

Abstract
Pathogen infection triggers host innate defenses which may result in the activation of regulated cell death (RCD) pathways such as apoptosis. Given a vital role in immunity, apoptotic effectors are often counteracted by pathogen-encoded antagonists. Mounting evidence indicates that programmed necrosis, which is mediated by the RIPK3/MLKL axis and termed necroptosis, evolved as a countermeasure to pathogen-mediated inhibition of apoptosis. Yet, it is unclear whether components of this emerging RCD pathway display signatures associated with pathogen conflict that are rare in combination but common to key host defense factors, namely, rapid evolution, viral homolog (virolog), and cytokine induction. We leveraged evolutionary sequence analysis that examines rates of amino acid replacement, which revealed: 1) strong and recurrent signatures of positive selection for primate and bat RIPK3 and MLKL, and 2) elevated rates of amino acid substitution on multiple RIPK3/MLKL surfaces suggestive of past antagonism with multiple, distinct pathogen-encoded inhibitors. Furthermore, our phylogenomics analysis across poxvirus genomes illuminated volatile patterns of evolution for a recently described MLKL viral homolog. Specifically, poxviral MLKLs have undergone numerous gene replacements mediated by duplication and deletion events. In addition, MLKL protein expression is stimulated by interferons in human and mouse cells. Thus, MLKL displays all three hallmarks of pivotal immune factors of which only a handful of factors like OAS1 exhibit. These data support the hypothesis that over evolutionary time MLKL functions—which may include execution of necroptosis—have served as a major determinant of infection outcomes despite gene loss in some host genomes.
Keywords: necroptosis, MLKL, RIPK3, poxvirus, phylogenetics, synteny
Introduction
Regulated cell death (RCD) programs are pivotal host defense responses against pathogens (Lamkanfi and Dixit 2010; Danthi 2016; Jorgensen et al. 2017). Consequently, pathogens have evolved a myriad of strategies to counteract these responses (Barber 2001; Best 2008), which includes the activation of apoptosis. Apoptosis is a well-characterized, noninflammatory form of RCD that was first classified by its distinct morphology defined by membrane blebbing, apoptotic bodies, and nuclear and cytoplasmic condensation (Kerr et al. 1972). Both extrinsic, as well as intrinsic cues, can trigger a signaling cascade that involves the activation of effector caspases to execute apoptosis (fig. 1A) (Galluzzi et al. 2018) which was characterized, in large part, using viral proteins (Strasser et al. 2000).
Fig. 1.

Cell death pathways and host defense. (A) Apoptosis is a regulated cell death pathway that acts as a key line of host defense against pathogens, which is executed by caspases via an extrinsic or intrinsic route (Galluzzi et al. 2018). (B) Necroptosis is an alternative regulated cell death pathway triggered by suppression of death receptor-mediated apoptosis by pathogens (Nailwal and Chan 2019) such as poxviruses. E3 (Koehler et al. 2017) and vMLKL (Petrie et al. 2019) are recently described poxvirus factors that counteract necroptotic signaling.
Recently, a RCD pathway defined as programmed necrosis, hereafter necroptosis, was discovered. Necroptosis is triggered when specific apoptotic signaling and effector functions are suppressed during pathogen infection. Given the conditions under which it is activated, necroptosis has often been considered a necessary “back-up” host response to pathogen-mediated inhibition of apoptosis (fig. 1B) (Pearson and Murphy 2017; Nailwal and Chan 2019). In contrast to apoptotic cell death, necroptosis is inflammatory and characterized by cell and organelle swelling followed by plasma membrane rupture (Vanden Berghe et al. 2014; Galluzzi et al. 2018; Green 2019). Apoptotic and necroptotic signaling also differ in their evolutionary dynamics over large timescales. Specifically, key effectors of cellular necroptotic signaling emerged later in evolution and display a “patchy” phylogenetic distribution (Dondelinger et al. 2016; Brault and Oberst 2017) which is in marked contrast to the more ancient and highly conserved components of apoptotic signaling (Koonin and Aravind 2002; Zmasek and Godzik 2013; Green and Fitzgerald 2016). Notably, the evolutionary dynamics for components of necroptotic signaling over more recent timescales is unknown but may inform determinants shaping contemporary infections.
Necroptosis can be initiated by at least three presumably independent receptors: 1) death receptor (DR) signaling, 2) the pathogen recognition receptor (PRR) Z-DNA Binding Protein 1 (ZBP1)/DNA Activator of Interferon (DAI)—hereafter ZBP1 (Upton et al. 2012; Upton and Kaiser 2017), and 3) Toll-like Receptor 3/4 (TLR3/TLR4) (He et al. 2011; Kaiser et al. 2013). Activation of DR signaling triggers Receptor Interacting Protein Kinase 1 (RIPK1) to self-oligomerize, which leads to the recruitment of Receptor Interacting Protein Kinase 3 (RIPK3) via physical RIP Homotypic Interaction Motif (RHIM) domain interactions. Activated RIPK3 subsequently binds and phosphorylates Mixed Lineage Kinase-Like (MLKL) (Sun et al. 2012; Murphy et al. 2013; Wang et al. 2014). Phosphorylation of MLKL results in a conformational change, enabling self-oligomerization of this factor to complete necroptosis through MLKL plasma membrane destabilization (Cai et al. 2014; Chen et al. 2014).
Importantly, DR initiation of necroptosis appears to function when caspase-8 activity is inhibited during apoptosis (Kaiser et al. 2011; Oberst et al. 2011). In contrast, ZBP1 triggers necroptosis upon binding foreign nucleic acids followed by activation of RIPK3 through RHIM-dependent interactions (Kaiser et al. 2008; Maelfait et al. 2017). In addition, TLR3 and TLR4 signaling can activate RIPK3 via TIR-domain-containing adaptor-inducing interferon-β (TRIF) also through physical RHIM domain interactions (He et al. 2011; Kaiser et al. 2013). The diverse and distinct means of activating necroptosis with convergence on the RIPK3/MLKL axis reflect the breadth of pathogens this response may protect against.
Consistently, a growing list of viral- and bacterial-encoded inhibitors that target discrete steps of necroptosis has emerged (Mack et al. 2008; Upton et al. 2008; Guo et al. 2015; Koehler et al. 2017; Pearson et al. 2017; Petrie et al. 2019; Fletcher-Etherington et al. 2020; Liu et al. 2021). However, whether this pathogen-mediated antagonism has shaped cellular factors essential for necroptotic signaling remains unknown. Strong selective pressure may be imposed on host factors to escape direct binding by pathogen-encoded antagonists. This pressure is often visible as elevated rates of amino acid substitution, a signature of rapid evolution and a hallmark of positive selection. Signatures of rapid evolution have been observed across orthologous sequences from closely related species (e.g., primates) for several key immune factors: PKR (Elde et al. 2009), OAS1 (Hancks et al. 2015; Mozzi et al. 2015), MxA (Mitchell et al. 2013), TRIM5α (Sawyer et al. 2005), APOBEC3G (Sawyer et al. 2005), and cGAS (Hancks et al. 2015; Mozzi et al. 2015). Studies of host-pathogen coevolution may reveal novel insights into cellular responses including evidence for undefined host components, genetic determinants shaping infection outcomes, and pathogen countermeasures of these defenses. We hypothesized that analysis for signatures characteristic of host-pathogen conflict would shed light on adaptive mechanisms shaping host and viral components associated with necroptotic outcomes. To test this, we performed evolutionary sequence analysis for host components using established bioinformatic methods that examine rates of amino acid replacement across orthologous sequences and phylogenomics analysis of poxvirus genomes for a recently reported homolog of MLKL in poxviruses (Petrie et al. 2019).
Consistent with pivotal roles in host defense across species, we report that the necroptotic axis—RIPK3 and MLKL—displays widespread and recurrent signatures of rapid evolution in primate and bat genomes. Interestingly, we found evidence for positive selection at a sequence in the RHIM domain of RIPK3 that is a known target site for a conserved bacterial protease named EspL (Pearson et al. 2017). EspL can also cleave ZBP1, TRIF, or RIPK1 in the RHIM domain to dramatically reduce protein levels. Unexpectedly, these factors lack evidence for positive selection at the homologous, demonstrated cleavage sites. In addition, a strong positive selection signature in the binding pocket of primate RIPK3 provides evidence for undefined pseudosubstrates mimicking MLKL. These mimics may resemble the poxvirus MLKL (Petrie et al. 2019) virolog encoded by viral species that infect vertebrates other than primates. Our phylogenomics analysis, which includes extensive synteny analysis, of poxvirus MLKL indicates that this antagonist has been subjected to repeated gene replacements mediated by duplications and deletion events over viral evolution with some poxviruses encoding up to three mimics. In summary, our data demonstrate that MLKL belongs to a small class of key immune factors, which includes OAS1 but not PKR or cGAS, that display all three hallmarks of pivotal immune factors—rare in combination: 1) rapidly evolving, 2) viral homolog, and 3) upregulation by cytokines such as interferons. Collectively, these findings suggest that RIPK3 and particularly MLKL functions, like the execution of necroptosis and perhaps additional roles such as MLKL-mediated regulation of vesicle trafficking (Yoon et al. 2017), have dramatically tipped infection outcomes over evolutionary time.
Results
Widespread Signatures of Rapid Evolution for RIPK3 and MLKL across Primate and Bat Genomes
To determine if known components of the necroptosis pathway display signatures of positive selection, we analyzed a matching set of twenty-one primate sequences and eight bat sequences for TRIF, ZBP1, RIPK1, RIPK3, and MLKL using a series of evolutionary analyses. Primates were selected for their biomedical relevance and because of the wealth of information for the human factors. Bats were selected as they are well-established reservoirs for a diverse array of viruses (Hayman 2016). To test for recurrent positive selection, we used a combination of maximum-likelihood based algorithms including Phylogenetic Analysis by Maximum Likelihood (PAML) (Yang 2007), which estimate rates of nonsynonymous amino acid replacements (dN) relative to synonymous (dS) amino acid substitutions (figs. 2–4). dN/dS values >1 are considered a “classic” hallmark of positive selection (Yang and Bielawski 2000). Rapid amino acid replacement in host defense factors has been functionally linked to escape from pathogen-encoded antagonists (Daugherty and Malik 2012; McLaughlin and Malik 2017) and in other cases restoration of antiviral activity such as in the case of TRIM5α targeting of retrovirus capsids (Sawyer et al. 2005). Due to strong selective pressure to manage infections, host variants that alter binding by pathogen factors are favored and selected for during evolution. This history can be observed as excessive amino acid replacements relative to silent substitutions—dN/dS > 1.
Fig. 2.

