

Abstract
This review makes a critical evaluation of 61 peer‐reviewed manuscripts that use a docking step in a virtual screening (VS) protocol to predict SARS‐CoV‐2 M‐pro (M‐pro) inhibitors in approved or investigational drugs. Various manuscripts predict different compounds, even when they use a similar initial dataset and methodology, and most of them do not validate their methodology or results. In addition, a set of known 150 SARS‐CoV‐2 M‐pro inhibitors extracted from the literature and a second set of 81 M‐pro inhibitors and 113 inactive compounds obtained from the COVID Moonshot project were used to evaluate the reliability of using docking scores as feasible predictors of the potency of a SARS‐CoV‐2 M‐pro inhibitor. Using two SARS‐CoV‐2 M‐pro structures and five protein‐ligand docking programs, we proved that the correlation between the pIC50 and docking scores is not good. Neither was any correlation found between the pIC50 and the ∆G calculated with an MM‐GBSA method. When a group of experimentally known inactive compounds was added, neither the docking scores or the ∆G were able to distinguish between compounds with or without M‐pro experimental inhibitory activity. Performances improved when covalent and noncovalent inhibitors were treated separately, but were not good enough to fully support using a docking score as a cutoff value for selecting new putative M‐pro inhibitors or predicting the relative bioactivity of a compound by comparison with a reference compound. The two sets of known SARS‐CoV‐2 M‐pro inhibitors presented here could be used for validating future VS protocols which aim to predict M‐pro inhibitors.
Keywords: drug repurposing, molecular docking, M‐pro inhibitors, SARS‐CoV‐2 3C protease, virtual screening
1. INTRODUCTION
It has been more than a year since the onset of the coronavirus disease 2019 (COVID‐19) global pandemic, which has affected 219 countries and territories around the world. According to the Johns Hopkins Coronavirus map tracker, 1 there have been more than 123 million cases worldwide. During the last year, more than 100 million people have recovered from the disease while over 2.7 million have died. 2 The pandemic has had a detrimental effect on healthcare systems worldwide and has affected almost all aspects of life. When the World Health Organization declared the COVID‐19 outbreak a global emergency, governments started to enforce border shutdowns, travel restrictions, quarantines, curfews and other restrictions, which in turn fuelled an impending economic crisis and recession. 3 The scientific community reacted to the COVID‐19 global pandemic in an outstandingly rapid and effective way, and thousands of articles were published in the first few months. Between January and May 2020, roughly 100 new articles on COVID‐19 were added every day to PubMed, 4 with a median time from submission to acceptance of only 6 days. 5 Although the nature of the COVID‐19 emergency warranted accelerated publishing, measures need to be taken to safeguard the integrity of the scientific evidence. 5 There is concern among some scientists that poor quality research prevents an effective evidence‐based response. 6 , 7
Coronaviruses (CoVs) are a group of viruses containing a linear, positive sense single‐stranded RNA genome. 8 Their genome size varies, but is larger than that of other types of virus. The genome is housed within an enveloping lipid bilayer in which three structural proteins—membrane (M), envelope (E) and spike (S)—are anchored. The E and M proteins are responsible for the shape of the virus, while the S protein mediates receptor attachment and viral and host cell membrane fusion. 8 The nucleocapsid (N) protein binds to the viral RNA and forms a ribonucleoprotein that is packaged in the virus envelope. In addition to these four structural proteins, the CoV genome also codes for several nonstructural proteins, synthesized as two very large polyproteins that are cleaved by two or three virus proteases, and several auxiliary proteins. CoVs are classified within four genera: Alphacoronavirus, Betacoronavirus, Gammacoronavirus and Deltacoronavirus. To date, seven strains of Alphacoronavirus and Betacoronavirus that infect humans have been identified. 9 But our main concern here lies within the Betacoronavirus genus which contains the severe acute respiratory syndrome (SARS) viruses. SARS‐like viruses emerged as human infectious diseases in 2002, and since then have caused several epidemics and outbreaks. Before COVID‐19, outbreaks of SARS in 2003 10 and Middle East Respiratory Syndrome (MERS) in 2012 11 had already been reported as serious diseases caused by Betacoronaviruses. The causative agent of COVID‐19 is SARS‐CoV‐2. 12 This virus has the capacity to affect multiple organs as well as the central nervous system and sometimes causes respiratory problems which might end up having fatal consequences. The SARS‐CoV‐2 genome consists of around 30,000 nucleotides and contains 12 functional open reading frames (ORFs): from 5′ to 3′, ORF1a, ORF1b, S, ORF3a, E, M, ORF6, ORF7a, ORF7b, ORF8, N, and ORF10. 13 , 14 Two‐thirds of the SARS‐CoV‐2 genome, located at the 5′ genomic region, codify the ORF1a and ORF1b genes, which encode the polyproteins pp1a and pp1ab. Polyprotein pp1ab contains the pp1a protein and its synthesis requires a −1 ribosomal frameshift. 15 Both polyproteins are further cleaved by the main protease (M‐pro, also known as 3CLpro) and papain‐like (PLpro) protease to produce 16 proteins that include both proteases, an RNA‐dependent RNA polymerase, a helicase, two nucleases and a methyltransferase, among other proteins. 15
Due to the lack of target‐specific drugs for treating COVID‐19, several strategies are being investigated. Among these strategies, several clinical trials are being developed to: (i) inhibit the RNA‐dependent RNA polymerase, 16 , 17 (ii) inhibit the viral M‐pro or PLpro proteases, 18 , 19 , 20 , 21 (iii) block the fusion with the host cells, 22 , 23 , 24 (iv) enhance the innate immune system, 25 (v) attenuate the inflammatory response, 26 (vi) control the symptoms, 27 and (vii) create pathogen‐specific artificial antigen‐presenting cells. 28 Given its importance in the replication of SARS‐CoV‐2, M‐pro has become a key target for the development of antiviral drugs. 29 M‐pro proteins from SARS‐CoV‐2 and SARS‐CoV show a high sequence (i.e., >96%) and structural similarity (i.e., an root mean‐squared deviation [RMSD] of 0.44 Å between the Cα of both structures). SARS‐CoV‐2 M‐pro has a length of 306 amino acids that form three different domains (domains I, II, and III). 30 The substrate‐binding site is located at a cleft between domain I (residues 8–101) and domain II (residues 102–184), which share a common fold consisting of antiparallel ß‐barrel structures (Figure 1). Domain III (residues 201–303) consists of an antiparallel globular cluster of five alpha‐helices and is involved in M‐pro dimerization, which is essential for M‐pro activity. 30 Unlike the conventional Ser(Cys)‐His‐Asp(Glu) triad found in other chymotrypsin‐like enzymes, SARS‐CoV‐2 M‐pro has a catalytic dyad formed by His41 and Cys145 (Figure 1) that catalyzes the hydrolysis of the peptide bond at highly specific sites of a polypeptide chain through a common nucleophilic‐type reaction. It was suggested that a water molecule might complete the catalytic triad by mediating crucial interactions between His41 and other important conserved residues, such as His164 and Asp187. 31 While noncovalent small inhibitors have the ability to interact with different residues that lie within the binding pocket of SARS‐CoV‐2 M‐pro, covalent inhibitors are found to mainly interact with the catalytic cysteine residue after an initial noncovalent mechanistic step that ensures the warhead is correctly oriented. 21 Thus, it is of great importance to consider covalent and noncovalent inhibitors as different, because their binding mode and mode of action are quite distinct. Most of the characterized SARS‐CoV‐2 M‐pro inhibitors were originally designed for other pathogens, such as SARS‐CoV, HIV and Hepatitis C viruses, which shows the importance of drug repurposing strategies.
Figure 1.
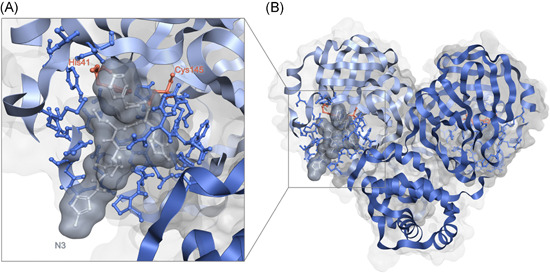
SARS‐CoV‐2 M‐Pro homodimer (PDBid 6LU7). Panel (B) shows an overview of the main structural features of M‐Pro and panel (A) displays in detail one of its binding pockets where the co‐crystallized ligand N3, in grey, fits. Each of the two protomers is a different tone of blue. In both panels, the residues that form the binding pocket (according to Gimeno et al. 29 ) are also highlighted in ball and stick format and the residues of the catalytic dyad (His41 and Cys145) are colored orange and labeled. This figure has been generated using the Flare software. 32 , 33 , 34
Drug repurposing, also known as drug repositioning, is a well‐known strategy in drug development, that is used to identify new medical uses for already approved or investigational drugs. This has enormous advantages compared to traditional drug development from scratch. Briefly, drug repurposing shortens time‐consuming steps in drug development, as safety assessment and testing are usually completed, while it is also known to substantially diminish the investment needed. This, together with the decreased risk of failure, makes drug repurposing an interesting approach when time presses, such as in a global pandemic like COVID‐19. 35 Given the high throughput of computational approaches, they are one of the mainstays of drug repurposing. Specifically, the most popular structure‐based in silico approach, protein‐ligand molecular docking, has been extensively used not only in drug discovery but also in drug repurposing. 36 , 37 Molecular docking is a receptor‐based VS method, that can be used to assess the possible binding poses of the ligands (e.g., a library of approved drugs) to a target (e.g., SARS‐CoV‐2 M‐pro). Thus, molecular docking makes it possible to interrogate thousands of drugs against the protein target at a relatively low computational cost, so it is a popular tool in computational and structural biology.
Several articles have reviewed the SARS‐CoV‐2 M‐pro inhibitors discovered to date. 20 , 21 , 38 , 39 , 40 , 41 , 42 , 43 In this review, we focus on the computational methods, especially protein‐ligand docking, which have been used to discover new SARS‐CoV‐2 M‐pro inhibitors. We check 61 peer‐reviewed manuscripts that use at least one docking step in a virtual screening (VS) protocol to predict SARS‐CoV‐2 M‐pro inhibitors in approved or investigational drugs. We put special emphasis on the validation of the docking process and whether the use of docking scores or other estimates of binding affinity are a reliable way to select new putative inhibitors of SARS‐CoV‐2 M‐pro. For this purpose, we have taken a set of 150 M‐pro inhibitors with known experimental bioactivity from the literature and a second set of 81 M‐pro known inhibitors and 113 inactive compounds from the COVID Moonshot project to evaluate the reliability of using docking scores as feasible predictors of the potency of a SARS‐CoV‐2 M‐pro inhibitor.
2. RECENT VIRTUAL SCREENINGS EFFORTS THAT USE PROTEIN‐LIGAND DOCKING TO PREDICT NEW SARS‐COV‐2 M‐PRO INHIBITORS
Computational approaches can contribute to drug discovery by reducing the cost and time of drug development and speeding up the analyses of the interactions between a target and candidate drugs. 37 Of all the VS methods, and specifically in drug repurposing, molecular docking is an attractive choice because it can predict the preferred orientation of a drug at the binding site of a target. 44 Molecular docking methods use two independent steps: pose generation and scoring. 45 The first step refers to the methods used to create different ligand poses within the binding site of the protein. The second, scoring, is required to rank the different poses and quantitatively estimate the pose quality. 45 Scoring functions attempt to estimate the binding affinity between the ligand and the protein. 45
We collected 61 articles that use a VS protocol based on protein‐ligand docking to predict drugs as SARS‐CoV‐2 M‐pro inhibitors. These articles were published in peer‐reviewed journals between the beginning of the COVID‐19 pandemic in early February 2020 and November 2020. It should be pointed out that preprints were not included. Table 1 outlines the most relevant information of these 61 articles. Further details and the drugs predicted in each can be found in Supporting Information File 1. As shown in Table 1, most of these articles used an initial database of approved drugs (e.g., the Drugbank database). A quarter of the articles also included a group of antiviral molecules, such as HIV or Hepatitis C protease inhibitors. The first crystallized structure of the SARS‐CoV‐2 M‐pro protein, the 6LU7 structure released on February 5, 2020 30 and crystallized with the covalent inhibitor N3 was largely used. Before this structure was available, eight articles used a structural model of the M‐pro constructed using the homologous protein from SARS‐CoV as a template. Subsequently, many other SARS‐CoV‐2 M‐pro structures were released and several articles used the M‐pro structure, such as 6W63, complexed with a noncovalent inhibitor. Most of the VS protocols reviewed here used AutoDock Vina or other docking methodologies available at the Schrödinger Drug Discovery suite (i.e., Glide HTVS, Glide SP and Glide XP; see Table 1). The majority of these VS protocols used only one docking program, although some of them used two or three. Generally, protocols using multiple docking programs are performed in a stepwise manner; i.e., the programs are used in a funnel approach to reduce the number of hits by using the different prediction algorithms and/or punctuation functions, which is indeed what happens when the increasingly exhaustive methodologies of the Schrödinger package Glide (Glide HTVS, SP, and XP) are used. Nonetheless, there is also room to use different programs in a parallel manner and then evaluate the equivalent poses predicted by each of them, as in our last contribution. 29 Usually, the docking score of the best pose of a reference compound (normally, a known or expected inhibitor) is generally used as a cutoff value for selecting new putative M‐pro inhibitors. If the best docking score of a compound is more negative than the docking score of the reference compound, it is usually assumed that the compound will be more active than the reference one. When a reference compound is not used, the most negative docking score values tend to be used as a cutoff to select a group of hit compounds and analyze them in further detail. The predicted interactions between the substrate‐binding site of the M‐pro protein and each selected compound are usually analyzed. In addition, some reviewed articles use molecular dynamics (MD) to evaluate the binding dynamics of the group of selected hits to the M‐pro structure. The molecular mechanics generalized Born surface area (MM‐GBSA) or the molecular mechanics Poisson–Boltzmann surface area (MM‐PBSA) algorithms were generally used to estimate the binding free energies of the putative inhibitors.
