

Abstract
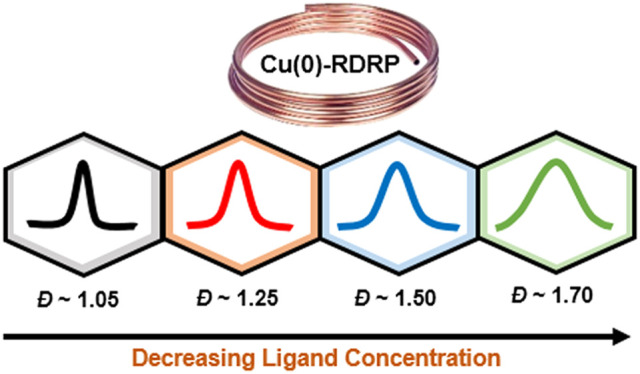
Cu(0)-reversible deactivation radical polymerization (RDRP) is a versatile polymerization tool, providing rapid access to well-defined polymers while utilizing mild reaction conditions and low catalyst loadings. However, thus far, this method has not been applied to tailor dispersity, a key parameter that determines the physical properties and applications of polymeric materials. Here, we report a simple to perform method, whereby Cu(0)-RDRP can systematically control polymer dispersity (Đ = 1.07–1.72), while maintaining monomodal molecular weight distributions. By varying the ligand concentration, we could effectively regulate the rates of initiation and deactivation, resulting in polymers of various dispersities. Importantly, both low and high dispersity PMA possess high end-group fidelity, as evidenced by MALDI-ToF-MS, allowing for a range of block copolymers to be prepared with different dispersity configurations. The scope of our method can also be extended to include inexpensive ligands (i.e., PMDETA), which also facilitated the polymerization of lower propagation rate constant monomers (i.e., styrene) and the in situ synthesis of block copolymers. This work significantly expands the toolbox of RDRP methods for tailoring dispersity in polymeric materials.
Keywords: Cu(0)-RDRP, Dispersity Control, Molecular Weight Distributions, Block Copolymers, Ligand Concentration
Introduction
Low and high molecular weight (MW) polymers display complementary properties critical for various applications. For example, higher MW materials typically possess greater mechanical strength and toughness and higher glass transition and thermal degradation temperatures.1−4 On the other hand, thanks to low chain entanglement and viscosity, low MW polymers can be easily processed by extrusion and injection molding technologies.4 As such, preparing polymers that contain a mixture of both high and low MW chains, more commonly referred to as a broad molecular weight distribution or a high dispersity, allows access to desirable materials combining the advantageous properties of both low and high molecular weight segments.5−7 In fact, increasing dispersity has been shown to significantly affect mechanical, thermal, and rheological properties and also how polymers self-assemble into nanostructured materials.7−17
To date, high dispersity polymers have commonly been obtained via free radical polymerization (FRP), a method where radicals uncontrollably react with monomers until irreversible termination occurs.18 Although FRP generates polymers with high dispersities (Đ = 1.5–2.0) that are suitable for various industrial applications, this approach has poor control over macromolecular structure, including the degree of polymerization, dispersity range, end-group functionality, chain architecture, and composition. Importantly, all chains are “dead”, which makes any subsequent polymerization or modification steps impossible, thus preventing access to block copolymers and other advanced materials.19,20 On the other hand, controlled radical polymerization (CRP) methodologies have been demonstrated to effectively regulate polymeric structures and produce block copolymers with high end-group fidelity. However, polymers prepared by CRPs typically have very low dispersities (Đ = 1.05–1.20), limiting their potential applications.21 This limitation has therefore resulted in the development of a whole raft of strategies whereby CRP and other polymerization strategies can be used to prepare polymers with any desired intermediate or high dispersity.5−7
One of the elegant approaches for tuning dispersity is the temporal regulation of initiation method developed by Fors and co-workers, where the initiator is fed in throughout a polymerization, so polymer chains start growing at different times.8,11,22−24 In addition, the groups of Boyer, Junkers, Frey, Leibfarth, Guironnet, and others have independently introduced a range of flow approaches, where dispersity can be carefully controlled by adjusting reaction parameters.25−34 These methods are often associated with excellent mathematical modeling and the ability to be automated, allowing desired molecular weight distributions to be obtained.25,28−30,35,36 Furthermore, several batch methods have been recently developed to tune polymer dispersity.37−43 These include reducing the catalyst concentration in atom transfer radical concentration (ATRP),17,39,44−46 mixing high and low activity chain transfer agents in reversible addition–fragmentation chain transfer (RAFT) polymerization,38,47 adjusting the solvent polarity and concentration of azide in reversible complexation-mediated polymerization,40 and using photochromic initiators to control dispersity in cationic polymerization.37 Other reported methods involve the modification of CRPs by either addition of a comonomer or a termination agent34,48,49 or blending prepurified polymers of either different molecular weight or dispersity.50,51 Despite these excellent recent breakthroughs, there are only limited techniques that can give access to well-defined polymers with controlled dispersity, hindering the widespread use of these materials.
One of the most versatile polymerization methods for preparing advanced polymeric materials is Cu(0)-reversible deactivation radical polymerization (RDRP),52−58 but this method has yet to be exploited to control polymer dispersity. This is a major limitation, given that Cu(0)-RDRP utilizes very low catalyst concentrations, typically has high polymerization rates, and furnishes polymers with very high end-group fidelity.59,60 This CRP method is also commonly associated with mild reaction conditions and simple removal of the catalyst postpolymerization.61−63 As such, access to polymers with tunable dispersity by Cu(0)-RDRP is highly desirable. To this end, we sought a new strategy to effectively tune dispersity. We present a system whereby, on variation of ligand concentration, a wide range of dispersity polymers can be obtained, with high end-group fidelity observed in all cases. Furthermore, in situ chain extensions could be achieved, even from high dispersity polymers, and the scope of the system could be expanded to incorporate a lower activity complex, which allows for dispersity control in both polyacrylates and polystyrene.
Experimental Section
Materials and Instrumentation
All chemicals were purchased from Sigma-Aldrich (Merck) and used as received unless otherwise stated. Tris(2-(dimethylamino)ethyl)amine (Me6TREN) was synthesized according to previously reported literature and distilled prior to use.64 Copper wire (gauge 0.25 mm) was purchased from Sigma-Aldrich.
