

Abstract
Heterologously expressed hERG channels represent a mainstay of in vitro drug safety screens intended to mitigate risk of cardiac IKr block and sudden cardiac death. This is true even as more channel types are adopted as part of the Comprehensive in vitro Proarrhythmia Assay (CiPA) intended to elevate specificity and thus enhance throughput of promising lead drugs. Until now, hERG1a homomeric channels have been used as a proxy for IKr despite a wealth of evidence showing that hERG1a/1b heteromers better represent native channels in terms of protein abundance and channel biophysical and pharmacological properties. Past efforts to create a stable hERG1a/1b cell line were met with unpredictable silencing of hERG1b expression despite stable integration of the gene into the HEK293 cell genome. Here we report a new cell line stably expressing hERG1a, with hERG1b reliably controlled by an inducible promoter sensitive to doxycycline. Co-immunoprecipitation, Western blot analysis and patch-clamp electrophysiology confirm the heteromeric composition of the expressed channels. Association with hERG1b was found to promote hERG1a protein levels and enhance membrane current levels. Optimal conditions for drug screening and experimental investigation were achieved at 24 h exposure to 100 ng/ml doxycycline. Differences in pharmacological sensitivity between homomeric and heteromeric channels were observed for dofetilide and ebastine, but not fluoxetine, as evaluated by their IC50 values. Using these values in the O’Hara-Rudy-CiPA in silico model revealed discrepancies in pro-arrhythmia risk, implying the hERG1a homomeric platform overestimates risk for these two drugs. Dofetilide block was use-dependent and faster for hERG1a/1b than hERG1a channels, whereas ebastine showed considerable block at rest and had a slower progression for hERG1a/1b channels. The hERG1a/1b cell line thus represents an advanced model for contemporary drug safety screening assays such as CiPA that employ IC50 values to estimate risk of proarrhythmia in computational models of ventricular cardiomyocytes. This novel technology fulfills an unmet need to enhance specificity and foster a safe yet expanded drug development pipeline.
Keywords: Kv11.1, KCNH2, KCNH, action potential, repolarization, arrhythmia, drug screening, CiPA, heart, qNET
INTRODUCTION
Cardiac IKr is the chief target of drugs causing acquired long QT syndrome and therefore a primary focus of safety in drug development (Vandenberg et al., 2012). IKr is blocked by an astounding array of compounds, some of which lead to prolonged repolarization and, more rarely, sudden cardiac death (Witchel & Hancox, 2000). Native IKr is composed of ERG1a and ERG1b, two subunits encoded by the Ether-a-go-go Related Gene, ERG, or KCNH2 (Jones et al., 2004; Lees-Miller et al., 1997; London et al., 1997; Sale et al., 2008). However, typical drug-screening assays featuring human ERG (hERG) utilize only hERG1a homomeric channels, despite documented differences compared to the hERG1a/1b channel of several-fold in drug sensitivities (Abi-Gerges et al., 2011; Harchi et al., 2018; Orvos et al., 2019).
Current hERG1a assays fall short of single-handedly predicting drug toxicity for a variety of reasons. Some hERG blockers such as fluoxetine, a selective serotonin reuptake inhibitor, are safe, possibly because of compensatory blocking effects on Ca2+ or Na+ channels (Pacher et al., 2000). Dofetilide, an antiarrhythmic drug with pro-arrhythmogenic potential, not only blocks IKr with high affinity (Jurkiewicz & Sanguinetti, 1993) but upon prolonged exposure can lead to an increase in late Na+ current, potentially exacerbating repolarization defects and enhancing risk of arrhythmia (Qiu et al., 2016). For this reason, ongoing efforts of the Comprehensive in vitro Proarrhythmia Assay (CiPA) initiative aim to improve drug safety by taking into account potencies of compounds affecting not only IKr but also L-type Ca2+ and late Na+ currents (Huang et al., 2017). Half-maximal concentrations (IC50’s) determined using heterologous expression systems will be combined into a computational model of a ventricular action potential comprising all relevant conductances as a function of time (Li et al., 2019). Because IC50 values represent the primary variable in the model, accurate measurements faithfully representing effects of drugs on native channels are critical.
Heteromeric association of hERG1a and hERG1b subunits occurs early in biogenesis (Liu et al., 2016; Phartiyal et al., 2007) with an avidity greater than that of homomeric association (McNally et al., 2017). Both subunits localize to T-tubules in ventricular myocytes, and co-IP evidence suggests native IKr channels are composed of both hERG1a and 1b subunits in vivo (E. M. C. Jones et al., 2004). Pharmacological studies in heterologous expression systems showed that some drugs exhibited similar potencies of block (IC50’s) of homomeric 1a and heteromeric 1a/1b channels, whereas other diverged by several-fold (Abi-Gerges et al., 2011; Sale et al., 2008). Unfortunately, a stable heteromeric cell line developed for such studies (Abi-Gerges et al., 2011) subsequently exhibited unpredictable silencing of the hERG1b subunit in follow-on studies, limiting utility of the cell line for academic or industrial applications.
Here we describe a new stable cell line in which hERG1a is constitutively expressed whereas hERG1b is under the control of a doxycycline-inducible promoter providing reliable expression under experimental control. This design uniquely allows observation of the quantitative effects of 1b induction as a function of doxycycline concentration or duration of application on hERG1a protein levels controlled within the same experiment. The expression quantitatively depends on doxycycline dose and duration of treatment, resulting in electrophysiological and pharmacological differences between homomeric and heteromeric channels. This new technology should be of broad use in drug safety testing and studies focused on the biology of cardiac hERG channels.
METHODS
Cell line development and maintenance
The hERG1a/1b-inducible cell line was created by transfecting an extant HEK293 line stably expressing hERG1a (Zhou et al., 1998) with 2.5 μg of a plasmid comprising the hERG1b cDNA cloned downstream from an inducible promoter in the pTet-One/Puro vector (Takara; Fig. 1). After 48 hours, confluent cells were split into four 10 cm dishes. After 48 additional hours, puromycin was added for cell selection. Medium containing puromycin was replaced every 4 days. After 3-5 days, cells not integrating the plasmid died and, in the following two weeks, drug resistant colonies appeared. Colonies were harvested and transferred individually into separate 24-well plates. Clones were selected and cultured with puromycin, grown to confluency, and split for further maintenance and testing.
Fig. 1.

Scheme showing the strategy used to develop hERG1a/1b cell line. A construct comprising the Tet-ON gene that, activated by doxycycline, promotes the expression of hERG1b gene, was transfected and selected using puromycin in the hERG1a stable cell line. Induction of hERG1b expression was achieved only when the extracellular medium contains doxycycline, which binds to the 3G protein and promotes hERG1b gene transcription and synthesis of hERG1b protein. PhPGK, human Phosphoglycerate Kinase 1 promoter; PTRE3GS, inducible promoter; 3G, Tet-ON transactivator; DOX, doxycycline.