Table 1.
Outlines of the 61 articles that predict drugs to be SARS‐CoV‐2 M‐pro inhibitors
| Compounds database | Counts | % articles |
|---|---|---|
| FDA‐approved | 27 | 44.26 |
| Approved | 36 | 59.02 |
| Drugbank | 21 | 34.43 |
| Selleckchem | 7 | 11.48 |
| ZINC | 12 | 19.67 |
| PubChem | 5 | 8.20 |
| ChEMBL | 6 | 9.84 |
| Antiviral | 15 | 24.59 |
| Others | 3 | 4.92 |
| PDB structure | ||
| 6LU7 | 39 | 63.93 |
| 6W63 | 6 | 9.84 |
| 6M03 | 4 | 6.56 |
| Model | 5 | 8.20 |
| Others | 8 | 13.11 |
| Docking software | ||
| AutoDock Vina | 26 | 42.62 |
| Glide HTVS | 13 | 21.31 |
| Glide SP | 13 | 21.31 |
| Glide XP | 14 | 22.95 |
| GOLD | 4 | 6.56 |
| Others | 13 | 21.31 |
| N° of docking programs | ||
| 1 | 37 | 60.66 |
| 2 | 12 | 19.67 |
| 3 | 12 | 19.67 |
| Molecular dynamics | ||
| Yes | 35 | 57.38 |
| No | 26 | 42.62 |
| Validation method | ||
| Actives enrichment in silico | 3 | 4.92 |
| In vitro | 2 | 3.28 |
| Re‐docking | 18 | 29.51 |
| No | 41 | 67.21 |
| Total of articlesa | 61 |
Note: Information is shown about the initial database, the PDB structure, the protein‐ligand docking method, whether they use molecular dynamics or not, and the validation method used (if any).
Abbreviations: FDA, Food and Drug Administration.
% Of articles does not necessarily add 100% because some articles may be included in more than one group.
The best way to validate the methodology and results of a VS protocol is to experimentally prove the bioactivity of some selected hits. Only two of the 61 articles reviewed confirmed their computational predictions in vitro. 29 , 46 When this is not possible, the methodology should be subject to a computational validation. 37 A common strategy for validating the docking step in some of the 61 articles was re‐docking or self‐docking. Re‐docking consists of removing the ligand from a protein‐ligand complex to dock it back and evaluate if a docking program can predict its initial binding pose. The RMSD between the best docking pose and the crystallographic one, or the lowest RMSD value of all the predicted poses, gives an estimation of the ability of the docking program to reproduce the co‐crystallized binding pose of the ligand. RMSD values under 2.0 Å are usually considered to be acceptable, but values close to zero are ideal. Although 18 of the 61 VS protocols analyzed used re‐docking to estimate the docking accuracy (see Table 1 and Supporting Information File 1), only two 47 , 48 converted the extracted ligand to SMILES format and then back to 3D. This is done to lose the binding coordinates of the ligand and evaluate the docking accuracy from scratch, similar to how the compounds are predicted. Because docking calculations are very sensitive to the starting geometries of ligands, 49 it may also be beneficial to re‐run the re‐docking with different starting conformations each time. 50 In addition to re‐docking, it is also recommended to use cross‐docking to try to predict the binding pose of a ligand using a different PDB structure. In this way, the possible effect of protein flexibility can be evaluated or the best protein structure identified when several are available. However, re‐docking and cross‐docking cannot suitably validate an entire VS process or each of its steps. In addition, if docking scores are used to rank the bioactivity of the predicted compounds, it needs to be shown that they correlate with the bioactivity. Without a proper validation, the predictions obtained are speculative and dubious. A VS protocol can be computationally validated by applying the same VS protocol to a set of active compounds and to a set of inactive or decoy compounds (when a group of inactive compounds is not available). The more accurate the methodology is, the more true positives (i.e., active compounds that pass the VS procedure or VS step) and true negatives (i.e., inactive molecules that do not pass the VS procedure or VS step) there will be. To quantify the performance of the entire methodology or each VS step, several metrics can be used. Of these, the most popular are the area under a ROC curve and the Enrichment Factor (EF), which quantify the extent to which a step of the VS can be enriched with active compounds. Only three of the 61 articles reviewed used a group of known M‐pro inhibitors to confirm that their VS procedure was able to enrich the predicted compounds with these inhibitors. 29 , 51 , 52 In one of the articles 52 12 M‐pro inhibitors, 10 weak M‐pro inhibitors and 114 DUDE‐generated decoys were screened to calculate the EF for each step of the VS and to choose the optimal scoring function for the final hit selection. Although some of the 61 articles used re‐docking as a validation step, more than the 67% did not apply a validation step (Table 1). This lack of validation of the structure‐based approaches to drug repurposing that target M‐pro and other SARS‐CoV‐2 proteins has been described elsewhere. 53 , 54 In a survey of the literature before 26 August, Bellera and coworkers 54 reviewed 96 articles that reported the use of molecular docking for identifying potential compounds against SARS‐CoV‐2 M‐pro. None of the 96 articles reported any level of experimental validation and 40.6% of the articles did not perform any kind of in silico validation. 54 We agree with Bellera and coworkers that “the preceding figures are surprising, considering the known fact that the performance of docking studies is highly dependent on the system under study, and the high false‐positive rate associated with docking‐based VS”. 54
An interesting approach used in some of the articles reviewed 48 , 52 , 55 , 56 , 57 was to add a pharmacophore‐based procedure before the docking step to enrich the database with some characteristics of the reported M‐pro inhibitors or M‐pro co‐crystallized ligands, or to consider only the compounds that can engage in specific interactions with the M‐pro active site. This strategy makes it possible to identify which of the initial number of compounds are most likely to be inhibitors, even though it requires prior knowledge of the interactions between the receptor and its ligands. Thus, it cannot be applied in early attempts, when little information is available, but it becomes a more powerful tool as more information arises.
Figure 2 shows that the 61 VS protocols analyzed predict very few M‐pro inhibitors in common. It could be argued that this lack of agreement about the potential inhibitors is due to differences in the initial set of compounds analyzed and the methods used. However, Figure 3 shows that when different protocols using similar initial datasets are compared, such as FDA‐approved drugs (Figure 3A) or approved and investigational drugs (Figure 3B), very few compounds are predicted by two protocols even if the same PDB structure and docking program have been used (Figure 3). Some of the compounds most commonly predicted as inhibitors of SARS‐CoV‐2 M‐pro in several of the 61 VS protocols reviewed are: the covalent inhibitors of the HIV‐protease ritonavir, saquinavir, indinavir, lopinavir and nelfinavir; other antiviral compounds, such as the hepatitis C protease inhibitors simeprevir and paritaprevir; and the HIV integrase inhibitor raltegravir. All of these compounds were predicted by more than five different articles (Figure 2). As these antiviral compounds were initially used in clinical trials to combat the SARS‐CoV‐2 pandemic, they are also often selected to be included among the final hits of the VS protocols analyzed. Ritonavir inhibits cytochrome P450‐3A4, and is often simultaneously used with lopinavir or other HIV or hepatitis C protease inhibitors since it increases their half‐life. Although ritonavir was found to inhibit the SARS‐CoV‐2 M‐pro in cell culture, 58 high concentrations are required to achieve significant inhibition, so it will be of limited clinical relevance. 58 The results of several clinical trials that have evaluated the role of lopinavir/ritonavir combination therapy in patients with COVID‐19 have shown little or no improvement. 59 , 60 , 61 Remdesivir was also predicted as an M‐pro inhibitor by 7 of the 61 articles. This compound was the first compound approved by the FDA to treat patients with severe symptoms of COVID‐19. However, remdesivir is an adenosine nucleoside triphosphate analog that inhibits the SARS‐CoV‐2 RNA‐dependent RNA polymerase and induces an irreversible chain terminator. So, it is surprising that it is predicted to be a SARS‐CoV‐2 M‐pro inhibitor. There is no experimental evidence to suggest that remdesivir binds to SARS‐CoV‐2 M‐pro. 62 In addition, remdesivir is a prodrug, converted to its active form by host enzymes, and this was often not taken into account in the VS protocols analyzed. To the best of our knowledge, only one study has docked remdesivir and its active form to SARS‐CoV‐2 M‐pro and suggested that both molecules have a comparable affinity against SARS‐CoV‐2 M‐pro. 62 More experiments are required to precisely determine if, in addition to inhibiting the RNA‐dependent RNA polymerase of SARS‐CoV‐2, remdesivir's mechanism of action includes the inhibition of M‐pro.
Figure 2.
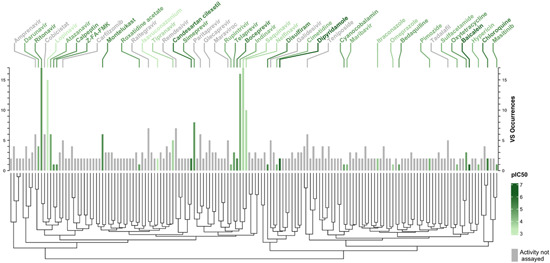
Dendrogram and bar plot of the compounds that are most predicted to be inhibitors of M‐pro in several VS. The dendrogram clusters the compounds in terms of their structural pairwise similarity established by the Tanimoto coefficients calculated from the ECFP6 circular fingerprint from each compound. The bar plot represents the number of times that each compound has been predicted as a putative M‐pro inhibitor in the reviewed articles (VS occurrences). The in vitro activity, when available, is expressed as pIC50 values and represented in a green scale. The only compounds displayed are those that are predicted in two or more different VS or for which experimental pIC50 is available. The names of the compounds with known activity and those with five or more VS occurrences are displayed. VS, virtual screening
Figure 3.
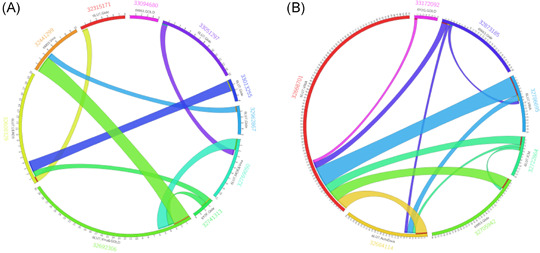
Circos plots showing the number of shared compounds among the predicted hits of different VS studies which use similar initial datasets. Panel (A) shows the number of compounds that are predicted simultaneously by two different articles that use a group of FDA‐approved drugs (between 1500 and 2800 drugs) as the initial dataset. Panel (B) shows the compounds predicted simultaneously by two articles that use a group of approved and investigational drugs (between 8500 and 13,563 drugs). The information shown is the PubMed ID of each article, the PDB structure and docking program used, the number of compounds predicted by each article and the number of common compounds. FDA, Food and Drug Administration; VS, virtual screening [Color figure can be viewed at wileyonlinelibrary.com]
Figure 2 also shows that the M‐pro inhibitors predicted by most articles are not the most active inhibitors. The IC50 values of lopinavir, saquinavir, indinavir and nelfinavir have been estimated to be 486, 63 between 31.4 and 411, 58 , 63 43.1, 46 and 234 µM 63 , respectively. In other studies, lopinavir, indinavir, nelfinavir, tipranavir and other HIV protease inhibitors have not shown any inhibitory activity between 20 64 and 200 µM. 58 It has been suggested that the reduction in viral activity shown by nelfinavir and lopinavir in cell cultures is due to the inherent cytostatic and/or cytotoxic effects of these compounds. 65 Dipyridamole, 46 , 66 baicalein, 66 candesartan cilexetil 46 , 67 (predicted in 2, 1, and 2 studies, respectively) have the lowest IC50 values of the most predicted M‐pro inhibitors, but in the three cases, their respective IC50 of 0.6, 0.94 and 2.78 µM 46 , 68 are a long way from the most potent SARS‐CoV‐2 M‐pro inhibitors, such as PF‐00835231, MI‐21 and MI‐23, which have an IC50 in the nanomolar range. 69 , 70 As depicted in the dendrogram in Figure 2, in structural terms the most predicted compounds in VS studies vary considerably, probably due to the high flexibility induced by ligand binding and to the size of the M‐pro's binding site. 31 , 71 Also, relatively similar compounds (e.g., ritonavir and lopinavir) can have a quite different pIC50, which makes M‐pro a challenging target in docking and SAR studies, as a wide variety of scaffolds, activity and selectivity cliffs might have to be taken into account. To discover new therapeutics for COVID‐19 it would be useful to make a comprehensive and systematic assessment of the chemical space of known SARS‐CoV‐2 inhibitors. 72 Although this approach opens up an interesting line in SARS research for the discovery of new SARS‐CoV‐2 M‐pro inhibitors, it falls outside the scope of the present work and, for the sake of brevity, no further discussion will be conducted.
