

Abstract
INTRODUCTION:
The tightly controlled process of gene expression requires messenger RNAs (mRNAs), which represent DNA-derived blueprints for polypeptides, to be translated by the protein-producing machinery of the cell, the ribosomes. Therefore, protein levels depend largely on cellular mRNA levels, and the control of mRNA decay is one of the most critical processes for setting the overall level of gene expression. Half-lives of mRNAs vary greatly between different transcripts, and regulation of the mRNA decay rate is intimately connected to the elongation phase of mRNA translation. To that end, codon optimality has been established as a key parameter for determining mRNA half-life in multiple eukaryotic organisms. It has also been established that the timely decay of short-lived mRNAs enriched with nonoptimal codons requires the Ccr4-Not complex. Ccr4-Not is an essential protein complex, with its best understood role in mRNA degradation, where it serves as the major cytoplasmic 3′-poly(A)-tail deadenylase that initiates decay of most mRNAs. By deadenylation and subsequent activation of the mRNA decapping machinery, the Ccr4-Not complex renders mRNAs accessible to the major degrading exonucleases, such as Xrn1 on the 5′ end and the exosome on the 3′ end. The molecular mechanism underlying codon optimality monitoring and coordination with mRNA decay by the Ccr4-Not complex has remained elusive.
RATIONALE:
Because nonoptimal codons affect decoding kinetics of the ribosome and mRNA degradation occurs largely cotranslationally, it is highly plausible that codon optimality is directly monitored on the ribosome. In addition, a direct physical link between the participating Ccr4-Not complex and the ribosome has been suggested previously, and the Not4 subunit of the complex, an E3 ligase, ubiquitinates the eS7 protein of the 40S ribosomal subunit in yeast. Therefore, we set out to gain insights into the connection between the Ccr4-Not complex and the translation machinery in the context of mRNA homeostasis by combining cryo–electron microscopy (cryo-EM), ribosome profiling, and biochemical analysis.
RESULTS:
We used affinity-purified native Ccr4-Not–ribosome complexes from Saccharomyces cerevisiae for analysis by cryo-EM and found that recruitment of Ccr4-Not to the ribosome occurs via the Not5 subunit. The N terminus of Not5—in particular, a three α-helix bundle—interacted specifically with the ribosomal E-site, and deletion of the Not5 N-terminus resulted in the loss of stable ribosome association of the Ccr4-Not complex. However, ubiquitination of the small ribosomal subunit protein eS7 through the Not4 subunit still occurred. The Not5 interaction involved the ribosomal protein eS25 of the small subunit, in addition to transfer RNA (tRNA) and ribosomal RNAs (rRNAs). We found that Ccr4-Not interacts with both initiating and elongating ribosomes. In either case, Not5 engaged the E-site only when the ribosome adopted a distinct conformation lacking accommodated tRNA in the A-site, indicative of impaired decoding kinetics. Ribosome profiling revealed that low-optimality codons were enriched in the A-site in the Ccr4-Not–bound elongating ribosomes. This observation explained the low A-site tRNA occupancy observed with cryo-EM and suggested a link to codon optimality monitoring. Consistently, using mRNA stability assays, we found that loss of Not5 resulted in the inability of the mRNA degradation machinery to sense codon optimality. The observed dysregulation of mRNA half-life was detected upon Not5 deletion, Not5 N-terminal deletion, eS25 deletion, and loss of eS7 ubiquitination by Not4, which apparently serves as an upstream prerequisite for further Ccr4-Not activity on the ribosome. In addition, mRNA decapping was found to be impaired in these mutants, which confirmed that, in this pathway, Ccr4-Not triggers decapping downstream of optimality monitoring.
CONCLUSION:
Our analysis elucidates a direct physical link between the mRNA decay–mediating Ccr4-Not complex and the ribosome. Dependent on preceding ubiquitination of eS7 by the Not4 subunit, the Ccr4-Not complex binds (via the Not5 subunit) specifically to the ribosomal E-site when the A-site lacks tRNA because of slow decoding kinetics. This state of the ribosome occurs in the presence of nonoptimal codons in the A-site, which explains the shorter half-lives of transcripts enriched in nonoptimal codons. Thus, our findings provide mechanistic insights into the coordination of translation efficiency with mRNA stability through the Ccr4-Not complex.
Control of messenger RNA (mRNA) decay rate is intimately connected to translation elongation, but the spatial coordination of these events is poorly understood. The Ccr4-Not complex initiates mRNA decay through deadenylation and activation of decapping. We used a combination of cryo–electron microscopy, ribosome profiling, and mRNA stability assays to examine the recruitment of Ccr4-Not to the ribosome via specific interaction of the Not5 subunit with the ribosomal E-site in Saccharomyces cerevisiae. This interaction occurred when the ribosome lacked accommodated A-site transfer RNA, indicative of low codon optimality. Loss of the interaction resulted in the inability of the mRNA degradation machinery to sense codon optimality. Our findings elucidate a physical link between the Ccr4-Not complex and the ribosome and provide mechanistic insight into the coupling of decoding efficiency with mRNA stability.