Maintenance of the stable cell line was achieved by culturing the cells at 37°C, 5% CO2 in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum. 5 μg/mL G418 and 0.25 μg/mL puromycin were freshly added to the medium for the maintenance of hERG1a and hERG1b, respectively. Antibiotic stock solution aliquots were kept at 4°C and added every other day when the medium was changed. To induce hERG1b protein expression, different concentrations of doxycycline (DOX) were added directly to the medium from a freshly prepared stock. Expression of hERG1b was then assessed by Western blot analysis at different DOX concentrations and post-treatment time points. For patch-clamp experiments cells were trypsinized and plated on coverslips at a low density for 24 hours to allow cells to recover; DOX was then added to the medium to induce hERG1b expression and recordings were performed at different time points.
Electrophysiology
Manual patch clamp under whole cell configuration was used to record hERG currents in HEK293 cells stably expressing hERG1a and the inducible hERG1b. All data obtained were from a minimum of two separate inductions. Borosilicate glass pipettes with a resistance of 2.5 – 5 MΩ when filled with “internal solution” were fabricated using an automatic puller (Sutter Instruments). External solution used for recording contained (in mM) 150 NaCl, 5.4 KCl, 1.8 CaCl2, 1 MgCl2, 15 Glucose, 15 HEPES, 1 Na-pyruvate, pH 7.4 (NaOH); internal solution contained 5 NaCl, 150 KCl, 2 CaCl2, 5 EGTA, 10 HEPES, 5 Mg-ATP, pH 7.3 (KOH). Recordings were done near physiological temperature (35°C ± 2°C) or room temperature (21°C ± 2°C) as noted. hERG currents were recorded in response to 4 s pulses from a holding potential of −80 mV, stepping up to +40 mV in 10 mV increments, followed by a repolarizing step to −60 mV for 5 s (Fig. 2A). Leak subtraction was done offline using Ohm’s Law based on passive leak current measured at −80 mV. The deactivating current was fit to a second order exponential equation to obtain both the slow and fast time constants (τ) of deactivation. Steady-state activation curves were generated by plotting the current at the end of the 4 s pulse from −80 to +40 mV in 10 mV increments from a holding potential of −80 mV. Tail currents were measured at −60 mV at the moment of voltage change (4 s pre-pulses from +40 mV to −60 mV). Tail currents plot was fit to a Boltzmann equation (I(t) = [(V − Vrev) * Imax]/(1 + exp(V − V1/2/k))) to obtain the voltage dependence of activation parameters.
Fig. 2.
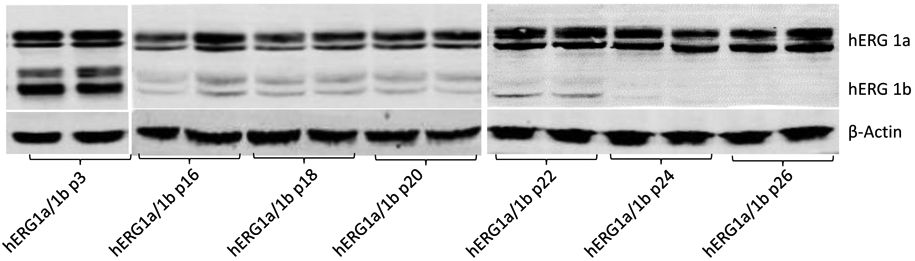
Silencing of hERG1b expression in a previous developed cell line. Representative Western blot showing the expression of mature and immature hERG1a (upper bands) and hERG1b (middle bands) in different passage numbers (p#) in an old cell line expressing both channels. Reduction or complete silencing of hERG1b expression but not hERG1a is evident as the passage number increases. β-actin was used as a loading control (lower band).
To study hERG current under conditions mimicking a ventricular action potential we used a waveform previously generated from a rabbit ventricular myocyte (Zhou et al., 1998). The voltage protocol was applied three times and then averaged to obtain hERG current. Recordings for this protocol were done at near-physiological temperature (35°C ± 2°C). Data were sampled at 1.15 kHz and 1152 samples were acquired per channel, with low-pass filter set to 100 kHz. Compensation was used to ensure less than ±5 mV voltage error when needed. The integral of hERG current during the action potential protocol was measured to determine the charge conducted through hERG channels normalized by the cell capacitance.
Drug sensitivity was measured by comparing the IC50 values obtained for dofetilide, fluoxetine or ebastine in homomeric versus heteromeric channels. Different concentrations of the drugs were used in cells treated with 0 or 100 ng/ml of DOX for 24 hours unless otherwise specified. hERG block was assessed by measuring hERG tail current at −60 mV from a pre-pulse of +20 mV before and after perfusing the drug for at least 10 minutes or until current was constant. The voltage step protocol was applied every 30 seconds. Dose-response curves were generated by plotting the steady-state current vs. drug concentration and fitted with the following Hill equation:
; where IC50 is the concentration causing half-maximal current block, Imax is the maximal current, [drug] is the concentration of the drug and nH is the Hill coefficient.
Action Potential Simulations
To compare the effects on risk markers of the different drug IC50 values obtained for hERG1a or hERG1a/1b channels we used the ActionPotential-Portal application (Williams & Mirams, 2015; https://cardiac.nottingham.ac.uk/ActionPotential/about). For all simulations we selected the version of the O’Hara-Rudy-CiPA model (Rudy et al., 2011) described by Dutta et al. (2017) with a pacing frequency of 0.5 Hz and a maximum pacing time of 120 minutes. From this model we obtained the net charge during the action potential (qNET) and changes in the action potential duration (ΔAPD) as parameters to assess pro-arrhythmic risk when comparing hERG1a or hERG1a/1b IC50 values. We exported the data to Excel and graphed the results using SigmaPlot 13.0 or Origin 2018a.
Western blot analysis
Cells were lysed using a buffer containing in mM: 150 Tris·NaCl, 25 Tris·HCl, 10 NaEGTA, 20 NaEDTA, and 5 glucose, supplemented with 1% Triton X-100, and a 1:100 dilution of protease inhibitor cocktail (#K272 from BioVison), sonicated, incubated for 1 h at 4°C in an automatic spinning wheel, and then centrifuged for 10 minutes at 12,500 rpm. Protein concentration was calculated using the Bradford assay (BIO-RAD, USA). 200 μg protein lysate was loaded into each lane of a 7.5% SDS-polyacrylamide gels and then transferred to polyvinylidene (PDVF) membranes for 1 h at 100 mV. Membranes were blocked with 5% milk and then incubated overnight at 4°C with a dilution of 1:1000 anti-pan hERG (#ALX-215-049-R100 from ENZO) and 1:5000 anti-β-actin (#A2228 from Sigma). Membranes were washed with TBS-TWEEN and incubated for at least 1 h at room temperature with secondary antibodies using a dilution of 1:10,000 (Alexa 488 or 647 from Thermofisher). Membranes were imaged using Chelidon-MP Imaging System (BIORAD).
