

Abstract
Rationale: ACE2 (angiotensin-converting enzyme 2), the entry receptor for severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), is expressed in type 2 alveolar epithelial cells (AT2) that may play key roles in postinjury repair. An imbalance between ACE2 and ACE has also been hypothesized to contribute to lung injury.
Objectives: To characterize the expression and distribution of ACE2 and ACE and to compare AT2 with endothelial cell expression in coronavirus disease (COVID-19)–related or –unrelated acute respiratory distress syndrome (ARDS) and controls.
Methods: Lung tissue stainings (using multiplex immunofluorescence) and serum concentrations of ACEs were determined retrospectively in two different cohorts of patients. AT2 and endothelial cells were stained in lung tissue for ProSPC (pro-surfactant protein C) and CD31, respectively.
Measurements and Main Results: Pulmonary ACE2 expression was increased in patients with COVID-19–related and –unrelated ARDS (0.06% of tissue area and 0.12% vs. 0.006% for control subjects; P = 0.013 and P < 0.0001, respectively). ACE2 was upregulated in endothelial cells (0.32% and 0.53% vs. 0.01%; P = 0.009 and P < 0.0001) but not in AT2 cells (0.13% and 0.08% vs. 0.03%; P = 0.94 and P = 0.44). Pulmonary expression of ACE was decreased in both COVID-19–related and –unrelated ARDS (P = 0.057 and P = 0.032). Similar increases in ACE2 and decreases in ACE were observed in sera of COVID-19 (P = 0.0054 and P < 0.0001) and non–COVID-19 ARDS (P < 0.0001 and P = 0.016). In addition, AT2 cells were decreased in patients with COVID-19–related ARDS compared with COVID-19–unrelated ARDS (1.395% vs. 2.94%, P = 0.0033).
Conclusions: ACE2 is upregulated in lung tissue and serum of both COVID-19–related and –unrelated ARDS, whereas a loss of AT2 cells is selectively observed in COVID-19–related ARDS.
Keywords: angiotensin-converting enzyme 2, angiotensin-converting enzyme, type 2 alveolar epithelial cells, COVID-19, ARDS
At a Glance Commentary
Scientific Knowledge on the Subject
ACE1 and 2 (angiotensin-converting enzyme 1 and 2) are thought to play important roles in COVID-19–related acute respiratory distress syndrome (ARDS). In particular, ACE2 is the entry receptor for severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and is expressed in type 2 alveolar epithelial (AT2) cells, which may play key roles in postinjury repair. However, the expression of ACEs and of AT2 cells in patients with ARDS, related or not to COVID-19, has never been quantified.
What This Study Adds to the Field
In this monocentric retrospective study, we found a significant shift in the expression of ACEs, from ACE to ACE2, that is seen both in the lung and in the serum of patients with ARDS, related or not to COVID-19, compared with control subjects. In contrast, a decrease in the expression of AT2 cells was selectively observed in COVID-19–related ARDS.
ACEs (angiotensin-converting enzymes) are parts of the renin–angiotensin (Ang) system (RAS) pathway that play important roles in the regulation of blood pressure as well as electrolyte and fluid homeostasis. ACE cleaves Ang I into Ang II, a vasoconstrictive peptide with profibrotic and proinflammatory effects; ACE2 degrades Ang II into smaller peptides (mainly Ang [1–7]), which, via the Mas receptor (1), antagonize the action of Ang II.
Severe acute respiratory syndrome coronaviruses (SARS-CoV) use ACE2 as host receptors for cell entry (2, 3). Alterations in the ACE and/or ACE2 pathways may also contribute to the pathophysiology of both SARS-CoV (2, 4) and SARS-CoV-2 (5) lung infection. Animal and experimental models of SARS-CoV showed that, after binding to ACE2, the viral complex is endocytosed and surface ACE2 is downregulated, probably through proteolytic cleavage (4). Although formally unproven, similar mechanisms are thought to take place during SARS-CoV-2 infection (3, 6). This decrease in ACE2 expression in tissues could lead to an imbalance in the ACE/Ang II–ACE2/Ang (1–7) ratio, which has been linked with acute lung and cardiovascular injury (7). In addition, ACE2 has been shown to exert protective effects against experimental acute respiratory distress syndrome (ARDS) caused by a variety of experimental insults (7–11). Altogether, current evidence suggests that ACE/ACE2 (im)balance could contribute to acute lung injury owing to SARS-CoV-2 infection and could influence coronavirus disease (COVID-19) course. However, so far, few human studies have assessed the role of ACE and ACE2 in patients with ARDS (12, 13), and none have compared the expression of ACE and ACE2 among COVID-19–related ARDS, ARDS from other causes, and control lung tissue.
Alveolar epithelial type II cells (AT2) play important roles in postinjury repair of the alveolar surface (14), frequently express ACE2 (15), and are thought to be a main target of SARS-CoV-2 (16). However, their expression has never been compared among lung samples from patients with COVID-19–related ARDS, patients with COVID-19–unrelated ARDS, and control patients without ARDS. We investigated distribution and intensity of expression of ACE2 and ACE and density of AT2 cells in lung samples from these three groups of patients—those with COVID-19–related ARDS, those with COVID-19–unrelated ARDS, and control patients without ARDS. We also measured ACE and ACE2 levels (and their respective metabolites) in sera from large cohorts of patients with severe SARS-CoV-2 infection, patients with ARDS unrelated to SARS-CoV-2 infection, and control subjects with no SARS-CoV-2 infection and no other respiratory disease.
Methods
More details regarding study protocols, patient selection, data collection, sampling processing, immunostaining, and imaging can be found in the online supplement.
Patients
This is a retrospective monocenter study that included pulmonary autopsy specimens from patients who died at the Cliniques universitaires Saint-Luc (Brussels, Belgium) between March 19 and May 4, 2020, with a diagnosis of COVID-19 and who fulfilled the Berlin definition for ARDS (17). The COVID-19 cases were defined as the combination of a positive RT-PCR for SARS-CoV-2 (details in the online supplement) in a nasopharyngeal swab or in BAL specimens and consistent abnormalities on chest X-ray or computed tomography. Among the 19 patients who died in our ICU with proven SARS-CoV-2 infection, autopsy was performed on 16 patients (84%). One specimen was rejected because of poor quality of the lung tissue, so postmortem samples from 15 patients with SARS-CoV-2 ARDS were analyzed in this report. COVID-19–unrelated ARDS and control human lung biopsies were selected from our biobank. Patients included in the COVID-19–unrelated ARDS group were retrospectively selected from a database of patients with a diagnosis of ARDS (17, 18) and for whom lung tissue had been sampled with open-lung biopsy between 2008 and 2020. To match the COVID-19 cohort, these patients were selected based on severity of ARDS (PaO2/FiO2 < 200 mm Hg) and on the presence of diffuse alveolar damage on histopathological examination. Samples of lung tissue from 13 patients were selected and included in the analysis. The control group consisted of lung tissue from 15 patients who had undergone lobectomy for a solitary lung tumor, with sections taken at distance from the tumor lesions, between 2018 and 2019. For COVID-19–unrelated ARDS and for control subjects, the negative status for COVID-19 was reasonably assumed for patients who had undergone lung biopsy before November 2019 and was proven by at least two negative RT-PCR tests on nasopharyngal swabs for patients who underwent lung biopsy after November 2019.
For quantification of serum ACE and ACE2 expression, we retrospectively reviewed all patients with PCR-proven severe SARS-CoV-2 infection, admitted in the ICU of Cliniques universitaires Saint-Luc (Brussels, Belgium) between March 12 and October 15, 2020 (n = 95), and for whom serum had been prospectively collected within 48 hours of ICU admission (owing to enrollment in another prospective observational study, n = 87). We included all patients aged >18 years for whom the reason for ICU admission was acute respiratory failure (n = 82). Sera from these patients with severe SARS-CoV-2 were compared first with sera from patients with moderate or severe ARDS unrelated to COVID-19, who had been prospectively enrolled in an observational study between 2016 and 2020 (unpublished data) and for whom serum had to be sampled, per protocol, within the 24 hours of the diagnosis of ARDS (n = 24). Sera from the two ARDS patient cohorts were then compared with serum samples from subjects with no evidence of either SARS-CoV-2 infection or respiratory disease (n = 18). Sera from the same patients were used for Ang (1–7) and Ang II quantification, but because of limited sample availability, measurements were performed in 35 out of 82 patients in the COVID-19 group (and in all control subjects and patients with COVID-19–unrelated ARDS).
