

Abstract
Hypoxic environment is essential for chondrocyte maturation and longitudinal bone growth. Although hypoxia-inducible factor 1 alpha (Hif-1α) has been known as a key player for chondrocyte survival and function, the function of Hif-2α in cartilage is mechanistically and clinically relevant but remains unknown. Here we demonstrated that Hif-2α was a novel inhibitor of chondrocyte maturation through downregulation of Runx2 stability. Mechanistically, Hif-2α binding to Runx2 inhibited chondrocyte maturation by Runx2 degradation through disrupting Runx2/Cbfβ complex formation. The Hif-2α-mediated-Runx2 degradation could be rescued by Cbfβ transfection due to the increase of Runx2/Cbfβ complex formation. Consistently, mesenchymal cells derived from Hif-2α heterozygous mice were more rapidly differentiated into hypertrophic chondrocytes than those of wild type mice in a micromass culture system. Collectively, these findings demonstrate that Hif-2α is a novel inhibitor for chondrocyte maturation by disrupting Runx2/Cbfβ complex formation and consequential regulatory activity.
Keywords: Hif-2α, Runx2, Cbfβ, Proteasomal degradation, chondrocyte maturation
INTRODUCTION
In vertebrates, the skeleton is formed through intramembranous and endochondral ossification. During endochondral ossification, mesenchymal cells condense and differentiate into hypertrophic chondrocytes by induction of key transcription factors such as Sox9 and Runx2 (Komori, 2018; Long & Ornitz, 2013; Stein et al., 2004). To date, Runx2 together with Runx3 have been known as a transcriptional activator of chondrocyte hypertrophy and maturation (Yoshida et al., 2004). Ablation or C-termini truncation of Runx2 leads to the extension of the hypertrophic chondrocyte zone without bone marrow formation (Choi et al., 2001; Komori et al., 1997; Kundu et al., 2002; Otto et al., 1997). In contrast, overexpression of Runx2 in cartilage accelerates chondrocyte maturation and endochondral bone formation lead to short limb formation and enhanced mineralization (Takeda et al., 2001; Ueta et al., 2001). In addition, core binding factor β (Cbfβ), a partner protein of Runx family transcription factors is essential for Runx2 protein stability and transcription activity. Chondrocyte-specific deletion of Cbfβ in mice shows delayed endochondral bone formation with a short appendage skeletal length and increased proliferative chondrocytes (Park et al., 2016). Taken together, Runx2/Cbfβ complex is essential for chondrocyte maturation and longitudinal bone growth.
The hypoxic environment is a prominent feature of the developmental growth plate in mammals, and this hypoxia occurs in the inner layer, not in the outer periphery of cartilage. Hypoxia-inducible factor (Hif)-1α and Hif-2α are key transcription factors that respond to hypoxia changes and signaling in chondrocytes (Semenza, 2012). They are all degraded by the proteasome-mediated degradation in normoxic conditions and, conversely, stabilized under hypoxia (Semenza, 2012). Mice deficient in Hif-1α or Hif-2α die from vascular defects and impaired erythropoiesis on days 10.5 and 12.5 of embryos, respectively (Iyer et al., 1998; Peng et al., 2000). A previous study has reported that Hif-1α is essential for chondrocyte growth inhibition and survival (Schipani et al., 2001). In addition, Hif-1α, conditionally inactivated in the limb bud mesenchyme using a Prx1 promoter-driven Cre, exhibits early chondrogenesis and joint dysplasia (Araldi et al., 2011). Indeed, Hif-1α is a positive regulator of Sox9, which is required for chondrocyte differentiation and cartilage formation (Tanimoto et al., 2003). Hif-1α is a positive regulator of chondrocyte maturation, survival and in vivo activity, at least in part of the regulation of collagen synthesis in chondrocytes (Stegen et al., 2019).
Previously, Hif-2α has been known as a Runx2 upstream regulator in osteoblasts (Tamiya et al., 2008) and is also expressed in the hypertrophic zone of the growth plate and in articular chondrocytes (Stewart et al., 2006). Hif-2α null mice die in the early embryonic stage (between E9.5 and E13.5) before cartilage anlage formation with varying phenotypes depending on the genetic background of the mice (Patel & Simon, 2008). Heterozygous Hif-2α or conditional deletion of Hif-2α in limb bud mesenchyme causes a temporary and modest delay in endochondral bone development (Araldi et al., 2011; Saito et al., 2010; Yang et al., 2010). However, the Hif-2α function of chondrocyte differentiation and maturation still controversial. As a catabolic factor, Hif-2α enhances osteoarthritis or accelerates articular chondrocyte differentiation into hypertrophic chondrocytes (Saito et al., 2010; Yang et al., 2010). Furthermore, Hif-2α upregulates Sox9 and Trap6, respectively, impairing osteoblast and partially stimulating osteoclast differentiation (Lee et al., 2019; Merceron et al., 2019). Considering that both Runx2 and Hif-2α are highly expressed in hypertrophic chondrocytes of the growth plate, we hypothesized that Hif-2α regulates Runx2 during chondrocyte differentiation and maturation. In this study, we found that Hif-2α blocks Runx2/Cbfβ complex formation, thereby playing an inhibitory role in chondrocyte maturation through accelerated Runx2 proteasomal degradation.
MATERIALS AND METHODS
Animals
Animal care and experiments were performed in accordance with the guidelines issued by the Institutional Animal Care and Use Committee of Kyungpook National University (KNU-2018–0020). Animals were housed 5 per cage in a specific pathogen-free environment on a 12-h light cycle at 22±2°C with standard chow (Greeenpia Technology, Yeoju-si, South Korea) and had free access to tap water.
Antibodies and reagents
Rabbit anti-HA, Rabbit anti-Hif-2α, goat anti-Runx2, monoclonal anti-actin and goat anti-Lamin B antibodies were purchased from Santa Cruz Biotechnology (CA, USA). Rabbit anti-Myc and monoclonal anti-Runx2 were purchased from Abcam (MA, USA). Monoclonal anti-Flag, monoclonal anti-Flag-HRP, HA-anti HA-agarose antibody and other reagents were purchased from Sigma-Aldrich (St. Louis, MO, USA).
Cells and micromass culture
ROS 17/2.8 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM, Gibco BRL; NY, USA) containing 10% fetal calf serum (FCS, Gibco BRL) and 1% antibiotics (Lonza, Rockland, ME, USA). ATDC5 cells were cultured in DMEM/F12 (1:1) hybrid medium (Lonza) supplemented with 5% fetal bovine serum (FBS, Gibco BRL), 10 μg/ml human transferrin (Sigma-Aldrich), and 3×10−8 M sodium selenite (Sigma-Aldrich). Vascular smooth muscle MOVAS cells (ATCC, VA, USA) were maintained in DMEM containing 10% FBS, 0.2 mg/ml G −418 (sigma) and 1% antibiotics.
For micromass culture (Stanton et al., 2004), primary mesenchymal cells were isolated from limbs of wild type and Hif-2a+/− mouse embryos at day 11.5 (E11.5) and digested in 10% (v/v) fetal bovine serum (FBS)/Puck’s solution A (0.4 g of KCl/l, 8 g of NaCl/l, 0.35 g of NaHCO3/l, 1 g of glucose/l) containing 10 mg of dispase/ml (1 unit/ml) for 2 hours at 37 °C. Dispase activity was neutralized with 2:3 DMEM/F12 media containing 10% FBS (GibcoBRL). The digested cells were filtered through 40 μm cell strainers and centrifuged at 1500 rpm for 3 min to obtain a cell suspension. The cells were resuspended in growth media at 2×107 cells/ml and spotted in 10 μl drops/well of a 12-well culture plate. After a 2h attachment, the micro-masses were fed with growth media that consisted of 2:3 DMEM/F12 medium (Sigma-Aldrich) containing 100 U/ml penicillin, 100 μg/ml streptomycin and 10% FBS. For chondrocyte differentiation, 10 mM β-glycerophosphate and 50 μg/ml ascorbic acid were added to the growth medium for 28 days. Media was replenished with fresh media every two days (Park et al., 2016).