Graphical Abstract
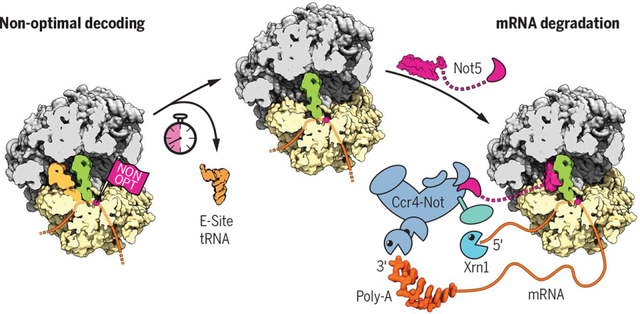
Ccr4-Not couples translation efficiency to mRNA degradation. When ribosomes encounter nonoptimal codons, low decoding efficiency leads to an increased likelihood of dissociation of the E-site tRNA before the cognate tRNA is accommodated in the A-site. As a result, the ribosomal E-site adopts a specific conformation, which is recognized by the Ccr4-Not complex through the N-terminus of its Not5 subunit, eventually triggering mRNA degradation by Xrn1.
The Ccr4-Not (carbon catabolite repressor 4–negative on TATA) complex is an essential and conserved protein complex comprising at least six core subunits arranged in a modular architecture. Although Ccr4-Not has roles in many aspects of regulating gene expression (e.g., chromatin remodeling, transcription, mRNA export, and RNA interference), its most defined and best understood role is in mRNA degradation, serving as the major cytoplasmic deadenylase other than Pan2-Pan3 (1, 2). Two subunits of Ccr4-Not (Caf1 and Ccr4) are poly(A)-specific nucleases that initiate decay of most mRNAs. Their activity in shortening the 3′-poly(A) tail is typically followed by removal of the 7-methylguanylate cap at the 5′ end by the Dcp2-Dcp1 holoenzyme and then subsequent 5′-to-3′ degradation by Xrnl (3). For most transcripts, decapping is strictly dependent on prior deadenylation. However, little is known about the spatial organization that allows the coordination of deadenylation with decapping by Ccr4-Not.
The control of mRNA decay is one of the most critical processes for setting the overall level of gene expression. Half-lives of mRNAs vary greatly between transcripts, and codon optimality has been established as a major parameter for determining mRNA half-life in eukaryotes (4–6). The timely decay of short-lived mRNAs enriched with nonoptimal codons involves the Ccr4-Not complex (7) and the activator of decapping Dhhl (8), but the molecular principles underlying codon optimality reading have remained enigmatic. Because nonoptimal codons affect decoding kinetics and mRNA degradation occurs cotranslationally (9), it can be reasoned that codon optimality is directly monitored on the ribosome. A physical link of the Ccr4-Not complex to the ribosome has been suggested previously (10, 11). The Not4 subunit of the complex, an E3 ligase, ubiquitinates proteins of the 40S ribosomal subunit (12, 13), and the Not5 subunit appears to be important for mRNA translatability and polyribosome levels in yeast (14). In this study, we set out to gain mechanistic insights into the connection between Ccr4-Not and the translation machinery in the context of mRNA homeostasis.
The Not5 subunit anchors Ccr4-Not to the ribosome
To look for a physical link between the ribosome and Ccr4-Not, we copurified endogenous ribosome-bound Ccr4-Not complexes from Saccharomyces cerevisiae, using tagged Not4 as bait. We performed sucrose density gradient centrifugation and isolated a monosome 80S and a polysome fraction, both stabilized by the antibiotic tigecycline to prevent ribosomal runoff. Single-particle cryo–electron microscopy (cryo-EM) analysis (table Sl) of the monosome fraction resulted in a structure of an 80S ribosome with a vacant A-site; a transfer RNA (tRNA) in the P-site; and additional, mostly α-helical, density in the E-site (Fig. 1A). The overall resolution of 2.8 Å (fig. S1, A to C) allowed us to identify the extra density as the N-terminal domain (NTD) of Not5 (residues 2 to 113). Not5 is a highly conserved component of Ccr4-Not (CNOT3 in humans) with known roles in controlling mRNA stability (15, 16). We were able to build an atomic model of the previously structurally uncharacterized Not5-NTD (Fig. 1B and fig. S1D). The remaining part of Not5 and also the entire Ccr4-Not complex were apparently flexibly linked to the NTD and therefore not resolved, with the exception of a small helical bundle extending from the NTD near the 40S head, which we could visualize but not model in a chemically cross-linked cryo-EM sample (fig. S1, E and F). Nevertheless, the presence of the entire Ccr4-Not complex in the cryo-EM sample was confirmed by SDS–polyacrylamide gel electrophoresis (SDS-PAGE) and mass spectrometry (Fig. 1C). The NTD of Not5 was composed of a three–α-helix bundle and a short linker, followed by a β turn directly leading into a short fourth α helix (Fig. 1, D and E). The three-helix bundle established a stable hydrophobic core, and the entire NTD bound to the ribosome in the E-site by precisely spanning the distance between the 60S and the 40S ribosomal subunits. The domain directly interacted with the P-site tRNA, the 25S ribosomal RNA (rRNA), 18S rRNA, and