Data Collection
Clinical data were retrospectively retrieved from Electronic Medical Recording (Qcare PDMS, HIM Health Information Management GmBH). All details appear in the online supplement.
Multiplex Fluorescence Immunohistochemistry in Lung Tissue
Formalin-fixed, paraffin-embedded tissue processing and Tyramide signal amplification–based multiplex fluorescence immunohistochemistry were performed for ACE2 and ACE, CD31, ProSPC (pro-surfactant protein C), CD45, Ki-67, and HTII-280, as detailed in the online supplement.
Immunofluorescent probes were selected based on their specificity as follows: CD31 for endothelial cells (also staining immune cells such as macrophages), ProSPC for AT2 cells, CD45 for leukocytes (all hematopoietic cells except erythrocytes and platelets), Ki-67 as a generic marker of cell proliferation, and HTII-280 for apical membrane of AT2 cells. Negative controls stained with control IgG from the adequate species were performed to assess nonspecific binding of secondary antibodies.
Imaging and Quantitative Evaluation of Immunostaining in Whole Tissue Sections
Multiplex immunostained slides were digitalized in fluorescence using a Pannoramic 250 FlashIII scanner (3DHistech) at ×20 magnification. Stainings were quantified on entire tissue sections with software applications using the image analysis tool Author version 2017.2 (Visiopharm). For the localization of ACE1 and ACE2 in endothelial or epithelial compartments, ACE1, ACE2, CD31, and proSPC-stained pixels were detected at high resolution (×20) using a thresholding classification method on each fluorescence channel. Results were expressed as percentage of ACE1 or ACE2 costained pixels among either CD31 or ProSPC-stained pixels. The same procedure was applied to investigate the localization of Ki-67 within AT2 cells. The detection parameters were kept constant for all slides. All analyses were performed by an experienced observer who was blinded to the clinical categories of the tissue samples. A further description of the quantified imaging appears in the online supplement.
Serum Assays
For ACE and ACE2 quantification, we performed ELISA according to the manufacturer’s instructions with Human ACE DuoSet ELISA (R&D, DY929) and Human ACE2 DuoSet ELISA (R&D, DY933-05). Ang (1–7) concentration was determined by ELISA according to the manufacturer’s instructions with MyoBioSource Ang1–7 ELISA Kit, MBS084052. Ang II was quantified using enzyme immunoassay technique with RayBio Human/Mouse/Rat Angiotensin II Enzyme Immunoassay Kit.
Statistical Analysis
Statistical analyses were performed using SPSS 21 (IBM SPSS Statistics for Windows, Version 21.0), and figures were created using Graphpad Prism 8 (GraphPad Software). Values were expressed as median (interquartile range [IQR]) or mean (SD) for continuous values and counts (per percent of group) for qualitative variables. Categorical and continuous variables were analyzed using the chi-square test and Mann-Whitney test, respectively. We compared tissue and serum expression of relevant biochemical variables between the three groups using Kruskall-Wallis test with a Dunn’s post hoc test to correct for multiple comparisons. To find factors influencing survival and to assess whether serum levels of ACE and ACE2 were related to outcomes, a Cox proportional hazards model was built as follows: all the variables significant in the univariate analysis (P < 0.25) were entered into a multivariate logistic regression with a backward elimination procedure based on likelihood ratios. A second, simplified model (details in the online supplement) was built to test the findings. The results were expressed as hazard ratios with 95% confidence intervals. All tests were two-sided, with the significance level set at 0.05.
Results
Expression of ACE2 Is Upregulated in Severe COVID-19–Related and –Unrelated ARDS
The demographic characteristics and use of adjuvant treatments of the 15 patients with COVID-19–related ARDS, 13 patients with COVID-19–unrelated ARDS, and 15 control subjects are summarized in Table 1.
Table 1.
Baseline Characteristics, Adjunctive Treatments, and Tissue Sampling Characteristics in Control Subjects and Patients with COVID-19–Unrelated ARDS and COVID-19–Related ARDS for Lung Tissue Analyses
| Variables | Control Subjects (n = 15) | Non–COVID-19 ARDS (n = 13) | COVID-19 ARDS (n = 15) |
|---|---|---|---|
| Age, yr | 56 (45–68) | 58 (40–70) | 62 (54–67) |
| Sex, M | 9 (60) | 7 (54) | 9 (60) |
| Body mass index, kg/m2 | 24.6 (21.6–26.2) | 25.9 (22.6–33.9) | 27.3 (23.6–30.8) |
| Hypertension | 2 (13) | 6 (46) | 7 (43) |
| Diabetes | 1 (6.5) | 2 (15) | 3 (20) |
| Active smoking | 5 (33) | 2 (15) | 1 (6.5) |
| RAS interacting drugs | 1 (6.5) | 2 (15) | 6 (40)* |
| Chronic obstructive pulmonary disease | 0 | 0 | 1 (6.5) |
| Active cancer | 14 (93) | 2 (15)* | 0* |
| APACHE II score | 19 (17–25) | 16 (10–21) | |
| SOFA score | 6.5 (5–9) | 4 (4–9) | |
| Cause of ARDS | |||
| COVID-19 | 0 | 15 (100)† | |
| Pneumonia | 7 (54) | 0† | |
| Sepsis, extrapulmonary origin | 4 (31) | 0† | |
| Other | 2 (15) | 0 | |
| Adjunctive treatments before tissue sample | |||
| Invasive mechanical ventilation | 13 (100) | 14 (93) | |
| Prone positioning | 5 (38) | 14 (93)† | |
| Inhaled nitric oxide | 4 (31) | 14 (93)† | |
| ECMO | 0 | 4 (27)† | |
| Vasopressors | 5 (38) | 14 (93)† | |
| Renal replacement therapy | 4 (31) | 4 (26) | |
| Corticosteroids | 0 | 0 | |
| Type of tissue sampling | |||
| Postmortem autopsy | 0 | 0* | 15 (100)*† |
| Open-lung biopsy | 0 | 13 (100) | 0 |
| Surgical lobectomy | 15 (100) | 0 | 0 |
| Duration between diagnosis and tissue sampling, d | 13 (8–17) | 16 (10–25)† | |
| Duration between MV initiation and tissue sampling, d | 11 (3–13) | 13 (9–24) | |
| Diffuse alveolar damage on pathological examination | 0 | 13 (100)* | 14 (93)* |
Definition of abbreviations: APACHE II = Acute Physiology and Chronic Health Evaluation II; ARDS = acute respiratory distress syndrome; COVID-19 = coronavirus disease; ECMO = extracorporeal membrane oxygenation; IQR = interquartile range; MV = mechanical ventilation; RAS = renin–angiotensin system; SOFA = Sequential Organ Failure Assessment.
Data are presented as count (%) or median (IQR).
P < 0.05 compared with control subjects.
P < 0.05 compared with ARDS.
ACE2 staining analysis revealed a marginal presence of ACE2 in control lungs (0.006% of total tissue area [IQR, 0.002–0.04]), mainly in proSPC expressing AT2 (0.03% of AT2 area [0.02–0.15]) (Figure 1B). ACE2 expression was increased (Figure 1E) in both COVID-19–related (0.06% of total tissue area [0.015–0.11]; P = 0.013) (Figures 1A and 1C) and COVID-19–unrelated ARDS (0.12% of total tissue area [IQR, 0.047–0.26]; P < 0.0001) (Figure 1D). The difference in ACE2 expression between COVID-19–related and –unrelated ARDS was not statistically significant (P = 0.55). Quantification in ProSPC-stained AT2 cells and CD31-stained endothelial cells was performed (i.e., CD31+, ACE2+, and proSPC+AEC2+ cells) and showed a selective upregulation of ACE2 in pulmonary endothelial cells in patients with COVID-19 and non–COVID-19 ARDS compared with control subjects (0.32% of endothelial area [0.05–0.52] and 0.53% [0.37–1.440] vs. 0.01% [0.005–0.02]; P = 0.009 and P < 0.0001) (Figure 1F) but no upregulation in AT2 cells (0.13% of AT2 area [0.01–0.42] and 0.08% [0.025–0.16] vs. 0.03% [0.02–0.15]; P = 0.94 and P = 0.44, respectively) (Figure 1G).
