

Abstract
The pervasion of three daily meals and snacks is a relatively new introduction to our shared experience, and is coincident with an epidemic rise in obesity and cardiometabolic disorders of overnutrition. The past two decades have yielded convincing evidence regarding the adaptive, protective effects of calorie restriction (CR) and intermittent fasting (IF) against cardiometabolic, neurodegenerative, proteostatic and inflammatory diseases. Yet, durable adherence to intensive lifestyle changes is rarely attainable. New evidence now demonstrates that restricting carbohydrate entry into the hepatocyte by itself mimics several key signaling responses and physiological outcomes of IF and CR. This discovery raises the intriguing proposition that targeting hepatocyte carbohydrate transport to mimic fasting and caloric restriction can abate cardiometabolic and perhaps other fasting-treatable diseases. Here, we review the metabolic and signaling fates of a hepatocyte carbohydrate, identify evidence to target the key mediators within these pathways, and provide rationale and data to highlight carbohydrate transport as a broad, proximal intervention to block the deleterious sequelae of hepatic glucose and fructose metabolism.
Keywords: Glucokinase, GLUT, ChREBP, Fructose, Obesity, non-alcoholic fatty liver disease, non-alcoholic steatohepatitis, intermittent fasting, caloric restriction, insulin resistance, diabetes, ketogenic diet, cardiovascular disease, heart failure, low-fat diet, glycemic index, trehalose, lactotrehalose, metabolism, NAFLD, mitochondrial pyruvate carrier, sirtuin, FOXO, nicotinamide adenine dinucleotide, fibroblast growth factor 21
Graphical Abstract
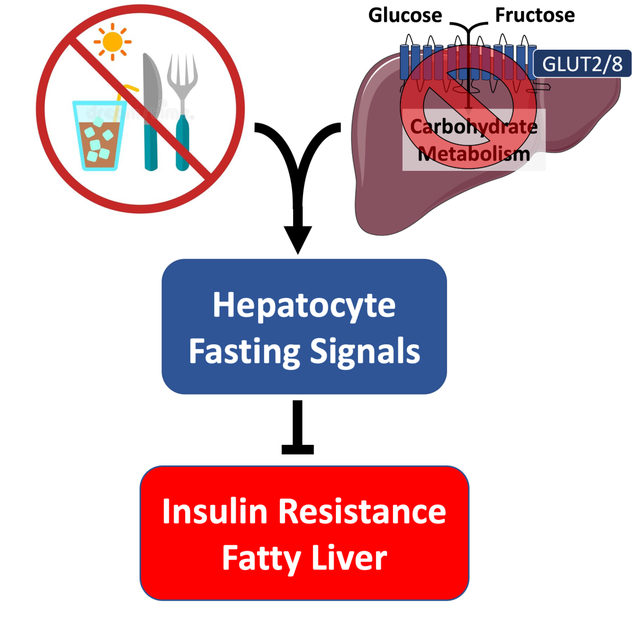
INTRODUCTION
Long before three daily meals and interceding snacks became part of modern industrialized life, the Ancient Romans viewed consumption of more than one meal daily to be a form of gluttony, and monastic practices of the Middle Ages forbade food consumption prior to morning Mass (Denise Winterman, BBC News Magazine, 2012). The act of “breaking fast”, only became a widespread, shared practice across social classes during the Industrial Revolution, initially as a means to sustain day laborers through long workdays in the mid-1800s.
In contrast with the practice of three meals, caloric restriction and intermittent fasting (CR and IF), have long been known to exert therapeutic effects on healthspan and lifespan in animals from worms to humans (1). In real-world clinical contexts, however, implementing and sustaining such lifestyle changes has proven to be difficult. As a result, we have yet to fully leverage this biology to mitigate a range of fasting-responsive human diseases. Recent provocative data now demonstrate that restricting carbohydrate entry into the hepatocyte recapitulates several key metabolic effects of CR and IF (2–14). This raises the possibility that fasting mimetic therapies that target hepatocyte carbohydrate metabolism may be viable treatments against metabolic disease, including non-alcoholic fatty liver disease, and type 2 diabetes mellitus (15). The purpose of this review is to briefly examine the metabolic fate of a carbohydrate in the hepatocyte, define downstream hepatocyte carbohydrate signaling, and then finally focus on promising nodes within this axis to leverage against metabolic disease.
Clinical utility of CR, IF, and reduced dietary carbohydrate
IF and CR exert manifold protective metabolic effects in rodents and in humans (16, 17). This includes broadly adaptive effects on hepatic steatosis and inflammation, insulin resistance, and cardiovascular disease (1, 16, 18–29). These effects occurred independently of whether the intervention consisted of chronic daily caloric restriction, or of a fast:fed intermittent fasting of 5:2 days, 16:8hr, or 18:6hr. Furthermore, IF exerts several key effects, even when controlled for caloric intake, and even in the absence of weight loss. For example, 18h intermittent fasting for five weeks reduce insulin resistance, blood pressure, oxidative stress and appetite without lowering weight in pre-diabetic men, when compared with men consuming an isocaloric diet instead over a 12h timespan (23, 29). Together, these observations suggest that IF and CR in humans improve cardiometabolic function.
In parallel, a significant body of data interrogating specific macronutrient withdrawal has come to light. Whereas low-fat foods surged in popularity over the preceding decades amidst an unprecedented rise in obesity in the United States, recent studies have examined the metabolic effects of dietary carbohydrate restriction. Carbohydrate-selective restriction overall appears to improve circulating lipids, glucose and insulin tolerance with equal or greater efficacy as compared with calorie restriction and low-fat intervention. This seems to hold true in varied populations. For example a randomized trial of ketogenic diet (KD) in both obese men and women demonstrated that the low carbohydrate, high-fat, high-protein diet was equally efficacious when compared to a low calorie, low-fat, high-carbohydrate diet with regard to lowering diastolic BP and improving glucose tolerance. In addition, the low-carbohydrate diet produced lower initial weight loss, and increased HDL and lowered TG in ketogenic diet-treated subjects when compared with those on a low-calorie, low-fat diet (30). In 132 severely obese subjects with a high prevalence of diabetes or metabolic syndrome, patients on a carbohydrate-restricted diet lost more weight, and exhibited improved insulin sensitivity and triglycerides when compared with subjects on a low-fat diet (31). In overweight, dyslipidemic subjects, KD was more effective than a calorie-restricted, low-fat diet at reducing endogenous lipogenesis, insulin resistance, body weight, adiposity, and dyslipidemia (32). In obese females without co-morbidities, a very low carbohydrate diet was more efficacious in reducing body weight, raising HDL and ketones without adversely affecting other cardiometabolic risk factors when compared with calorie-restricted low-fat diet (33). Similarly, when compared with calorie restriction and Mediterranean diet interventions, low-carbohydrate diets produced the most weight loss, and had the most favorable effects on lipid profiles in moderately obese human subjects over a 2-year period (34). Moreover, in children with NAFLD, low-glycemic index foods reduced systolic blood pressure, plasma ALT, and insulin resistance indices (HOMA-IR) after 3- and 6-months treatment (35). In mice, ketogenic diets reduce circulating lipids, intrahepatic lipids, and insulin resistance markers (36), and improve cardiac function during heart failure (37). In balance of these findings, low-carbohydrate diets in some contexts do not outperform other forms of caloric restriction (38). Nevertheless, data from human and mouse models indicate the efficacy of carbohydrate-specific withdrawal, and substantiate further investigation into how carbohydrates regulate metabolic function, and the extent to which targeting carbohydrate metabolism is a therapeutically viable option.
The fate of a carbohydrate in the hepatocyte
The liver sits at the nexus of portal and venous circulations. In this position, the hepatocyte negotiates an organism’s present and immediate future systemic energetic status. In light of promising data to suggest that dietary carbohydrate is an important determinant of cardiometabolic risk, significant efforts have focused on carbohydrate metabolism in the liver, and its subsequent host effects.
Carbohydrate entry into the hepatocyte occurs via at least two primary facilitative transporters in the glucose transporter (GLUT) family of solute carrier proteins (Figure 1) (39, 40). These are GLUT2 (encoded by the Slc2a2 gene) and GLUT8 (encoded by the Slc2a8 gene), each of which transports glucose and fructose, among other carbohydrates (39, 40). GLUT2 is a high-capacity, low-affinity glucose, fructose, and galactose transporter (39). Its rapid transport kinetics are tuned such that its substrate concentrations quickly equilibrate with the extracellular fed, fasting, or diabetic milieu. GLUT2 is the most highly expressed liver GLUT, and accordingly, its activity comprises the majority of carbohydrate flux into the hepatocyte (39). In contrast GLUT8 is a high-affinity, low-capacity facilitative carrier of glucose, fructose, galactose, and possibly trehalose (39, 41–45). GLUT8 mediates only about 20–25% of hepatocyte glucose transport (44), and it differs from GLUT2 most prominently in that it localizes both to plasma membrane and intracellular organellar membranes (43, 44, 46–48), although its intracellular functions are not fully understood. Together, these two transporters mediate the preponderance of carbohydrate entry into the hepatocyte cytoplasm.
Figure 1.