Co-immunoprecipitation
Co-immunoprecipitation of hERG1a and hERG1b subunits was done as previously described (Phartiyal et al., 2007). Briefly, control and DOX-treated cell lysates were precleared with 30 μl protein G sepharose beads for 1 h at 4°C. Lysates were centrifuged at 10,000 g for 1 min at 4°C to remove the beads. hERG1a specific antibody (hERG1a (D1Y2J) Rabbit mAb #12889 from Cell Signaling) was added and incubated overnight at 4°C. Immunoprecipitates were washed three times with lysis buffer and eluted into sample buffer to perform Western blot experiments to detect hERG1a/1b complex with a hERG1 A12 antibody (#ALX-804-652-R300, Enzo Life Sciences).
Statistics
hERG currents were analyzed with Clampfit 10.4 software (Molecular Devices). SigmaPlot 13.0 (Systat Software) or Origin 2019 (OriginLab) was used to compare results among cells treated for 0, 24, and 48 h with 100 ng/ml DOX using either t-test or one-way ANOVA analysis. Results were considered statistically significant when p < 0.05. Mycoplasma contamination of the cell line was not tested during the study. No blinding or randomization was conducted for the experiments. No data were excluded as outliers.
RESULTS
Unpredictable hERG1b silencing in a conventional stable cell line
Extended utilization of a constitutively-expressing hERG1a/1b cell line generated previously (Abi-Gerges et al., 2011) yielded erratic reductions in hERG1b expression. Reduced expression was evident from passage 16, and complete hERG1b silencing occurred in later passages (24-26) as shown in Fig. 2. In separate platings, loss of hERG1b was not strictly linked to passage number, sometimes occurring within the first few passages, nor could it be attributed to culture conditions such as plating densities. These challenges prompted the design of a cell line permitting parallel pharmacological testing of homomeric hERG1a and heteromeric hERG1a/1b channels on a timeline suitable for screening assays and too short to allow extensive cell division that might evoke the hERG1b silencing process. The mechanism of hERG1b silencing was not investigated; we speculate it may be related to a role of hERG1b in cell cycle control (Crociani et al., 2003).
Inducible expression of hERG1b with DOX is time- and dose-dependent
The design of the cell line in which hERG1a is expressed constitutively and hERG1b is induced under control of a doxycycline (DOX) promoter is diagrammed in Fig. 1 (see Methods). Application of DOX yielded quantitative induction of hERG1b in the background of hERG1a at 24, 48 and 72 h (Fig. 3A). No expression of hERG1b was observed for up to 72 h under culture conditions in the absence of DOX (Fig. 3B). DOX did not affect levels of stably expressed hERG1a alone (Fig. 3C). Quantification shows that hERG1b expression was achieved at low concentrations of DOX (50 ng/ml) and increased in a dose-dependent manner (Fig. 3D, E). Expression of mature and immature hERG1b reached its approximate maximum at 48 h after cells were treated with different concentrations of DOX (Fig. S1). These observations indicate that the inducible hERG1b plasmid stably integrated into the cell line genome and its expression is DOX dose dependent.
Fig. 3.
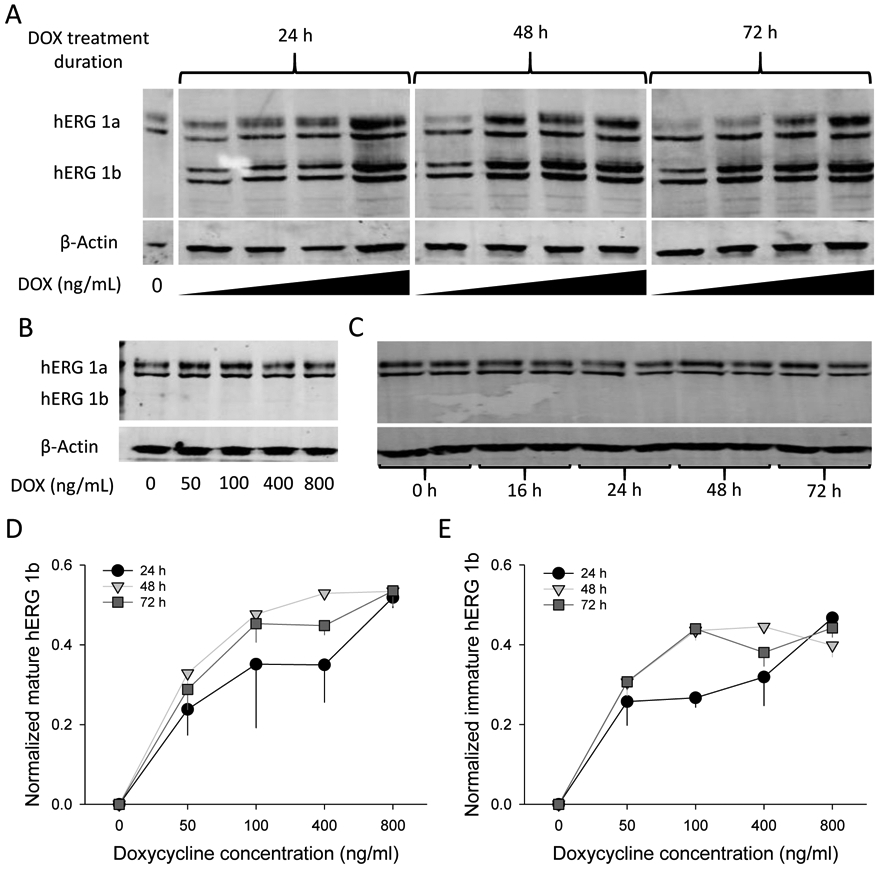
hERG1b inducible expression is DOX dose dependent. A) Representative blot of cells treated with increasing concentrations of DOX for 0, 24, 48, and 72 h. Both mature and immature bands are revealed for hERG1a (upper bands) and hERG1b (lower bands). Beta-actin was used as a loading control. DOX concentrations used for the experiments were 0, 50, 100, 400 and 800 ng/ml, from left to right, corresponding to each lane. B) Representative blot from hERG1a stable cell line treated with different concentrations of DOX for 24h to investigate the effects of DOX on hERG1a expression alone. C) Representative blot of hERG1a/1b cell line kept under culture conditions without DOX treatment and analyzed at different time points to determine if any “leak” expression was present. D and E) Quantification of mature and immature hERG1b, respectively after 24, 48 and 72 h treatment with DOX. hERG1b mature and immature bands were normalized to maximal hERG1b total signal for each independent blot (mature + immature signals). β-actin was used a loading control and no significant differences between lanes were observed for its signal intensity. N = 4.