the N-terminal tail of the ribosomal protein eS25, suggesting that binding requires a fully assembled 80S ribosome with tRNA in the P-site (Fig. 1, E to G). To rule out the possibility of an artifact induced by tigecycline, we repeated the structure determination in absence of antibiotics and confirmed the presence of the NTD in the same position and conformation (fig. S1, G to I). Moreover, the same overall architecture was also observed for the complexes after chemical cross-linking or when isolated from the polysome fraction (see below). To test whether the NTD is required for ribosome association of the Ccr4-Not complex in vivo, we generated a yeast strain carrying a Not5 construct lacking the NTD. Analysis of this mutant strain using sucrose density gradient centrifugation revealed that association of the Ccr4-Not complex with ribosomes was decreased, as evident from the signal for the Not5 subunit (Fig. 1H and fig. S2, A and B). Most of the mutant complex was observed in the top fraction of the gradient, with low signal remaining throughout the gradient. Thus, the NTD of Not5 makes a key contribution to the association of the Ccr4-Not complex with the ribosome. The remaining association with the translation machinery is likely to reflect a second binding mode of Ccr4-Not that could involve the interaction of the Not4 subunit with ribosomal proteins for ubiquitylation. Consistent with this idea, ubiquitination of eS7 still occurred in the Not5-ΔNTD strain (Fig. 1I and fig. S2, A and B).
Fig. 1. The N terminus of Not5 binds to the ribosomal E-site.

(A) Cryo-EM density map and (B) atomic model of the Not5–80S complex. (C) SDS-PAGE analysis and mass spectrometry of the cryo-EM sample (asterisk: TEV-protease). (D) Overview of Not5-NTD, interacting ribosomal features, and tRNAiMet (tCM: tRNA clamp-motif). Residues that establish the hydrophobic core of the three-helix bundle are shown as sticks. (E) Topology of the Not5-NTD. Key residues are highlighted (pink: hydrophobic core, green: interaction with tRNA, blue: interaction with eS25, beige: interaction with 18S rRNA, gray: interaction with 25S rRNA). Single-letter abbreviations for the amino acid residues are as follows: A, Ala; C, Cys; D, Asp; E, Glu; F, Phe; G, Gly; H, His; I, Ile; K, Lys; L, Leu; M, Met; N, Asn; P, Pro; Q, Gln; R, Arg; S, Ser; T, Thr; V, Val; W, Trp; and Y, Tyr. (F) Atomic model of the 40S subunit, P-site tRNA, and Not5-NTD, as seen from the ribosomal intersubunit space. Locations of head (h), beak (bk), platform (pf), shoulder (sh), left foot (lf), and right foot (rf) of the 40S subunit are indicated. (G) Atomic model of the 60S subunit, P-site tRNA, and Not52−113, as seen from the ribosomal intersubunit space. Locations of the central protuberance (cp) and other characteristic features are indicated. (H) Density gradient profiles of Not5-HA and Not5-ΔNTD-HA strains and corresponding Western blot. (I) Western blot of eS7-HA in the fractions of the sucrose density gradients from (H).
Not5 targets initiating and elongating ribosomes
We initially used the native Ccr4-Not:80S complexes from the monosome fraction of the sucrose gradient and expected to find a mixture of different mRNAs and tRNAs. To our surprise, the codon present within the P-site was well resolved and identified as AUG (Fig. 2A). Moreover, the tRNA could be unambiguously identified as the initiator tRNAiMet, not the elongator tRNAMet (Fig. 2B and fig. S2C). These data suggest an association of the Ccr4-Not complex with late-initiation complexes. To confirm this, we probed by Northern blotting for individual tRNAs in 80S fractions before and after Not4 pull-down assay and in the presence or absence of tigecycline or cycloheximide (CHX) (Fig. 2C). As expected, we observed a strong enrichment of tRNAiMet and not tRNAAla (as control) in the ribosomal samples upon pull-down assay independent of tigecycline, whereas copurifications in presence of CHX yielded significantly fewer ribosomes. A structural feature of tRNAiMet that is also shared by other tRNAs is a short D-loop (17) (fig. S2D), which in our structure directly interacted with the three-helix bundle of Not5 (Fig. 2, B and D). This may suggest some degree of specificity of Not5 conferred by proximity and probing of the tRNA D-loop. Finally, interaction with late-initiation complexes was also consistent with the results of selective ribosome profiling after coimmunoprecipitation using Not4 as bait (Not4-IP) or the ribosomal protein uL30 as control (Fig. 2E): We observed a broad association of Not4 over the entire open reading frame (ORF), yet with a twofold enrichment of ribosomes on the initiation codon compared with the control (Fig. 2F).