1H NMR spectra were recorded on a Bruker-300 Ultrashield at 25 °C using deuterated chloroform as the solvent. Chemical shifts are given in parts per million and are referenced to residual solvent proton signals. Size-exclusion chromatography (SEC) was performed using an Agilent 390-LC MDS instrument, equipped with differential refractive index (DRI) and dual wavelength UV detectors. The system was equipped with 2× PLgel mixed C columns (300 × 7.5 mm) and a PLgel 5 μm guard column. The eluent was N,N-dimethylacetamide (HPLC grade, with 0.03% w/v LiBr) run with a 1 mL/min flow rate at 40 °C. A molecular weight calibration curve was produced using commercial narrow molecular weight distribution poly(methyl methacrylate) standards with molecular weights ranging from 5000 to 1.5 × 106. Samples were passed through a column of basic alumina to remove Cu species and subsequently filtered through 0.45 μm filters prior to injection. Experimental molar mass (Mn(SEC) and dispersity (Đ) values of synthesized polymers were determined by conventional calibration using Agilent SEC software. UV–vis absorbance spectra were recorded on a JASCO V-730 spectrophotometer equipped with STR-773 water thermostated cell holder and stirrer. Spectra were typically recorded from 400 to 1000 nm at a rate of 400 nm min–1 at 25 °C. Matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-ToF-MS) data were recorded on an Autoflex speed time-of-flight mass spectrometer (Bruker Daltonics, Bremen, Germany) equipped with a Bruker smartbeamTM-II laser (355 nm wavelength) in reflection mode; ∼1000 spectra added up for processing, accumulated at a scan rate of 2 kHz in the mass range of 600–7000 m/z; mass calibration to <5 ppm accuracy with commercial poly(ethylene glycol) (PEG) calibrant. Solutions in THF (HPLC grade) with dithranol as the matrix (20 mg/mL), sodium trifluoroacetate as the cationization agent (1.0 mg/mL), and sample (20 mg/mL) were prepared. Ten microliters of matrix solution was mixed with 2 μL of cationization agent solution and 10 μL of sample solution, and 1.0 μL of the mixture was applied to the target plate prior to measurement.
General Procedure 1: Cu(0)-RDRP of MA with Various Concentrations of Me6TREN
In a 5 mL vial, 1 mL of DMSO, 1 mL of MA (11.2 mmol, 100 equiv), and Me6TREN were added. EBiB (16.4 μL, 0.112 mmol, 1 equiv) was transferred into the reaction vessel via a microliter syringe. Concurrently, in a separate vial, a stirrer bar wrapped with 5 cm of copper wire was immersed in 37% HCl, stirred for 15 min, washed sequentially with water and acetone, and dried. The stirrer bar was then placed into the reaction vessel before it was sealed with a rubber septum and deoxygenated by bubbling with nitrogen for 15 min. The reaction mixture was stirred at 200 rpm, and the reaction proceeded at 25 °C. Samples were taken and analyzed via 1H NMR and SEC. A stock solution of 17.9 μL of Me6TREN was prepared in 1 mL of DMSO and used for all reactions. For example, reactions were performed with 0.02 equiv of ligand (33 μL of stock solution, 2.2 μmol), 0.005 equiv of ligand (8.3 μL of stock solution, 0.56 μmol), 0.0025 equiv of ligand (4.2 μL of stock solution, 0.28 μmol), and 0.00125 equiv of ligand (2.1 μL of stock solution, 0.14 μmol) to prepare PMA with dispersities of 1.07, 1.16, 1.31, and 1.58.
General Procedure 2: Chain Extension of Poly(methyl acrylate) (PMA) Macroinitiator
The PMA macroinitiator was prepared in a 5 mL vial, using 2 mL of DMSO, 2 mL of MA (22.4 mmol, 100 equiv), and 2.6 μL of Me6TREN stock solution (0.00075 equiv, 0.17 μmol). EBiB (32.6 μL, 0.224 mmol, 1 equiv) was transferred into the reaction vessel via a microliter syringe. Concurrently, in a separate vial, a stirrer bar wrapped with 5 cm of copper wire was immersed in 37% HCl, stirred for 15 min, washed sequentially with water and acetone, and dried. The stirrer bar was then placed into the reaction vessel, before it was sealed with a rubber septum, and deoxygenated by bubbling with nitrogen for 15 min. The reaction mixture was stirred at 200 rpm, and the reaction proceeded at 25 °C. Once the reaction was complete, the PMA was then purified first by dilution in ethyl acetate and extraction three times with sodium bromide aqueous solution, thus removing copper salts. Magnesium sulfate was then added to the remaining reaction mixture to remove water and the mixture filtered. The organic phase was subsequently diluted with acetone and then concentrated by blowing with air. This process was repeated a total of three times, thus removing any unreacted monomer, and the final polymer was isolated by drying in a vacuum oven at 25 °C overnight. Subsequently, a chain extension reaction was performed. In a 5 mL vial, the PMA macroinitiator (153 mg, degree of polymerization (DP) = 51, 0.034 mmol, 1 equiv) was dissolved in 0.5 mL of DMSO. Me6TREN (various amounts depending on target dispersity) and MA (0.3 mL, 3.4 mmol, 100 equiv) were added. Concurrently, in a separate vial, a stirrer bar wrapped with 5 cm of copper wire was immersed in 37% HCl, stirred for 15 min, washed sequentially with water and acetone, and dried. The stirrer bar was then placed into the reaction vessel, before it was sealed with a rubber septum, and deoxygenated by bubbling with nitrogen for 15 min. The reaction mixture was stirred at 200 rpm, and the reaction proceeded at 25 °C. Samples were taken and analyzed via 1H NMR and SEC.
General Procedure 3: In Situ Chain Extension of PMA Macroinitiator
A stock solution of 4.7 μL of PMDETA was prepared in 1 mL of DMSO. In a 5 mL vial, 12.5 μL of the stock solution (0.275 μmol, 0.0025 equiv), 0.99 mL of DMSO, and MA (1 mL, 11.2 mmol, 100 equiv) were added. EBiB (16.4 μL, 0.112 mmol, 1 equiv) was transferred into the reaction vessel via a microliter syringe. Concurrently, in a separate vial, a stirrer bar wrapped with 5 cm of copper wire was immersed in 37% HCl, stirred for 15 min, washed sequentially with water and acetone, and dried. The stirrer bar was then placed into the reaction vessel, sealed with a rubber septum, and deoxygenated by bubbling with nitrogen for 15 min. The reaction mixture was stirred at 200 rpm to proceed at 25 °C. Samples were taken and analyzed via 1H NMR and SEC. Once a near quantitative conversion had been reached, 1.5 mL of DMSO, 1.5 mL of MA, and 0.13 mL of PMDETA stock solution (1.22 μL) were mixed in a separate vial. The vial was sealed with a rubber septum and deoxygenated by bubbling with nitrogen for 15 min. Two milliliters of the mixture was transferred into the vial in which polymerization was conducted via a gastight syringe. Consequently, the components of the reaction mixture were as follows: 2 mL of DMSO, PMA macroinitiator (0.112 mmol, 1 equiv), MA (1 mL, 11.2 mmol, 100 equiv), and PMDETA (0.47 μL, 2.2 μmol, 0.02 equiv). The reaction was allowed to proceed at 25 °C, with a stirring rate of 200 rpm.