In the following sections we describe a group of known SARS‐CoV‐2 M‐pro inhibitors and inactive compounds, and show how their bioactivity can be used to validate a protein‐ligand docking step.
3. KNOWN SARS‐COV‐2 M‐PRO INHIBITORS
Figure S1 and Supporting Information File 2 show the 2D structure, bioactivity and other details of 150 known SARS‐CoV‐2 M‐pro inhibitors extracted from the literature. This set of inhibitors includes only those compounds whose inhibitory capacity, mainly expressed as the IC50 value, against M‐pro from SARS‐CoV‐2 has been determined in vitro. The bioactivities of these compounds have been obtained since the second trimester of 2020 and show how quickly the scientific community has responded to the challenge of the COVID‐19 pandemic, which appeared abruptly at the beginning of 2020. Most of these 150 compounds are found in other reviews that describe recent progress made in searching for SARS‐CoV‐2 M‐pro inhibitors. 20 , 21 , 38 , 39 , 40 , 41 , 42 , 43 Among these M‐pro inhibitors there are SARS‐CoV M‐pro inhibitors such as the compound MAC‐5576; HIV‐protease inhibitors such as atazanavir, darunavir, indinavir, lopinavir, nelfinavir, ritonavir and saquinavir; Hepatitis C protease inhibitors such as boceprevir, narlaprevir, simeprevir and telaprevir; Caspase‐3 inhibitors such as Z‐DEVD‐FMK and Z‐FA‐FMK; Calpain and other protease inhibitors; and inhibitors obtained from high‐throughput screening experiments. Most of the HIV, SARS‐CoV or hepatitis C protease inhibitors can make a covalent bond with the catalytic Cys145 of SARS‐CoV‐2 M‐pro. Among the 150 SARS‐CoV‐2 M‐pro inhibitors reviewed here, there are compounds with moderate or low inhibitory activity; compounds, such as carmofur, ebselen, disulfiram, PX‐2, shikonin and tideglusib, whose inhibition potencies decreased in the presence of reducing agents, such as DTT, and are considered to be nonspecific inhibitors 73 , 74 ; compounds such as shikonin, which quench the signal used to evaluate its activity and therefore may represent a false positive of the analysis 73 ; and compounds such as the HIV‐protease inhibitors lopinavir, indinavir, nelfinavir saquinavir, and tipranavir, which have not shown M‐pro inhibitory activity in some concentrations analyzed. 58 , 64 , 75 , 76 To test the hypothesis that inhibiting the enzymatic activity of M‐pro will inhibit SARS‐CoV‐2 replication, a cellular antiviral assay is usually performed and an EC50 value calculated. This EC50 value is defined as the concentration of the compound that reduces the viral‐induced cytopathic effect by 50% of with respect to the virus control. Several SARS‐CoV‐2 M‐pro inhibitors have EC50 values that are slightly more than 10 times their IC50 values. 76 Some of the 150 known SARS‐CoV‐2 M‐pro inhibitors, such as carmofur, darunavir, disulfiram, ebselen, MG‐132, PX‐2, shikonin and tideglusib, cannot inhibit SARS‐CoV‐2 replication in cell cultures or do inhibit it due to their cytotoxicity. 73 , 74 , 77 , 78 , 79 This shows that not all the compounds that inhibit the SARS‐CoV‐2 M‐pro in vitro will be useful in vivo.
Boceprevir, calpain inhibitors I, II, and XII, carmofur, cinanserin, disulfiram, ebselen, GC376, N3, narlaprevir, PX‐12, shikonin, telaprevir and tideglusib are some of the covalent inhibitors described for the SARS‐CoV‐2 M‐pro (see Figure S1 and Supporting Information File 2). The role of covalent inhibitors in drug discovery is controversial. Some of the risks they pose are toxicity, immune‐mediated drug hypersensitivity, nonspecificity, and hyperreactivity that might lead to further drug‐induced toxicity (e.g., hepatotoxicity, mutagenicity, or carcinogenicity). 80 However, three potential benefits are that the high biochemical efficiency may lead to lower doses and reduced off‐target effects, lower drug resistance and fewer cases of decreased potency due to competing endogenous substrates. 80 Reversible or irreversible covalent inhibitors that have some kind of specificity for a target or group of targets must first be recognized by the target binding site to place a reactive functional group, usually referred to as a warhead, in an appropriate position to form a covalent bond between the inhibitor and its target. 21 , 81 The formation of the covalent bond depends not only on the correct structural complementarity between the protein and the inhibitor, but also on the appropriate chemical reactivity of the inhibitor and the protein environment that stabilizes the covalent complex. 82 The protonation state of catalytic Cys145 and His41 and His172 is crucial for the activity variation caused by pH changes and the M‐pro catalytic mechanism 83 , 84 , 85 and may be crucial to the action of covalent inhibitors. The majority of SARS‐CoV‐2 covalent inhibitors are large substrate mimetics with an electrophilic warhead that covalently binds with the catalytic Cys145 sulfur atom. 21 But other more nucleophilic cysteines, such as Cys44 located near the substrate binding pocket, are additional sites for covalent linkage. 85 Table 2 shows such covalent warheads, as acrylamide, chloroacetamide, vinylsulfonamide, nitrile, a Michael acceptor group, alpha‐ketoamide, aldehyde, disulfide and several methyl ketone groups, which have been used here to identify putative M‐pro covalent inhibitors. Whereas some covalent warheads, such as acrylamide and chloromethyl ketones, form irreversible bonds with their biological targets, others, such as nitriles or electron‐deficient ketones, have been found to form a reversible covalent bond. 21 , 80 For irreversible covalent inhibitors, if a sufficient period of time is allowed, inhibition is expected to be complete. 86 Thus, conventional IC50 measurements are of limited value for characterizing the potency of irreversible inhibitors because incubation for a different period of time would provide a different IC50 value. 86 Of the 150 SARS‐CoV‐2 M‐pro inhibitors reviewed here, 73 present a covalent warhead (Supporting Information File 2) and are plausibly covalent inhibitors.
Table 2.
Covalent warheads that can be used to identify putative covalent inhibitors. It shows the SMARTS that can be used to identify each warhead and some examples of SARS‐CoV‐2 inhibitors that contain each warhead
| Warhead | SMARTS | Examples |
|---|---|---|
| acrylamide | [C;H2:1]=[C;H1]C(N)=O | |
| chloroacetamide | Cl[C;H2:1]C(N)=O | |
| vinylsulfonamide | NS(=O)([C;H1]=[C;H2:1])=O | |
| nitrile | N#[C:1]‐[*] | Cimetidine, isavuconazole |
| Michael_acceptors | C=!@CC=[O,S] | Cinanserin, MPI2, MPI9, N3 |
| alpha‐ketoamide | C(=O)(C=O)N | Boceprevir, narlaprevir, telaprevir |
| aldehyde | [CX3H1](=O) | Leupeptin, 2413716‐71‐7 |
| bisulfite_adduct_of_aldehyde | C(O)S(=[OX1])([O])(=[OX1]) | GC376 |
| urea_carbonyl | [NX3][CX3](=[OX1])([NX3,nX3]) | Carmofur |
| bis(dialkylaminethiocarbonyl)disulfide | [CX3](=[SX1])SS[CX3](=[SX1]) | Disulfiram |
| carbamoylsulfanyl | [NX3,nX3][C,c](=[OX1])([SX2,sx2]) | Tideglusib |
| disulfide | [SX2][SX2] | PX‐12 |
| Hydroxymethy_lketone | [CX3H0](=[OX1])[CH2][OH] | PF‐00835231 |
| Alkoxymethyl_ketone | [CX3H0](=[OX1])[CH2][OX2H0] | GW‐0742, 2434601‐94‐0 |
| Acyloxymethyl_ketone | [CX3H0](=[OX1])[CH2][OX2H0][CX3H0](=O) | 2434601‐94‐0, 2434601‐96‐2 |
| Fluoro, Chloro‐methyl_ketone | [CX3H0](=[OX1])[CH2][Cl,F] | Z‐DEVD‐FMK, Z‐FA‐FMK |
Among the most potent inhibitors of the SARS‐CoV‐2 M‐pro, the compounds PF‐00835231, MPI3, UAWJ248, MPI4 and MPI5 have a pIC50 higher than 7.5, and an IC50 in the nanomolar range. PF‐008325231 is a hydroxymethyl ketone covalent inhibitor selected as a development candidate for SARS‐CoV, the clinical advancement of which was suspended in 2003 due to the end of the SARS‐CoV outbreak. 69 It is the most potent SARS‐CoV‐2 M‐pro inhibitor known to date, with an IC50 of 0.00027 µM. 69 It is a highly selective pan‐coronavirus M‐pro inhibitor that can also inhibit SARS‐CoV‐2 replication in cells. 69 Preclinical research is ongoing in the use of PF‐00835231 as a possible treatment for COVID‐19. 69 Pfizer has initiated a clinical trial with PF‐07304814, a prodrug which metabolizes into PF‐008325231, against the M‐pros of SARS‐CoV‐2 and other coronaviruses. GC376 is one of the SARS‐CoV‐2 M‐pro covalent inhibitors that is most commonly used as a reference compound or positive control. Its IC50 activity ranges between 0.03 and 0.89 µM 64 , 73 , 74 , 78 , 87 , 88 , 89 , 90 , 91 , 92 , 93 and this activity is consistent both in the absence and presence of the reducing agent DTT. 74 GC376 was developed as an inhibitor of the main protease of the feline coronavirus FCoV, and it also showed activity against the M‐pro from MERS and SARS‐CoV viruses. 88 GC376 is a prodrug and its bisulphite adduct is converted to an aldehyde that has been proved to form a covalent interaction with the catalytic Cys145 of the SARS‐CoV‐2 M‐pro. 88 Several GC376 analogs or derived compounds have shown greater inhibitory activity than GC376. 21 MPI3, MPI4, and MPI5 are reversible covalent inhibitors derived from the GC376 inhibitor. 91 MPI3, MPI4, and MPI5 display an IC50 value of 0.0085, 0.015, and 0.033 µM, respectively. 91 Although MPI3 is ranked second in potency as a SARS‐CoV‐2 M‐pro inhibitor, its inhibition of SARS‐CoV‐2 replication is much lower than other compounds, which may be explained by the compound's low cell stability. 91 UAWJ248 is a GC376 analog with an alpha‐ketoamide reactive warhead that binds irreversibly to Cys145. 94 The IC50 of UAWJ248 is 0.012 µM, 3.2‐fold more potent than GC376. 94
Another set of SARS‐CoV‐2 M‐pro inhibitors and inactive compounds can be obtained from the COVID Moonshot project 95 , 96 set up to develop M‐pro inhibitors. It focused on turning early fragment‐screening results into potent compounds. 96 Drug‐designers the world over have proposed more than 10,000 M‐pro inhibitors, and over 1000 compounds have been either ordered or synthesized and tested for activity against SARS‐CoV‐2 M‐pro using two complementary biochemical assays: a fluorescence based assay and a RapidFire Mass Spectrometry assay. 96 The Fluorescence M‐pro inhibition assay consists of a screening in duplicate at 20 and 50 µM. Compounds with a value greater than 50% M‐pro inhibition at 50 µM are confirmed by a dose response assay and the value of IC50 is calculated. 96 For the RapidFire Mass Spectrometry assay, compounds are triaged by testing the % of inhibition at a final concentration of 20 and 50 µM, and IC50 values are calculated from dose response curves. 96 From the COVID Moonshot activity data available on the August 18, 2020, we selected those compounds that have a pIC50 difference between the two activity assays of less than two units. In addition, further quality control steps were carried out: (i) a compound was not selected if the percentage of inhibition at 20 µM was higher than the percentage at 50 µM or (ii) if the percentage of inhibition at 50 µM and the IC50 of a compound were both simultaneously higher than 50. Finally, compounds with undefined chirality were discarded. Figure S2 and Supporting Information File 3 show the final list of the 81 SARS‐CoV‐2 M‐pro inhibitors selected from the COVID Moonshot activity data. The pIC50 values of these 81 compounds are between 4.14 and 7.28, and 36 of them contain at least one of the covalent warheads defined in Table 2 (Supporting Information File 3) and are putative covalent inhibitors of the SARS‐CoV‐2 M‐pro.
The activity data available from the COVID Moonshot initiative makes it possible to select a group of inactive compounds against the SARS‐CoV‐2 M‐pro. Although some articles have reported inactive compounds against SARS‐CoV‐2 M‐pro, such as the DDP‐4 inhibitors linagliptin, other gliptins and structural analogues, 97 this kind of information is not usually available. From the COVID Moonshot activity data available on 18 August 2020, we selected the compounds with a percentage of inhibition less than 2% at 50 µM in the two different assays simultaneously (fluorescence and RapidFire). Compounds with undefined chirality were discarded. Thus, a group of 113 inactive compounds against SARS‐CoV‐2 were selected (see Figure S3 and Supporting Information File 4). Of these 113 inactive compounds, 29 present at least one of the covalent warheads from Table 2.