Fig. 2. Not5 interacts with initiating and elongating ribosomes.

(A) Cryo-EM density and atomic model of the codon-anticodon interaction. (B) Cryo-EM density and atomic model of tRNAiMet. The position of Not5-NTD is outlined in pink. AC, anticodon. (C) Northern blot against tRNAiMet (top) and tRNAAla (bottom). The same amount of ribosomes was loaded before (80S) and after (Pull) affinity purification. The samples were prepared without antibiotics (noAB), in the presence of tigecycline (TIG), or in the presence of cycloheximide (CHX). (D) D-loop recognition through Not5. (E and F) Selective ribosome profiling of Ccr4-Not associated ribosomes (Not4 IP) and total ribosomes (uL30 IP). (G and H) Cryo-EM densities of the Not5 binding site in the initiator and elongating ribosome (low-pass filtered to 5 Å). (I) Peptidyl transferase center (PTC) of the Not5–80S complex with ester bonded methionine (blue). (J) PTC of the elongating Not5-bound ribosome (nascent chain: blue).
We next asked whether the Ccr4-Not complex also binds to elongating ribosomes and determined cryo-EM structures from heavy fractions of a sucrose gradient (between three and six ribosomes). We also observed a population of ribosomes with Not5 bound in the E-site, tRNA present in the P-site, and a vacant A-site. This structure resembled our observations with Not5 bound to the late-initiation complex (Fig. 2, G and H). However, the local resolution of the tRNA was not high enough to identify the tRNA unambiguously, suggesting the presence of a mix of different tRNAs species. In contrast to the initiation structure, there was clear density for a nascent chain extending from the peptidyl transferase center into the ribosomal exit tunnel, confirming the elongating state of the ribosomes (Fig. 2, I and J). Thus, Not5 links the Ccr4-Not complex to both late-initiating and elongating ribosomes within the ORF.
Molecular interactions of the Not5-NTD
One interaction of the NTD with the ribosome is established through the P-site tRNA. It involves multiple hydrogen bonds between the backbone of the tRNA D-loop and helix α2 of Not5 (Fig. 3A and fig. S3A), whereas the backbone of the D-arm hydrogen bonds with helices α2 and α3 of Not5 (Fig. 3, B and C, and fig. S3B). Another interaction involves residues K103 to L110 of Not5, which we termed the tRNA clamp-motif (tCM) (Fig. 3D and fig. S3, C to E) because it locks the phosphate backbone of the tRNA anticodon stem loop. This interaction is further stabilized by the N-terminal tail of the ribosomal protein eS25 (Fig. 3, D and E). This tail of eS25 is usually flexible; however, in the presence of Not5 it is stabilized and can be observed extending from the globular part of eS25 at the head of the small ribosomal subunit to the ribosomal P-site (Figs. 1F and 3C). There, residues K25 and K29 of eS25 form hydrogen bonds with the carbonyl groups of F105 and A109 of Not5, respectively. Thereby, eS25 holds the tCM of Not5 in place, and together the tCM, eS25, and 18S rRNA form a groove that accommodates the tRNA backbone (Fig. 3E). W27 of eS25 pins the flexible N-terminal tail to this location through a stacking interaction with G1575 of the 18S rRNA between rRNA helices h29 and h42. G1575 also stabilizes the β turn of Not5 through a stacking interaction between the ribose and F105 of Not5 (Fig. 3D). Residues K25, W27, and K29 of eS25 belong to the highly conserved KKKWSK motif, and a human K33E mutation (corresponding to yeast K25E) has been found in thyroid carcinoma cells (18). The central residue F105 of Not5 is coordinated simultaneously by 18S rRNA through the side chain, by eS25 through the carbonyl oxygen, and by the tRNA through the backbone nitrogen. Furthermore, interactions of the Not5-NTD with the 18S rRNA involve rRNA helices h23 and h24 (Fig. 3F and fig. S3F). Interaction with the 25S rRNA involves helix α2 of Not5 and the extension of rRNA helix H69 (Fig. 3G and fig. S3G) as well as helix α1 of Not5 and rRNA helix H74 (Fig. 3H and fig. S3H). The human homolog of Not5, CNOT3, contains a highly conserved NTD (fig. S3I), suggesting a conserved function of CNOT3 as an anchor to the human ribosome. Multiple cancer mutations cluster in the NTD of CNOT3 (18), with one of the most frequent mutations being R57W/E (K58 in yeast) (19). K58 directly interacts with the phosphate backbone of 25S rRNA in our structure (Fig. 3G), and analogous interaction can be predicted for R57 of the human homolog. Mutation of K58 (or R57 in human) to tryptophan or glutamate would not allow hydrogen bond formation and would therefore destabilize the interaction. Thus, the Not5-NTD engages in a highly specific and complex binding mode in the ribosomal E-site involving 40S and 60S ribosomal rRNA as well as eS25 and a P-site tRNA with a small D-loop.