Figure 1.
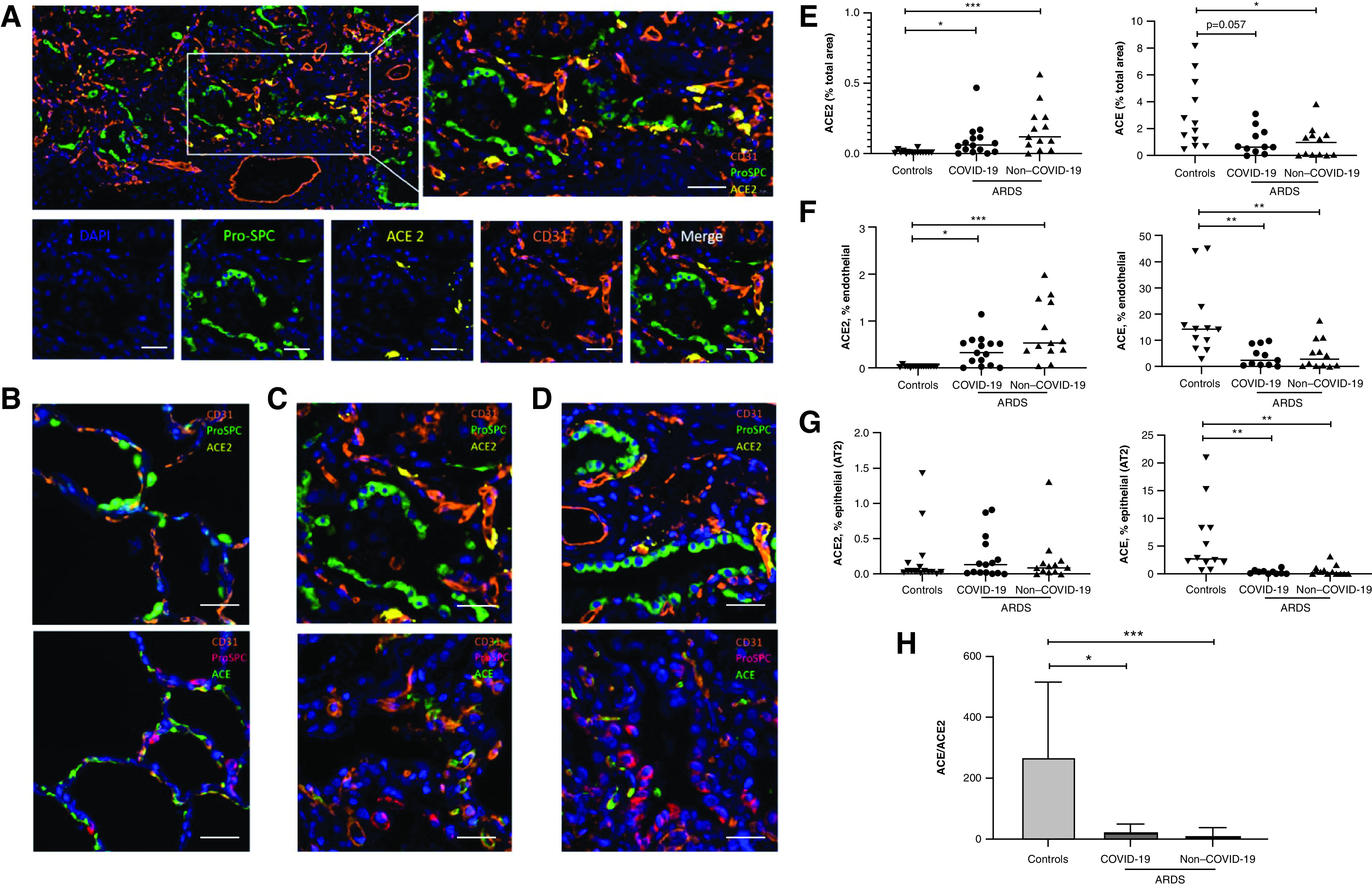
Expression of ACE2 (angiotensin-converting enzyme 2) is upregulated and expression of ACE is downregulated in severe coronavirus disease (COVID-19)–related and –unrelated acute respiratory distress syndrome (ARDS). (A) One representative picture of a lung section from a patient with COVID-19–related ARDS, with corresponding high-magnification panels, is shown for distal lung tissue, costained with ProSPC (pro-surfactant protein C) (type 2 alveolar epithelial cells, green), CD31 (endothelial cells, orange), and ACE2 (yellow) and counterstained with DAPI (blue). (B–D) Sections from control subjects (B), patients with COVID-19–related ARDS (C), and patients with COVID-19–unrelated ARDS (D) were costained with ACE2 (upper panels), ACE (lower panels), CD31, and ProSPC and counterstained with DAPI (blue). One representative picture is shown for each staining. Scale bars, 50 μm. (E–G, left) Quantification of the total ACE2 area (percentage of total tissue area) (E), of the area coexpressing CD31 and ACE2 (percentage of the total CD31+ area) (F), and of the area coexpressing ProSPC and ACE2 (percentage of the total proSPC area) (G), comparing control subjects (n = 15) with patients with COVID-19–related ARDS (n = 14) and patients with COVID-19–unrelated ARDS (n = 13). (E–G, right) Quantification of the total ACE area (percentage of total tissue area) (E), of the area coexpressing CD31 and ACE (percentage of the total CD31+ area) (F), of the area coexpressing ProSPC and ACE (percentage of the total proSPC area) (G), comparing control subjects (n = 12) with patients with COVID-19–related ARDS (n = 11) and patients with COVID-19–unrelated ARDS (n = 12). Because of technical issues with the staining, data regarding ACE2/ACE could be analyzed in 15/12 out of 15 control subjects, 14/11 out of 15 COVID-19–related ARDS, and 13/12 out of 13 COVID-19–unrelated ARDS. (H) Relative expression of ACE and ACE2 in control subjects (n = 12), patients with COVID-19–related ARDS (n = 11), and patients with COVID-19–unrelated ARDS (n = 12). The ACE/ACE2 ratio was calculated as the ratio of the total areas stained for ACE and ACE2. Data were expressed as means ± SD. Each dataset was compared separately using Kruskall-Wallis test with a Dunn’s post hoc test. *P < 0.05, **P < 0.005, and ***P < 0.0005. AT2 = type 2 alveolar epithelial cells.
In contrast to ACE2, pulmonary expression of ACE (Figure 1C, lower panel, with detailed immunostaining provided in Figure E1 in the online supplement) was decreased in COVID-19–related (0.64% [0.27–1.64] of total tissue area) and COVID-19–unrelated ARDS (0.71% [0.057–1.54]) (Figure 1D) compared with controls (2.11% [0.84–5.14]; P = 0.057 and P = 0.0323, respectively) (Figure 1B), although this did not reach statistical significance for COVID-19–related ARDS lungs (Figure 1E). Colocalization studies indicated that ACE downregulation concerned both endothelial (in which ACE was predominantly expressed in controls) and AT2 epithelial cells (Figures 1F and 1G). In addition, costaining for ACE, ACE2, CD31, and CD45 (pan-leukocyte marker) was performed on a sample of four control subjects and five patients with ARDS. Examination for this representative sample revealed no colocalization of ACE2 or ACE with CD45, confirming that double positive CD31+ ACE2+ (or CD31+ ACE+) cells were endothelial cells (Figures E2 and E3).
The ACE–ACE2 shift in patients with ARDS, both COVID-19–related and –unrelated, was witnessed by a drop of the ACE/ACE2 ratio compared with control subjects (7.99 [3.4–36.7] and 1.7 [0.35–17.5] vs. 131 [91.2–514.2]; P = 0.016 and P = 0.0002, respectively), whereas the difference between ARDS groups was not statistically significant (P = 0.92) (Figure 1G). No correlation of this altered ratio (or its components) with classical risk factors of severe COVID-19 (age, male sex, tobacco use, arterial hypertension, diabetes, obesity) was observed within our lung samples from patients with COVID-19 (Figure E4).