Transient transfection and knock-down experiments
ATDC5, ROS 17/2.8 cells, primary chondrocytes or MOVAS cells were plated at a density of 2 × 105 cells/well on 6 well plates for transfection experiments. Cells were transfected with a total of 4 μg DNA, including 2 μg pCMV-HA-Hif-2α (Tamiya et al., 2008), 1 μg pcDNA3.1-myc-Cbfβ and 1 μg pCS4–2flag-Runx2 by using Lipofectamine 2000 (Invitrogen, CA, USA), and cells were grown for 24 h. All transfections were performed in triplicate. The Sh-Control-GFP and Sh-Hif-2α-GFP constructs (kindly provided by Dr. Je-Yoel Cho, Seoul National University, Seoul, South Korea) were infected to the ATDC5 cells.
Western blot analysis
Whole cells were lysed by radioimmunoprecipitation assay (RIPA) buffer (10 mM Tris-HCl [pH 7.4], 0.15 M NaCl, 0.5% SDS, 1% NP-40, 1% Na-deoxycholate, 1 mM EDTA, 1 mM phenylmethylsulfonyl fluoride [PMSF], 1 μg/ml pepstatin, and 1 μg/ml leupeptin), and the protein concentration was measured by Bradford protein assay kit (Bio-Rad, CA, USA). Proteins were separated on SDS-polyacrylamide gels and transferred onto polyvinylidene difluoride (PVDF) membranes. Membrane was blocked with 5% skim-milk and immunoblotted with primary antibody overnight at 4°C. Proteins were detected by using an enhanced chemiluminescence detection system (GE Healthcare, Uppsala, Sweden).
Total RNA isolation and gene expression analysis
Total RNA was isolated from cells by using TRIzol reagent (Invitrogen) according to the manufacturers’ instructions. Complementary DNA (cDNA) was synthesized from 1 μg of total RNA by using Superscript II RT (Invitrogen) according to the manufacturer’s instructions.
Quantitative real-time PCR was performed using an ABI 7500 real-time PCR system and the relative quantification of Gapdh mRNA levels were analyzed by using SYBR green PCR master mix (Applied Biosystems, Foster City, CA, USA). Primers were designed by using PrimerExpress software (Applied Biosystems). The sequences of primers are as follows: Hif-2, 5′-GCTTCCTTCGGACACATAAGCT-3′ and 5′-GAAACCCTCCAAGGCTTTCAG-3′; Runx2, 5′-ACATGGCCAGATTCACAGTGG-3′ and 5′-TGGTGCCCGTTAGCAATTG-3′;
Immunofluorescent staining
ATDC5 cells on chamber slide were fixed with 4% paraformaldehyde for 1 min. Cells were permeabilized with 0.1% Triton X-100 and blocked with 1% BSA then incubated with primary antibody for 90 min. After washing the cells 3 times with PBS-T, FITC-conjugated secondary antibodies and DAPI were added to the cells. Fluorescence images were analyzed with a fluorescence microscope (Leica, Wetzlar, Germany). The fluorescence intensity was analyzed by the Leica or image J program.
Immunohistochemistry
E15.5 mice forelimbs were fixed in 4% formaldehyde solution (Junsei, Tokyo, Japan) at 4°C for 24 h. After fixation, the sample was gradually dehydrated in ethanol and embedded in paraffin, and then the paraffin block was sectioned at 3 μm. Deparaffinized sections were quenched in hydrogen peroxide treatment (3% H2O2 in 100% methanol, 30 min), retrieved in Tris-EGTA (TEG) buffer (pH 9.0) for 30 min at 60°C oven and incubated in 50 mM ammonium chloride (NH4Cl) for 30 min at RT. After blocking with 1% BSA, the sections were incubated overnight at 4°C with anti-Runx2 or anti-Cbfβ antibodies then incubated for 1 h with the appropriate HRP conjugated anti-IgG antibody then developed using Dako Liquid DAB+ substrate-chromogen system (Dako, CA, USA). For histological analysis of micromass culture, cell aggregates were sectioned at day 28, rinsed 3 times in PBS, and fixed in 4% (v/v) paraformaldehyde for 1 h. Specimens were gradually dehydrated in ethanol, embedded in paraffin and sectioned at 3μm for histological procedures. Images were analyzed with a Leica fluorescence microscope (Leica).
Ubiquitination assay
ATDC5 cells were plated in 100 mm plates at a density of 2 × 106 cells/well then transfected with a total of 12 μg DNA, including 4 μg of the pCMV-HA-Hif-2α (Tamiya et al., 2008), 4 μg of pcDNA3.1-flag-Ubiquitin and 4 μg of pCS4-myc-Runx2 using Lipofectamine 2000, and were grown for 24 h then treated with 20 μm MG132 for 2 h. Whole cells were lysed with lysis buffer [20 mM HEPES (Sigma-Aldrich), pH 7.9, 300 mM KCl (Sigma-Aldrich), the 10% NP-40 (Sigma-Aldrich), 1mM DTT(Invitrogen) and proteinase inhibitors]. Cell lysates (400 μg) were bound with anti-Myc antibody overnight at 4 °C with gentle agitation, then 0.1 g/ml of Separose A beads (GE Healthcare) were incubated in cell lysates at 4 °C for 2 h with gentle stirring. The mixture was washed with lysis buffer. Proteins were eluted by boiling in 50μl protein loading buffer. Ubiquitination levels were confirmed by Western blotting with anti-Flag antibody.
Co-IP assay
ATDC5 cells were plated at a density of 2 × 106 cells/well in 100 mm plates for transfection experiments. Cells were transfected with a total of 6 μg DNA, including 2 μg of the pCMV-HA-Hif-2α (Tamiya et al., 2008), 2 μg of pcDNA3.1-myc-Cbfβ or 2 μg of pCS4–2Flag-Runx2 or pCS4–2Flag-delta Runx2 (Runx2 Runt domain deleted gene (Lu et al., 1995) [Kindly provided by YOSHIAKI ITO, Cancer Science Institute of Singapore, Singapore] cloned to pCS4–2Flag) by using Lipofectamine 2000 (Invitrogen), and were grown for 16 h. Total cell were lysed with lysis buffer (150mM Sodium chloride, 10mM Tris pH 7.4, 1 mM EDTA, 1mM EGTA, pH 8.0, 1 % Triton-X, 0.5 % NP 40, 2 Mm sodium ortho-vanadate and proteinase inhibitor). Protein concentration was measured by Bradford protein assay kit. Cell lysates (400 μg) were bound with rabbit anti-Myc (Abcam), monoclonal anti-Flag (Sigma-Aldrich) or rabbit anti-HA (Santa Cruz) antibody overnight at 4 °C with gentle agitation then 0.1 g of Separose A beads were incubated in lysis buffer at 4 °C for 2 h with gentle stirring. The mixture was washed with lysis buffer for 5 times. Proteins were eluted by boiling in 50 μl protein loading buffer. Co-IP was confirmed by western blot with anti-Flag or anti-Myc antibody.
Alcian Blue staining
Chondrocyte differentiation was detected by Alcian Blue staining. After washing 3 times with the PBS, micromass sections were fixed in 4% formalin for 1 h at RT. After adding 1% Alcian blue for 1 h, the sections were rinsed with 0.1M HCl then washed with distilled water.
Von Kossa staining
Calcium deposition was detected by von Kossa staining. After washing 3 times with the PBS, micro-mass sections were fixed in 4% formalin for 1 h at RT. After adding 1% silver nitrate around 5 min under long wave ultraviolet light, the sections were thoroughly rinsed with distilled water.
Statistical analyses
Statistical analyses were performed using Sigma Plot software 10.0 (Systat Software Inc, Chicago, USA). Data are expressed as mean ± SD. Difference between groups means were assessed by Student’s t-test. *p<0.05 was considered statistically significant.
RESULTS
Hif-2α inhibits Runx2 protein expression in chondrocytes
Runx2 has been known as a positive regulator for chondrocyte maturation. To investigate Hif-2α if could affect the Runx2 expression, we first examined Runx2 protein level after transient transfection with Hif-2α in osteoblasts and chondrocytes. As previously reported (Tamiya et al., 2008), overexpression of Hif-2α increased Runx2 expression in osteoblast-like ROS 17/2.8 cells (Figure 1A). However, in the chondrogenic cell line ATDC5, Runx2 protein levels were dose-dependently reduced by Hif-2α (Figure 1B). In addition, overexpressed Hif-2α reduced Runx2 and Mmp13 expression at various time points in ATDC5 (Figure 1C) as well as primary chondrocytes (Supplementary Figure 1A–B). More interestingly, vascular smooth muscle MOVAS cell line endogenously expressed Hif-2α but not Runx2, and forced expression of Runx2 or Hif-2α did not affect each other (Supplementary Figure 1C). Conversely, knockdown of Hif-2α with Hif-2α-specific shRNA increased Runx2 (Figure 1D). However, overexpression or knockdown of Hif-2α did not affect Runx2 mRNA expression in the chondrocytes (Figure 1E and F). Moreover, Hif-2α reduced Runx2 expression in the nucleus and knockdown of Hif-2α by Sh-Hif-2α increased Runx2 in chondrocytes (Figure 2A–D). These results indicate that Hif-2α inhibits Runx2 protein expression at the post-transcriptional level in chondrocytes.