Fig. 3. Not5–80S complex interface.

(A) Interactions of the Not5-NTD with the D-loop of tRNAiMet. Hydrogen bonds and ion bridges are shown as black dashed lines. (B) Interactions of Not5 with the D-stem of tRNAiMet. (C) Overview of the interaction sites of the Not5-NTD and the initiator-80S ribosome. (D) Interactions between the tCM of Not5, tRNAiMet, eS25, and 18S rRNA. Hydrogen bonds and ion bridges are shown as black dashed lines. Hydrophobic interactions are shown as gray dotted lines. (E) Surface representation of the interface shown in (D) and atomic model of the anticodon stem loop of tRNAiMet. (F) Interactions of Not5-NTD with 18S rRNA. (G and H) Interactions of Not5 with 25S rRNA. (I) Schematic summary of the binding interface of the tCM of Not5 with the initiator 80S (solid arrows: polar interactions; dashed arrows: hydrophobic interactions).
A-site occupation determines E-site preference for either Not5 or eIF5A
We observed Not5 binding to ribosomes in the posttranslocation state, in which ribosomes usually carry a deacylated tRNA in the E-site and a peptidyl-tRNA in the P-site but no A-site tRNA (–/PP/EE). Not5 can apparently bind to this posttranslocation-state conformation as soon as also the E-site is vacant (–/PP/–) (Fig. 4A). In this state, the small ribosomal subunit is rotated toward the E-site and the L1-stalk cannot adopt the far inside conformation, which is necessary to accommodate eIF5A because of a potential clash with the 40S subunit (fig. S3J). By contrast, upon accommodation of the A-site tRNA (AA/PP/–) (fig. S3, K to N), the 40S ribosomal subunit undergoes a conformational change in the opposite direction, known as subunit rolling (20) (Fig. 4, B and C). Together, these movements result in characteristic remodeling of the E-site. In the presence of CHX, we observed enrichment of the (AA/PP/–) state and found eIF5A bound to the E-site, as previously described (21).
Fig. 4. eIF5A and Not5 compete for the E-site in an A-site–dependent manner.

(A) Density (top) and model (bottom) of the Not5-bound 40S subunit of the (–/PP/–) 80S ribosome (only 18S rRNA model shown). (B) Density (top) and model (bottom) of the eIF5A-bound 40S subunit of the (AA/PP/–) 80S ribosome (only 18S rRNA model shown). (C) Superposition of the densities (top) and the models (bottom) from (A) and (B). Subunit rolling is indicated. Structures in (A) to (C) were aligned with respect to the 60S subunits. (D) The tCM of Not5 is contacted by the anticodon stem of the P-site tRNA in the (–/PP/–) situation (also see Fig. 3D). (E) Conformational changes of the P-site tRNA upon A-site accommodation would cause clashes (red crosses). (F) Length distribution of mRNA fragments during selective ribosome profiling using Not4 and uL30 as baits. (G) Illustration of A-site–E-site coupling. Arrows represent quantified trajectories of Ca atoms (protein) and P atoms (RNA) during transition from (–/PP/–) to (AA/PP/–) conformations. Ribosomal A- and E-sites are schematically indicated with gray ellipsoids. (H) Schematic summary of subunit rolling and the competition between eIF5A and Not5.
Not5 and eIF5A binding to a vacant E-site appear to be mutually exclusive, not only because of a direct steric clash between the two factors, but also as a result of different conformational requirements for E-site binding. Comparison of the Not5-bound (–/PP/–) ribosome with the eIF5A-bound (AA/PP/–) ribosome shows that, upon A-site accommodation, the anticodon stem loop of the P-site tRNA has moved toward the E-site. This involves nucleotides 41 to 43, which are clamped by the tCM of Not5, and this subtle movement of the tRNA backbone (~1.6 Å) would cause clashes with the tCM upon A-site accommodation (Fig. 4, D and E). This explains how Not5 can probe the shape of the E-site and stably and specifically bind to the E-site in the absence of an accommodated A-site tRNA. In agreement with this observation, we found ribosomes simultaneously interacting with A-site tRNA and Not5-NTD only when in a nonrolled and non-accommodated conformation, resembling a tRNA sampling situation (fig. S4C). Also, we generally observed only small populations of Ccr4-Not:80S complexes in the presence of CHX, which stabilizes the unfavored, A-site tRNA-accommodated pretranslocation state.