Results and Discussion
We first explored the possibility of increasing polymer dispersity by reducing the concentration of ligand used in Cu(0)-RDRP. Initial experiments were performed with methyl acrylate as the monomer, ethyl-α-bromoisobutyrate (EBiB) as the initiator, dimethyl sulfoxide (DMSO) as the solvent, Cu(0) wire as the copper source, and Me6TREN as the ligand (Scheme S1). Model reaction conditions were selected with a target DP of 100, 5 cm of Cu(0) wire, and a ligand concentration 18% that of the initiator ([MA]:[EBiB]:[Me6TREN] = 100:1:0.18).65,66 As expected, after 3 h of polymerization, well-defined PMA was obtained with a very low dispersity (96% conversion, Đ = 1.06, Figure S1 and Table S1, entry 1). Kinetic analysis revealed typical features of a well-controlled polymerization with rapid initiator consumption, more than 70% conversion achieved in just 20 min and low dispersity values throughout the synthesis (Figures S2 and S3 and Table S2). We next reduced the amount of Me6TREN from 18 to 6% ([MA]:[EBiB]:[Me6TREN] = 100:1:0.06), but a similarly low dispersity was obtained (Đ = 1.05, Figure S4 and Table S1, entry 2). Even on lowering the Me6TREN concentration to 2% ([MA]:[EBiB]:[Me6TREN] = 100:1:0.02), polymerization yielded PMA with a dispersity of 1.07 (Figure 1a, Figures S5 and S6 and Table S1, entry 3). Although, under the aforementioned conditions, polymer dispersity could not be increased, it is important to highlight that low dispersity polymers can be obtained with much lower amounts of ligand than previously reported.53 This finding is particularly useful for large-scale synthesis or industrial applications as Me6TREN is a relatively expensive ligand ($167 per mL).
Figure 1.

Dispersity control of PMA synthesized with various concentrations of ligand (2, 0.5, 0.25, and 0.1% with regard to initiator) by Cu(0)-RDRP. In (a), size-exclusion chromatography illustrates monomodal SEC traces with dispersities ranging from 1.07 to 1.59. In (b), the linear inverse relationship between the ligand concentration and the dispersity is presented, along with an equation to predict the concentration of ligand required to obtain intermediate dispersities. [L]0 is the equivalent of ligand with respect to initiator at time zero.
Importantly, when we further decreased the ligand concentration by a factor of 4 (from 2 to 0.5%, [MA]/[EBiB]/[Me6TREN] = 100:1:0.005), a slightly broader molecular weight distribution was obtained (Đ = 1.16, Figure 1a and Table S1, entry 4). This encouraged us to continue decreasing the concentration of ligand to 0.25% ([MA]:[EBiB]:[Me6TREN] = 100:1:0.0025) and then to 0.125% ([MA]:[EBiB]:[Me6TREN] = 100:1:0.00125), which resulted in final values of 1.31 and 1.59, respectively (Figure 1a and Figure S7 and Table S1, entries 5–7). A further decrease in the ligand concentration, for example, to ([MA]:[EBiB]:[Me6TREN] = 100:1:0.000625), did yield an even higher dispersity PMA (Đ = 1.76), although this was accompanied by a pronounced deviation between theoretical and experimental molecular weights (Figure S7 and Table S1, entry 8). These results clearly illustrate that dispersity can be effectively tuned by simply changing the ligand concentration within a suitable range. In all cases, the molecular weight distributions obtained were monomodal with minimal tailing observed. Interestingly, we discovered a fairly linear inverse relationship between the ligand concentration and the resulting dispersity value with the equation Đ = 0.0007/[L]0 + 1.03 (for ligand equivalents between 0.02 and 0.00125 with regard to initiator, Figure 1b). This equation could be a useful tool to predict the required concentration of ligand when a specific intermediate dispersity value is targeted.
To gain an insight into why a low concentration of Me6TREN (0.125% with regard to initiator) resulted in a high dispersity polymer (Đ = 1.59), we performed detailed kinetics analysis of the polymerization (Figure 2 and Figure S8 and Table S3). The living nature of the polymerization was evidenced by a linear dependence of ln([M]0/[M]t) with time, but a significant contrast was observed in comparison to when higher ligand concentrations were employed (Figure 2c). The Mn and Đ values remained high throughout the polymerization, with slow initiation occurring as evidenced by 1H nuclear magnetic resonance spectroscopy. In particular, full initiator consumption had not occurred until more than 40% conversion had been reached (Figure 2b and Table S4). During this period, a gradual decrease in Mn was observed (Mn,2h = 6800, Mn,4h = 5800, Mn,6h = 5500, Mn,8h = 5200), which was attributed to the continuous formation of new polymer chains as a result of slow initiator consumption. Once all initiator had been consumed, existing chains then started to propagate, and hence, there was subsequently an increase in the Mn (Mn,12h = 5600 and Mn,24h = 7100, Figure 2d,e). It is noted that full initiator consumption should be achieved in order to obtain comparable DPs and similar dispersity values regardless of the targeted conversions. Together, these results suggest that the ligand concentration plays an important role in determining the rate of initiator consumption at the beginning of the polymerization. Although with our method all of the initiator is added prepolymerization, a phenomenon is observed that is similar to that in previous methods where the initiator is slowly fed in throughout the polymerization.24 Thus, polymer chains form and start to grow at different times, resulting in various chain lengths and a broad molecular weight distribution. The higher dispersity values were attributed to a lower rate of deactivation which would, in theory, result in a longer lifetime of radicals before they are capped, thus the potential for chains to grow to a wider range of lengths during each activation/deactivation cycle.67,68
Figure 2.

Kinetics for the synthesis of high dispersity PMA using a low concentration of ligand in Cu(0)-RDRP. (a) Scheme of the polymerization reaction. (b) Cascade of 1H NMR spectra for the different kinetic time points. (c) Conversion vs time plot. (d) Mn and dispersity vs conversion plot. (e) Illustrates SEC data for this polymerization.
To confirm whether slow deactivation was playing a role in increasing the dispersity, we performed UV–vis experiments to measure the concentration of the effective deactivator complex (CuBr2/Me6TREN) generated in our reactions. In these experiments, we employed Cu(0) wire, EBiB, DMSO, and the various concentrations of Me6TREN that we had previously used to tune the dispersity (i.e., 2, 0.5, 0.25, and 0.125% with regard to initiator or 1.11, 0.278, 0.139, and 0.0695 mM, respectively). It is noted that no external CuBr2 was added in these experiments, in agreement with the protocol we used for the actual polymerizations. UV–vis measurements showed that the intensity of the CuBr2/Me6TREN absorbance decreased with decreasing amounts of ligand (Figure S9). Therefore, in line with our initial hypothesis, lower ligand concentrations led to lower deactivation rates and, as a result, an increased polymer dispersity.