Figure 1. Negative regulation of Hif-2α on Runx2 expression in chondrocytes.
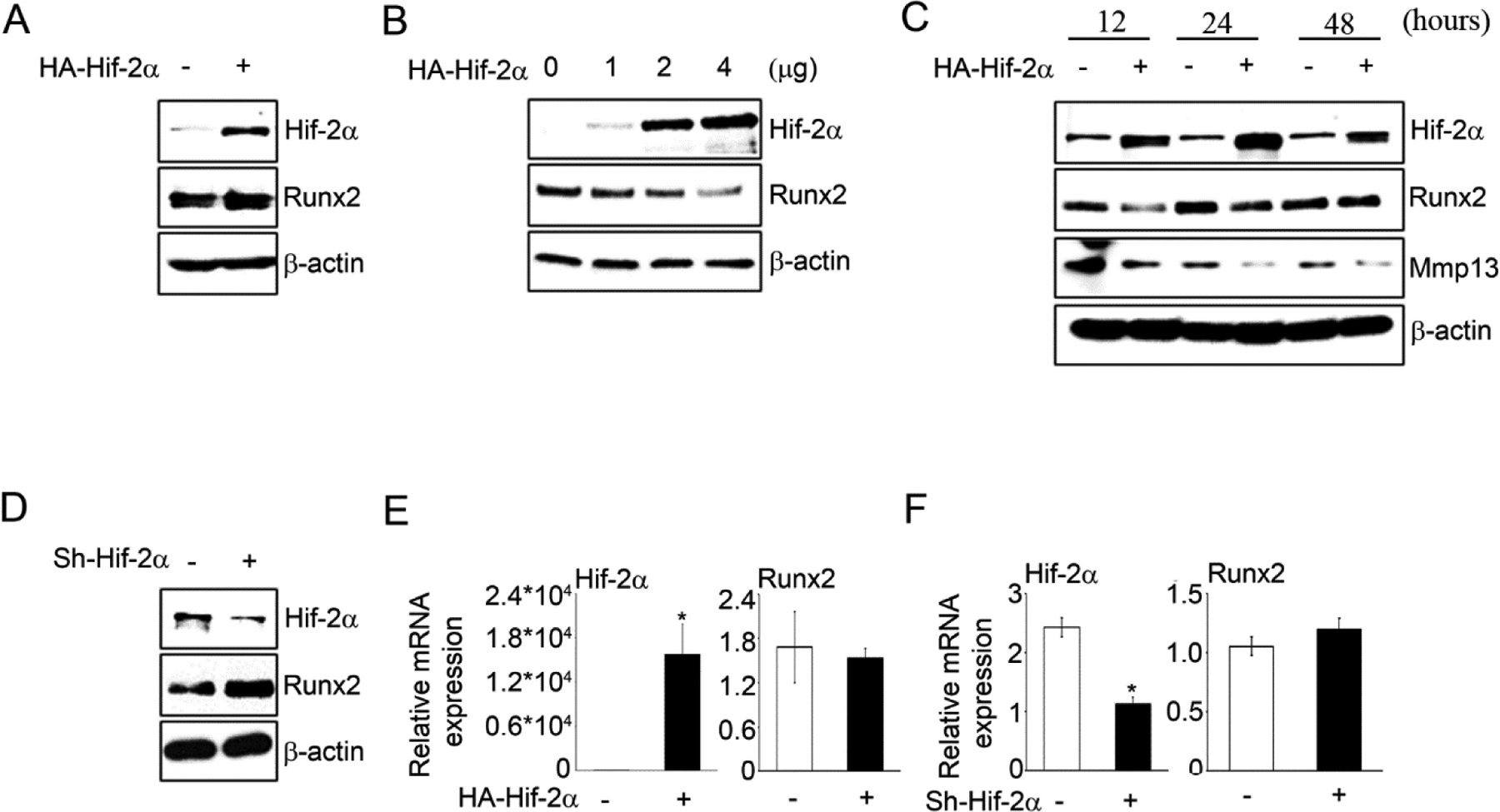
(A) Runx2 protein levels were assessed in ROS17/2.8 osteosarcoma cells after transfection of HA-Hif-2α for 24 h. (B) Runx2 was evaluated by transfection with HA-Hif-2α 1, 2, 4μg in ATDC5 cells. (C) Runx2 and Mmp13 were assessed in ATDC5 cells time dependently after transfection of HA-Hif-2α for 12, 24 and 48 h, respectively. (D) Runx2 protein levels were assessed in ATDC5 cells after sh-control-GFP or sh-Hif-2α-GFP infection. (E-F) Runx2 mRNA expression was determined under transfection of HA-Hif-2α or infection of after sh-control-GFP or sh-Hif-2α-GFP. *P < 0.01 vs. control (n=3)
Figure 2. Hif-2α inhibits endogenous Runx2 level in chondrocytes.
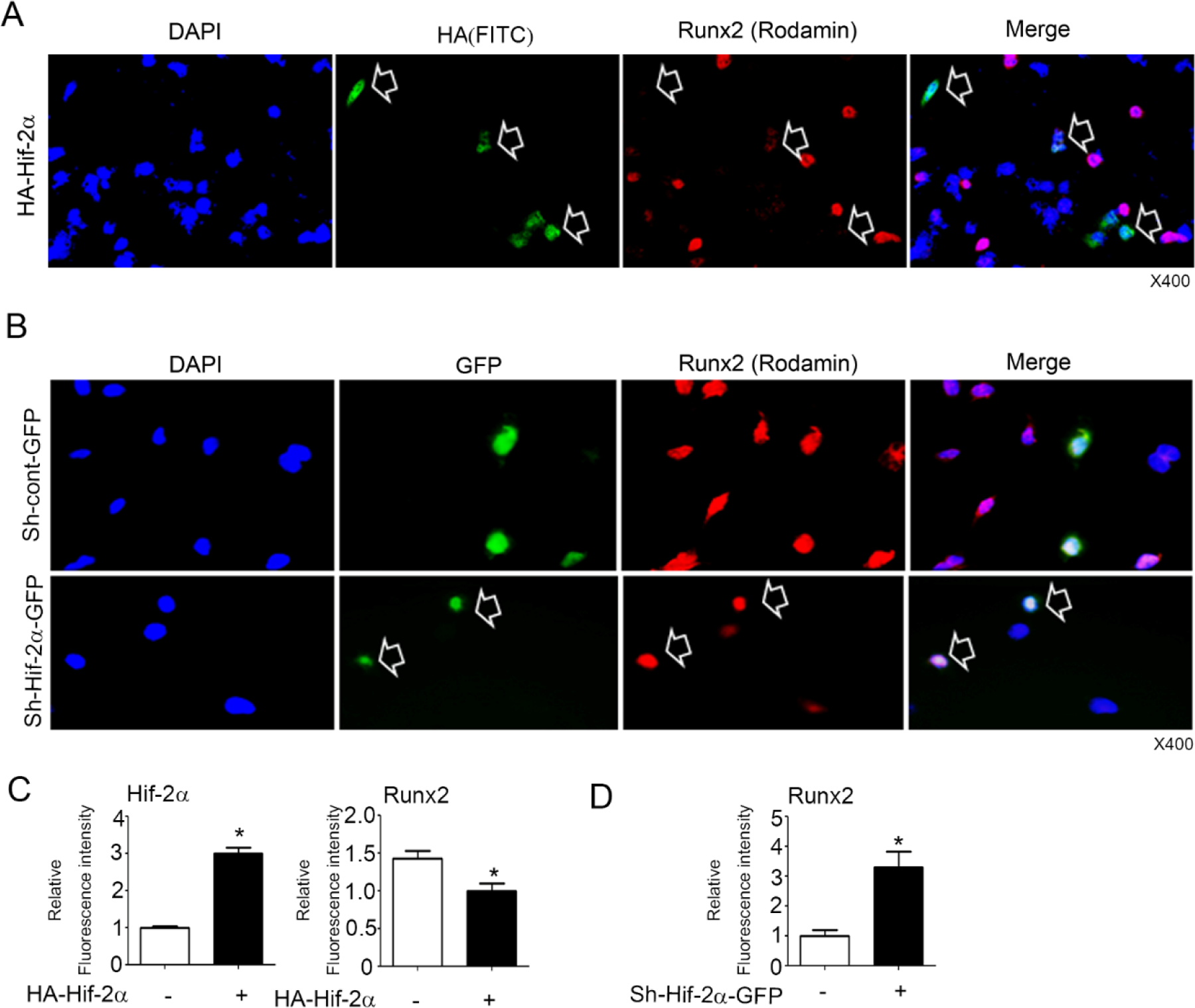
(A) Runx2 was determined by immunofluorescence staining after transient transfection with HA-Hif-2α in ATDC5 cells. (B) Runx2 was determined after infection with sh-control-GFP or sh-Hif-2α-GFP in ATDC5 cells. Arrow indicates up- and down-regulated Hif-2α and Runx2 levels in single cells as green and red colors, respectively. Blue is the DAPI staining of the nucleus. X400. (C-D) The fluorescence intensity was analyzed by the Leica and Image J program (n=4–12 for each group).
