

Abstract
The methyltransferase-like (METTL) proteins constitute a family of seven-beta-strand methyltransferases with S-adenosyl methionine-binding domains that modify DNA, RNA, and proteins. Methylation by METTL proteins contributes to the epigenetic, and in the case of RNA modifications, epitranscriptomic regulation of a variety of biological processes. Despite their functional importance, most investigations of the substrates and functions of METTLs within metazoans have been restricted to model vertebrate taxa. In the present work, we explore the evolutionary mechanisms driving the diversification and functional differentiation of 33 individual METTL proteins across Metazoa. Our results show that METTLs are nearly ubiquitous across the animal kingdom, with most having arisen early in metazoan evolution (i.e., occur in basal metazoan phyla). Individual METTL lineages each originated from single independent ancestors, constituting monophyletic clades, which suggests that each METTL was subject to strong selective constraints driving its structural and/or functional specialization. Interestingly, a similar process did not extend to the differentiation of nucleoside-modifying and protein-modifying METTLs (i.e., each METTL type did not form a unique monophyletic clade). The members of these two types of METTLs also exhibited differences in their rates of evolution. Overall, we provide evidence that the long-term evolution of METTL family members was driven by strong purifying selection, which in combination with adaptive selection episodes, led to the functional specialization of individual METTL lineages. This work contributes useful information regarding the evolution of a gene family that fulfills a variety of epigenetic functions, and can have profound influences on molecular processes and phenotypic traits.
Keywords: methyltransferase, METTL, epigenetics, phylogenetics, selection, metazoan
Introduction
Methyltransferase enzymes catalyze the transfer of methyl groups to DNA, RNA, proteins, and other biomolecules (Cheng and Blumenthal 1999). An increasing interest in these proteins has been driven by epigenetics, formally defined as, “the study of phenomena and mechanisms that cause chromosome-bound, heritable changes to gene expression that are not dependent on changes to the DNA sequence” (Deans and Maggert 2015). In particular, work examining DNA methylation, as well as the enzymes responsible for this modification, has been conducted for decades across a broad range of contexts and model systems (Holliday 2006). In addition, other studies have recognized the structural and functional importance of methyltransferases that modify specific residues in proteins such as histones (Couture and Trievel 2006; Ng et al. 2009). More recently, there has been a growing interest in the post-transcriptional modification of RNA molecules, a concept known as RNA epigenetics or epitranscriptomics (He 2010; Saletore et al. 2012). Although over 170 different RNA modifications have been recorded (Machnicka et al. 2013), relatively little is known about the enzymes responsible for these modifications.
Depending on their protein structure, most methyltransferases are categorized into three large superfamilies: seven-beta-strand methyltransferases, SET methyltransferases, and SPOUT methyltransferases (Petrossian and Clarke 2009). Among the seven-beta-strand methyltransferases, the methyltransferase-like (METTL) gene family (table 1) encodes proteins characterized by a conserved S-adenosyl methionine (SAM or AdoMet) binding domain that is formed by part of the seven-beta-strand structure (Martin and McMillan 2002; Petrossian and Clarke 2009). In this study, we examine the METTL family because these enzymes have been demonstrated to modify DNA/RNA nucleosides as well as protein residues (Ignatova et al. 2019), leading to changes in gene expression and phenotype that can have profound effects on an organism’s condition. For example, METTL3 and METTL14 are responsible for the formation of N6-methyladenosine (m6A) in RNA (Liu et al. 2014). In eukaryotes, m6A is a very common RNA modification, and has been shown to play a role in mammalian temperature stress response by promoting translation initiation of heat shock response genes (Zhou et al. 2015). As another example, it has been demonstrated that METTL21D methylates a lysine residue of valosin-containing protein (VCP), and may be linked to diseases, including cancer, in humans (Thiele et al. 2011; Kernstock et al. 2012). However, the precise targets and functions of several METTL proteins remain unresolved. In addition to structure, methyltransferases can be characterized by the type of biomolecule that they modify. Here, METTLs are characterized into three types: those that modify DNA/RNA, those that modify proteins, and those for which the biomolecule type they modify is currently unknown.
Table 1.
METTL Genes Separated by Type (i.e., DNA/RNA-modifying, protein-modifying, and unknown).
| Gene Name | NCBI ID | Aliases | Preferred Name | Target |
|---|---|---|---|---|
| DNA/RNA-modifying METTLs | ||||
| METTL1 | 4234 | C12orf1, TRM8, TRMT8, YDL201w | tRNA (guanine-N(7)-)-methyltransferase | m7G (tRNA, mRNA, miRNA) |
| METTL2 (METTL2A/B) | 339175/55798 | METL, PSENIP1 | tRNA N(3)-methylcytidine methyltransferase METTL2; methyltransferase-like protein 2 | m3C (tRNA) |
| METTL3 | 56339 | IME4, M6A, MT-A70, Spo8, hMETTL3 | N6-adenosine-methyltransferase catalytic subunit | m6A (mRNA) |
| METTL4 | 64863 | HsT661 | N(6)-adenine-specific methyltransferase METTL4 | m6A (DNA)/m6Am (snRNA) |
| METTL5 | 29081 | HSPC133, MRT72 | rRNA N6-adenosine-methyltransferase METTL5 | m6A (rRNA) |
| METTL6 | 131965 | None | tRNA N(3)-methylcytidine methyltransferase METTL6 | m3C (tRNA) |
| METTL8 | 79828 | TIP | mRNA N(3)-methylcytidine methyltransferase METTL8 | m3C (mRNA) |
| METTL14 | 57721 | None | N6-adenosine-methyltransferase noncatalytic subunit | m6A (mRNA) |
| METTL15 | 196074 | METT5D1 | 12S rRNA N4-methylcytidine (m4C) methyltransferase | m4C (rRNA) |
| METTL16 | 79066 | METT10D | RNA N6-adenosine-methyltransferase METTL16 | m6A (ncRNA, pre-mRNA, mRNA) |
| METTL19 | 152992 | TRMT44, C4orf23, TRM44 | Probable tRNA (uracil-O(2)-)-methyltransferase | 2′-O-methyluridine (tRNA) |
| METTL25B | 51093 | RRNAD1, C1orf66, CGI-41 | Ribosomal RNA adenine dimethylase domain containing 1 | m6A (rRNA) |
|
| ||||
| Protein-modifying METTLs | ||||
| METTL9 | 51108 | CGI-81, DREV, DREV1, PAP1 | Methyltransferase-like protein 9 | Unknown |
| METTL10 | 399818 | EEF1AKMT2, C10orf138, Efm4 | EEF1A lysine methyltransferase 2 | Lysine (EEF1A) |
| METTL11A | 28989 | NTMT1, AD-003, C9orf32, HOMT1A, NRMT, NRMT1, NTM1A | N-terminal Xaa-Pro-Lys N-methyltransferase 1 | N-terminal |
| METTL11B | 149281 | C1orf184, HOMT1B, NTM1B | Alpha N-terminal protein methyltransferase 1B | N-terminal |
| METTL12 | 751071 | CSKMT, CS-KMT, U99HG | Citrate synthase-lysine N-methyltransferase CSKMT, mitochondrial | Lysine (citrate synthase) |
| METTL13 | 51603 | EEF1AKNMT, 5630401D24Rik, CGI-01, DFNB26, DFNB26M, DFNM1, KIAA0859, feat | eEF1A lysine and N-terminal methyltransferase | Lysine (EEF1A) |
| METTL18 | 92342 | AsTP2, C1orf156, HPM1 | Histidine protein methyltransferase 1 homolog | Histidine |
| METTL20 | 254013 | ETFBKMT, C12orf72, ETFB-KMT | Electron transfer flavoprotein beta subunit lysine methyltransferase | Lysine (ETFβ) |
| METTL21A | 151194 | FAM119A, HCA557b, HSPA-KMT | Protein N-lysine methyltransferase METTL21A | Lysine (HSPA) |
| METTL21B | 25895 | EEF1AKMT3, FAM119B | EEF1A lysine methyltransferase 3 | Lysine (EEF1A) |
| METTL21C | 196541 | C13orf39 | Protein-lysine methyltransferase METTL21C | Lysine (VCP) |
| METTL21D | 79609 | VCPKMT, C14orf138, VCP-KMT | Protein-lysine methyltransferase METTL21D | Lysine (VCP) |
| METTL21E | 403183 | 4832428D23Rik, Gm991 | Protein-lysine methyltransferase METTL21E | Lysine (VCP) |
| METTL22 | 79091 | C16orf68 | Methyltransferase-like protein 22 | Lysine (Kin17) |
| METTL23 | 124512 | C17orf95, MRT44 | Methyltransferase-like protein 23 | Unknown |
|
| ||||
| METTLs of unknown function | ||||
| METTL7A | 25840 | AAM-B, AAMB | Methyltransferase-like protein 7A | Unknown |
| METTL7B | 196410 | ALDI | Methyltransferase-like protein 7B | Unknown |
| METTL17 | 64745 | METT11D1 | Methyltransferase-like protein 17, mitochondrial | Unknown |
| METTL25 | 84190 | C12orf26 | Methyltransferase-like protein 25 | Unknown |
| METTL26 | 84326 | C16orf13, JFP2 | Methyltransferase-like 26 | Unknown |
| METTL27 | 155368 | WBSCR27 | Methyltransferase-like protein 27 | Unknown |
Note.—The gene names, NCBI IDs, aliases, and preferred names are vertebrate METTL genes.
We also chose to examine the METTL family because it has been studied considerably less than other families of methyltransferases, such as the DNA methyltransferase (DNMT) family. The structures, activities, and functions of DNMTs have been studied across all domains of life (Lyko 2018; Bhattacharyya et al. 2020). In contrast, the examination of METTL proteins has remained extremely limited in nonmodel organisms despite the potential epigenetic functions of METTLs and their resulting influence on biological processes and phenotypic characteristics. Within Metazoa, studies of METTL proteins are often restricted to humans, mice, and rats. Thus, there remains a knowledge gap regarding the function of METTLs within other metazoan taxa, particularly within nonvertebrate phyla. Exploring how METTL proteins have evolved throughout Metazoa is important for understanding: 1) the biological functions of different METTLs, 2) the contribution of METTL functional specialization to the process of diversification among metazoan groups, and 3) the relative importance of different types of METTLs and their gene regions that vary in evolutionary conservation. The present work uses extensive data mining to collect available METTL sequences across metazoan taxa. From these, a select number of sequences from nine representative taxa across eight different metazoan phyla are used for multiple phylogenetic and evolutionary analyses. Lastly, evidence of diversifying selection episodes is assessed using vertebrate METTL sequences. By investigating METTL proteins across Metazoa, this work provides insight into the evolution of this epigenetically relevant gene family.
Results and Discussion
METTLs Are Widespread across Metazoa
In this study, we examined 33 genes that encode proteins within the METTL family (table 1). These were broadly characterized into three types: 1) those that target and modify the nucleosides of DNA or RNA molecules, 2) those that target and modify the residues of proteins, and 3) those with unknown modification targets. Overall, we assessed 12 METTLs that have been demonstrated to modify DNA or RNA nucleosides, 15 METTLs that have been demonstrated to modify protein residues, and 6 METTLs of currently unknown function (table 1). A detailed search of these methyltransferase-like genes was conducted within the NCBI GenBank database (https://www.ncbi.nlm.nih.gov/genbank). Importantly, this search was restricted to NCBI and may be limited by sequence data availability and annotation. Thus, any failure to detect a specific METTL gene within a particular phylum does not necessarily indicate that the gene is lost in that phylum. Nonetheless, the search found that METTL genes are present in most of the major metazoan phyla examined here including Porifera, Cnidaria, Brachiopoda, Mollusca, Platyhelminthes, Nematoda, Priapulida, Arthropoda, Echinodermata, Hemichordata, and Chordata (fig. 1). Chordates possess the largest number of METTL genes (i.e., 33 out of 33 examined in this current study), however, the diversification of METTL genes does not appear to be restricted to chordates and other deuterostomes. Indeed, among protostomes, our search detected that Mollusca possesses 28 unique METTL genes and Arthropoda has 26 unique METTLs. Basally to triploblastic animals, diploblastic cnidarians have 25 METTLs and parazoan sponges, which lack specialized tissues, have 19 METTLs. The widespread presence of METTLs in ancient metazoan lineages such as Cnidaria and Porifera suggests that they arose quite early in the evolutionary history of Metazoa. Furthermore, it appears that the early functional diversification of these various METTL types was necessary to enable critical cellular functionality, and therefore, the evolution of these phyla (Carroll et al. 2013).
Fig. 1.

METTLs in major metazoan phyla according to NCBI search results. The metazoan cladogram is modified from the Tree of Life web project (http://tolweb.org/tree/phylogeny.html). The matrix displays the detection (gray), probable detection but with uncertain identification (striped-gray), or failure of detection (white) by the search for each METTL gene within each phylum. The number of METTL genes out of 33 total is noted in parenthesis with asterisks indicating phyla in which probable METTL sequences were either located but could not be definitively identified as specific METTL genes, or were located outside of our search in the NCBI GenBank database (e.g., Ensembl Metazoa). METTLs are listed in order of decreasing number of representatives in each phylum (left to right).