A key parameter associated with controlled radical polymerization is the ability to maintain high end-group fidelity, as this allows for efficient block formation, access to advanced polymeric architectures, as well as quantitative end-group modification. To assess this, low (Đ = 1.08) and high dispersity PMA (Đ = 1.53) were synthesized, with the aforementioned conditions (Figure S10 and Table S5). We then MALDI-ToF-MS of our two dispersity extremes (Figure 3a,b). In both cases, a single polymer distribution could be observed, with excellent correlation between the observed molecular weight and the expected values for PMA oligomers initiated by the expected ATRP initiator fragment and terminated with an active bromine, with each peak separated by the mass of one monomer unit. Of particular note are the isotopic splitting patterns of the respective molecular ions observed for both the low and the high dispersity polymer, which further support the high chain-end fidelity obtained through our approach (Figure S11). Thus, regardless of the targeted dispersity, excellent preservation of the active bromine could be maintained. In addition, the increase in dispersity for the second polymer could also be evidenced by the extended mass range of the distribution and an increase in the number of polymeric species (Figure 3b).
Figure 3.
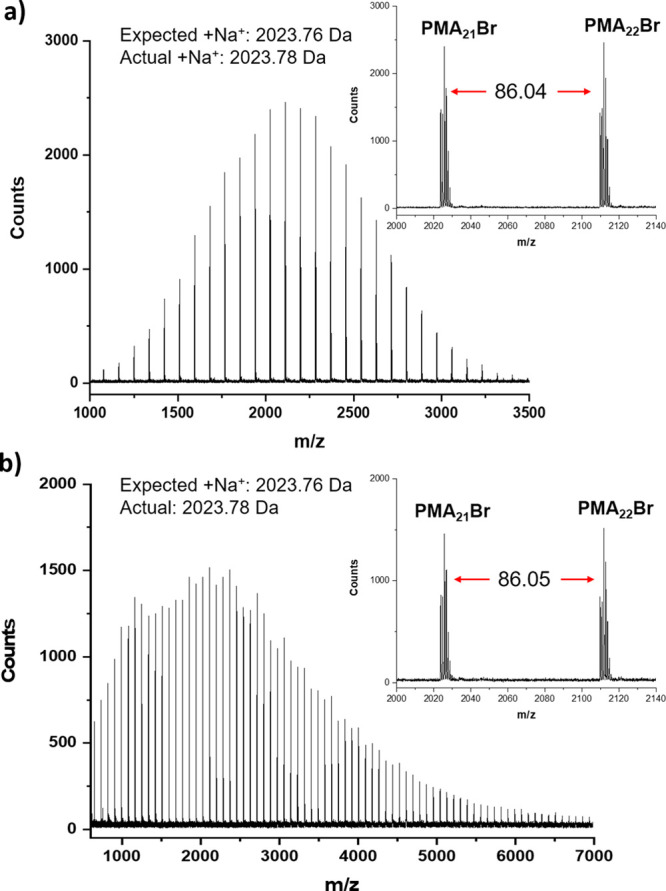
MALDI-ToF-MS spectra of (a) low dispersity (Đ = 1.08) PMA prepared with 0.02 equiv of ligand and (b) high dispersity (Đ = 1.53) PMA prepared with 0.00075 equiv of ligand.
Considering the high end-group fidelity achieved, the next step was the preparation of well-defined diblock copolymers. Low dispersity diblocks are widely reported with Cu(0)-RDRP, but blocks incorporating medium and higher dispersities are yet to be realized.59,60,69,70 We therefore prepared a high dispersity PMA macroinitiator, by selecting the lowest concentration of ligand ([MA]:[EBiB]:[CuBr2]:[Me6TREN] = 100:1:0:0.00075) and, as desired, obtained a final dispersity of 1.55 (DP51 by 1H NMR, Figure S12 and Table S6, entry 1). A chain extension experiment was then performed using a high concentration of ligand to target a low dispersity diblock ([MA]:[PMA-Br]:[Me6TREN] = 100:1:0.18). A clear shift in the molecular weight distribution to higher molecular weight and a low final dispersity were observed (ĐDB = 1.16, Figure 4a and Table S6, entry 2). By decreasing the concentration of ligand, excellent control over diblock dispersity could also be achieved, with ratios of [MA]:[PMA-Br]:[Me6TREN] equal to 100:1:0.005 and 100:1:0.0025, yielding diblocks with final dispersities of 1.31 and 1.47, respectively (Figure 4b,c and Table S6, entries 3 and 4). These results demonstrate that the dispersity of both homopolymers and diblocks can be carefully controlled by judicious selection of a suitable ligand concentration.
Figure 4.

Chain extensions of a high dispersity PMA macroinitiator. By varying the concentration of ligand, a diblock of (a) low, (b) medium, and (c) high dispersity can be obtained.
We subsequently expanded the scope of the system to prepare diblocks, where the second block was composed of either a hydrophobic or hydrophilic monomer. Butyl acrylate (BA) and poly(ethylene glycol) methyl ether acrylate (PEGA) were selected as the monomers and polymerizations were conducted with high concentrations of ligand, using a high dispersity PMA macroinitiator (Scheme S2). P(MA-b-BA) and P(MA-b-PEGA) diblock copolymers were obtained with monomodal molecular weight distributions, large shifts in the molecular weight distributions, and final dispersity values of less than 1.20 (Figure 5 and Figures S13 and S14 and Table S7). Overall, these data show that the high end-group fidelity of this polymerization system can be successfully exploited to provide diblock copolymers with both tunable dispersity and hydrophobicity.
Figure 5.
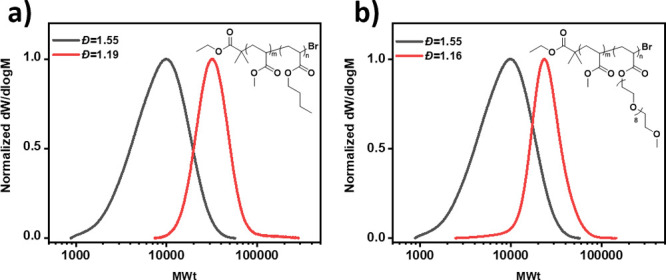
Dispersity controlled diblock copolymers of (a) P(MA-b-BA) and (b) P(MA-b-PEGA) prepared by Cu(0)-RDRP.