The rapid evolution of the necroptotic axis, RIPK3 and MLKL, across primate lineages. (A) Summary of rapid evolution results produced by NSsites analysis implemented in PAML for M7 × M8 (F3x4) Likelihood ratio test statistics (2δ) for necroptotic factors across a set of twenty-one matching primate species. (B) Western blot analysis demonstrates that MLKL protein is upregulated by IFNγ treatment in HeLa and HT29 cell lines and by IFNα as well as IFNγ treatment in murine embryonic fibroblasts. (C) dN/dS values estimated using FreeRatio analysis implemented in PAML across primate evolution. Rapidly evolving lineages (red branches) are defined by dN/dS >1, or greater than or equal to 3 nonsynonymous amino acid changes relative to zero synonymous amino acid changes.
Using codon-based models implemented in PAML, we detected robust gene-wide rapid evolution signatures for primate MLKL (M7 vs. M8 [F3x4] P < 5.400 × 10−10), RIPK3 (M7 vs. M8 [F3x4] P < 3.056 × 10−9), and to a lesser extent, ZBP1 (M7 vs. M8 [F3x4] P < 5.767 × 10−3), but not RIPK1 (M7 vs. M8 [F3x4] P < 0.066) or TRIF (M7 vs. M8 [F3x4] P < 0.065) (fig. 2A, supplementary file S1, Supplementary Material online). Similar signatures were observed for bat factors: MLKL (M7 vs. M8 [F3x4] P < 1.647 × 10−10), RIPK3 (M7 vs. M8 [F3x4] P < 3.745 × 10−16), and to a lesser extent, ZBP1 (M7 vs. M8 [F3x4] P < 9.351 × 10−3) and TRIF (M7 vs. M8 [F3x4] P < 2.000 × 10−3), but not RIPK1 (M7 vs. M8 [F3x4] P < 0.138) (fig. 4A, supplementary file S1, Supplementary Material online). The robust positive selection signature for the main effectors of the necroptosis pathway, RIPK3, and MLKL, in primate and bat genomes, is comparable to other key host defense factors—PKR, cGAS, and OAS1—which are also upregulated by cytokines like interferons. Indeed, MLKL protein is also upregulated by interferons (Rusinova et al. 2013; Knuth et al. 2019; Sarhan et al. 2019). In agreement, we observe upregulation of MLKL protein in human cell lines by IFNγ and mouse embryonic fibroblasts with IFNα and IFNγ (fig. 2B).
Fig. 4.

The rapid evolution of bat factors associated with necroptosis. (A) Summary of rapid evolution results produced using NSsites implemented in PAML for M7 × M8 (F3x4) Likelihood ratio test statistics (2δ) for necroptotic factors across a set of eight matching bat species. (B) dN/dS values estimated using Free Ratio analysis in PAML across bat phylogeny. Rapidly evolving lineages (red branches) defined by dN/dS >1, or greater than or equal to 3 nonsynonymous amino acid changes relative to zero synonymous amino acid changes. (C) Rapidly evolving sites (triangles) identified—for bat TRIF (orange), ZBP1 (pink), RIPK1 (red), RIPK3 (green), and MLKL (blue)—were predicted using NSsites in PAML (yellow triangle), FUBAR (blue triangle) and MEME (green triangle). Sites with P-values <0.05 or posterior probabilities (P) >0.95 are listed above the protein cartoons, while sites with P-values <0.01 or posterior probabilities (P) >0.99 are also bolded and have an asterisk. Amino acid alignment position refers to the Indian flying fox reference sequences. (D) Location of rapidly evolving sites (shown in red) on the b(at)RIPK3 (green) and bMLKL (light blue) homology models. The homology models were predicted using Swiss-Model (Waterhouse et al. 2018) and aligned to mRIPK3: mMLKL (silver) cocrystal structure (Xie et al. 2013).
To assay when in evolution and to what extent selection pressure has shaped components of necroptotic signaling, we estimated dN/dS values using PAML for each factor across bat and primate phylogenies (figs. 2C and 4B). Consistent with the gene-wide tests of evolution (figs. 2A and 4A), we observed recurrent and widespread signatures of positive selection across primate (fig. 2C) and bat (fig. 4B) evolution for RIPK3 and MLKL with a notable, albeit lesser signal for primate ZBP1. Specifically, numerous recent and ancestral primate and bat lineages, including branches in each major group, show substitution patterns that are characteristic of genetic conflict for both RIPK3 and MLKL. Strikingly, especially strong signatures are evident for RIPK3 in the lineage preceding the divergence of Hominidae (10 nonsynonymous changes: 0 synonymous changes) and for MLKL in the lineage prior to Hominidae divergence (dN/dS = 4.49) (fig. 2C). In contrast, only one lineage with a rapid evolution signature was detected for both primate TRIF and RIPK1. Likewise, only one rapidly evolving lineage was detected for bat TRIF and ZBP1 (fig. 4B). No lineages for bat RIPK1 show evidence for positive selection. Thus, repeated innovation in factors associated with necroptosis activation has been focused on the downstream axis of RIPK3/MLKL during primate and bat evolution.
To uncover where strong selection pressure may have been imposed within these proteins essential for necroptosis, dN/dS values were estimated for individual amino acid positions using three distinct but commonly implemented methods: PAML (Yang 2007), MEME (Murrell et al. 2012), and FUBAR (Murrell et al. 2013) (figs. 3A and 4C and supplementary file S1, Supplementary Material online). The distribution and the number of rapidly evolving sites are thought to reflect the number of protein surfaces in genetic conflict with other factors, such as pathogen-encoded inhibitors (Daugherty and Malik 2012). In agreement with these host factors being associated with broad-acting immune defenses, rapidly evolving sites are distributed throughout primate (fig. 3A) as well as bat (fig. 4C) MLKL, RIPK3, ZBP1, RIPK1, and TRIF. In support of our preceding analysis, primate MLKL (20 total sites/11 by multiple methods) and RIPK3 (27 total sites/7 by multiple methods) have the most sites identified (fig. 3A). The majority (70%) of primate MLKL positively selected sites are localized to the kinase-like domain, with 12/14 sites within this domain occurring between amino acid positions 370–470 (fig. 3A). Similarly, bat RIPK3 (20 total sites/5 by multiple methods) and MLKL (19 total sites/5 by multiple methods) have the most sites identified by multiple methods (fig. 4C). In contrast to primate MLKL, rapidly evolving sites for bat MLKL are largely absent from the C-terminus and tend to be sequestered inside or adjacent to the N-terminal bundle and brace (NBB).
Fig. 3.

Widespread signatures of positive selection for cellular factors associated with necroptosis. (A) Rapidly evolving sites (triangles) identified—for primate TRIF (orange), ZBP1 (pink), RIPK1 (red), RIPK3 (green), and MLKL (blue)—were predicted using NSsites in PAML (yellow triangle), FUBAR (blue triangle) and MEME (green triangle). Sites with P-values <0.05 or posterior probabilities (P) >0.95 are listed above the protein cartoons, while sites with P-values <0.01 or posterior probabilities (P) >0.99 are also bolded and have an asterisk. Amino acid position refers to the human reference sequences. (B) Protein modeling for hRIPK3 and hMLKL highlights that multiple, discrete surfaces have rapid evolution signatures (red). H(uman)RIPK3 (green) homology model and the hMLKL (light blue) predicted structure (Murphy et al. 2013) are shown. The homology model was predicted using Swiss-Model (Waterhouse et al. 2018) and aligned to predicted structures m(ouse)RIPK3 (silver), mMLKL (silver), and mRIPK3: mMLKL (silver) (Xie et al. 2013). (C) Prominent rapid evolution (red) at the mRIPK3 hydrophobic pocket. (D) Rapid evolution exclusive to the RIPK3 RHIM domain proximal to the demonstrated EspL cleavage motif (Q*G**N—highlighted in black). Sequences spanning the EspL cleavage motif for 12 primates for RIPK3 (green), RIPK1 (red), TRIF (orange), and ZBP1 (pink) RHIM domains. The EspL cleavage site is underlined and displays a black arrow above the amino acid. Sites (triangles) with P-values <0.05 or posterior probabilities (P) >0.95 are listed above the protein cartoons. Sites with P-values <0.01 or posterior probabilities (P) >0.99 are bolded and have an asterisk. Amino acid positions refer to the human reference sequences. Note: PAML, MEME, and FUBAR analysis were performed for RIPK1 without drill monkey since this region was excluded due to trimming in preparation of sequences.
By mapping positively selected sites onto a previously published cocrystal structure of mouse RIPK3 and MLKL (Xie et al. 2013), it becomes evident that these sites represent distinct surfaces for primate (fig. 3B) and bat (fig. 4D) RIPK3/MLKL. As a control, we analyzed space-filling models for primate RIPK3 and MLKL. These data demonstrate that rapidly evolving sites localize to external surfaces, which would be expected if they represent interfaces for protein-protein interactions (supplementary fig. S1, Supplementary Material online). Notably, there is a cluster of positively selected sites for primate RIPK3 that surrounds MLKL and corresponds to the hydrophobic pocket where RIPK3 and MLKL interact via hydrophobic interactions (fig. 3C). Rapid evolution is also observed at the bat RIPK3: MLKL interface (fig. 4D). One potential explanation for these signatures is pathogen-encoded RIPK3 pseudosubstrates, as this pocket has been demonstrated previously to be necessary for RIPK3:MLKL interactions and for necroptosis to occur (Xie et al. 2013; Petrie et al. 2019).
Rapid Evolution Is Exclusive to RIPK3 at a Bacterial Protease Cleavage Site Common to RHIM Domains
RHIM-RHIM homotypic interactions between RIPK3 and established activators RIPK1, TRIF, and ZBP1 are required for necroptosis signal transduction. RIPK3 (AA: 450–466) (Sun et al. 2002; Mompean et al. 2018), RIPK1 (AA: 531–547) (Mompean et al. 2018), and TRIF (AA: 679–695) (Kaiser and Offermann 2005) each have one RHIM domain whereas ZBP1 has two (AA: 195–219; 253–277) (Shanmugam et al. 2021); numbering is relative to human sequence and defined using UniProt (2019) as well as the indicated citations. The critical nature of these interactions is also supported by the identification of both viral- and bacterial-encoded inhibitors that target the RHIM domains of these proteins. These include cytomegalovirus M45 and herpes simplex virus ICP6 and ICP10, which all encode RHIM domains and target host RHIM-dependent interactions (Upton et al. 2008; Guo et al. 2015). A recent report has shown that an orthopoxvirus protein named viral inducer of RIPK3 degradation (vIRD) targets RIPK3 protein via the RHIM domain, but not other RHIM domain-containing proteins, for degradation (Liu et al. 2021). Of particular interest is EspL, a bacterial protease, which is encoded by diverse species including enteropathogenic Escherichia coli (EPEC), Salmonella enterica, and Yersinia pestis (Pearson et al. 2017). Notably, EPEC EspL has been shown to cleave a specific motif (Q*G**N) in the RHIM domains of human and mouse TRIF, ZBP1, RIPK3, and RIPK1 to suppress necroptosis, which is activated by EPEC NleB1-mediated inhibition of death receptor apoptosis (Pearson et al. 2013; Pearson et al. 2017). Unexpectedly, rapid evolution is evident at the EspL cleavage site in the primate RIPK3 RHIM domain but not at the known cleavage sites in the RHIM domains across primate TRIF, ZBP1, and RIPK1 (fig. 3D). Specifically, RIPK3 has two rapidly evolving sites identified (462D and 463N) by PAML immediately adjacent to the determined RHIM EspL cleavage motif—GDN/NYL; where “/” indicates cleavage, and underline denotes rapidly evolving sites (fig. 3D). A comparable signature is not present for the bat factors. While bat ZBP1 has two rapidly evolving sites in one of the two RHIM domains (202K, 211V) and RIPK3 RHIM has one (455F) relative to the Indian flying fox, a giant fruit bat, they are distal to the cleavage site. Given the demonstrated activities of the EPEC factors EspL and NleB1, this evolutionary signature may suggest RIPK3 anti-bacterial functions independent of RHIM domain interactions essential for necroptosis and RIPK1-dependent RIPK3 kinase-independent apoptosis (Mandal et al. 2014; Newton et al. 2014) was the basis of this selection.