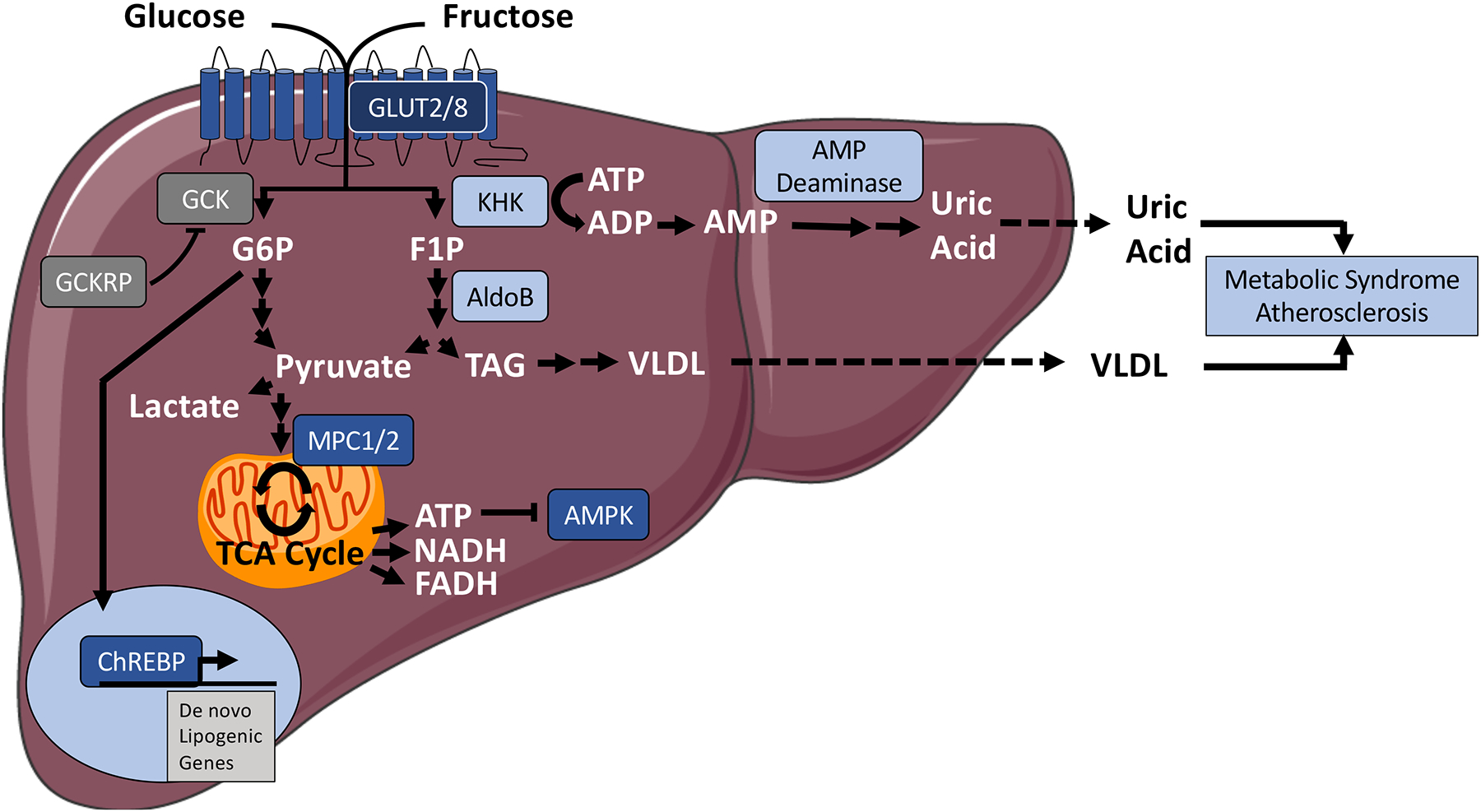
Post-prandial glucose and fructose fates inside the hepatocyte. In the fed state, glucose and fructose are transported into the hepatocyte via the glucose transporter, and then catabolized to glucose-6-phosphate or fructose-1-phosphate, by glucokinase and ketohexokinase respectively. Left, Glucose-6-phosphate is catabolized via glycolysis to pyruvate. Glucose-6-phosphate also induces de novo lipogenic gene transcription by activating the transcription factor, Carbohydrate response element binding protein. Glucokinase regulatory protein regulates glucose catabolism overall by suppressing glucokinase activity. Right, The ketohexokinase reaction generates fructose-1-phosphate, from which aldolase B initiates fructolytic conversion toward pyruvate, triacylglycerol, and ultimately VLDL synthesis. Pyruvate from glycolysis and fructolysis is catabolized to lactate, or it is transported via the mitochondrial pyruvate carrier to undergo oxidative metabolism in the mitochondrion. ATP production inhibits a key fasting regulator kinase, AMP-activated protein kinase. The ketohexokinase reaction also generates ADP, which is further catabolized to AMP. This provides substrate, which is shunted toward uric acid production via AMP deaminase. Dark grey enzymes are selective to the glucose-metabolic pathway. Light blue-colored enzymes represent fructose-selective catabolic pathways. Dark blue-colored enzymatic pathways represent common points of intervention in carbohydrate metabolism. Abbreviations: AldoB, aldolase B; AMPK, AMP-activated protein kinase; ChREBP, carbohydrate response element binding protein; CR, caloric restriction; FOXO, forkhead box transcriptiion factor O; G6P, glucose-6-phosphate; F1P, fructose-1-phosphate; GCK, glucokinase; GCKRP, glucokinase regulatory protein; GLUT, glucose transporter; KHK, ketohexokinase; MPC, mitochondrial pyruvate carrier; PPAR, peroxisome proliferator antigen receptor; TAG, triacylglycerol; TCA cycle, tricarboxylic acid cycle; VLDL, very low density lipoprotein.
Glucokinase (GCK) and glucokinase regulatory protein (GCKRP) represent the first regulatory step following facilitative diffusion into the hepatocyte (49). GCK (hexokinase IV) phosphorylates glucose to glucose-6-phosphate (G6P), which attenuates glucose excursion to the cell exterior. Indeed, this is a highly regulated step, mediated in large part by GCK regulatory peptide (GCKRP). This enzyme binds and inhibits GCK to attenuate GCK-mediated glucose phosphorylation and subsequent hepatocyte glycolytic metabolism (50, 51). This regulation is particularly critical, because glucose conversion to G6P also might be a rate-limiting step for glycolysis in hepatocytes (49).
Glucose-6-phosphate subsequently has both signaling and catabolic functions (Figure 1) within the hepatocyte (52). In the post-prandial hepatocyte, G6P undergoes glycolysis, and is converted to pyruvate to generate fatty acids via de novo lipogenesis, or it is converted to UDP-glucose, and stored as glycogen (53). First, following G6P conversion to pyruvate through glycolysis, pyruvate is either converted to lactate by lactate dehydrogenase, or transported into the mitochondrial matrix via the mitochondrial pyruvate carrier (MPC) (54). After entry into the mitochondrion, mitochondrial pyruvate metabolism is important for de novo lipogenesis. Pyruvate can be carboxylated to produce oxaloacetate (anaplerotic metabolism) or oxidized to acetyl-CoA (catabolic metabolism), and both play a role in de novo lipogenesis (54–56). Moreover, pyruvate carboxylation is important for producing new glucose via gluconeogenesis and cholesterol synthesis (54, 56, 57). Second, from a metabolic signaling standpoint, ATP generated by G6P catabolism suppresses AMPK activity, to contribute to hepatic steatosis and obesity (7, 8, 58). In addition, G6P and potentially other metabolites activate a key carbohydrate sensor in hepatocytes, the carbohydrate response element binding protein (ChREBP) (59). Activated ChREBP transcriptionally activates genes of de novo lipogenesis (Figure 1) (60).
Although glucose and fructose both ultimately activate ChREBP, fructose and glucose entering the hepatocyte take initially divergent metabolic pathways. These key differences in initial fructose and catabolism are considered to mediate their distinct physiological sequelae (5, 6, 61–63). Whereas GCK phosphorylates free glucose to G6P, ketohexokinase (KHK) phosphorylates free fructose to fructose-1-phosphate (F1P) upon entry into the hepatocyte. Aldolase B catabolizes F1P to generate dihydroxyacetone phosphate (DHAP) and (after the triose kinase or triose phosphate isomerase reactions) glyceraldehyde-3-phosphate (GA3P). GA3P is the common metabolite in glycolysis at which glucose and fructose metabolism converge.
At least two additional points of divergence exist when comparing fructose metabolism with glucose metabolism. First, KHK is an ATP-dependent enzyme, and AMP generated in the KHK reaction provides substrate for AMP-deaminase, a committed enzymatic step in uric acid synthesis (61). Both decreased intracellular [phosphate] and F1P allosterically activate AMP deaminase to exacerbate uric acid generation, which predisposes the host to cardiometabolic syndrome unless cleared by the kidney or intestine (64–67). A second divergence between fructose and glucose metabolism is that carbon from F1P is used to produce the triacylglycerol backbone, glycerol-3-phosphate (G3P). This occurs via concerted catalysis of aldolase B and glyceraldehyde phosphate dehydrogenase (GAPDH). TAGs are exported systemically in the form of VLDL, which by itself portends cardiometabolic risk (68). Overall, the divergence between fructose and glucose metabolism may explain added risk in response to intracellular fructose (6).
Carbohydrate-Induced Pathways as Targets for Metabolic Intervention
Our current understanding of hepatocyte carbohydrate metabolism and signaling gives us glimpses into therapeutic targets against metabolic disease (Table 1). We have alluded to the hepatic and extrahepatic metabolic effects of hepatocyte GLUT deletion (11, 44, 69, 70), and will discuss this specific therapeutic approach in greater detail in a subsequent section.
TABLE 1.
Summary of outcomes upon targeting glucose intermediary metabolism. Outcomes described (if reported in the cited study) are: glucose homeostasis, lipid homeostasis, and other important readouts.
| Study Ref. | Target | Targeting Method | Disease Model | Outcome Measures (targeted relative to untargeted control) |
|---|---|---|---|---|
| 69 | GLUT2 | Liver-specific KO | Fast (24h)-refeed versus fast alone |
|
| 7 | GLUTs (non-specific) | Trehalose | 60% fructose diet (10 days) |
|
| 9 | GLUTs (non-specific) | Lactotrehalose | 60% fructose diet (10 days) |
|
| 11 | GLUT8 | GLUT8 Antisense oligonucleotide | Ad libitum fed vs. 12–24h fasting |
|
| 11 | GLUT8 | Whole-body KO | Ad libitum fed vs. 12–24h fasting |
|
| 44 | GLUT8 | Whole-body KO | 60% fructose diet (24wk) |
|
| 70 | GLUT8 | Whole-body KO | 60% fructose diet (24wk) |
|
| 72 | GCK | Hepatocyte-specific overexpression | Chow vs. 42% High-fat diet (12wk) |
|
| 74 | GCK | Hepatocyte-specific KO | No perturbation |
|
| 73 | GCK | Hepatocyte-specific haploinsufficiency |
|
|
| 79 | GCKR-GCK interaction | Small molecule inhibitors: AMG-1694 AMG-3969 | ob/ob, db/db, or 60% fat diet (12wk) |
|
| 6 | KHK | Liver-specific KHK siRNA | 60% high-fat diet + 30% glucose water or fructose water (10wk) |
|
| 80 | KHK-A/C KHK-A |
Whole-body KHK deletion Whole-body KHK-A deletion |
AldoB KO ± Fructose water AldoB KO ± Fructose water |
|
| 81 | KHK-A/C KHK-A |
Whole-body KHK-A/C deletion Whole-body KHK-A deletion |
15–30% fructose water (25wk) |
|
| 86 | MPC1 | Acute AAV8-Cre-mediated MPC1 deletion |
|
|
| 87 | MPC1/2 | MSDC-0602 | ob/ob or 60% high-fat diet (10–12wk) |
|
| 90 | MPC1/2 | MSDC-0602 | HTF-C (16wk) |
|
| 90 | MPC2 | MPC2 LKO | HTF-C (16wk) |
|
Glucokinase may be an attractive target, because it is a proximal enzyme that links glucose influx with glycolysis and downstream metabolism, such as glycogen synthesis and lipogenesis. This is highlighted by data that pharmacological GCK activation as a method to treat type 2 diabetes mellitus produces hypoglycemia, hyperlipidemia and hepatic steatosis (71). Genetic GCK overexpression similarly increased hepatic triglyceride deposition, and induced hyperglycemia, hyperinsulinemia, insulin resistance and glucose intolerance in mice (72). Conversely, liver-specific GCK-deficient mice have decreased hepatic glycogen content (73, 74), and reduced de novo lipogenic gene expression in liver, including pyruvate kinase and fatty acid synthase (73). However, GCK heterozygous mice develop fasting hyperglycemia (73) and liver-specific GCK-knockout mice also exhibited impaired glucose tolerance. Thus, targeting GCK to treat NAFLD may not be ideal, since many patients with NAFLD are also insulin resistant and/or diabetic, and inhibiting GCK has the potential to exacerbate these conditions.