Association of hERG1a and DOX-induced hERG1b
To determine whether DOX-induced hERG1b forms a stable complex with hERG1a, we used hERG1a-specific antibodies to immunoprecipitate hERG1a. Association with hERG1b was revealed by probing for both subunits with a pan-hERG antibody via Western blot (Fig. 4). The results clearly show that, despite prior and constitutive expression of hERG1a, coassembly with hERG1b readily occurs upon its induction. Based on findings of previous studies (Phartiyal et al., 2007), this association likely occurs early in biogenesis. Maturation of both subunits was robust, and indeed hERG1a levels were enhanced upon hERG1b induction (Fig. 5). Trafficking effects were not thoroughly characterized but channel protein stability was not clearly different between homomeric and heteromeric channels (Fig. S2).
Fig. 4.
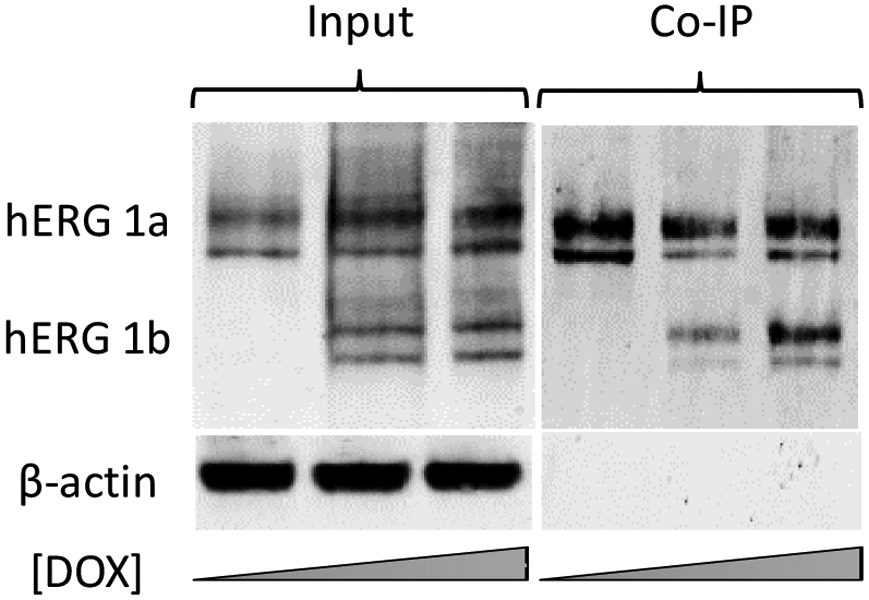
hERG1a and hERG1b coimmunoprecipitate when hERG1b expression is induced. Cells were treated for 24 hours with increasing concentrations of DOX and probed for hERG expression and interaction between hERG1a and 1b subunits. Western blot showing expression of hERG1a and the induction of hERG1b using 50 and 100 ng/ml of DOX (Input). Coimmunoprecipitation of hERG1a with hERG1b was revealed by probing for both subunits using a pan-hERG antibody in cells treated or not with DOX (Co-IP).
Fig. 5.

Mature hERG1a protein increases as hERG1b expression is induced. A) Representative blot for hERG1a, same as in Fig. 2, with only hERG1a signal and after 48 h of treatment with DOX. Lanes are 50, 100, 400 and 800 ng/ml from left to right, respectively. B) Quantification of immature and mature hERG1a protein in cells treated for 48 h with increasing DOX concentrations. C) Time dependence of immature and mature hERG1a protein expression in cells treated with 100 ng/ml of DOX. hERG1a mature and immature bands were normalized to maximum hERG1a total signal for each independent blot (mature + immature signals). β-actin was used a loading control and no significant differences between lanes were observed for its signal intensity.Statistical significance (p ≤ 0.05) was evaluated using t-test comparison with respect to time 0 h. N = 4.
Heteromeric channel properties induced by DOX treatment
Patch-clamp electrophysiology revealed the hallmark functional properties of heteromeric channels when hERG1b was induced. Deactivating tail currents, expected to accelerate with the contribution of hERG1b subunits (London et al., 1997; Sale et al., 2008), exhibited faster deactivation time constants (τ) in DOX-treated cells compared with untreated cells (Fig. 6A-C). This effect was true at 24 and 48 h after DOX treatment. No differences were observed in voltage dependence of activation between controls and cells treated with DOX (Table S1). Current densities of both steady-state and tail currents reached maximum effect at 24 h treatment (Fig. 6D-E). An increase in hERG tail current, a proxy for density of functional channels at the membrane, supports the conclusion that hERG1b induction enhances channel maturation or stability (cf. Fig. 5). This effect, together with previously established acceleration of activation and recovery from inactivation kinetics (Sale et al., 2008), result in the enhanced current evoked by a voltage-clamp protocol designed to mimic a ventricular action potential (Fig. 6F, G). Because heteromeric currents more faithfully represent cardiac IKr (Larsen & Olesen, 2010; Sanguinetti & Jurkiewicz, 1990; Weerapura et al., 2002), the 1b-inducible cell line is a notably improved model over the homomeric hERG1a cell-based assay currently in wide use.
Fig. 6.

hERG1b expression results in ion channels with heteromeric properties. Cells were treated with 0 or 100 ng/ml of DOX for 24 and 48 h, followed by current recording using patch-clamp under whole-cell configuration. A) Representative traces in a non-treated cell (0 h), and cells treated with 100 ng/ml DOX for 24 h and 48 h. Voltage step protocol used is shown on top of the 0 h trace. B) IV plot for hERG steady-state currents in cells treated with DOX 100 ng/ml for 0, 24 and 48 h. C) IV plot for peak tail currents under same conditions as B. D) Slow and fast E) deactivation time constants calculated by fitting the tail current elicited at −60 mV after a pre-pulse potential of +40 mV. Statistical significance was tested using a One-way ANOVA (* p < 0.05). N = 8, 8, 5 cells for 0, 24 and 48 h, respectively. F) Representative traces of hERG current during an action potential clamp protocol (red trace) in one cell treated with DOX, for 24 h, and another without treatment. G) Charge conducted by hERG channels in cells treated with 0 or 100 ng/ml DOX for 24 h, as determined by integrating the current during the action potential clamp protocol. One-way ANOVA test was used to determine significant changes (* p < 0.05). N = 8, 8 for 0 and DOX, respectively. All currents were recorded at 35°C ± 2°C.