Preferential association of Ccr4-Not to ribosomes with empty A-sites was further confirmed by ribosome profiling, which has previously been shown to yield shorter (~21-nucleotide oligomer) fragments from ribosomes with an empty A-site (22). Using Not4 as bait for selective profiling, we indeed observed enrichment of these short mRNA fragments (Fig. 4F). Despite slight variations of the exact fragment length distribution in different experiments, the effect of enrichment of short reads in Ccr4-Not–bound ribosomes was consistent (fig. S3O). The allosteric coupling between the A- and E-sites of the 80S ribosome was illustrated by quantified trajectories of ribosomal proteins (Cα atoms) and RNA (P atoms) that describe a transition between the Not5-bound posttranslocation state and the A-site tRNA accommodated pretranslocation state (Fig. 4G).
Thus, the NTD of Not5 can probe the ribosomal E-site in a highly selective way, resulting in the Ccr4-Not complex most efficiently interacting with ribosomes with a vacant A-site, as schematically summarized in Fig. 4H. Consequently, the Ccr4-Not complex and eIF5a may facilitate a differential readout of the translational state of the ribosome via E-site probing: The Ccr4-Not complex would be recruited with preference to ribosomes displaying low decoding efficiency and thereby would increase the likelihood of mRNA deadenylation, decapping, and degradation (23). By contrast, the competing eIF5A would preferentially interact with ribosomes that remain longer in the pretranslocation state because of slow peptidyl transferase kinetics, thereby allowing for stabilization of a productive peptidyl transferase center geometry and effectively counteracting Ccr4-Not recruitment.
Not5 senses codon optimality for mRNA decay dependent on Not4
Codon optimality is a major determinant of mRNA degradation, with the proportion of nonoptimal codons within the transcript determining its half-life (4). In essence, a nonoptimal codon is one whose functional tRNA concentration is low, resulting in prolonged vacancy of the ribosomal A-site. Because eviction of the E-site tRNA does not require A-site accommodation (24), prolonged vacancy of the A-site results in an increased probability of a ribosome adopting the (–/PP/–) state, which can be recognized by Not5. Thus, a ribosome with an empty E-site in the posttranslocation-state conformation could be specifically sensed by Not5 as a proxy for the presence of a nonoptimal codon in the A-site. To test this hypothesis, we performed selective ribosome profiling upon coimmunoprecipitation of Ccr4-Not:ribosome complexes, again using Not4 as bait. We aimed at reducing the background of ribosomes, which are copurified merely because of association with a Not4-bound ribosome within the same polysome. To that end, we included an incubation step during the Not4-IP, resulting in polysome dissociation [most likely through endogenous ribonucleases (RNases)] after adding tigecycline to prevent runoff (fig. S5, A to D). The data revealed a significant inverse correlation between enrichment of messages upon Not4-IP and their associated codon stabilization coefficient (CSC) or degree of codon optimality, respectively (Fig. 5A). An even stronger correlation was observed at the codon level: Enrichment of specific codons at the A-site position upon Not4-IP was inversely correlated with CSC and tRNA adaptation index (tAI) (Fig. 5B). Listing the A-site codons according to their enrichment through Not4-IP resulted in an almost perfect separation into optimal and nonoptimal codons (Fig. 5C and fig. S5E). Thus, the presence of Ccr4-Not on the ribosome correlates strongly with low codon optimality of the associated mRNAs and in particular with the presence of nonoptimal codons in the ribosomal A-site. This strongly supports our hypothesis that the Ccr4-Not complex is recruited by ribosomes during slowed decoding of nonoptimal codons in the A-site.
Fig. 5. Not5-NTD and eS7 ubiquitination by Not4 are involved in degradation of nonoptimal mRNAs.

(A) Monosome-enriched selective ribosome profiling of Ccr4-Not associated ribosomes and total ribosomes. Enrichment of messages through Not4-IP is inversely correlated with codon stabilization coefficient (CSC) and codon optimality of the messages. (B) Enrichment of specific codons present in the A-site through Not4-IP is inversely correlated with their CSC and the corresponding tAI. (C) Individual codons present in the A-site listed according to their enrichment through Not4-IP. (D) Half-life determination of optimal and nonoptimal PGK1pG reporter mRNAs through Northern blotting at various time points after transcriptional shutoff (T) in either wild-type or not5Δ strains. (E) HIS3 reporter mRNA half-life measurements in different cell lines (not5Δ, WT, not4Δ, eS7a/bΔ, and not5AeS7a/bΔ) in combination with expression of different constructs from plasmids (pNot5, pNot5-ΔN, peS7a, and peS7a-4KR) on the basis of Northern blotting (fig. S6; data are means ± SDs, N = 3 replicates). (F) Quantification of the association of Not5 with the translation machinery and stabilization of a nonoptimal PGK1pG reporter mRNA in relation to CHX concentration.