The set of 81 M‐pro inhibitors from the COVID Moonshot project and the 150 M‐pro inhibitors extracted from the literature and presented above could be used to validate a VS procedure and/or a docking step that aims to predict new SARS‐CoV‐2 M‐pro inhibitors. The COVID Moonshot set of active compounds is less diverse than the set taken from the literature, but their inhibitory activities were estimated using the same procedure and laboratory, and all the inhibitors were confirmed by two different assays (fluorescence and RapidFire). In the section below, these sets of compounds are used to check whether protein‐ligand docking methods can rank these compounds in order of their potency and if they can discriminate between active and inactive compounds.
4. DOCKING METHODOLOGY NEEDS TO BE VALIDATED BEFORE USE
Although molecular docking programs can usually generate ligand poses that are similar to the crystallographic conformation for several targets, 98 protein‐ligand docking has several limitations. One of these is that, for practical reasons, most docking methods use a rigid protein approximation, where the ligand is treated as flexible, but the protein conformation is restricted. 45 , 99 Some methods, such as ensemble docking which takes place in an ensemble of protein conformations, 100 enable protein flexibility to be analysed. Another limitation is that, contrary to what is expected, the poses and docking scores obtained by docking can be heavily dependent on the ligand input structure, 49 so small changes in the ligand input conformation can lead to large differences in the docked poses. 49 One of the major limitations of the scoring functions used by docking programs is that they do not always make a useful prediction of ligand binding affinity. 36 , 98 Thus, the best energy score is not a reliable criterion for selecting the best solution in common docking applications. 101 For these reasons, docking scores should not be used as a measure or threshold for compound activity, but rather to enrich the library by discarding those compounds that do not fit inside the binding site. 37 It should also be noted that the docking scores of different docking programs cannot be directly compared because the scores are dependent on the force fields and protocols used for each program. Another limitation of molecular docking is how they deal with water molecules present in the binding site during the docking process 45 , 102 which can lead to errors when trying to ascertain the possible interaction between the ligand and the target. Finally, the ability of a docking program to separate decoys from active compounds is usually strongly dependent on the protein structure used and the similarity between the screened ligands and the co‐crystallized ones. 36
We used the aforementioned sets of 150 M‐pro inhibitors extracted from the literature and 81 M‐pro inhibitors from the COVID Moonshot project to check whether docking scores can be used to predict the potency of a SARS‐CoV‐2 M‐pro inhibitor. To test this hypothesis we performed a protein‐ligand docking of both sets of compounds with two different 3D structures of the SARS‐CoV‐2 M‐pro protein, the 6LU7 30 and 6WQF, 31 and three docking programs, i.e, FRED, 103 Glide 104 and Autodock Vina. 105 For Glide, we used three distinct docking methodologies: Glide HTVS, Glide SP and Glide XP. So in total we considered 5 different docking methods, which appeared to be the most common in the reviewed articles (see Table 1). Briefly, the three methodological approaches from the Schrödinger package Glide differ from each one. Essentially, Glide HTVS and Glide SP use the same docking algorithm, but their main difference is that Glide HTVS minimizes the number of intermediate conformations during the docking procedure and the accuracy of the final torsional refinement and sampling. 106 On the other hand, Glide XP uses a more exhaustive and sophisticated scoring function and sampling process, and is stricter in terms of ligand‐receptor shape complementarity. 107
We used the 6LU7 structure 30 because it is the most used structure in molecular docking studies of the SARS‐CoV‐2 M‐pro (see Table 1). The 6WQF structure of unliganded SARS‐CoV‐2 M‐pro was also used as it was suggested that this structure may provide more relevant information at physiological temperatures, which could be helpful in protein‐ligand molecular docking studies. 31 Both M‐pro structures (6LU7 and 6WQF) were obtained from the Protein Data Bank and were superimposed using the alpha carbons (Cα) as reference points. The 6LU7 ligand (N3) was removed from the structure before the protein was prepared. Protein preparation was performed with different tools, depending on the docking software used. For Glide, the structures were prepared with the Protein Preparation Wizard 108 with the following settings: (a) N and C termini were capped; (b) hydrogens were added after original hydrogens had been removed; (c) water molecules were removed; (d) Epik 109 was used to generate tautomers and protonation states with neutral pH; (e) H‐bond assignment was optimized with PROPKA 110 ; and (f) the structure was minimized with the default force field. The grid for docking was generated using Glide. The center coordinates corresponded to the centroid of N3 (−10.36, 12.46, 68.7) and the box size was set to 35 × 35 × 35 Å. In the case of FRED, the utility MakeReceptor 103 was used to (a) define a box, (b) set a shape potential and (c) define the inner and outer contours of the grid based on those used to create the grid for Glide. The preparations needed for AutoDock Vina were performed with AutoDockTools 111 which consisted of removing all waters and adding polar hydrogens. The grid was based on the same coordinates and size as the previous programs, and the M‐pro inhibitors were converted into isomeric SMILES format. Only achiral molecules or molecules with a fully defined chirality were used. Before the docking procedure with Autodock Vina and the three methods of Glide, Omega 112 was used to generate the 3D conformation with the lowest energy and Ligprep 113 was used to generate all the possible protonation states in the pH range 7.2 ± 1.0 and the default number of tautomers. The docking program FRED had its own pipeline that included (i) fixpka, tautomer and molcharge from the QUACPAC program 114 which set the ionization states, enumerated the tautomeric forms and assigned atomic partial charges, and (ii) Omega 112 which generated one conformation per compound and discarded unspecified chiralities.
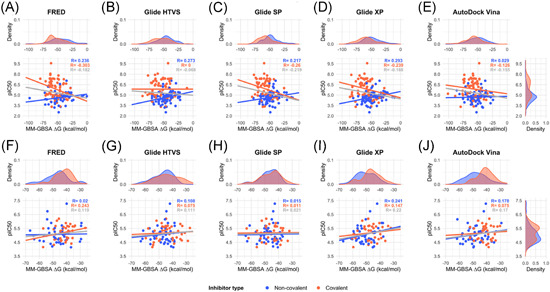
The most negative docking scores obtained for each compound and each protein‐ligand methodology were correlated with the inhibitory potency, measured as pIC50 values. Figure 4 and Figure S4 show that for both datasets, the correlations were far from satisfactory. The correlations improved if covalent and noncovalent inhibitors were treated separately (see Figure 4 and Figure S4). For the 6LU7 structure, and considering solely the covalent inhibitors extracted from the literature, correlations were negative, as one would expect because the more negative the docking score, the higher pIC50 value (Figure 4A–E). The best correlation had a Pearson's r coefficient of −0.335 (p value 0.003) for the covalent inhibitors when AutoDock Vina was used (Figure 4E). However, the Pearson correlation coefficients for the noncovalent inhibitors were positive or around zero (Figure 4A–E). For the set of 81 M‐pro inhibitors from the COVID MoonShot project (Figure 4F–J), most of the correlations were unexpectedly found to be positive. Conclusions were similar when the 6WQF structure was used (Figure S4). This lack of correlation between the bioactivities and docking scores for this target was previously reported 115 , 116 and questions the reliability of using a docking score as a cutoff value for selecting new putative M‐pro inhibitors or predicting the relative bioactivity of a compound by comparison with reference compounds or even with other predicted compounds.
Figure 4.
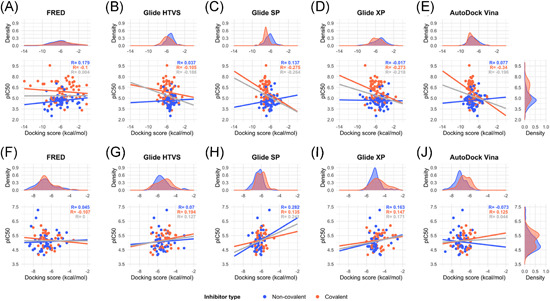
Correlation between the highest docking scores from five different docking methods (FRED, Glide HTVS, Glide SP, Glide XP and AutoDock Vina) and the experimental inhibitory activity, measured as pIC50, of a group of 150 M‐pro inhibitors extracted from the literature (A–E) and 81 M‐pro inhibitors from the COVID Moonshot project (F–J). The protein from the 6LU7 PDB structure was used for docking. Data has been adjusted to a linear regression and the correlation coefficient is displayed in gray. Covalent and noncovalent inhibitors are also represented separately in orange and blue, respectively [Color figure can be viewed at wileyonlinelibrary.com]
To overcome the limitations of docking programs, they can be combined with other methods. Standard molecular dynamics or binding free energy estimations, such as MM‐GBSA and MM‐PBSA enable the protein flexibility to be explored. MM‐GBSA or MM‐PBSA are popular methods for estimating ligand‐binding affinities with a modest computational effort. 117 Their results are considered to be more accurate than those of most docking scoring functions 118 and they have been widely used in virtual screening protocols to find M‐pro inhibitors (see Supporting Information File 1). Because these methods are not suitable for screening large libraries of compounds, 36 they are usually used to analyze compounds selected by molecular docking. The best docking poses of a selected group of hits can be the input of the MM‐GBSA or MM‐PBSA algorithms which can be used to estimate their ligand‐binding affinities, expressed as a ∆G value. The combination of molecular docking and MM‐GBSA or MM‐PBSA re‐scoring has proven to be a promising strategy for identifying the correct binding poses and correctly ranking the binding affinities of a series of ligands. 118 However, the success of these methods depends on the target structure 117 and they cannot be used to predict true drug candidates in drug design without experimental verification because their accuracy is relatively limited. 118 To assess how successfully these methods predict the bioactivity ranking of the two sets of known M‐pro inhibitors presented above, we used the MM‐GBSA minimization available at Prime 119 to re‐score the best docking pose obtained with the five docking methods and two SARS‐CoV‐2 M‐pro structures, and to estimate the ∆G value of ligand‐binding affinity for each compound. Figure 5 and Figure S5 show the correlation between the pIC50 and the estimated ∆G values. A negative correlation is expected, because the higher the pIC50 of a compound is, the lower the ∆G value. Figure 5 and Figure S5 show that most correlations were positive, even though noncovalent and covalent inhibitors were treated separately. Moderate negative correlations were obtained for covalent inhibitors from the bibliography (Figure 5A–E and Figure S5A–E). The best Pearson r coefficient (with a value of −0.303) was obtained with the covalent inhibitors from the bibliography docked to the 6LU7 structure with the FRED program (Figure 5A). Thus, for the inhibitors and M‐pro structures assayed here, the MM‐GBSA algorithm was not able to correct the lack of correlation obtained with the docking programs.
Figure 5.
Correlation between the binding energy, calculated as the ∆G of the MM‐GBSA method, and the experimental inhibitory activity, measured as pIC50, of a group of 150 M‐pro inhibitors extracted from the literature (A–E) and 81 M‐pro inhibitors from the COVID Moonshot project (F–J). The protein from the 6LU7 PDB structure was used for docking. The poses with the highest docking scores from each of the five different docking methods (FRED, Glide HTVS, Glide SP, Glide XP, and AutoDock Vina) were used as the input to the MM‐GBSA program to estimate the binding energy. Data has been adjusted to a linear regression and the correlation coefficient is displayed in gray. Covalent and noncovalent inhibitors are also represented separately in orange and blue, respectively. MM‐GBSA, molecular mechanics generalized Born surface area [Color figure can be viewed at wileyonlinelibrary.com]
Although the docking scores or ∆G values estimated with the MM‐GBSA algorithm of known SARS‐CoV‐2 M‐pro inhibitors do not correlate with their potency, we wanted to find out whether these parameters made it possible to discriminate between SARS‐CoV‐2 M‐pro inhibitors and compounds with no SARS‐CoV‐2 M‐pro inhibitory activity. For this reason, we added a group of inactive compounds to the set of COVID Moonshot inhibitors, that is to say, compounds with no activity against M‐pro in the two assays used in the COVID Moonshot project. As inactive compounds do not have IC50 values, we used ROC curves to test whether M‐pro inhibitors could be distinguished from inactive compounds by either the docking score or ∆G estimated with MM‐GBSA. Again, we used the five aforementioned docking methods and the two SARS‐CoV‐2 M‐pro structures. ROC curves are used to assess the performance of a classification method. They represent true positive versus false positive rates and the area under the ROC curve (AUC) measures the performance across all possible classification thresholds. AUC values range from 0 to 1, and represent the probability that a random positive example is positioned before a random negative example, when they are sorted by decreasing docking scores or ∆G values. A random two‐class classification has an AUC value of 0.5. Figure 6 and Figure S6 show the ROC curves and AUC values for the COVID Moonshot M‐pro inhibitors and inactive compounds presented above when using the two M‐pro structures. Most of the AUC values obtained were near to 0.5 (Figure 6 and Figure S6), which means that M‐pro inhibitors generally cannot be distinguished from inactive compounds when using docking scores or ∆G values estimated by the MM‐GBSA method. In some cases, the predictions improved if noncovalent and covalent inhibitors were treated separately (Figure 6 and Figure S6). The best AUC obtained was 0.744 when the 6WQF M‐pro structure and the FRED docking poses of the noncovalent inhibitors were used to estimate the ∆G values with the MM‐GBSA method (Figure S6F). For the 6LU7 structure, the best AUC was obtained with the Glide XP docking poses of the noncovalent inhibitors and ∆G values estimated with the MM‐GBSA method (Figure 6I).