Differences in drug sensitivities for heteromeric versus homomeric channels
Previous work shows that drug sensitivity of hERG channels is affected by subunit composition and temperature (Abi-Gerges et al., 2011; Harchi et al., 2018; Perissinotti et al., 2018). To validate these effects in the inducible cell line, we tested sensitivities of three known hERG blockers: dofetilide, ebastine and fluoxetine (Fig. 7). We recorded hERG current before and after perfusion of each drug at different concentrations. Recordings were done at 35°C ± 2°C (see Methods section). We found differences in IC50 values of approximately three-fold for dofetilide between hERG1a and hERG1a/1b channels (Fig. 7B). Dose-response curves at room temperature showed no significant difference from those at higher temperatures despite faster deactivation kinetics (Fig. S3). Ebastine exhibited an approximately two-fold difference (Fig. 7B) while no statistically significant difference was observed for fluoxetine (Fig. 7C).
Figure 7.

hERG homomeric and heteromeric channels have different drug sensitivities and time course of drug action. All points were peak tail current measurements obtained from cells expressing hERG1a or hERG1a/1b channels measured at 35°C ± 2°C. Tail currents were measured at −60 mV following a 3-s prepulse to +20 mV. A) Dofetilide dose-response curve. B) Ebastine dose-response curve. C) Fluoxetine dose-response curve. IC50 values are shown in each panel for 1a (red) and 1a/1b (blue) cells. Each point of the curve was obtained from at least 3 different cells (n > 3) from two or more separate inductions. D) Normalized peak tail current in the presence of dofetilide (100 nM) plotted as a function of the number of sweeps and fitted to a single exponential curve to estimate the time course (τ) of block for hERG1a (red) and hERG1a/1b (blue). E) Normalized peak tail current in the presence of ebastine (100 nM) plotted as a function of the number of sweeps and fitted to a single exponential curve to estimate the time course (τ) of block for hERG1a (red) and hERG1a/1b (blue). F) Normalized peak tail current in the presence of fluoxetine (1 μM) plotted as a function of the number of sweeps and fitted to a single exponential curve to estimate the time course (τ) of block for hERG1a (red) and hERG1a/1b (blue). Drugs were perfused for 2 minutes (gray area) before running the first sweep. For this experiments cells were treated or not with 100 ng/ml of DOX for at least 24 hours.
Differences in kinetics of drug action were also observed. We compared the onset of block of dofetilide, ebastine and fluoxetine for hERG1a and hERG1a/1b channels. Interestingly, we found that block by dofetilide (at 100 nM), an open channel blocker showing use-dependence (Ficker et al., 1998), developed faster in heteromeric channels (Fig. 7D). This may be expected based on faster activation kinetics of hERG1a/1b compared to hERG1a (Sale et al., 2008). The opposite behavior was observed for ebastine, in which block occurred more slowly for heteromeric vs. homomeric channels (Fig. 7E). Moreover, 60% of homomeric channels were blocked by ebastine by the time of the first pulse, suggesting this drug can block resting channels. No significant differences were observed for fluoxetine (Fig. 7F). Future experiments will be required to determine mechanistic differences in block by these compounds and how they are affected by the biophysical properties of the two channel types.
Evaluation of Torsadogenic risk using IC50 values determined for hERG1a and hERG1a/1b cells using an in silico model
To illustrate the consequences of hERG subunit composition on arrhythmia risk, the O’Hara-Rudy CiPA (Dutta et al., 2017) model was selected in the ActionPotential-Portal application to run simulations in the presence of hERG block by dofetilide, ebastine or fluoxetine. Two main parameters were evaluated to assess risk: the conventional changes in APD (ΔAPD) and changes to the net charge (qNet) carried by ionic currents during the action potential, which was previously shown to effectively assess Torsadogenic risk in a CiPA set of 12 drugs (Dutta et al., 2017).
Simulations based on IC50 values and Hill coefficients from our dose response curves at near-physiological temperature revealed hERG1a- and hERG1a/1b-specific differences in measures of proarrhythmia (Fig. 8). Consistent with elevated IC50’s for dofetilide and ebastine, APD showed less prolongation and reduced propensity for early afterdepolarizations (EADs) for hERG1a/1b compared to hERG1a at each concentration (Fig. 8A, B, D, E). For both channels, the value of qNet diminished as a function of drug concentration, reflecting a reduction in repolarizing outward currents. Negative qNet values reflect a net positive current associated with early afterdepolarizations. For dofetilide and ebastine, repolarizing capacity decreased more rapidly in simulations where the IC50 value for hERG1a was used rather than that for hERG1a/1b. This simulation indicates a higher predicted risk when hERG1a IC50 value was utilized in the model for these drugs (Fig. 8C, F). Notable differences were not observed for fluoxetine (Fig. 8G, H, 8I) as expected from similar IC50’s for hERG1a and 1a/1b. In general, reduction of qNET and prolongation of APD with increasing concentrations of dofetilide or ebastine are more drastic using hERG1a compared to hERG1a/1b IC50 values, an effect that is evident even when low drug concentrations are evaluated (see insets in qNet and ΔAPD graphs). Our results suggest that even small discrepancies in the IC50 values obtained from in vitro assays can result in dramatic changes when introduced to an in silico model, a result evident only when studying both hERG1a and hERG1a/1b channels.
Figure 8.

Comparison of pro-arrhythmic parameters using IC50 values from hERG1a versus hERG1a/1b cells. Simulations were done using the web ActionPotential Portal application by introducing IC50 values determined in hERG1a (red) or hERG1a/1b cells (blue) for each drug at physiological temperature (see Methods for details). A) Action potential simulations with increasing concentrations of dofetilide. B) Changes in the action potential duration (ΔAPD) in percentage estimated with different concentrations of dofetilide. Insert, amplification for lower concentrations of dofetilide. C) qNet changes during the action potential at different concentrations of dofetilide. Insert, amplification for lower concentrations of dofetilide. D) Action potential simulations with increasing concentrations of ebastine. E) Changes in the action potential duration (ΔAPD) in percentage estimated with different concentrations of ebastine. Insert, amplification for lower concentrations of ebastine. F) qNet changes during the action potential at different concentrations of ebastine. Insert, amplification for lower concentrations of ebastine. G) Action potential simulations with increasing concentrations of fluoxetine. H) Changes in the action potential duration (ΔAPD) in percentage estimated with different concentrations of fluoxetine. Insert, amplification for lower concentrations of fluoxetine. I) qNet changes during the action potential at different concentrations of fluoxetine. Insert, amplification for lower concentrations of fluoxetine.