AT2 Cells Are Downregulated in COVID-19–Related ARDS
The immunostaining for proSPC (Figures 2A–2C, upper panels) that reflects the number of AT2 cells was decreased in COVID-19–related ARDS compared with COVID-19–unrelated ARDS (1.395% of total tissue area [0.1950–2.11] vs. 2.94% [2.18–4.65]; P = 0.0033) (Figure 2D). A second staining with anti–HTII-280 (Figures 2A–2C, middle panels), a biomarker specific of the apical plasma membrane of human AT2 cells (19), confirmed the decrease in AT2 cells (Figure 2F) in patients with COVID-19–related ARDS (0.44% of total tissue area [0.25–0.61]) in comparison with COVID-19–unrelated ARDS (1.2% [0.83–1.7]; P = 0.0028) but also with control subjects (1.76% [0.85–1.9]; P = 0.0004). In contrast, no change in CD31, reflecting the number of endothelial cells, was observed (Figure 2E).
Figure 2.
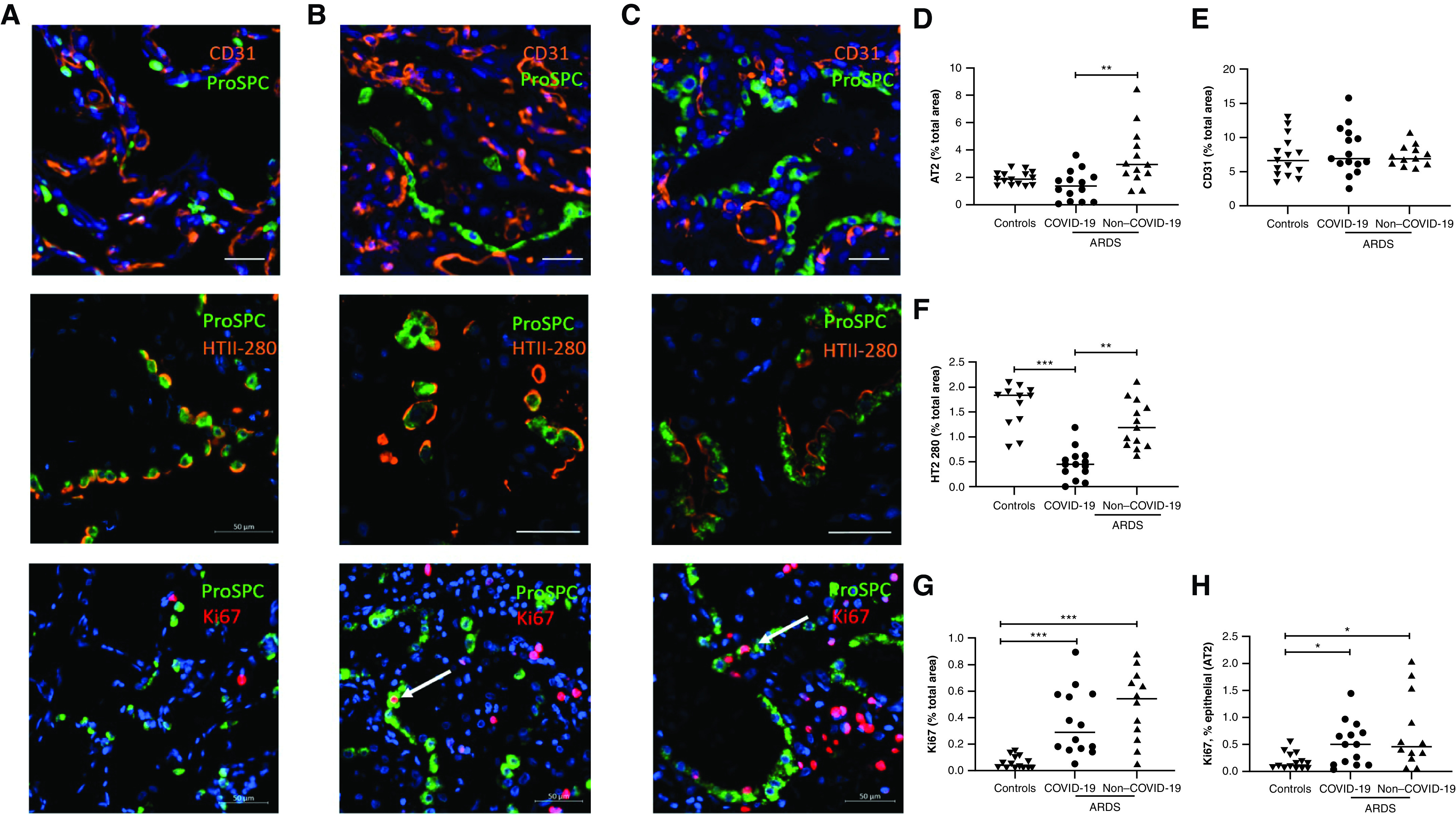
Type 2 alveolar epithelial cells (AT2) are downregulated in coronavirus disease (COVID-19)–related acute respiratory distress syndrome (ARDS). (A–C) Sections from control subjects (A), patients with COVID-19–related ARDS (B), and patients with COVID-19–unrelated ARDS (C) were costained with CD31 (endothelial cells, orange) (upper panel), HTII-280 (middle panel), or Ki-67 (proliferative marker, red) (lower panel), and ProSPC (surfactant protein C; AT2, green) and counterstained with DAPI (blue). Arrows show representative examples of AT2 cells expressing Ki-67. Scale bars, 50 μm. (D and E). Quantification of the AT2 area (total ProSPC area/total tissue area) (D) and of the area CD31+ (total CD31 area/total tissue area) (E), comparing control subjects (n = 15) with patients with COVID-19–related ARDS (n = 15) and patients with COVID-19–unrelated ARDS (n = 13). (F) Quantification of the area expressing HTII-280 (total HTII-280 area/total tissue area), comparing control subjects (n = 12) with patients with COVID-19–related ARDS (n = 14) and patients with COVID-19–unrelated ARDS (n = 13). (G and H) Quantification of the area expressing Ki-67+ (total Ki67 area/total tissue area) (G) and of the area coexpressing Ki-67 and ProSPC (percentage of the total area ProSPC+) (H), comparing control subjects (n = 14) with patients with COVID-19–related ARDS (n = 14) and patients with COVID-19–unrelated ARDS (n = 12). Because of technical issues with the staining, data regarding HTII-280/Ki-67 could be analyzed in 12/14 out of 15 control subjects, 14/14 out of 15 COVID-19–related ARDS, and 13/12 out of 13 COVID-19–unrelated ARDS. Each dataset was compared separately using a Kruskall-Wallis test with a Dunn’s post hoc test. *P < 0.05, **P < 0.005, and ***P < 0.0005.
Finally, Ki-67 staining was used to assess all proliferation (Figures 2A–2C, lower panels). Ki-67 expression was increased in both COVID-19–unrelated and COVID-19–related ARDS compared with controls: 0.54% (of total lung tissue) and 0.29% versus 0.04% (0.02–0.11) (P < 0.0001 and P = 0.0005) for whole lung tissue and 0.46% (of AT2 area) and 0.51% versus 0.13% (0.06–0.32) (P = 0.014 and P = 0.046) specifically for AT2 cells (Figure 2A–2C, lower panels). There was, however, no difference in this proliferative index between samples from lungs with COVID-19–unrelated ARDS and COVID-19–related ARDS: 0.54% of total lung tissue for COVID-19–unrelated samples (0.25–0.70) versus 0.29% for COVID-19–related samples (0.16–0.58) (P = 0.23). There was no difference in the Ki-67 staining specifically between AT2 cells in the two settings: 0.46% of AT2 area (0.25–1.38) in COVID-19–unrelated ARDS samples and 0.51% of AT2 area (0.12–0.67) in the COVID-19–related ARDS samples (P = 0.63) (Figures 2G and 2H).
