

Abstract
Technological advances in HLA laboratory testing undoubtedly improved the sensitivity and specificity of HLA antibody assessment but not without introducing a set of challenges regarding data interpretation. In particular, the introduction of solid-phase single-antigen bead (SAB) antibody assessment brought the belief that mean fluorescence intensity (MFI) was a quantifiable value. As such, MFI levels heavily influenced HLA antibody reporting, monitoring, and clinical practice. However, given that SAB testing was neither intended for nor approved to be quantifiable, is the use of MFI in current clinical and laboratory practice valid? What, if anything, does this numerical value actually reveal about the pathogenic potential of the antibody? What are the pitfalls and caveats associated with reporting MFI? Herein, we travel the road to HLA antibody assessment and explore the reliability of MFI values to make clinical decisions.
Introduction
Times have certainly changed. A transplant candidate with a calculated panel-reactive antibody value of 100% who lives in Georgia was recently offered a virtual cross-match–negative, HLA-nonidentical kidney from a deceased donor in California. With no detectable donor-specific antibodies (DSAs) in a current serum sample (<30 days) and no recent intervening events, the offer was accepted. In contrast to historic practice, the patient proceeded directly to transplantation and the final (physical) crossmatch was performed retrospectively. From an HLA centric viewpoint, this scenario is astonishing for two reasons. First, the patient was offered a kidney that likely would never have been allocated before implementation of the new kidney allocation system on December 14, 2014. Second, the kidney was transplanted into a highly sensitized candidate without a prospective physical crossmatch. Not long ago, a prospective crossmatch was a figurative line-in-the-sand that could not be ignored. Historically, a positive complement-dependent cytotoxicity (CDC) crossmatch was a contraindication to kidney transplantation. The assay had many imperfections; it was not particularly specific (some patient allografts with positive crossmatches had surprisingly good survival) or sensitive (some patient allografts with negative crossmatches still failed), but the CDC test was the best available. The challenge was to develop a more reliable testing methodology to identify and define clinically relevant HLA antibodies.
Under Construction: The Journey Begins
Advances in techniques and technology paved the way to improved testing. Enhancement of the CDC assay with antihuman globulin made it possible to detect lower levels of antibodies than had been previously feasible. Next, the advent of flow cytometry led to the discovery and clinical implementation of the flow cytometric cross-match. Once again, sensitivity to detect even lower levels of antibodies was enhanced with the added benefit of simultaneous and independent assessment of both T cell and B cell crossmatches (1). The game-changing moment in the detection and identification of HLA antibodies was the introduction of microparticle technology. Coated with HLA class I and/or class II molecules, microparticles (cell-sized latex or polystyrene beads) became surrogate lymphocyte targets. Today, the current iterations of those early tests are the FlowPRA™ and SAB assays (One Lambda, Inc., Canoga Park, CA). The former is a screening test, one that qualitatively detects the presence/absence of HLA antibodies. The latter is a semiquantitative test used to determine antibody specificity. These two tests provided a platform to detect and identify HLA antibodies with sensitivity and specificity that were previously inconceivable. However, increasing sensitivity and specificity came with its own sets of challenges.
Hard Hats Required: Controversies in HLA Antibody Assessment
While multiplex Luminex technology (Luminex Corporation, Austin, TX) has provided a specific and sensitive platform to identify HLA antibodies, it is not flawless. A major point of contention revolves around results from SAB testing being reported as a numerical value referred to as mean fluorescence intensity (MFI). It is natural to think of a number as a quantitative assessment, but MFI values were never intended to quantify antibodies, nor was the Luminex-based test approved as a quantitative assay by the US Food and Drug Administration (2). Instead, MFI values reflect a given bead’s relative fluorescence without reference to a standard. It is important to recognize that relative fluorescence can be affected by many variables. Nevertheless, MFI values have consistently been used as a quasiquantitative assessment of antibody strength by both laboratorians and clinicians. The tendency is to correlate MFI values with clinical outcomes and to serially monitor their fluctuations as a measure of clinical status. Decreasing MFI values after a patient has been treated for antibody-mediated rejection (AMR) is interpreted as a proper clinical response, while no change (or an increase in value) is considered a therapy failure. Is it even reasonable to expect a single test designed to identify antibody specificity to accurately assess antibody levels/strength and/or pathogenicity? What are the implications of relying solely on MFI values to manage patient care?