Figure 6.
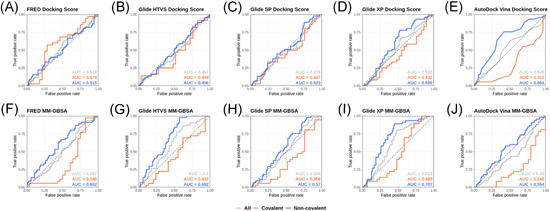
ROC curves obtained from the docking scores of five different docking methods (FRED, Glide HTVS, Glide SP, Glide XP and AutoDock Vina) (A–E) and the ∆G calculated with the MM‐GBSA program (F–J) of a group of known SARS‐CoV‐2 M‐pro inhibitors and compounds with no activity against M‐pro from the COVID Moonshot initiative. The protein from the 6LU7 PDB structure was used for docking. Covalent, noncovalent and the whole set of inhibitors are represented separately. The area under the curve (AUC) values are displayed for each group. MM‐GBSA, molecular mechanics generalized Born surface area [Color figure can be viewed at wileyonlinelibrary.com]
To test if there is a correlation between the pIC50 of a group of SARS‐CoV‐2 M‐pro inhibitors and their ∆G values without using docking, from the COVID Moonshot activity data we selected a group of 40 noncovalent inhibitors that were co‐crystallized with the SARS‐CoV‐2 M‐pro (Supporting Information File 5). The 3‐D structures of the protein‐ligand complex of these compounds, available from Fragalysis, 120 were used to estimate the protein‐ligand affinity, expressed as ∆G values, using the KDeep server. 121 In this server, the structure of the protein and the ligand is represented using 8 pharmacophoric‐like features (i.e., hydrophobic, aromatic, hydrogen‐bond donor and acceptor, positive and negative ionizable, metallic and total excluded volume) and protein‐ligand affinity is predicted using a Deep Convolutional Neural Network pre‐trained with the PDBbind v.2016 database. 121 Figure 7 shows that the correlation between the pIC50 and ∆G values is good, with a Pearson correlation coefficient of −0.688 (p value 9.32E‐07). However, this correlation decreased when the ligands were transformed to SMILES format to lose the binding coordinates of the ligand and were docked with the five docking programs used above (Figure 8 and Figure S7). The best correlation, with a Pearson coefficient value of −0.359 (p value 0.023), was obtained with the FRED docking program, a long way from the correlation of −0.688 obtained with the crystallographic poses. This shows that for the two SARS‐CoV‐2 M‐pro structures and the five docking methods used here, these methodologies were not able to predict the crystallographic pose as the best docking pose, as it has recently been shown. 122 Again this reinforces the idea that it is essential that docking results be validated before use.
Figure 7.
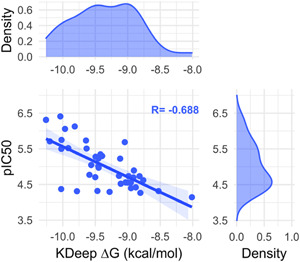
Correlation between the pIC50 and the ∆G values estimated with the KDeep server from the crystallized protein‐ligand complexes of a group of 40 noncovalent SARS‐CoV‐2 M‐pro inhibitors that were co‐crystallized with the SARS‐CoV‐2 M‐pro protein [Color figure can be viewed at wileyonlinelibrary.com]
Figure 8.
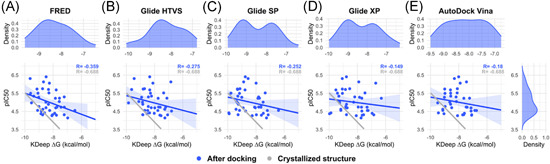
Correlation between the pIC50 and the ∆G values estimated with the KDeep server from the best docking poses from five different docking methods (FRED, Glide HTVS, Glide SP, Glide XP and AutoDock Vina) (A–E) and the 6LU7 M‐pro structure of a group of 40 noncovalent SARS‐CoV‐2 M‐pro inhibitors that were co‐crystallized with the SARS‐CoV‐2 M‐pro protein [Color figure can be viewed at wileyonlinelibrary.com]
5. OUTLOOK
Although the SARS‐CoV‐2 pandemic needed an accelerated process of drug development, measures are required to safeguard this process. Dotolo and coworkers put this idea into words with the proverb “haste makes waste”, and remarked that “what is true for popular wisdom is doubly true for scientific research”. 53 Overconfident statements and conclusions that are not fully supported by experimental evidence are frequently found in articles that use protein‐ligand docking to find SARS‐CoV‐2 M‐pro inhibitors. 54 The methods used and the results obtained by computational approaches must be validated so that these methods are not devalued and efforts focus on the most promising compounds. If docking scores are used to rank or select a set of compounds or to compare the potency of two inhibitor compounds, it must first be shown that there is a correlation between docking scores and activity or potency, for example expressed as the IC50. If there is no such correlation, there is no reason to apply this methodology. A VS protocol can be computationally validated by applying the same VS protocol to a set of both active and inactive compounds. Therefore, the methodology is expected to be able to selectively identify the known active compounds from the validation set. The use of inactive compounds for this validation is crucial because they act as a negative control. Of course, the validation of a VS protocol with active and inactive compounds requires prior knowledge of the target, so it is not applicable if information is scarce, as is the case when targets are new or poorly understood. However, it must be used as an efficient validation tool when more information is available. The set of SARS‐CoV‐2 M‐pro inhibitors and inactive compounds reviewed here could be helpful in validating future screenings.
The flexibility and plasticity of the SARS‐CoV‐2 M‐pro make it a challenging target for small‐molecule inhibitor design. 71 Thus, techniques that have been applied to other targets are not always effective. When using any methodology, such as protein‐ligand docking, it is important to consider not only its well‐known advantages, but also its weaknesses. Binding free energies solely based on docking poses are, in general, insufficiently reliable. 116 Here, in two large groups of SARS‐CoV‐2 M‐pro inhibitors, using two different SARS‐CoV‐2 M‐pro structures and five protein‐ligand docking methods we have shown that there is not a good correlation between the bioactivity and such affinity scores, as the docking scores or the ∆G calculated with the MM‐GBSA method. In addition, these affinity scores cannot distinguish between compounds with or without M‐pro inhibitory activity, especially if covalent and noncovalent inhibitors are analyzed together. This is not to suggest that protein‐ligand docking is useless. On the contrary, it is indeed an invaluable tool for reducing the initial compounds in a virtual screening when used in the initials steps, for plausible pose prediction and for semiquantitative assessment of the inhibitory power of a ligand. 116 There are some successful examples of the use of protein‐ligand docking as an initial step to identify new SARS‐CoV‐2 M‐pro inhibitors, 46 , 63 , 123 , 124 , 125 but none of them used protein‐ligand docking alone. Consensus docking, i.e., the use of different protein‐ligand docking programs, has proved to be also helpful. 29 , 123 When designing a new VS procedure to find SARS‐CoV‐2 M‐pro inhibitors it should be considered if either covalent or noncovalent inhibitors (or both) are expected. The covalent warheads provided herein can be used to differentiate between potentially covalent and noncovalent M‐pro inhibitors and to treat both sets separately. Covalent docking 126 , 127 is an alternative when the aim is to find covalent inhibitors, and it has been used successfully to find M‐pro inhibitors. 128 , 129 In addition to docking, other computational methods have been used to identify new SARS‐CoV‐2 M‐pro inhibitors. These methods include pharmacophores 76 , 130 , 131 ; combined quantum mechanics/molecular mechanics (QM/MM) methods 132 , 133 ; a Docking Consensus Approach and Common Hits Approach that combine molecular dynamics simulations with molecular docking and pharmacophores, respectively 131 ; interactive molecular dynamics simulation in virtual reality 134 ; Quantitative Structure‐activity Relationship models, 115 and binding free energies calculations. 82 , 116 , 135 Among the various methods to calculate protein‐ligand binding affinities, Free Energy Perturbation (FEP) has been used successfully to predict new M‐pro inhibitors. 46 , 124 , 136 Advances in computer power, enhanced sampling algorithms, and increase of force field accuracy has led to newer FEP implementations that has enabled accurate and reliable calculations of protein‐ligand binding free energies. 137 Only when combined with other methods, should the results of automated protein‐ligand docking be considered worthy for further experimental testing.
Supporting information
Supplementary file 1. Details of the 61 reviewed articles and the drugs they predicted to be SARS‐CoV‐2 M‐pro inhibitors. When available, information is provided about the methodology other experimental aspects, database identifiers for the predicted drugs (CAS number, UNII, DrugBank ID, PubChem ID, ChEMBL and ZINC), their IUPAC names and SMILES.
Supplementary file 2. Details of the 150 known SARS‐CoV‐2 M‐pro inhibitors extracted from the bibliography.
Supplementary file 3. Details of the 81 SARS‐CoV‐2 M‐pro inhibitors from the COVID Moonshot project used for validation.
Supplementary file 4. Details of the 113 compounds from the COVID Moonshot project that are inactive against SARS‐CoV‐2 M‐pro and which were used for validation.
Supplementary file 5. Details of the 40 SARS‐CoV‐2 M‐pro inhibitors from the COVID Moonshot project crystallized with the SARS‐CoV‐2 M‐pro and whose inhibitory activity is available.
Supporting information.
ACKNOWLEDGMENTS
We thank OpenEye Scientific Software and Cresset for kindly providing us with a software bursary to use their programs. This manuscript has been edited by the English language service of our university. This project has received funding from the European Union's Horizon 2020 research and innovation program under the Marie Skłodowska‐Curie grant agreement No. 713679 and from the Universitat Rovira i Virgili (grant 2019PFR‐URV‐B2‐69).
Biographies
Guillem Macip Sancho is a PhD student at the Cheminformatics & Nutrition Research Group of the URV currently focused in finding inhibitors of SARS‐CoV‐2 main protease (Mpro). His academic studies previous to this position were a Bachelor Degree in Biochemistry by the Autonomous University of Barcelona (UAB) and a Master's Degree in Bioinformatics and Biostatistics by the Open University of Catalonia—University of Barcelona (UOC‐UB). As a student trainee in the Department of Biophysics at the UAB he has worked in the analysis of Mycoplasma genitalium membrane proteome in order to find therapeutic targets against its infection.
Pol Garcia‐Segura is an undergraduate student of a double BSc. in Biochemistry and Molecular Biology and Biotechnology at Rovira i Virgili University (URV). He has been working as a trainee student in the Cheminformatics & Nutrition Research Group at URV for a couple of years, performing protein‐ligand docking simulations, genomic sequences analysis and data analysis.
Júlia Mestres‐Truyol is an undergraduate student of a double BSc. in Biochemistry and Molecular Biology and Biotechnology at Rovira i Virgili University (URV). She has been part of the Cheminformatics & Nutrition Research Group at URV for two years as a trainee student, where she has worked on protein‐ligand molecular docking, lead optimization and drug development as well as data analysis.
Bryan P. Saldivar‐Espinoza has a bachelor degree in telecommunications engineering and he is a PhD student at the Cheminformatics & Nutrition Research Group of the URV. He has worked applying Machine learning on medical images for tuberculosis and anemia diagnosis. Similarly, he has applied machine learning methods to text mining in a public policy context and process optimization in a business operation framework.
María José Ojeda‐Montes holds a Bsc. in Biotechnology and a Msc. in Nutrition and Metabolism, both by Universitat Rovira i Virgili (URV), and a Msc. in Biostatistics and Bioinformatics by Universitat Oberta de Catalunya (UOC). She obtained the PhD in 2017 in the Cheminformatics & Nutrition Research Group of the URV. Her PhD thesis was focused on the improvement of virtual screening performance using ligand‐based and receptor‐based methods with the aim of identifying bioactive inhibitors for interesting targets in the medicinal chemistry field, such as DPP‐IV involved in Type 2 Diabetes. Then, she was working in Intelligent Pharma company as a computational chemist consultant for different pharma clients and European projects. María José has joined the Molecular Modeling Group of the Swiss Institute Bioinformatics (SIB) as a Postdoctoral Computational Chemist in September 2020 developing and providing methods for computer‐aided drug design, which are applied to academic and industrial drug discovery.
Aleix Gimeno acquired a bachelor in Biochemistry and Molecular Biology at the university Universitat Rovira i Virgili (URV, Tarragona) in 2013. In 2013, he was also an exchange student of the Erasmus program at the Institute of Immunology from the Universitetet i Oslo (Norway). In 2014, he acquired a master of Bioinformatics at the Universitat Autònoma de Barcelona. He was awarded with the CSA Trust Grant in 2015. He obtained the PhD in 2017 at the Cheminformatics & Nutrition Research Group of the URV. Currently, he is a member of the Joint IRB‐BSC‐CRG Program in Computational Biology, Institute for Research in Biomedicine (IRB Barcelona), at The Barcelona Institute of Science and Technology.