There were several sequences that were determined to be probable METTL sequences, but could not be definitively identified as specific METTL genes based on the data available (fig. 1, represented as striped-gray boxes). These sequences were tentatively recognized as METTLs via orthologous gene searches and Basic Local Alignment Search Tool (BLAST) searches but did not have any predicted gene identity or function recorded in NCBI. Gene searches in NCBI only identified putative METTL orthologs within one species of annelid, the leech Helobdella robusta (NCBI: txid6412). For example, searches for orthologs to human METTL1 identified one gene in H. robusta (HELRODRAFT_91834), however, it is described as a, “hypothetical protein” and the reference sequence is defined as, “Helobdella robusta hypothetical protein partial mRNA” (XM_009013229.1). NCBI BLAST results of this sequence include hits to METTL1 sequences in other metazoan phyla, although with relatively low query coverage and percent identity. All search results of putative METTL orthologs within Annelida were similar “hypothetical” proteins in H. robusta. Upon including putative METTL protein sequences from H. robusta in a separate phylogenetic analysis, each sequence clustered with its prospective METTL orthologs from other metazoan taxa (supplementary fig. S5, Supplementary Material online). Thus, it is probable that METTLs occur in Annelida, however, based on the current data available in NCBI, we hesitate to state with certainty the precise identity of these H. robusta sequences.
Our search in NCBI was not able to locate any methyltransferase-like genes within the phylum Ctenophora. This is not to say that the methylation of DNA, RNA, or protein molecules does not occur in ctenophores. DNA methylation has been detected in the promoter and gene body regions of ctenophore genomes, and ctenophores have been shown to possess DNMT1 (Dabe et al. 2015). DNMT1, a highly conserved member of the DNMT family of methyltransferases, acts to methylate cytosines in DNA and has been demonstrated to maintain methylation patterns following DNA replication (Kangaspeska et al. 2008; Lyko 2018). Ctenophore genomes also contain a DNMT2 gene (Dabe et al. 2015). Although a member of the DNMT family, DNMT2 is a RNA methyltransferase also known as tRNA aspartic acid methyltransferase 1 (TRDMT1) (Goll et al. 2006; Lyko 2018). Thus, methylation in ctenophores may be driven by methyltransferases outside of the METTL family, such as by DNMTs. Alternatively, METTLs may be present within ctenophores, but they were undetected in our search due to a limited availability of genomic resources. For instance, Ensembl Metazoa (https://metazoa.ensembl.org/index.html) detected at least one putative METTL in the ctenophore Mnemiopsis leidyi, which was identified as a potential ortholog of METTL1 (ML09559a). Upon including this putative METTL1 sequence from M. leidyi in a separate phylogenetic analysis, it clustered with METTL1 sequences from other metazoan taxa, although with low support (supplementary fig. S5, Supplementary Material online). This indicates that METTLs likely occur in Ctenophora. Nevertheless, evidence of this remains scant due to limited sequence data for multiple ctenophore species across available databases. Indeed, neither this nor similar METTL sequences were detected in Ctenophora by our NCBI search used for this study (fig. 1). As sequence data continue to increase in quality and quantity, METTLs may later be identified within Ctenophora and we expect additional METTLs will be found within other phyla as well (e.g., Annelida).
METTL2, a methyltransferase that forms N3-methylcytidine (m3C) in tRNAs (Xu et al. 2017), is found in the most phyla examined here (fig. 1). Additionally, humans possess two paralogs of METTL2 that share 99% amino acid sequence identity, METTL2A and METTL2B (Arimbasseri et al. 2016; Lentini et al. 2020). Interestingly, the majority of METTLs found present in at least nine metazoan phyla are known to modify RNA (e.g., METTL1, METTL2, METTL3, METTL5, METTL6, METTL14, and METTL15). The ubiquity of RNA methyltransferases underscores the importance of RNA modifications in metazoan evolution. It is probable that RNA molecules required more than only four canonical nucleosides, and that the evolution of chemically distinct modified nucleosides enabled their multiple, necessary functions (Grosjean 2009).
Phylogeny of METTLs Part I: Overview
Following our data search, we opted to limit our analyses to nine preselected metazoan taxa (the sponge Amphimedon queenslandica, the stony coral Acropora millepora, the sea hare Aplysia californica, the priapulid Priapulus caudatus, the shrimp Litopenaeus vannamei, the sea star Acanthaster planci, the acorn worm Saccoglossus kowalevskii, the lancelet Branchiostoma belcheri, and humans Homo sapiens). This allowed us to analyze a reasonable quantity of data while including taxa that represented a diverse set of metazoan phyla. Furthermore, each of these taxa is the subject of numerous ecology and evolution studies and was selected due to its genome availability on NCBI. Thus, the phylogenetic and subsequent evolutionary analyses, with the exception of the episodic selection analysis that is limited to vertebrate METTLs, only include taxa from the phyla Porifera, Cnidaria, Mollusca, Priapulida, Arthropoda, Echinodermata, Hemichordata, and Chordata. Representatives from the phyla Ctenophora, Brachiopoda, Annelida, Platyhelminthes, and Nematoda are not included.
Phylogenetic reconstructions were performed based on an alignment of METTL protein sequences (alignment available in Supplementary Material online) from the nine preselected metazoan taxa. Escherichia coli prokaryotic rRNA dimethyltransferase (KsgA) was selected as the outgroup. KsgA, which was first identified in E. coli (Helser et al. 1972), dimethylates adenosines in the terminal helix (helix 45) near the 3′ end of the small subunit rRNA (Van Knippenberg et al. 1984). Escherichia coli KsgA is a member of the KsgA/Dim1 methyltransferase family, and like METTL proteins, it contains a seven-beta-strand sheet structure (Tu et al. 2009). KsgA, as well as the modifications it catalyzes, are nearly universally conserved throughout evolution (Xu et al. 2008), and have been described in archaea (O’Farrell et al. 2006), eubacteria (Helser et al. 1972; Van Buul et al. 1983), eukaryotes (Lafontaine et al. 1994; Housen et al. 1997), and in eukaryotic organelles (Tokuhisa et al. 1998; Seidel-Rogol et al. 2003). Using both in vivo and in vitro analyses, one study showed that archaeal and eukaryotic orthologs of KsgA were capable of complimenting for KsgA function in bacteria, demonstrating that the recognition elements and methyltransferase activity of the KsgA/Dim1 family have evolved little since the three domains diverged (O’Farrell et al. 2006). Thus, E. coli KsgA was selected as a methyltransferase outside of, but that shares structural similarities with, the METTL family, and has orthologs present throughout all domains of life but has demonstrated little evolutionary change. The selection of E. coli KsgA as the outgroup for our metazoan METTL phylogeny is further supported by a separate phylogenetic analysis that included several METTL orthologs from eukaryotic taxa outside of Metazoa (supplementary fig. S5, Supplementary Material online). This included METTL protein sequences from the choanoflagellate Salpingoeca rosetta and the yeast Saccharomyces cerevisiae, all of which grouped with their respective METTL lineages rather than by taxa or with E. coli KsgA.
The obtained tree showed that overall, the amino acid sequences cluster together by METTL lineage rather than by the taxa to which they belong (fig. 2; supplementary fig. S1, Supplementary Material online). Nearly all METTL lineages were each represented by a distinct monophyletic clade supported with high confidence values (e.g., all METTL1 orthologs from different metazoan taxa formed a monophyletic clade with 100% bootstrap). This observation suggests that the evolution of METTLs has been largely driven by selective constraints associated with the particular functional identity of each METTL protein, likely involved in specific outcomes of structural or functional specialization. Prior studies examining the structures, interactions, and functions of various METTLs have provided insight into their probable targets for methylation (e.g., Cloutier et al. 2013; Xu et al. 2017; Ignatova et al. 2019). Accordingly, METTL proteins tend to cluster together by their modification targets (i.e., DNA/RNA nucleosides versus protein residues), although with several exceptions.
Fig. 2.

Maximum likelihood tree describing the evolutionary relationships across metazoan METTL proteins. Bootstrap values (1,000 replicates) are displayed at each internal node. Branch labels displayed in blue indicate METTLs that have been shown to target DNA or RNA nucleosides, labels in red are METTLs shown to target protein residues, and labels in purple represent METTLs whose function and targets are unknown. The METTL proteins comprise nine different taxa representing eight different phyla: A sponge (Amphimedon queenslandica, Porifera), a stony coral (Acropora millepora, Cnidaria), a sea hare (Aplysia californica, Mollusca), a priapulid (Priapulus caudatus, Priapulida), a shrimp (Litopenaeus vannamei, Arthropoda), a sea star (Acanthaster planci, Echinodermata), an acorn worm (Saccoglossus kowalevskii, Hemichordata), a lancelet (Branchiostoma belcheri, Chordata), and human (Homo sapiens, Chordata). Subtrees with the same METTL gene from multiple taxa have been collapsed for figure readability.
Despite generally grouping by type (i.e., METTLs that modify nucleic acids versus METTLs that modify proteins), it seems clear that not all of the DNA/RNA-modifying METTLs (fig. 2, labeled in blue) exclusively possess a single, unique monophyletic origin and therefore do not appear to have evolved from a shared common ancestor. All of the protein-modifying METTLs (fig. 2, labeled in red) do not appear to share a single, unique monophyletic origin with one another either. Indeed, METTLs that target nucleosides for methylation are distributed across multiple clades within the phylogeny, and are occasionally grouped with METTLs that modify protein residues. This pattern could be explained by convergent molecular evolution that resulted in the emergence of METTLs with similar targets due to comparable selective pressures and modification requirements (Losos 2011). Alternatively, the diversification of the METTL family may have occurred very early in, or possibly prior to, the evolution of metazoans; this may have been driven by different selective pressures and constraints or even stochastic processes (Stayton 2015). The phylogenetic relationships among the various METTL types that target nucleic acids, proteins, or whose targets remain undetermined is further elaborated below.
Phylogeny of METTLs Part II: DNA and RNA Modifiers
DNA- and RNA-modifying METTLs target nucleosides for modification, resulting in the formation of methylcytidines, methylguanosines, methyladenosines, or methyluridines. Almost all of these METTLs appear to primarily target RNA molecules, although there is evidence that METTL4 may act to methylate DNA (Kweon et al. 2019; Zhang et al. 2020). METTL15, METTL6, METTL2, and METTL8 are all associated with the formation of methylcytidine. METTL15 falls most basally to the E. coli KsgA outgroup (fig. 2) and forms N4-methylcytidine (m4C) in RNA (Van Haute et al. 2019). The remaining proteins that methylate cytidine, METTL2, METTL6, and METTL8, form a highly supported cluster (100% bootstrap). Both METTL2 and METTL6 form m3C in specific tRNA molecules, whereas METTL8 forms m3C in mRNA (Xu et al. 2017). Furthermore, of the METTL genes, our NCBI search detected that METTL2 and METTL6 are present across the greatest number of major metazoan phyla, whereas METTL8 was only found to be present in the phylum Chordata (fig. 1). METTL1, the only METTL protein known to contribute methylguanosine, forms N7-methylguanosine (m7G) in tRNA, mRNA, and miRNA (Okamoto et al. 2014; Pandolfini et al. 2019; Zhang et al. 2019). In the phylogeny, METTL1 groups with methylcytidine-forming proteins as well as with METTL7A and METTL7B, whose precise function is unknown, although with very low confidence (bootstrap <10%) (fig. 2).
Several other nucleic acid-modifying METTLs target adenosine to form methyladenosine. These include METTL3, METTL4, METTL5, METTL14, METTL16, and METTL25B, which all function to form m6A. However, despite this shared function, these METTLs do not form a single cluster in the METTL phylogeny (fig. 2). Perhaps the best characterized of these are METTL3 and METTL14, which together form a heterocomplex with WTAP (Wilms’ Tumor1-Associating Protein) to form m6A in mRNA (Fu et al. 2014; Liu et al. 2014; Meyer and Jaffrey 2017). METTL3, METTL14, and WTAP are considered to be m6A writers (i.e., factors that encode the chemical modification) (Lewis et al. 2017). Although METTL3 is the primary m6A-forming enzyme, METTL14 does not demonstrate enzymatic activity and instead appears to bind substrate RNA and augment METTL3 activity (Śledź and Jinek 2016; Wang P et al. 2016; Wang X et al. 2016; Meyer and Jaffrey 2017). Unsurprisingly, METTL3 and METTL14 cluster together in the METTL phylogeny (86% bootstrap) (fig. 2).
The precise function(s) of METTL4, which clusters closest with METTL14 (95% bootstrap), is less clear than that of the METTL3−METTL14 heterocomplex. There is evidence that METTL4 modifies RNA, DNA, or both nucleic acids. Studies have demonstrated that METTL4 forms m6A in DNA (Kweon et al. 2019; Zhang et al. 2020), whereas others have shown that it forms N6,2′O-dimethyladenosine (m6Am) in snRNA (Chen et al. 2020; Goh et al. 2020). Interestingly, a recent study demonstrated that the human METTL3−METTL14 complex was active in vitro as a DNA N6-adenine methyltransferase, although it is unclear if it functions in vivo (Woodcock et al. 2019).
Evidence suggests that METTL5, METTL16, and METTL25B also function in the formation of m6A. METTL5 and METTL16 cluster near to each other in the METTL phylogeny (92% bootstrap). METTL5 forms a heterodimeric complex with TRMT112 (tRNA methyltransferase subunit 11-2) to provide metabolic stability in the formation of m6A in 18S rRNA (van Tran et al. 2019; Leismann et al. 2020). The METTL5−TRMT112 complex has structural similarities to that of a m6A DNMT, and has been hypothesized to possess an RNA-binding mode unique to other m6A methyltransferases (van Tran et al. 2019). METTL16, which forms m6A in ncRNA, pre-mRNAs (Warda et al. 2017), and mRNA (Nance et al. 2020) appears to be structurally distinct from METTL5 (van Tran et al. 2019) as well as the METTL3−METTL14 heterocomplex (Ruszkowska et al. 2018). Furthermore, because METTL16 has been documented as both a nuclear protein and as a cytoplasmic methyltransferase, it has been suggested that its RNA binding targets may differ by the location of METTL16 within the cell (Nance et al. 2020). The function of METTL25B is less clear, although Gene Ontology (GO) annotations related to METTL25B infers it possesses rRNA (adenine-N6, N6-)-dimethyltransferase activity. Lastly, the function of METTL19, which does not cluster with other RNA-modifying METTLs in the phylogeny (fig. 2), remains uncertain, but it has been reported to be a likely tRNA uracil-O(2)-methyltransferase that forms 2′-O-methyluridine (Leschziner et al. 2011). Overall, the majority of METTLs that modify nucleic acids appear to target primarily RNA, rather than DNA, nucleosides. Although some of these DNA/RNA-modifying METTLs are closely related phylogenetically, they do not share a single monophyletic origin and are instead interspersed with METTLs that modify protein residues.