Be Prepared to Stop: Limitations
The limitations of MFI are immediately apparent based on where and how thresholds are set to determine whether a bead is positive or negative. If the threshold is set too low, false-positives may preclude a patient from receiving a compatible organ; alternatively, if the threshold is set too high, there may be increased risk that a patient will experience early and/or irreversible AMR. Indeed, there have been reports in which patients, transplanted across extremely low DSA levels (100 MFI), experienced poor graft survival (3). In contrast, patients transplanted across extremely high DSA values (>10 000 MFI) had not only negative crossmatches against their donors but also excellent graft outcomes (4). Admittedly, the majority of published studies report that low and high MFI levels have some degree of correlation with good and poor graft outcomes, respectively, but the dynamic range of MFI values is broad and inconsistent between centers. Because the spectrum of graft outcome based solely on MFI is so wide, setting rigid cutoff values is unrealistic. Indeed, there is no single value that corresponds to an unequivocal threshold above or below which organ transplantation is contraindicated or risk free, respectively. As a result, cutoff MFI values can vary significantly by the organ transplanted and/or a transplant program’s practice.
MFI variability is also compounded by vendor, antigen source, test reagents, and intralaboratory variability (5). Intralaboratory variability alone can exceed 50% for SAB analysis, resulting from technologist and day-to-day variability (e.g. ambient laboratory temperatures, otherwise acceptable differences in pH ranges of routine reagents, etc.) (6). Indeed, the SAB assay is particularly sensitive to technologist-dependent actions (including pipetting, washing, vortex speeds, centrifuging, etc.), which influence assay performance and the reported MFI value. Still another concern regarding MFI values stems from interferences due to antibody-dependent complement activation. Complement split products covalently bind to antigen–antibody complexes on the beads and sterically block binding sites for secondary (reporter) antibodies (7). These events can artifactually diminish MFI values to well below a cutoff threshold. Because the prevalence of sensitized patient sera exhibiting interference is >70% (8), it is likely that published studies reporting extremely low MFI DSAs associated with poor outcomes were, in fact, compromised by interfering factors. Other mechanisms of assay interference include IgM HLA antibodies, wherein such antibodies compete with or block the binding of IgG HLA antibodies (9). Factors intrinsic to the manufacturing and coating of the microparticles also muddle the meaning of an MFI value. False-positive reactivity can occur due to antibody binding to misfolded HLA proteins, commonly referred to as denatured antigens. Reactivity is attributed to binding of antibodies to so-called cryptic epitopes, which under normal circumstances are considered to be antibody inaccessible (10). Such antibodies have a high prevalence among waitlist candidates (11–40%) (11,12), likely explaining why some high-MFI DSAs were not associated with poor graft outcome. The limited data published on this topic suggest that reactivity to denatured proteins does not contribute to poor graft outcome (11). However, Visentin et al reported that sera from some patients contain antibodies to both native and denatured HLA antigens of the same specificity or specificities (13). Thus, unless and until reactivity to native and denatured HLA antigens can be easily discerned, it is premature to consider reactivity to denatured antigens as clinically innocuous.
Rerouting: New Directions
With the introduction of more sensitive and specific approaches to determine donor–recipient compatibility, the HLA community has reaffirmed the power of pattern recognition. In the mid 1960s–1970s, the term “cross-reactive groups” (CREGs) was used to describe the serologic phenomenon of multiple unique HLA antigens that reacted with a single antibody. Rodey and Fuller reported that it was more common for patients to produce a few antibodies to shared determinants among a group of antigens than to generate multiple individual specificities (14). With 20:20 hindsight this makes perfect sense. In the following example, a patient received a kidney transplant from a donor mismatched for a single antigen, namely, HLA-A2. After the transplant failed (secondary to polyomavirus infection) and immunosuppression was stopped, the patient developed DSAs against HLA-A2. Additionally, reactivity against other antigens in the HLA-A2 CREG (HLA-A23, -A24, -A68, -A69, -B57, and -B58) also appeared (Figure 1). While the same observations are seen today, the terminology has changed: epitopes, eplets, and conformational variances are the current versions of CREGs. In this case, the reactivity observed can be attributed to antibodies directed against just two epitopes: the 62GE epitope expressed by HLA-A2, -B57, and -B58 and the 127K epitope expressed by HLAA2, -A23, -A24, -A68, and -A69 (http://www.epitopes.net/). By considering antibody reactivity to be epitope directed (i.e. to shared sequences) rather than to individual HLA antigens, what previously would have been interpreted to be third-party antibodies (i.e. non-DSAs) would actually be considered as DSAs. Clearly, the mindset of how to interpret bead reactivity needs to change from antigens to epitopes.