Expression of ACE2 Is Increased and Expression of ACE Is Decreased in the Serum of Patients with Severe COVID-19 and Non–COVID-19 ARDS
Demographic characteristics, use of adjuvant treatments, and outcomes of 82 patients with severe COVID-19, 24 patients with non–COVID-19 ARDS, and 18 control subjects are summarized in Table 2. In line with findings in the lungs, circulating ACE levels were significantly lower (30.4 ng/ml [11.4–59.8] and 62.01 [36.2–79] vs. 113.2 [89–147.8]; P < 0.0001 and P = 0.016, respectively) (Figure 3A) whereas ACE2 concentrations were significantly higher in patients with severe COVID-19 and in COVID-19–unrelated ARDS compared with control subjects (695.7 pg/ml [224.5–1565] and 1,434 [688.2–2115] vs. 302.7 [24.8–398.8]; P = 0.0054 and P < 0.0001, respectively) (Figure 3B). These differences led to decreased ACE/ACE2 ratios in both ARDS groups (28.37 [7.69–73.95] and 45.15 [23.68–76.81] vs. 395.8 [220.2–1122]; P < 0.0001 and P = 0.001, respectively) (Figure 3E). We also found a statistically significant increase in Ang (1–7) concentrations in a subset of patients with severe COVID-19 (whose characteristics are summarized in Table E2) and in patients with non–COVID-19 ARDS compared with control subjects (208 pg/ml [136–295] and 170 [122–236] vs. 24 [15–40]; P < 0.0001 and P = 0.0002, respectively), whereas concentrations of Ang II were below detection levels in most samples from both patients with ARDS and control subjects (Figures 3C and 3D).
Table 2.
Baseline Characteristics, Adjunctive Treatments, and Outcomes for Patients with COVID-19–Unrelated ARDS, Patients with Severe COVID-19, and Control Subjects for Serum ACE (1/2) Assays
| Variables | Control Subjects (n = 18) | ARDS (n = 24) | COVID-19 (n = 82) |
|---|---|---|---|
| Age, yr | 60.5 (55–66) | 59.5 (48–74.5) | 62.5 (54–68.5) |
| Sex, M | 11 (65) | 18 (75) | 64 (78) |
| Body mass index, kg/m2 | 26.4 (24–32) | 27.7 (23.9–31) | 27.6 (24.5–31) |
| Hypertension | 47 (58) | 12 (50) | 51 (61) |
| Diabetes | 3 (16) | 3 (12) | 19(24) |
| Active smoking | 4 (22) | 4 (16) | 3 (4)* |
| RAS interacting drugs | 12 (66) | 8 (33) | 33 (40) |
| Chronic obstructive pulmonary disease | 1 (5) | 2 (8) | 2 (3) |
| Active cancer | 0 | 3 (12) | 8 (10) |
| APACHE II score | 20 (15–26) | 14 (12–18)† | |
| SOFA score | 7 (4–10) | 4 (3–5)† | |
| Cause of ARDS | |||
| COVID-19 | 0 | 82 (100)† | |
| Pneumonia | 11 (46) | 0 | |
| Sepsis, extrapulmonary origin | 8 (33) | 0 | |
| Other | 5 (20) | 0 | |
| Delay ICU admission, sampling, d | 2 (1–3) | 1 (0–1)† | |
| Delay hospital admission, sampling, d | 3 (2–5) | 2 (1–4)† | |
| Mode of ventilation (day of sampling) | |||
| HFNC/NIPPV | 0 | 77 (94)† | |
| Invasive MV | 24 (100) | 5 (6)† | |
| PaO2/FiO2 ratio (day of sampling), mm Hg | 130 (104–170) | 85 (61–117)† | |
| Adjunctive treatments | |||
| Invasive mechanical ventilation | 24 (100) | 51 (62)† | |
| Prone positioning | 8 (33) | 46 (56)† | |
| ECMO | 0 | 14 (17)† | |
| Vasopressors | 13 (54) | 42 (52) | |
| RRT | 7 (29) | 12 (15) | |
| Corticosteroids | 3 (12) | 24 (30) | |
| 30-d mortality | 8 (33) | 28 (35) | |
| ICU mortality | 8 (33) | 29 (38) | |
| ICU LOS | 12.5 (8–22) | 9 (3–23) | |
| VFDays D28 | 5.5 (0–20) | 3.2 (0–28) |
Definition of abbreviations: ACE = angiotensin-converting enzyme; APACHE II = Acute Physiology and Chronic Health Evaluation II; ARDS = acute respiratory distress syndrome; COVID-19 = coronavirus disease; ECMO = extracorporeal membrane oxygenation; HFNC = high-flow nasal canula; IQR = interquartile range; LOS = length of stay; MV = mechanical ventilation; NIPPV = noninvasive positive pressure ventilation; RAS = renin–angiotensin system; RRT = renal replacement therapy; SOFA = Sequential Organ Failure Assessment; VFDays D28 = ventilatory-free days at Day 28.
Data are presented as count (%) or median (IQR).
P < 0.05 compared with control subjects.
P < 0.05 compared with ARDS.
Figure 3.

Quantification of ACE (angiotensin-converting enzyme), ACE2, angiotensin II (Ang II), and Ang (1–7) serum concentrations and of the relative expression of ACE to ACE2 in patients with coronavirus disease (COVID-19)–unrelated acute respiratory distress syndrome (ARDS), patients with severe COVID-19, and control subjects. (A and B) Serum concentrations of ACE (A) and ACE2 (B) were determined using ELISA in control patients (n = 18), patients with severe COVID-19 (n = 82), and patients with COVID-19–unrelated ARDS (n = 24). (C) Serum concentrations of Ang II were compared using enzyme immunoassay in control patients (n = 18) and those with severe COVID-19 (n = 35). Most values were below the detection threshold (0.3 pg/ml according to manufacturer’s instructions). (D) Serum concentrations of Ang (1–7) were compared using ELISA in control patients (n = 18), patients with severe COVID-19 (n = 35), and patients with ARDS (n = 24). Because of sample availability, Ang (1–7) and Ang II could be quantified in only 35/82 patients with severe COVID-19, and no quantification of Ang II could be made in COVID-19–unrelated ARDS. (E) ACE/ACE2 ratio was calculated as the ratio of the concentrations of ACE and ACE2 (means ± SD). Each dataset was compared separately using Kruskall-Wallis test with a Dunn’s post hoc test. *P < 0.05, **P < 0.005, and ***P < 0.0005.
In a multivariate analysis conducted on the 82 patients with severe COVID-19, neither ACE, ACE2, nor their ratio were associated with ICU mortality or with a composite of mortality or invasive mechanical ventilation using two different models (details in Tables E3–E6).
Discussion
To the best of our knowledge, this study is the first to compare the expression of ACE and ACE2 in the lung tissue and serum of patients with severe COVID-19, patients with ARDS, and control subjects. A significant increase in ACE2 was observed, mainly driven by its induction on endothelial cells, both in COVID-19–related and –unrelated ARDS. In contrast, ACE was downregulated in the same patients both in endothelial and type 2 epithelial cells. This change in the ACE/ACE2 ratio documents a dramatic shift from ACE to ACE2 in all ARDS, whether owing to SARS-CoV-2 or not.
Similar increases in ACE2 and decreases in ACE were observed in the serum of patients with COVID-19 and ARDS and were associated with increases in Ang (1–7).
In our study, a low tissue expression of ACE2 (albeit higher in patients with COVID-19–related or –unrelated ARDS) was observed, with the ACE2-positive area accounting for less than 0.2% of the total tissue area. The exact distribution of pulmonary ACE2 remains to be definitely determined, as several studies have attempted to map ACE2 expression, relying mainly on mRNA data, but have returned contradictory results (20–23). Few publications have addressed the expression of ACE2 at the protein level (15, 24–26). In the largest series to date, pulmonary ACE2 expression was very limited, being mainly confined to a small subset of AT2 cells (15). In our control sections, ACE2 appeared mostly in AT2 cells, with only occasional localization to endothelial and airway lining cells. Furthermore, this study is one of the few efforts so far to quantify ACE expression and distribution in lung tissue, whether diseased or healthy. In our control sections, ACE appeared predominantly in endothelial cells as it usually does in microvascular-rich tissues.