Adrià Cereto‐Massagué acquired a bachelor in Biotechnology by the Universitat Rovira i Virgili (URV, Tarragona) in 2011, a MSc in Nutrition and Metabolism in 2012, and PhD in Cheminformatics in 2017 by the same university. Currently, he is the lead bioinformatician of the Center for Omic Sciences (COS) in Reus (Catalonia, Spain), where he works on data analysis and integration from different omics sources. In his beginning as a researcher, he was introduced to the fields of cheminformatics, structure‐activity relationship (SAR) and machine learning, in which he developed tools aid in the process of virtual screening and target fishing.
Santiago Garcia‐Vallvé received his Bachelor degree in Chemistry in 1995 and received his PhD in Biochemistry in 1999 at the Universitat Rovira i Virgili (URV) in Tarragona, Spain. Since 2000 he is member of the Department of Biochemistry and Biotechnology at URV, where in 2003 he won a permanent position. From 1995 to 2009 his research included the development of several databases, servers and tools in the fields of bioinformatics, sequence analysis and molecular evolution for, among others, the prediction of horizontally transferred genes, highly expressed genes and phylogenetic analysis. In 2001, he was a visitor of the Computational Genomics Group at the European Bioinformatics Institute (EBI) in Cambridge, UK. At present, he is member of the Cheminformatics & Nutrition Research Group at URV. His current research interests include the use of cheminformatics tools to detect new bioactive compounds for a given target and the development of new tools to improve virtual screening performance and predict the bioactivity of a compound. He has been involved in the development of several virtual screening procedures for predicting anti‐inflammatory (i.e., IKK‐2 inhibitors) or anti‐diabetic (i.e., DPP‐IV inhibitors and PPAR‐gamma partial agonists) compounds and tools for target fishing. In summer 2016, he was a visitor at the Bender Group at the Department of Chemistry, Cambridge University, UK. Since 2020, part of the research of the Cheminformatics & Nutrition Research Group is focused on SARS‐CoV‐2 and finding inhibitors of the main protease (Mpro).
Gerard Pujadas is graduate in Chemistry by the Universitat de Barcelona (1992) and PhD in Chemistry by the Universitat Rovira i Virgili (URV, Tarragona) in 1998. In his beginning as a researcher, he was introduced to the fields of Bioinformatics and protein structure analysis and its relationship with sequence evolution. In 2001 he made a post‐doctoral stay at Prof. Richard Haser's lab at the Institut de Biologie et Chimie des Protéines (Lyon, France). After this post‐doctoral stay, he returned to the URV where in September 2002 he won a permanent position at the Biochemistry and Biotechnology Department. Since then, his main contributions to science has been the development of: (1) several virtual screening workflows for predicting molecules that act as anti‐inflammatories (i.e., IKK‐2 inhibitors) or anti‐diabetics (i.e., DPP‐IV inhibitors and PPAR‐gamma partial agonists); (2) tools for validating virtual screening workflows (i.e., DecoyFinder); and (3) tools for validating the coordinates of drug‐target complexes (i.e., VHELIBS). Since April 2014, he leads the research group on “Cheminformatics & Nutrition” at the URV that aims to: (1) develop potent antidiabetic compounds from lead molecules identified with our virtual screening workflows; and (2) develop new tools and databases for improving virtual screening workflows performance.
Macip G, Garcia‐Segura P, Mestres‐Truyol J, et al. Haste makes waste: a critical review of docking‐based virtual screening in drug repurposing for SARS‐CoV‐2 main protease (M‐pro) inhibition. Med Res Rev. 2022;42:744‐769. 10.1002/med.21862
Guillem Macip, Pol Garcia‐Segura, Júlia Mestres‐Truyol contributed equally to this study.
Contributor Information
Santiago Garcia‐Vallvé, Email: santi.garcia-vallve@urv.cat.
Gerard Pujadas, Email: gerard.pujadas@gmail.com.
REFERENCES
- 1. Dong E, Du H, Gardner L. An interactive web‐based dashboard to track COVID‐19 in real time. Lancet Infect Dis. 2020;20(5):533‐534. 10.1016/S1473-3099(20)30120-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Worldometers Coronavirus Update (Live) . Accessed March 22, 2021. https://www.worldometers.info/coronavirus/
- 3. Nicola M, Alsafi Z, Sohrabi C, et al. The socio‐economic implications of the coronavirus pandemic (COVID‐19): a review. Int J Surg. 2020;78:185‐193. 10.1016/j.ijsu.2020.04.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Odone A, Galea S, Stuckler D, Signorelli C. University Vita‐Salute San Raffaele COVID‐19 literature monitoring working group. The first 10 000 COVID‐19 papers in perspective: are we publishing what we should be publishing? Eur J Public Health. 2020;30(5):849‐850. 10.1093/eurpub/ckaa170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Palayew A, Norgaard O, Safreed‐Harmon K, Andersen TH, Rasmussen LN, Lazarus JV. Pandemic publishing poses a new COVID‐19 challenge. Nat Hum Behav. 2020;4(7):666‐669. 10.1038/s41562-020-0911-0 [DOI] [PubMed] [Google Scholar]
- 6. Guha R, Willighagen E, Zdrazil B, Jeliazkova N. What is the role of cheminformatics in a pandemic? J Cheminform. 2021;13:16. 10.1186/s13321-021-00491-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Glasziou PP, Sanders S, Hoffmann T. Waste in covid‐19 research. Br Med J. 2020;369:m1847. 10.1136/bmj.m1847 [DOI] [PubMed] [Google Scholar]
- 8. Masters PS. The molecular biology of coronaviruses. Adv Virus Res. 2006;66:193‐292. 10.1016/S0065-3527(06)66005-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mishra SK, Tripathi T. One year update on the COVID‐19 pandemic: where are we now? Acta Trop. 2021;214:105778. 10.1016/j.actatropica.2020.105778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ksiazek TG, Erdman D, Goldsmith CS, et al. A novel coronavirus associated with severe acute respiratory syndrome. N Engl J Med. 2003;348(20):1953‐1966. 10.1056/NEJMoa030781 [DOI] [PubMed] [Google Scholar]
- 11. Zaki AM, van Boheemen S, Bestebroer TM, Osterhaus ADME, Fouchier RAM. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med. 2012;367(19):1814‐1820. 10.1056/NEJMoa1211721 [DOI] [PubMed] [Google Scholar]
- 12. Coronaviridae Study Group of the International Committee on Taxonomy of Viruses . The species Severe acute respiratory syndrome‐related coronavirus: classifying 2019‐nCoV and naming it SARS‐CoV‐2. Nat Microbiol. 2020;5(4):536‐544. 10.1038/s41564-020-0695-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wu F, Zhao S, Yu B, et al. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579(7798):265‐269. 10.1038/s41586-020-2008-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kim D, Lee J‐Y, Yang J‐S, Kim JW, Kim VN, Chang H. The architecture of SARS‐CoV‐2 transcriptome. Cell. 2020;181(4):914‐921. 10.1016/j.cell.2020.04.011.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chen Y, Liu Q, Guo D. Emerging coronaviruses: genome structure, replication, and pathogenesis. J Med Virol. 2020;92(4):418‐423. 10.1002/jmv.25681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang M, Cao R, Zhang L, et al. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019‐nCoV) in vitro. Cell Res. 2020;30(3):269‐271. 10.1038/s41422-020-0282-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Furuta Y, Komeno T, Nakamura T. Favipiravir (T‐705), a broad spectrum inhibitor of viral RNA polymerase. Proc Jpn Acad Ser B Phys Biol Sci. 2017;93(7):449‐463. 10.2183/pjab.93.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Amin SA, Banerjee S, Gayen S, Jha T. Protease targeted COVID‐19 drug discovery: what we have learned from the past SARS‐CoV inhibitors? Eur J Med Chem. 2021;215:113294. 10.1016/j.ejmech.2021.113294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Petushkova AI, Zamyatnin AA. Papain‐like proteases as coronaviral drug targets: current inhibitors, opportunities, and limitations. Pharmaceuticals. 2020;13(10):277. 10.3390/ph13100277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Banerjee R, Perera L, Tillekeratne LMV. Potential SARS‐CoV‐2 main protease inhibitors. Drug Discov Today. 2021;26(3):804‐816. 10.1016/j.drudis.2020.12.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Xiong M, Su H, Zhao W, Xie H, Shao Q, Xu Y. What coronavirus 3C‐like protease tells us: from structure, substrate selectivity, to inhibitor design. Med Res Rev. 2021;41:1965‐1998. 10.1002/med.21783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Monteil V, Kwon H, Prado P, et al. Inhibition of SARS‐CoV‐2 infections in engineered human tissues using clinical‐grade soluble human ACE2. Cell. 2020;181(4):905‐913.e7. 10.1016/j.cell.2020.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gautret P, Lagier J‐C, Parola P, et al. Hydroxychloroquine and azithromycin as a treatment of COVID‐19: results of an open‐label non‐randomized clinical trial. Int J Antimicrob Agents. 2020;56(1):105949. 10.1016/j.ijantimicag.2020.105949 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 24. Nojomi M, Yassin Z, Keyvani H, et al. Effect of Arbidol (Umifenovir) on COVID‐19: a randomized controlled trial. BMC Infect Dis. 2020;20(1):954. 10.1186/s12879-020-05698-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lee WS, Wheatley AK, Kent SJ, DeKosky BJ. Antibody‐dependent enhancement and SARS‐CoV‐2 vaccines and therapies. Nat Microbiol. 2020;5(10):1185‐1191. 10.1038/s41564-020-00789-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhou G, Zhao Q. Perspectives on therapeutic neutralizing antibodies against the Novel Coronavirus SARS‐CoV‐2. Int J Biol Sci. 2020;16(10):1718‐1723. 10.7150/ijbs.45123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Stasi C, Fallani S, Voller F, Silvestri C. Treatment for COVID‐19: an overview. Eur J Pharmacol. 2020;889:173644. 10.1016/j.ejphar.2020.173644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tu Y‐F, Chien C‐S, Yarmishyn AA, et al. A Review of SARS‐CoV‐2 and the ongoing clinical trials. Int J Mol Sci. 2020;21(7):2657. 10.3390/ijms21072657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gimeno A, Mestres‐Truyol J, Ojeda‐Montes MJ, et al. Prediction of novel inhibitors of the main protease (M‐pro) of SARS‐CoV‐2 through consensus docking and drug reposition. Int J Mol Sci. 2020;21(11):3793. 10.3390/ijms21113793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jin Z, Du X, Xu Y, et al. Structure of Mpro from SARS‐CoV‐2 and discovery of its inhibitors. Nature. 2020;582(7811):289‐293. 10.1038/s41586-020-2223-y [DOI] [PubMed] [Google Scholar]
- 31. Kneller DW, Phillips G, O'neill HM, et al. Structural plasticity of SARS‐CoV‐2 3CL Mpro active site cavity revealed by room temperature X‐ray crystallography. Nat Commun. 2020;11(1):3202. 10.1038/s41467-020-16954-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Flare , version 4.0.2., Cresset®, Litlington, Cambridgeshire, UK.