Phylogeny of METTLs Part III: Protein Modifiers
Multiple METTL proteins are responsible for the methylation of eukaryotic elongation factor 1 alpha (EEF1A). In addition to its role in protein synthesis as a translation factor that delivers aminoacyl-tRNA to the ribosome, EEF1A has been ascribed to a wide variety of other functions in eukaryotes beyond protein synthesis (Mateyak and Kinzy 2010). METTL10, METTL21B, and METTL13 are responsible for the methylation of lysine residues within EEF1A (Li et al. 2014; Shimazu et al. 2014; Hamey et al. 2017; Malecki et al. 2017; Jakobsson et al. 2018). Interestingly, these METTLs do not cluster together within the phylogeny (fig. 2). For example, METTL10 clusters closest with METTL19 (86% bootstrap), which is predicted to form 2′-O-methyluridine in RNA. On the other hand, METTL21B appears to share a close common ancestor with METTL21A (100% bootstrap), which also methylates lysine, although its target is within the molecular chaperone heat shock protein 70 (Hsp70) (Shimazu et al. 2014). METTL13 is unique in that it possesses two distinct methyltransferase domains and methylates the N-terminal as well as a lysine residue (Lys55) within EEF1A (Jakobsson et al. 2018; Liu et al. 2019).
Other METTL proteins methylate lysine residues of VCP, also known as p97, an ATP-driven chaperone involved in numerous, independent cellular processes that is highly conserved across eukaryotes (Kernstock et al. 2012; Meyer et al. 2012). METTL21C and METTL21E, which form a highly supported cluster (100% bootstrap), are both responsible for lysine methylation in VCP (Wiederstein et al. 2018; Wang et al. 2019). Although METTL21D, which is also known to methylate lysine residues of VCP (Kernstock et al. 2012), forms a cluster with other METTL21 lineages (98% bootstrap), it is not as closely related to the other VCP methyltransferases as they are to each other. Our search detected METTL21C and METTL21E only in chordates, whereas METTL21D is present in at least eight different metazoan phyla (fig. 1). Thus, METTL21D may have evolved prior to METTL21C and METTL21E, although their functions appear to be conserved.
METTL22, which methylates a lysine residue of Kin17 (Cloutier et al. 2013), appears to share a common monophyletic origin with a handful of other lysine methyltransferases, METTL21A-E (fig. 2). Kin17 functions in DNA replication and repair as well as RNA metabolism (Kannouche et al. 2000; Despras et al. 2003; Masson et al. 2003; Angulo et al. 2005). METTL22 may impact these processes because methylation of Kin17 affects its distribution between chromatin and the cytoplasm (Cloutier et al. 2014). METTL20 targets and trimethylates lysines of the electron transfer flavoprotein β-subunit (ETFβ) (Rhein et al. 2014; Małecki et al. 2015). Unlike most lysine methyltransferases, METTL20 is localized to the mitochondria where it appears to modulate the function of ETF. This difference in target and function may partially explain why METTL20 fails to cluster with other lysine protein methyltransferases (fig. 2). METTL12 is yet another lysine methyltransferase, although it targets citrine synthase (CS) (Małecki et al. 2017; Rhein et al. 2017). CS is localized within the mitochondrial matrix of eukaryotic cells and catalyzes the first step in the Krebs cycle (Wiegand and Remington 1986). Interestingly, METTL12 was not phylogenetically close to any other lysine methyltransferases (fig. 2).
Not all protein-modifying METTL targets are restricted to lysine residues. For instance, METTL18 appears to monomethylate a histidine residue of the ribosomal protein RPL3, and potentially other proteins, to form a 3-methylhistidine (Webb, Zurita-Lopez, et al. 2010; Cloutier et al. 2013). METTL18 is currently the only METTL for which there is evidence that its primary target is histidine, although it shares a common phylogenetic origin with several lysine methyltransferases (fig. 2). Other protein-modifiers such as METTL11A and METTL11B appear to target N-terminal residues. METTL11A and METTL11B are homologs and cluster closely together (100% bootstrap) (fig. 2), and whereas our search found that METTL11A is present in several metazoan phyla, METTL11B is only found in chordates (fig. 1). METTL11A recognizes the N-terminal X-Pro-Lys sequence motif (Webb, Lipson, et al. 2010) and likely acts upon multiple protein targets (Petkowski et al. 2012; Faughn et al. 2018). Furthermore, METTL11A is capable of mono-, di-, or trimethylating N-terminal residues (Tooley et al. 2010). Although it recognizes the same sequence motifs, METTL11B appears to be an N-terminal monomethylase that increases the activity of METTL11A in chordates (Petkowski et al. 2013; Faughn et al. 2018). METTL11A and METTL11B appear to be closely related to METTL9 (94% bootstrap) (fig. 2). Although the precise identities of METTL9 target molecules is currently unknown, METTL9 has been demonstrated to interact with multiple protein partners, including the membrane protein Calnexin precursor (CANX) (Ignatova et al. 2019).
Lastly, METTL23 is localized in both the nucleus and cytoplasm, and is predicted to contain a domain for either an adenosine−methionine or lysine methyltransferase (Reiff et al. 2014). Although its precise target has not been definitely identified, METTL23 has been shown to associate with a transcription factor subunit, GABPA (GA-binding protein transcription factor, alpha subunit), and a molecular chaperone, GroEL (Bernkopf et al. 2014; Reiff et al. 2014). Here, METTL23 appears to be phylogenetically related to several lysine methyltransferases that are associated with various molecular chaperones and other stress responsive proteins (e.g., Hsp70, VCP, and possibly Kin17), including METTL21A-E and METTL22 (fig. 2). Future studies investigating molecular responses to stress may benefit from examining these METTLs and their potential contributions to post-translational modifications of stress-activated proteins. Overall, although the target substrates of many of the protein-modifying METTLs have been identified, much less is known regarding the specific functional purpose behind each modification. It is also worth noting that although there has been a sizable effort to examine enzymes that methylate histones (Clarke 2013), the majority of protein-modifying METTLs discussed here appear to act upon the lysine residues of nonhistone proteins. Regardless, all of the protein-modifying METTLs do not share a single, unique monophyletic origin.
Phylogeny of METTLs Part IV: Unknown Modifiers
The functions and biomolecule target types of several METTLs are currently unknown. This includes METTL7A, METTL7B, METTL17, METTL25, METTL26, and METTL27. The phylogenetic reconstruction may provide some insight into the potential function and target of these METTLs (fig. 2, labeled in purple), although this should be interpreted with caution. METTL7A has been reported to be an integral membrane protein, and METTL7B has been shown to interact closely with TMEM126A, a mitochondrial membrane protein (Zehmer et al. 2009; Ignatova et al. 2019). Furthermore, both appear to function in lipid droplet formation in the endoplasmic reticulum (Turró et al. 2006; Zehmer et al. 2009). Here we found that METTL7A and METTL7B are closely related to one another (100% bootstrap), and appear to be most phylogenetically related to RNA-modifying METTLs that create m3C, although with relatively low confidence (70% bootstrap, fig. 2).
METTL17 has been shown to physically and functionally interact with estrogen receptors (ERs), ERɑ and ERβ, and acts as a coactivator, modulating their transcriptional activities (Du et al. 2015). This suggests that METTL17 is a protein modifier that targets ER proteins. However, another study proposed that METTL17 may function to form methylcytidine in RNA (Shi et al. 2019). This study found that METTL17 interacted with 12S mitochondrial ribosomal RNA (mt-rRNA) and small subunits of mitochondrial ribosome (MSSU). In particular, METTL17 appeared to regulate m4C and m5C modifications in 12S mt-rRNA, and its presence or absence altered mitochondrial ribosome function (Shi et al. 2019). Thus, it is unclear if METTL17 modifies protein residues or RNA nucleosides. In the phylogeny, METTL17 was most closely related to METTL18, a protein histidine methyltransferase, though with low support (fig. 2).
Lastly, very little is known about METTL25, METTL26, and METTL27, although all three are predicted to have the characteristic seven-beta-strand structure METTL motif and possess S-adenosylmethionine-dependent methyltransferase (SAM) activity. METTL25 possibly functions in RNA methylation as it is phylogenetically very similar to METTL25B (100% bootstrap, fig. 2), which forms m6A in rRNA. METTL26 forms a lowly supported cluster with METTL11A, METTL11B, and METTL9, which may suggest its function as a protein-modifying METTL. Based on the phylogeny, there is no clear indication of the potential function or target of METTL27, which forms a lowly supported cluster with a protein-modifying METTL (METTL12), several RNA-modifying METTLs (METTL2, METTL6, and METTL8), and other METTLs of unknown function (METTL7A and METTL7B) (fig. 2). The phylogenetic tree may provide some indication of the potential substrates for the METTLs of currently unknown functions. However, given that the DNA/RNA-modifying METTLs and protein-modifying METTLs are not separated into distinct clades, we must once again stress that these findings should be interpreted with caution. Additional studies will be required to identify the specific purpose of METTL7A, METTL7B, METTL17, METTL25, METTL26, and METTL27, but the phylogeny does provide some insight into their evolution.
Phylogeny of METTLs Part V: Conserved Domains
All conserved domains (CDs) that were identified within each METTL fell within one of two CD superfamilies: the AdoMet MTases superfamily or the MT-A70 superfamily (table 2; supplementary tables S1 and S2, Supplementary Material online). Only METTL3, METTL14, and METTL4 contain CDs within the MT-A70 superfamily (CDD accession number cl01947), whereas all other METTLs contain CDs within the AdoMet MTases superfamily (CDD accession number cl17173). S-adenosyl-l-methionine (SAM or AdoMet) methyltransferases (MTases) use SAM/AdoMet as a substrate for methyl transfer (Schubert et al. 2003). MT-A70 refers specifically to the 70-kDa SAM-binding subunit first identified in human mRNA (N6-adenosine)-methyltransferase (i.e., human METTL3) (Bokar et al. 1997; Bujnicki et al. 2002). Thus, the CD Search successfully identified the conserved SAM-binding domains formed by the central seven-beta-strand motif that is structurally conserved across all METTLs.
Table 2.
Conserved Domains within Each METTL Gene Based on Results from CD-Search and Batch CD-Search (NCBI).
| Superfamily | CD | Gene |
|---|---|---|
| AdoMet MTases superfamily | COG2263 | METTL5 |
| DREV | METTL9 | |
| DUF938 | METTL26 | |
| Methyltransf_11 | METTL7A, METTL7B | |
| Methyltransf_12 | METTL2, METTL6 | |
| Methyltransf_16 | METTL21A, METTL21D | |
| Methyltransf_25 | METTL2, METTL6, METTL7A, METTL8, METTL10, METTL12, METTL13, METTL27 | |
| Methyltransf_31 | METTL10 | |
| Methyltransf_32 | METTL25B | |
| Methyltransf_4 | METTL1 | |
| Methyltransf_PK | METTL11A | |
| Nnt1 | METTL20 | |
| PRK00050 | METTL15 | |
| No specific CD (superfamily only) | METTL11B, METTL16, METTL17, METTL18, METTL19, METTL21B, METTL21C, METTL21E, METTL22, METTL23, METTL25 | |
|
| ||
| MT-A70 superfamily | MT-A70 | METTL3, METTL14 |
| No specific CD (superfamily only) | METTL4 | |
For several METTLs, no specific CD was recognized (i.e., only the CD superfamily was identified). A second phylogenetic reconstruction was performed using only the CD-containing regions of the protein sequences (alignment available in Supplementary Material online). Interestingly, although some METTLs that share CDs cluster together (e.g., METTL3, METTL14, and METTL4), this is not true for all METTLs with shared CDs (fig. 3; supplementary fig. S2, Supplementary Material online). For instance, eight different METTLs share the CD Methyltransf_25 (table 2), but these METTLs form separate clusters in the phylogenetic reconstruction (fig. 3). Thus, even within relatively well-conserved areas of the amino acid sequence, there exists enough sequence-level diversity to support the specialization of multiple METTLs with different and highly specific targets and functions.
Fig. 3.

Maximum likelihood phylogeny reconstructed based on the CDs of each METTL protein across Metazoa. Bootstrap values (1,000 replicates) are displayed at each node. Branch label colors are identical to figure 2.
Overall, the full sequence phylogeny and the CD phylogeny were similar, which indicates that METTL evolution was largely determined by the selective constraints operating on these CDs. However, there were some notable differences between the full sequence and the CD phylogenies. For instance, in the phylogeny represented by full protein sequences (fig. 2), METTL17 was most closely related to METTL18, a protein histidine methyltransferase, whereas in the CD phylogeny (fig. 3), METTL17 forms a lowly conserved cluster with several RNA-modifying METTLs: METTL6, METTL2, METTL8, and METTL1 (62% bootstrap). As previously mentioned, there is conflicting evidence regarding whether METTL17 acts on protein residues or RNA nucleosides. Another difference between the phylogenetic reconstructions worth noting is the relationship of METTL27 to other METTLs, which remained unclear in the full sequence phylogeny (fig. 2). In the CD phylogeny, however, METTL27 is most closely related to METTL11A and METTL11B (78% bootstrap, fig. 3), which modify protein N-terminal residues. Thus, restricting the protein sequences to only the CD regions provided some additional insight into the potential targets of METTLs of unknown function.