Hif-2α enhances Runx2 polyubiquitination
To elucidate the molecular mechanism of Hif-2α-mediated post-transcriptional Runx2 inhibition, Runx2 polyubiquitination assays were performed in ATDC5 cells by transfection with HA-Hif-2α, Myc-Runx2 and Flag-Ub expression vectors with or without MG132. Hif-2α significantly enhanced Runx2 polyubiquitination compared to MG132-treated control cells, indicating that Hif-2α accelerated Runx2 degradation via Runx2 polyubiquitination (Figure 3A). Our previous studies have shown that Runx2 post-transcriptional stability increases chondrocyte differentiation and maturation and osteoblast differentiation primarily due to the formation of a complex with Cbfβ in chondrocytes and osteoblasts. Cbfβ has no nuclear localization signal, and translocation of Cbfβ into the nucleus mediated by heterodimerization with Runx2 upregulates Runx2 transcription activity in the chondrocytes and osteoblasts (Lim et al., 2015; Park et al., 2016). To test whether Hif-2α can affect the cellular distribution or expression level of Cbfβ, Hif-2α or Hif-2α shRNA were expressed in ATDC5 cells. Forced expression of Hif-2α distinctly inhibited nuclear localization of Cbfβ from the cytoplasm (Figure 3B–C) and knockdown of Hif-2α by shRNA promoted nuclear localization of Cbfβ (Figure 3D). Total Cbfβ protein levels were not changed by Hif-2α overexpression or knockdown in chondrocytes (Figure 3B and 3D). Furthermore, Hif-2α-mediated Runx2 inhibition was rescued by Cbfβ transient transfection in chondrocytes (Figure 3E). These results indicate that Hif-2α enhanced Runx2 polyubiquitination-mediated proteasomal degradation in chondrocytes due to the failure of Cbfβ and Runx2 complex formation.
Figure 3. Hif-2α enhances Runx2 polyubiquitination.
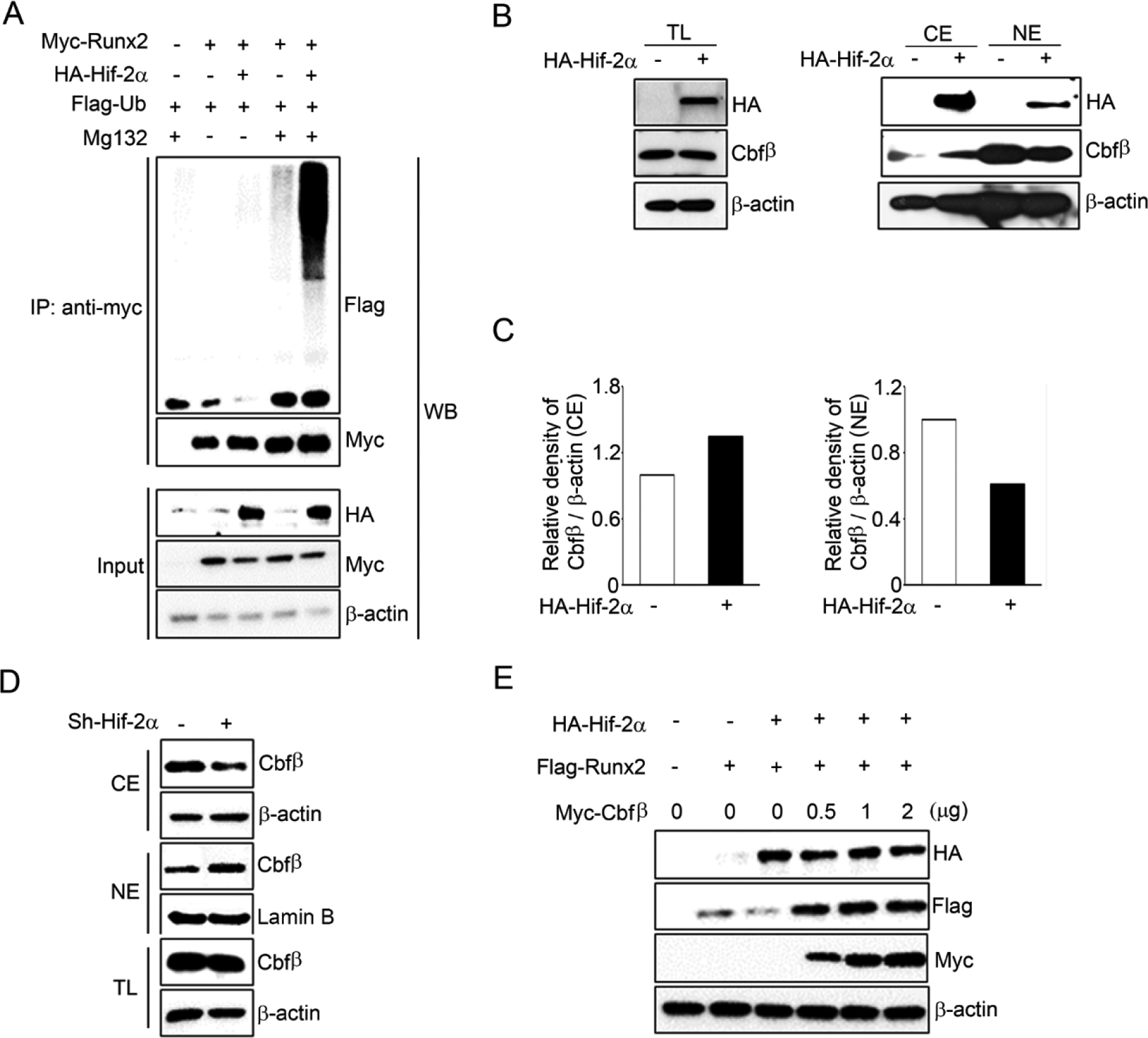
(A) Polyubiquitination assay was performed after transfection with expression vectors HA-Hif-2α, Myc-Runx2, and Flag-Ub in ATDC5 cells. Cells were treated with or without 10 μM MG132. (B) Expression and subcellular location of Cbfβ was determined by western blotting after transfection with HA-Hif-2α for 24 hours at total lysates (TL), nuclear (NE) and cytoplasmic (CE) extracts. (C) Relative density of Cbfβ was normalized with β-actin in CE and NE fractions and analyzed by image J program. (D) ATDC5 cells were infected with sh-Hif-2α or sh-control lentiviral particles and Cbfβ was examined by western blot analysis using TL, NE and CE extracts. (E) Cells were co-transfected with vectors expressing Hif-2α, Runx2, Cbfβ and each protein levels were analyzed by Western blotting with anti-HA, anti-Flag, and anti-Myc antibodies, respectively.