We next tested whether the Not5 subunit of the Ccr4-Not complex affects optimality-dependent mRNA degradation and evaluated the decay rates of reporters previously developed to monitor effects of codon optimality on mRNA half-life. Loss of Not5 indeed stabilized nonoptimal mRNAs and, to a lesser extent, also destabilized optimal mRNAs, whereas the semi-optimal mRNAs were stabilized (Fig. 5D and fig. S5F). Thus, loss of the entire Not5 largely disables a cell′s ability to degrade mRNAs according to codon usage. Because our structures suggest a central role for the NTD of Not5, we tested whether codon optimality-dependent mRNA degradation could be maintained in the absence of this domain. We performed mRNA half-life measurements in Not5 deletion cells that expressed nothing, full-length Not5, or Not5 lacking the NTD from plasmids. The N-terminally truncated Not5 failed to rescue the deletion phenotype of global dysregulation of mRNA decay, in contrast to full-length Not5 (Fig. 5E and fig. S6A). Moreover, deletion of eS25, the central ribosomal interaction partner of Not5, also stabilized nonoptimal mRNAs (fig. S5G). Thus, the interaction of the Not5-NTD with the ribosomal E-site is required for communicating codon optimality to the mRNA degradation machinery. Consistent results have also been reported for the mammalian system, in which deletion of the NTD of CNOT3 led to a stabilizing effect for several mRNAs (25).
In addition to the interaction of Not5-NTD with the E-site, the Ccr4-Not complex establishes a second interaction with the ribosome, which is mediated through the Not4 E3 ligase by ubiquitination of the small subunit protein eS7. As presented earlier (Fig. 1I), the activity of the Not5-NTD is not required for efficient eS7 ubiquitination. However, we wondered whether eS7 ubiquitination by Not4 affects the function of Not5 in sensing codon optimality. Deletion of Not4 strongly stabilized nonoptimal and semi-optimal reporters, similar to the deletion of the Not5-NTD (Fig. 5E and fig. S6B). This effect could be clearly ascribed to a loss of eS7 ubiquitination, because mutation of the four lysine residues targeted by Not4 (eS7–4KR) resulted in the same phenotype, with respect to codon optimality, as that produced by Not4 deletion. The phenotypes of neither the Not4 deletion nor the eS7a-4KR mutation were additive with the Not5-NTD deletion (Fig. 5E and fig. S6, B and C). Thus, eS7-ubiquitination by Not4 is part of the same pathway as Not5 but occurs upstream of E-site probing. Consistent with this finding, Not5 binding to ribosomes was dependent on prior eS7 ubiquitination, because deletion of Not4 or eS7a-4KR mutation substantially reduced Not5 association with (poly)-ribosomes (fig. S5H).
The codon optimality–dependent mRNA degradation pathway has previously been demonstrated to strictly depend on Dhh1 (8), which activates mRNA decapping downstream of Ccr4-Not action (7). We determined whether the Not5:E-site interaction or eS7 ubiquitination also affects the presence of Dhh1 on the translation machinery. Either deletion of the Not5-NTD or disabling of eS7 ubiquitination was indeed sufficient to prevent Dhh1 association with ribosomes (fig. S5, H and I) and also to cause a decapping defect (fig. S7). This observation confirmed the previously determined sequence of events during codon optimality–dependent mRNA decay (7).
Stabilization of mRNA can also be triggered by CHX treatment, which was ascribed to a steric protection of the mRNAs through accumulation of stalled ribosomes (26). According to our results, CHX should indeed have a stabilizing effect on mRNAs, but rather through a mechanism of stalling ribosomes with an altered E-site conformation that disfavors Not5 binding, effectively impairing codon optimality sensing. To test this, we employed increasing concentrations of CHX and analyzed polysome association of Not5, as well as the half-life of a nonoptimal mRNA reporter in vivo. In agreement with our hypothesis, treatment of cells with CHX resulted in a dose-dependent depletion of Ccr4-Not from ribosomes (fig. S5J) and in a dose-dependent stabilization of mRNA (Fig. 5F). Together, these in vivo data support a model in which E-site probing by the Not5-NTD represents the key event for sensing codon optimality in the A-site and for coupling it to mRNA degradation. On the basis of our results, we propose a general model for codon optimality–dependent mRNA degradation (Fig. 6).
Fig. 6. Model of cotranslational mRNA degradation.

Upon subunit association, eS7 is ubiquitinated by Not4. Ccr4-Not can then be anchored to the ubiquitinated (–/PP/–) ribosome through the Not5-NTD. In the case of successful decoding, the ribosome adopts the rolled (AA/PP/–) conformation, disfavoring Ccr4-Not association. Efficient peptidyl transfer leads to translocation, resulting in a classical posttranslocation state (–/PP/EE). If peptidyl transfer is slow, eIF5A binds to the E-site, promoting PTC activity and rescuing translation. When decoding efficiency is low because of a nonoptimal codon in the A-site, there is an increased probability that the E-site tRNA dissociates before the correct tRNA is accommodated in the A-site, resulting in a (–/PP/–) posttranslocation-state ribosome. This complex is recognized by the Not5-NTD, leading to deadenylation and Dhh1-dependent mRNA degradation. It is unclear whether the mRNA also enters the degradation pathway in the case of the Ccr4-Not–bound initiator ribosome.