DISCUSSION
We have established and characterized a cell line constitutively expressing hERG1a with controlled induction of hERG1b expression as a platform for drug safety screening and pharmacological and biological studies of hERG and cardiac IKr. By inducing hERG1b expression, the system overcomes the challenges of unpredictable hERG1b silencing in a previously developed hERG1a/1b stable cell line (Abi-Gerges et al., 2011). Induction of hERG1b expression by DOX is dose- and time-dependent, with optimal expression of protein levels and function achieved after 24 h with 100 ng/ml DOX as determined by Western blot and electrophysiological analysis. Heteromerization of hERG channels was shown by coimmunoprecipitating both subunits and evident as electrophysiological and pharmacological differences arose when hERG1b expression was induced. Interestingly, we found that hERG1b co-expression enhanced expression of hERG1a subunits and the number of functional hERG channels at the plasma membrane. We confirmed previous studies that, compared with hERG1a channels, hERG1a/1b channels are less potently blocked by dofetilide and ebastine (Harchi et al., 2018; Perissinotti et al., 2018). Additionally, simulations used to predict Torsadogenic risk of drugs showed that even small differences in IC50 values can result in markedly distinct outcomes. Because cardiac IKr is produced by heteromeric hERG1a/1b channels and has been previously shown to exhibit drug sensitivities different from homomeric hERG1a channels (Abi-Gerges et al., 2011; Harchi et al., 2018; Perissinotti et al., 2018; Sale et al., 2008), this technology responds to calls for greater accuracy in drug safety screening moving forward as proposed in the CiPA initiative (Wallis et al., 2018).
Previous studies have explored mechanisms of assembly of heteromeric hERG1a/1b channels. Association occurs early in biogenesis, during translation of hERG subunits, through their heterotypic N-terminal domains (Phartiyal et al., 2007). The effect is cotranslational, as mRNA transcripts encoding both subunits coimmunoprecipitate with the nascent hERG1a protein (Liu et al., 2016). When hERG1b subunits are present, preferential formation of heteromers over homomers results (McNally et al., 2017). The present study shows that even a system constitutively expressing hERG1a homomers can nimbly shift to produce heteromers upon hERG1b induction. Thus, the cell line represents a research tool for further studies enriching our understanding of hERG subunit interactions.
Functional consequences of coexpression of hERG1a and1b subunits are an increased probability of opening (Po) compared to hERG1a homomers resulting from accelerated activation and recovery from inactivation, which in concert lead to increased current amplitudes during an action potential despite the concomitant acceleration of deactivation (Sale et al., 2008). Another study revealed similar single-channel conductance (γ) values of ~12 pS for the two isoforms (Wilson et al., 2019). Here we demonstrate heteromerization also promotes plasma membrane channel density (N) as reflected in enhanced fraction of mature hERG1a protein and larger peak currents. Thus, a more complete description of effects of heteromerization on conductance (G = Ν·Po·γ) emerges as fundamental characteristics of cardiac IKr.
The importance of heteromeric assembly of hERG1a and 1b is demonstrated in emerging studies of new mechanisms of long QT syndrome (LQTS) (Mehta et al., 2014; Mesquita et al., 2019). For example, the LQTS R25W mutation in hERG1b reduces levels of hERG1a protein and its expression at the plasma membrane (Jones et al., 2016). Some hERG1a disease mutants exert dominant negative effects on trafficking only when coexpressed with hERG1b subunit (Puckerin et al., 2016). Deleting ERG1b in mice results in loss of IKr in adult ventricular myocytes (J. P. Lees-Miller et al., 2003). These studies all support the conclusion that hERG1b promotes hERG1a maturation or functional expression under physiological and pathological conditions. Underlying mechanisms of this regulation will require additional investigation.
Although the expression of hERG1b protein is steadily time-dependent in the presence of DOX, we detected an unexpected reduction in current levels at 48 h compared to 24 h. The basis for this discrepancy was not determined. Possible explanations may lie in differences in cell culture confluency between cells destined for patch clamp and those used for Western blot. Alternatively, mature protein visible via Western blot may represent channels in compartments in addition to the surface membrane, such as those undergoing endocytosis (Guo et al., 2011).
These results suggest the optimal point for studying the pharmacology of heteromeric channels in the cell line is 24 h post treatment with DOX. They also reflect the novel suitability of the cell line for studying mechanisms regulating channel density at the plasma membrane for future investigations that lie beyond the scope of the present study.
The current study provides evidence for a reliably stable assay producing hERG1a/1b currents more closely mimicking native IKr with properties in accordance with previous studies using transient transfections (McPate et al., 2009; Orvos et al., 2019; Sale et al., 2008). IC50 values for dofetilide are nearly identical to those in rabbit ventricular (13 nM vs. 12 nM, respectively) yet 3-fold different from values for hERG1a homomeric channels (Orvos et al., 2019). With respect to ebastine, the drug showing the greatest differences in IC50 between hERG1a and hERG1a/1b channels, one previous study on hERG1a/1b channels transiently transfected shows the drug more potently blocked hERG1a vs. hERG1a/1b by six-fold (Harchi et al., 2018), whereas we found a somewhat reduced difference of three-fold. These values are in contrast to an earlier studies conducted at room temperature in high throughput planar patch configuration, in which relative ebastine potency was reversed for the two channel types (Abi-Gerges et al., 2011), suggesting more work is needed to test the inducible hERG1a/1b cell line on multiple platforms. Altogether, our in vitro hERG block assessment using this cell line suggests that, compared with the standard hERG1a cell line, hERG1a/1b channels better reflect native IKr, and therefore as a component of a comprehensive safety test will provide more accurate IC50 data required to estimate proarrhythmic potential of drugs destined for therapeutic use. Moreover, the inducible cell line will comport better with other elements of a comprehensive assay, including iPSC-derived cardiomyocytes in which IKr is known to be conducted by heteromeric hERG1a/1b channels (Jones et al., 2014).
Drug development and safety screening protocols proposed by the CiPA initiative assesses hERG block in vitro to calculate an IC50 value using a hERG1a cell line as the standard model, while the presence of hERG1b subunit has not been considered. Here we have shown that IC50 values obtained in hERG1a and hERG1a/1b led to different Torsadogenic risk predictions as measured by qNet and ΔAPD (Fig. 8). A limitation of the approach is that the model used is based on hERG1a, which compared to hERG1a/1b yields lower Po and net repolarizing charge during an action potential (Sale et al., 2008). Incorporating hERG1a/1b gating behavior would further separate proarrhythmic outcomes for the two channel types and enhance accuracy of predictive models. Further improvements may result by incorporating differences in kinetics of drug action, which were noted but not rigorously examined within the scope of the current study. Based on these results, further experiments with the hERG1a/1b cell line should not only improve drug screening but also current and future action potential models predicting proarrhythmia.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank the members of the Robertson lab for valuable discussion.