Figure 1:
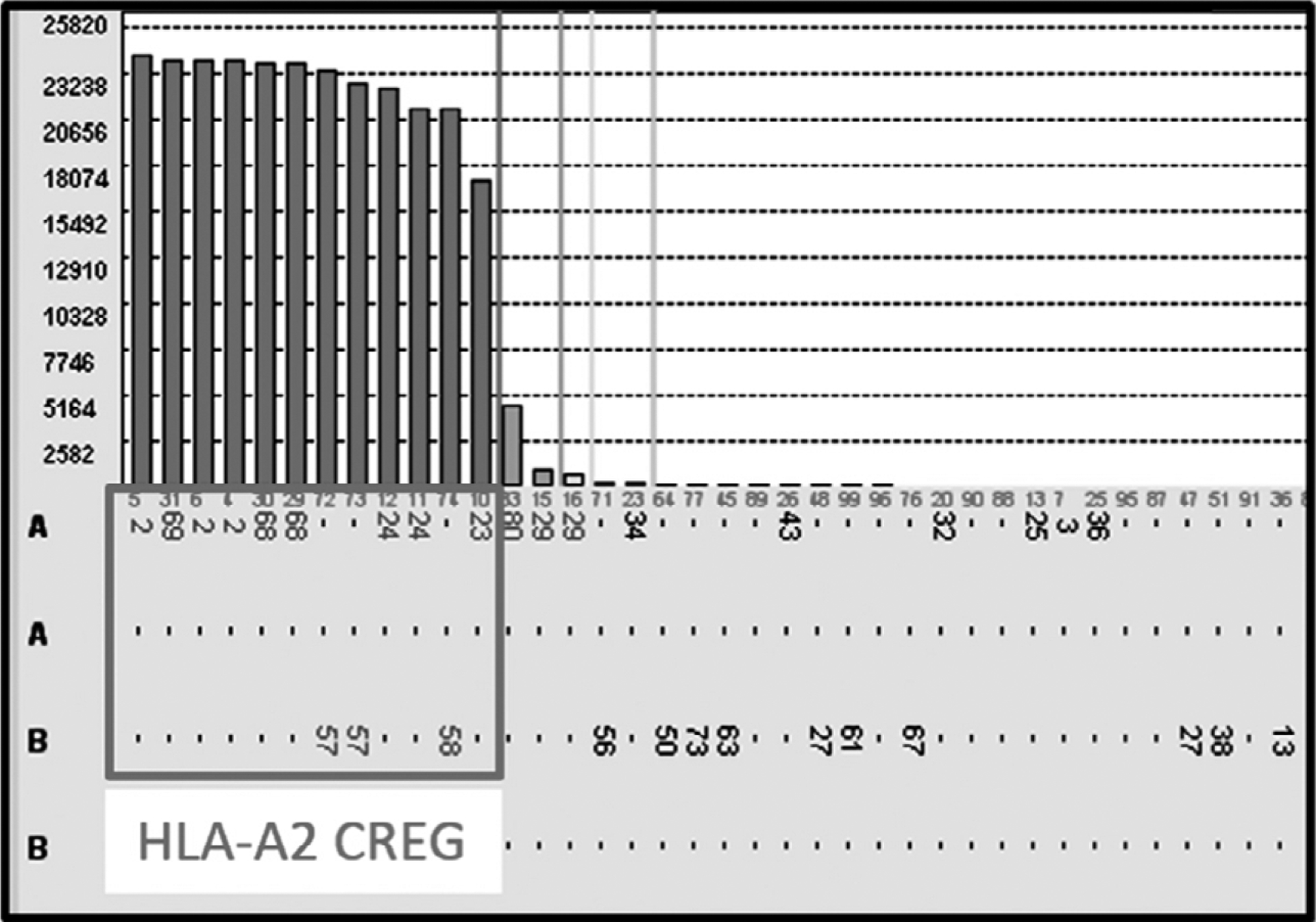
Antibody reactivity against the HLA-A2 cross-reactive groups (A2, A23, A24, A68, A69, B57, and B58) antigens in a patient with a failed kidney transplant from a donor with a single antigen mismatch (HLA-A2).
Bumps in the Road: Conformation—An Integral Component to Antibody Pathogenicity?
While there has been a strong interest in the quantification of HLA antibodies, there has been virtually no attention given to how or whether antigen conformation affects antibody affinity, avidity, and resulting MFI values. The following example illustrates that it is reasonable to consider that the degree and strength of antibody binding is influenced by antigen conformation. As shown in Figure 2(A), an antibody to Bw6 was identified on class I SAB with MFI values >17 000 except for the B*46:01- and B*73:01-bearing beads, which had MFI values of ~2000 and ~1500, respectively. Of note, several HLA-C locus–bearing beads displayed low-level MFI values similar to those of B*46:01 and B*73:01. As illustrated in Figure 2(B), all Bw6-positive beads, including B*46:01 and B*73:01, bear the same amino acid sequence at residues 77–83 (LRNLRG; the recognized Bw6 epitope), but both B*46:01 and B*73:01 have valine at position 75 (V75) while all other Bw6-bearing antigens have glutamic acid at position 75. Interestingly, the HLA-C locus antigen beads seen in Figure 2 all contained the Bw6 epitope as well as V75 (Figure 2C). The decreased MFI values for beads containing the Bw6/V75 epitope suggest that the presence of V75 alters the Bw6 epitope conformation in such a way that the corresponding antibody is restricted in its binding ability. Does this correspond to a reduced risk for recipients who possess Bw6 antibodies who are transplanted with an organ that has the alternative Bw6 epitope? No data are currently available to make an accurate prediction.