Hif-2α interferes with the formation of the Runx2/Cbfβ complex in chondrocytes
The formation of heterodimeric complexes between Runx2 and Cbfβ protects Runx2 from proteasomal degradation in osteoblasts and chondrocytes (Lim et al., 2015; Park et al., 2016). To determine whether Hif-2α interferes with the formation of Runx2/Cbfβ complexes, immunoprecipitation assays were performed by transfecting chondrocytes with HA-Hif-2α, Flag-Runx2, and Myc-Cbfβ. Hif-2α dose-dependently reduced the formation of the complex between Cbfβ and Runx2 (Figure 4A). These results indicate that Hif-2α-mediated Runx2 inhibition was due to blocking of Cbfβ and Runx2 heterodimerization. Since the Runt domain of Runx2 is the binding site of Cbfβ, we asked if Hif-2α could bind to the Runt domain of Runx2 to compete with Cbfβ binding to Runx2. Therefore, immunoprecipitation assays were performed on ATDC5 cells by transfecting with HA-Hif-2α, Flag-Runx2, and Flag- Runt domain deleted Runx2 (Runx2 delta-Runt) expression vectors. Interestingly, Hif-2α bound to full length of Runx2 but not Runx2 delta-Runt (Figure 4B). Furthermore, Hif-2α and Runx2 were mainly co-localized in the nucleus, however, Runx2 delta-Runt was localized in the perinuclear area (Figure 4C). Taken together, Hif-2α bound to the Runt domain of Runx2 directly, which resulted in competing out Cbfβ binding to Runx2 and accelerating Runx2 degradation by interruption of Runx2/Cbfβ complex formation.
Figure 4. Inhibition of Runx2/Cbfβ complex formation by Hif-2α binding to Runt domain of Runx2.
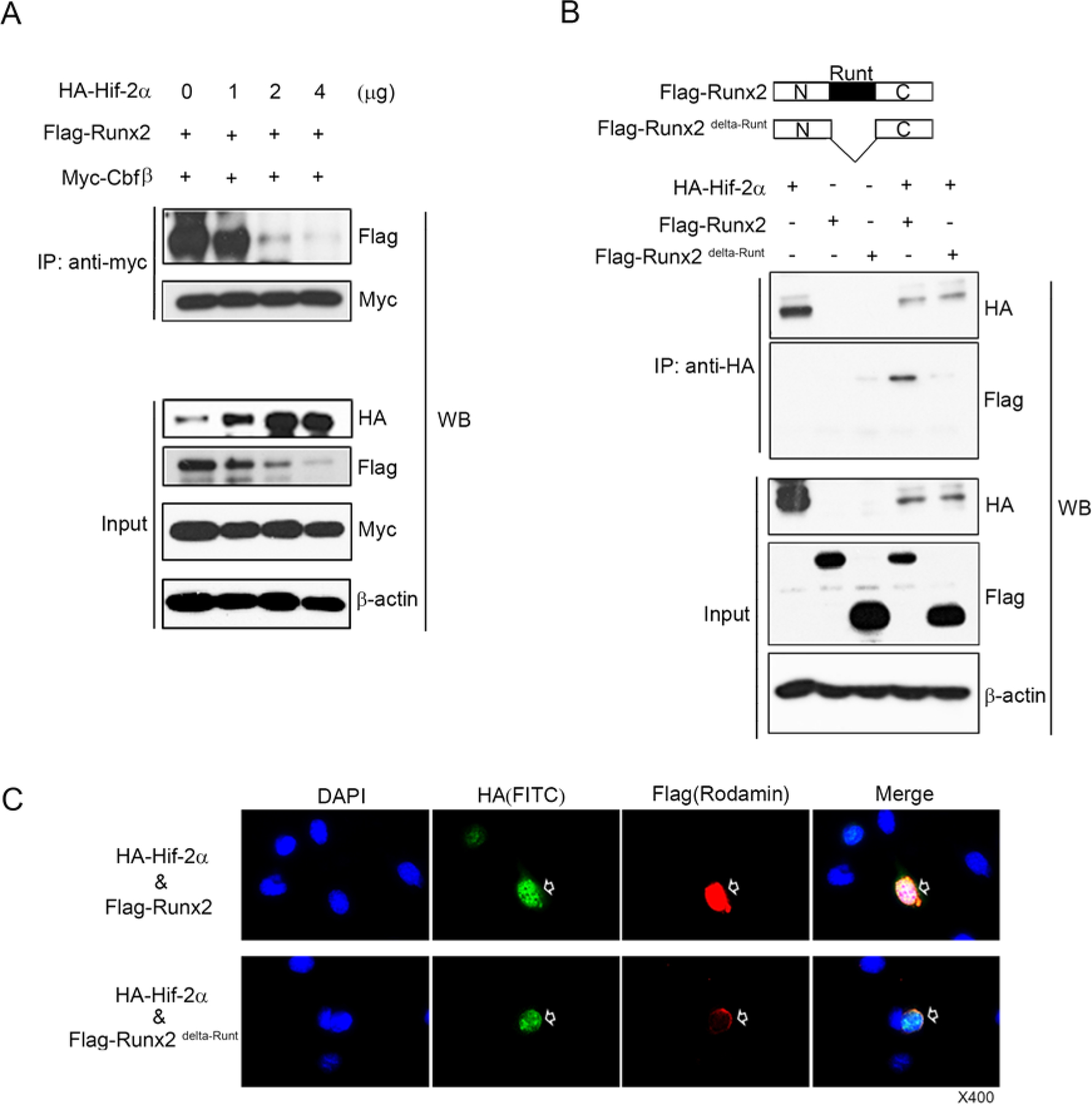
(A) ATDC5 cells were co-transfected with vectors expressing Flag-Runx2, Myc-Cbfβ and HA-Hif-2α. Total extracts were immunoprecipitated with anti-Myc antibody and Runx2 was detected by western blotting (WB) with anti-Flag antibody. (B) ATDC5 cells were co-transfected with Flag-Runx2 and Flag-Runx2delta-Runt expression vectors. Total extracts were immunoprecipitated with anti-HA antibody-tagged bead, and Runx2 and Runx2 delta-Runt were detected by western blotting with anti-Flag-HRP antibody. (C) Subcellular localization of Hif-2α, Flag-Runx2, and Flag- Runx2 delta-Runt protein in chondrocytes after co-transfection of HA-Hif-2α with Flang-Runx2 or Flag- Runx2 delta-Runt expression vectors. Hif-2α and Runx2 were mainly co-localized in the nucleus, however, Runx2 delta-Runt was localized in the perinuclear area.
Hif-2α is a novel inhibitor for chondrocyte maturation through inhibition of Runx2
Based on Hif-2α inhibition of Cbfβ nuclear translocation and Runx2 expression in vitro, we investigated whether Runx2 and Cbfβ expression was diminished in Hif-2α heterozygote mice in vivo. As expected, Cbfβ nuclear localization was increased by dark spot staining in the nucleus and Runx2 expression was also increased in the Hif-2α+/− scapular chondrocytes (Figure 5A). Next, we performed ex vivo chondrocyte differentiation assays using mesenchymal cells derived from WT and Hif-2a+/− limb bud at E11.5. Mesenchymal cells derived from Hif-2α heterozygous mice were more rapidly differentiated into hypertrophic chondrocyte than those of wild type mice in a micromass culture system in the Alcian Blue, von Kossa, and immunofluorescence staining (Figure 5B–C). Indeed, heterozygous Hif-2α mice slightly accelerated endochondral bone formation of long bone cartilage compared to WT mice (Supplementary Figure 2). These results indicate that Hif-2α is a novel inhibitor for chondrocyte maturation through inhibition of Runx2.
Figure 5. Accelerated chondrocyte differentiation in Hif-2α+/− mice.