Data regarding changes in ACE and ACE2 expression in lung tissue during ARDS or during coronavirus infection is limited. Nonetheless, previous work in murine models showed that ACE2 expression was decreased during ARDS (8) or following SARS-CoV infection (presumably because of viral-induced endocytosis or increased cleavage by the metalloproteinase ADAM17) (4), whereas ACE pulmonary expression was unaltered (4). This evidence seems in apparent contradiction with our study, showing an increase in ACE2 expression (and no change of ACE2 expression by AT2 cells) and a decrease of ACE expression. However, recent work comparing ACE2 expression in patients with COVID-19 or influenza and control subjects showed similar results, reporting a significant increase of ACE2 expression in the first two groups (27), more specifically within the endothelial cell compartment. Discrepancies between experimental animal models and human data might be explained by ACE2 species-related regulatory mechanisms. Thus, analysis of multiple single-cell RNA sequencing datasets has revealed that, within specific subsets of human epithelial cells, ACE2 expression is stimulated by IFN (28, 29), and human epithelial cells treated with IFN-α2 and IFN-γ show an upregulation of ACE2 expression (28). In contrast, this response is absent in mice (28), suggesting a species-specific mechanism. Despite conflicting evidence, robust IFN-I response has been linked with severe forms of SARS-CoV-2 infection (30–33) or with ARDS of various origins (34–37). Of note, the loss of ACE2 expression observed in murine models during acute lung injury or SARS-CoV infection seems to take place early in the pathogenesis, whereas the dysregulated IFN-I response, and subsequent ACE2 upregulation, could appear later in the disease course (33). As lung tissue sampling occurred late in our study (after a median of 16 days in patients with COVID-19 vs. 13 days in patients with ARDS), the observed upregulation of ACE2 expression could therefore be the consequence of a prolonged exposure to a dysregulated IFN-I response.
In light of the increase of ACE2 in both COVID-19–related and –unrelated ARDS, the shift in the ACE/ACE2 balance seems to be a generic response of the lung to acute injury rather than a specific feature of severe COVID-19. Likewise, the shift of the ACE/ACE2 balance is probably a time-sensitive phenomenon, as illustrated by the interplay with IFN-I, and could potentially be influenced by many confounding factors.
We also observed an increase in serum ACE2 combined with a decrease in ACE in patients with COVID-19–related and COVID-19–unrelated ARDS compared with control subjects, which we could not connect with any changes in clinical outcomes.
In addition, Ang (1–7) was increased in a subset of patients with COVID-19 and ARDS compared with control subjects, probably reflecting an increase in the conversion of Ang II (whose concentrations could not reliably be determined in the present study) by ACE2, whose lung tissue expression and serum concentrations were shown to be increased. However, the increase of Ang (1–7) might also reflect a more global RAS dysregulation, as it was suggested recently that conversion from Ang II to Ang (1–7) in the circulation was mainly independent from ACE2 (38). ACE and ACE2 concentrations have been recently characterized in patients with severe COVID-19 with consistent results. Reindl and colleagues found an increase in enzymatically active plasma ACE2 concentrations in patients with severe COVID-19 from early to late time points during the disease course, with no difference in early ACE2 concentrations between severe COVID-19 and a control cohort of patients with severe influenza under invasive mechanical ventilation (39). In two other retrospective studies, ACE2 activity was shown to be markedly increased in critically ill patients with COVID-19 compared with healthy control subjects (40, 41). On the other hand, ACE activity was found to be decreased in patients with severe and nonsevere COVID-19 compared with control subjects, with the lowest level in the severe group (42). However, conflicting results have been reported regarding the main RAS peptides, Ang II and Ang (1–7). Whereas an increase in plasma Ang II levels in patients with COVID-19 was found in early reports (43, 44), the opposite was shown in recent work using gold-standard assays (40). Finally, Ang (1–7) was recently shown to be increased in severe COVID-19 compared with controls (40, 45) or compared with mild COVID-19 (39), supporting our results. Other mechanisms that could influence the expected correlation between ACE expression and Ang II/Ang (1–7) levels are the cleavage/shedding of ACE2 (by ADAM17), dynamic changes in ACE/ACE2 expression and activity, and influence of receptor occupancy by the virus.
The second main finding of our study is a selective decrease of AT2 cells in COVID-19–related ARDS compared with both controls and COVID-19–unrelated ARDS. Animal models have shown that AT2 cells play a major role in regeneration, through proliferation and differentiation into alveolar type I cells (14, 46). In man, regeneration processes after lung injury are still poorly characterized, but proliferation of AT2 cells has been shown during both COVID-19–related and COVID-19–unrelated ARDS (47, 48). However, single-cell analysis of the lungs of a COVID-19 autopsy cohort recently revealed that AT2 cells were significantly decreased in patients with COVID-19 compared with control subjects, suggesting extensive virally induced cell death (16). We found increased expression of indicators of cell proliferation in AT2 cells (increased coexpression of ProSPC and Ki-67) to a similar extent in both COVID-19–related and COVID-19–unrelated ARDS. This suggests that the decrease in AT2 cells in COVID-19 ARDS cannot be attributed to defective AT2 proliferation but likely also involves accelerated cell death. Increased apoptosis or necroptosis of AT2 cells, possibly virally induced, has been reported (49) and could take part in the observed decrease in AT2 cells. However, it cannot be excluded that this finding could relate, at least in part, to technical issues related to postmortem protein degradation or to a difference in kinetics, as proliferation of AT2 cells mainly takes place during late stages of ARDS (47).
Some limitations of our study need to be acknowledged. First, whereas COVID-19 samples were obtained through autopsy, ARDS and control tissues were taken using open-lung biopsy and surgical lobectomy, respectively, and the different nature of the specimens (and kinetics in disease processes) may have influenced some findings. The delay between death and tissue sampling was, however, kept short, and the quality of the lung tissue was rigorously evaluated by an experienced pathologist before inclusion in the analysis. Second, regarding tissue quantification, because of the limited number of patients in each group, the study was underpowered to detect small but relevant differences between COVID-19–related and –unrelated ARDS. For the same reason, it cannot be excluded that, within the COVID-19–unrelated ARDS group, the different etiologies may have influenced the expression of ACE/ACE2. Third, the use of adjunctive treatment was different between COVID-19–related and –unrelated ARDS, with more prone positioning, nitric oxide, extracorporeal membrane oxygenation, and an unusually high proportion of vasopressors in the COVID-19 group despite similar baseline severity as measured with Acute Physiology and Chronic Health Evaluation II and Sequential Organ Failure Assessment scores. Many of those differences are probably related to the fact that all patients with COVID-19–related ARDS evolved toward greater severity and eventually died, whereas 50% of patients with COVID-19–unrelated ARDS ultimately survived. It is unclear whether this higher severity at the time of tissue sampling in the COVID-19–related ARDS group could have influenced our results. Besides, because we compared COVID-19–related ARDS to a historical cohort of COVID-19–unrelated ARDS, we cannot exclude that temporal changes in ARDS management could partly explain this different use of adjunctive treatments and, somehow, could have influenced our findings. Fourth, regarding ACE/ACE2 serum measurements, sampling methodologies were slightly different between COVID-19–related and –unrelated ARDS, resulting in a difference in the delay between ICU admission and serum sampling (median of 1 d in the COVID-19 group vs. 2 d in the COVID-19–unrelated ARDS group). Differences in disease severity (use of invasive mechanical ventilation and PaO2/FiO2 ratio) at the time of sampling may also have influenced serum concentrations. Furthermore, because of limited sample availability, Ang (1–7) quantification could only be performed in a subset of all patients with COVID-19. However, baseline characteristics, use of adjunctive treatment, and outcomes were similar in this subset of patients and in the population of all patients with COVID-19. Fifth, the retrospective nature of the study prevented measurements of ACE2 enzymatic activity and of Ang II and Ang (1–7) in optimized conditions (in fresh plasma samples). Therefore, no direct correlation between enzyme levels and activities could be made, as large discrepancies between AngII/Ang (1–7) concentrations measured by ELISA or radioimmunoassays have been reported (50). However, as highlighted above, similar findings regarding ACE/ACE2 activities and Ang II/Ang (1–7) concentrations were found in recent reports and, in this study, were consistent with lung tissue data. Finally, the observational nature of this study prevents investigation of causative mechanisms.
Despite these limitations, this study is the first to compare lung tissue expression of both ACE and ACE2 using immunohistochemistry in well-characterized and relatively large cohorts of COVID-19–related or –unrelated ARDS and controls. In addition, we used a cutting-edge, semiautomated quantification technique on entire tissue sections, which limited observer bias and increased the accuracy of quantification. The retrospective analysis of ACE and ACE2 expression in lung tissue was also combined with measurements of serum ACE and ACE2 (and Ang [1–7]) concentrations in prospective series of patients, with consistent results.