Figure 2:
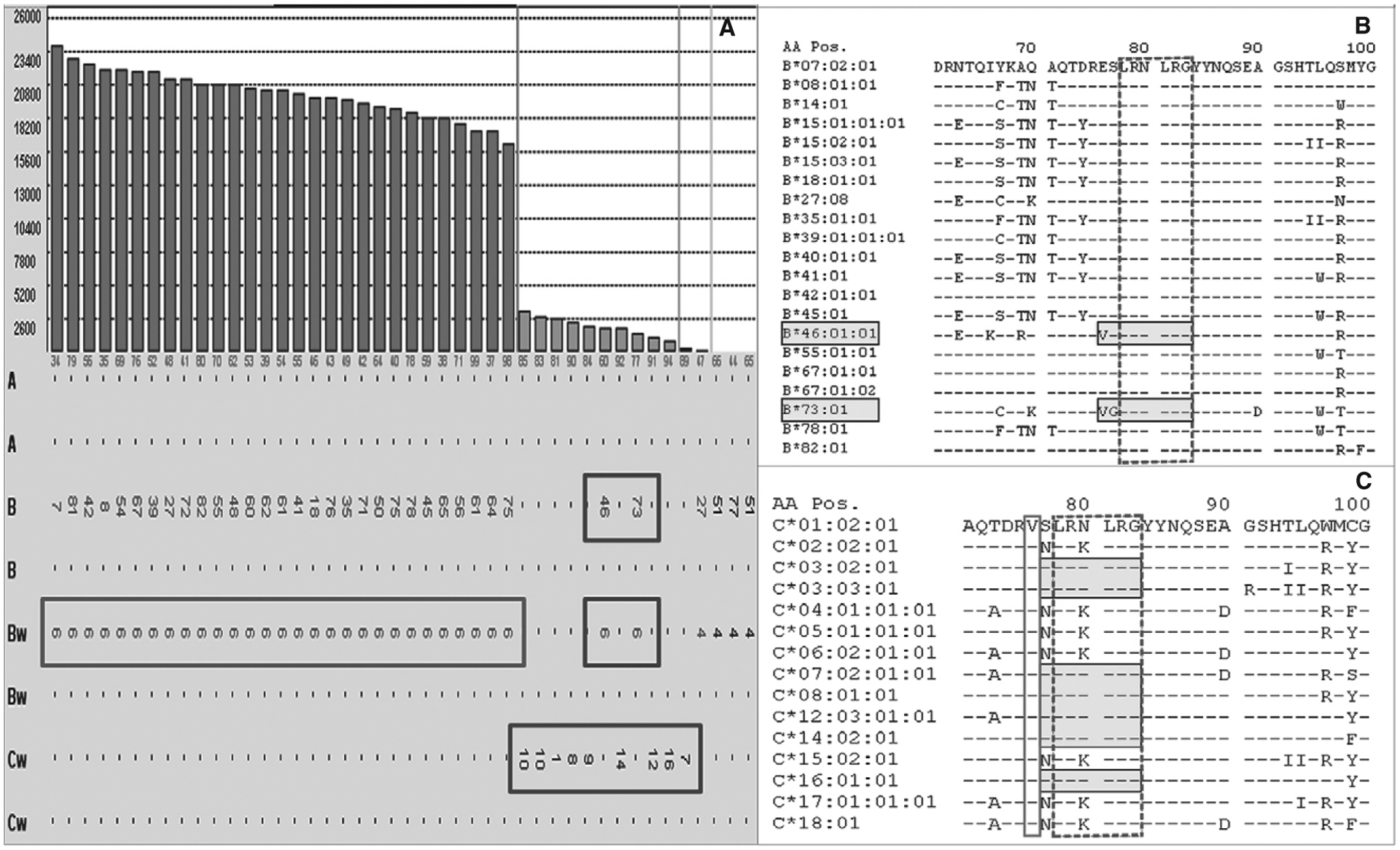
(A) Antibody to the Bw6 epitope with strong positive reactivity with the exception to B*46:01- and B*73:01-bearing beads. Several HLA-C locus-bearing beads display the same low-level MFI values as the B46 and B73. (B) The Bw6-positive beads, including B46 and B73 (shaded boxes), contain the LRNLRG sequence (Bw6 epitope) at residues 77–83 (dash-lined box). Both B*46:01 and B*73:01 differ from the remaining Bw6-bearing antigens by a valine instead of a glutamic acid at position 75 (V75) (B). (C) Of note, the selected HLA-C locus antigen beads (shaded boxes) all contain the Bw6 epitope (dash-lined box) as well as the V75 (solid-lined box) identical to B46 and B73.