Poxviral Antagonism of the RIPK3: MLKL Interface Serves as a Model for Antagonistic Coevolution
The rapid evolution signature of RIPK3 near the hydrophobic pocket points toward antagonism potentially by pathogen-encoded pseudosubstrates (fig. 3C). Indeed, a viral copy of MLKL (vMLKL) that is encoded by numerous poxviruses has been recently identified (Petrie et al. 2019). Yet, the origins and evolution of these vMLKLs remain unknown. We identified twenty-seven distinct copies of vMLKL in the sequence database across the genomes of Avipoxvirus, Clade II, along with two other poxvirus species that infect bats (eptesipoxvirus [EPTV] and hypsugopoxvirus [HYPV]) (fig. 5D and supplementary file S2, Supplementary Material online). While these poxviruses are not thought to infect primates, poxviruses have a broad species range and frequently spill over into new hosts. Studies of vMLKL and comparative analysis may serve as a model to understand the evolution of mimics, RIPK3/MLKL interactions, and RIPK3 antagonism by pseudosubstrates.
Fig. 5.

vMLKL mimics are encoded in Avipoxvirus, Clade II, eptesipoxvirus, and hypsugopoxvirus genomes. (A) Schematic of human MLKL (light blue) and myxoma v(irus)MLKL (dark blue) protein. (B) Protein homology model for myxoma vMLKL (dark blue)—Swiss-Model—aligned to published, predicted structures for hMLKL (light blue) and mMLKL (silver). (C) Heatmap comparing percent identities of vMLKL copies across poxvirus species used in this study. (D) Cladogram depicting distinct species containing a vMLKL copy with Avipoxvirus (green), Clade II (blue), and other poxvirus species (yellow, pink, and red). Note that eptesipoxvirus and hypsugopoxvirus contain multiple copies of vMLKL and are labeled based on their genome position—A (yellow), B (pink), C (red). Eptesipoxvirus (black dashed box) contains vMLKL copies located on the inverted terminal repeats (ITR) represented by A (yellow) and C (red). (E) Location of individual and multicopy vMLKLs across poxvirus genomes. Multicopy vMLKLs are highlighted (yellow, pink, and red) in eptesipoxvirus and hypsugopoxvirus. Diagrams created in CoGeBlast represent scaled locations of the vMLKL copies for each poxvirus genome. Genome accession numbers (top left corner) and genome length in nucleotides (top right corner) are present for each genome. (F) Multiple copies of vMLKL in the eptesipoxvirus genome are supported by relative depth analysis. The distribution of reads that align with the ITR and non-ITR regions is shown. Zoomed panels (black boxes) display relative depth of EPTV vMLKLs (black) and the two immediately flanking upstream and downstream ORFs (gray). The ITR relative depth mean is 1.852 and the median is 1.899. The non-ITR relative depth mean is 1.000 and the median is 1.025. (G) Repertoire of poxvirus apoptosis and necroptosis antagonists explained, in part, by the evolutionary distribution of vMLKL. Only proteins predicted to be functional are shown. Two exceptions are the E3 proteins of myxoma virus and rabbit fibroma virus which have N-terminal truncations (*). Additional analysis is present in supplementary file S2, Supplementary Material online.
vMLKL lacks the N-terminal bundle and brace (NBB) domain but maintains the C-terminal kinase-like domain, to which RIPK3 binds to phosphorylate the host cellular homolog (Petrie et al. 2019) (fig. 5A). Ectopic expression of two vMLKLs from poxvirus species that primarily infect mice was demonstrated to competitively inhibit both mouse and human RIPK3 in binding and cell culture necroptosis assays (Petrie et al. 2019). Superposition of a homology model of myxoma virus (MYXV) MLKL onto published crystal structures for mouse MLKL (Xie et al. 2013) and human MLKL (Murphy et al. 2014) indicated striking structural overlap with human and mouse MLKL (fig. 5B) despite a broad range of amino acid divergence (16–25% amino acid identity of the virus homologs to mouse and human MLKLs).
In agreement with the strong structural overlap, characterization of vMLKL sequences indicate conservation of key functional residues for human and mouse MLKL including ATP binding, hydrogen-bonding, and protein stability (supplementary fig. S2A, Supplementary Material online) (Sun et al. 2012; Murphy et al. 2013; Xie et al. 2013; Cai et al. 2014; Murphy et al. 2014; UniProt 2019). Next, we compared our primate sequence data (fig. 2), which consisted of more species and in turn better resolution, with our vMLKL data set. Several amino acid sites displaying positive selection for primate MLKL (fig. 3) overlapped or were in close proximity to sites for hydrogen bonding and protein stability, as demonstrated by the amino acid alignment (supplementary fig. S2A, Supplementary Material online). Of note, there are several intriguing examples of shared identity between the primate positively selected sites and the viral copies. One example is human MLKL F386 (F373 mouse), which has been shown to be necessary for RIPK3/MLKL complex formation (Petrie et al. 2019). This position exclusively has tyrosine, phenylalanine, or histidine amino acid variants across the sampled primates, with this pattern of aromatic residues evident and conserved to Aves MLKL orthologs (supplementary fig. S2B, Supplementary Material online). Consistently, poxvirus virologs have also maintained aromatic (phenylalanine or tyrosine) amino acids at the homologous position (supplementary fig. S2A, Supplementary Material online). These data implicate site 386 in host defense as well as counteraction by these viral mimics (Davies et al. 2020).
To further understand the evolutionary relationships between poxvirus vMLKLs, we performed a detailed phylogenomics analysis across corresponding viral genomes. Full-length vMLKLs (excluding partial Vulture gryphus poxvirus and Hawaiian goosepox virus sequences) display 18–100% amino acid identity relative to each other and cluster in a manner resembling known poxviral species relationships (fig. 5C). Unexpectedly, this analysis identified two distinct vMLKL copies, which differ in amino acid identity, in the genomes of both eptesipoxvirus (EPTV vMLKL-A vs. EPTV vMLKL-B: 45% amino acid identity) and hypsugopoxvirus (HYPV vMLKL-A vs. HYPV vMLKL-B: 43% amino acid identity). Analysis of the genomic context for each vMLKL was conducted using CoGeBlast, which allows for individual hit visualization. The results here are consistent with distinct genomic locations for these genes (fig. 5E). Specifically, a majority of vMLKLs are located primarily on the right arm of the poxvirus genome with EPTV having two copies that map to two different loci in this region. In addition, both HYPV and EPTV have one additional copy that maps to the left arm of the poxvirus genome. Interestingly, two of the vMLKLs in EPTV are present on the inverted terminal repeat (ITR), potentially representing more recent acquisitions. Likewise, fowlpox virus-282E4 vMLKL appears to be at a distinct inverted locus relative to other Avipoxvirus species on the left arm of the genome (fig. 5E).
To confirm additional vMLKL copy numbers present in the eptesipoxvirus genome, we next performed relative depth analysis, with approximately 1,000-fold genome coverage (fig. 5F and supplementary fig. S3, Supplementary Material online), using the raw reads from genome sequencing. By mapping reads across the unique genome and one reference ITR, we found that EPTV—A/C has a relative depth of two and EPTV—B has a relative depth of one, which supports the presence of three copies in the eptesipoxvirus genome (fig. 5F). As a control, we also conducted a similar analysis using reads from a recent myxoma virus sequencing project (Kerr et al. 2019) and found that myxoma virus MLKL relative depth remained near one, indicating the presence of only one genomic copy (supplementary fig. S3, Supplementary Material online). These data demonstrate that vMLKLs have different evolutionary histories and are likely not orthologous.
Phylogenetic Distribution of vMLKLs Can Account for Variable Conservation of E3 across Species
Poxviruses encode numerous immunomodulators to subdue and circumvent host defenses including apoptosis. As a majority of poxviruses encode several characterized anti-apoptotic proteins (Bratke et al. 2013; Haller et al. 2014) (fig. 5G, supplementary fig. S4 and file S2, Supplementary Material online) like the serpin class of caspase pseudosubstrates (Taylor and Barry 2006; Nichols et al. 2017), these viruses would be expected to trigger necroptosis. Indeed, the model poxvirus vaccinia has been shown recently to trigger necroptosis through activation of ZBP1 in cultured cells and to counteract this response using the Z—nucleic acid binding (Zα) domain of the E3 protein (Koehler et al. 2017). Although E3 is considered an essential host-range gene that antagonizes several host defenses, some poxviruses that encode a repertoire of anti-apoptotic proteins lack E3 or possess truncated E3 ORFs. For example, the Avipoxvirus family does not possess any orthologs of E3 (fig. 5G and supplementary file S2, Supplementary Material online) and several Clade II poxviruses, like the well-characterized myxoma virus, are also predicted to have N-terminally truncated E3 copies which lack the Zα domain that is important for antagonism of ZBP1 (Bratke et al. 2013) (fig. 5G and supplementary file S2, Supplementary Material online). Relatedly, Avipoxvirus, Clade II, and poxvirus species that infect bats do not encode vIRD (Liu et al. 2021). To gain insights into how a subset of poxviruses may circumvent necroptosis given the presumed ability to trigger apoptosis, we compared the breadth of known anti-apoptotic proteins, including serpins, across the poxvirus phylogeny relative to vMLKL (fig. 5G). As vMLKLs are present in Avipoxvirus, hypsugopoxvirus, myxoma virus and rabbit fibroma virus—the maintenance of this pseudosubstrate may account for the tolerance of these species to a missing or truncated E3 protein.