Altogether, in a retrospective analysis of human lung tissue and of prospectively collected serum of severe COVID-19 and ARDS, we observed a significant shift in the expression of ACEs from ACE to ACE2 that is seen both in the lung and in serum from patients with ARDS and suggest that this pathological trait is part of the generic response of the lung to acute injury rather than a specific feature of severe COVID-19. In contrast, a decrease in AT2 cells may distinguish COVID-19–related from COVID-19–unrelated ARDS, whose underlying mechanism may possibly involve increased cell death induced by the SARS-CoV-2. Putative effects of the decreased AT2 cells on the risk of pulmonary fibrosis should, in particular, be addressed. Those hypothesis-generating results should be confirmed in contemporary series of patients with similar ARDS severity, tissue sampling mode, and use of adjunctive treatments.
Acknowledgments
Acknowledgment
The authors thank Suzanne Renard, Leslie Gielens, Marie-France Dujardin, and Caroline Berghe (Intensive Care Department, Cliniques universitaires St-Luc, Brussels) for their help with data and sample collection. They thank Xavier Wittebole, Christine Collienne, Philippe Hantson, Diego Castanares-Zapatero, Melanie Dechamps, Luc Jacquet, Olivier Van Caenegem, Fatima Laarbaui, Sophie Pierard, and Christophe Beauloye (same department) for their help with the enrollment of patients. They also thank Alexandre Persu for precious advice and help with the enrollment of control subjects.
Footnotes
L.G. is supported by the Fonds National de la Recherche Scientifique (grant F5/4/140/5), Belgium. C.P. is a postdoctoral specialist of the Fonds National de la Recherche Scientifique (grant 1.R016.18), Belgium. This work was supported by a specific grant from the Fondation Louvain (Université catholique de Louvain) for COVID-19 research.
Author Contributions: L.G. drafted the work, collected the data, and was a major contributor in writing the manuscript. M.L. contributed to data collection, performed technical analysis, and extensively revised the manuscript. C.B. contributed to data collection, performed image analysis, and extensively revised the manuscript. D.H. contributed to the acquisition of tissue material and to technical analysis and extensively revised the manuscript. G.S. contributed to the acquisition of the tissue material and extensively revised the manuscript. J.P.P. collected the data and extensively revised the manuscript. V.M. contributed to the acquisition of the tissue material and extensively revised the manuscript. T.P.-B. contributed to data collection, was a contributor in writing the manuscript, and extensively revised the manuscript. P.-F.L. contributed to the acquisition of the data and extensively revised the manuscript. C.P. contributed to drafting of the work, was a major contributor in writing the manuscript, and extensively revised the manuscript. All authors read and approved the final manuscript.
This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1164/rccm.202012-4461OC on August 27, 2021
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Gheblawi M, Wang K, Viveiros A, Nguyen Q, Zhong JC, Turner AJ, et al. Angiotensin-converting enzyme 2: SARS-CoV-2 receptor and regulator of the renin-angiotensin system: celebrating the 20th anniversary of the discovery of ACE2. Circ Res . 2020;126:1456–1474. doi: 10.1161/CIRCRESAHA.120.317015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Li W, Moore MJ, Vasilieva N, Sui J, Wong SK, Berne MA, et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature . 2003;426:450–454. doi: 10.1038/nature02145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell . 2020;181:271–280.e8. doi: 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kuba K, Imai Y, Rao S, Gao H, Guo F, Guan B, et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat Med . 2005;11:875–879. doi: 10.1038/nm1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature . 2020;579:270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lu R, Zhao X, Li J, Niu P, Yang B, Wu H, et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet . 2020;395:565–574. doi: 10.1016/S0140-6736(20)30251-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wösten-van Asperen RM, Lutter R, Specht PA, Moll GN, van Woensel JB, van der Loos CM, et al. Acute respiratory distress syndrome leads to reduced ratio of ACE/ACE2 activities and is prevented by angiotensin-(1-7) or an angiotensin II receptor antagonist. J Pathol. 2011;225:618–627. doi: 10.1002/path.2987. [DOI] [PubMed] [Google Scholar]
- 8. Imai Y, Kuba K, Rao S, Huan Y, Guo F, Guan B, et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature . 2005;436:112–116. doi: 10.1038/nature03712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Li Y, Zeng Z, Cao Y, Liu Y, Ping F, Liang M, et al. Angiotensin-converting enzyme 2 prevents lipopolysaccharide-induced rat acute lung injury via suppressing the ERK1/2 and NF-κB signaling pathways. Sci Rep . 2016;6:27911. doi: 10.1038/srep27911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zou Z, Yan Y, Shu Y, Gao R, Sun Y, Li X, et al. Angiotensin-converting enzyme 2 protects from lethal avian influenza A H5N1 infections. Nat Commun . 2014;5:3594. doi: 10.1038/ncomms4594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Klein N, Gembardt F, Supé S, Kaestle SM, Nickles H, Erfinanda L, et al. Angiotensin-(1-7) protects from experimental acute lung injury. Crit Care Med . 2013;41:e334–e343. doi: 10.1097/CCM.0b013e31828a6688. [DOI] [PubMed] [Google Scholar]
- 12. Annoni F, Orbegozo D, Rahmania L, Irazabal M, Mendoza M, De Backer D, et al. Angiotensin-converting enzymes in acute respiratory distress syndrome. Intensive Care Med . 2019;45:1159–1160. doi: 10.1007/s00134-019-05600-6. [DOI] [PubMed] [Google Scholar]
- 13. Wösten-van Asperen RM, Bos AP, Bem RA, Dierdorp BS, Dekker T, van Goor H, et al. Imbalance between pulmonary angiotensin-converting enzyme and angiotensin-converting enzyme 2 activity in acute respiratory distress syndrome. Pediatr Crit Care Med . 2013;14:e438–e441. doi: 10.1097/PCC.0b013e3182a55735. [DOI] [PubMed] [Google Scholar]
- 14. Barkauskas CE, Cronce MJ, Rackley CR, Bowie EJ, Keene DR, Stripp BR, et al. Type 2 alveolar cells are stem cells in adult lung. J Clin Invest . 2013;123:3025–3036. doi: 10.1172/JCI68782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ortiz ME, Thurman A, Pezzulo AA, Leidinger MR, Klesney-Tait JA, Karp PH, et al. Heterogeneous expression of the SARS-coronavirus-2 receptor ACE2 in the human respiratory tract. EBioMedicine . 2020;60:102976. doi: 10.1016/j.ebiom.2020.102976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Delorey TM, Ziegler CGK, Heimberg G, Normand R, Yang Y, Segerstolpe Å, et al. COVID-19 tissue atlases reveal SARS-CoV-2 pathology and cellular targets. Nature . 2021;595:107–113. doi: 10.1038/s41586-021-03570-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ranieri VM, Rubenfeld GD, Thompson BT, Ferguson ND, Caldwell E, Fan E, et al. ARDS Definition Task Force. Acute respiratory distress syndrome: the Berlin Definition. JAMA . 2012;307:2526–2533. doi: 10.1001/jama.2012.5669. [DOI] [PubMed] [Google Scholar]
- 18. Gerard L, Bidoul T, Castanares-Zapatero D, Wittebole X, Lacroix V, Froidure A, et al. Open lung biopsy in nonresolving acute respiratory distress syndrome commonly identifies corticosteroid-sensitive pathologies, associated with better outcome. Crit Care Med . 2018;46:907–914. doi: 10.1097/CCM.0000000000003081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gonzalez RF, Allen L, Gonzales L, Ballard PL, Dobbs LG. HTII-280, a biomarker specific to the apical plasma membrane of human lung alveolar type II cells. J Histochem Cytochem . 2010;58:891–901. doi: 10.1369/jhc.2010.956433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bunyavanich S, Do A, Vicencio A. Nasal gene expression of angiotensin-converting enzyme 2 in children and adults. JAMA . 2020;323:2427–2429. doi: 10.1001/jama.2020.8707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Han T, Kang J, Li G, Ge J, Gu J. Analysis of 2019-nCoV receptor ACE2 expression in different tissues and its significance study. Ann Transl Med . 2020;8:1077. doi: 10.21037/atm-20-4281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liao M, Liu Y, Yuan J, Wen Y, Xu G, Zhao J, et al. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat Med . 2020;26:842–844. doi: 10.1038/s41591-020-0901-9. [DOI] [PubMed] [Google Scholar]