- 33. Cheeseright T, Mackey M, Rose S, Vinter A. Molecular field extrema as descriptors of biological activity: definition and validation. J Chem Inf Model. 2006;46(2):665‐676. 10.1021/ci050357s [DOI] [PubMed] [Google Scholar]
- 34. Bauer MR, Mackey MD. Electrostatic complementarity as a fast and effective tool to optimize binding and selectivity of protein‐ligand complexes. J Med Chem. 2019;62(6):3036‐3050. 10.1021/acs.jmedchem.8b01925 [DOI] [PubMed] [Google Scholar]
- 35. Pushpakom S, Iorio F, Eyers PA, et al. Drug repurposing: progress, challenges and recommendations. Nat Rev Drug Discov. 2019;18(1):41‐58. 10.1038/nrd.2018.168 [DOI] [PubMed] [Google Scholar]
- 36. Pinzi L, Rastelli G. Molecular docking: shifting paradigms in drug discovery. Int J Mol Sci. 2019;20(18):4331. 10.3390/ijms20184331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gimeno A, Ojeda‐Montes MJ, Tomás‐Hernández S, et al. The light and dark sides of virtual screening: what is there to know? Int J Mol Sci. 2019;20(6):1375. 10.3390/ijms20061375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cui W, Yang K, Yang H. Recent progress in the drug development targeting SARS‐CoV‐2 main protease as treatment for COVID‐19. Front Mol Biosci. 2020;7:616341. 10.3389/fmolb.2020.616341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pillaiyar T, Wendt LL, Manickam M, Easwaran M. The recent outbreaks of human coronaviruses: a medicinal chemistry perspective. Med Res Rev. 2021;41(1):72‐135. 10.1002/med.21724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wang X, Guan Y. COVID‐19 drug repurposing: a review of computational screening methods, clinical trials, and protein interaction assays. Med Res Rev. 2021;41(1):5‐28. 10.1002/med.21728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cannalire R, Cerchia C, Beccari AR, Di Leva FS, Summa V. Targeting SARS‐CoV‐2 proteases and polymerase for COVID‐19 treatment: state of the art and future opportunities. J Med Chem. 2020. 10.1021/acs.jmedchem.0c01140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Xu J, Xue Y, Zhou R, Shi P, Li H, Zhou J. Drug repurposing approach to combating coronavirus: potential drugs and drug targets. Med Res Rev. 2021;41(3):1375‐1426. 10.1002/med.21763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chen CC, Yu X, Kuo CJ, et al. Overview of antiviral drug candidates targeting coronaviral 3C‐like main proteases. FEBS J. 2021;288(17):5089‐5121. 10.1111/febs.15696 [DOI] [PubMed] [Google Scholar]
- 44. Lengauer T, Rarey M. Computational methods for biomolecular docking. Curr Opin Struct Biol. 1996;6(3):402‐406. 10.1016/s0959-440x(96)80061-3 [DOI] [PubMed] [Google Scholar]
- 45. Pantsar T, Poso A. Binding affinity via docking: fact and fiction. Molecules. 2018;23(8):1899. 10.3390/molecules23081899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Li Z, Li X, Huang Y‐Y, et al. Identify potent SARS‐CoV‐2 main protease inhibitors via accelerated free energy perturbation‐based virtual screening of existing drugs. Proc Natl Acad Sci USA. 2020;117(44):27381‐27387. 10.1073/pnas.2010470117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Fiorucci D, Milletti E, Orofino F, Brizzi A, Mugnaini C, Corelli F. Computational drug repurposing for the identification of SARS‐CoV‐2 main protease inhibitors. J Biomol Struct Dyn. 2020;39(16):5121‐6248. 10.1080/07391102.2020.1796805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kanhed AM, Patel DV, Teli DM, Patel NR, Chhabria MT, Yadav MR. Identification of potential Mpro inhibitors for the treatment of COVID‐19 by using systematic virtual screening approach. Mol Divers. 2021;25(1):383‐401. 10.1007/s11030-020-10130-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Feher M, Williams CI. Effect of input differences on the results of docking calculations. J Chem Inf Model. 2009;49(7):1704‐1714. 10.1021/ci9000629 [DOI] [PubMed] [Google Scholar]
- 50. Mohan V, Gibbs A, Cummings M, Jaeger E, DesJarlais R. Docking: successes and Challenges. Curr Pharm Des. 2005;11(3):323‐333. 10.2174/1381612053382106 [DOI] [PubMed] [Google Scholar]
- 51. Meyer‐Almes F‐J. Repurposing approved drugs as potential inhibitors of 3CL‐protease of SARS‐CoV‐2: virtual screening and structure based drug design. Comput Biol Chem. 2020;88:107351. 10.1016/j.compbiolchem.2020.107351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Mandour YM, Zlotos DP, Alaraby Salem M. A multi‐stage virtual screening of FDA‐approved drugs reveals potential inhibitors of SARS‐CoV‐2 main protease. J Biomol Struct Dyn. 2020:1‐12. 10.1080/07391102.2020.1837680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Dotolo S, Marabotti A, Facchiano A, Tagliaferri R. A review on drug repurposing applicable to COVID‐19. Brief Bioinform. 2020;22(September):726‐741. bbaa288 10.1093/bib/bbaa288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bellera CL, Llanos M, Gantner ME, et al. Can drug repurposing strategies be the solution to the COVID‐19 crisis? Expert Opin Drug Discov. 2021;16(6):605‐612. 10.1080/17460441.2021.1863943 [DOI] [PubMed] [Google Scholar]
- 55. Arun KG, Sharanya CS, Abhithaj J, Francis D, Sadasivan C. Drug repurposing against SARS‐CoV‐2 using E‐pharmacophore based virtual screening, molecular docking and molecular dynamics with main protease as the target. J Biomol Struct Dyn. 2020;39:1‐12. 10.1080/07391102.2020.1779819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Mitra K, Ghanta P, Acharya S, Chakrapani G, Ramaiah B, Doble M. Dual inhibitors of SARS‐CoV‐2 proteases: pharmacophore and molecular dynamics based drug repositioning and phytochemical leads. J Biomol Struct Dyn. 2020;39(16):6324‐6337. 10.1080/07391102.2020.1796802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. J A, Francis D, C SS, K GA, C S, Variyar EJ. Repurposing simeprevir, calpain inhibitor IV and a cathepsin F inhibitor against SARS‐CoV‐2 and insights into their interactions with Mpro. J Biomol Struct Dyn. 2020:1‐12. 10.1080/07391102.2020.1813200 [DOI] [PubMed] [Google Scholar]
- 58. Mahdi M, Mótyán JA, Szojka ZI, Golda M, Miczi M, Tőzsér J. Analysis of the efficacy of HIV protease inhibitors against SARS‐CoV‐2′s main protease. Virol J. 2020;17:190. 10.1186/s12985-020-01457-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Li Y, Xie Z, Lin W, et al. Efficacy and safety of lopinavir/ritonavir or arbidol in adult patients with mild/moderate COVID‐19: an exploratory randomized controlled trial. Med. 2020;1(1):105‐113.e4. 10.1016/j.medj.2020.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Cao B, Wang Y, Wen D, et al. A trial of lopinavir–ritonavir in adults hospitalized with severe covid‐19. N Engl J Med. 2020;382(19):1787‐1799. 10.1056/NEJMoa2001282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. RECOVERY Collaborative G. Lopinavir–ritonavir in patients admitted to hospital with COVID‐19 (RECOVERY): a randomised, controlled, open‐label, platform trial. Lancet. 2020;396(10259):1345‐1352. 10.1016/S0140-6736(20)32013-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Chakraborti S, Bheemireddy S, Srinivasan N. Repurposing drugs against the main protease of SARS‐CoV‐2: mechanism‐based insights supported by available laboratory and clinical data. Mol Omi. 2020;16(5):474‐491. 10.1039/d0mo00057d [DOI] [PubMed] [Google Scholar]
- 63. Vatansever EC, Yang KS, Drelich AK, et al. Bepridil is potent against SARS‐CoV‐2 in vitro. Proc Natl Acad Sci USA. 2021;118(10):e2012201118. 10.1073/pnas.2012201118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Hung HC, Ke YY, Huang SY, et al. Discovery of M protease inhibitors encoded by SARS‐CoV‐2. Antimicrob Agents Chemother. 2020;64(9):e00872‐20. 10.1128/AAC.00872-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Hattori S, Higshi‐Kuwata N, Raghavaiah J, et al. GRL‐0920, an Indole Chloropyridinyl Ester, Completely Blocks SARS‐CoV‐2 Infection. Weiss LM, ed. MBio. 2020;11(4):e01833‐20. 10.1128/mBio.01833-20 [DOI] [PMC free article] [PubMed]
- 66. Mittal L, Kumari A, Srivastava M, Singh M, Asthana S. Identification of potential molecules against COVID‐19 main protease through structure‐guided virtual screening approach. J Biomol Struct Dyn. 2020;39(10):1‐19. 10.1080/07391102.2020.1768151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Cavasotto CN, Di Filippo JI. In silico Drug Repurposing for COVID‐19: targeting SARS‐CoV‐2 Proteins through Docking and Consensus Ranking. Mol Inform. 2021;40(1):e2000115. 10.1002/minf.202000115 [DOI] [PubMed] [Google Scholar]
- 68. Su HX, Yao S, Zhao WF, et al. Anti‐SARS‐CoV‐2 activities in vitro of Shuanghuanglian preparations and bioactive ingredients. Acta Pharmacol Sin. 2020;41(9):1167‐1177. 10.1038/s41401-020-0483-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Hoffman RL, Kania RS, Brothers MA, et al. Discovery of Ketone‐based covalent inhibitors of coronavirus 3CL proteases for the potential therapeutic treatment of COVID‐19. J Med Chem. 2020;63(21):12725‐12747. 10.1021/acs.jmedchem.0c01063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Qiao J, Li Y‐S, Zeng R, et al. SARS‐CoV‐2 Mpro inhibitors with antiviral activity in a transgenic mouse model. Science. 2021;371(6536):1374‐1378. 10.1126/science.abf1611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Bzówka M, Mitusińska K, Raczyńska A, Samol A, Tuszyński JA, Góra A. Structural and evolutionary analysis indicate that the SARS‐CoV‐2 Mpro is a challenging target for small‐molecule inhibitor design. Int J Mol Sci. 2020;21(9):3099. 10.3390/ijms21093099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kumar A, Loharch S, Kumar S, Ringe RP, Parkesh R. Exploiting cheminformatic and machine learning to navigate the available chemical space of potential small molecule inhibitors of SARS‐CoV‐2. Comput Struct Biotechnol J. 2021;19:424‐438. 10.1016/j.csbj.2020.12.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Gurard‐Levin ZA, Liu C, Jekle A, et al. Evaluation of SARS‐CoV‐2 3C‐like protease inhibitors using self‐assembled monolayer desorption ionization mass spectrometry. Antiviral Res. 2020;182:104924. 10.1016/j.antiviral.2020.104924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Ma C, Hu Y, Townsend JA, et al. Ebselen, disulfiram, carmofur, PX‐12, tideglusib, and shikonin are nonspecific promiscuous SARS‐CoV‐2 main protease inhibitors. ACS Pharmacol Transl Sci. 2020;3(6):1265‐1277. 10.1021/acsptsci.0c00130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Jan J‐T, Cheng TR, Juang Y‐P, et al. Identification of existing pharmaceuticals and herbal medicines as inhibitors of SARS‐CoV‐2 infection. Proc Natl Acad Sci USA. 2021;118(5):e2021579118. 10.1073/pnas.2021579118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Pathak N, Chen Y‐T, Hsu Y‐C, et al. Uncovering flexible active site conformations of SARS‐CoV‐2 3CL proteases through protease pharmacophore clusters and COVID‐19 drug repurposing. ACS Nano. 2021;15(1):857‐872. 10.1021/acsnano.0c07383 [DOI] [PubMed] [Google Scholar]
- 77. Bhimraj A, Morgan RL, Shumaker AH, et al. Infectious diseases society of america guidelines on the treatment and management of patients with coronavirus disease 2019 (COVID‐19). Clin Infect Dis. 2020. ciaa478 10.1093/cid/ciaa478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Ma C, Sacco MD, Hurst B, et al. Boceprevir, GC‐376, and calpain inhibitors II, XII inhibit SARS‐CoV‐2 viral replication by targeting the viral main protease. Cell Res. 2020;30(8):678‐692. 10.1038/s41422-020-0356-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Rawson JMO, Duchon A, Nikolaitchik OA, Pathak VK, Hu W‐S. Development of a cell‐based luciferase complementation assay for identification of SARS‐CoV‐2 3CLpro inhibitors. Viruses. 2021;13(2):173. 10.3390/v13020173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Bauer RA. Covalent inhibitors in drug discovery: from accidental discoveries to avoided liabilities and designed therapies. Drug Discov Today. 2015;20(9):1061‐1073. 10.1016/j.drudis.2015.05.005 [DOI] [PubMed] [Google Scholar]
- 81. Lonsdale R, Burgess J, Colclough N, et al. Expanding the armory: predicting and tuning covalent warhead reactivity. J Chem Inf Model. 2017;57(12):3124‐3137. 10.1021/acs.jcim.7b00553 [DOI] [PubMed] [Google Scholar]
- 82. Mondal D, Warshel A. Exploring the mechanism of covalent inhibition: simulating the binding free energy of α‐ketoamide inhibitors of the main protease of SARS‐CoV‐2. Biochemistry. 2020;59(48):4601‐4608. 10.1021/acs.biochem.0c00782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Kneller DW, Phillips G, O'neill HM, et al. Room‐temperature X‐ray crystallography reveals the oxidation and reactivity of cysteine residues in SARS‐CoV‐2 3CL Mpro: insights into enzyme mechanism and drug design. IUCrJ. 2020;7(Pt 6):1028‐1035. 10.1107/S2052252520012634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Kneller DW, Phillips G, Weiss KL, et al. Unusual zwitterionic catalytic site of SARS–CoV‐2 main protease revealed by neutron crystallography. J Biol Chem. 2020;295(50):17365‐17373. 10.1074/jbc.AC120.016154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Verma N, Henderson JA, Shen J. Proton‐coupled conformational activation of SARS coronavirus main proteases and opportunity for designing small‐molecule broad‐spectrum targeted covalent inhibitors. J Am Chem Soc. 2020;142(52):21883‐21890. 10.1021/jacs.0c10770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Singh J, Petter RC, Baillie TA, Whitty A. The resurgence of covalent drugs. Nat Rev Drug Discov. 2011;10(4):307‐317. 10.1038/nrd3410 [DOI] [PubMed] [Google Scholar]
- 87. Rathnayake AD, Zheng J, Kim Y, et al. 3C‐like protease inhibitors block coronavirus replication in vitro and improve survival in MERS‐CoV–infected mice. Sci Transl Med. 2020;12(557):eabc5332. 10.1126/scitranslmed.abc5332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Vuong W, Khan MB, Fischer C, et al. Feline coronavirus drug inhibits the main protease of SARS‐CoV‐2 and blocks virus replication. Nat Commun. 2020;11:4282. 10.1038/s41467-020-18096-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Fu L, Ye F, Feng Y, et al. Both boceprevir and GC376 efficaciously inhibit SARS‐CoV‐2 by targeting its main protease. Nat Commun. 2020;11:4417. 10.1038/s41467-020-18233-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Wang Y‐C, Yang W‐H, Yang C‐S, et al. Structural basis of SARS‐CoV‐2 main protease inhibition by a broad‐spectrum anti‐coronaviral drug. Am J Cancer Res. 2020;10(8):2535‐2545. [PMC free article] [PubMed] [Google Scholar]
- 91. Yang KS, Ma XR, Ma Y, et al. A quick route to multiple highly potent SARS‐CoV‐2 main protease inhibitors. ChemMedChem. 2021;16(6):942‐948. 10.1002/cmdc.202000924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Zhu W, Xu M, Chen CZ, et al. Identification of SARS‐CoV‐2 3CL protease inhibitors by a quantitative high‐throughput screening. ACS Pharmacol Transl Sci. 2020;3(5):1008‐1016. 10.1021/acsptsci.0c00108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Iketani S, Forouhar F, Liu H, et al. Lead compounds for the development of SARS‐CoV‐2 3CL protease inhibitors. Nat Commun. 2021;12(1):2016. 10.1038/s41467-021-22362-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Sacco MD, Ma C, Lagarias P, et al. Structure and inhibition of the SARS‐CoV‐2 main protease reveal strategy for developing dual inhibitors against M pro and cathepsin L. Sci Adv. 2020;6(50):eabe0751. 10.1126/sciadv.abe0751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. COVID Moonshot. Accessed March 22, 2021. https://covid.postera.ai/covid
- 96. Achdout H, Aimon A, Bar‐David E, et al. COVID moonshot: open science discovery of SARS‐CoV‐2 main protease inhibitors by combining crowdsourcing, high‐throughput experiments, computational simulations, and machine learning. bioRxiv. 2020;2020 10.1101/2020.10.29.339317 [DOI] [Google Scholar]
- 97. Nar H, Schnapp G, Hucke O, Hardman TC, Klein T. Action of dipeptidyl peptidase‐4 inhibitors on SARS‐CoV‐2 main protease. ChemMedChem. 2021;16(9):1425‐1426. 10.1002/cmdc.202000921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Warren GL, Andrews CW, Capelli A‐M, et al. A critical assessment of docking programs and scoring functions. J Med Chem. 2006;49(20):5912‐5931. 10.1021/jm050362n [DOI] [PubMed] [Google Scholar]
- 99. Erickson JA, Jalaie M, Robertson DH, Lewis RA, Vieth M. Lessons in molecular recognition: the effects of ligand and protein flexibility on molecular docking accuracy. J Med Chem. 2004;47(1):45‐55. 10.1021/jm030209y [DOI] [PubMed] [Google Scholar]
- 100. Amaro RE, Baudry J, Chodera J, et al. Ensemble docking in drug discovery. Biophys J. 2018;114(10):2271‐2278. 10.1016/j.bpj.2018.02.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Ramírez D, Caballero J. Is it reliable to take the molecular docking top scoring position as the best solution without considering available structural data? Molecules. 2018;23(5):1038. 10.3390/molecules23051038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Sethi A, Joshi K, Sasikala K, Alvala M. Molecular docking in modern drug discovery: principles and recent applications. In: Gaitonde V, Karmakar P, Trivedi A, eds. Drug Discovery and Development ‐ New Advances. IntechOpen 2020. 10.5772/intechopen.85991 [DOI] [Google Scholar]
- 103. OEDOCKING 3.4.0.2: OpenEye Scientific Software, Santa Fe, NM, USA, 2019.