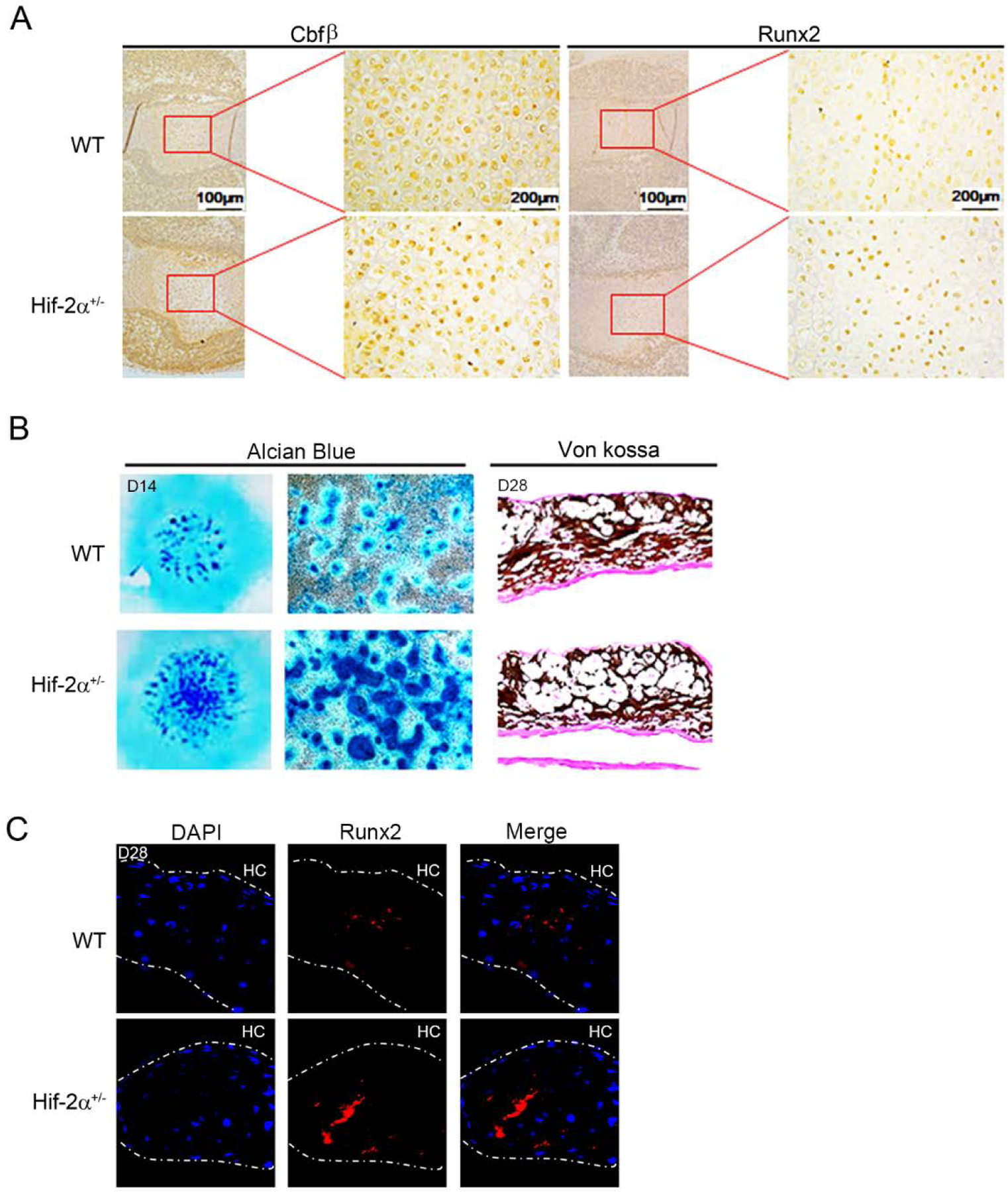
(A) Expression of Cbfβ and Runx2 were determined in Hif-2a+/− and WT scapular chondrocytes at embryonic day 15.0 by immunohistochemistry. (B) For micromass culture, mesenchymal cells were obtained on day 11.5 of embryos from WT and Hif-2a+ mice. Chondrocyte differentiation was assessed with Alcian Blue and von Kossa staining. (C) Runx2 (red) was detected in Hif-2a+/− and WT micromass cultured cells by immunofluorescence staining. Nucleus was stained by DAPI (blue). HC, hypertrophic chondrocytes. Magnification, x40.
DISCUSSION
In this study, we uncovered Hif-2α as a novel modulator that inhibits chondrocyte maturation by promoting Runx2 proteasome degradation by interfering with the formation of the Runx2/Cbfβ complex in chondrocytes (Figure 6).
Figure 6. Schema of Hif-2α as a negative regulator of Runx2 during chondrocyte maturation.
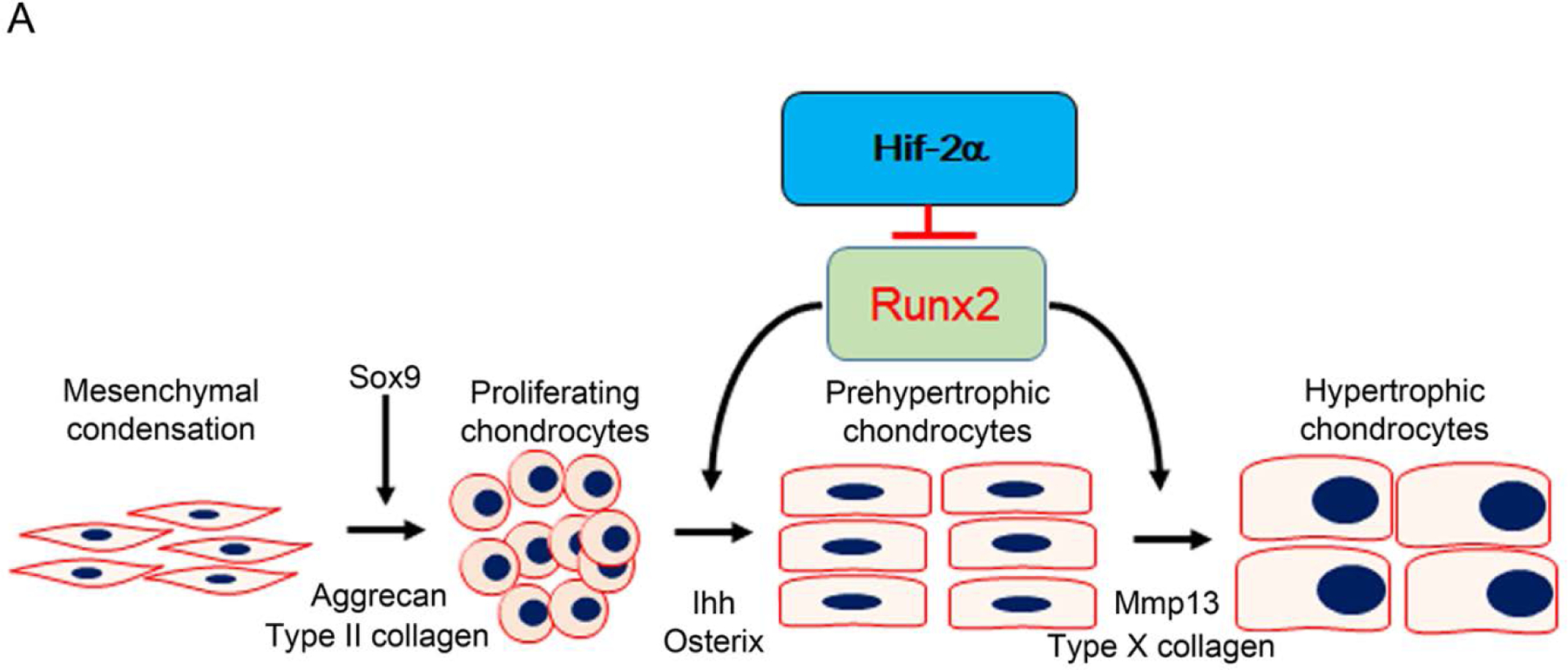
During endochondral ossification, Runx2/Cbfβ is necessary for chondrocyte differentiation and maturation in growth plates. Hif-2α inhibits chondrocyte maturation through interfering Runx2/Cbfβ complex formation to degrade Runx2.
Notably, our study showed that Hif-2α binds to the Runt domain of Runx2 and interferes with the formation of the Runx2/Cbfβ complex. Heterodimerization between Runx2 and Cbfβ is important for Runx2 stability and transcriptional activity (Kundu et al., 2002; Yoshida et al., 2002). Loss of Cbfβ in chondrocytes or osteoblasts induces rapid proteasomal degradation of Runx2, which delays endochondral bone formation and reduces cortical bone thickness (Lim et al., 2015; Park et al., 2016). Hif-2α did not regulate Runx2 or Cbfβ transcription, but inhibited the Runx2 and Cbfβ heterodimerization and Cbfβ nuclear translocation in chondrocytes. As a novel binding factor for the Runt domain of Runx2, Hif-2α can compete with Cbfβ and other transcription factors such as Hdac4 (Vega et al., 2004), Twist-1(Hinoi et al., 2006), and Sox9 (Zhou et al., 2006) in chondrocytes. Thus, Hif-2α may act as a fine-tuner of a variety of physiologically mature hypertrophic chondrocyte functions, including biomineralization, apoptosis, trans-differentiation, and vasculature invasion in the ossification center.