These observations underscore how challenging it can be to determine donor–recipient compatibility. In the past, computer software programs like HLAMatchmaker (http://www.epitopes.net/) compared only linear sequences of different HLA molecules to identify what are termed shared epitopes. The most current version of HLAMatch-maker and the epitope registry (http://www.epregistry.com.br/) factor in discontinuous polymorphic regions (eplets) that have recently become a major component of compatibility determination in many laboratories. The following case depicts another example of how differences in protein sequences distal to a functional epitope can affect conformation and how such changes can alter antibody–antigen binding:
Class I SAB testing revealed antibodies to B*15:02(B75) (~6000 MFI) and B*51:02 (~5000 MFI); however, reactivity to B*15:11 (B75) (<1000 MFI) and B*51:01 (~1000 MFI) would likely be considered negative based on MFI values alone (Figure 3A). Examining amino acid sequences, B*51:01 and B*51:02 differ by a single amino acid at residue 171 (exon 3, α-helix) (Figure 3B). Given that residue 171 is positioned on the helix underside (and spatially inaccessible to antibody), the difference at this position infers a change in epitope conformation that diminishes antibody affinity for the target antigen. Sequence differences between B*15:02 and B*15:11 are more numerous and located on both the floor of the groove and the α-helix (Figure 3C). Therefore, predicting which change contributes to the observed difference in reactivity is not simple, because variation in MFI may be due to a combinatorial effect of more than one of the amino acid differences. Nonetheless, regardless of whether changes occur at a single or multiple positions, the result is a physical alteration that can affect MFI values.
Figure 3:
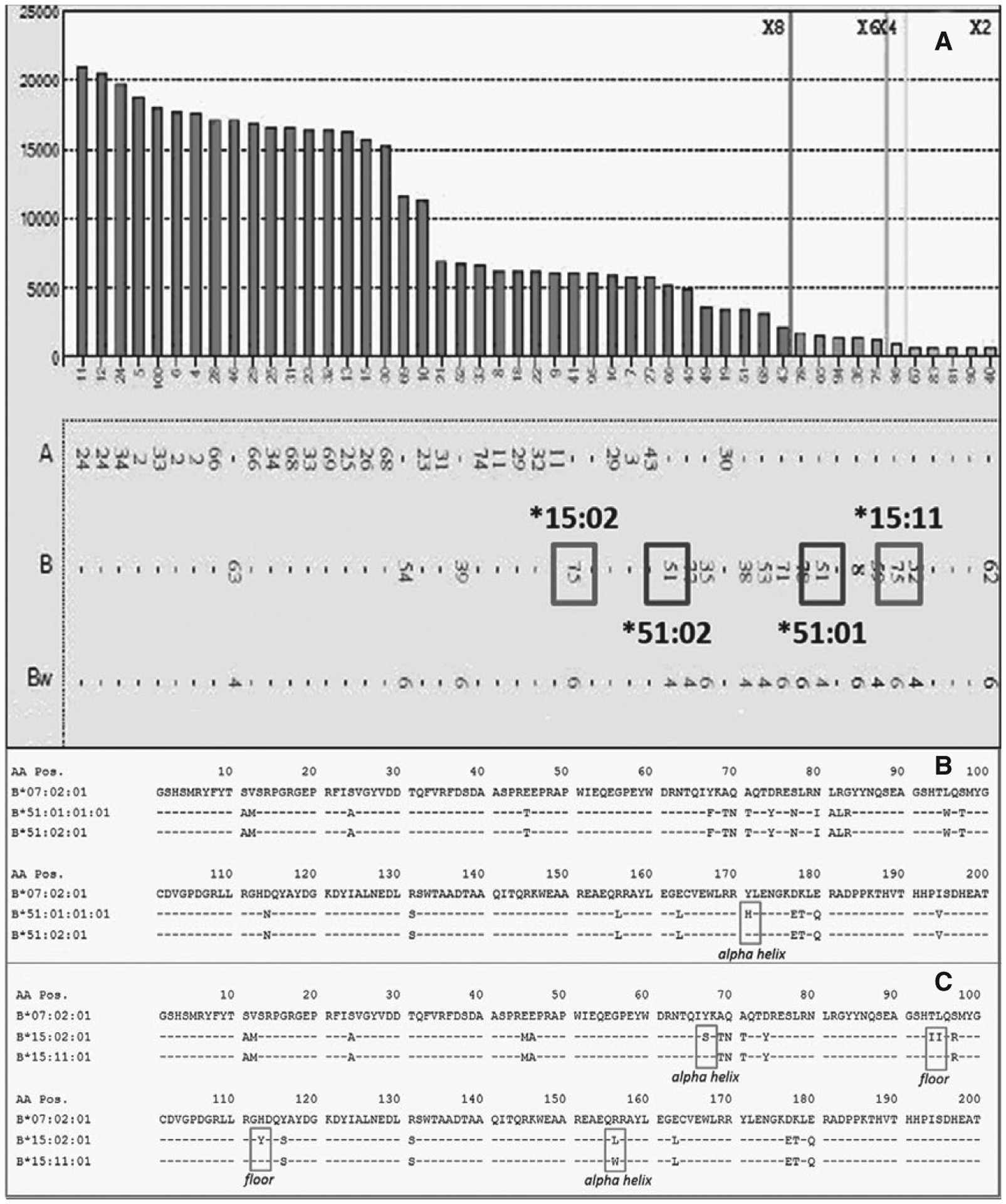
(A) Antibodies with positive reactivity to beads bearing B*15:02(B75) and B*51:02 but negative reactivity to B*15:11(B75)- and B*51:01-bearing beads. (B) Amino acid sequences reveal that B*51:01 and B*51:02 differ by a single amino acid at residue 171 (exon 3, α-helix), while sequence differences between B*15:02 and B*15:11 are numerous, located on both the floor of the groove and α-helix (C). Amino acid differences highlighted in boxes in (B) and (C).