GCKR encodes the GCK regulatory protein, GCKRP. This protein inhibits glucose trapping and metabolism by binding and inhibiting GCK. Therefore it is unsurprising that the GCKR locus is associated with NAFLD in humans (68, 75, 76). More specifically, polymorphisms that prevent GCKRP-GCK binding (e.g. P466L) result in lower plasma glucose, and increased hepatic steatosis, circulating triglycerides and cholesterol in human subjects (68, 77, 78). Similarly, pharmacologically disrupting the GCKRP and GCK interaction lowered circulating glucose and increased respiratory exchange ratio (RER) in Zucker diabetic rats. The data indicate a switch from fat oxidation to a glucose oxidative predilection (79), although direct measurements of circulating or intrahepatic lipid contents are not reported. Overall, the data point to an important glucose homeostatic function for the interaction between GCK and GCKRP. However, understanding how to optimally target this segment of hepatocyte glucose metabolic pathway is required, due to the delicate, inverse relationship between GCK activity and hepatic steatosis.
In regard to fructose metabolism, recent promising pre-clinical and clinical demonstrations suggest that proximal fructose metabolism blockade improves hepatic and peripheral energy metabolism. KHK regulates the first committed step in fructose metabolism upon entry into the hepatocyte cytoplasm. There are two major KHK isoforms, a minor A-isoform, and a major C isoform. KHK-A and KHK-C deletion together, germline whole-body KHK-C deletion, and liver-specific KHK-C knockdown each block fructose-induced metabolic dysfunction in multiple models (6, 80, 81). These data suggest that targeting dual KHK-A/C isoforms, or single-isoform KHK-C alone is a viable therapeutic strategy against metabolic disease. This is underscored by recent phase 2 clinical trial data, which demonstrate that pharmacologic KHK inhibition reduces hepatic steatosis, as measured by magnetic resonance imaging proton density fat fraction in patients with NAFLD (82). In contrast, patients with aldolase B deficiency have increased intrahepatic lipid content (83), and genetic aldolase B inhibition in mice phenocopies hereditary fructose intolerance in humans. Data by Lanaspa and colleagues recently implicated the product of KHK enzymatic action, fructose-1-phosphate (F1P), as a potential mechanistic link that explains differential outcomes due to aldolase B and KHK targeting (80). They showed that aldolase B deletion exacerbated fructose-induced hepatic TG and uric acid accumulation. In contrast deleting KHK-C reversed these detrimental effects of aldolase B on fructose-induced metabolism, indicating that F1P availability in hepatocytes may be deleterious (80). Thus, aldolase B targeting has not been a major focus for clinical development against metabolic disease (84, 85).
Anaerobic metabolism of both glucose and fructose generates pyruvate, which can enter the mitochondrion via a carrier-mediated mechanism. The mitochondrial pyruvate carrier (MPC) is composed of two proteins, MPC1 and MPC2, which form a heterodimer in the inner mitochondrial membrane. Liver-specific MPC1 or MPC2 deletion or knockdown improved basal hyperglycemia and glucose tolerance in high-fat diet-fed or genetically obese db/db mice (86). It is likely that these effects were mediated, at least in part, by blocking pyruvate entry into the gluconeogenic pathway, since that requires pyruvate carboxylation in the mitochondrial matrix (86). Acute pharmacological MPC blockade by the novel PPARγ-sparing thiazolidinedione, MSDC-0602, also attenuated diet-induced insulin resistance, glucose tolerance (87) and hepatic steatosis, inflammation, and fibrosis in mice (56, 88–90). In patients with NASH with or without insulin resistance, MSDC-0602 lowered glycated hemoglobin, circulating insulin, and serum transaminases, but failed to affect NASH histology (91). Moreover, although such data are not yet published in liver, MPC inhibition by MSDC-0160 in neurons induces autophagic flux and compensatory branched chain amino acid catabolism (92). Together, the data suggest that pyruvate transport is the mitochondrial extension of glycolytic flux, and inhibiting substrate catabolism at any of these catabolic steps induces compensatory changes that benefit host metabolism. In addition, the broad, adaptive effects of acute inhibition, and corroborating genetic data highlight an important role for the hepatic MPC in peripheral and hepatic glucose and lipid homeostasis, and hepatic inflammation.
Restricting hepatocyte carbohydrate entry mimics the broader effects of fasting
The above data demonstrate tremendous advances in understanding hepatocyte glucose and fructose intermediary metabolism and signaling. To add to this, preventing hepatocyte carbohydrate entry prior to commitment into glycolysis, fructolysis and downstream signaling merits its own consideration as a viable target to prevent and treat metabolic disease. Our group and others recently examined some of the molecular intermediaries that convey the effects of hepatocyte carbohydrate restriction. In this section, we briefly review the rationale to translate this particular therapeutic approach, define intermediaries and mechanisms that are activated upon blocking carbohydrate entry, and delineate future considerations for study in hepatocyte glucose fasting.
Rationale to target hepatocyte carbohydrate transport
A primary goal of fasting physiology is to provide glucose and ketones to the brain (17). To that end, the hepatocyte switches to anabolism to generate glucose via glycogenolysis and gluconeogenesis, and to generate ketones from fatty acids. It then follows that the absence or paucity of glucose entering the hepatocyte activates homeostatic pathways that signal fasted status to the periphery. These peripheral carbon sources for the latter processes are fatty acids from peripheral adipose stores, and alanine derived from skeletal muscle (17). Aoki, Cahill and colleagues provided some of the early human data to suggest that glucose determines the systemic (and hepatocyte) fasting response in classical experiments (93). In this experiment, Glucose feeding was sufficient to maintain circulating insulin, and suppress ketogenesis and peripheral lipolysis even in the context of profound caloric restriction. Even after three weeks of starvation, 150 g / day glucose (~70% calorie restriction) by itself raised serum insulin, and suppressed hepatic ketogenesis and urea nitrogen excretion. The data underscore that, even during starvation and severe caloric restriction, minimal amounts of glucose are sufficient to abrogate the fasting response. A substantial body of data have accumulated to demonstrate that both endocrine and cell-autonomous hepatocyte regulation direct ketogenesis, gluconeogenesis, glycogenolysis, and the peripheral lipolysis and glucose-alanine cycling that fuel these processes. Our focus here will remain on cell-autonomous substrate regulation of hepatocyte glucose transport, because this process is quite directly amenable to small-molecule therapeutics.
Cellular Consequences of Hepatocyte Carbohydrate Restriction
Blocking carbohydrate entry activates autophagy primarily by AMPK-dependent (7, 94, 95) and potentially AMPK-independent pathways (Figure 2) (96). This activates homeostatic processes to correct for this perturbation. First, the hepatocyte activates autophagic flux to recycle damaged or misfolded proteins as a means to free substrate to meet subsequent energy demands (7, 8, 13, 14, 97, 98) in part by direct AMPK stimulatory phosphorylation of Beclin 1 (99) and unc52-like kinase 1 (ULK1) (94, 95, 100). Secondly, glucose deprivation activates ER and oxidative stress-responsive pathways, and this includes GRP78 (101, 102), NRF2/KEAP1, and AMPK alpha 2 (103) pathways (104). Distally, glucose deprivation activates several key transcriptional regulators, including TFEB (105–107), PGC1α (108–110), and PPARα (111). These factors activate lysosomal biogenesis and fat oxidative transcriptional programs to coordinate the metabolic switch from glucose to autophagy-derived macromolecules and fat. In addition, glucose-stimulated ATP production regulates the redox state through NAD+/NADH levels to activate the deacetylase sirtuin family member, SIRT1, which deacetylates and activates PGC1α and the stress-responsive factor, FOXO1 (112). Finally, we identified non-canonical fasting mediators of hepatocyte glucose-specific restriction (10, 113), including the lipoxygenase, Aloxe3 and the arginine ureahydrolase, Arg2. Both genes are induced by macronutrient withdrawal, fasting, and trehalose and LT treatment in vivo and in vitro, and mediate aspects of the hepatocyte fasting response (10, 113).
Figure 2.
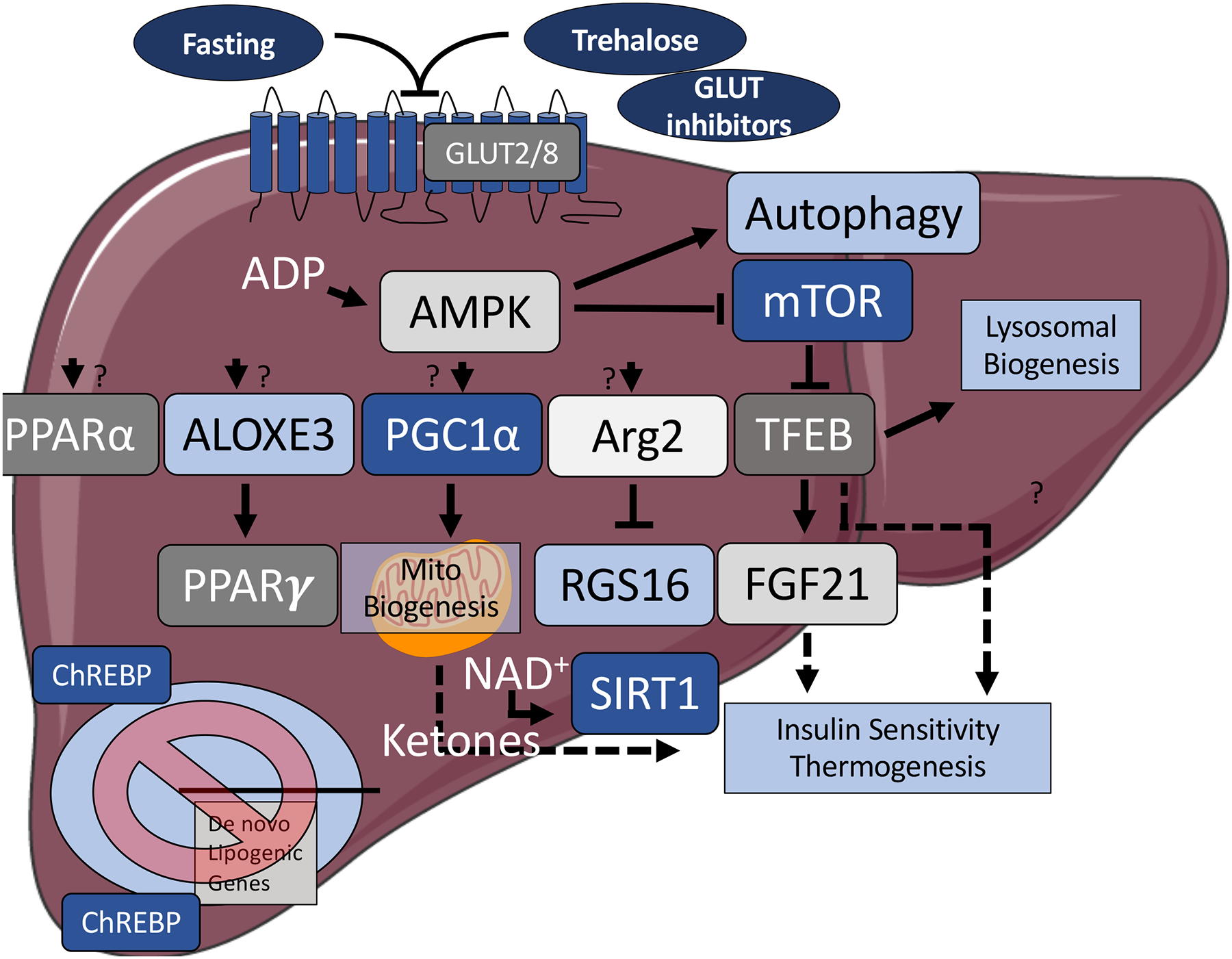
Some of the key signaling pathways that are activated upon hepatocyte glucose withdrawal, or GLUT blockade. Fasting, and treatment with glucose transporter inhibitors reduce hepatocyte ATP production, and activate key fasting-mimetic signaling pathways, which are metabolically protective to the host. These pathways include induction of autophagy to recycle aging or damaged organelles and proteins to be used as fuel, inhibition of mechanistic target of rapamycin (mTOR) and carbohydrate response element binding protein, and activation of AMP-activated protein kinase. These serve as key proximal fasted-state intracellular sensors. Downstream fasting-like signals upon suppressed carbohydrate entry include peroxisome proliferator antigen receptors α and γ, arachidonate lipoxygenase 3, peroxisome proliferator antigen receptor gamma coactivator 1α;, arginase 2 and sirtuin 1, transcription factor EB, and release of fibroblast growth factor 21 and ketones into the peripheral circulation. The full mechanisms by carbohydrate withdrawal activates these signals, and the full consequences of their activation, however, remain subjects of continued investigation. Dashed arrows: secreted factor. Abbreviations: ALOXE3, arachidonate lipoxygenase 3; AMPK, AMP-activated protein kinase; Arg2, arginase 2; ChREBP, carbohydrate response element binding protein; FGF21, fibroblast growth factor 21; GLUT, glucose transporter; mTOR, mechanistic target of rapamycin; NAD+, nicotinamide adenine dinucleotide; PGC1α, peroxisome proliferator antigen receptor gamma coactivator 1α; PPARα, peroxisome proliferator antigen receptor; RGS16, regulator of G-protein signaling 16; SIRT1, sirtuin1; TFEB, transcription factor EB.