Molecular Evolution and Selection in METTL Proteins
Despite shared similarities in protein structure (i.e., an SAM-binding domain and seven-beta-strand motif), METTL evolution throughout Metazoa appears to have been largely driven by functional constraints that led to the specialization of each METTL lineage. To further examine the evolutionary mechanisms underlying their differentiation, variation within each METTL lineage was evaluated using two separate alignments (nucleotide and protein) of all METTLs from each of the nine preselected metazoan taxa (table 3) (alignments available in Supplementary Material online). Although variation in METTLs with only one representative taxa/sequence in the selected data set was not assessed (i.e., METTL8, METTL11B, METTL21B, METTL21C, and METTL21E), the obtained results showed that METTL27, a METTL of unknown function, displayed the greatest variation (mean amino acid distance, dAA = 0.69 ± 0.03) (table 3). METTL27 also displayed slightly higher levels of codon bias (ENC = 48.47). However, codon bias was relatively low overall, with average ENC values per METTL ranging from 45.13 (METTL21B) to 54.92 (METTL7B). The second most variation in protein sequences was observed in METTL12 (dAA = 0.63 ± 0.03), a lysine methyltransferase found in several different metazoan phyla (fig. 1) that was not closely related to other lysine methyltransferases in the phylogenetic reconstruction (fig. 2). The METTL that exhibited the least diversity was METTL14 (dAA = 0.32 ± 0.02), an m6A writer that forms a heterocomplex with METTL3 and WTAP. The next lowest levels of variation were in METTL1, METTL2, METTL3, METTL5, and METTL6 (average dAA = 0.38 ± 0.02). Thus, the six METTLs that showed the least amount of variation are each associated with the methylation of RNA nucleosides, indicating that high sequence conservation may be necessary to preserve their function.
Table 3.
Selection Results, Variance Estimations,a and Best-Fit ML Models for All METTLs and Each Individual METTL Gene.
| Gene | R | Z-test | d NT ± SE | d AA ± SE | d S ± SE | d N ± SE | ENC | ModelNT | ModelAA |
|---|---|---|---|---|---|---|---|---|---|
| All | 0.5 | 5.8*** | 0.65 ± 0.01 | 0.82 ± 0.02 | 0.74 ± 0.005 | 0.62 ± 0.02 | 52.45 | GTR + G + I | LG + G |
| METTL1 | 0.8 | 23.38*** | 0.36 ± 0.01 | 0.32 ± 0.02 | 0.83 ± 0.02 | 0.23 ± 0.02 | 53.60 | GTR + G + I | WAG + G |
| METTL2 | 0.7 | 22.02*** | 0.4 ± 0.01 | 0.39 ± 0.02 | 0.79 ± 0.01 | 0.28 ± 0.02 | 51.09 | GTR + G + I | LG + G |
| METTL3 | 0.7 | 27.28*** | 0.4 ± 0.01 | 0.4 ± 0.02 | 0.82 ± 0.01 | 0.28 ± 0.01 | 53.89 | GTR + G | LG + G |
| METTL4 | 0.6 | 12.93*** | 0.49 ± 0.01 | 0.55 ± 0.02 | 0.78 ± 0.02 | 0.4 ± 0.02 | 50.78 | T92 + G + I | LG + G + I |
| METTL5 | 0.8 | 18.17*** | 0.4 ± 0.01 | 0.42 ± 0.02 | 0.81 ± 0.02 | 0.28 ± 0.02 | 53.16 | T92 + G | LG + G |
| METTL6 | 0.8 | 20.0*** | 0.39 ± 0.01 | 0.4 ± 0.02 | 0.81 ± 0.02 | 0.27 ± 0.02 | 51.25 | GTR + G | LG + G |
| METTL7A | 0.6 | 12.02*** | 0.52 ± 0.01 | 0.61 ± 0.02 | 0.78 ± 0.02 | 0.44 ± 0.02 | 54.10 | K2 + G + I | LG + G + I |
| METTL7B | 0.6 | 10*** | 0.53 ± 0.01 | 0.61 ± 0.03 | 0.8 ± 0.02 | 0.45 ± 0.02 | 54.92 | JC + I | LG + I |
| METTL8 | Vertebrates only | 50.53 | |||||||
| METTL9 | 0.7 | 14.15*** | 0.49 ± 0.01 | 0.54 ± 0.02 | 0.81 ± 0.01 | 0.39 ± 0.02 | 52.31 | K2 + G + I | LG + G + I |
| METTL10 | 0.7 | 12.85*** | 0.47 ± 0.01 | 0.55 ± 0.02 | 0.75 ± 0.02 | 0.38 ± 0.02 | 54.78 | T92 + G + I | LG + G + I |
| METTL11A | 0.7 | 18.84*** | 0.45 ± 0.01 | 0.49 ± 0.02 | 0.8 ± 0.01 | 0.35 ± 0.02 | 51.79 | K2 + G + I | LG + G + I |
| METTL11B | Vertebrates only | 49.58 | |||||||
| METTL12 | 0.6 | 9.02*** | 0.54 ± 0.01 | 0.63 ± 0.03 | 0.76 ± 0.02 | 0.46 ± 0.02 | 53.23 | K2 + I | LG + G + I |
| METTL13 | 0.7 | 24.82*** | 0.48 ± 0.01 | 0.56 ± 0.01 | 0.78 ± 0.01 | 0.39 ± 0.01 | 50.84 | GTR + G + I | LG + G |
| METTL14 | 0.7 | 29.64*** | 0.37 ± 0.01 | 0.32 ± 0.02 | 0.83 ± 0.01 | 0.23 ± 0.01 | 53.61 | GTR + G | LG + G |
| METTL15 | 0.7 | 18.11*** | 0.45 ± 0.01 | 0.48 ± 0.02 | 0.79 ± 0.01 | 0.34 ± 0.02 | 51.84 | HKY + G + I | LG + G + I |
| METTL16 | 0.6 | 18.45*** | 0.49 ± 0.01 | 0.57 ± 0.02 | 0.79 ± 0.01 | 0.41 ± 0.01 | 53.99 | HKY + G + I | LG + G + I |
| METTL17 | 0.7 | 17.89*** | 0.53 ± 0.01 | 0.61 ± 0.02 | 0.79 ± 0.01 | 0.44 ± 0.01 | 54.27 | GTR + G | LG + G + I |
| METTL18 | 0.7 | 13.1*** | 0.5 ± 0.01 | 0.57 ± 0.02 | 0.78 ± 0.02 | 0.41 ± 0.02 | 52.54 | T92 + G + I | LG + G + I |
| METTL19 | 0.7 | 13.24*** | 0.49 ± 0.01 | 0.56 ± 0.02 | 0.77 ± 0.01 | 0.41 ± 0.02 | 51.68 | GTR + G + I | LG + G + I |
| METTL20 | 0.7 | 14.86*** | 0.45 ± 0.01 | 0.48 ± 0.03 | 0.77 ± 0.01 | 0.35 ± 0.02 | 52.27 | K2 + G + I | LG + G + I |
| METTL21A | 0.6 | 17.06*** | 0.48 ± 0.01 | 0.54 ± 0.02 | 0.78 ± 0.01 | 0.39 ± 0.02 | 54.38 | K2 + G + I | LG + G |
| METTL21B | Vertebrates only | 45.13 | |||||||
| METTL21C | Vertebrates only | 54.33 | |||||||
| METTL21D | 0.7 | 13.23*** | 0.47 ± 0.01 | 0.54 ± 0.02 | 0.76 ± 0.02 | 0.38 ± 0.02 | 54.89 | T92 + G + I | LG + G + I |
| METTL21E | Chordates only | 53.19 | |||||||
| METTL22 | 0.6 | 15.33*** | 0.5 ± 0.01 | 0.58 ± 0.02 | 0.79 ± 0.01 | 0.42 ± 0.02 | 53.77 | T92 + G + I | LG + G + I |
| METTL23 | 0.6 | 18.63*** | 0.45 ± 0.01 | 0.5 ± 0.02 | 0.78 ± 0.01 | 0.35 ± 0.02 | 52.42 | K2 + G + I | LG + G |
| METTL25 | 0.6 | 16.26*** | 0.5 ± 0.01 | 0.58 ± 0.02 | 0.76 ± 0.01 | 0.42 ± 0.01 | 50.58 | T92 + G + I | LG + G + I |
| METTL25B | 0.7 | 17.41*** | 0.5 ± 0.01 | 0.56 ± 0.02 | 0.79 ± 0.01 | 0.4 ± 0.01 | 51.20 | GTR + G + I | LG + G + I |
| METTL26 | 0.7 | 13.38*** | 0.44 ± 0.01 | 0.49 ± 0.03 | 0.8 ± 0.02 | 0.34 ± 0.02 | 50.61 | K2 + I | LG + G + I |
| METTL27 | 0.5 | 4.12*** | 0.55 ± 0.02 | 0.69 ± 0.03 | 0.68 ± 0.04 | 0.5 ± 0.03 | 48.47 | JC | LG |
Note.—SE, standard error; R, average transition/transversion ratio; dNT, mean nucleotide distance; dAA, mean amino acid distance; dS, synonymous substitution distance; dN, nonsynonymous substitution distance; ENC, effective number of codons (codon bias) ranging from 61 (i.e., no bias) to 20 (i.e., maximum bias); ModelNT, best-fit maximum likelihood (ML) DNA model; ModelAA, best-fit ML protein model.
All SE values were calculated using the Bootstrap method with 1,000 replications.
P value < 0.001 in Z-test of purifying selection (i.e., HA: dN < dS).
Many conserved multigene families are thought to be subject to concerted evolution, in which gene family members evolve as a unit (i.e., in concert) via gene conversion or unequal crossing-over (Eirín-López et al. 2012). In the case of concerted evolution, we might expect to observe roughly the same level of synonymous variation (dS) and nonsynonymous variation (dN) because gene conversion and crossover processes should affect synonymous and nonsynonymous sites equally (Nei and Rooney 2005). However, the nucleotide variation underlying the diversity observed among METTLs is primarily synonymous. For all METTLs, dS was significantly greater than dN and all Z-tests for selection (i.e., HA: dN < dS) were highly significant (P value < 0.001) (table 3). Therefore, strong purifying selection appears to have operated on the different METTLs. This finding was additionally supported by the results from Fisher’s exact tests of selection (supplementary tables S3 and S4, Supplementary Material online). Indications of strong purifying selection have been reported in many other protein coding gene families, including highly conserved histone and ubiquitin gene families (Nei et al. 2000; Piontkivska et al. 2002; Rooney et al. 2002; Eirín-López et al. 2004; González-Romero et al. 2008, 2010, 2012). These studies argued that, rather than concerted evolution, these gene families are subject to the birth-and-death model of evolution in which gene family members evolve independently via gene duplication followed by their maintenance or loss (Nei and Hughes 1992). Indeed, the birth-and-death concept may be the primary mechanism directing the long-term evolution of most multigene families (Eirín-López et al. 2012). Although detecting birth-and-death was not a goal of the present work, future studies of this gene family might investigate intra- and interspecific gene duplication and the presence of pseudogenes, which would further support that METTLs are subject to this model of evolution.
Rates of METTL Evolution
In order to further explore functional constraints across METTLs that vary in target and function, the present study conducted estimations of the rates of evolution in METTL proteins. Results show that several RNA-modifying METTLs exhibited a relatively slow rate of evolution (fig. 4). In particular, METTL1, the only METTL known to form methylguanosine, had the slowest rate of protein evolution at 3.91 × 10−4 amino acid substitutions/site/million years (My) (supplementary table S5, Supplementary Material online). METTL14 and METTL3, which together form a heterocomplex to form m6A in RNA, also had relatively slow rates of evolution. In fact, the six METTLs with the slowest evolution rates (i.e., METTL1, METTL14, METTL2, METTL6, METTL3, and METTL5) all function to methylate RNA molecules (fig. 4; supplementary table S5, Supplementary Material online). These same METTLs are also found in multiple metazoan phyla, including basal metazoans (fig. 1). Thus, the formation of m7G, m3C, and m6A modifications in RNA appears to be a fundamental epitranscriptomic mechanism that has been preserved throughout metazoan evolution. In contrast, METTL12, otherwise known as citrate synthase-lysine N-methyltransferase (CSKMT), exhibited the fastest rate of evolution with 7.95 × 10−4 amino acid substitutions/site/My (fig. 4; supplementary table S5, Supplementary Material online). Methylation modifications have been identified in numerous diverse, structurally complex, and specialized proteins (Paik and Kim 1971; Clarke 2013). Therefore, a comparatively rapid evolution and diversification of protein-modifying methyltransferases may have been necessary to target these proteins and fulfill a wide variety of highly specific roles. Protein-modifying METTLs being subjected to different selective pressures than those that modify DNA/RNA nucleosides may explain their different rates of evolution. Also exhibiting relatively rapid rates of evolution were METTL7A and METTL17, although the precise functions of these proteins are unknown. All remaining METTLs that exhibited a relatively intermediate rate of evolution were a combination of RNA-modifying METTLs, protein-modifying METTLs, and METTLs of unknown function (fig. 4). These METTLs exhibited highly similar rates of evolution at an average of 6.64 × 10−4 ± 3.48 × 10−5 amino acid substitutions/site/My (supplementary table S5, Supplementary Material online).
Fig. 4.