FUNDING
This work was supported by NIH grants 1R01HL131403 and 5R01NS081320 (GAR), and T32HL007936 fellowships (EBR-P and WAS-S).
References
- Abi-Gerges N, Holkham H, Jones EMC, Pollard CE, Valentin JP, & Robertson GA (2011). HERG subunit composition determines differential drug sensitivity. British Journal of Pharmacology, 164(2 B), 419–432. 10.1111/j.1476-5381.2011.01378.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crociani O, Guasti L, Balzi M, Becchetti A, Wanke E, Olivotto M, Wymore RS, & Arcangeli A (2003). Cell cycle-dependent expression of HERG1 and HERG1B isoforms in tumor cells. Journal of Biological Chemistry, 278(5), 2947–2955. 10.1074/jbc.M210789200 [DOI] [PubMed] [Google Scholar]
- Dutta S, Chang KC, Beattie KA, Sheng J, Tran PN, Wu WW, Wu M, Strauss DG, Colatsky T, & Li Z (2017). Optimization of an in silico cardiac cell model for proarrhythmia risk assessment. Frontiers in Physiology, 8(AUG), 1–15. 10.3389/fphys.2017.00616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ficker E, Jarolimek W, Kiehn J, Baumann A, Brown AM, Johann K, Baumann A, & Brown AM (1998). Molecular determinants of dofetilide block of HERG K+ channels. Circulation Research, 82(3), 386–395. 10.1161/01.RES.82.3.386 [DOI] [PubMed] [Google Scholar]
- Guo J, Li X, Shallow H, Xu J, Yang T, Massaeli H, Li W, Sun T, Pierce GN, & Zhang S (2011). Involvement of caveolin in probucol-induced reduction in hERG plasma-membrane expression. Molecular Pharmacology, 79(5), 806–813. 10.1124/mol.110.069419 [DOI] [PubMed] [Google Scholar]
- Harchi A. El, Melgari D, Zhang H, & Hancox JC (2018). Investigation of hERG1b Influence on hERG Channel Pharmacology at Physiological Temperature. Journal of Pharmacology and Pharmacotherapeutics, 9(2), 92–103. 10.4103/jpp.JPP [DOI] [Google Scholar]
- Huang H, Pugsley MK, Fermini B, Curtis MJ, Koerner J, Accardi M, & Authier S (2017). Cardiac voltage-gated ion channels in safety pharmacology: Review of the landscape leading to the CiPA initiative. Journal of Pharmacological and Toxicological Methods, 87, 11–23. 10.1016/j.vascn.2017.04.002 [DOI] [PubMed] [Google Scholar]
- Jones DK, Liu F, Dombrowski N, Joshi S, & Robertson GA (2016). Dominant negative consequences of a hERG 1b-specific mutation associated with intrauterine fetal death. Progress in Biophysics and Molecular Biology, 120(1-3), 67–76. 10.1016/j.pbiomolbio.2016.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DK, Liu F, Vaidyanathan R, Eckhardt LL, Trudeau MC, & Robertson GA (2014). hERG 1b is critical for human cardiac repolarization. Proceedings of the National Academy of Sciences of the United States of America, 111(50), 18073–18077. 10.1073/pnas.1414945111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones EMC, Roti Roti EC, Wang J, Delfosse SA, & Robertson GA (2004). Cardiac IKr channels minimally comprise hERG 1a and 1b subunits. Journal of Biological Chemistry, 279(43), 44690–44694. 10.1074/jbc.M408344200 [DOI] [PubMed] [Google Scholar]
- Jurkiewicz NK, & Sanguinetti MC (1993). Rate-dependent prolongation of cardiac action potentials by a methanesulfonanilide class III antiarrhythmic agent: Specific block of rapidly activating delayed rectifier K+ current by dofetilide. Circulation Research, 72(1), 75–83. 10.1161/01.res.72.1.75 [DOI] [PubMed] [Google Scholar]
- Larsen AP, & Olesen S-P (2010). Differential expression of hERG1 channel isoforms reproduces properties of native IKr and modulates cardiac action potential characteristics. PLoS ONE, 5(2), 1–9. 10.1371/journal.pone.0009021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lees-Miller JP, Guo J, Somers JR, Roach DE, Sheldon RS, Rancourt DE, & Duff HJ (2003). Selective knockout of mouse ERG1 B potassium channel eliminates I(Kr) in adult ventricular myocytes and elicits episodes of abrupt sinus bradycardia. Mol Cell Biol, 23(6), 1856–1862. 10.1128/mcb.23.6.1856-1862.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lees-Miller, James P, Kondo C, Wang L, & Duff HJ (1997). Electrophysiological characterization of an alternatively processed ERG K+ channel in mouse and human hearts. Circulation Research, 81(5), 719–726. 10.1161/01.RES.81.5.719 [DOI] [PubMed] [Google Scholar]
- Li Z, Ridder BJ, Han X, Wu WW, Sheng J, Tran PN, Wu M, Randolph A, Johnstone RH, Mirams GR, Kuryshev Y, Kramer J, Wu C, Crumb WJ, & Strauss DG (2019). Assessment of an In Silico Mechanistic Model for Proarrhythmia Risk Prediction Under the CiPA Initiative. Clinical Pharmacology and Therapeutics, 105(2), 466–475. 10.1002/cpt.1184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Jones DK, de Lange WJ, & Robertson GA (2016). Cotranslational association of mRNA encoding subunits of heteromeric ion channels. Proc Natl Acad Sci U S A, 113(17), 4859–4864. 10.1073/pnas.1521577113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- London B, Trudeau MC, Newton KP, Beyer AK, Copeland NG, Gilbert DJ, Jenkins NA, Satler CA, & Robertson GA (1997). Two isoforms of the mouse ether-a-go-go-related gene coassemble to form channels with properties similar to the rapidly activating component of the cardiac delayed rectifier K+ current. In Circulation Research (Vol. 81, Issue 5, pp. 870–878). 10.1161/01.RES.81.5.870 [DOI] [PubMed] [Google Scholar]
- McNally BA, Pendon ZD, & Trudeau MC (2017). hERG1a and hERG1b potassium channel subunits directly interact and preferentially form heteromeric channels. Journal of Biological Chemistry, 292(52), 21548–21557. 10.1074/jbc.M117.816488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPate MJ, Zhang H, Cordeiro JM, Dempsey CE, Witchel HJ, & Hancox JC (2009). hERG1a/1b heteromeric currents exhibit amplified attenuation of inactivation in variant 1 short QT syndrome. Biochemical and Biophysical Research Communications, 386(1), 111–117. 10.1016/j.bbrc.2009.05.134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta A, Sequiera GL, Ramachandra CJA, Sudibyo Y, Chung Y, Sheng J, Wong KY, Tan TH, Wong P, Liew R, & Shim W (2014). Re-trafficking of hERG reverses long QT syndrome 2 phenotype in human iPS-derived cardiomyocytes. Cardiovascular Research, 102(3), 497–506. 10.1093/cvr/cvu060 [DOI] [PubMed] [Google Scholar]