Carbohydrate withdrawal therefore exerts important molecular changes within the hepatocyte. This suggests that methods to target hepatocyte cytosolic carbohydrate excursion via GLUT2 and GLUT8 have translational value. In the liver-specific GLUT2 knockout model (LG2KO), liver fluorodeoxyglucose uptake was blunted when compared with uptake in WT mice (69), but increased skeletal muscle glucose uptake renders basal glycemia in these mice unchanged when compared with wild-type mice. Moreover, although the role of hepatocyte GLUT2 in obese or diabetic models has not been reported, LG2KO mice had decreased fasting cholesterol biosynthesis and were protected from fasting-induced steatosis. This phenotype was attributed to lower glucose substrate entry to fuel lipid droplet deposition. These adaptations were observed despite paradoxically higher expression of ChREBP and its target lipogenic and glycolytic genes, and despite progressive pancreatic β-cell dysfunction over time in LG2KO mice versus WT mice (69). Accordingly, GLUT8 deletion attenuated radiolabeled hepatocyte fructose uptake, fructose-induced ChREBP activation, de novo lipogenic gene expression, and triacylglyerol synthesis when compared with wild-type cultured primary murine hepatocytes (44). In addition, germline whole-body GLUT8-deficient mice were protected from high-fat and high-fructose diet-induced hepatic steatosis, dyslipidemia and glucose intolerance without any obvious chronic maladaptive metabolic sequelae reported, in comparison with WT littermates. These effects were mediated in part by enhanced fasting-induced hepatic PPARα activity (11, 44, 70, 114).
Pharmacological studies of restricted hepatocyte carbohydrate entry largely recapitulate the metabolic effects of hepatocyte GLUT deficiency. In particular the disaccharide glucose mimetics trehalose and lactotrehalose (LT) are useful probes to define the effects of hepatocyte GLUT blockade (7, 8, 13, 14, 98, 115–118). Both disaccharides block enterohepatic glucose transport (GLUT) (7, 8, 13, 14, 98), thus offering a window into acute effects of enterohepatic GLUT blockade. Trehalose and LT reduced fructose-induced hepatic steatosis and induced hepatocyte fasting response signals, PGC1α, TFEB and FGF21. Extrahepatic cardiometabolic effects of these compounds are broad, including peripheral insulin sensitization (119), activating peripheral thermogenesis, reduced adipocyte hypertrophy (120, 121), in addition to reduced atherosclerotic plaque lesion area (27, 122) and pathological cardiac remodeling in response to injury (123, 124), that mimicked ketogenic dietary effects on heart failure (37). Treating liver-specific GLUT8-deficient mice with oral trehalose did not increase the efficacy of trehalose on diet-induced hepatic steatosis, which implies some mechanistic overlap between GLUT8 deletion and trehalose action (98). Restricting carbohydrate entry into the hepatocyte thus exerts hepatic and extrahepatic therapeutic adaptative effects. The complex interactions between hepatocyte glucose transport and extrahepatic tissues therefore justify much deeper exploration into both the potential limitations and the physiologic and signaling adaptations of hepatocyte GLUT-specific targeting.
By corollary, detailing the intracellular consequences of hepatocyte carbohydrate restriction may illuminate novel therapies against both hepatic and extrahepatic metabolic disease. For example, hepatocyte glucose restriction induces ALOXE3, a lipoxygenase that metabolizes arachidonic acid intermediaries to enhance peripheral insulin sensitivity, thermogenesis and reduce hepatic steatosis in genetic and diet-induced obese models (113). In addition, Arg2 is upregulated during fasting, and we postulate that the purpose of this is to handle the excess liver nitrogen load to fuel gluconeogenesis (17). Forced Arg2 expression in hepatocytes improved insulin resistance, heat generation and hepatic insulin sensitivity in genetic and diet-induced obese models. These actions depended upon an inverse relationship with the hepatocyte regulator of G-protein signaling (RGS) protein, RGS16, although more detailed mechanisms of this axis remain to be fully explored. Elucidating new pathways that hepatocyte glucose restriction activates will continue to offer novel leverage points for new metabolic therapies.
FUTURE DIRECTIONS AND CONCLUSIONS
The metabolic efficacy of IF and CR are widely recognized across basic scientific, medical, and popular domains (1, 16, 22), but the mechanisms and full breadth of utility for therapeutic hepatocyte glucose transport restriction are only beginning to be elucidated. In addition to unbiased screening approaches, we can look to the paradigm process of fasting itself to point us to mechanisms under carbohydrate control. On this basis, hepatocyte glucose transport restriction and its effects on other core fasting-regulated processes remain prime opportunities for future investigation (Figure 2). This includes, in particular, hepatocyte glucose-specific withdrawal and its effects on NAD+ metabolism and circadian rhythm (1, 22, 125–127). Lastly, it should be recognized first that the observations regarding effects hepatocyte glucose restriction might not merely represent a subset of the generalized fasting response. Rather, there are likely to be pathways unique to glucose-specific restriction that are key leverage points for therapy. Secondly, glucose restriction might share pathways with other adaptive processes, such as cold thermogenesis (128–131). Notably, if hepatocyte glucose restriction incites a distinct adaptive response, this opens the exciting possibility that this pathway can be utilized additively or synergistically with other distinct leverage points in fasting-like signaling. Certainly, in the face of the ongoing epidemics of obesity, diabetes and NAFLD, the future certainly has never looked sweeter.
ACKNOWLEDGEMENTS
Grant support: The Doris Duke Charitable Foundation Clinical Scientist Development Award, NIH/NCCIH 1R21AT010520, NIH/NHLBI 1R01HL147968-01A1, the AGA-Allergan Pilot Research Award in Non-Alcoholic Fatty Liver Disease, and the AASLD Pilot Research Award (to BJD), and R01 DK104735 (to BNF).