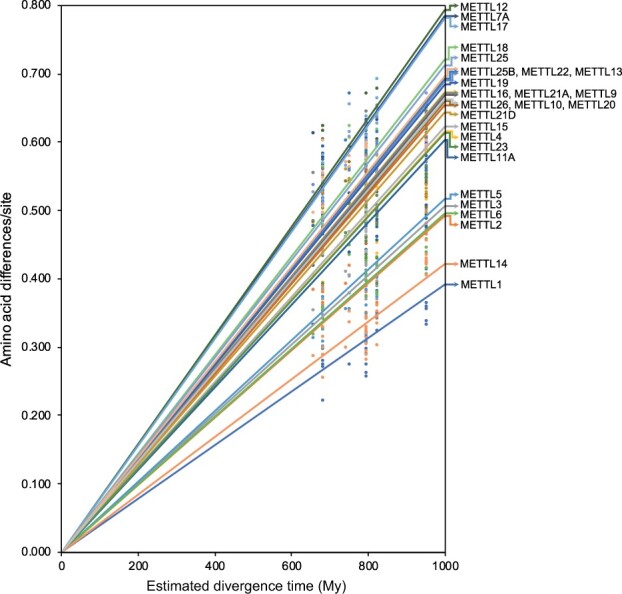
Estimated rates of evolution of METTL proteins. Rates were only estimated for METTLs in which amino acid differences per site could be calculated by comparing the protein between three or more of the preselected metazoan phyla.
Episodic Selection within METTL Lineages
We wanted to explore what drives the variation in evolutionary rates that we calculated across the METTL family, and we searched for indications of adaptive selective episodes that may have occurred during METTL evolution. Since METTL lineages each have a monophyletic origin (fig. 2), additional evolutionary analyses using only vertebrate METTLs were implemented to further explore how selection drove the differentiation of METTL types (alignment available in Supplementary Material online). Vertebrates were selected because all members of the METTL family are present within this subphylum of metazoans. Only two representative orthologous sequences per METTL were included in the analysis due to computational requirements and constraints. Accordingly, we examined ortholog sequences of all METTLs from each of two classes of vertebrates: Mammalia and Amphibia. METTL7B was not included in this analysis as orthologous gene searches in NCBI failed to clearly identify METTL7B orthologs in amphibians. The obtained results revealed that METTL proteins diverged from a homogeneous evolution pattern as expected by the global molecular clock hypothesis (ln L with clock = −10239.1, ln L without clock = −9715.854, P value < 0.001). This result was also supported when testing the molecular clock hypothesis using nucleotide sequences of the vertebrate METTLs (ln L with clock = −109637.787, ln L without clock = −108792.321, P value < 0.001). These findings, along with the estimated rates of evolution reported above, reaffirm that various METTL proteins appear to have evolved at different rates from one another.
Given the observed heterogeneous rates of evolution across the METTL family, the presence of diversifying (i.e., adaptive) selection episodes was examined across vertebrate METTLs, uncovering traces of diversifying selection (ω > 1) on specific branches of the phylogeny (P value < 0.05) (fig. 5A). The terminal branch leading to METTL6, which forms m3C in tRNA (Xu et al. 2017), was significant (P value < 0.01) (fig. 5A). The terminal branch of METTL9, a probable protein methyltransferase (Ignatova et al. 2019), also showed significant traces of diversifying selection (P value < 0.001) (fig. 5A). Several subtrees that included closely related METTLs (i.e., METTL11A and METTL11B; METTL21A, METTL21B, METTL21C, METTL21D, and METTL21E; METTL25 and METTL25B) were collapsed into a single branch for figure readability.
Fig. 5.

Episodes of diversifying selection shaping the evolution of METTL proteins in vertebrates. For figure readability, “METTL” has been removed from the names of each tip label (e.g., “METTL1” is simply displayed as “1”). (A) The strength of selection at significant branches is represented in red (ω > 5), gray (ω = 1), and blue (ω = 0), with the proportion of sites within each class represented by the color width. Thicker branches have been classified as undergoing episodic diversifying selection at corrected P value < 0.001 (thicker branches) and P value < 0.01 (thinner branches). (B) The physical positions of adaptive selection episodes involved in the diversification of METTL genes. Numbers of synonymous (blue bars) and nonsynonymous (red bars) substitutions at codon positions that are subject to significant episodes of diversifying selection in vertebrates (P value < 0.05). (C) Phylogenetic location of the mutations involved in such episodes. Branches in red account for higher numbers of nonsynonymous mutations, branches in blue indicate higher numbers of synonymous mutations, and branches in green represent cases with the same numbers of nonsynonymous and synonymous mutations. Light blue, light red, and light purple backgrounds behind each branch indicate METTLs that modify DNA/RNA, proteins, or unknown targets, respectively. Subtrees that include several closely related METTLs (i.e., METTL11A and METTL11B; METTL21A, METTL21B, METTL21C, METTL21D, and METTL21E; METTL25 and METTL25B) were collapsed into a single branch for figure readability.
Several individual sites subject to diversifying selection in METTLs were also identified using the mixed effects model of evolution (MEME) model (Murrell et al. 2012) (fig. 5B and C), a method we have previously used to explore the evolution of high mobility group (HMG) proteins (González-Romero et al. 2015) and sex-determining proteins (Eirín-López and Sánchez 2015). As expected, the number and identities of these sites varied depending on whether analyses included all METTL types, or if analyses discriminated among METTLs based on their target molecule type (i.e., those that target DNA or RNA nucleosides versus those that target protein residues). Upon analyzing all METTL sequences available for vertebrates regardless of type (alignment available in Supplementary Material online), 19 codon sites were identified, most of which were subjected to episodic positive selection (fig. 5B). Four sites of predominantly positive selection (codons 89, 114, 747, and 785) were common to the majority of METTLs, spanning a variety of different targets and functions (fig. 5C). Analysis of these codon positions within the context of the METTL phylogeny suggests that mutations at these sites led to the differentiation of multiple METTL lineages (fig. 5C).
Evidence of positive selection at codon 89 was indicated at several internal nodes of the phylogeny, as well as the terminal branches leading to METTL1, METTL3, and METTL14 (fig. 5C). Codon 89 was also identified as a site of positive selection when the MEME analysis was restricted to only METTLs that modify DNA or RNA nucleosides (fig. 6A). Analysis of codon 114 indicated instances of both positive and negative (purifying) selection (fig. 5C). Specifically, although the majority of METTL lineages were subject to positive selection at codon 114, a few terminal branches (e.g., METTL3 and METTL14) indicate the presence of more synonymous than nonsynonymous mutations. Codon 114 was additionally identified by both MEME analyses that separately analyzed DNA/RNA-modifying METTLs and protein-modifying METTLs (fig. 6). Thus, episodic selection at codon 114 may have been particularly important to the diversification of METTLs, both within and across different METTL types. Mutations at codon 747 were also common to many different METTL lineages within the phylogeny. Interestingly, this position is located within the CD regions of all METTLs that possess CDs belonging to the AdoMet MTases superfamily (supplemental table 2, Supplementary Material online). Codon 747 is not, however, located within the CDs belonging to the MT-A70 superfamily, which are found in METTL3, METTL14, and METTL4. Codon 785 had the most substitutions of the sites identified from the MEME analysis of all vertebrate METTLs (fig. 5B), and appears to have contributed to the differentiation of many of the METTL lineages. This site was also identified as a site of positive selection for the MEME analysis of only DNA/RNA-modifying METTLs, and therefore, appears to have played a significant role in the diversification of METTLs that bind to nucleosides. Although only a few prominent sites identified from the MEME analysis of all METTLs are discussed here, additional substitutions at over a dozen more positions were identified (fig. 5B and C).
Fig. 6.

Episodes of adaptive selection identified upon separately analyzing vertebrate DNA/RNA-modifying METTLs and vertebrate protein-modifying METTLs. (A) The physical positions of adaptive selection episodes involved in the diversification of METTLs that modify DNA/RNA nucleosides (top) and METTLs that modify protein residues (bottom). Numbers of synonymous (blue bars) and nonsynonymous (red bars) substitutions at codon positions that are subject to significant episodes of diversifying selection in vertebrates (P value < 0.05). (B) Phylogenetic location of the mutations involved in such episodes for DNA/RNA-modifying METTLs, indicated by a light blue background, and (C) protein-modifying METTLs, indicated by a light red background. For figure readability, “METTL” has been removed from the names of each tip label (e.g., “METTL1” is simply displayed as “1”). Branches in red account for higher numbers of nonsynonymous mutations, branches in blue indicate higher numbers of synonymous mutations, and branches in green represent cases with the same numbers of nonsynonymous and synonymous mutations.
Several additional sites subject to diversifying selection that were not recognized in the indiscriminate MEME analysis of all vertebrate METTLs were identified when performing MEME analyses that were separated by METTL type (fig. 6A). The analysis of only METTLs that modify DNA or RNA nucleosides identified additional sites of predominantly positive selection at codons 33, 53, 365, 1295, 1520, 1793, and 1825 (fig. 6A). Sites at codons 89, 114, 414, and 785 were previously identified in the METTL analysis that included all vertebrate METTLs (fig. 5B). Upon examining these sites within the context of the phylogeny, a mutation at codon 89 shows evidence of positive selection on the internal node leading to METTL2, METTL6, METTL8, and METTL15 (fig. 6B). However, additional substitutions at codons 33, 53, 114, 785, 1520, and 1825 were necessary for the differentiation of these METTLs (fig. 6B), all of which are responsible for the formation of methylcytidine in RNA (Xu et al. 2017; Van Haute et al. 2019). Although the evolution of METTLs that target DNA/RNA nucleosides included several sites that were primarily subjected to positive selection, there were also evidence of neutral evolution and purifying selection for certain METTL lineages at a few sites (i.e., codon 114, 1793, and 1825) (fig. 6B).
Although sites at codons 114 and 541 had been identified in the METTL analysis that included all vertebrate METTLs (fig. 5B), by separately analyzing only METTLs that modify protein residues, MEME identified additional sites of predominantly positive selection at codons 69, 72, 91, 243, 719, 768, and 997 (fig. 6A and C). Nearly all of the site mutations identified using MEME in protein-modifying METTLs were subjected to positive selection. However, there was evidence of purifying selection at codon 69 (METTL20), codon 719 (METTL23), and codon 768 (METTL10 and METTL22) (fig. 6C). There were also evidence of equal synonymous and nonsynonymous substitutions (i.e., neutral evolution) for METTL22 at codons 72 and 997 when analyzed within the context of the phylogeny (fig. 6C).
Conclusions
METTLs are widespread throughout the animal kingdom, with some having emerged quite early in the evolution of Metazoa. Individual METTL lineages formed independent monophyletic clades, and whereas various METTLs grouped together roughly by methylation target type, those with similar targets did not appear to have evolved from the same, recent common ancestor (e.g., all METTLs that modify nucleosides did not share an exclusive, single monophyletic origin). Evidence indicates that the long-term evolution of the METTL family is primarily driven by strong purifying selection and exhibits heterogeneous rates of evolution across the different METTL lineages. Functional specialization of the various METTLs seems to have occurred via episodes of adaptive selection at specific evolutionary times and codon sites. Lastly, given their presence in basal metazoans, comparatively low sequence-level variation, and slow estimated rates of evolution, several METTLs that target and methylate RNA nucleosides seem to be more conserved than those that modify protein residues, possibly due to their biological necessity and particular functional constraints. In view of the ubiquity of METTLs throughout metazoans, it is clear that these proteins fulfill a diverse set of essential biological functions across an expansive range of animal taxa, and that the epigenetic mechanisms associated with this gene family contributed to metazoan evolution and complexity. We therefore stress the importance of investigating these methyltransferases in species beyond the traditional metazoan model systems. This present work provides important insight into the evolution of this gene family and constitutes a valuable resource for studying the epigenetic functions of METTL proteins, and their impacts on biological processes and phenotype.
Materials and Methods
Molecular Data Mining
Extensive data mining was performed using the GenBank database (https://www.ncbi.nlm.nih.gov/genbank) to collect METTL sequences (available as of June 2020). METTLs across metazoan taxa were located using orthologous gene searches as well as the BLAST in NCBI. Each METTL gene (NCBI IDs provided in table 1) was used to search for NCBI orthologs and similar genes. These are calculated using protein sequence similarity, local synteny information, and similarity of protein architectures (CDD domains defined by NCBI SPARCLE) by NCBI’s Eukaryotic Genome Annotation pipeline and Gene database. Each METTL gene listed in table 1 was searched in BLAST using the somewhat similar sequences (BlastN) algorithm and limiting the organism search set to Porifera (NCBI: txid6040), Ctenophora (NCBI: txid10197), Cnidaria (NCBI: txid6073), Brachiopoda (NCBI: txid7568), Mollusca (NCBI: txid6447), Annelida (NCBI: txid6340), Platyhelminthes (NCBI: txid6157), Priapulida (NCBI: txid33467), Nematoda (NCBI: txid6231), Arthropoda (NCBI: txid6656), Hemichordata (NCBI: txid10219), and Echinodermata (NCBI: txid7586). For any phyla for which search results only located sequences that were similar to a METTL, but did not have any predicted gene identity or function recorded in NCBI, they were noted as probable METTL sequences for that phylum.