Repeated Replacements of vMLKL over Poxvirus Evolution
Our data illustrate that multiple vMLKL copies can exist in a poxvirus genome and that not all of the copies across the genomes may be orthologous. To determine the relationship of poxvirus vMLKLs, we performed synteny analysis using five flanking upstream and downstream genes across the entire phylogeny (fig. 6 and supplementary file S3, Supplementary Material online). These data show that while each vMLKL within Avipoxvirus and Clade II are flanked largely by the same upstream and downstream genes, vMLKLs between the groups are flanked by different genes. This arrangement would support the notion that Clade II and Avipoxvirus vMLKLs are distinct copies. Furthermore, the lack of vMLKL in Yatapoxvirus species (Yaba monkey tumor virus and Yaba-like disease virus), which belong to Clade II, revealed that this loss is likely due to a deletion event in these lineages consisting of multiple upstream and downstream genes (fig. 6 and supplementary file S3, Supplementary Material online).
Fig. 6.

Diverse evolutionary histories for vMLKL are indicated by high-resolution synteny analysis. Analysis of upstream and downstream flanking genes indicates vMLKLs (black) reside at different locations across poxvirus genomes. Protein homology was determined using reciprocal BLAST hits for the 5 genes upstream (left of vMLKL) and 5 genes downstream (right of vMLKL) of vMLKL. While a gene window of 11 is shown here, an expanded analysis is found in supplementary file S3, Supplementary Material online. Genes oriented to the right represent ORFs on the top strand, while genes oriented to the left are on the bottom strand. The letter “U” represents a gene that is either unique to the individual species or unique to the group of poxviruses that have maintained a vMLKL copy. Gene blocks that have homologous genes (colored borders) that are outside the scope of this figure have arrows pointing to the left (representing upstream) or to the right (representing downstream). Two Yatapoxvirus species (Yaba monkey tumor virus and Yaba-like disease virus) were included as they belong to Clade II but lack vMLKL.  represents a deletion event. Species defined as “Other” (red) are either unclassified (hypsugopoxvirus) or part of vespertilionpoxviruses (eptesipoxvirus).
represents a deletion event. Species defined as “Other” (red) are either unclassified (hypsugopoxvirus) or part of vespertilionpoxviruses (eptesipoxvirus).
This analysis also increased resolution for the multiple vMLKL copies in eptesipoxvirus and hypsugopoxvirus. First, this analysis indicated that EPTV-B (EPTV-WA-166) and HYPV-B (QDJ95132.1) are orthologous to each other and are likely orthologous to the Clade II copies, as evidenced by some but not complete overlap in syntenic gene neighbors (fig. 6). Second, EPTV-A (EPTV-WA-006) and HYPV-A (QDJ94987.1) share gene neighbors indicating that these sequences are also likely orthologs. EPTV-A shares 100% identity with EPTV-C (EPTV-WA-186) with both annotated as being located in the inverted terminal repeats (ITRs). Consistently, EPTV-C has the same gene neighbors as EPTV-A albeit inverted. The copies are labeled corresponding to their location starting from the left arm of the poxvirus genome—A, B, C.
To corroborate the findings from the above synteny analysis, we performed whole-genome comparisons using CoreGenes (https://coregenes.ngrok.io/). These data were visualized with RIdeogram (Zhaodong Hao 2020) and focused on the copies encoded by Avipoxvirus, Clade II, and the two poxvirus species that primarily infect bats. Indeed, we found that canarypox virus vMLKL and myxoma virus vMLKL localize to distinct viral genomic locations that lack regions of detectable synteny (supplementary fig. S5A, Supplementary Material online). Furthermore, these data are consistent with loss of the locus encoding Clade II vMLKL in a Yatapoxvirus ancestor (supplementary fig. S5B, Supplementary Material online). Lastly, we compared eptesipoxvirus with myxoma virus (supplementary fig. S5C, Supplementary Material online). The upstream regions of synteny for vMLKL indicate that MYXV vMLKL is orthologous to the EPTV-B vMLKL copy, which is further supported by MYXV vMLKL not being located in the ITRs where EPTV-A/C reside. These data demonstrate that vMLKLs are at distinct loci in poxvirus genomes, which are the likely result of repeated duplications followed by deletions of the source locus during evolution.
Phylogenetic Analysis Reveals Origination of vMLKL Copy from One Horizontal Gene Transfer Event
The distinct copies of vMLKLs may have originated due to either 1) a single transfer event from a host genome to an ancestral poxvirus followed by diversification or 2) multiple independent transfer events from presumably distinct host genomes to different poxviruses. To address this, we performed a phylogenetic analysis using amino acid sequences for vMLKLs and 38 diverse host MLKLs. If distinct vMLKLs were acquired via independent events, placement of viral sequences should vary across the tree in a manner where they pair with different host sequences. Such topology could reflect the source genome or that of a related, perhaps ancestral, animal host. In contrast, if vMLKLs originated from a single transfer event, the tree topology would group these sequences into one clade. Phylogenetic analysis using PhyML (Lefort et al. 2017) revealed that vMLKL virologs cluster as one group distinct from animal vMLKLs (fig. 7). These data support a single transfer event from an ancestral host genome as the most parsimonious origin for vMLKL (fig. 7 and supplementary file S4, Supplementary Material online). Consistently, the topology of the poxvirus branches mirrors that of known species relationships. For example, all of the Avipoxvirus species cluster together. Likewise, the Clade II species cluster together broadly, as well as at a finer level such as in the Leporipoxviruses (e.g., MYXV and RFV). The tree topology also complements the synteny analysis with HYPV-B and EPTV-B being most closely related along with EPTV-A/C and HYPV-A vMLKL clustering distinctly.
Fig. 7.

Phylogenetic analysis supports a single vMLKL gene transfer event from an ancestral host. A maximum-likelihood phylogenetic tree was generated using PhyML (Lefort et al. 2017) of MAFFT aligned diverse host and poxviral MLKL amino acid sequences (supplementary file S4, Supplementary Material online). Consistent with a single transfer event, Avipoxvirus (green), Clade II (blue), and other poxvirus sequences (yellow, pink, and red) cluster together. Branch lengths represent amino acid changes per site. 100 bootstrap replicates were performed; branch support >70% (*) or 90% (**) are indicated.
Still, it remains unclear whether Clade II vMLKLs represent a distinct or orthologous locus to Avipoxvirus vMLKL and which vMLKL locus represents the ancestral locus. Indeed, our data for Clade II vMLKL could be explained, in part, by either 1) duplication of the Avipoxvirus vMLKL locus followed by a deletion event of the original locus in the Clade II ancestor or 2) that Avipoxvirus and Clade II vMLKL are orthologous but divergence no longer allows for syntenic comparison between these species. We further assessed the likelihood of these evolutionary histories by analyzing patterns of amino acid variation shared between vMLKLs and mammalian MLKLs, which cluster closest to vMLKLs (fig. 7). We evaluated innovations specific to vMLKLs but not host MLKLs across our alignment. We identified seventeen amino acid residues largely conserved between Avipoxvirus vMLKLs and mammalian MLKLs (fig. 8) but not among Clade II, EPTV, and HYPV vMLKLs. Conversely, different variants at the same sites are shared across Clade II, EPTV, and HYPV vMLKLs but not Avipoxvirus or mammalian MLKLs. These data support the hypothesis that Avipoxvirus vMLKL is the original locus and that Clade II vMLKL is a distinct copy but derived from the Avipoxvirus vMLKL presumably through duplication. Collectively, these data illustrate a volatile history of vMLKL defined by repeated sampling of new loci since its acquisition by an ancestral poxvirus from an infected host.
Fig. 8.
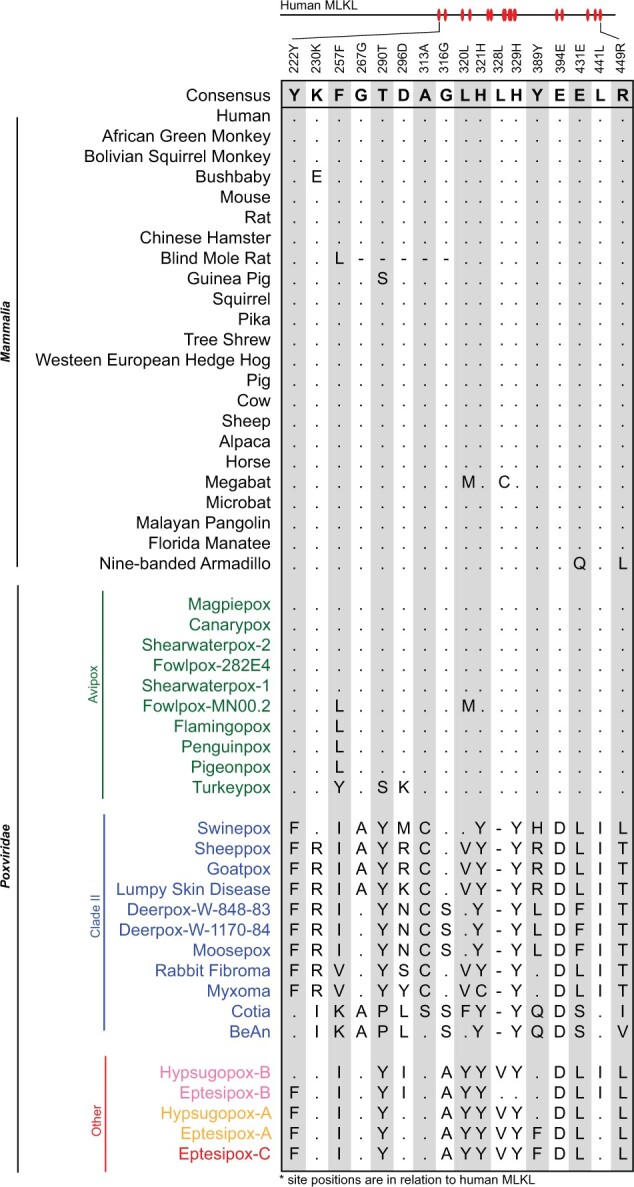
Marker amino acids delineate serial duplications; Avipoxvirus vMLKL is likely the original locus, and Clade II vMLKL is an independent, but derived copy. A set of informative amino acid positions were identified in the alignment of mammalian and vMLKL sequences (supplementary file S4, Supplementary Material online). Residues identical to consensus are shown as “.” while “-” represents a gap in the sequence alignment. Amino acid site positions (shown above the alignment) are relative to human MLKL.
Discussion
Studies of host-pathogen coevolution can reveal new insights that forge infection outcomes. These include vulnerabilities in host defense and determinants that shape pathogen-host range and zoonoses. In addition, analysis of host and pathogen effectors can illuminate new evolutionary paradigms such as the evolutionary trajectories of viral mimics. Here, we focused on components essential for the emerging regulated cell death program of necroptosis; these factors also have roles in overlapping as well as potentially independent host defense pathways (Mandal et al. 2014; Newton et al. 2014; Lawlor et al. 2015; Newton 2015) and cell biology (Yoon et al. 2017). Our findings, particularly the robust signatures of positive selection in primate and bat RIPK3/MLKL, resemble that of key immune factors like cGAS, PKR, and OAS1 (Elde et al. 2009; Hancks et al. 2015). These features are consistent with critical roles for RIPK3/MLKL functions in immune defense during primate and bat evolution. In addition, the maintenance and repeated replacements of vMLKL throughout poxvirus evolution showcase the need to actively counteract RIPK3 functions via competitive binding of its active site (Petrie et al. 2019).