To further expand the potential of our method, we were interested to investigate an alternative ligand. We selected PMDETA, as it is an inexpensive, commercially available, and widely used ligand ($100 for 250 mL).65,71 We kept all other reaction conditions consistent with previous experiments and performed our first reaction with a ratio of [MA]:[EBiB]:[PMDETA] of 100:1:0.02. Similarly to Me6TREN, we also obtained PMA homopolymer with a low final dispersity of 1.18 when using this high concentration of PMDETA (Figure 6a and Table S8, entry 1). On subsequent reduction of the PMDETA concentration to 0.5 and 0.25% with respect to initiator ([MA]:[EBiB]:[PMDETA] of 100:1:0.005 and 100:1:0.0025), the molecular weight distributions could be efficiently tuned with final dispersity values of 1.40 and 1.72 (Figure 6a and Table S8, entries 2 and 3). In all cases, very high monomer conversions were observed and the initiator was fully consumed, thus resulting in good agreement between theoretical and experimental molecular weights (Figure S15 and Table S8). We performed in situ chain extensions by adding a second aliquot of monomer once high conversions had been reached (>90%). High to low and medium to low dispersity diblocks were successfully obtained (Figure 6b,c and Figures S15 and S16 and Table S9). The possibility to perform in situ chain extensions substantially simplifies our method, as the need for macroinitiator purifications can be avoided. It is noted that further decreasing the PMDETA concentration results in even higher dispersity values. For example, with 0.125% of PMDETA, a final dispersity of 2.04 can be obtained ([MA]:[EBiB]:[PMDETA] of 100:1:0.00125), but there was a significant loss of initiator efficiency and some shouldering in the molecular weight distribution (Figure S17 and Table S8, entry 4). As such, we propose a minimum concentration of 0.125% for Me6TREN and 0.25% for PMDETA to ensure monomodal molecular weight distributions, full initiator consumption, and high end-group fidelity.
Figure 6.

PMDETA as a ligand to tailor dispersity with Cu(0)-RDRP. SEC data illustrate (a) monomodal traces with dispersities ranging from 1.18 to 1.72 for PMA synthesis, (b,c) in situ chain extension data from medium and high dispersity PMA, and (d) monomodal traces with dispersities ranging from 1.16 to 1.52 for PS synthesis.
One of the great advantages of PMDETA’s compatibility with our approach is that it may allow us to facilitate the polymerization of low propagation rate constant (kp) monomers and tailor the dispersity of the resulting polymers. We selected styrene (S) as the monomer and performed polymerization with ethyl-2-bromopropionate (EBP) as the initiator and a toluene/acetonitrile mixture (9:1) as the solvent medium (Scheme S3).72 The initial reaction was performed at 60 °C, with a ratio of [S]:[EBP]:[PMDETA] of 100:1:0.36. With this high concentration of ligand, a low dispersity PS was obtained (Đ = 1.16, Figure 6d and Figure S18 and Table S10, entry 1). On systematically lowering the concentration of ligand to 6, 3.5, and 1% with respect to initiator, the dispersity of polystyrene could be accurately tuned, yielding final dispersity values of 1.24, 1.32, and 1.52 (Figure 6d and Table S10, entries 2–4). These preliminary data suggest that our developed strategy can also be applied to different monomer classes (acrylates and styrene), thus expanding the scope of the materials accessible by this approach. However, we anticipate that controlling the dispersity of methacrylate monomers would be more challenging, as lowering the ligand concentration would potentially lead to cessation of the polymerization, and as such, different reaction conditions would need to be optimized.
Conclusions
To summarize, we have successfully developed a simple Cu(0)-RDRP method that can effectively tune polymer dispersity (Đ = 1.07–1.72). The key to our approach is to change the ligand concentration, which allows the rates of initiation and deactivation to be carefully regulated, thus controlling the dispersity of the resulting polymer. High end-group fidelity is maintained during all polymerizations, which can subsequently be exploited in the preparation of a wide range of dispersity-controlled diblock copolymers. Importantly, we illustrate that the lower activity copper complex formed with PMDETA is advantageous in increasing polymerization conversions, thus allowing for in situ diblock copolymers to be realized. Furthermore, the scope of obtainable materials can be extended to incorporate block copolymers of various hydrophobicities and polystyrene. Therefore, our method significantly expands the toolbox of approaches to control dispersity, allowing facile access to polymeric materials with not only high end-group fidelity but also tailored dispersity.
Acknowledgments
A.A. gratefully thanks ETH Zürich for financial support. N.P.T. acknowledges the award of a DECRA Fellowship from the ARC (DE180100076). This project has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (DEPO: Grant Agreement No. 949219). T.S. acknowledges Mitsubishi Chemical Corporation for generous support. We also acknowledge Laurent Bigler and Urs Stadler (University of Zurich) for MALDI-ToF-MS access.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acspolymersau.1c00030.
Additional synthetic procedures, polymerization reaction schemes, 1H NMR, SEC, and UV–vis data (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. T.S. and N.P.T. contributed equally to the work.
The authors declare no competing financial interest.
Supplementary Material
References
- Johnson B. Effect of molecular weight distribution on physical properties of natural and synthetic polymers. Ind. Eng. Chem. 1948, 40, 351–356. 10.1021/ie50458a038. [DOI] [Google Scholar]