In addition to the pathological function of Hif-2α as a catabolic factor associated with both enhanced chondrocyte hypertrophy and apoptosis in the articular chondrocytes (Saito et al., 2010; Yang et al., 2010), this study shows that Hif-2α plays as a new negative regulator for chondrocyte maturation in growth plates. Establishing the correct interaction of Hif-2α function between articular chondrocytes and growth plate chondrocytes is an important question that requires further investigation. Hypoxia increases VEGF through upregulation of Hif-1α, and increased VEGF inhibits osteogenic differentiation but promotes chondrogenic differentiation. This suggests that Hif-1α may have the opposite function in regulating osteoblast or chondrocyte activity (Hwang, Noh, Hong, & Lee, 2020). This study also suggests that Hif-2α may have various functions in maintaining the homeostasis of articular chondrocytes and chondrocyte maturation of growth plate.
Longitudinal bone growth is controlled by various endocrine, autocrine, and paracrine factors, which eventually leads to growth plate closure with loss of proliferating chondrocytes (Hallett, Ono, & Ono, 2019; Karimian, Chagin, & Savendahl, 2011). Among transcription factors, Runx2 is a crucial executor that promotes chondrocyte maturation and mineralization to influence longitudinal bone growth. Mmp13, a downstream target of Runx2, is an important factor in chondrocyte hypertrophy and maturation (D’Angelo et al., 2000; Nagai & Aoki, 2002) and in this study, Hif-2α inhibited Mmp13 expression through Runx2 degradation. We presented the importance of Hif-2α-Runx2 pathway in chondrocyte maturation in growth plates. Based on the fact that PTHrP inhibits Runx2 expression through inducing PKA signaling pathway and cyclin-D1-mediated proteasome degradation (Li et al., 2004; Zhang et al., 2009), further studies are required to investigate whether the Hif-2α-Runx2 pathway could affect the PTHrP pathway in chondrocytes.
Collectively, our results show that Hif-2α is a novel inhibitor for chondrocyte maturation by interfering with the formation of the Runx2/Cbfβ complex.
Supplementary Material
Supplementary Figure 1. Hif-2α inhibits Runx2 in primary chondrocytes, not in the Runx2 unexpressed MOVAS cells (A) Primary chondrocytes were transiently transfected for 24 h with Hif-2α expression vector. Runx2 protein levels were assessed by western blotting. (B) Expression of Runx2 mRNA was determined by real time Q-PCR. (C) Vascular smooth muscle MOVAS cells were transiently transfected with expression vectors of Hif-2α or Runx2 for 24 h and their protein levels were measured by western blotting using anti-Hif-2α and anti-Runx2 antibodies, respectively. MOVAS cells endogenously expressed Hif-2α but not Runx2. The forced expression of Runx2 or Hif-2α did not affect each other in MOVAS cells.
Supplementary Figure 2. Enhanced chondrocyte maturation in Hif-2a+/− mice. Humerus (E15.0) and femur (E15.5) were stained by Hematoxylin & Eosin in Hif-2a+/− and WT mice. Magnification, x40 (boxed) and x100 (enlarged photos).
ACKNOWLEDGEMENTS
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (NRF-2018R1D1A1B07043598), (NRF-2017R1A2A2A05001305), (NRF-2017R1A5A2015391), and Korea Mouse Phenotyping Project (NRF-2014M3A9D5A01073658).
Funding information
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (NRF-2018R1D1A1B07043598), Korea government (MSIT) (NRF-2017R1A2A2A05001305) and Korea Mouse Phenotyping Project (NRF-2014M3A9D5A01073658).
Footnotes
Disclosure Statement: All the authors have nothing to disclose.
CONFLICT OF INTEREST STATEMENT
The authors declare that they have no conflict of interests.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are openly available in [repository name “JCP-Hif2a-Runx2-Figures” and “JCP-Hif2a-Runx2-Figures-Raw data”] at https://drive.google.com/drive/folders/15O7G0gX8ypbl9qFUUkizPEd6v3ycJyXC?usp=sharing and https://drive.google.com/drive/folders/18UjNmtxL8LjM9fcgU0ovSlN2WBioVYEA?usp=sharing.
REFERENCES
- Araldi E, Khatri R, Giaccia AJ, Simon MC, & Schipani E (2011). Lack of HIF-2alpha in limb bud mesenchyme causes a modest and transient delay of endochondral bone development. Nat Med, 17(1), 25–26; author reply 27–29. doi: 10.1038/nm0111-25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JY, Pratap J, Javed A, Zaidi SK, Xing L, Balint E, … Stein GS (2001). Subnuclear targeting of Runx/Cbfa/AML factors is essential for tissue-specific differentiation during embryonic development. Proc Natl Acad Sci U S A, 98(15), 8650–8655. doi: 10.1073/pnas.151236498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Angelo M, Yan Z, Nooreyazdan M, Pacifici M, Sarment DS, Billings PC, & Leboy PS (2000). MMP-13 is induced during chondrocyte hypertrophy. J Cell Biochem, 77(4), 678–693. [PubMed] [Google Scholar]
- Hallett SA, Ono W, & Ono N (2019). Growth Plate Chondrocytes: Skeletal Development, Growth and Beyond. Int J Mol Sci, 20(23). doi: 10.3390/ijms20236009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinoi E, Bialek P, Chen YT, Rached MT, Groner Y, Behringer RR, … Karsenty G (2006). Runx2 inhibits chondrocyte proliferation and hypertrophy through its expression in the perichondrium. Genes Dev, 20(21), 2937–2942. doi: 10.1101/gad.1482906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang OK, Noh YW, Hong JT, & Lee JW (2020). Hypoxia Pretreatment Promotes Chondrocyte Differentiation of Human Adipose-Derived Stem Cells via Vascular Endothelial Growth Factor. Tissue Eng Regen Med, 17(3), 335–350. doi: 10.1007/s13770-020-00265-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer NV, Kotch LE, Agani F, Leung SW, Laughner E, Wenger RH, … Semenza GL (1998). Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1 alpha. Genes Dev, 12(2), 149–162. doi: 10.1101/gad.12.2.149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karimian E, Chagin AS, & Savendahl L (2011). Genetic regulation of the growth plate. Front Endocrinol (Lausanne), 2, 113. doi: 10.3389/fendo.2011.00113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komori T (2018). Runx2, an inducer of osteoblast and chondrocyte differentiation. Histochem Cell Biol, 149(4), 313–323. doi: 10.1007/s00418-018-1640-6 [DOI] [PubMed] [Google Scholar]
- Komori T, Yagi H, Nomura S, Yamaguchi A, Sasaki K, Deguchi K, … Kishimoto T (1997). Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell, 89(5), 755–764. doi: 10.1016/s0092-8674(00)80258-5 [DOI] [PubMed] [Google Scholar]
- Kundu M, Javed A, Jeon JP, Horner A, Shum L, Eckhaus M, … Liu PP (2002). Cbfbeta interacts with Runx2 and has a critical role in bone development. Nat Genet, 32(4), 639–644. doi: 10.1038/ng1050 [DOI] [PubMed] [Google Scholar]
- Lee SY, Park KH, Yu HG, Kook E, Song WH, Lee G, … Ryu JH (2019). Controlling hypoxia-inducible factor-2alpha is critical for maintaining bone homeostasis in mice. Bone Res, 7, 14. doi: 10.1038/s41413-019-0054-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li TF, Dong Y, Ionescu AM, Rosier RN, Zuscik MJ, Schwarz EM, … Drissi H (2004). Parathyroid hormone-related peptide (PTHrP) inhibits Runx2 expression through the PKA signaling pathway. Exp Cell Res, 299(1), 128–136. doi: 10.1016/j.yexcr.2004.05.025 [DOI] [PubMed] [Google Scholar]