Proposed Model for vMLKL Acquisition
The findings from our evolutionary analysis for vMLKL can be represented by a model consisting of a series of duplications and subsequent deletions of the source locus for this virolog over time (fig. 9A). Our data paired with those of Petrie et al., which also suggested a host origin for vMLKL (Petrie et al. 2019), support the notion that this gene was initially acquired via one horizontal gene transfer event (fig. 7). As vMLKL mirrors a truncated copy of cellular MLKL lacking introns, we hypothesize that this transfer likely occurred via an RNA intermediate. The N-terminal truncation of vMLKL could have transpired either during or after integration. The copy at the original site of integration has been maintained through evolutionary time in Avipoxvirus species and represents locus 1. Following the Avipoxvirus ancestral divergence from a common ancestor shared with Clade II and HYPV/EPTV species, locus 1 appears to have been duplicated to produce locus 2. We hypothesize that locus 1 was subsequently deleted while locus 2 was maintained in the shared ancestor Clade II/EPTV/HYPV, and remains present today in most contemporary genomes of Clade II poxviruses and EPTV-B/HYPV-B. Yet, another possibility is—due to extensive rearrangements resulting in the degradation of any detectable synteny—that locus 1 across Avipoxvirus represents the Clade II/EPTV/HYPV vMLKL. In Yatapoxviruses, locus 2 vMLKL was lost in what appears to be a multigene deletion event (fig. 6 and supplementary fig. S5, Supplementary Material online). Following divergence of the EPTV/HYPV ancestor from Clade II poxviruses, locus 2 was duplicated to a region in the ITR (locus 3) with a subsequent duplication occurring in the other ITR (locus 4). Both vMLKL copies in the EPTV ITRs are supported by our relative depth analysis (fig. 5F and supplementary fig. S3, Supplementary Material online). A similar analysis was attempted but not possible with the hypsugopoxvirus genome data. Thus, vMLKL resides at four distinct loci in poxvirus genomes across the phylogeny. Given a large number of vMLKLs and their diverse histories, these viral genes might represent a novel means to study the origin and evolution of poxvirus promoters and regulatory elements.
Fig. 9.

Model for pathogen-conflict driven evolution of viral and host MLKL. (A) Model for the acquisition and evolution of poxvirus MLKL. Data indicate that vMLKL was likely acquired by a vertebrate host presumably through a single horizontal gene transfer event to a shared poxvirus ancestor (fig. 7). This copy at the original site of integration, located on the right arm, has been maintained in Avipoxvirus (green) genomes to the present day (fig. 5). Following divergence from Avipoxvirus (figs. 6 and 7, supplementary fig. S5, Supplementary Material online), a duplication occurred followed by a deletion event in the common ancestor of Clade II (dark blue) and species classified as “Other” (red). This second copy also on the right arm is present in all Clade II species except in Yatapoxvirus lineages where it is lost (fig. 6). Subsequently, the second copy was duplicated to the ITR in the common ancestor of eptesipoxvirus and hypsugopoxvirus where it remains to present. While hypsugopoxvirus ITR sequence was not included in the final genome assembly, our synteny analysis (fig. 6) suggests vMLKL is also located on an ITR (dashed box) in this lineage. “” represents predicted deletion events. Dashed arrows represent the progression of evolutionary time (left to right). Scaled genome locations for present-day vMLKL copies are represented on the right. (B) Model for the adaptative escape of RIPK3 from EspL-like bacterial proteases supported by rapid evolutionary analysis. EspL cleaves RIPK1, RIPK3, TRIF and ZBP1 (Pearson et al. 2017). Through adaptation at the known EspL cleavage motif in the RHIM domain, RIPK3 may escape cleavage by EspL-like proteases to activate host immune responses. (C) MLKL displays all three key hallmarks that are often common to key host defense factors but as a subset, which OAS1 (Darby et al. 2014; Hancks et al. 2015; Mozzi et al. 2015), MISTRAV, and MISTR1 (Sorouri et al. 2020) also harbor. The factors noted are all rapidly evolving, possess a viral homolog/virolog, and are regulated by immune signals (includes upregulation and downregulation).
Positive Selection Signatures in the Components of Necroptotic Signaling Are Localized to the RIPK3/MLKL Axis
The signatures of rapid evolution for primate (figs. 2 and 3) and bat (fig. 4) RIPK3 and MLKL resemble “molecular arms races” dynamics with pathogen-encoded inhibitors (Daugherty and Malik 2012). The broad distribution of rapidly evolving sites in both RIPK3 and MLKL—several of which are detected by multiple methods (figs. 3 and 4)—is indicative of multiple, undefined antagonists that have existed over evolutionary time. This analysis across the core circuitry of necroptosis enabled two key observations. First, strong selective pressure has been primarily exerted on the downstream components of the pathway where upstream signaling converges (figs. 2–4). Presently, it is not possible to discriminate between the contributions of RIPK3 and MLKL necroptotic and additional activities to these signatures. However, the rapid evolution at the RIPK3:MLKL interface combined with poxvirus vMLKLs signals strong, evolutionary selective pressures exerted on this interaction.
Second, our evolutionary analysis across “necroptotic” factors allowed the identification of rampant positive selection exclusive to the RHIM domain of RIPK3, but not the RHIM domains of the upstream activators ZBP1, TRIF, or RIPK1 (fig. 3). This is extremely striking given the rapid evolution of primate RIPK3, but not the other factors, at amino acid positions that define a demonstrated cleavage site (fig. 3D) for the conserved bacterial protease EspL, which is capable of universally cleaving RHIM. These data might signify a novel “arms race” with bacteria and primates, which are only beginning to come into focus (Barber and Elde 2014). Precedent exists for pathogen proteases imposing strong selective pressure on cellular factors. Specifically, rapid evolution in primate MAVS proximal to the hepatitis protease NS3/4A cleavage site (EREVPR/HRPSPG, “/” cleavage, V506 rapidly evolving site, relative to human) shapes cleavage of this target and its ability to induce interferon (Patel et al. 2012).
This evolutionary pattern might imply a RIPK3 activity, which is counteracted by EspL-like proteases, that is independent of activation by RHIM homotypic interactions (e.g., ZBP1/TRIF/RIPK1) and RIPK1-dependent RIPK3-kinase-independent apoptosis (Mandal et al. 2014; Newton et al. 2014) (fig. 9B). One possible explanation for this signature is the presence of a yet undefined upstream activator of RIPK3 that is triggered by pathogenic bacteria that escapes EspL cleavage-induced degradation due to lack of a RHIM domain. An alternative explanation would be direct activation of RIPK3 by bacteria when both death receptor-initiated apoptosis is blocked by NleB1 (Pearson et al. 2013) and other RHIM proteins are degraded by EspL-like proteases (Pearson et al. 2017). Distinctly, it is unclear whether the rapid evolution in primate RIPK3 would influence antagonism by vIRD (Liu et al. 2021) as the residues in the RHIM domain essential for vIRD binding were not determined. Importantly, we cannot exclude that other functions of these factors independent of necroptosis signaling and execution contributed and/or account for the positive selection observed. Nevertheless, a pathway perspective combined with evolutionary insights can seed new avenues of investigation.
RIPK3/MLKL as a Model for the Maintenance of Compatible, Functional Interactions
Our study describes volatile evolution defined by serial replacement of vMLKL loci in poxvirus genomes by gene duplication to new loci. This work complements the recent discovery of this mimic, which showed two vMLKLs are capable of counteracting necroptosis in cell culture (Petrie et al. 2019). Our integrated comparison of established poxvirus immunomodulators with phylogenomics analysis of vMLKL appears to resolve the long-standing conundrum of how some poxvirus species persist with N-terminally truncated or completely absent E3 ORFs (fig. 5G). In addition, the lack of vMLKLs in orthopoxviruses can be explained, in part, by the newly described vIRD encoded by this genus (Liu et al. 2021). The resolution of the evolutionary history of vMLKL here may serve as a resource in guiding the functional analysis of host and viral MLKL variants and their impact on RIPK3 binding and activity. It has not escaped our attention that the RIPK3/MLKL axis resembles another well-defined host defense axis—PKR/eIF2α—which also consists of a kinase and its substrate, that is counteracted by a different poxvirus pseudosubstrate (K3L) (Elde et al. 2009). However, a major difference between the two systems is that RIPK3 and MLKL are both rapidly evolving, in contrast to PKR being rapidly evolving and eIF2α being ultraconserved to yeast. This is interesting given the PKR/eIF2α system has been useful in understanding how a rapidly evolving factor maintains its interaction with a conserved factor while evolving to escape antagonism (Elde et al. 2009). Perhaps RIPK3/MLKL may be powerful in understanding how two key host defense factors maintain their essential interaction to drive necroptosis when both need to escape pathogen-mediated inhibition.
Evolutionary Signatures of Host–Pathogen Conflict Associated with MLKL Resemble Pivotal Host Defense Factors
MLKL is well-known as the executioner of necroptosis but may have additional roles including functions outside of host defense (Yoon et al. 2017). MLKL: 1) is (up)regulated by immune signals (fig. 2B) (Rusinova et al. 2013; Knuth et al. 2019; Sarhan et al. 2019), 2) has a viral homolog/virolog (figs. 5–8) (Petrie et al. 2019) and 3) is rapidly evolving in bats and primates (figs. 2–4). While several key host defense factors typically display two of these signatures, to our knowledge, only a select few are published that display all three of these hallmarks (fig. 9C): OAS1 (Darby et al. 2014; Hancks et al. 2015; Mozzi et al. 2015), and the recently described MISTRAV and MISTR1 (Sorouri et al. 2020). OAS1 is one of the first interferon-stimulated genes identified and functions in host defense against numerous, diverse viruses (Chakrabarti et al. 2011). MISTR proteins appear to interface with electron transport chain complexes to regulate stress responses and cellular adaptation during infection (Sorouri et al. 2020). This collection of signatures points to MLKL activities as being pivotal over evolution.
Notably, a potential paradox stands of how a factor like MLKL that displays signatures associated with host proteins that determine infection outcomes can be lost repeatedly over evolution (Dondelinger et al. 2016). Based on the literature, we consider that loss of important cellular factors, particularly host factors vital for an immune defense that is also rapidly evolving, may not be so unique. For instance, OAS1 enzymatic loss-of-function has occurred independently in primate lineages (Carey et al. 2019). Another example is the persistence of a high-frequency allele of TRIM5α which impairs retroviral restriction, circulating in the human population (Sawyer et al. 2006).