Divided Highway: Antigens Versus Antibodies
It is important to realize that detecting DSAs is predicated on a two-sided equation. On one side is the patient’s serum (antibody); on the other, the antigen it recognizes. When a DSA is identified, it is crucial to understand that microparticles have been coated with an amount of antigen designed to detect low levels of antibody, not to mimic the quantity of antigen on lymphocytes or tissues. Thus, it is plausible that a DSA that binds a bead in vitro may fail to bind to the corresponding antigen in vivo because its expression is too low. However, does this mean that the DSA identified in vitro will always be harmless? The logical conclusion is no, because HLA expression in vivo can increase (e.g. after an inflammatory event) and the transplanted organ may then be susceptible to antibody binding. Supporting this notion are studies from Wiebe et al, who have reported that the most common de novo antibody produced posttransplantation are directed to HLA-DQ antigens (15) despite low expression of the DQ loci (16). This infers that donor kidneys can express sufficient HLA-DQ to induce an antibody response. In vitro, DSAs with MFI values below threshold may only appear that way because the DSAs are of low concentration. As such, the antibody could be diluted over many beads sharing the same epitope, none of which would be sufficiently strong to produce an adequate fluorescent signal. In vivo, a lower level of HLA antigen expression compared with beads could provide a target to which the DSAs can bind. For these reasons, the “pathogenicity” of an antibody cannot and should not be assumed based strictly on MFI levels.
Merge: “Functional” Methods to Assess Antibody Pathogenicity
Several studies have focused on the complement fixing characteristics of HLA antibodies, anticipating that those properties would be associated with their pathogenicity. SAB assays were modified to assess which, if any, HLA antibodies in patient sera bound specific complement components (C1q, C3d, C4d) as a surrogate of complement activation in vivo. Published studies have been inconsistent, with some reporting strong associations between complement fixation and poor allograft outcome (17–19), while others found no correlations (20,21). Importantly, one of the largest studies evaluating complement-fixating DSAs with poor graft survival found clinical significance only in the posttransplantation period (17). Pretransplantation correlation was not seen in that study or in several others (20,21).
In vitro, IgG1 and IgG3 fix complement, while IgG2 and IgG4 do not (or do so to a much lesser degree). Recent studies report associations between IgG subclass (in particular, IgG3) and poor clinical outcomes, at least in the posttransplantation setting (22,23). Pretransplantation IgG subclass analysis appears to be of little predictive value (24,25). Interestingly, Schaub et al reported that not all sera containing strong complement fixing antibodies (IgG1/IgG3) fix complement in vitro (24). Other studies have shown that IgG1 DSAs, the most prevalent subclass, often correlate with normal graft function (23,24), suggesting that IgGs that fix complement in vitro may not do so in vivo. There are multiple explanations for this possibility, including the in vivo presence of complement-inhibiting molecules such as heparin sulfate (26) and/or increased levels of decay-accelerating factor (27). Additionally, after the engagement of antigen, it has been reported that hexamerization of IgG Fc fragments is required to initiate the activation of the complement cascade (28). Thus, if insufficient IgG molecules bind or antibody alignment is incongruous, hexamer formation and subsequent C1q recruitment may not proceed regardless of the presence of strong complement fixing subclasses. Last, it has been clearly demonstrated that AMR need not be driven by complement fixation. Complement-independent AMR does occur with possible mechanisms including smooth muscle and endothelial cell activation and antibody-dependent cellular cytotoxicity via natural killer cells (29).
Repaving the Road: Guides to Antibody Assessment
Difficult and problematic cases cannot always be resolved with additional testing and/or sophisticated algorithms. Sometimes, there are no other tests to perform. For example, identical antibody presentations (e.g. antibodies to HLA-A*02 well below the established cutoff value) in three different individuals being considered for transplantation with an HLA-A*02–positive donor do not mean the same pathway will be taken for all three subjects. In one situation, the patient is a pediatric candidate whose father is the potential donor. The second patient is an adult repeat-graft candidate who previously rejected a kidney from an HLA-A*02–positive donor and is about to be relisted for a deceased donor organ. The third subject is a 25-year-old male, nontransfused, primary graft recipient, with no known sensitizing events. Should these three patients be considered the same because they all have the same HLA antibody pattern? In our opinion, the answer is “no.” Each subject has his/her unique story that must be considered separately. There is no single “correct” approach for any of these theoretical subjects, and how to proceed is center dependent. It is for cases such as these that a team approach can be extremely beneficial (30). Challenging cases are a perfect opportunity for face-to-face discussions among laboratorians, surgeons, clinicians, and coordinators. Such exchanges serve multiple purposes, from outlining the immunological complexities of a particular donor–recipient pair to discussing issues that typically one or more of the participants do not consider. It is through such interactions that institutional practices are fine-tuned to best fit patient needs.