Abbreviations:
- AldoB
aldolase B
- ALOXE3
arachidonate lipoxygenase 3
- AMPK
AMP-activated protein kinase
- Arg2
arginase 2
- ChREBP
carbohydrate response element binding protein
- CR
caloric restriction
- FOXO
forkhead box transcriptiion factor O
- GCK
glucokinase
- GCKRP
glucokinase regulatory protein
- GLUT
glucose transporter
- IF
intermittent fasting
- KHK
ketohexokinase
- MPC
mitochondrial pyruvate carrier
- mTOR
mechanistic target of rapamycin
- NAD+
nicotinamide adenine dinucleotide
- NAFLD
non-alcoholic fatty liver disease
- PGC1α
peroxisome proliferator antigen receptor gamma coactivator 1α
- PPAR
peroxisome proliferator antigen receptor
- RGS
regulator of G-protein signaling 16
- SIRT1
sirtuin1
- TFEB
transcription factor EB
Footnotes
Conflicts of Interest: None
REFFERENCES
- 1.Mattson MP, Longo VD, Harvie M. Impact of intermittent fasting on health and disease processes. Ageing research reviews. 2017;39:46–58. Epub 2016/10/31. doi: 10.1016/j.arr.2016.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Benhamed F, Denechaud PD, Lemoine M, Robichon C, Moldes M, Bertrand-Michel J, Ratziu V, Serfaty L, Housset C, Capeau J, Girard J, Guillou H, Postic C. The lipogenic transcription factor ChREBP dissociates hepatic steatosis from insulin resistance in mice and humans. J Clin Invest. 2012;122(6):2176–94. Epub 2012/05/02. doi: 10.1172/JCI41636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schugar RC, Crawford PA. Low-carbohydrate ketogenic diets, glucose homeostasis, and nonalcoholic fatty liver disease. Current opinion in clinical nutrition and metabolic care. 2012;15(4):374–80. doi: 10.1097/MCO.0b013e3283547157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kosinski C, Jornayvaz FR. Effects of Ketogenic Diets on Cardiovascular Risk Factors: Evidence from Animal and Human Studies. Nutrients. 2017;9(5):517. doi: 10.3390/nu9050517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Softic S, Stanhope KL, Boucher J, Divanovic S, Lanaspa MA, Johnson RJ, Kahn CR. Fructose and hepatic insulin resistance. Critical Reviews in Clinical Laboratory Sciences. 2020:1–15. doi: 10.1080/10408363.2019.1711360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Softic S, Gupta MK, Wang G-X, Fujisaka S, O’Neill BT, Rao TN, Willoughby J, Harbison C, Fitzgerald K, Ilkayeva O, Newgard CB, Cohen DE, Kahn CR. Divergent effects of glucose and fructose on hepatic lipogenesis and insulin signaling. The Journal of Clinical Investigation. 2017;127(11):4059–74. doi: 10.1172/JCI94585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.DeBosch BJ, Heitmeier MR, Mayer AL, Higgins CB, Crowley JR, Kraft TE, Chi M, Newberry EP, Chen Z, Finck BN, Davidson NO, Yarasheski KE, Hruz PW, Moley KH. Trehalose inhibits solute carrier 2A (SLC2A) proteins to induce autophagy and prevent hepatic steatosis 2016;9(416):ra21–ra. doi: 10.1126/scisignal.aac5472 %J Science Signaling. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mardones P, Rubinsztein DC, Hetz C. Mystery solved: Trehalose kickstarts autophagy by blocking glucose transport. Science Signaling. 2016;9(416):fs2. doi: 10.1126/scisignal.aaf1937. [DOI] [PubMed] [Google Scholar]
- 9.Zhang Y, Shaikh N, Ferey JL, Wankhade UD, Chintapalli SV, Higgins CB, Crowley JR, Heitmeier MR, Stothard AI, Mihi B, Good M, Higashiyama T, Swarts BM, Hruz PW, Shankar K, Tarr PI, DeBosch BJ. Lactotrehalose, an Analog of Trehalose, Increases Energy Metabolism Without Promoting Clostridioides difficile Infection in Mice. Gastroenterology. 2020;158(5):1402–16.e2. doi: 10.1053/j.gastro.2019.11.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang YHC, Fortune HM, Chen P, Stothard AI, Mayer AL, Swarts BM and DeBosch BJ. Hepatocyte arginase 2 is sufficient to convey the therapeutic metabolic effects of fasting. Nature Communications. 2019;10. doi: 10.1038/s41467-019-09642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mayer AL, Higgins CB, Feng EH, Adenekan O, Zhang Y, DeBosch BJ, Pietka TA, Beatty WL. Enhanced Hepatic PPARα Activity Links GLUT8 Deficiency to Augmented Peripheral Fasting Responses in Male Mice. Endocrinology. 2018;159(5):2110–26. doi: 10.1210/en.2017-03150 %J Endocrinology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ortega-Prieto P, Postic C. Carbohydrate Sensing Through the Transcription Factor ChREBP. Frontiers in Genetics. 2019;10(472). doi: 10.3389/fgene.2019.00472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang Y, DeBosch BJ. Using trehalose to prevent and treat metabolic function: effectiveness and mechanisms. Current Opinion in Clinical Nutrition and Metabolic Care. 2019;22(4):303–10. Epub 4/30/2019. doi: 10.1097/mco.0000000000000568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang YaDB. Microbial and Metabolic Impacts of Trehalose and Trehalose Analogues. Gut Microbes. 2020;In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Madeo F, Carmona-Gutierrez D, Hofer SJ, Kroemer G. Caloric Restriction Mimetics against Age-Associated Disease: Targets, Mechanisms, and Therapeutic Potential. Cell Metab. 2019;29(3):592–610. Epub 2019/03/07. doi: 10.1016/j.cmet.2019.01.018. [DOI] [PubMed] [Google Scholar]
- 16.de Cabo R, Mattson MP. Effects of Intermittent Fasting on Health, Aging, and Disease. N Engl J Med. 2019;381(26):2541–51. Epub 2019/12/28. doi: 10.1056/NEJMra1905136. [DOI] [PubMed] [Google Scholar]
- 17.Cahill GF Jr. Fuel metabolism in starvation. Annu Rev Nutr. 2006;26:1–22. Epub 2006/07/20. doi: 10.1146/annurev.nutr.26.061505.111258. [DOI] [PubMed] [Google Scholar]
- 18.WAN R, CAMANDOLA S, MATTSON MP. Intermittent fasting and dietary supplementation with 2-deoxy-d-glucose improve functional and metabolic cardiovascular risk factors in rats 2003;17(9):1133–4. doi: 10.1096/fj.02-0996fje. [DOI] [PubMed] [Google Scholar]
- 19.Hatori M, Vollmers C, Zarrinpar A, DiTacchio L, Bushong Eric A, Gill S, Leblanc M, Chaix A, Joens M, Fitzpatrick James AJ, Ellisman Mark H, Panda S. Time-Restricted Feeding without Reducing Caloric Intake Prevents Metabolic Diseases in Mice Fed a High-Fat Diet. Cell Metabolism. 2012;15(6):848–60. doi: 10.1016/j.cmet.2012.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chaix A, Lin T, Le HD, Chang MW, Panda S. Time-Restricted Feeding Prevents Obesity and Metabolic Syndrome in Mice Lacking a Circadian Clock. Cell Metabolism. 2019;29(2):303–19.e4. doi: 10.1016/j.cmet.2018.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chaix A, Zarrinpar A, Miu P, Panda S. Time-Restricted Feeding Is a Preventative and Therapeutic Intervention against Diverse Nutritional Challenges. Cell Metabolism. 2014;20(6):991–1005. doi: 10.1016/j.cmet.2014.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Longo VD, Panda S. Fasting, Circadian Rhythms, and Time-Restricted Feeding in Healthy Lifespan. Cell Metabolism. 2016;23(6):1048–59. doi: 10.1016/j.cmet.2016.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sutton EF, Beyl R, Early KS, Cefalu WT, Ravussin E, Peterson CM. Early Time-Restricted Feeding Improves Insulin Sensitivity, Blood Pressure, and Oxidative Stress Even without Weight Loss in Men with Prediabetes. Cell Metabolism. 2018;27(6):1212–21.e3. doi: 10.1016/j.cmet.2018.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barnosky AR, Hoddy KK, Unterman TG, Varady KA. Intermittent fasting vs daily calorie restriction for type 2 diabetes prevention: a review of human findings. Translational Research. 2014;164(4):302–11. doi: 10.1016/j.trsl.2014.05.013. [DOI] [PubMed] [Google Scholar]
- 25.Carter S, Clifton PM, Keogh JB. Effect of Intermittent Compared With Continuous Energy Restricted Diet on Glycemic Control in Patients With Type 2 Diabetes: A Randomized Noninferiority Trial. JAMA Network Open. 2018;1(3):e180756–e. doi: 10.1001/jamanetworkopen.2018.0756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Trepanowski JF, Kroeger CM, Barnosky A, Klempel MC, Bhutani S, Hoddy KK, Gabel K, Freels S, Rigdon J, Rood J, Ravussin E, Varady KA. Effect of Alternate-Day Fasting on Weight Loss, Weight Maintenance, and Cardioprotection Among Metabolically Healthy Obese Adults: A Randomized Clinical Trial. JAMA Internal Medicine. 2017;177(7):930–8. doi: 10.1001/jamainternmed.2017.0936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sergin I, Evans TD, Zhang X, Bhattacharya S, Stokes CJ, Song E, Ali S, Dehestani B, Holloway KB, Micevych PS, Javaheri A, Crowley JR, Ballabio A, Schilling JD, Epelman S, Weihl CC, Diwan A, Fan D, Zayed MA, Razani B. Exploiting macrophage autophagy-lysosomal biogenesis as a therapy for atherosclerosis. Nat Commun. 2017;8:15750. Epub 2017/06/08. doi: 10.1038/ncomms15750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wilkinson MJ, Manoogian ENC, Zadourian A, Lo H, Fakhouri S, Shoghi A, Wang X, Fleischer JG, Navlakha S, Panda S, Taub PR. Ten-Hour Time-Restricted Eating Reduces Weight, Blood Pressure, and Atherogenic Lipids in Patients with Metabolic Syndrome. Cell Metabolism. 2019. doi: 10.1016/j.cmet.2019.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jamshed H, Beyl AR, Della Manna LD, Yang SE, Ravussin E, Peterson MC. Early Time-Restricted Feeding Improves 24-Hour Glucose Levels and Affects Markers of the Circadian Clock, Aging, and Autophagy in Humans. Nutrients. 2019;11(6). doi: 10.3390/nu11061234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Foster GD, Wyatt HR, Hill JO, McGuckin BG, Brill C, Mohammed BS, Szapary PO, Rader DJ, Edman JS, Klein S. A Randomized Trial of a Low-Carbohydrate Diet for Obesity. New England Journal of Medicine. 2003;348(21):2082–90. doi: 10.1056/NEJMoa022207. [DOI] [PubMed] [Google Scholar]
- 31.Samaha FF, Iqbal N, Seshadri P, Chicano KL, Daily DA, McGrory J, Williams T, Williams M, Gracely EJ, Stern L. A Low-Carbohydrate as Compared with a Low-Fat Diet in Severe Obesity. New England Journal of Medicine. 2003;348(21):2074–81. doi: 10.1056/NEJMoa022637. [DOI] [PubMed] [Google Scholar]
- 32.Volek JS, Phinney SD, Forsythe CE, Quann EE, Wood RJ, Puglisi MJ, Kraemer WJ, Bibus DM, Fernandez ML, Feinman RD. Carbohydrate Restriction has a More Favorable Impact on the Metabolic Syndrome than a Low Fat Diet. Lipids. 2009;44(4):297–309. doi: 10.1007/s11745-008-3274-2. [DOI] [PubMed] [Google Scholar]