While gene loss is often considered indicative of dispensability, it is also likely that it reflects genetic incompatibilities. More recently, genetic losses in diverse contexts have begun to be appreciated as a type of “gain” facilitating adaptation. It is now known that deletion of specific enhancers is linked to the acquisition of specific human traits (McLean et al. 2011). In the context of host defense, a prime example is where lentiviruses switched the means by which they inhibit the rapidly evolving primate Tetherin. Namely, a human-specific deletion in Tetherin resulted in the loss of the ability of Nef from primate lentiviruses to antagonize it. As an adaptive countermeasure, HIV-1 Vpu protein evolved to inhibit human Tetherin (Lim et al. 2010). An additional, perhaps classic example is the delta 32 allele of CCR5 (Dean et al. 1996), that promotes resistance to HIV-1, which may have been selected due to selective pressure imposed by smallpox (Galvani and Slatkin 2003). Moreover, loss of the centromere histone protein CenH3 in multiple independent insect lineages, which is associated with a shift to holocentricity, serves as an example of where an essential factor is dispensable (Drinnenberg et al. 2014). Thus, emerging evidence supports the adaptive potential of genetic loss not only in immune defense but also in different aspects of cell biology and across species. Similar dynamics, albeit poorly understood, might be at play for RIPK3 and MLKL. Specifically, while Aves species lack RIPK3—vMLKL is present in all known species of Avipoxvirus (fig. 5D). These data suggest, in part, that at least RIPK3 can be substituted for by another kinase, which may be quickly targeted by pathogens. Regardless, this collection of three hallmarks—rapid evolution, virolog, and induction by cytokines—may serve as a guide to identify other pivotal host defense factors from the numerous uncharacterized genes including hundreds of interferon-stimulated genes lacking known functions. Collectively, we interpret this rare combination of signatures that are associated with OAS1 and now MLKL as strong evidence that its activities have shaped infection outcomes over millions of years.
Materials and Methods
Protein Modeling
Published crystal structures of m(ouse) RIPK3 (PDB:4M66), mMLKL (PBD:4M68), and cocrystal structure for mRIPK3:mMLKL (PDB:4M69) were used for modeling following pairwise alignment with h(uman)MLKL (PDB:4BTF) or the predicted structure of hRIPK3 (Murphy et al. 2013; Xie et al. 2013). Swiss-Model was used to predict the structure of hRIPK3, b(at) RIPK3, bMLKL, and myxoma v(iral)MLKL (Waterhouse et al. 2018). UCSF Chimera (https://www.cgl.ucsf.edu/chimera/, last accessed July 2021) was used to perform analysis and visualize structures including rapidly evolving sites (Pettersen et al. 2004).
Positive Selection Analysis
Nucleotide sequences were obtained from NCBI and Ensembl databases (supplementary file S1, Supplementary Material online). Multiple sequence alignments (MSA) were performed using MAFFT iterative refinement method FFT-NS-i and visualized using Geneious Prime 2020.1.2 (https://www.geneious.com/, last accessed July 2021) (Katoh et al. 2019). Indels were removed through manual trimming. dN/dS lineage estimates were obtained using sample primate gene MSA and phylogenetic tree (created using previously described primate lineage relationships (Perelman et al. 2011)). These served as input for FreeRatio analysis implemented in PAML (Yang 2007). Nucleotide substitution (NS) site analysis was performed with PAML using both F3x4 and F61 codon frequency models. MEME and FUBAR analysis were performed using Datamonkey (https://www.datamonkey.org/, last accessed July 2021) to predict positively selected amino acid sites (Murrell et al. 2012, 2013; Weaver et al. 2018). Supplementary file S1, Supplementary Material online contains additional findings from this analysis.
Identification of Poxvirus vMLKL
Virologs to host MLKL were identified using tBLASTn and/or BLASTp (Altschul et al. 1990). Sequences were selected based on returned BLAST parameters (total score, e-value, and percent identity) and the phylogenetic clustering of the sequences using PhyML (Lefort et al. 2017). Scaled individual hit visualization was constructed using CoGeBlast (Lyons et al. 2008).
Relative Depth Analysis
Eptesipox raw sequencing reads, published in Tu et al., were obtained directly from Dr. Chris Upton (University of Victoria, Victoria BC, Canada) and Dr. Yoshinori Nakazawa (CDC, Atlanta, Georgia) (Tu et al. 2017). NC_035460.1 was used as the eptesipoxvirus reference sequence to map the reads. Myxoma virus raw sequencing reads were obtained from the European Nucleotide Archive (https://www.ebi.ac.uk/ena/browser/home, last accessed April 2021), from Project PRJNA513218 (Kerr et al. 2019). Accession IDs used for this analysis are SRR8402032, SRR8402033, SRR8402034, and SRR8402035. As a control, AF170726.2 was used as the myxoma virus reference sequence to map the reads. Raw sequencing reads were processed using Trimmomatic-0.39 (Bolger et al. 2014) to remove adaptors. Tanoti short read aligner (https://bioinformatics.cvr.ac.uk/software/tanoti/, last accessed April 2021) was then used to map reads to the reference genome. SAMtools (Li et al. 2009) was used to convert between file formats, merge files and calculate coverage per base pair. Bins of 500 nucleotides were created for relative depth analysis, which was calculated by dividing the bins by the mean of the non-ITR genomic region. Data were visualized using gplots (Warnes et al. 2020).
Phylogenetic Analysis
Multiple Sequence Alignments were performed using MAFFT, with iterative refinement method L-INS-i, on amino acid sequences retrieved from NCBI and Ensembl databases (supplementary file S4, Supplementary Material online) (Katoh et al. 2019). Maximum-Likelihood trees were inferred by PhyML using 100 bootstrap replicates (Lefort et al. 2017).
Percent Amino Acid Identity Heatmap
Multiple Sequence Alignments were performed using MAFFT, with iterative refinement method L-INS-i, on amino acid sequences retrieved from the NCBI database (supplementary file S2, Supplementary Material online) (Katoh et al. 2019). The R package gplots were used to create the heatmap found in figure 5C (R Core Team 2013; Warnes et al. 2020).
Poxvirus RCD-Associated Host Range Gene Identification
Host range genes were identified using BLASTp and tBLASTn. Sequences were confirmed using reciprocal BLAST hits. Protein families were found using genomic database annotations and published results for included poxvirus genomes. Domain classification for homologs and protein families was confirmed using NCBI conserved genome database (Lu et al. 2020). Results were compared to previous homolog studies performed by Bratke et al. 2013 and Haller et al. 2014, and can be found in Supplementary file S2, Supplementary Material online (Bratke et al. 2013; Haller et al. 2014). Proteins truncated at the N- and C-termini were not included in figure 5G but can be found in Supplementary file S2, Supplementary Material online with appropriate labels. An exception is necroptosis inhibitor E3, which is predicted to be N-terminally truncated in Leporipoxviruses (Bratke et al. 2013). Genes residing in the inverted terminal repeats (ITR) were counted as separate genes and thus two instances in order to stay consistent with the figure 5 initial analysis, where the eptesipoxvirus ITR is included and analyzed. The p28/N1R and Ankyrin-repeat families were annotated using NCBI database annotations. The presence of these domains was confirmed using NCBI conserved genome database or via BLASTp (Lu et al. 2020). This analysis includes all proteins possessing a p28/N1R-like or Ankyrin-repeat domain and does not predict if these proteins are functional and/or likely truncated.
High Resolution Synteny Analysis
Reciprocal BLAST hits (RBH) were performed for coding sequences upstream and downstream of vMLKL genomic locations across poxvirus species used in this study. Results can be found in supplementary file S2, Supplementary Material online. Genomic database annotations and the NCBI conserved genome database were used to confirm results (Lu et al. 2020). Gene blocks representing Ankyrin-repeat containing proteins without one distinct hit, but conserved homology across poxvirus species are colored navy.
Whole Genome Comparison
Coregenes 5.0 (https://coregenes.ngrok.io/) was used to compare selected poxvirus genomes (Contreras-Moreira and Vinuesa 2013). Matches were identified using bidirectional best hit with an e-value = 1 × 105. RIdeogram (Zhaodong Hao 2020) was used to visualize the output obtained from Coregenes 5.0.
Cell Lines
HT29 and HeLa cell lines were obtained from ATCC. Mouse embryonic fibroblasts (MEFs) were produced in-house using day E15 embryos. All cell lines were cultured in Corning DMEM with L-Glutamine, 4.5 g/L Glucose, and Sodium Pyruvate supplemented with 10% FBS and 1X Gibco Antibiotic-Antimycotic solution. Cell lines were maintained at 37 °C in a humidified incubator at 5% CO2.
Cell Culture Treatments
Cells were treated with either human IFNα (1000 U/mL [PBL Assay Science, USA]) or human IFNγ (1000 U/mL [ThermoFisher, USA]) diluted in DMEM and incubated for 24 h. After 24 h, cell lysates were harvested for western blots as described below.
Western Blot Analysis
Cells were collected using RIPA Lysis and Extraction Buffer (ThermoFisher, 89901) supplemented with Protease and Phosphatase Inhibitor Cocktail (Abcam, ab201119). Protein concentrations were measured by Bradford assay. For HT29 and HeLa, 5 µg of protein lysates were loaded onto an SDS-PAGE. For MEFs, 10 µg of protein lysates were separated by SDS-PAGE. Proteins were transferred to 0.45 µM Immobilon-P PVDF membrane (Millipore, IPVH00010) at 200 mA for 90 min. Membranes were blocked with blocking buffer (5% milk in TBST) for 30 min at RT. Membranes were incubated in primary antibodies at 4 °C overnight. The following primary antibodies were used: human MLKL (GeneTex, GTX107538), mouse MLKL (Cell Signaling Technology, 37705S), STAT1 D1K9Y Rabbit mAb (Cell Signaling Technology, 14994S), STAT1 Antibody #9172 (Cell Signaling Technology, 9172S), p-STAT1 Tyr701 58D6 Rabbit mAb (Cell Signaling Technology, 9167S), and β-Actin (Abcam, ab49900). Membranes were washed three times with TBST for 5 min and incubated with Goat Anti-Rabbit IgG (Bio-Rad, 170-6515) for 1 h at RT. Membranes were washed three times with TBST and incubated with Clarity Max Western ECL Substrate (Bio-Rad, 1705062). Following incubation, blots were imaged using the Chemidoc MP Imager (Bio-Rad).
Supplementary Material
Supplementary data are available at Molecular Biology and Evolution online.
Supplementary Material
Acknowledgments
This work was supported by an R00 Pathway to Independence Award from the National Institute of General Medical Sciences ([5R00GM119126-03] to D.C.H.) and a Recruitment of First-Time, Tenure-Track Faculty from Cancer Prevention & Research Institute of Texas Award (D.C.H). C.P. is funded by the National Institutes of Health, Molecular Microbiology (Training Grant No. [T32 AI007520]). We express gratitude to other members of the Hancks Lab along with Dr. Anant Gharpure and Dr. Sherry Haller for feedback, discussion, and comments on the manuscript. We also thank Dr. Chris Upton and Dr. Yoshinori Nakazawa as well as Dr. Chiara Chiapponi and Dr. Davide Lelli for their assistance with the eptesipoxvirus and hypsugopoxvirus data, respectively.
