. Brief Bioinform. 2021;22:1346‐1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Uchiyama R, Tsutsui H. Caspases as the key effectors of inflammatory responses against bacterial infection. Arch Immunol Ther Exp. 2015;63:1‐13. [DOI] [PubMed] [Google Scholar]
- 32. Vlach J, Bender AT, Przetak M, et al. Discovery of M5049: a novel selective toll‐like receptor 7/8 inhibitor for treatment of autoimmunity. J Pharmacol Exp Ther. 2021;376:397‐409. [DOI] [PubMed] [Google Scholar]
- 33. Nishiga M, Wang DW, Han Y, Lewis DB, Wu JC. COVID‐19 and cardiovascular disease: from basic mechanisms to clinical perspectives. Nat Rev Cardiol. 2020:1‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Conti P, Younes A. Coronavirus COV‐19/SARS‐CoV‐2 affects women less than men: clinical response to viral infection. J Biol Regul Homeost Agents. 2020;34:71. [DOI] [PubMed] [Google Scholar]
- 35. Aboudounya MM, Heads RJ. COVID‐19 and toll‐like receptor 4 (TLR4): SARS‐CoV‐2 may bind and activate TLR4 to increase ACE2 expression, facilitating entry and causing hyperinflammation. Mediators Inflamm. 2021;2021:8874339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Keddie S, Ziff O, Chou MKL, et al. Laboratory biomarkers associated with COVID‐19 severity and management. Clin Immunol. 2020;221:108614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kermali M, Khalsa RK, Pillai K, Ismail Z, Harky A. The role of biomarkers in diagnosis of COVID‐19–a systematic review. Life Sci. 2020;254:117788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kim JS, Lee JY, Yang JW, et al. Immunopathogenesis and treatment of cytokine storm in COVID‐19. Theranostics. 2021;11:316‐329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pelaia C, Tinello C, Vatrella A, De Sarro G, Pelaia G. Lung under attack by COVID‐19‐induced cytokine storm: pathogenic mechanisms and therapeutic implications. Ther Adv Respir Dis. 2020;14:1753466620933508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zeng Y, Zhou X, Gang Y, et al. A fatal outcome from SARS‐CoV‐2 infection: One case report of a young man with multiple organ damage. Radiol Infect Dis. 2020;7:208‐212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Giménez VMM, Inserra F, Tajer CD, et al. Lungs as target of COVID‐19 infection: protective common molecular mechanisms of vitamin D and melatonin as a new potential synergistic treatment. Life Sci. 2020;254:117808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ojo AS, Balogun SA, Williams OT, Ojo OS. Pulmonary Fibrosis in COVID‐19 Survivors: Predictive Factors and Risk Reduction Strategies. Pulm Med. 2020;2020:6175964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tao S‐L, Wang X‐m, Feng Y‐g, et al. Is the presence of lung injury in COVID‐19 an independent risk factor for secondary lung cancer? Med Hypotheses. 2020;143:110074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rapkiewicz AV, Mai X, Carsons SE, et al. Megakaryocytes and platelet‐fibrin thrombi characterize multi‐organ thrombosis at autopsy in COVID‐19: a case series. EClinicalMedicine. 2020;24:100434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Uzzan M, Corcos O, Martin JC, Treton X, Bouhnik Y. Why is SARS‐CoV‐2 infection more severe in obese men? The gut lymphatics–lung axis hypothesis. Med Hypotheses. 2020;144:110023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Noris M, Benigni A, Remuzzi G. The case of complement activation in COVID‐19 multiorgan impact. Kidney Int. 2020;98:314‐322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kaur M, Tiwari S, Jain R. Protein based biomarkers for non‐invasive Covid‐19 detection. Sens Biosensing Res. 2020;29:100362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chaudhary R, Garg J, Houghton DE, et al. Thromboinflammatory biomarkers in COVID‐19: systematic review and meta‐analysis of 17,052 patients. Mayo Clin Proc Innov Qual Outcomes. 2021;5:388‐402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Malik P, Patel U, Deep Mehta NP, Kelkar R, Muhammad Akrmah JLG, Sacks H. Biomarkers and outcomes of COVID‐19 hospitalisations: systematic review and meta‐analysis. BMJ Evid Based Med. 2021:26(3):107‐108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Santoso A, Pranata R, Wibowo A, Al‐Farabi MJ, Huang I, Antariksa B. Cardiac injury is associated with mortality and critically ill pneumonia in COVID‐19: A meta‐analysis. Am J Emerg Med. 2021;44:352‐357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tersalvi G, Vicenzi M, Calabretta D, Biasco L, Pedrazzini G, Winterton D. Elevated troponin in patients with coronavirus disease 2019: possible mechanisms. J Card Fail. 2020;26:470‐475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ahmed AR, Ebad CA, Stoneman S, Satti MM, Conlon PJ. Kidney injury in COVID‐19. World J Nephrol. 2020;9:18‐32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Leemans JC, Kors L, Anders H‐J, Florquin S. Pattern recognition receptors and the inflammasome in kidney disease. Nat Rev Nephrol. 2014;10:398‐414. [DOI] [PubMed] [Google Scholar]
- 54. Henderson LA, Canna SW, Schulert GS, et al. On the Alert for Cytokine Storm: Immunopathology in COVID‐19. Arthritis Rheumatol. 2020;72:1059‐1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Herr C, Mang S, Mozafari B, et al. Distinct patterns of blood cytokines beyond a cytokine storm predict mortality in COVID‐19. J Inflamm Res. 2021;14:4651‐4667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hu B, Huang S, Yin L. The cytokine storm and COVID‐19. J Med Virol. 2021;93(1):250‐256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Pooladanda V, Thatikonda S, Godugu C. The current understanding and potential therapeutic options to combat COVID‐19. Life Sci. 2020;254:117765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Gupta R, Hussain A, Misra A. Diabetes and COVID‐19: evidence, current status and unanswered research questions. Eur J Clin Nutr. 2020;74:864‐870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Rebe C, Vegran F, Berger H, Ghiringhelli F. STAT3 activation: a key factor in tumor immunoescape. JAKSTAT. 2013;2:e23010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ye G, Pan Z, Pan Y, et al. Clinical characteristics of severe acute respiratory syndrome coronavirus 2 reactivation. J Infect. 2020;80(5):e14‐e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ponti G, Maccaferri M, Ruini C, Tomasi A, Ozbend T. Biomarkers associated with COVID‐19 disease progression. Crit Rev Clin Lab Sci. 2020: 1‐11. 10.1002/jmv.27405. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data are available from the corresponding author upon reasonable request.