In conclusion, the road toward HLA antibody detection and identification is still (and may always be) under construction. Will there ever be a test for HLA antibodies with 100% specificity and sensitivity, devoid of the artifacts, limitations, and barriers described in this review? Certainly, the desire for a “perfect” test is strong, but even if this “perfect” test was engineered, one that could determine and quantify antibody specificity with unequivocal accuracy, how would those results be applied in a clinical setting? Even the ideal test would provide only a single result, a snapshot in time that may not reflect the patient’s complete history. Is it reasonable to believe that a single time point could, with certainty, predict a clinical outcome? Can the outcome of a glucose tolerance test be predicted from a single blood glucose level? Without clinical context and history (not to mention biological variability), the amount of information that can be extrapolated from a single value is limited. Moreover, the quantification of HLA antibody provides only a limited scope of its pathophysiology; a better understanding of the processes that drive alloantibody production in an individual is necessary to best characterize the pathogenicity of a given antibody. Even as technology advances, old questions will almost certainly remain as new questions will inevitably arise. And so, the journey continues….
Abbreviations:
- AMR
antibody-mediated rejection
- CDC
complement-dependent cytotoxicity
- CREG
cross-reactive group
- DSA
donor-specific antibody
- MFI
mean fluorescence intensity
- SAB
single-antigen bead
Footnotes
Disclosure
The authors of this manuscript have no conflicts of interest to disclose as described by the American Journal of Transplantation.
References
- 1.Bray RA, Lebeck LK, Gebel HM. The flow cytometric cross-match. Dual-color analysis of T cell and B cell reactivities. Transplantation 1989; 48: 834–840. [PubMed] [Google Scholar]
- 2.Archdeacon P, Chan M, Neuland C, et al. Summary of FDA antibody-mediated rejection workshop. Am J Transplant 2011; 11: 896–906. [DOI] [PubMed] [Google Scholar]
- 3.Singh N, Djamali A, Lorentzen D, et al. Pretransplant donor-specific antibodies detected by single-antigen bead flow cytometry are associated with inferior kidney transplant outcomes. Transplantation 2010; 90: 1079–1084. [DOI] [PubMed] [Google Scholar]
- 4.Phanish MK. Immunological risk assessment and human leukocyte antigen antibody testing in kidney transplantation. Indian J Nephrology 2016; 26: 80–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gebel HM, Bray RA. HLA antibody detection with solid phase assays: Great expectations or expectations too great? Am J Transplant 2014; 14: 1964–1975. [DOI] [PubMed] [Google Scholar]
- 6.Reed EF, Rao P, Zhang Z, et al. Comprehensive assessment and standardization of solid phase multiplex-bead arrays for the detection of antibodies to HLA. Am J Transplant 2013; 13: 1859–1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weinstock C, Schnaidt M. The complement-mediated prozone effect in the Luminex single-antigen bead assay and its impact on HLA antibody determination in patient sera. Int J Immunogenet 2013; 40: 171–177. [DOI] [PubMed] [Google Scholar]
- 8.Tambur AR, Herrera ND, Haarberg KM, et al. Assessing antibody strength: Comparison of MFI, C1q, and titer information. Am J Transplant 2015; 15: 2421–2430. [DOI] [PubMed] [Google Scholar]
- 9.Kosmoliaptsis V, Bradley JA, Peacock S, Chaudhry AN, Taylor CJ. Detection of immunoglobulin G human leukocyte antigen-specific alloantibodies in renal transplant patients using single-antigen-beads is compromised by the presence of immunoglobulin M human leukocyte antigen-specific alloantibodies. Transplantation 2009; 87: 813–820. [DOI] [PubMed] [Google Scholar]
- 10.El-Awar N, Terasaki PI, Nguyen A, et al. Epitopes of HLA antibodies found in sera of normal healthy males and cord blood. Clin Transpl 2008; 23: 199–214. [PubMed] [Google Scholar]
- 11.Visentin J, Guidicelli G, Bachelet T, et al. Denatured class I human leukocyte antigen antibodies in sensitized kidney recipients: Prevalence, relevance, and impact on organ allocation. Transplantation 2014; 98: 738–744. [DOI] [PubMed] [Google Scholar]