Data Availability
The data in this article are available in the supplementary files S1–S4, Supplementary Material online and were obtained through publicly available databases. All analytical software are also publicly available.
References
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ.. 1990. Basic local alignment search tool. J Mol Biol. 215(3):403–410. [DOI] [PubMed] [Google Scholar]
- Barber GN. 2001. Host defense, viruses and apoptosis. Cell Death Differ. 8(2):113–126. [DOI] [PubMed] [Google Scholar]
- Barber MF, Elde NC.. 2014. Escape from bacterial iron piracy through rapid evolution of transferrin. Science 346(6215):1362–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best SM. 2008. Viral subversion of apoptotic enzymes: escape from death row. Annu Rev Microbiol. 62:171–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B.. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30(15):2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bratke KA, McLysaght A, Rothenburg S.. 2013. A survey of host range genes in poxvirus genomes. Infect Genet Evol. 14:406–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brault M, Oberst A.. 2017. Controlled detonation: evolution of necroptosis in pathogen defense. Immunol Cell Biol. 95(2):131–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Z, Jitkaew S, Zhao J, Chiang HC, Choksi S, Liu J, Ward Y, Wu LG, Liu ZG.. 2014. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat Cell Biol. 16(1):55–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey CM, Govande AA, Cooper JM, Hartley MK, Kranzusch PJ, Elde NC.. 2019. Recurrent loss-of-function mutations reveal costs to OAS1 antiviral activity in primates. Cell Host Microbe. 25(2):336–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarti A, Jha BK, Silverman RH.. 2011. New insights into the role of RNase L in innate immunity. J Interferon Cytokine Res. 31(1):49–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Li W, Ren J, Huang D, He WT, Song Y, Yang C, Li W, Zheng X, Chen P, et al. 2014. Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death. Cell Res. 24(1):105–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contreras-Moreira B, Vinuesa P.. 2013. GET_HOMOLOGUES, a versatile software package for scalable and robust microbial pangenome analysis. Appl Environ Microbiol. 79(24):7696–7701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danthi P. 2016. Viruses and the diversity of cell death. Annu Rev Virol. 3(1):533–553. [DOI] [PubMed] [Google Scholar]
- Darby AC, McInnes CJ, Kjær KH, Wood AR, Hughes M, Martensen PM, Radford AD, Hall N, Chantrey J.. 2014. Novel host-related virulence factors are encoded by squirrelpox virus, the main causative agent of epidemic disease in red squirrels in the UK. PLoS One. 9(7):e96439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daugherty MD, Malik HS.. 2012. Rules of engagement: molecular insights from host-virus arms races. Annu Rev Genet. 46:677–700. [DOI] [PubMed] [Google Scholar]
- Davies KA, Fitzgibbon C, Young SN, Garnish SE, Yeung W, Coursier D, Birkinshaw RW, Sandow JJ, Lehmann WIL, Liang L-Y, et al. 2020. Distinct pseudokinase domain conformations underlie divergent activation mechanisms among vertebrate MLKL orthologues. Nat Commun. 11(1):3060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean M, Carrington M, Winkler C, Huttley GA, Smith MW, Allikmets R, Goedert JJ, Buchbinder SP, Vittinghoff E, Gomperts E, et al. 1996. Genetic restriction of HIV-1 infection and progression to AIDS by a deletion allele of the CKR5 structural gene. Hemophilia growth and development study, multicenter AIDS cohort study, multicenter hemophilia cohort study, San Francisco City Cohort, ALIVE Study. Science 273(5283):1856–1862. [DOI] [PubMed] [Google Scholar]
- Dondelinger Y, Hulpiau P, Saeys Y, Bertrand MJM, Vandenabeele P.. 2016. An evolutionary perspective on the necroptotic pathway. Trends Cell Biol. 26(10):721–732. [DOI] [PubMed] [Google Scholar]
- Drinnenberg IA, deYoung D, Henikoff S, Malik HS.. 2014. Recurrent loss of CenH3 is associated with independent transitions to holocentricity in insects. Elife 3:e03676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elde NC, Child SJ, Geballe AP, Malik HS.. 2009. Protein kinase R reveals an evolutionary model for defeating viral mimicry. Nature 457(7228):485–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher-Etherington A, Nobre L, Nightingale K, Antrobus R, Nichols J, Davison AJ, Stanton RJ, Weekes MP.. 2020. Human cytomegalovirus protein pUL36: a dual cell death pathway inhibitor. Proc Natl Acad Sci USA. 117(31):18771–18779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, Alnemri ES, Altucci L, Amelio I, Andrews DW, et al. 2018. Molecular mechanisms of cell death: recommendations of the nomenclature committee on cell death 2018. Cell Death Differ. 25(3):486–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvani AP, Slatkin M.. 2003. Evaluating plague and smallpox as historical selective pressures for the CCR5-Delta 32 HIV-resistance allele. Proc Natl Acad Sci USA. 100(25):15276–15279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green DR. 2019. The coming decade of cell death research: five riddles. Cell 177(5):1094–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green DR, Fitzgerald P.. 2016. Just so stories about the evolution of apoptosis. Curr Biol. 26(13):R620–R627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H, Omoto S, Harris PA, Finger JN, Bertin J, Gough PJ, Kaiser WJ, Mocarski ES.. 2015. Herpes simplex virus suppresses necroptosis in human cells. Cell Host Microbe. 17(2):243–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haller SL, Peng C, McFadden G, Rothenburg S.. 2014. Poxviruses and the evolution of host range and virulence. Infect Genet Evol. 21:15–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancks DC, Hartley MK, Hagan C, Clark NL, Elde NC.. 2015. Overlapping patterns of rapid evolution in the nucleic acid sensors cGAS and OAS1 suggest a common mechanism of pathogen antagonism and escape. PLoS Genet. 11(5):e1005203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayman DT. 2016. Bats as viral reservoirs. Annu Rev Virol. 3(1):77–99. [DOI] [PubMed] [Google Scholar]
- He S, Liang Y, Shao F, Wang X.. 2011. Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3-mediated pathway. Proc Natl Acad Sci USA. 108(50):20054–20059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen I, Rayamajhi M, Miao EA.. 2017. Programmed cell death as a defence against infection. Nat Rev Immunol. 17(3):151–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser WJ, Offermann MK.. 2005. Apoptosis induced by the toll-like receptor adaptor TRIF is dependent on its receptor interacting protein homotypic interaction motif. J Immunol. 174(8):4942–4952. [DOI] [PubMed] [Google Scholar]
- Kaiser WJ, Sridharan H, Huang C, Mandal P, Upton JW, Gough PJ, Sehon CA, Marquis RW, Bertin J, Mocarski ES.. 2013. Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J Biol Chem. 288(43):31268–31279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser WJ, Upton JW, Long AB, Livingston-Rosanoff D, Daley-Bauer LP, Hakem R, Caspary T, Mocarski ES.. 2011. RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature 471(7338):368–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser WJ, Upton JW, Mocarski ES.. 2008. Receptor-interacting protein homotypic interaction motif-dependent control of NF-kappa B activation via the DNA-dependent activator of IFN regulatory factors. J Immunol. 181(9):6427–6434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Rozewicki J, Yamada KD.. 2019. MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinform. 20(4):1160–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr JF, Wyllie AH, Currie AR.. 1972. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 26(4):239–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr PJ, Eden JS, Di Giallonardo F, Peacock D, Liu J, Strive T, Read AF, Holmes EC.. 2019. Punctuated evolution of myxoma virus: rapid and disjunct evolution of a recent viral lineage in Australia. J Virol. 93(8):e01994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knuth AK, Rosler S, Schenk B, Kowald L, van Wijk SJL, Fulda S.. 2019. Interferons transcriptionally up-regulate MLKL expression in cancer cells. Neoplasia 21(1):74–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koehler H, Cotsmire S, Langland J, Kibler KV, Kalman D, Upton JW, Mocarski ES, Jacobs BL.. 2017. Inhibition of DAI-dependent necroptosis by the Z-DNA binding domain of the vaccinia virus innate immune evasion protein, E3. Proc Natl Acad Sci USA. 114(43):11506–11511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonin EV, Aravind L.. 2002. Origin and evolution of eukaryotic apoptosis: the bacterial connection. Cell Death Differ. 9(4):394–404. [DOI] [PubMed] [Google Scholar]
- Lamkanfi M, Dixit VM.. 2010. Manipulation of host cell death pathways during microbial infections. Cell Host Microbe. 8(1):44–54. [DOI] [PubMed] [Google Scholar]
- Lawlor KE, Khan N, Mildenhall A, Gerlic M, Croker BA, D’Cruz AA, Hall C, Kaur Spall S, Anderton H, Masters SL, et al. 2015. RIPK3 promotes cell death and NLRP3 inflammasome activation in the absence of MLKL. Nat Commun. 6:6282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefort V, Longueville JE, Gascuel O.. 2017. SMS: smart model selection in PhyML. Mol Biol Evol. 34(9):2422–2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R.. 2009. The sequence alignment/map format and SAMtools. Bioinformatics. 25(16):2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim ES, Malik HS, Emerman M.. 2010. Ancient adaptive evolution of tetherin shaped the functions of Vpu and Nef in human immunodeficiency virus and primate lentiviruses. J Virol. 84(14):7124–7134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Nailwal H, Rector J, Rahman MM, Sam R, McFadden G, Chan FK.. 2021. A class of viral inducer of degradation of the necroptosis adaptor RIPK3 regulates virus-induced inflammation. Immunity 54(2):247–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu S, Wang J, Chitsaz F, Derbyshire MK, Geer RC, Gonzales NR, Gwadz M, Hurwitz DI, Marchler GH, Song JS, et al. 2020. CDD/SPARCLE: the conserved domain database in 2020. Nucleic Acids Res. 48(D1):D265–D268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons E, Pedersen B, Kane J, Alam M, Ming R, Tang H, Wang X, Bowers J, Paterson A, Lisch D, et al. 2008. Finding and comparing syntenic regions among Arabidopsis and the outgroups papaya, poplar, and grape: CoGe with rosids. Plant Physiol. 