- Lim KE, Park NR, Che XG, Han MS, Jeong JH, Kim SY, … Choi JY (2015). Core Binding Factor beta of Osteoblasts Maintains Cortical Bone Mass via Stabilization of Runx2 in Mice. Journal of Bone and Mineral Research, 30(4), 715–722. doi: 10.1002/jbmr.2397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long F, & Ornitz DM (2013). Development of the endochondral skeleton. Cold Spring Harb Perspect Biol, 5(1), a008334. doi: 10.1101/cshperspect.a008334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Maruyama M, Satake M, Bae SC, Ogawa E, Kagoshima H, … Ito Y (1995). Subcellular localization of the alpha and beta subunits of the acute myeloid leukemia-linked transcription factor PEBP2/CBF. Mol Cell Biol, 15(3), 1651–1661. doi: 10.1128/mcb.15.3.1651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merceron C, Ranganathan K, Wang E, Tata Z, Makkapati S, Khan MP, … Schipani E (2019). Hypoxia-inducible factor 2alpha is a negative regulator of osteoblastogenesis and bone mass accrual. Bone Res, 7, 7. doi: 10.1038/s41413-019-0045-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai H, & Aoki M (2002). Inhibition of growth plate angiogenesis and endochondral ossification with diminished expression of MMP-13 in hypertrophic chondrocytes in FGF-2-treated rats. Journal of Bone and Mineral Metabolism, 20(3), 142–147. doi: 10.1007/s007740200020 [DOI] [PubMed] [Google Scholar]
- Otto F, Thornell AP, Crompton T, Denzel A, Gilmour KC, Rosewell IR, … Owen MJ (1997). Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell, 89(5), 765–771. doi: 10.1016/s0092-8674(00)80259-7 [DOI] [PubMed] [Google Scholar]
- Park NR, Lim KE, Han MS, Che X, Park CY, Kim JE, … Choi JY (2016). Core Binding Factor beta Plays a Critical Role During Chondrocyte Differentiation. J Cell Physiol, 231(1), 162–171. doi: 10.1002/jcp.25068 [DOI] [PubMed] [Google Scholar]
- Patel SA, & Simon MC (2008). Biology of hypoxia-inducible factor-2alpha in development and disease. Cell Death Differ, 15(4), 628–634. doi: 10.1038/cdd.2008.17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng J, Zhang L, Drysdale L, & Fong GH (2000). The transcription factor EPAS-1/hypoxia-inducible factor 2alpha plays an important role in vascular remodeling. Proc Natl Acad Sci U S A, 97(15), 8386–8391. doi: 10.1073/pnas.140087397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito T, Fukai A, Mabuchi A, Ikeda T, Yano F, Ohba S, … Kawaguchi H (2010). Transcriptional regulation of endochondral ossification by HIF-2alpha during skeletal growth and osteoarthritis development. Nat Med, 16(6), 678–686. doi: 10.1038/nm.2146 [DOI] [PubMed] [Google Scholar]
- Schipani E, Ryan HE, Didrickson S, Kobayashi T, Knight M, & Johnson RS (2001). Hypoxia in cartilage: HIF-1alpha is essential for chondrocyte growth arrest and survival. Genes Dev, 15(21), 2865–2876. doi: 10.1101/gad.934301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza GL (2012). Hypoxia-inducible factors in physiology and medicine. Cell, 148(3), 399–408. doi: 10.1016/j.cell.2012.01.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanton LA, Sabari S, Sampaio AV, Underhill TM, & Beier F (2004). p38 MAP kinase signalling is required for hypertrophic chondrocyte differentiation. Biochem J, 378(Pt 1), 53–62. doi: 10.1042/BJ20030874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stegen S, Laperre K, Eelen G, Rinaldi G, Fraisl P, Torrekens S, … Carmeliet G (2019). HIF-1 alpha metabolically controls collagen synthesis and modification in chondrocytes. Nature, 565(7740), 511-+. doi: 10.1038/s41586-019-0874-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein GS, Lian JB, van Wijnen AJ, Stein JL, Montecino M, Javed A, … Pockwinse SM (2004). Runx2 control of organization, assembly and activity of the regulatory machinery for skeletal gene expression. Oncogene, 23(24), 4315–4329. doi: 10.1038/sj.onc.1207676 [DOI] [PubMed] [Google Scholar]
- Stewart AJ, Houston B, & Farquharson C (2006). Elevated expression of hypoxia inducible factor-2alpha in terminally differentiating growth plate chondrocytes. J Cell Physiol, 206(2), 435–440. doi: 10.1002/jcp.20481 [DOI] [PubMed] [Google Scholar]
- Takeda S, Bonnamy JP, Owen MJ, Ducy P, & Karsenty G (2001). Continuous expression of Cbfa1 in nonhypertrophic chondrocytes uncovers its ability to induce hypertrophic chondrocyte differentiation and partially rescues Cbfa1-deficient mice. Genes Dev, 15(4), 467–481. doi: 10.1101/gad.845101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamiya H, Ikeda T, Jeong JH, Saito T, Yano F, Jung YK, … Choi JY (2008). Analysis of the Runx2 promoter in osseous and non-osseous cells and identification of HIF2A as a potent transcription activator. Gene, 416(1–2), 53–60. doi: 10.1016/j.gene.2008.03.003 [DOI] [PubMed] [Google Scholar]
- Tanimoto K, Yoshiga K, Eguchi H, Kaneyasu M, Ukon K, Kumazaki T, … Nishiyama M (2003). Hypoxia-inducible factor-1alpha polymorphisms associated with enhanced transactivation capacity, implying clinical significance. Carcinogenesis, 24(11), 1779–1783. doi: 10.1093/carcin/bgg132 [DOI] [PubMed] [Google Scholar]
- Ueta C, Iwamoto M, Kanatani N, Yoshida C, Liu Y, Enomoto-Iwamoto M, … Komori T (2001). Skeletal malformations caused by overexpression of Cbfa1 or its dominant negative form in chondrocytes. J Cell Biol, 153(1), 87–100. doi: 10.1083/jcb.153.1.87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vega RB, Matsuda K, Oh J, Barbosa AC, Yang X, Meadows E, … Olson EN (2004). Histone deacetylase 4 controls chondrocyte hypertrophy during skeletogenesis. Cell, 119(4), 555–566. doi: 10.1016/j.cell.2004.10.024 [DOI] [PubMed] [Google Scholar]
- Yang S, Kim J, Ryu JH, Oh H, Chun CH, Kim BJ, … Chun JS (2010). Hypoxia-inducible factor-2alpha is a catabolic regulator of osteoarthritic cartilage destruction. Nat Med, 16(6), 687–693. doi: 10.1038/nm.2153 [DOI] [PubMed] [Google Scholar]
- Yoshida CA, Furuichi T, Fujita T, Fukuyama R, Kanatani N, Kobayashi S, … Komori T (2002). Core-binding factor beta interacts with Runx2 and is required for skeletal development. Nat Genet, 32(4), 633–638. doi: 10.1038/ng1015 [DOI] [PubMed] [Google Scholar]
- Yoshida CA, Yamamoto H, Fujita T, Furuichi T, Ito K, Inoue K, … Komori T (2004). Runx2 and Runx3 are essential for chondrocyte maturation, and Runx2 regulates limb growth through induction of Indian hedgehog. Genes Dev, 18(8), 952–963. doi: 10.1101/gad.1174704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Xie R, Hou W, Wang B, Shen R, Wang X, … Chen D (2009). PTHrP prevents chondrocyte premature hypertrophy by inducing cyclin-D1-dependent Runx2 and Runx3 phosphorylation, ubiquitylation and proteasomal degradation. J Cell Sci, 122(Pt 9), 1382–1389. doi: 10.1242/jcs.040709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou G, Zheng Q, Engin F, Munivez E, Chen Y, Sebald E, … Lee B (2006). Dominance of SOX9 function over RUNX2 during skeletogenesis. Proc Natl Acad Sci U S A, 103(50), 19004–19009. doi: 10.1073/pnas.0605170103 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Hif-2α inhibits Runx2 in primary chondrocytes, not in the Runx2 unexpressed MOVAS cells (A) Primary chondrocytes were transiently transfected for 24 h with Hif-2α expression vector. Runx2 protein levels were assessed by western blotting. (B) Expression of Runx2 mRNA was determined by real time Q-PCR. (C) Vascular smooth muscle MOVAS cells were transiently transfected with expression vectors of Hif-2α or Runx2 for 24 h and their protein levels were measured by western blotting using anti-Hif-2α and anti-Runx2 antibodies, respectively. MOVAS cells endogenously expressed Hif-2α but not Runx2. The forced expression of Runx2 or Hif-2α did not affect each other in MOVAS cells.
Supplementary Figure 2. Enhanced chondrocyte maturation in Hif-2a+/− mice. Humerus (E15.0) and femur (E15.5) were stained by Hematoxylin & Eosin in Hif-2a+/− and WT mice. Magnification, x40 (boxed) and x100 (enlarged photos).
Data Availability Statement
The data that support the findings of this study are openly available in [repository name “JCP-Hif2a-Runx2-Figures” and “JCP-Hif2a-Runx2-Figures-Raw data”] at https://drive.google.com/drive/folders/15O7G0gX8ypbl9qFUUkizPEd6v3ycJyXC?usp=sharing and https://drive.google.com/drive/folders/18UjNmtxL8LjM9fcgU0ovSlN2WBioVYEA?usp=sharing.