- Nunes R. W.; Martin J. R.; Johnson J. F. Influence of molecular weight and molecular weight distribution on mechanical properties of polymers. Polym. Eng. Sci. 1982, 22, 205–228. 10.1002/pen.760220402. [DOI] [Google Scholar]
- Carmean R. N.; Becker T. E.; Sims M. B.; Sumerlin B. S. Ultra-high molecular weights via aqueous reversible-deactivation radical polymerization. Chem. 2017, 2, 93–101. 10.1016/j.chempr.2016.12.007. [DOI] [Google Scholar]
- Fetters L.; Lohse D.; Richter D.; Witten T.; Zirkel A. Connection between polymer molecular weight, density, chain dimensions, and melt viscoelastic properties. Macromolecules 1994, 27, 4639–4647. 10.1021/ma00095a001. [DOI] [Google Scholar]
- Whitfield R.; Truong N. P.; Messmer D.; Parkatzidis K.; Rolland M.; Anastasaki A. Tailoring polymer dispersity and shape of molecular weight distributions: methods and applications. Chem. Sci. 2019, 10, 8724–8734. 10.1039/C9SC03546J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junkers T. Polymers in the Blender. Macromol. Chem. Phys. 2020, 221, 2000234. 10.1002/macp.202000234. [DOI] [Google Scholar]
- Gentekos D. T.; Sifri R. J.; Fors B. P. Controlling polymer properties through the shape of the molecular-weight distribution. Nat. Rev. Mater. 2019, 4, 761–774. 10.1038/s41578-019-0138-8. [DOI] [Google Scholar]
- Rosenbloom S. I.; Gentekos D. T.; Silberstein M. N.; Fors B. P. Tailor-made thermoplastic elastomers: customisable materials via modulation of molecular weight distributions. Chem. Sci. 2020, 11, 1361–1367. 10.1039/C9SC05278J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sifri R. J.; Padilla-Vélez O.; Coates G. W.; Fors B. P. Controlling the shape of molecular weight distributions in coordination polymerization and its impact on physical properties. J. Am. Chem. Soc. 2020, 142, 1443–1448. 10.1021/jacs.9b11462. [DOI] [PubMed] [Google Scholar]
- Gentekos D. T.; Jia J.; Tirado E. S.; Barteau K. P.; Smilgies D.-M.; DiStasio R. A. Jr; Fors B. P. Exploiting molecular weight distribution shape to tune domain spacing in block copolymer thin films. J. Am. Chem. Soc. 2018, 140, 4639–4648. 10.1021/jacs.8b00694. [DOI] [PubMed] [Google Scholar]
- Nadgorny M.; Gentekos D. T.; Xiao Z.; Singleton S. P.; Fors B. P.; Connal L. A. Manipulation of molecular weight distribution shape as a new strategy to control processing parameters. Macromol. Rapid Commun. 2017, 38, 1700352. 10.1002/marc.201700352. [DOI] [PubMed] [Google Scholar]
- Doncom K. E.; Blackman L. D.; Wright D. B.; Gibson M. I.; O’Reilly R. K. Dispersity effects in polymer self-assemblies: a matter of hierarchical control. Chem. Soc. Rev. 2017, 46, 4119–4134. 10.1039/C6CS00818F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynd N. A.; Meuler A. J.; Hillmyer M. A. Polydispersity and block copolymer self-assembly. Prog. Polym. Sci. 2008, 33, 875–893. 10.1016/j.progpolymsci.2008.07.003. [DOI] [Google Scholar]
- George S.; Champagne-Hartley R.; Deeter G.; Campbell D.; Reck B.; Urban D.; Cunningham M. Amphiphilic block copolymers as stabilizers in emulsion polymerization: effects of the stabilizing block molecular weight dispersity on stabilization performance. Macromolecules 2015, 48, 8913–8920. 10.1021/acs.macromol.5b01853. [DOI] [Google Scholar]
- Li T.-H.; Yadav V.; Conrad J. C.; Robertson M. L. Effect of Dispersity on the Conformation of Spherical Polymer Brushes. ACS Macro Lett. 2021, 10, 518–524. 10.1021/acsmacrolett.0c00898. [DOI] [PubMed] [Google Scholar]
- Yadav V.; Jaimes-Lizcano Y. A.; Dewangan N. K.; Park N.; Li T.-H.; Robertson M. L.; Conrad J. C. Tuning bacterial attachment and detachment via the thickness and dispersity of a pH-responsive polymer brush. ACS Appl. Mater. Interfaces 2017, 9, 44900–44910. 10.1021/acsami.7b14416. [DOI] [PubMed] [Google Scholar]
- Wang Z.; Yan J.; Liu T.; Wei Q.; Li S.; Olszewski M.; Wu J.; Sobieski J.; Fantin M.; Bockstaller M. R.; Matyjaszewski K. Control of dispersity and grafting density of particle brushes by variation of ATRP catalyst concentration. ACS Macro Lett. 2019, 8, 859–864. 10.1021/acsmacrolett.9b00405. [DOI] [PubMed] [Google Scholar]
- Matyjaszewski K.; Davis T. P.. Handbook of Radical Polymerization; John Wiley & Sons: Hoboken, NJ, 2002. [Google Scholar]
- Mahabadi H.; O’driscoll K. Termination rate constant in free-radical polymerization. J. Polym. Sci., Polym. Chem. Ed. 1977, 15, 283–300. 10.1002/pol.1977.170150203. [DOI] [Google Scholar]
- Stevens M. P.Polymer Chemistry; Oxford University Press: New York, 1990; Vol. 2. [Google Scholar]
- Hawker C. J. Molecular weight control by a″ living″ free-radical polymerization process. J. Am. Chem. Soc. 1994, 116, 11185–11186. 10.1021/ja00103a055. [DOI] [Google Scholar]
- Rosenbloom S.; Sifri R.; Fors B. Achieving Molecular Weight Distribution Shape Control and Broad Dispersities using RAFT Polymerizations. Polym. Chem. 2021, 12, 4910–4915. 10.1039/D1PY00399B. [DOI] [Google Scholar]
- Kottisch V.; Gentekos D. T.; Fors B. P. Shaping” the Future of Molecular Weight Distributions in Anionic Polymerization. ACS Macro Lett. 2016, 5, 796–800. 10.1021/acsmacrolett.6b00392. [DOI] [PubMed] [Google Scholar]
- Gentekos D. T.; Dupuis L. N.; Fors B. P. Beyond dispersity: Deterministic control of polymer molecular weight distribution. J. Am. Chem. Soc. 2016, 138, 1848–1851. 10.1021/jacs.5b13565. [DOI] [PubMed] [Google Scholar]
- Liu K.; Corrigan N.; Postma A.; Moad G.; Boyer C. A Comprehensive Platform for the Design and Synthesis of Polymer Molecular Weight Distributions. Macromolecules 2020, 53, 8867–8882. 10.1021/acs.macromol.0c01954. [DOI] [Google Scholar]
- Corrigan N.; Manahan R.; Lew Z. T.; Yeow J.; Xu J.; Boyer C. Copolymers with Controlled Molecular Weight Distributions and Compositional Gradients through Flow Polymerization. Macromolecules 2018, 51, 4553–4563. 10.1021/acs.macromol.8b00673. [DOI] [Google Scholar]