- Mesquita FCP, Arantes PC, Kasai-Brunswick TH, Araujo DS, Gubert F, Monnerat G, Silva dos Santos D, Neiman G, Leitão IC, Barbosa RAQ, Coutinho JL, Vaz IM, dos Santos MN, Borgonovo T, Cruz FES, Miriuka S, Medei EH, Campos de Carvalho AC, & Carvalho AB (2019). R534C mutation in hERG causes a trafficking defect in iPSC-derived cardiomyocytes from patients with type 2 long QT syndrome. Scientific Reports, 9(1), 1–9. 10.1038/s41598-019-55837-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orvos P, Kohajda Z, Szlovák J, Gazdag P, Árpádffy-Lovas T, Tóth D, Geramipour A, Tálosi L, Jost N, Varró A, & Virág L (2019). Evaluation of Possible Proarrhythmic Potency: Comparison of the Effect of Dofetilide, Cisapride, Sotalol, Terfenadine, and Verapamil on hERG and Native i Kr Currents and on Cardiac Action Potential. Toxicological Sciences, 168(2), 365–380. 10.1093/toxsci/kfy299 [DOI] [PubMed] [Google Scholar]
- Pacher P, Magyar J, Szigligeti P, Banyasz T, Pankucsi C, Korom Z, Ungvari Z, Kecskemeti V, Nanasi PP, Bányász T, Pankucsi C, Korom Z, Ungvári Z, Kecskeméti V, & Nánási PP (2000). Electrophysiological effects of fluoxetine in mammalian cardiac tissues. Naunyn Schmiedebergs Arch Pharmacol, 361(1), 67–73. 10.1007/s002109900154 [DOI] [PubMed] [Google Scholar]
- Perissinotti LL, De Biase PM, Guo J, Yang PC, Lee MC, Clancy CE, Duff HJ, & Noskov SY (2018). Determinants of isoform-specific gating kinetics of hERG1 channel: Combined experimental and simulation study. Frontiers in Physiology, 9(APR), 1–20. 10.3389/fphys.2018.00207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phartiyal P, Jones EMC, & Robertson GA (2007). Heteromeric assembly of human ether-à-go-go-related gene (hERG) 1a/1b channels occurs cotranslationally via N-terminal interactions. Journal of Biological Chemistry, 282(13), 9874–9882. 10.1074/jbc.M610875200 [DOI] [PubMed] [Google Scholar]
- Puckerin A, Aromolaran KA, Chang DD, Zukin RS, Colecraft HM, Boutjdir M, & Aromolaran AS (2016). HERG 1a LQT2 C-terminus truncation mutants display hERG 1b-dependent dominant negative mechanisms. Heart Rhythm, 13(5), 1121–1130. 10.1016/j.hrthm.2016.01.012 [DOI] [PubMed] [Google Scholar]
- Qiu XS, Chauveau S, Anyukhovsky EP, Rahim T, Jiang YP, Harleton E, Feinmark SJ, Lin RZ, Coronel R, Janse MJ, Opthof T, Rosen TS, Cohen IS, & Rosen MR (2016). Increased late sodium current contributes to the electrophysiological effects of chronic, but not acute, dofetilide administration. Circulation: Arrhythmia and Electrophysiology, 9(4), 1–10. 10.1161/CIRCEP.115.003655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudy Y, Hara TO, O’Hara T, Virág L, Varró A, & Rudy Y (2011). Simulation of the undiseased human cardiac ventricular action potential: Model formulation and experimental validation. PLoS Computational Biology, 7(5), 1–29. 10.1371/journal.pcbi.1002061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sale H, Wang J, O’Hara TJ, Tester DJ, Phartiyal P, He JQ, Rudy Y, Ackerman MJ, & Robertson GA (2008). Physiological Properties of hERG 1a/1b Heteromeric Currents and a hERG 1b-Specific Mutation Associated With Long-QT Syndrome. Circulation Research, 103(7), e81–e95. doi: 10.1161/CIRCRESAHA.108.185249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanguinetti MC, & Jurkiewicz NK (1990). Two components of cardiac delayed rectifier K+ current: Differential sensitivity to block by class III antiarrhythmic agents. Journal of General Physiology, 96(1), 195–215. 10.1085/jgp.96.1.195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenberg JI, Perry MD, Perrin MJ, Mann SA, Ke Y, & Hill AP (2012). hERG K+ Channels: Structure, Function, and Clinical significance. Physiological Reviews, 92(3), 1393–1478. 10.1152/physrev.00036.2011 [DOI] [PubMed] [Google Scholar]
- Wallis R, Benson C, Darpo B, Gintant G, Kanda Y, Prasad K, Strauss DG, & Valentin JP (2018). CiPA challenges and opportunities from a non-clinical, clinical and regulatory perspectives. An overview of the safety pharmacology scientific discussion. Journal of Pharmacological and Toxicological Methods, 93(June), 15–25. 10.1016/j.vascn.2018.06.005 [DOI] [PubMed] [Google Scholar]
- Weerapura M, Nattel S, Chartier D, Caballero R, & Hébert TE (2002). A comparison of currents carried by HERG, with and without coexpression of MiRP1, and the native rapid delayed rectifier current. Is MiRP1 the missing link? Journal of Physiology, 540(1), 15–27. 10.1111/jphysiol.2001.013296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams G, & Mirams GR (2015). A web portal for in-silico action potential predictions. Journal of Pharmacological and Toxicological Methods, 75, 10–16. 10.1016/j.vascn.2015.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson SL, Dempsey CE, Hancox JC, & Marrion NV (2019). Identification of a proton sensor that regulates conductance and open time of single hERG channels. Scientific Reports, 9(1), 19825. 10.1038/s41598-019-56081-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witchel HJ, & Hancox JC (2000). Familial and acquired long QT syndrome and the cardiac rapid delayed rectifier potassium current. Clinical and Experimental Pharmacology and Physiology, 27(10), 753–766. 10.1046/j.1440-1681.2000.03337.x [DOI] [PubMed] [Google Scholar]
- Zhou Z, Gong Q, Ye B, Fan Z, Makielski JC, Robertson GA, & January CT (1998). Properties of HERG channels stably expressed in HEK 293 cells studied at physiological temperature. Biophys J, 74(1), 230–241. 10.1016/S0006-3495(98)77782-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.