- 33.Brehm BJ, Seeley RJ, Daniels SR, D’Alessio DA. A Randomized Trial Comparing a Very Low Carbohydrate Diet and a Calorie-Restricted Low Fat Diet on Body Weight and Cardiovascular Risk Factors in Healthy Women. The Journal of Clinical Endocrinology & Metabolism. 2003;88(4):1617–23. doi: 10.1210/jc.2002-021480. [DOI] [PubMed] [Google Scholar]
- 34.Shai I, Schwarzfuchs D, Henkin Y, Shahar DR, Witkow S, Greenberg I, Golan R, Fraser D, Bolotin A, Vardi H, Tangi-Rozental O, Zuk-Ramot R, Sarusi B, Brickner D, Schwartz Z, Sheiner E, Marko R, Katorza E, Thiery J, Fiedler GM, Blüher M, Stumvoll M, Stampfer MJ. Weight Loss with a Low-Carbohydrate, Mediterranean, or Low-Fat Diet. New England Journal of Medicine. 2008;359(3):229–41. doi: 10.1056/NEJMoa0708681. [DOI] [PubMed] [Google Scholar]
- 35.Mager DR, Iñiguez IR, Gilmour S, Yap J. The Effect of a Low Fructose and Low Glycemic Index/Load (FRAGILE) Dietary Intervention on Indices of Liver Function, Cardiometabolic Risk Factors, and Body Composition in Children and Adolescents With Nonalcoholic Fatty Liver Disease (NAFLD). Journal of Parenteral and Enteral Nutrition. 2015;39(1):73–84. doi: 10.1177/0148607113501201. [DOI] [PubMed] [Google Scholar]
- 36.Luukkonen PK, Dufour S, Lyu K, Zhang X-M, Hakkarainen A, Lehtimäki TE, Cline GW, Petersen KF, Shulman GI, Yki-Järvinen H. Effect of a ketogenic diet on hepatic steatosis and hepatic mitochondrial metabolism in nonalcoholic fatty liver disease. Proceedings of the National Academy of Sciences. 2020;117(13):7347. doi: 10.1073/pnas.1922344117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McCommis KS, Kovacs A, Weinheimer CJ, Shew TM, Koves TR, Ilkayeva OR, Kamm DR, Pyles KD, King MT, Veech RL, DeBosch BJ, Muoio DM, Gross RW, Finck BN. Ketogenic Diet Prevents Heart Failure from Defective Mitochondrial Pyruvate Metabolism. bioRxiv. 2020:2020.02.21.959635. doi: 10.1101/2020.02.21.959635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sacks FM, Bray GA, Carey VJ, Smith SR, Ryan DH, Anton SD, McManus K, Champagne CM, Bishop LM, Laranjo N, Leboff MS, Rood JC, de Jonge L, Greenway FL, Loria CM, Obarzanek E, Williamson DA. Comparison of Weight-Loss Diets with Different Compositions of Fat, Protein, and Carbohydrates. New England Journal of Medicine. 2009;360(9):859–73. doi: 10.1056/NEJMoa0804748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mueckler M, Thorens B. The SLC2 (GLUT) family of membrane transporters. Mol Aspects Med. 2013;34(2–3):121–38. doi: 10.1016/j.mam.2012.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thorens B, Mueckler M. Glucose transporters in the 21st Century 2010;298(2):E141–E5. doi: 10.1152/ajpendo.00712.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mayer AL, Higgins CB, Heitmeier MR, Kraft TE, Qian X, Crowley JR, Hyrc KL, Beatty WL, Yarasheski KE, Hruz PW, DeBosch BJ. SLC2A8 (GLUT8) is a mammalian trehalose transporter required for trehalose-induced autophagy. Scientific Reports. 2016;6:38586. doi: 10.1038/srep3858610.1038/srep38586https://www.nature.com/articles/srep38586#supplementary-informationhttps://www.nature.com/articles/srep38586#supplementary-information . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Carayannopoulos MO, Chi MM-Y, Cui Y, Pingsterhaus JM, McKnight RA, Mueckler M, Devaskar SU, Moley KH. GLUT8 is a glucose transporter responsible for insulin-stimulated glucose uptake in the blastocyst 2000;97(13):7313–8. doi: 10.1073/pnas.97.13.7313 %J Proceedings of the National Academy of Sciences. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Augustin R, Riley J, Moley KH. GLUT8 Contains a [DE]XXXL[LI] Sorting Motif and Localizes to a Late Endosomal/Lysosomal Compartment 2005;6(12):1196–212. doi: 10.1111/j.1600-0854.2005.00354.x. [DOI] [PubMed] [Google Scholar]
- 44.Debosch BJ, Chen Z, Saben JL, Finck BN, Moley KH. Glucose transporter 8 (GLUT8) mediates fructose-induced de novo lipogenesis and macrosteatosis. J Biol Chem. 2014;289(16):10989–98. Epub 2014/02/13. doi: 10.1074/jbc.M113.527002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cui L, Ranalletta M, Gorovits N, Charron MJ, Busik JV, de-Mouzon SH. Regulation of Hepatic GLUT8 Expression in Normal and Diabetic Models. Endocrinology. 2003;144(5):1703–11. doi: 10.1210/en.2002-220968. [DOI] [PubMed] [Google Scholar]
- 46.Ibberson Mark, U M, Bernard Thorens. GLUTX1 a novel mammalian glucose transporter expressed in the central nervous system and insulin-sensitive tissues. J Biol Chem. 2000;275(7):4607–12. [DOI] [PubMed] [Google Scholar]
- 47.Membrez M, Hummler E, Beermann F, Haefliger J-A, Savioz R, Pedrazzini T, Thorens B. GLUT8 Is Dispensable for Embryonic Development but Influences Hippocampal Neurogenesis and Heart Function 2006;26(11):4268–76. doi: 10.1128/MCB.00081-06 %J Molecular and Cellular Biology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schmidt S, Joost HG, Schurmann A. GLUT8, the enigmatic intracellular hexose transporter. Am J Physiol Endocrinol Metab. 2009;296(4):E614–8. Epub 2009/01/30. doi: 10.1152/ajpendo.91019.2008. [DOI] [PubMed] [Google Scholar]
- 49.Matschinsky FM, Wilson DF. The Central Role of Glucokinase in Glucose Homeostasis: A Perspective 50 Years After Demonstrating the Presence of the Enzyme in Islets of Langerhans. Frontiers in Physiology. 2019;10(148). doi: 10.3389/fphys.2019.00148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Baltrusch S, Francini F, Lenzen S, Tiedge M. Interaction of Glucokinase With the Liver Regulatory Protein Is Conferred by Leucine-Asparagine Motifs of the Enzyme. Diabetes. 2005;54(10):2829. doi: 10.2337/diabetes.54.10.2829. [DOI] [PubMed] [Google Scholar]
- 51.Raimondo A, Rees MG, Gloyn AL. Glucokinase regulatory protein: complexity at the crossroads of triglyceride and glucose metabolism. Curr Opin Lipidol. 2015;26(2):88–95. Epub 2015/02/19. doi: 10.1097/MOL.0000000000000155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Adeva-Andany MM, Pérez-Felpete N, Fernández-Fernández C, Donapetry-García C, Pazos-García C. Liver glucose metabolism in humans. Biosci Rep. 2016;36(6):e00416. doi: 10.1042/BSR20160385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Thorens B. GLUT2, glucose sensing and glucose homeostasis. Diabetologia. 2015;58(2):221–32. doi: 10.1007/s00125-014-3451-1. [DOI] [PubMed] [Google Scholar]
- 54.McCommis Kyle S, Finck Brian N. Mitochondrial pyruvate transport: a historical perspective and future research directions. Biochemical Journal. 2015;466(3):443–54. doi: 10.1042/BJ20141171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mahmood S, Birkaya B, Rideout TC, Patel MS. Lack of mitochondria-generated acetyl-CoA by pyruvate dehydrogenase complex downregulates gene expression in the hepatic de novo lipogenic pathway. Am J Physiol Endocrinol Metab. 2016;311(1):E117–27. Epub 2016/05/12. doi: 10.1152/ajpendo.00064.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Finck BN. Targeting Metabolism, Insulin Resistance, and Diabetes to Treat Nonalcoholic Steatohepatitis. Diabetes. 2018;67(12):2485–93. doi: 10.2337/dbi18-0024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Softic S, Cohen DE, Kahn CR. Role of Dietary Fructose and Hepatic De Novo Lipogenesis in Fatty Liver Disease. Digestive Diseases and Sciences. 2016;61(5):1282–93. doi: 10.1007/s10620-016-4054-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Garcia D, Hellberg K, Chaix A, Wallace M, Herzig S, Badur MG, Lin T, Shokhirev MN, Pinto AFM, Ross DS, Saghatelian A, Panda S, Dow LE, Metallo CM, Shaw RJ. Genetic Liver-Specific AMPK Activation Protects against Diet-Induced Obesity and NAFLD. Cell Rep. 2019;26(1):192–208 e6. Epub 2019/01/04. doi: 10.1016/j.celrep.2018.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Abdul-Wahed A, Guilmeau S, Postic C. Sweet Sixteenth for ChREBP: Established Roles and Future Goals. Cell Metabolism. 2017;26(2):324–41. doi: 10.1016/j.cmet.2017.07.004. [DOI] [PubMed] [Google Scholar]
- 60.Kim M-S, Krawczyk SA, Doridot L, Fowler AJ, Wang JX, Trauger SA, Noh H-L, Kang HJ, Meissen JK, Blatnik M, Kim JK, Lai M, Herman MA. ChREBP regulates fructose-induced glucose production independently of insulin signaling. The Journal of Clinical Investigation. 2019;126(11):4372–86. doi: 10.1172/JCI81993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jensen T, Abdelmalek MF, Sullivan S, Nadeau KJ, Green M, Roncal C, Nakagawa T, Kuwabara M, Sato Y, Kang D-H, Tolan DR, Sanchez-Lozada LG, Rosen HR, Lanaspa MA, Diehl AM, Johnson RJ. Fructose and sugar: A major mediator of non-alcoholic fatty liver disease. Journal of hepatology. 2018;68(5):1063–75. Epub 2018/02/02. doi: 10.1016/j.jhep.2018.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tappy L, Le KA. Metabolic effects of fructose and the worldwide increase in obesity. Physiol Rev. 2010;90(1):23–46. Epub 2010/01/21. doi: 10.1152/physrev.00019.2009. [DOI] [PubMed] [Google Scholar]