Ultimately, sequences were selected from nine representative taxa across eight different metazoan phyla for phylogenetic and evolution analyses. These taxa included the sponge (phylum Porifera) Amphimedon queenslandica (NCBI: txid400682), the stony coral (phylum Cnidaria) Acropora millepora (NCBI: txid45264), the sea hare (phylum Mollusca) Aplysia californica (NCBI: txid6500), the priapulid (phylum Priapulida) Priapulus caudatus (NCBI: txid3762), the shrimp (phylum Arthropoda) Litopenaeus vannamei (NCBI: txid6689), the sea star (phylum Echinodermata) Acanthaster planci (NCBI: txid133434), the acorn worm (phylum Hemichordata) Saccoglossus kowalevskii (NCBI: txid10224), the lancelet (phylum Chordata) Branchiostoma belcheri (NCBI: txid7741), and humans (phylum Chordata) Homo sapiens (NCBI: txid9606). A total of 207 nucleotide coding sequences (CDS) and their corresponding amino acid sequences were collected across 33 METTLs (supplementary tables S6 and S7, Supplementary Material online). Sequence alignments for both CDS nucleotide sequences and amino acid sequences were performed using MAFFT version 7.309 (Katoh 2002; Katoh and Standley 2013) in Geneious version 9.1.8 (https://www.geneious.com). All METTL CDS nucleotide sequences, using a translation alignment, and all METTL amino acid sequences were aligned. Both the CDS nucleotide and amino acid alignments were aligned using the E-INS-I strategy algorithm and the BLOSUM62 scoring matrix with a 1.53 gap open penalty. CDs were identified within each of 207 CDS nucleotide sequences and 207 protein sequences using the CD-Search and Batch CD-Search tools (Marchler-Bauer and Bryant 2004; Marchler-Bauer et al. 2011) in NCBI. All sequences were searched across the CD Database (CDD) (Lu et al. 2020) using an expect value (E-value) threshold of 0.01.
Phylogenetic Analyses
The ribosomal RNA small subunit methyltransferase A (KsgA) from Escherichia coli O157: H7 strain Sakai (NCBI: txid386585) was included as an outgroup for the phylogenetic analyses. Best-fit substitution models for nucleotide and protein sequence alignments were calculated using a maximum likelihood approach in MEGA version 10.1.8 (Tamura et al. 2013), and included all used sites (i.e., no data were excluded). Maximum likelihood phylogenetic trees were reconstructed using IQ-TREE version 1.6.12 (Nguyen et al. 2015) using the best-fit substitution models determined from MEGA. Accordingly, the LG model (Le and Gascuel 2008) corrected for discrete Gamma model (Yang 1994) with four rate categories (LG + G) was used for the reconstruction of METTL protein phylogenies across the nine preselected metazoan taxa. Similarly, a protein tree was also reconstructed for all METTL proteins across the nine preselected metazoan taxa but using only CD amino acid sequences identified using the Batch CD-Search tools (Marchler-Bauer and Bryant 2004; Marchler-Bauer et al. 2011) in NCBI. The reliability of the reconstructed topologies was contrasted by an ultra-fast bootstrap approximation (UFBoot) (Hoang et al. 2018) with 1,000 replicates. Additional phylogenetic reconstructions were performed using the CDS nucleotide alignment as well as for individual METTL lineage protein sequences (supplementary figs. S3 and S4, Supplementary Material online).
Molecular Evolution and Selection Analyses
Several molecular evolutionary analyses were performed in MEGA version 10.1.8 (Tamura et al. 2013). Sequences from each METTL gene were analyzed based on CDS nucleotide and amino acid alignments of all METTL sequences from each of the nine preselected metazoan taxa (alignments available in Supplementary Materials online). The nucleotide alignment includes 9,684 nucleotide sites (3,228 codons) and the protein alignment includes 3,124 amino acid sites. The transition/transversion ratio (R) was calculated for all METTLs except for those in which only one sequence across the nine preselected taxa was available (i.e., METTL8, METTL11B, METTL21B, METTL21C, and METTL21E). Due to their smaller variance (Nei and Kumar 2000), nucleotide (dNT) and protein (dAA) sequence distances were estimated using uncorrected differences (P distances) for each METTL in which sequences were obtained from more than one representative taxa (i.e., all METTLs with the exception of METTL8, METTL11B, METTL21B, METTL21C, and METTL21E). These estimations, along with their standard errors, were calculated using bootstrap variance estimation (1,000 replicates) and the proportion of different nucleotide sites (i.e., transitions + transversions). Nucleotide and amino acid distances were computed using uniform rates among sites. Gaps and missing data were removed prior to the analysis if a site had higher than 95% of ambiguous sites (i.e., partial deletion). The rates of protein evolution were estimated for each METTL based on the distances obtained, in those cases in which there were representatives across at least three of the preselected metazoan taxa. METTLs that did not fulfill this requirement and were therefore excluded from the rate of evolution analysis included METTL7B, METTL8, METTL11B, METTL21B, METTL21C, METTL21E, and METTL27. Evolution rates were estimated by correlating pairwise protein divergences between pairs of metazoan taxa with their corresponding divergence as defined in the TimeTree database (Kumar et al. 2017) (supplementary table S8, Supplementary Material online). Linear regression analyses were implemented using Microsoft Excel version 16.42.
The footprint of selection on METTL genes was studied using two major approaches. First, descriptive analyses of nucleotide variation and the mode of evolution displayed by METTLs were carried out using the CDS nucleotide alignment of METTLs from the nine preselected taxa (alignment available in Supplementary Material online). Accordingly, the numbers of synonymous (dS) and nonsynonymous (dN) substitutions per site, as well as their standard errors, were computed using bootstrap variance estimations (1,000 replications) and the modified Nei−Gojobori method (Nei and Gojobori 1986) with the corresponding transition/transversion ratio (R), uniform rates among sites, and partial deletion (95% coverage cutoff) of gaps and missing data. Codon-based Z-tests were performed to gauge the presence and nature of selection by comparing the estimated number of synonymous (dS) versus nonsynonymous (dN) nucleotide differences per site. In this case, estimations of nucleotide substitutions were performed using uncorrected differences (P distances) considering heterogeneity in transition/transversion ratio, as these encompass smaller variance than other methods (Nei and Kumar 2000). In addition, the large number of taxa analyzed further contributes to reducing the potential effects of multiple substitutions by breaking long branches (Lartillot and Philippe 2008). For these tests, neutral evolution was set as the null hypothesis (H0: dN = dS) with purifying selection set as the alternative hypothesis (HA: dN < dS). The variance of the difference between dN and dS was estimated using the bootstrap method with 1,000 replications and a modified Nei−Gojobori method (Nei and Gojobori 1986) with the corresponding transition/transversion ratio (R). Codon-based Fisher’s exact tests of selection were also performed (supplementary tables S3 and S4, Supplementary Material online), but with positive selection set as the alternative hypothesis (HA: dS < dN). The amount of codon usage bias and the presence of molecular clocks were investigated using the programs DnaSP version 5 (Librado and Rozas 2009) and HyPhy (Pond et al. 2005), respectively.
Second, the presence of lineages displaying evidence of diversifying (adaptive) selection episodes (ω > 1) was examined across vertebrate METTL evolution by using the branch-site random effects likelihood model (Pond and Frost 2005). To this end, a total of 1,856 codon positions were examined using a maximum likelihood phylogeny that was reconstructed using vertebrate METTL nucleotide coding regions as a reference (alignment available in Supplementary Material online). The alignment included two ortholog sequences (i.e., one from Mammalia and one from Amphibia) of each METTL that were downloaded from GenBank (supplementary table S9, Supplementary Material online). In this case, nucleotide substitution models incorporating multiple substitutions were used as indicated in table 3. For this instance, the best-fit model of evolution was defined as a general time reversible model (Tavaré 1986) with a discrete Gamma model that allows for a proportion of invariable sites (Gu et al. 1995) (GTR + G + I). No prior assumptions about which lineages have been subject to diversifying selection were made. The proportion of sites inferred to be evolving under diversifying selection at each branch was estimated using likelihood ratio tests, resulting in a P value for episodic selection. The strength of selection was partitioned for descriptive purposes into three categories (ω > 5, ω = 1, and ω = 0), using three different significance levels (P value < 0.001, P value < 0.01, and P value < 0.05, respectively) to assess the obtained results. Additionally, the presence of selection at individual sites was assessed by using MEME, modeling variable ω (dN/dS) across lineages at individual sites (Murrell et al. 2012). Codons subject to significant episodes of diversifying selection (P value < 0.05) were detected using MEME, and analyzed in the context of the METTL phylogeny, providing information on internal branches accumulating higher numbers of nonsynonymous mutations. In addition to conducting this analysis across all vertebrate METTLs, additional analyses using MEME were performed separately for vertebrate DNA/RNA-modifying METTLs only and for vertebrate protein-modifying METTLs only. All analyses in this section were carried out using the HyPhy program (Pond et al. 2005) and the Datamonkey web server (Poon et al. 2009; Delport et al. 2010).
Supplementary Material
Supplementary data are available at Molecular Biology and Evolution online.
Supplementary Material
Acknowledgments
This work was supported by grants from the National Science Foundation (Grant No. EF-1921402) awarded to J.E.L. J.M.W. was supported by the College of Arts, Science and Education Distinguished Postdoctoral Program at Florida International University, and by the NSF Postdoctoral Research Fellowships in Biology Program under Grant No. DBI-2010791. This material is also based upon work supported by the National Science Foundation under Grant No. HRD-1547798. This NSF Grant was awarded to Florida International University as part of the Centers of Research Excellence in Science and Technology (CREST) Program. This is contribution number 266 from the Coastlines and Oceans Division, and contribution number 1036 from the Freshwater Resources Division of the Institute of Environment at Florida International University.
Data Availability
The data underlying this article are available within the National Center for Biotechnology Information (NCBI) GenBank database (https://www.ncbi.nlm.nih.gov/genbank), and all GenBank accession numbers are listed in supplementary tables S6, S7, and S9, Supplementary Material online. Alignments analyzed in the present work are available in the article’s Supplementary Material online.
References
- Angulo JF, Mauffirey P, Pinon-Lataillade G, Miccoli L, Biard DSF.. 2005. Putative roles of kin17, a mammalian protein binding curved DNA, in transcription. In: Ohyama T, editor. DNA conformation and transcription. Molecular Biology Intelligence Unit. Boston (MA): Springer. p. 75–89. [Google Scholar]
- Arimbasseri AG, Iben J, Wei F-Y, Rijal K, Tomizawa K, Hafner M, Maraia RJ.. 2016. Evolving specificity of tRNA 3-methyl-cytidine-32 (m3C32) modification: a subset of tRNAsSer requires N6-isopentenylation of A37. RNA 22(9):1400–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernkopf M, Webersinke G, Tongsook C, Koyani CN, Rafiq MA, Ayaz M, Müller D, Enzinger C, Aslam M, Naeem F, et al. 2014. Disruption of the methyltransferase-like 23 gene METTL23 causes mild autosomal recessive intellectual disability. Hum Mol Genet. 23(15):4015–4023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya M, De S, Chakrabarti S.. 2020. Origin and evolution of DNA methyltransferases (DNMT) along the tree of life: a multi-genome survey. bioRxiv 2020.04.09.033167.
- Bokar JA, Shambaugh ME, Polayes D, Matera AG, Rottman FM.. 1997. Purification and cDNA cloning of the AdoMet-binding subunit of the human mRNA (N6-adenosine)-methyltransferase. RNA 3(11):1233–1247. [PMC free article] [PubMed] [Google Scholar]
- Bujnicki JM, Feder M, Radlinska M, Blumenthal RM.. 2002. Structure prediction and phylogenetic analysis of a functionally diverse family of proteins homologous to the MT-A70 subunit of the human mRNA:m(6)A methyltransferase. J Mol Evol. 55(4):431–444. [DOI] [PubMed] [Google Scholar]
- Carroll SB, Grenier JK, Weatherbee SD.. 2013. From DNA to diversity: molecular genetics and the evolution of animal design. Hoboken (NJ): John Wiley & Sons.
- Cheng X, Blumenthal R.. 1999. S-adenosylmethionine-dependent methyltransferases: structures and functions. Singapore: World Scientific.