- 12.Otten HG, Verhaar MC, Borst HP, et al. The significance of pre-transplant donor-specific antibodies reactive with intact or denatured human leucocyte antigen in kidney transplantation. Clin Exp Immunol 2013; 173: 536–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Visentin J, Guidicelli G, Moreau JF, Lee JH, Taupin JL. Deciphering allogeneic antibody response against native and denatured HLA epitopes in organ transplantation. Eur J Immunol 2015; 45: 2111–2121. [DOI] [PubMed] [Google Scholar]
- 14.Rodey GE, Fuller TC. Public epitopes and the antigenic structure of the HLA molecules. Crit Rev Immunol 1987; 7: 229–267. [PubMed] [Google Scholar]
- 15.Wiebe C, Nevins TE, Robiner WN, Thomas W, Matas AJ, Nickerson PW. The synergistic effect of class II HLA epitope-mismatch and nonadherence on acute rejection and graft survival. Am J Transplant 2015; 15: 2197–2202. [DOI] [PubMed] [Google Scholar]
- 16.Fernandez-Vina MA, Klein JP, Haagenson M, et al. Multiple mismatches at the low expression HLA loci DP, DQ, and DRB3/4/5 associate with adverse outcomes in hematopoietic stem cell transplantation. Blood 2013; 121: 4603–4610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Loupy A, Lefaucheur C, Vernerey D, et al. Complement-binding anti-HLA antibodies and kidney-allograft survival. N Engl J Med 2013; 369: 1215–1226. [DOI] [PubMed] [Google Scholar]
- 18.Bartel G, Wahrmann M, Schwaiger E, et al. Solid phase detection of C4d-fixing HLA antibodies to predict rejection in high immunological risk kidney transplant recipients. Transpl Int 2013; 26: 121–130. [DOI] [PubMed] [Google Scholar]
- 19.Comoli P, Cioni M, Tagliamacco A, et al. Acquisition of C3d-binding activity by de novo donor-specific hla antibodies correlates with graft loss in nonsensitized pediatric kidney recipients. Am J Transplant 2016; 16: 2106–2116. [DOI] [PubMed] [Google Scholar]
- 20.Thammanichanond D, Wiwattanathum P, Mongkolsuk T, et al. Role of pretransplant complement-fixing donor-specific antibodies identified by C1q assay in kidney transplantation. Transpl Proc 2016; 48: 756–760. [DOI] [PubMed] [Google Scholar]
- 21.Honger G, Wahrmann M, Amico P, Hopfer H, Bohmig GA, Schaub S. C4d-fixing capability of low-level donor-specific HLA antibodies is not predictive for early antibody-mediated rejection. Transplantation 2010; 89: 1471–1475. [DOI] [PubMed] [Google Scholar]
- 22.Lefaucheur C, Viglietti D, Bentlejewski C, et al. IgG donor-specific anti-human HLA antibody subclasses and kidney allograft antibody-mediated injury. J Am Soc Nephrol 2016; 27: 293–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kaneku H, O’Leary JG, Taniguchi M, Susskind BM, Terasaki PI, Klintmalm GB. Donor-specific human leukocyte antigen antibodies of the immunoglobulin G3 subclass are associated with chronic rejection and graft loss after liver transplantation. Liver Transpl 2012; 18: 984–992. [DOI] [PubMed] [Google Scholar]
- 24.Schaub S, Honger G, Koller MT, Liwski R, Amico P. Determinants of C1q binding in the single antigen bead assay. Transplantation 2014; 98: 387–393. [DOI] [PubMed] [Google Scholar]
- 25.Honger G, Hopfer H, Arnold ML, Spriewald BM, Schaub S, Amico P. Pretransplant IgG subclasses of donor-specific human leukocyte antigen antibodies and development of antibody-mediated rejection. Transplantation 2011; 92: 41–47. [DOI] [PubMed] [Google Scholar]
- 26.Zaferani A, Vives RR, van der Pol P, et al. Factor h and prop-erdin recognize different epitopes on renal tubular epithelial heparan sulfate. J Biol Chem 2012; 287: 31471–31481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brodsky SV, Nadasdy GM, Pelletier R, et al. Expression of the decay-accelerating factor (CD55) in renal transplants—a possible prediction marker of allograft survival. Transplantation 2009; 88: 457–464. [DOI] [PubMed] [Google Scholar]
- 28.Diebolder CA, Beurskens FJ, de Jong RN, et al. Complement is activated by IgG hexamers assembled at the cell surface. Science 2014; 343: 1260–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thomas KA, Valenzuela NM, Reed EF. The perfect storm: HLA antibodies, complement, FcgammaRs, and endothelium in transplant rejection. Trends Mol Med 2015; 21: 319–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bettinotti MP, Zachary AA, Leffell MS. Clinically relevant interpretation of solid phase assays for HLA antibody. Curr Opin Organ Transplant 2016; 21: 453–458. [DOI] [PMC free article] [PubMed] [Google Scholar]