- Corrigan N.; Almasri A.; Taillades W.; Xu J.; Boyer C. Controlling molecular weight distributions through photoinduced flow polymerization. Macromolecules 2017, 50, 8438–8448. 10.1021/acs.macromol.7b01890. [DOI] [Google Scholar]
- Junkers T.; Vrijsen J. H. Designing molecular weight distributions of arbitrary shape with selectable average molecular weight and dispersity. Eur. Polym. J. 2020, 134, 109834. 10.1016/j.eurpolymj.2020.109834. [DOI] [Google Scholar]
- Rubens M.; Junkers T. Comprehensive control over molecular weight distributions through automated polymerizations. Polym. Chem. 2019, 10, 6315–6323. 10.1039/C9PY01013K. [DOI] [Google Scholar]
- Rubens M.; Junkers T. A predictive framework for mixing low dispersity polymer samples to design custom molecular weight distributions. Polym. Chem. 2019, 10, 5721–5725. 10.1039/C9PY01012B. [DOI] [Google Scholar]
- Morsbach J.; Müller A. H.; Berger-Nicoletti E.; Frey H. Living polymer chains with predictable molecular weight and dispersity via carbanionic polymerization in continuous flow: Mixing rate as a key parameter. Macromolecules 2016, 49, 5043–5050. 10.1021/acs.macromol.6b00975. [DOI] [Google Scholar]
- Reis M. H.; Varner T. P.; Leibfarth F. A. The Influence of Residence Time Distribution on Continuous-Flow Polymerization. Macromolecules 2019, 52, 3551–3557. 10.1021/acs.macromol.9b00454. [DOI] [Google Scholar]
- Walsh D. J.; Schinski D. A.; Schneider R. A.; Guironnet D. General route to design polymer molecular weight distributions through flow chemistry. Nat. Commun. 2020, 11, 3094. 10.1038/s41467-020-16874-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia R.; Tu Y.; Glauber M.; Huang Z.; Xuan S.; Zhang W.; Zhou N.; Li X.; Zhang Z.; Zhu X. Fine control of the molecular weight and polymer dispersity via a latent monomeric retarder. Polym. Chem. 2021, 12, 349–355. 10.1039/D0PY01569E. [DOI] [Google Scholar]
- Liu H.; Xue Y.-H.; Zhu Y.-L.; Gu F.-L.; Lu Z.-Y. Inverse Design of Molecular Weight Distribution in Controlled Polymerization via a One-Pot Reaction Strategy. Macromolecules 2020, 53, 6409–6419. 10.1021/acs.macromol.0c01383. [DOI] [Google Scholar]
- Vrijsen J. H.; Rubens M.; Junkers T. Simple and secure data encryption via molecular weight distribution fingerprints. Polym. Chem. 2020, 11, 6463–6470. 10.1039/D0PY01071E. [DOI] [Google Scholar]
- Liu D.; Sponza A. D.; Yang D.; Chiu M. Modulating Polymer Dispersity with Light: Cationic Polymerization of Vinyl Ethers Using Photochromic Initiators. Angew. Chem., Int. Ed. 2019, 58, 16210–16216. 10.1002/anie.201908775. [DOI] [PubMed] [Google Scholar]
- Whitfield R.; Parkatzidis K.; Truong N. P.; Junkers T.; Anastasaki A. Tailoring Polymer Dispersity by RAFT Polymerization: A Versatile Approach. Chem. 2020, 6, 1340–1352. 10.1016/j.chempr.2020.04.020. [DOI] [Google Scholar]
- Whitfield R.; Parkatzidis K.; Rolland M.; Truong N. P.; Anastasaki A. Tuning Dispersity by Photoinduced Atom Transfer Radical Polymerisation: Monomodal Distributions with ppm Copper Concentration. Angew. Chem., Int. Ed. 2019, 58, 13323–13328. 10.1002/anie.201906471. [DOI] [PubMed] [Google Scholar]
- Wang C.-G.; Chong A. M. L.; Goto A. One Reagent with Two Functions: Simultaneous Living Radical Polymerization and Chain-End Substitution for Tailoring Polymer Dispersity. ACS Macro Lett. 2021, 10, 584–590. 10.1021/acsmacrolett.1c00179. [DOI] [PubMed] [Google Scholar]
- Wallace M. A.; Sita L. R. Temporal Control over Two-and Three-State Living Coordinative Chain Transfer Polymerization for Modulating the Molecular Weight Distribution Profile of Polyolefins. Angew. Chem. 2021, 133, 19823. 10.1002/ange.202105937. [DOI] [PubMed] [Google Scholar]
- Wang H. S.; Parkatzidis K.; Harrisson S.; Truong Phuoc N.; Anastasaki A. Controlling Dispersity in Aqueous Atom Transfer Radical Polymerization: Rapid and Quantitative Synthesis of One-Pot Block Copolymers. Chem. Sci. 2021, 10.1039/D1SC04241F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkatzidis K.; Rolland M.; Truong N. P.; Anastasaki A. Tailoring polymer dispersity by mixing ATRP initiators. Polym. Chem. 2021, 12, 5583. 10.1039/D1PY01044A. [DOI] [Google Scholar]
- Plichta A.; Zhong M.; Li W.; Elsen A. M.; Matyjaszewski K. Tuning dispersity in diblock copolymers using ARGET ATRP. Macromol. Chem. Phys. 2012, 213, 2659–2668. 10.1002/macp.201200461. [DOI] [Google Scholar]
- Rolland M.; Lohmann V.; Whitfield R.; Truong N. P.; Anastasaki A. Understanding dispersity control in photo-atom transfer radical polymerization: Effect of degree of polymerization and kinetic evaluation. J. Polym. Sci. 2021, 10.1002/pol.20210319. [DOI] [Google Scholar]
- Rolland M.; Truong N. P.; Whitfield R.; Anastasaki A. Tailoring Polymer Dispersity in Photoinduced Iron-Catalyzed ATRP. ACS Macro Lett. 2020, 9, 459–463. 10.1021/acsmacrolett.0c00121. [DOI] [PubMed] [Google Scholar]
- Parkatzidis K.; Truong N. P.; Antonopoulou M. N.; Whitfield R.; Konkolewicz D.; Anastasaki A. Tailoring polymer dispersity by mixing chain transfer agents in PET-RAFT polymerization. Polym. Chem. 2020, 11, 4968–4972. 10.1039/D0PY00823K. [DOI] [Google Scholar]
- Yadav V.; Hashmi N.; Ding W.; Li T.-H.; Mahanthappa M. K.; Conrad J. C.; Robertson M. L. Dispersity control in atom transfer radical polymerizations through addition of phenylhydrazine. Polym. Chem. 2018, 9, 4332–4342. 10.1039/C8PY00033F. [DOI] [Google Scholar]
- Liu X.; Wang C. G.; Goto A. Polymer Dispersity Control by Organocatalyzed Living Radical Polymerization. Angew. Chem. 2019, 131, 5654–5659. 10.1002/ange.201814573. [DOI] [PubMed] [Google Scholar]
- Lynd N. A.; Hillmyer M. A. Influence of polydispersity on the self-assembly of diblock copolymers. Macromolecules 2005, 38, 8803–8810. 10.1021/ma051025r. [DOI] [Google Scholar]