- Chen H, Gu L, Orellana EA, Wang Y, Guo J, Liu Q, Wang L, Shen Z, Wu H, Gregory RI, et al. 2020. METTL4 is an snRNA m6Am methyltransferase that regulates RNA splicing. Cell Res. 30(6):544–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke SG. 2013. Protein methylation at the surface and buried deep: thinking outside the histone box. Trends Biochem Sci. 38(5):243–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cloutier P, Lavallée-Adam M, Faubert D, Blanchette M, Coulombe B.. 2013. A newly uncovered group of distantly related lysine methyltransferases preferentially interact with molecular chaperones to regulate their activity. PLoS Genet. 9(1):e1003210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cloutier P, Lavallée-Adam M, Faubert D, Blanchette M, Coulombe B.. 2014. Methylation of the DNA/RNA-binding protein Kin17 by METTL22 affects its association with chromatin. J Proteomics 100:115–124. [DOI] [PubMed] [Google Scholar]
- Couture J-F, Trievel RC.. 2006. Histone-modifying enzymes: encrypting an enigmatic epigenetic code. Curr Opin Struct Biol. 16(6):753–760. [DOI] [PubMed] [Google Scholar]
- Dabe EC, Sanford RS, Kohn AB, Bobkova Y, Moroz LL.. 2015. DNA Methylation in Basal Metazoans: insights from Ctenophores. Integr Comp Biol. 55(6):1096–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deans C, Maggert KA.. 2015. What do you mean “Epigenetic”? Genetics 199(4):887–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delport W, Poon AFY, Frost SDW, Kosakovsky Pond SL.. 2010. Datamonkey 2010: a suite of phylogenetic analysis tools for evolutionary biology. Bioinformatics 26(19):2455–2457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Despras E, Miccoli L, Créminon C, Rouillard D, Angulo JF, Biard DSF.. 2003. Depletion of KIN17, a human DNA replication protein, increases the radiosensitivity of RKO cells. Radiat Res. 159(6):748–758. [DOI] [PubMed] [Google Scholar]
- Du P, Yuan B, Cao J, Zhao J, Ding L, Chen L, Ying S, Jiang L, Lin J, Xu X, et al. 2015. Methyltransferase-like 17 physically and functionally interacts with estrogen receptors. IUBMB Life. 67(11):861–868. [DOI] [PubMed] [Google Scholar]
- Eirín-López JM, González-Tizón AM, Martínez A, Méndez J.. 2004. Birth-and-death evolution with strong purifying selection in the histone H1 multigene family and the origin of orphon H1 genes. Mol Biol Evol. 21(10):1992–2003. [DOI] [PubMed] [Google Scholar]
- Eirín-López JM, Rebordinos L, Rooney AP, Rozas J.. 2012. The birth-and-death evolution of multigene families revisited. Genome Dyn. 7:170–196. [DOI] [PubMed] [Google Scholar]
- Eirín-López JM, Sánchez L.. 2015. The comparative study of five sex-determining proteins across insects unveils high rates of evolution at basal components of the sex determination cascade. Dev Genes Evol. 225(1):23–30. [DOI] [PubMed] [Google Scholar]
- Faughn JD, Dean WL, Schaner Tooley CE.. 2018. The N-terminal methyltransferase homologs NRMT1 and NRMT2 exhibit novel regulation of activity through heterotrimer formation. Protein Sci. 27(9):1585–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Dominissini D, Rechavi G, He C.. 2014. Gene expression regulation mediated through reversible m6A RNA methylation. Nat Rev Genet. 15(5):293–306. [DOI] [PubMed] [Google Scholar]
- Goh YT, Koh CWQ, Sim DY, Roca X, Goh WSS.. 2020. METTL4 catalyzes m6Am methylation in U2 snRNA to regulate pre-mRNA splicing. Nucleic Acids Res. 48(16):9250–9261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goll MG, Kirpekar F, Maggert KA, Yoder JA, Hsieh C-L, Zhang X, Golic KG, Jacobsen SE, Bestor TH.. 2006. Methylation of tRNAAsp by the DNA methyltransferase homolog Dnmt2. Science 311(5759):395–398. [DOI] [PubMed] [Google Scholar]
- González-Romero R, Ausió J, Méndez J, Eirín-López JM.. 2008. Early evolution of histone genes: prevalence of an “Orphon” H1 lineage in protostomes and birth-and-death process in the H2A family. J Mol Evol. 66(5):505–518. [DOI] [PubMed] [Google Scholar]
- González-Romero R, Eirín-López JM, Ausió J.. 2015. Evolution of high mobility group nucleosome-binding proteins and its implications for vertebrate chromatin specialization. Mol Biol Evol. 32(1):121–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Romero R, Rivera-Casas C, Ausió J, Méndez J, Eirín-López JM.. 2010. Birth-and-death long-term evolution promotes histone H2B variant diversification in the male germinal cell line. Mol Biol Evol. 27(8):1802–1812. [DOI] [PubMed] [Google Scholar]
- González-Romero R, Rivera-Casas C, Frehlick LJ, Méndez J, Ausió J, Eirín-López JM.. 2012. Histone H2A (H2A.X and H2A.Z) variants in Molluscs: molecular characterization and potential implications for chromatin dynamics. PLoS One 7(1):e30006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosjean H. 2009. DNA and RNA modification enzymes: structure, mechanism, function and evolution. Boca Raton (FL): CRC Press.
- Gu X, Fu YX, Li WH.. 1995. Maximum likelihood estimation of the heterogeneity of substitution rate among nucleotide sites. Mol Biol Evol. 12(4):546–557. [DOI] [PubMed] [Google Scholar]
- Hamey JJ, Wienert B, Quinlan KGR, Wilkins MR.. 2017. METTL21B is a novel human lysine methyltransferase of translation elongation factor 1A: discovery by CRISPR/Cas9 knockout. Mol Cell Proteomics 16(12):2229–2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He C. 2010. Grand challenge commentary: RNA epigenetics? Nat Chem Biol. 6(12):863–865. [DOI] [PubMed] [Google Scholar]
- Helser TL, Davies JE, Dahlberg JE.. 1972. Mechanism of kasugamycin resistance in Escherichia coli. Nat New Biol. 235(53):6–9. [DOI] [PubMed] [Google Scholar]
- Hoang DT, Chernomor O, von Haeseler A, Minh BQ, Vinh LS.. 2018. UFBoot2: improving the ultrafast bootstrap approximation. Mol Biol Evol. 35(2):518–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holliday R. 2006. Epigenetics: a historical overview. Epigenetics 1(2):76–80. [DOI] [PubMed] [Google Scholar]
- Housen I, Demonté D, Lafontaine D, Vandenhaute J.. 1997. Cloning and characterization of the KlDIM1 gene from Kluyveromyces lactis encoding the m2(6)A dimethylase of the 18S rRNA. Yeast 13(8):777–781. [DOI] [PubMed] [Google Scholar]
- Ignatova VV, Jansen PWTC, Baltissen MP, Vermeulen M, Schneider R.. 2019. The interactome of a family of potential methyltransferases in HeLa cells. Sci Rep. 9(1):6584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakobsson ME, Małecki JM, Halabelian L, Nilges BS, Pinto R, Kudithipudi S, Munk S, Davydova E, Zuhairi FR, Arrowsmith CH, et al. 2018. The dual methyltransferase METTL13 targets N terminus and Lys55 of eEF1A and modulates codon-specific translation rates. Nat Commun. 9(1):3411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kangaspeska S, Stride B, Métivier R, Polycarpou-Schwarz M, Ibberson D, Carmouche RP, Benes V, Gannon F, Reid G.. 2008. Transient cyclical methylation of promoter DNA. Nature 452(7183):112–115. [DOI] [PubMed] [Google Scholar]
- Kannouche P, Mauffrey P, Pinon-Lataillade G, Mattei MG, Sarasin A, Daya-Grosjean L, Angulo JF.. 2000. Molecular cloning and characterization of the human KIN17 cDNA encoding a component of the UVC response that is conserved among metazoans. Carcinogenesis 21(9):1701–1710. [DOI] [PubMed] [Google Scholar]
- Katoh K, Misawa K, Kuma K-I, Miyata T.. 2002. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30(14):3059–3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Standley DM.. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kernstock S, Davydova E, Jakobsson M, Moen A, Pettersen S, Mælandsmo GM, Egge-Jacobsen W, Falnes PØ.. 2012. Lysine methylation of VCP by a member of a novel human protein methyltransferase family. Nat Commun. 3:1038. [DOI] [PubMed] [Google Scholar]
- Kumar S, Stecher G, Suleski M, Hedges SB.. 2017. TimeTree: a resource for timelines, timetrees, and divergence times. Mol Biol Evol. 34(7):1812–1819. [DOI] [PubMed] [Google Scholar]
- Kweon S-M, Chen Y, Moon E, Kvederaviciutė K, Klimasauskas S, Feldman DE.. 2019. An adversarial DNA N6-methyladenine-sensor network preserves polycomb silencing. Mol Cell. 74(6):1138–1147.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafontaine D, Delcour J, Glasser AL, Desgrès J, Vandenhaute J.. 1994. The DIM1 gene responsible for the conserved m6(2)Am6(2)A dimethylation in the 3’-terminal loop of 18 S rRNA is essential in yeast. J Mol Biol. 241(3):492–497. [DOI] [PubMed] [Google Scholar]
- Lartillot N, Philippe H.. 2008. Improvement of molecular phylogenetic inference and the phylogeny of Bilateria. Philos Trans R Soc Lond B Biol Sci. 363(1496):1463–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leismann J, Spagnuolo M, Pradhan M, Wacheul L, Vu MA, Musheev M, Mier P, Andrade-Navarro MA, Graille M, Niehrs C, et al. 2020. The 18S ribosomal RNA m A methyltransferase Mettl5 is required for normal walking behavior in Drosophila. EMBO Rep. 21(7):e49443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lentini JM, Alsaif HS, Faqeih E, Alkuraya FS, Fu D.. 2020. DALRD3 encodes a protein mutated in epileptic encephalopathy that targets arginine tRNAs for 3-methylcytosine modification. Nat Commun. 11(1):2510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leschziner GD, Coffey AJ, Andrew T, Gregorio SP, Dias-Neto E, Calafato M, Bentley DR, Kinton L, Sander JW, Johnson MR.. 2011. Q8IYL2 is a candidate gene for the familial epilepsy syndrome of partial epilepsy with pericentral spikes (PEPS). Epilepsy Res. 96(1 − 2):109–115. [DOI] [PubMed] [Google Scholar]
- Le SQ, Gascuel O.. 2008. An improved general amino acid replacement matrix. Mol Biol Evol. 25(7):1307–1320. [DOI] [PubMed] [Google Scholar]
- Lewis CJT, Pan T, Kalsotra A.. 2017. RNA modifications and structures cooperate to guide RNA–protein interactions. Nat Rev Mol Cell Biol. 18(3):202–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Librado P, Rozas J.. 2009. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25(11):1451–1452. [DOI] [PubMed] [Google Scholar]
- Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang L, Jia G, Yu M, Lu Z, Deng X, et al. 2014. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol. 10(2):93–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Hausmann S, Carlson SM, Fuentes ME, Francis JW, Pillai R, Lofgren SM, Hulea L, Tandoc K, Lu J, et al. 2019. METTL13 methylation of eEF1A increases translational output to promote tumorigenesis. Cell 176(3):491–504.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Gonzalez PA, Sasvari Z, Kinzy TG, Nagy PD.. 2014. Methylation of translation elongation factor 1A by the METTL10-like See1 methyltransferase facilitates tombusvirus replication in yeast and plants. Virology 448:43–54. [DOI] [PubMed] [Google Scholar]
- Losos JB. 2011. Convergence, adaptation, and constraint. Evolution 65(7):1827–1840. [DOI] [PubMed] [Google Scholar]
- Lu S, Wang J, Chitsaz F, Derbyshire MK, Geer RC, Gonzales NR, Gwadz M, Hurwitz DI, Marchler GH, Song JS, et al. 2020. CDD/SPARCLE: the conserved domain database in 2020. Nucleic Acids Res. 48(D1):D265–D268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyko F. 2018. The DNA methyltransferase family: a versatile toolkit for epigenetic regulation. Nat Rev Genet. 19(2):81–92. [DOI] [PubMed] [Google Scholar]
- Machnicka MA, Milanowska K, Osman Oglou O, Purta E, Kurkowska M, Olchowik A, Januszewski W, Kalinowski S, Dunin-Horkawicz S, Rother KM, et al. 2013. MODOMICS: a database of RNA modification pathways–2013 update. Nucleic Acids Res. 41(Database issue):D262–D267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malecki J, Aileni VK, Ho AYY, Schwarz J, Moen A, Sørensen V, Nilges BS, Jakobsson ME, Leidel SA, Falnes PØ.. 2017. The novel lysine specific methyltransferase METTL21B affects mRNA translation through inducible and dynamic methylation of Lys-165 in human eukaryotic elongation factor 1 alpha (eEF1A). Nucleic Acids Res. 45(8):4370–4389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Małecki J, Ho AYY, Moen A, Dahl H-A, Falnes PØ.. 2015. Human METTL20 is a mitochondrial lysine methyltransferase that targets the β subunit of electron transfer flavoprotein (ETFβ) and modulates its activity. J Biol Chem. 290(1):423–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Małecki J, Jakobsson ME, Ho AYY, Moen A, Rustan AC, Falnes PØ.. 2017. Uncovering human METTL12 as a mitochondrial methyltransferase that modulates citrate synthase activity through metabolite-sensitive lysine methylation. J Biol Chem. 292(43):17950–17962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchler-Bauer A, Bryant SH.. 2004. CD-Search: protein domain annotations on the fly. Nucleic Acids Res. 32(Web Server issue):W327–W331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchler-Bauer A, Lu S, Anderson JB, Chitsaz F, Derbyshire MK, DeWeese-Scott C, Fong JH, Geer LY, Geer RC, Gonzales NR, et al. 2011. CDD: a conserved domain database for the functional annotation of proteins. Nucleic Acids Res. 39(Database issue):D225–D229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin JL, McMillan FM.. 2002. SAM (dependent) I AM: the S-adenosylmethionine-dependent methyltransferase fold. Curr Opin Struct Biol. 12(6):783–793. [DOI] [PubMed] [Google Scholar]
- Masson C, Menaa F, Pinon-Lataillade G, Frobert Y, Chevillard S, Radicella JP, Sarasin A, Angulo JF.. 2003. Global genome repair is required to activate KIN17, a UVC-responsive gene involved in DNA replication. Proc Natl Acad Sci U S A. 100(2):616–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateyak MK, Kinzy TG.. 2010. eEF1A: thinking outside the ribosome. J Biol Chem. 285(28):21209–21213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer H, Bug M, Bremer S.. 2012. Emerging functions of the VCP/p97 AAA-ATPase in the ubiquitin system. Nat Cell Biol. 14(2):117–123. [DOI] [PubMed] [Google Scholar]
- Meyer KD, Jaffrey SR.. 2017. Rethinking m6A readers, writers, and erasers. Annu Rev Cell Dev Biol. 33:319–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murrell B, Wertheim JO, Moola S, Weighill T, Scheffler K, Kosakovsky Pond SL.. 2012. Detecting individual sites subject to episodic diversifying selection. PLoS Genet. 8(7):e1002764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nance DJ, Satterwhite ER, Bhaskar B, Misra S, Carraway KR, Mansfield KD.. 2020. Characterization of METTL16 as a cytoplasmic RNA binding protein. PLoS One 15(1):e0227647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei M, Gojobori T.. 1986. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol Biol Evol. 3(5):418–426. [DOI] [PubMed] [Google Scholar]
- Nei M, Hughes AL.. 1992. Balanced polymorphism and evolution by the birth-and-death process in the MHC loci. In: Tsuji K, Aizawa M, Sasazuki T, editors. 11th Histocompatibility Workshop and Conference. Oxford: Oxford University Press. Available from: https://ci.nii.ac.jp/naid/10015427478/.