- 104. Schrödinger Release 2020‐3 . Glide, Schrödinger. New York, NY: LLC, 2020. [Google Scholar]
- 105. Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31(2):455‐461. 10.1002/jcc.21334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Friesner RA, Banks JL, Murphy RB, et al. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem. 2004;47(7):1739‐1749. 10.1021/jm0306430 [DOI] [PubMed] [Google Scholar]
- 107. Friesner RA, Murphy RB, Repasky MP, et al. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein‐ligand complexes. J Med Chem. 2006;49(21):6177‐6196. 10.1021/jm051256o [DOI] [PubMed] [Google Scholar]
- 108. Schrödinger Release 2020‐3 . Protein Preparation Wizard, Schrödinger. New York, NY, USA: LLC; 2020. [Google Scholar]
- 109. Schrödinger Release 2020‐3 . Epik, Schrödinger. New York, NY, USA: LLC; 2020. [Google Scholar]
- 110. Olsson MHM, Søndergaard CR, Rostkowski M, Jensen JH. PROPKA3: consistent treatment of internal and surface residues in empirical pKa predictions. J Chem Theory Comput. 2011;7(2):525‐537. 10.1021/ct100578z [DOI] [PubMed] [Google Scholar]
- 111. Morris GM, Huey R, Lindstrom W, et al. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem. 2009;30(16):2785‐2791. 10.1002/jcc.21256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. OMEGA 3.1.1.2: OpenEye Scientific Software, Santa Fe, NM, USA, 2019.
- 113. Schrödinger Release 2020‐3 . LigPrep, Schrödinger. New York, NY: LLC; 2020. [Google Scholar]
- 114. QUACPAC 2.0.2.2 : OpenEye Scientific Software, Santa Fe, NM, USA, 2019.
- 115. Alves VM, Bobrowski T, Melo‐Filho CC, et al. QSAR modeling of SARS‐CoV M pro inhibitors identifies sufugolix, cenicriviroc, proglumetacin, and other drugs as candidates for repurposing against SARS‐CoV‐2. Mol Inform. 2021;40(1):2000113. 10.1002/minf.202000113 [DOI] [PubMed] [Google Scholar]
- 116. Macchiagodena M, Pagliai M, Karrenbrock M, Guarnieri G, Iannone F, Procacci P. Virtual double‐system single‐box: a nonequilibrium alchemical technique for absolute binding free energy calculations: application to ligands of the SARS‐CoV‐2 main protease. J Chem Theory Comput. 2020;16(11):7160‐7172. 10.1021/acs.jctc.0c00634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Genheden S, Ryde U. The MM/PBSA and MM/GBSA methods to estimate ligand‐binding affinities. Expert Opin Drug Discov. 2015;10(5):449‐461. 10.1517/17460441.2015.1032936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Wang E, Sun H, Wang J, et al. End‐point binding free energy calculation with MM/PBSA and MM/GBSA: strategies and applications in drug design. Chem Rev. 2019;119(16):9478‐9508. 10.1021/acs.chemrev.9b00055 [DOI] [PubMed] [Google Scholar]
- 119. Schrödinger Release 2020‐3 . Prime, Schrödinger. New York, NY, USA: LLC; 2020. [Google Scholar]
- 120. Mpro : Fragalysis. Accessed March 22, 2021. https://fragalysis.diamond.ac.uk/viewer/react/preview/target/Mpro
- 121. Jiménez J, Škalič M, Martínez‐Rosell G, De Fabritiis G. KDEEP: protein‐ligand absolute binding affinity prediction via 3D‐convolutional neural networks. J Chem Inf Model. 2018;58(2):287‐296. 10.1021/acs.jcim.7b00650 [DOI] [PubMed] [Google Scholar]
- 122. Zev S, Raz K, Schwartz R, Tarabeh R, Gupta PK, Major DT. Benchmarking the ability of common docking programs to correctly reproduce and score binding modes in SARS‐CoV‐2 protease Mpro. J Chem Inf Model. 2021;61(6):2957‐2966. 10.1021/acs.jcim.1c00263 [DOI] [PubMed] [Google Scholar]
- 123. Ghahremanpour MM, Tirado‐Rives J, Deshmukh M, et al. Identification of 14 known drugs as inhibitors of the main protease of SARS‐CoV‐2. ACS Med Chem Lett. 2020;11(12):2526‐2533. 10.1021/acsmedchemlett.0c00521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Zhang C‐H, Stone EA, Deshmukh M, et al. Potent noncovalent inhibitors of the main protease of SARS‐CoV‐2 from molecular sculpting of the drug perampanel guided by free energy perturbation calculations. ACS Cent Sci. 2021;7(3):467‐475. 10.1021/acscentsci.1c00039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Gupta A, Rani C, Pant P, et al. Structure‐based virtual screening and biochemical validation to discover a potential inhibitor of the SARS‐CoV‐2 main protease. ACS Omega. 2020;5(51):33151‐33161. 10.1021/acsomega.0c04808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Scarpino A, Ferenczy GG, Keserű GM. Comparative evaluation of covalent docking tools. J Chem Inf Model. 2018;58(7):1441‐1458. 10.1021/acs.jcim.8b00228 [DOI] [PubMed] [Google Scholar]
- 127. Kumalo H, Bhakat S, Soliman M. Theory and applications of covalent docking in drug discovery: merits and pitfalls. Molecules. 2015;20(2):1984‐2000. 10.3390/molecules20021984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Shitrit A, Zaidman D, Kalid O, et al. Conserved interactions required for inhibition of the main protease of severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2). Sci Rep. 2020;10:20808. 10.1038/s41598-020-77794-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Amendola G, Ettari R, Previti S, et al. Lead discovery of SARS‐CoV‐2 main protease inhibitors through covalent docking‐based virtual screening. J Chem Inf Model. 2021;61(4):2062‐2073. 10.1021/acs.jcim.1c00184 [DOI] [PubMed] [Google Scholar]
- 130. Kouznetsova VL, Huang DZ, Tsigelny IF. Potential SARS‐CoV‐2 protease Mpro inhibitors: Repurposing FDA‐approved drugs. Phys Biol. 2021;18(2):025001. 10.1088/1478-3975/abcb66 [DOI] [PubMed] [Google Scholar]
- 131. Battisti V, Wieder O, Garon A, Seidel T, Urban E, Langer T. A computational approach to identify potential novel inhibitors against the coronavirus SARS‐CoV‐2. Mol Inform. 2020;39(10):e2000090. 10.1002/minf.202000090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Arafet K, Serrano‐Aparicio N, Lodola A, et al. Mechanism of inhibition of SARS‐CoV‐2 Mpro by N3 peptidyl Michael acceptor explained by QM/MM simulations and design of new derivatives with tunable chemical reactivity. Chem Sci. 2021;12(4):1433‐1444. 10.1039/d0sc06195f [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Ramos‐Guzmán CA, Ruiz‐Pernía JJ, Tuñón I. Multiscale simulations of SARS‐CoV‐2 3CL protease inhibition with aldehyde derivatives. Role of protein and inhibitor conformational changes in the reaction mechanism. ACS Catal. 2021;11(7):4157‐4168. 10.1021/acscatal.0c05522 [DOI] [PubMed] [Google Scholar]
- 134. Deeks HM, Walters RK, Barnoud J, Glowacki DR, Mulholland AJ. Interactive molecular dynamics in virtual reality is an effective tool for flexible substrate and inhibitor docking to the SARS‐CoV‐2 main protease. J Chem Inf Model. 2020;60(12):5803‐5814. 10.1021/acs.jcim.0c01030 [DOI] [PubMed] [Google Scholar]
- 135. Jukič M, Janežič D, Bren U. Ensemble docking coupled to linear interaction energy calculations for identification of coronavirus main protease (3CLpro) Non‐Covalent Small‐Molecule Inhibitors. Molecules. 2020;25(24):5808. 10.3390/molecules25245808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Ngo ST, Tam NM, Pham MQ, Nguyen TH. Benchmark of popular free energy approaches revealing the inhibitors binding to SARS‐CoV−2 Mpro. J Chem Inf Model. 2021;61(5):2302‐2312. 10.1021/acs.jcim.1c00159 [DOI] [PubMed] [Google Scholar]
- 137. Wang L, Chambers J, Abel R. Protein‐ligand binding free energy calculations with FEP. In: Bonomi M, Camilloni C, eds. Biomolecular Simulations. Methods in Molecular Biology. Vol 2022. Humana; 2019:201‐232. 10.1007/978-1-4939-9608-7_9 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary file 1. Details of the 61 reviewed articles and the drugs they predicted to be SARS‐CoV‐2 M‐pro inhibitors. When available, information is provided about the methodology other experimental aspects, database identifiers for the predicted drugs (CAS number, UNII, DrugBank ID, PubChem ID, ChEMBL and ZINC), their IUPAC names and SMILES.
Supplementary file 2. Details of the 150 known SARS‐CoV‐2 M‐pro inhibitors extracted from the bibliography.
Supplementary file 3. Details of the 81 SARS‐CoV‐2 M‐pro inhibitors from the COVID Moonshot project used for validation.
Supplementary file 4. Details of the 113 compounds from the COVID Moonshot project that are inactive against SARS‐CoV‐2 M‐pro and which were used for validation.
Supplementary file 5. Details of the 40 SARS‐CoV‐2 M‐pro inhibitors from the COVID Moonshot project crystallized with the SARS‐CoV‐2 M‐pro and whose inhibitory activity is available.
Supporting information.