148(4):1772–1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mack C, Sickmann A, Lembo D, Brune W.. 2008. Inhibition of proinflammatory and innate immune signaling pathways by a cytomegalovirus RIP1-interacting protein. Proc Natl Acad Sci USA. 105(8):3094–3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maelfait J, Liverpool L, Bridgeman A, Ragan KB, Upton JW, Rehwinkel J.. 2017. Sensing of viral and endogenous RNA by ZBP1/DAI induces necroptosis. EMBO J. 36(17):2529–2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal P, Berger SB, Pillay S, Moriwaki K, Huang C, Guo H, Lich JD, Finger J, Kasparcova V, Votta B, et al. 2014. RIP3 induces apoptosis independent of pronecrotic kinase activity. Mol Cell. 56(4):481–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin RN Jr, Malik HS.. 2017. Genetic conflicts: the usual suspects and beyond. J Exp Biol. 220(1):6–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean CY, Reno PL, Pollen AA, Bassan AI, Capellini TD, Guenther C, Indjeian VB, Lim X, Menke DB, Schaar BT, et al. 2011. Human-specific loss of regulatory DNA and the evolution of human-specific traits. Nature 471(7337):216–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell PS, Emerman M, Malik HS.. 2013. An evolutionary perspective on the broad antiviral specificity of MxA. Curr Opin Microbiol. 16(4):493–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mompean M, Li W, Li J, Laage S, Siemer AB, Bozkurt G, Wu H, McDermott AE.. 2018. The structure of the necrosome RIPK1-RIPK3 core, a human hetero-amyloid signaling complex. Cell 173(5):1244–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mozzi A, Pontremoli C, Forni D, Clerici M, Pozzoli U, Bresolin N, Cagliani R, Sironi M.. 2015. OASes and STING: adaptive evolution in concert. Genome Biol Evol. 7(4):1016–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy JM, Czabotar PE, Hildebrand JM, Lucet IS, Zhang JG, Alvarez-Diaz S, Lewis R, Lalaoui N, Metcalf D, Webb AI, et al. 2013. The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity 39(3):443–453. [DOI] [PubMed] [Google Scholar]
- Murphy JM, Lucet IS, Hildebrand JM, Tanzer MC, Young SN, Sharma P, Lessene G, Alexander WS, Babon JJ, Silke J, et al. 2014. Insights into the evolution of divergent nucleotide-binding mechanisms among pseudokinases revealed by crystal structures of human and mouse MLKL. Biochem J. 457(3):369–377. [DOI] [PubMed] [Google Scholar]
- Murrell B, Moola S, Mabona A, Weighill T, Sheward D, Kosakovsky Pond SL, Scheffler K.. 2013. FUBAR: a fast, unconstrained Bayesian approximation for inferring selection. Mol Biol Evol. 30(5):1196–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murrell B, Wertheim JO, Moola S, Weighill T, Scheffler K, Kosakovsky Pond SL.. 2012. Detecting individual sites subject to episodic diversifying selection. PLoS Genet. 8(7):e1002764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nailwal H, Chan FK.. 2019. Necroptosis in anti-viral inflammation. Cell Death Differ. 26(1):4–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton K. 2015. RIPK1 and RIPK3: critical regulators of inflammation and cell death. Trends Cell Biol. 25(6):347–353. [DOI] [PubMed] [Google Scholar]
- Newton K, Dugger DL, Wickliffe KE, Kapoor N, de Almagro MC, Vucic D, Komuves L, Ferrando RE, French DM, Webster J, et al. 2014. Activity of protein kinase RIPK3 determines whether cells die by necroptosis or apoptosis. Science 343(6177):1357–1360. [DOI] [PubMed] [Google Scholar]
- Nichols DB, De Martini W, Cottrell J.. 2017. Poxviruses utilize multiple strategies to inhibit apoptosis. Viruses 9(8):215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberst A, Dillon CP, Weinlich R, McCormick LL, Fitzgerald P, Pop C, Hakem R, Salvesen GS, Green DR.. 2011. Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature 471(7338):363–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel MR, Loo YM, Horner SM, Gale M Jr., Malik HS.. 2012. Convergent evolution of escape from hepaciviral antagonism in primates. PLoS Biol. 10(3):e1001282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson JS, Giogha C, Muhlen S, Nachbur U, Pham CL, Zhang Y, Hildebrand JM, Oates CV, Lung TW, Ingle D, et al. 2017. EspL is a bacterial cysteine protease effector that cleaves RHIM proteins to block necroptosis and inflammation. Nat Microbiol. 2:16258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson JS, Giogha C, Ong SY, Kennedy CL, Kelly M, Robinson KS, Lung TW, Mansell A, Riedmaier P, Oates CV, et al. 2013. A type III effector antagonizes death receptor signalling during bacterial gut infection. Nature 501(7466):247–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson JS, Murphy JM.. 2017. Down the rabbit hole: is necroptosis truly an innate response to infection? Cell Microbiol. 19:e12750. [DOI] [PubMed] [Google Scholar]
- Perelman P, Johnson WE, Roos C, Seuanez HN, Horvath JE, Moreira MA, Kessing B, Pontius J, Roelke M, Rumpler Y, et al. 2011. A molecular phylogeny of living primates. PLoS Genet. 7(3):e1001342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrie EJ, Sandow JJ, Lehmann WIL, Liang LY, Coursier D, Young SN, Kersten WJA, Fitzgibbon C, Samson AL, Jacobsen AV, et al. 2019. Viral MLKL homologs subvert necroptotic cell death by sequestering cellular RIPK3. Cell Rep. 28(13):3309–3319. [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE.. 2004. UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem. 25(13):1605–1612. [DOI] [PubMed] [Google Scholar]
- R Core Team. 2013. R: a language and environment for statistical computing. Vienna (Austria: ): R Core Team. [Google Scholar]
- Rusinova I, Forster S, Yu S, Kannan A, Masse M, Cumming H, Chapman R, Hertzog PJ.. 2013. Interferome v2.0: an updated database of annotated interferon-regulated genes. Nucleic Acids Res. 41:D1040–D1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarhan J, Liu BC, Muendlein HI, Weindel CG, Smirnova I, Tang AY, Ilyukha V, Sorokin M, Buzdin A, Fitzgerald KA, et al. 2019. Constitutive interferon signaling maintains critical threshold of MLKL expression to license necroptosis. Cell Death Differ. 26(2):332–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawyer SL, Wu LI, Akey JM, Emerman M, Malik HS.. 2006. High-frequency persistence of an impaired allele of the retroviral defense gene TRIM5a in humans. Curr Biol. 16(1):95–100. [DOI] [PubMed] [Google Scholar]
- Sawyer SL, Wu LI, Emerman M, Malik HS.. 2005. Positive selection of primate TRIM5a identifies a critical species-specific retroviral restriction domain. Proc Natl Acad Sci USA. 102(8):2832–2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanmugam N, Baker M, Sanz-Hernandez M, Sierecki E, Gambin Y, Steain M, Pham CLL, Sunde M.. 2021. Herpes simplex virus encoded ICP6 protein forms functional amyloid assemblies with necroptosis-associated host proteins. Biophys Chem. 269:106524. [DOI] [PubMed] [Google Scholar]
- Sorouri M, Chang T, Jesudhasan P, Pinkham C, Elde NC, Hancks DC.. 2020. Signatures of host-pathogen evolutionary conflict reveal MISTR-A conserved MItochondrial STress Response network. PLoS Biol. 18(12):e3001045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strasser A, O’Connor L, Dixit VM.. 2000. Apoptosis signaling. Annu Rev Biochem. 69:217–245. [DOI] [PubMed] [Google Scholar]
- Sun L, Wang H, Wang Z, He S, Chen S, Liao D, Wang L, Yan J, Liu W, Lei X, et al. 2012. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 148(1–2):213–227. [DOI] [PubMed] [Google Scholar]
- Sun X, Yin J, Starovasnik MA, Fairbrother WJ, Dixit VM.. 2002. Identification of a novel homotypic interaction motif required for the phosphorylation of receptor-interacting protein (RIP) by RIP3. J Biol Chem. 277(11):9505–9511. [DOI] [PubMed] [Google Scholar]
- Taylor JM, Barry M.. 2006. Near death experiences: poxvirus regulation of apoptotic death. Virology 344(1):139–150. [DOI] [PubMed] [Google Scholar]
- Tu SL, Nakazawa Y, Gao J, Wilkins K, Gallardo-Romero N, Li Y, Emerson GL, Carroll DS, Upton C.. 2017. Characterization of Eptesipoxvirus, a novel poxvirus from a microchiropteran bat. Virus Genes. 53(6):856–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UniProt. 2019. UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 47:D506–D515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upton JW, Kaiser WJ.. 2017. DAI another way: necroptotic control of viral infection. Cell Host Microbe. 21(3):290–293. [DOI] [PubMed] [Google Scholar]
- Upton JW, Kaiser WJ, Mocarski ES.. 2008. Cytomegalovirus M45 cell death suppression requires receptor-interacting protein (RIP) homotypic interaction motif (RHIM)-dependent interaction with RIP1. J Biol Chem. 283(25):16966–16970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upton JW, Kaiser WJ, Mocarski ES.. 2012. DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe. 11(3):290–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanden Berghe T, Linkermann A, Jouan-Lanhouet S, Walczak H, Vandenabeele P.. 2014. Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol. 15(2):135–147. [DOI] [PubMed] [Google Scholar]
- Wang H, Sun L, Su L, Rizo J, Liu L, Wang LF, Wang FS, Wang X.. 2014. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell. 54(1):133–146. [DOI] [PubMed] [Google Scholar]
- Warnes GR, Bolker B, Bonebakker L, Gentleman R, Huber W, Liaw A, Lumley T, Maechler M, Magnusson A, Moeller S, Schwartz M, et al. 2020. gplots: various R programming tools for plotting data. R package version 3.0.3. 3(1):1. Available from: https://github.com/talgalili/gplots
- Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, Heer FT, de Beer TAP, Rempfer C, Bordoli L, et al. 2018. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 46(W1):W296–W303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver S, Shank SD, Spielman SJ, Li M, Muse SV, Kosakovsky Pond SL.. 2018. Datamonkey 2.0: a modern web application for characterizing selective and other evolutionary processes. Mol Biol Evol. 35(3):773–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie T, Peng W, Yan C, Wu J, Gong X, Shi Y.. 2013. Structural insights into RIP3-mediated necroptotic signaling. Cell Rep. 5(1):70–78. [DOI] [PubMed] [Google Scholar]
- Yang Z. 2007. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 24(8):1586–1591. [DOI] [PubMed] [Google Scholar]
- Yang Z, Bielawski JP.. 2000. Statistical methods for detecting molecular adaptation. Trends Ecol Evol. 15(12):496–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon S, Kovalenko A, Bogdanov K, Wallach D.. 2017. MLKL, the protein that mediates necroptosis, also regulates endosomal trafficking and extracellular vesicle generation. Immunity 47(1):51–65. [DOI] [PubMed] [Google Scholar]
- Hao Z, Lv D, Ge Y, Shi J, Weijers D, Yu G, Chen J.. 2020. RIdeogram: drawing SVG graphics to visualize and map genome-wide data on the idiograms. PeerJ Comput Sci. 6:e251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zmasek CM, Godzik A.. 2013. Evolution of the animal apoptosis network. Cold Spring Harb Perspect Biol. 5(3):a008649. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data in this article are available in the supplementary files S1–S4, Supplementary Material online and were obtained through publicly available databases. All analytical software are also publicly available.