- Nei M, Kumar S.. 2000. Molecular evolution and phylogenetics. Oxford: Oxford University Press. [Google Scholar]
- Nei M, Rogozin IB, Piontkivska H.. 2000. Purifying selection and birth-and-death evolution in the ubiquitin gene family. Proc Natl Acad Sci U S A. 97(20):10866–10871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei M, Rooney AP.. 2005. Concerted and birth-and-death evolution of multigene families. Annu Rev Genet. 39:121–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng SS, Yue WW, Oppermann U, Klose RJ.. 2009. Dynamic protein methylation in chromatin biology. Cell Mol Life Sci. 66(3):407–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen L-T, Schmidt HA, von Haeseler A, Minh BQ.. 2015. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 32(1):268–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Farrell HC, Pulicherla N, Desai PM, Rife JP.. 2006. Recognition of a complex substrate by the KsgA/Dim1 family of enzymes has been conserved throughout evolution. RNA 12(5):725–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto M, Fujiwara M, Hori M, Okada K, Yazama F, Konishi H, Xiao Y, Qi G, Shimamoto F, Ota T, et al. 2014. tRNA modifying enzymes, NSUN2 and METTL1, determine sensitivity to 5-fluorouracil in HeLa cells. PLoS Genet. 10(9):e1004639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paik WK, Kim S.. 1971. Protein methylation. Science 174(4005):114–119. [DOI] [PubMed] [Google Scholar]
- Pandolfini L, Barbieri I, Bannister AJ, Hendrick A, Andrews B, Webster N, Murat P, Mach P, Brandi R, Robson SC, et al. 2019. METTL1 promotes let-7 MicroRNA processing via m7G methylation. Mol Cell. 74(6):1278–1290.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petkowski JJ, Bonsignore LA, Tooley JG, Wilkey DW, Merchant ML, Macara IG, Schaner Tooley CE.. 2013. NRMT2 is an N-terminal monomethylase that primes for its homologue NRMT1. Biochem J. 456(3):453–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petkowski JJ, Schaner Tooley CE, Anderson LC, Shumilin IA, Balsbaugh JL, Shabanowitz J, Hunt DF, Minor W, Macara IG.. 2012. Substrate specificity of mammalian N-terminal α-amino methyltransferase NRMT. Biochemistry 51(30):5942–5950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrossian T, Clarke S.. 2009. Bioinformatic identification of novel methyltransferases. Epigenomics 1(1):163–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piontkivska H, Rooney AP, Nei M.. 2002. Purifying selection and birth-and-death evolution in the histone H4 gene family. Mol Biol Evol. 19(5):689–697. [DOI] [PubMed] [Google Scholar]
- Pond SLK, Frost SDW.. 2005. A genetic algorithm approach to detecting lineage-specific variation in selection pressure. Mol Biol Evol. 22(3):478–485. [DOI] [PubMed] [Google Scholar]
- Pond SLK, Frost SDW, Muse SV.. 2005. HyPhy: hypothesis testing using phylogenies. Bioinformatics 21(5):676–679. [DOI] [PubMed] [Google Scholar]
- Poon AFY, Frost SDW, Kosakovsky Pond SL.. 2009. Detecting signatures of selection from DNA Sequences Using Datamonkey. Methods Mol Biol. 537:163–183. [DOI] [PubMed] [Google Scholar]
- Reiff RE, Ali BR, Baron B, Yu TW, Ben-Salem S, Coulter ME, Schubert CR, Hill RS, Akawi NA, Al-Younes B, et al. 2014. METTL23, a transcriptional partner of GABPA, is essential for human cognition. Hum Mol Genet. 23(13):3456–3466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhein VF, Carroll J, Ding S, Fearnley IM, Walker JE.. 2017. Human METTL12 is a mitochondrial methyltransferase that modifies citrate synthase. FEBS Lett. 591(12):1641–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhein VF, Carroll J, He J, Ding S, Fearnley IM, Walker JE.. 2014. Human METTL20 methylates lysine residues adjacent to the recognition loop of the electron transfer flavoprotein in mitochondria. J Biol Chem. 289(35):24640–24651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooney AP, Piontkivska H, Nei M.. 2002. Molecular evolution of the nontandemly repeated genes of the histone 3 multigene family. Mol Biol Evol. 19(1):68–75. [DOI] [PubMed] [Google Scholar]
- Ruszkowska A, Ruszkowski M, Dauter Z, Brown JA.. 2018. Structural insights into the RNA methyltransferase domain of METTL16. Sci Rep. 8(1):5311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saletore Y, Meyer K, Korlach J, Vilfan ID, Jaffrey S, Mason CE.. 2012. The birth of the epitranscriptome: deciphering the function of RNA modifications. Genome Biol. 13(10):175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert HL, Blumenthal RM, Cheng X.. 2003. Many paths to methyltransfer: a chronicle of convergence. Trends Biochem Sci. 28(6):329–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidel-Rogol BL, McCulloch V, Shadel GS.. 2003. Human mitochondrial transcription factor B1 methylates ribosomal RNA at a conserved stem-loop. Nat Genet. 33(1):23–24. [DOI] [PubMed] [Google Scholar]
- Shimazu T, Barjau J, Sohtome Y, Sodeoka M, Shinkai Y.. 2014. Selenium-based S-adenosylmethionine analog reveals the mammalian seven-beta-strand methyltransferase METTL10 to be an EF1A1 lysine methyltransferase. PLoS One 9(8):e105394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Z, Xu S, Xing S, Yao K, Zhang L, Xue L, Zhou P, Wang M, Yan G, Yang P, et al. 2019. Mettl17, a regulator of mitochondrial ribosomal RNA modifications, is required for the translation of mitochondrial coding genes. FASEB J. 33(11):13040–13050. [DOI] [PubMed] [Google Scholar]
- Śledź P, Jinek M. 2016. Structural insights into the molecular mechanism of the m6A writer complex. Elife 5:e18434. . doi: 10.7554/eLife.18434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stayton CT. 2015. What does convergent evolution mean? The interpretation of convergence and its implications in the search for limits to evolution. Interface Focus 5(6):20150039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Stecher G, Peterson D, Filipski A, Kumar S.. 2013. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 30(12):2725–2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavaré S. 1986. Some probabilistic and statistical problems in the analysis of DNA sequences. Lectures Math Life Sci 17:57–86. [Google Scholar]
- Thiele W, Novac N, Mink S, Schreiber C, Plaumann D, Fritzmann J, Cremers N, Rothley M, Schwager C, Regiert T, et al. 2011. Discovery of a novel tumour metastasis-promoting gene, NVM-1. J Pathol. 225(1):96–105. [DOI] [PubMed] [Google Scholar]
- Tokuhisa JG, Vijayan P, Feldmann KA, Browse JA.. 1998. Chloroplast development at low temperatures requires a homolog of DIM1, a yeast gene encoding the 18S rRNA dimethylase. Plant Cell 10(5):699–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tooley CES, Petkowski JJ, Muratore-Schroeder TL, Balsbaugh JL, Shabanowitz J, Sabat M, Minor W, Hunt DF, Macara IG.. 2010. NRMT is an alpha-N-methyltransferase that methylates RCC1 and retinoblastoma protein. Nature 466(7310):1125–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Tran N, Ernst FGM, Hawley BR, Zorbas C, Ulryck N, Hackert P, Bohnsack KE, Bohnsack MT, Jaffrey SR, Graille M, et al. 2019. The human 18S rRNA m6A methyltransferase METTL5 is stabilized by TRMT112. Nucleic Acids Res. 47(15):7719–7733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu C, Tropea JE, Austin BP, Court DL, Waugh DS, Ji X.. 2009. Structural basis for binding of RNA and cofactor by a KsgA methyltransferase. Structure 17(3):374–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turró S, Ingelmo-Torres M, Estanyol JM, Tebar F, Fernández MA, Albor CV, Gaus K, Grewal T, Enrich C, Pol A.. 2006. Identification and characterization of associated with lipid droplet protein 1: a novel membrane-associated protein that resides on hepatic lipid droplets. Traffic 7(9):1254–1269. [DOI] [PubMed] [Google Scholar]
- Van Buul CP, Damm JB, Van Knippenberg PH.. 1983. Kasugamycin resistant mutants of Bacillus stearothermophilus lacking the enzyme for the methylation of two adjacent adenosines in 16S ribosomal RNA. Mol Gen Genet. 189(3):475–478. [DOI] [PubMed] [Google Scholar]
- Van Haute L, Hendrick AG, D’Souza AR, Powell CA, Rebelo-Guiomar P, Harbour ME, Ding S, Fearnley IM, Andrews B, Minczuk M.. 2019. METTL15 introduces N4-methylcytidine into human mitochondrial 12S rRNA and is required for mitoribosome biogenesis. Nucleic Acids Res. 47(19):10267–10281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Knippenberg PH, Van Kimmenade JM, Heus HA.. 1984. Phylogeny of the conserved 3’ terminal structure of the RNA of small ribosomal subunits. Nucleic Acids Res. 12(6):2595–2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Zhang B, Ratliff AC, Arrington J, Chen J, Xiong Y, Yue F, Nie Y, Hu K, Jin W, et al. 2019. Methyltransferase-like 21e inhibits 26S proteasome activity to facilitate hypertrophy of type IIb myofibers. FASEB J. 33(8):9672–9684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Doxtader KA, Nam Y.. 2016. Structural basis for cooperative function of Mettl3 and Mettl14 methyltransferases. Mol Cell 63(2):306–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Feng J, Xue Y, Guan Z, Zhang D, Liu Z, Gong Z, Wang Q, Huang J, Tang C, et al. 2016. Structural basis of N(6)-adenosine methylation by the METTL3-METTL14 complex. Nature 534(7608):575–578. [DOI] [PubMed] [Google Scholar]
- Warda AS, Kretschmer J, Hackert P, Lenz C, Urlaub H, Höbartner C, Sloan KE, Bohnsack MT.. 2017. Human METTL16 is a -methyladenosine (mA) methyltransferase that targets pre-mRNAs and various non-coding RNAs. EMBO Rep. 18(11):2004–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb KJ, Lipson RS, Al-Hadid Q, Whitelegge JP, Clarke SG.. 2010. Identification of protein N-terminal methyltransferases in yeast and humans. Biochemistry. 49(25):5225–5235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb KJ, Zurita-Lopez CI, Al-Hadid Q, Laganowsky A, Young BD, Lipson RS, Souda P, Faull KF, Whitelegge JP, Clarke SG.. 2010. A novel 3-methylhistidine modification of yeast ribosomal protein Rpl3 is dependent upon the YIL110W methyltransferase. J Biol Chem. 285(48):37598–37606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiederstein JL, Nolte H, Günther S, Piller T, Baraldo M, Kostin S, Bloch W, Schindler N, Sandri M, Blaauw B, et al. 2018. Skeletal muscle-specific methyltransferase METTL21C Trimethylates p97 and regulates autophagy-associated protein breakdown. Cell Rep. 23(5):1342–1356. [DOI] [PubMed] [Google Scholar]
- Wiegand G, Remington SJ.. 1986. Citrate synthase: structure, control, and mechanism. Annu Rev Biophys Chem. 15:97–117. [DOI] [PubMed] [Google Scholar]
- Woodcock CB, Yu D, Hajian T, Li J, Huang Y, Dai N, Corrêa IR, Wu T, Vedadi M, Zhang X, et al. 2019. Human MettL3–MettL14 complex is a sequence-specific DNA adenine methyltransferase active on single-strand and unpaired DNA in vitro. Cell Discov. 5:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Liu X, Sheng N, Oo KS, Liang J, Chionh YH, Xu J, Ye F, Gao Y-G, Dedon PC, et al. 2017. Three distinct 3-methylcytidine (mC) methyltransferases modify tRNA and mRNA in mice and humans. J Biol Chem. 292(35):14695–14703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z, O’Farrell HC, Rife JP, Culver GM.. 2008. A conserved rRNA methyltransferase regulates ribosome biogenesis. Nat Struct Mol Biol. 15(5):534–536. [DOI] [PubMed] [Google Scholar]
- Yang Z. 1994. Maximum likelihood phylogenetic estimation from DNA sequences with variable rates over sites: approximate methods. J Mol Evol. 39(3):306–314. [DOI] [PubMed] [Google Scholar]
- Zehmer JK, Bartz R, Bisel B, Liu P, Seemann J, Anderson RGW.. 2009. Targeting sequences of UBXD8 and AAM-B reveal that the ER has a direct role in the emergence and regression of lipid droplets. J Cell Sci. 122(Pt 20):3694–3702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L-S, Liu C, Ma H, Dai Q, Sun H-L, Luo G, Zhang Z, Zhang L, Hu L, Dong X, et al. 2019. Transcriptome-wide mapping of internal N7-methylguanosine methylome in Mammalian mRNA. Mol Cell 74(6):1304–1316.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Hou Y, Wang Y, Gao T, Ma Z, Yang Y, Zhang P, Yi F, Zhan J, Zhang H, et al. 2020. Regulation of adipocyte differentiation by METTL4, a 6 mA. Sci Rep. 10(1):8285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Wan J, Gao X, Zhang X, Jaffrey SR, Qian S-B.. 2015. Dynamic m6A mRNA methylation directs translational control of heat shock response. Nature 526(7574):591–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this article are available within the National Center for Biotechnology Information (NCBI) GenBank database (https://www.ncbi.nlm.nih.gov/genbank), and all GenBank accession numbers are listed in supplementary tables S6, S7, and S9, Supplementary Material online. Alignments analyzed in the present work are available in the article’s Supplementary Material online.