- 63.Hannou SA, Haslam DE, McKeown NM, Herman MA. Fructose metabolism and metabolic disease. The Journal of Clinical Investigation. 2018;128(2):545–55. doi: 10.1172/JCI96702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.DeBosch BJ, Kluth O, Fujiwara H, Schurmann A, Moley K. Early-onset metabolic syndrome in mice lacking the intestinal uric acid transporter SLC2A9. Nat Commun. 2014;5:4642. Epub 2014/08/08. doi: 10.1038/ncomms5642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Preitner F, Bonny O, Laverrière A, Rotman S, Firsov D, Da Costa A, Metref S, Thorens B. Glut9 is a major regulator of urate homeostasis and its genetic inactivation induces hyperuricosuria and urate nephropathy. Proceedings of the National Academy of Sciences. 2009;106(36):15501. doi: 10.1073/pnas.0904411106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Johnson RJ, Nakagawa T, Sanchez-Lozada LG, Shafiu M, Sundaram S, Le M, Ishimoto T, Sautin YY, Lanaspa MA. Sugar, Uric Acid, and the Etiology of Diabetes and Obesity. Diabetes. 2013;62(10):3307. doi: 10.2337/db12-1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Johnson RJ, Bakris GL, Borghi C, Chonchol MB, Feldman D, Lanaspa MA, Merriman TR, Moe OW, Mount DB, Sanchez Lozada LG, Stahl E, Weiner DE, Chertow GM. Hyperuricemia, Acute and Chronic Kidney Disease, Hypertension, and Cardiovascular Disease: Report of a Scientific Workshop Organized by the National Kidney Foundation. American Journal of Kidney Diseases. 2018;71(6):851–65. doi: 10.1053/j.ajkd.2017.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sookoian S, Pirola CJ, Valenti L, Davidson NO. Genetic pathways in nonalcoholic fatty liver disease: Insights from systems biology. Hepatology. 2020;n/a(n/a). doi: 10.1002/hep.31229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Seyer P, Vallois D, Poitry-Yamate C, Schütz F, Metref S, Tarussio D, Maechler P, Staels B, Lanz B, Grueter R, Decaris J, Turner S, da Costa A, Preitner F, Minehira K, Foretz M, Thorens B. Hepatic glucose sensing is required to preserve β cell glucose competence. The Journal of Clinical Investigation. 2013;123(4):1662–76. doi: 10.1172/JCI65538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.DeBosch BJ, Chen Z, Finck BN, Chi M, Moley KH. Glucose transporter-8 (GLUT8) mediates glucose intolerance and dyslipidemia in high-fructose diet-fed male mice. Mol Endocrinol. 2013;27(11):1887–96. Epub 2013/09/14. doi: 10.1210/me.2013-1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Matschinsky FM, Zelent B, Doliba N, Li C, Vanderkooi JM, Naji A, Sarabu R, Grimsby J. Glucokinase activators for diabetes therapy: May 2010 status report. Diabetes Care. 2011;34 Suppl 2(Suppl 2):S236–S43. doi: 10.2337/dc11-s236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ferre T, Riu E, Franckhauser S, Agudo J, Bosch F. Long-term overexpression of glucokinase in the liver of transgenic mice leads to insulin resistance. Diabetologia. 2003;46(12):1662–8. Epub 2003/11/14. doi: 10.1007/s00125-003-1244-z. [DOI] [PubMed] [Google Scholar]
- 73.Wang R, Gao H, Xu W, Li H, Mao Y, Wang Y, Guo T, Wang X, Song R, Li Z, Irwin DM, Niu G, Tan H. Differential expression of genes and changes in glucose metabolism in the liver of liver-specific glucokinase gene knockout mice. Gene. 2013;516(2):248–54. doi: 10.1016/j.gene.2012.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hayashi H, Sato Y, Li Z, Yamamura K-i, Yoshizawa T, Yamagata K. Roles of hepatic glucokinase in intertissue metabolic communication: Examination of novel liver-specific glucokinase knockout mice. Biochemical and Biophysical Research Communications. 2015;460(3):727–32. doi: 10.1016/j.bbrc.2015.03.097. [DOI] [PubMed] [Google Scholar]
- 75.Speliotes EK, Yerges-Armstrong LM, Wu J, Hernaez R, Kim LJ, Palmer CD, Gudnason V, Eiriksdottir G, Garcia ME, Launer LJ, Nalls MA, Clark JM, Mitchell BD, Shuldiner AR, Butler JL, Tomas M, Hoffmann U, Hwang S-J, Massaro JM, O’Donnell CJ, Sahani DV, Salomaa V, Schadt EE, Schwartz SM, Siscovick DS, Nash CRN, Consortium G, Investigators M, Voight BF, Carr JJ, Feitosa MF, Harris TB, Fox CS, Smith AV, Kao WHL, Hirschhorn JN, Borecki IB, Consortium G. Genome-Wide Association Analysis Identifies Variants Associated with Nonalcoholic Fatty Liver Disease That Have Distinct Effects on Metabolic Traits. PLOS Genetics. 2011;7(3):e1001324. doi: 10.1371/journal.pgen.1001324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Anstee QM, Day CP. The genetics of NAFLD. Nature Reviews Gastroenterology & Hepatology. 2013;10(11):645–55. doi: 10.1038/nrgastro.2013.182. [DOI] [PubMed] [Google Scholar]
- 77.Rees MG, Wincovitch S, Schultz J, Waterstradt R, Beer NL, Baltrusch S, Collins FS, Gloyn AL. Cellular characterisation of the GCKR P446L variant associated with type 2 diabetes risk. Diabetologia. 2012;55(1):114–22. doi: 10.1007/s00125-011-2348-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Stender S, Grarup N, Hansen T. Genetic Aspects of Non-alcoholic Fatty Liver Disease (NAFLD). In: Krag A, Hansen T, editors. The Human Gut-Liver-Axis in Health and Disease. Cham: Springer International Publishing; 2019. p. 195–206. [Google Scholar]
- 79.Lloyd DJ, St Jean DJ, Kurzeja RJM, Wahl RC, Michelsen K, Cupples R, Chen M, Wu J, Sivits G, Helmering J, Komorowski R, Ashton KS, Pennington LD, Fotsch C, Vazir M, Chen K, Chmait S, Zhang J, Liu L, Norman MH, Andrews KL, Bartberger MD, Van G, Galbreath EJ, Vonderfecht SL, Wang M, Jordan SR, Véniant MM, Hale C. Antidiabetic effects of glucokinase regulatory protein small-molecule disruptors. Nature. 2013;504(7480):437–40. doi: 10.1038/nature12724. [DOI] [PubMed] [Google Scholar]
- 80.Lanaspa MA, Andres-Hernando A, Orlicky DJ, Cicerchi C, Jang C, Li N, Milagres T, Kuwabara M, Wempe MF, Rabinowitz JD, Johnson RJ, Tolan DR. Ketohexokinase C blockade ameliorates fructose-induced metabolic dysfunction in fructose-sensitive mice. The Journal of Clinical Investigation. 2018;128(6):2226–38. doi: 10.1172/JCI94427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ishimoto T, Lanaspa MA, Le MT, Garcia GE, Diggle CP, MacLean PS, Jackman MR, Asipu A, Roncal-Jimenez CA, Kosugi T, Rivard CJ, Maruyama S, Rodriguez-Iturbe B, Sánchez-Lozada LG, Bonthron DT, Sautin YY, Johnson RJ. Opposing effects of fructokinase C and A isoforms on fructose-induced metabolic syndrome in mice. Proceedings of the National Academy of Sciences. 2012;109(11):4320. doi: 10.1073/pnas.1119908109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Calle R, Bergman A, Somayaji V, Chidsey K, Kazierad D. PS-110-Ketohexokinase inhibitor PF-06835919 administered for 6 weeks reduces whole liver fat as measured by magnetic resonance imaging-proton density fat fraction in subjects with non-alcoholic fatty liver disease. Journal of Hepatology. 2019;70(1):e69–e70. doi: 10.1016/S0618-8278(19)30122-7. [DOI] [Google Scholar]
- 83.Simons N, Debray F-G, Schaper NC, Kooi ME, Feskens EJM, Hollak CEM, Lindeboom L, Koek GH, Bons JAP, Lefeber DJ, Hodson L, Schalkwijk CG, Stehouwer CDA, Cassiman D, Brouwers MCGJ. Patients With Aldolase B Deficiency Are Characterized by Increased Intrahepatic Triglyceride Content. The Journal of Clinical Endocrinology & Metabolism. 2019;104(11):5056–64. doi: 10.1210/jc.2018-02795. [DOI] [PubMed] [Google Scholar]
- 84.Oppelt SA, Sennott EM, Tolan DR. Aldolase-B knockout in mice phenocopies hereditary fructose intolerance in humans. Molecular Genetics and Metabolism. 2015;114(3):445–50. doi: 10.1016/j.ymgme.2015.01.001. [DOI] [PubMed] [Google Scholar]
- 85.Buziau AM, Schalkwijk CG, Stehouwer CDA, Tolan DR, Brouwers MCGJ. Recent advances in the pathogenesis of hereditary fructose intolerance: implications for its treatment and the understanding of fructose-induced non-alcoholic fatty liver disease. Cellular and Molecular Life Sciences. 2019. doi: 10.1007/s00018-019-03348-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gray LR, Sultana MR, Rauckhorst AJ, Oonthonpan L, Tompkins SC, Sharma A, Fu X, Miao R, Pewa AD, Brown KS, Lane EE, Dohlman A, Zepeda-Orozco D, Xie J, Rutter J, Norris AW, Cox JE, Burgess SC, Potthoff MJ, Taylor EB. Hepatic Mitochondrial Pyruvate Carrier 1 Is Required for Efficient Regulation of Gluconeogenesis and Whole-Body Glucose Homeostasis. Cell Metab. 2015;22(4):669–81. Epub 2015/09/08. doi: 10.1016/j.cmet.2015.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chen Z, Vigueira PA, Chambers KT, Hall AM, Mitra MS, Qi N, McDonald WG, Colca JR, Kletzien RF, Finck BN. Insulin resistance and metabolic derangements in obese mice are ameliorated by a novel peroxisome proliferator-activated receptor gamma-sparing thiazolidinedione. J Biol Chem. 2012;287(28):23537–48. Epub 2012/05/25. doi: 10.1074/jbc.M112.363960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.McCommis KS, Finck BN. Treating Hepatic Steatosis and Fibrosis by Modulating Mitochondrial Pyruvate Metabolism. Cellular and Molecular Gastroenterology and Hepatology. 2019;7(2):275–84. doi: 10.1016/j.jcmgh.2018.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Colca JR, McDonald WG, McCommis KS, Finck BN. Treating fatty liver disease by modulating mitochondrial pyruvate metabolism. Hepatology Communications. 2017;1(3):193–7. doi: 10.1002/hep4.1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.McCommis KS, Hodges WT, Brunt EM, Nalbantoglu I, McDonald WG, Holley C, Fujiwara H, Schaffer JE, Colca JR, Finck BN. Targeting the mitochondrial pyruvate carrier attenuates fibrosis in a mouse model of nonalcoholic steatohepatitis. Hepatology. 2017;65(5):1543–56. doi: 10.1002/hep.29025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Harrison SA, Alkhouri N, Davison BA, Sanyal A, Edwards C, Colca JR, Lee BH, Loomba R, Cusi K, Kolterman O, Cotter G, Dittrich HC. Insulin sensitizer MSDC-0602K in non-alcoholic steatohepatitis: A randomized, double-blind, placebo-controlled phase IIb study. Journal of Hepatology. 2020;72(4):613–26. doi: 10.1016/j.jhep.2019.10.023. [DOI] [PubMed] [Google Scholar]
- 92.Ghosh A, Tyson T, George S, Hildebrandt EN, Steiner JA, Madaj Z, Schulz E, Machiela E, McDonald WG, Escobar Galvis ML, Kordower JH, Van Raamsdonk JM, Colca JR, Brundin P. Mitochondrial pyruvate carrier regulates autophagy, inflammation, and neurodegeneration in experimental models of Parkinson’s disease. Sci Transl Med. 2016;8(368):368ra174. Epub 2016/12/09. doi: 10.1126/scitranslmed.aag2210. [DOI] [PubMed] [Google Scholar]
- 93.Aoki TT, Müller WA, Brennan MF, Cahill GF Jr. Metabolic effects of glucose in brief and prolonged fasted man. The American Journal of Clinical Nutrition. 1975;28(5):507–11. doi: 10.1093/ajcn/28.5.507. [DOI] [PubMed] [Google Scholar]