- 23. Li MY, Li L, Zhang Y, Wang XS. Expression of the SARS-CoV-2 cell receptor gene ACE2 in a wide variety of human tissues. Infect Dis Poverty . 2020;9:45. doi: 10.1186/s40249-020-00662-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hamming I, Timens W, Bulthuis ML, Lely AT, Navis G, van Goor H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol . 2004;203:631–637. doi: 10.1002/path.1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ren X, Glende J, Al-Falah M, de Vries V, Schwegmann-Wessels C, Qu X, et al. Analysis of ACE2 in polarized epithelial cells: surface expression and function as receptor for severe acute respiratory syndrome-associated coronavirus. J Gen Virol. 2006;87:1691–1695. doi: 10.1099/vir.0.81749-0. [DOI] [PubMed] [Google Scholar]
- 26. Lee IT, Nakayama T, Wu CT, Goltsev Y, Jiang S, Gall PA, et al. ACE2 localizes to the respiratory cilia and is not increased by ACE inhibitors or ARBs. Nat Commun . 2020;11:5453. doi: 10.1038/s41467-020-19145-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ackermann M, Verleden SE, Kuehnel M, Haverich A, Welte T, Laenger F, et al. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in covid-19. N Engl J Med . 2020;383:120–128. doi: 10.1056/NEJMoa2015432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ziegler CGK, Allon SJ, Nyquist SK, Mbano IM, Miao VN, Tzouanas CN, et al. HCA Lung Biological Network. Electronic address: lung-network@humancellatlas.org; HCA Lung Biological Network. SARS-CoV-2 receptor ACE2 is an interferon-stimulated gene in human airway epithelial cells and is detected in specific cell subsets across tissues. Cell . 2020;181:1016–1035.e19. doi: 10.1016/j.cell.2020.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sajuthi SP, DeFord P, Li Y, Jackson ND, Montgomery MT, Everman JL, et al. Type 2 and interferon inflammation regulate SARS-CoV-2 entry factor expression in the airway epithelium. Nat Commun . 2020;11:5139. doi: 10.1038/s41467-020-18781-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee JS, Shin EC. The type I interferon response in COVID-19: implications for treatment. Nat Rev Immunol. 2020;20:585–586. doi: 10.1038/s41577-020-00429-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Blanco-Melo D, Nilsson-Payant BE, Liu WC, Uhl S, Hoagland D, Møller R, et al. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell . 2020;181:1036–1045.e9. doi: 10.1016/j.cell.2020.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lucas C, Wong P, Klein J, Castro TBR, Silva J, Sundaram M, et al. Yale IMPACT Team. Longitudinal analyses reveal immunological misfiring in severe COVID-19. Nature . 2020;584:463–469. doi: 10.1038/s41586-020-2588-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Galani IE, Rovina N, Lampropoulou V, Triantafyllia V, Manioudaki M, Pavlos E, et al. Untuned antiviral immunity in COVID-19 revealed by temporal type I/III interferon patterns and flu comparison. Nat Immunol . 2021;22:32–40. doi: 10.1038/s41590-020-00840-x. [DOI] [PubMed] [Google Scholar]
- 34. Lee JS, Park S, Jeong HW, Ahn JY, Choi SJ, Lee H, et al. Immunophenotyping of COVID-19 and influenza highlights the role of type I interferons in development of severe COVID-19. Sci Immunol . 2020;5:eabd1554. doi: 10.1126/sciimmunol.abd1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nick JA, Caceres SM, Kret JE, Poch KR, Strand M, Faino AV, et al. Extremes of interferon-stimulated gene expression associate with worse outcomes in the acute respiratory distress syndrome. PLoS One . 2016;11:e0162490. doi: 10.1371/journal.pone.0162490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Grunwell JR, Stephenson ST, Mohammad AF, Jones K, Mason C, Opolka C, et al. Differential type I interferon response and primary airway neutrophil extracellular trap release in children with acute respiratory distress syndrome. Sci Rep . 2020;10:19049. doi: 10.1038/s41598-020-76122-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Makris S, Paulsen M, Johansson C. Type I interferons as regulators of lung inflammation. Front Immunol . 2017;8:259. doi: 10.3389/fimmu.2017.00259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Serfozo P, Wysocki J, Gulua G, Schulze A, Ye M, Liu P, et al. Ang II (angiotensin II) conversion to angiotensin-(1-7) in the circulation is POP (prolyloligopeptidase)-dependent and ACE2 (angiotensin-converting enzyme 2)-independent. Hypertension. 2020;75:173–182. doi: 10.1161/HYPERTENSIONAHA.119.14071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Reindl-Schwaighofer R, Hodlmoser S, Eskandary F, Poglitsch M, Bonderman D, Strassl R, et al. ACE2 elevation in severe COVID-19. Am J Respir Crit Care Med . 2021;203:1191–1196. doi: 10.1164/rccm.202101-0142LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. van Lier D, Kox M, Santos K, van der Hoeven H, Pillay J, Pickkers P. Increased blood angiotensin converting enzyme 2 activity in critically ill COVID-19 patients. ERJ Open Res . 2021;7:00848-02020. doi: 10.1183/23120541.00848-2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Patel SK, Juno JA, Lee WS, Wragg KM, Hogarth PM, Kent SJ, et al. Plasma ACE2 activity is persistently elevated following SARS-CoV-2 infection: implications for COVID-19 pathogenesis and consequences. Eur Respir J . 2021;57:2003730. doi: 10.1183/13993003.03730-2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhu Z, Cai T, Fan L, Lou K, Hua X, Huang Z, et al. The potential role of serum angiotensin-converting enzyme in coronavirus disease 2019. BMC Infect Dis . 2020;20:883. doi: 10.1186/s12879-020-05619-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Liu Y, Yang Y, Zhang C, Huang F, Wang F, Yuan J, et al. Clinical and biochemical indexes from 2019-nCoV infected patients linked to viral loads and lung injury. Sci China Life Sci . 2020;63:364–374. doi: 10.1007/s11427-020-1643-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wu Z, Hu R, Zhang C, Ren W, Yu A, Zhou X. Elevation of plasma angiotensin II level is a potential pathogenesis for the critically ill COVID-19 patients. Crit Care . 2020;24:290. doi: 10.1186/s13054-020-03015-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Valle Martins AL, da Silva FA, Bolais-Ramos L, de Oliveira GC, Ribeiro RC, Pereira DAA, et al. Increased circulating levels of angiotensin-(1-7) in severely ill COVID-19 patients. ERJ Open Res . 2021;7:00114-02021. doi: 10.1183/23120541.00114-2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hogan BL, Barkauskas CE, Chapman HA, Epstein JA, Jain R, Hsia CC, et al. Repair and regeneration of the respiratory system: complexity, plasticity, and mechanisms of lung stem cell function. Cell Stem Cell . 2014;15:123–138. doi: 10.1016/j.stem.2014.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Abadie Y, Bregeon F, Papazian L, Lange F, Chailley-Heu B, Thomas P, et al. Decreased VEGF concentration in lung tissue and vascular injury during ARDS. Eur Respir J . 2005;25:139–146. doi: 10.1183/09031936.04.00065504. [DOI] [PubMed] [Google Scholar]
- 48. Chen J, Wu H, Yu Y, Tang N. Pulmonary alveolar regeneration in adult COVID-19 patients. Cell Res . 2020;30:708–710. doi: 10.1038/s41422-020-0369-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Li S, Zhang Y, Guan Z, Li H, Ye M, Chen X, et al. SARS-CoV-2 triggers inflammatory responses and cell death through caspase-8 activation. Signal Transduct Target Ther . 2020;5:235. doi: 10.1038/s41392-020-00334-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chappell MC, Pirro NT, South AM, Gwathmey TM. Concerns on the specificity of commercial ELISAs for the measurement of angiotensin (1-7) and angiotensin II in human plasma. Hypertension . 2021;77:e29–e31. doi: 10.1161/HYPERTENSIONAHA.120.16724. [DOI] [PMC free article] [PubMed] [Google Scholar]