- Whitfield R.; Truong N.; Anastasaki A. Precise Control of Both Dispersity and Molecular Weight Distribution Shape by Polymer Blending. Angew. Chem., Int. Ed. 2021, 60, 19383–19388. 10.1002/anie.202106729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anastasaki A.; Nikolaou V.; Nurumbetov G.; Wilson P.; Kempe K.; Quinn J. F.; Davis T. P.; Whittaker M. R.; Haddleton D. M. Cu (0)-mediated living radical polymerization: a versatile tool for materials synthesis. Chem. Rev. 2016, 116, 835–877. 10.1021/acs.chemrev.5b00191. [DOI] [PubMed] [Google Scholar]
- Anastasaki A.; Nikolaou V.; Haddleton D. M. Cu (0)-mediated living radical polymerization: recent highlights and applications; a perspective. Polym. Chem. 2016, 7, 1002–1026. 10.1039/C5PY01916H. [DOI] [Google Scholar]
- Peng C.-H.; Zhong M.; Wang Y.; Kwak Y.; Zhang Y.; Zhu W.; Tonge M.; Buback J.; Park S.; Krys P.; Konkolewicz D.; Gennaro A.; Matyjaszewski K. Reversible-deactivation radical polymerization in the presence of metallic copper. Activation of alkyl halides by Cu0. Macromolecules 2013, 46, 3803–3815. 10.1021/ma400150a. [DOI] [Google Scholar]
- Lligadas G.; Percec V. Ultrafast SET-LRP of methyl acrylate at 25° C in alcohols. J. Polym. Sci., Part A: Polym. Chem. 2008, 46, 2745–2754. 10.1002/pola.22607. [DOI] [Google Scholar]
- Lligadas G.; Grama S.; Percec V. Recent developments in the synthesis of biomacromolecules and their conjugates by single electron transfer–living radical polymerization. Biomacromolecules 2017, 18, 1039–1063. 10.1021/acs.biomac.7b00197. [DOI] [PubMed] [Google Scholar]
- Krys P.; Wang Y.; Matyjaszewski K.; Harrisson S. Radical generation and termination in SARA ATRP of methyl acrylate: effect of solvent, ligand, and chain length. Macromolecules 2016, 49, 2977–2984. 10.1021/acs.macromol.6b00345. [DOI] [Google Scholar]
- Feng X.; Maurya D. S.; Bensabeh N.; Moreno A.; Oh T.; Luo Y.; Lejnieks J.; Galia M.; Miura Y.; Monteiro M. J.; Lligadas G.; Percec V. Replacing Cu (II) Br2 with Me6-TREN in Biphasic Cu (0)/TREN Catalyzed SET-LRP Reveals the Mixed-Ligand Effect. Biomacromolecules 2020, 21, 250–261. 10.1021/acs.biomac.9b01282. [DOI] [PubMed] [Google Scholar]
- Boyer C.; Derveaux A.; Zetterlund P. B.; Whittaker M. R. Synthesis of multi-block copolymer stars using a simple iterative Cu (0)-mediated radical polymerization technique. Polym. Chem. 2012, 3, 117–123. 10.1039/C1PY00384D. [DOI] [Google Scholar]
- Soeriyadi A. H.; Boyer C.; Nyström F.; Zetterlund P. B.; Whittaker M. R. High-order multiblock copolymers via iterative Cu (0)-mediated radical polymerizations (SET-LRP): toward biological precision. J. Am. Chem. Soc. 2011, 133, 11128–11131. 10.1021/ja205080u. [DOI] [PubMed] [Google Scholar]
- Hatano T.; Rosen B. M.; Percec V. SET-LRP of vinyl chloride initiated with CHBr3 and catalyzed by Cu (0)-wire/TREN in DMSO at 25° C. J. Polym. Sci., Part A: Polym. Chem. 2010, 48, 164–172. 10.1002/pola.23774. [DOI] [Google Scholar]
- Nguyen N. H.; Rosen B. M.; Lligadas G.; Percec V. Surface-dependent kinetics of Cu (0)-wire-catalyzed single-electron transfer living radical polymerization of methyl acrylate in DMSO at 25 C. Macromolecules 2009, 42, 2379–2386. 10.1021/ma8028562. [DOI] [Google Scholar]
- Burns J. A.; Houben C.; Anastasaki A.; Waldron C.; Lapkin A. A.; Haddleton D. M. Poly (acrylates) via SET-LRP in a continuous tubular reactor. Polym. Chem. 2013, 4, 4809–4813. 10.1039/c3py00833a. [DOI] [Google Scholar]
- Ciampolini M.; Nardi N. Five-coordinated high-spin complexes of bivalent cobalt, nickel, and copper with tris (2-dimethylaminoethyl) amine. Inorg. Chem. 1966, 5, 41–44. 10.1021/ic50035a010. [DOI] [Google Scholar]
- Whitfield R.; Anastasaki A.; Nikolaou V.; Jones G. R.; Engelis N. G.; Discekici E. H.; Fleischmann C.; Willenbacher J.; Hawker C. J.; Haddleton D. M. Universal conditions for the controlled polymerization of acrylates, methacrylates, and styrene via Cu (0)-RDRP. J. Am. Chem. Soc. 2017, 139, 1003–1010. 10.1021/jacs.6b11783. [DOI] [PubMed] [Google Scholar]
- Anastasaki A.; Waldron C.; Wilson P.; McHale R.; Haddleton D. M. The importance of ligand reactions in Cu (0)-mediated living radical polymerisation of acrylates. Polym. Chem. 2013, 4, 2672–2675. 10.1039/c3py00270e. [DOI] [Google Scholar]
- Zapata-González I.; Hutchinson R. A.; Payne K. A.; Saldívar-Guerra E. Mathematical modeling of the full molecular weight distribution in ATRP techniques. AIChE J. 2016, 62, 2762–2777. 10.1002/aic.15232. [DOI] [Google Scholar]
- Matyjaszewski K.; Jakubowski W.; Min K.; Tang W.; Huang J.; Braunecker W. A.; Tsarevsky N. V. Diminishing catalyst concentration in atom transfer radical polymerization with reducing agents. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 15309–15314. 10.1073/pnas.0602675103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones G. R.; Whitfield R.; Anastasaki A.; Risangud N.; Simula A.; Keddie D. J.; Haddleton D. M. Cu (0)-RDRP of methacrylates in DMSO: importance of the initiator. Polym. Chem. 2018, 9, 2382–2388. 10.1039/C7PY01196B. [DOI] [Google Scholar]
- Percec V.; Guliashvili T.; Popov A. V.; Ramirez-Castillo E.; Coelho J. F.; Hinojosa-Falcon L. Accelerated synthesis of poly (methyl methacrylate)-b-poly (vinyl chloride)-b-poly (methyl methacrylate) block copolymers by the CuCl/tris (2-dimethylaminoethyl) amine-catalyzed living radical block copolymerization of methyl methacrylate initiated with α, ω-di (iodo) poly (vinyl chloride) in dimethyl sulfoxide at 90° C. J. Polym. Sci., Part A: Polym. Chem. 2005, 43, 1649–1659. 10.1002/pola.20616. [DOI] [Google Scholar]
- Tang W.; Kwak Y.; Braunecker W.; Tsarevsky N. V.; Coote M. L.; Matyjaszewski K. Understanding atom transfer radical polymerization: effect of ligand and initiator structures on the equilibrium constants. J. Am. Chem. Soc. 2008, 130, 10702–10713. 10.1021/ja802290a. [DOI] [PubMed] [Google Scholar]
- Whitfield R.; Anastasaki A.; Jones G. R.; Haddleton D. M. Cu (0)-RDRP of styrene: balancing initiator efficiency and dispersity. Polym. Chem. 2018, 9, 4395–4403. 10.1039/C8PY00814K. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.