- 94.Russell RC, Yuan HX, Guan KL. Autophagy regulation by nutrient signaling. Cell Res. 2014;24(1):42–57. Epub 2013/12/18. doi: 10.1038/cr.2013.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13(2):132–41. Epub 2011/01/25. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Williams T, Forsberg LJ, Viollet B, Brenman JE. Basal autophagy induction without AMP-activated protein kinase under low glucose conditions. Autophagy. 2009;5(8):1155–65. doi: 10.4161/auto.5.8.10090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Cantó C, Auwerx J. Calorie Restriction: Is AMPK a Key Sensor and Effector? Physiology. 2011;26(4):214–24. doi: 10.1152/physiol.00010.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zhang Y, Shaikh N, Ferey JL, Wankhade UD, Chintapalli SV, Higgins CB, Crowley JR, Heitmeier MR, Stothard AI, Mihi B, Good M, Higashiyama T, Swarts BM, Hruz PW, Shankar K, Tarr PI, DeBosch BJ. Lactotrehalose, an Analog of Trehalose, Increases Energy Metabolism Without Promoting Clostridioides difficile Infection in Mice. Gastroenterology. 2020;158(5):1402–16 e2. Epub 2019/12/16. doi: 10.1053/j.gastro.2019.11.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhang D, Wang W, Sun X, Xu D, Wang C, Zhang Q, Wang H, Luo W, Chen Y, Chen H, Liu Z. AMPK regulates autophagy by phosphorylating BECN1 at threonine 388. Autophagy. 2016;12(9):1447–59. doi: 10.1080/15548627.2016.1185576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ha J, Guan K-L, Kim J. AMPK and autophagy in glucose/glycogen metabolism. Mol Aspects Med. 2015;46:46–62. doi: 10.1016/j.mam.2015.08.002. [DOI] [PubMed] [Google Scholar]
- 101.Mote PL, Tillman JB, Spindler SR. Glucose regulation of GRP78 gene expression. Mechanisms of Ageing and Development. 1998;104(2):149–58. doi: 10.1016/S0047-6374(98)00064-5. [DOI] [PubMed] [Google Scholar]
- 102.Graham NA, Tahmasian M, Kohli B, Komisopoulou E, Zhu M, Vivanco I, Teitell MA, Wu H, Ribas A, Lo RS, Mellinghoff IK, Mischel PS, Graeber TG. Glucose deprivation activates a metabolic and signaling amplification loop leading to cell death. Molecular Systems Biology. 2012;8(1):589. doi: 10.1038/msb.2012.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Mizunoe Y, Kobayashi M, Sudo Y, Watanabe S, Yasukawa H, Natori D, Hoshino A, Negishi A, Okita N, Komatsu M, Higami Y. Trehalose protects against oxidative stress by regulating the Keap1–Nrf2 and autophagy pathways. Redox Biology. 2018;15:115–24. doi: 10.1016/j.redox.2017.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Qiu S-L, Xiao Z-C, Piao C-M, Xian Y-L, Jia L-X, Qi Y-F, Han J-H, Zhang Y-y, Du J. AMP-activated Protein Kinase α2 Protects against Liver Injury from Metastasized Tumors via Reduced Glucose Deprivation-induced Oxidative Stress. Journal of Biological Chemistry. 2014;289(13):9449–59. doi: 10.1074/jbc.M113.543447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zhang Y, Higgins CB, Mayer AL, Mysorekar IU, Razani B, Graham MJ, Hruz PW, DeBosch BJ. TFEB-dependent induction of thermogenesis by the hepatocyte SLC2A inhibitor trehalose AU - Zhang, Yiming. Autophagy. 2018;14(11):1959–75. doi: 10.1080/15548627.2018.1493044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Settembre C, De Cegli R, Mansueto G, Saha PK, Vetrini F, Visvikis O, Huynh T, Carissimo A, Palmer D, Klisch TJ, Wollenberg AC, Di Bernardo D, Chan L, Irazoqui JE, Ballabio A. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat Cell Biol. 2013;15(6):647–58. Epub 2013/04/23. doi: 10.1038/ncb2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Napolitano G, Ballabio A. TFEB at a glance. Journal of Cell Science. 2016;129(13):2475. doi: 10.1242/jcs.146365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Piccinin E, Villani G, Moschetta A. Metabolic aspects in NAFLD, NASH and hepatocellular carcinoma: the role of PGC1 coactivators. Nature Reviews Gastroenterology & Hepatology. 2019;16(3):160–74. doi: 10.1038/s41575-018-0089-3. [DOI] [PubMed] [Google Scholar]
- 109.Puigserver P. Tissue-specific regulation of metabolic pathways through the transcriptional coactivator PGC1-α. International Journal of Obesity. 2005;29(1):S5–S9. doi: 10.1038/sj.ijo.0802905. [DOI] [PubMed] [Google Scholar]
- 110.Finck BN, Kelly DP. PGC-1 coactivators: inducible regulators of energy metabolism in health and disease. The Journal of Clinical Investigation. 2006;116(3):615–22. doi: 10.1172/JCI27794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Bougarne N, Weyers B, Desmet SJ, Deckers J, Ray DW, Staels B, De Bosscher K. Molecular Actions of PPARα in Lipid Metabolism and Inflammation. Endocrine Reviews. 2018;39(5):760–802. doi: 10.1210/er.2018-00064. [DOI] [PubMed] [Google Scholar]
- 112.Vaquero A, Reinberg D. Calorie restriction and the exercise of chromatin. Genes & Development. 2009;23(16):1849–69. doi: 10.1101/gad.1807009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Higgins CB, Zhang Y, Mayer AL, Fujiwara H, Stothard AI, Graham MJ, Swarts BM, DeBosch BJ. Hepatocyte ALOXE3 is induced during adaptive fasting and enhances insulin sensitivity by activating hepatic PPARγ. JCI Insight. 2018;3(16). doi: 10.1172/jci.insight.120794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Adastra KL, Frolova AI, Chi MM, Cusumano D, Bade M, Carayannopoulos MO, Moley KH. Slc2a8 Deficiency in Mice Results in Reproductive and Growth Impairments. Biology of Reproduction. 2012;87(2). doi: 10.1095/biolreprod.111.097675 %J Biology of Reproduction. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Walmagh M, Zhao R, Desmet T. Trehalose Analogues: Latest Insights in Properties and Biocatalytic Production. Int J Mol Sci. 2015;16(6):13729–45. Epub 2015/06/18. doi: 10.3390/ijms160613729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Yoshizane C, Mizote A, Yamada M, Arai N, Arai S, Maruta K, Mitsuzumi H, Ariyasu T, Ushio S, Fukuda S. Glycemic, insulinemic and incretin responses after oral trehalose ingestion in healthy subjects. Nutrition Journal. 2017;16(1):9. doi: 10.1186/s12937-017-0233-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Danielson ND, Collins J, Stothard AI, Dong QQ, Kalera K, Woodruff PJ, DeBosch BJ, Britton RA, Swarts BM. Degradation-resistant trehalose analogues block utilization of trehalose by hypervirulent Clostridioides difficile. Chemical Communications. 2019;55(34):5009–12. doi: 10.1039/C9CC01300H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Sarkar S, Davies JE, Huang Z, Tunnacliffe A, Rubinsztein DC. Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and alpha-synuclein. J Biol Chem. 2007;282(8):5641–52. Epub 2006/12/22. doi: 10.1074/jbc.M609532200. [DOI] [PubMed] [Google Scholar]
- 119.Lim Y-M, Lim H, Hur KY, Quan W, Lee H-Y, Cheon H, Ryu D, Koo S-H, Kim HL, Kim J, Komatsu M, Lee M-S. Systemic autophagy insufficiency compromises adaptation to metabolic stress and facilitates progression from obesity to diabetes. Nature Communications. 2014;5:4934. doi: 10.1038/ncomms593410.1038/ncomms5934https://www.nature.com/articles/ncomms5934#supplementary-informationhttps://www.nature.com/articles/ncomms5934#supplementary-information . [DOI] [PubMed] [Google Scholar]
- 120.<Arai et al. - 2010. - Trehalose prevents adipocyte hypertrophy and mitigates insulin resistance.pdf>. [DOI] [PubMed]
- 121.Arai C, Suyama A, Arai S, Arai N, Yoshizane C, Koya-Miyata S, Mizote A, Endo S, Ariyasu T, Mitsuzumi H, Ushio S. Trehalose itself plays a critical role on lipid metabolism: Trehalose increases jejunum cytoplasmic lipid droplets which negatively correlated with mesenteric adipocyte size in both HFD-fed trehalase KO and WT mice. Nutrition & Metabolism. 2020;17(1). doi: 10.1186/s12986-020-00443-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Sahebkar A, Hatamipour M, Tabatabaei SA. Trehalose administration attenuates atherosclerosis in rabbits fed a high-fat diet. Journal of Cellular Biochemistry. 2019;120(6):9455–9. doi: 10.1002/jcb.28221. [DOI] [PubMed] [Google Scholar]
- 123.Sciarretta S, Yee D, Nagarajan N, Bianchi F, Saito T, Valenti V, Tong M, Del Re DP, Vecchione C, Schirone L, Forte M, Rubattu S, Shirakabe A, Boppana VS, Volpe M, Frati G, Zhai P, Sadoshima J. Trehalose-Induced Activation of Autophagy Improves Cardiac Remodeling After Myocardial Infarction. Journal of the American College of Cardiology. 2018;71(18):1999. doi: 10.1016/j.jacc.2018.02.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Mayer A, Higgins CB, Ferey JL, Zhang Y, Weinheimer CJ, Kovacs A, Matkovich SJ, DeBosch BJ. Hepatocyte TFEB attenuates the pathological cardiac response to cardiac injury. JCI Insight. 2020;Manuscript Accepted. [Google Scholar]
- 125.Imai S, Yoshino J. The importance of NAMPT/NAD/SIRT1 in the systemic regulation of metabolism and ageing. Diabetes, obesity & metabolism. 2013;15 Suppl 3(0 3):26–33. doi: 10.1111/dom.12171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Stromsdorfer KL, Yamaguchi S, Yoon MJ, Moseley AC, Franczyk MP, Kelly SC, Qi N, Imai S-I, Yoshino J. NAMPT-Mediated NAD(+) Biosynthesis in Adipocytes Regulates Adipose Tissue Function and Multi-organ Insulin Sensitivity in Mice. Cell reports. 2016;16(7):1851–60. Epub 2016/08/04. doi: 10.1016/j.celrep.2016.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Wei M, Brandhorst S, Shelehchi M, Mirzaei H, Cheng CW, Budniak J, Groshen S, Mack WJ, Guen E, Di Biase S, Cohen P, Morgan TE, Dorff T, Hong K, Michalsen A, Laviano A, Longo VD. Fasting-mimicking diet and markers/risk factors for aging, diabetes, cancer, and cardiovascular disease. Science Translational Medicine. 2017;9(377):eaai8700. doi: 10.1126/scitranslmed.aai8700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Liang H, Ward WF. PGC-1alpha: a key regulator of energy metabolism. Adv Physiol Educ. 2006;30(4):145–51. Epub 2006/11/17. doi: 10.1152/advan.00052.2006. [DOI] [PubMed] [Google Scholar]
- 129.Fisher FM, Kleiner S, Douris N, Fox EC, Mepani RJ, Verdeguer F, Wu J, Kharitonenkov A, Flier JS, Maratos-Flier E, Spiegelman BM. FGF21 regulates PGC-1alpha and browning of white adipose tissues in adaptive thermogenesis. Genes Dev. 2012;26(3):271–81. Epub 2012/02/04. doi: 10.1101/gad.177857.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.van der Lans AA, Hoeks J, Brans B, Vijgen GH, Visser MG, Vosselman MJ. Cold acclimation recruits human brown fat and increases nonshivering thermogenesis. J Clin Invest. 2013;123. doi: 10.1172/JCI68993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Yamaguchi S, Franczyk MP, Chondronikola M, Qi N, Gunawardana SC, Stromsdorfer KL, Porter LC, Wozniak DF, Sasaki Y, Rensing N, Wong M, Piston DW, Klein S, Yoshino J. Adipose tissue NAD+ biosynthesis is required for regulating adaptive thermogenesis and whole-body energy homeostasis in mice. Proceedings of the National Academy of Sciences. 2019;116(47):23822. doi: 10.1073/pnas.1909917116. [DOI] [PMC free article] [PubMed] [Google Scholar]