Discussion
We discovered a physical link between the Ccr4-Not complex and the ribosome mediated by the Not5 subunit, which specifically binds to ribosomes in the posttranslocation state with simultaneous vacancy of E- and A-sites. Along with the observed strong correlation of Ccr4-Not association to the ribosome with the presence of nonoptimal codons in the A-site, this finding demonstrates that the Ccr4-Not complex is specifically recruited to the translation machinery, when decoding is slowed. In vivo analysis of mRNA half-life showed that recruitment of Ccr4-Not to the ribosome through Not5 is crucial for the discriminative degradation of mRNAs with different codon optimality. Moreover, the interaction of Ccr4-Not and the ribosome is necessary for the presence of Dhh1 on the translation machinery. Decapping of nonoptimal messages was previously reported to depend strictly on Dhh1 and to occur downstream of deadenylation (7). We also discovered that ubiquitination of eS7 by Not4 is an upstream prerequisite for Not5 binding and the downstream events.
In addition to elongating ribosomes, we also found that a large fraction of the Ccr4-Not complex bound to late-initiation 80S complexes. From a structural perspective, these complexes are ideal targets for Ccr4-Not because they are fully assembled 80S ribosomes in the posttranslocation state with tRNA in the P-site and empty A- and E-sites. With respect to codon optimality sensing, however, the location seems puzzling. One possibility is that, independent of codon optimality, mRNA decay could also be triggered in this situation, owing to a flawed transition from translation initiation to elongation. This is supported by our finding that these initiation complexes were mainly present in the 80S monosome and not in the polysome fractions. Another possibility is that we were observing a later stage in the mRNA degradation pathway: Considering the closed-loop mRNA model, one could speculate that Ccr4-Not is handed over from the 3′ end to the 5′ end of the mRNA after deadenylation. Such a handover could be beneficial, because anchoring the complex to the E-site of an 80S ribosome located on the start codon would be an ideal way to assemble and activate the decapping machinery within spatial proximity to the 5′ cap while probably inhibiting further initiation. This idea is further supported by recent RNA-binding studies of deadenylation and decapping factors, showing a distributed allocation on the 5′ and 3′ ends of the transcripts (27). Nonetheless, Ccr4-Not function during initiation is likely to extend beyond mRNA decay (28).
Fundamentally, our study demonstrates that the ribosomal conformation, and specifically the E-site, is used as an allosteric indicator of both slow peptidyl transfer kinetics and slow decoding kinetics (movie S1). In either case, translation elongation slows, resulting in transient stabilization of a distinct ribosomal conformation with an accessible E-site. In the case of slow peptidyl transfer, this can be recognized by eIF5A, which promotes peptidyl transfer activity and hence rescues translation. In the case of slow decoding kinetics, this can be recognized by Ccr4-Not, thereby leading to an increased probability of mRNA degradation. This may represent a universal mechanism that can explain how mRNA decay is coupled to translation efficiency in eukaryotes.
Materials and methods
Yeast strains and genetic methods
Gene disruption and C-terminal tagging were performed by established recombination techniques as previously described (29, 30). The S. cerevisiae strains used in this study are listed in table S2.
Plasmids
All recombinant DNA techniques were performed according to standard procedures using Escherichia coli DH5a for cloning and plasmid propagation. All cloned DNA fragments generated by polymerase chain reaction (PCR) amplification were verified by sequencing. Plasmids used in this study are listed in table S3.
Native complex purification
A S. cerevisiae strain with genomically Flag-TEV-ProteinA–tagged Not4 (Not4-FTPA) was cultured in 10 l YPD medium supplemented with 5 μg/ml ampicillin and 10 μg/ml tetracycline and harvested by centrifugation at OD600 of 0.9. The pellet was washed in water and lysis buffer [20 mM HEPES pH 7.4, 100 mM KOAc, 10 mM Mg(OAc)2, 1 mM DTT, 0.5 mM PMSF, 100 μg/ml tigecycline, protease inhibitor cocktail tablet (Roche)] and frozen in liquid nitrogen. Lysis was performed by grinding the frozen cell pellet in a Freezer Mill (6970 EFM). The ground powder was thawed in 15 ml of lysis buffer, and the lysate was cleared by centrifugation at 12,000g for 15 min at 4°C. Cleared lysate was distributed and layered on top of six sucrose density gradients [10 to 50% sucrose in 20 mM HEPES pH 7.4, 100 mM KOAc, 10 mM Mg(OAc)2, 1 mM DTT, 10 μg/ml tigecycline in 25-mm–by–89-mm polyallomer tubes, SW32, Beckman Coulter] and centrifuged at 125,755g for 3 hours at 4°C. The gradients were subsequently fractionated from top to bottom using a Gradient Master (BioComp). The fractions corresponding to the 80S peak (monosome sample) or corresponding to polysomes in the range of 2 to 5 ribosomes (polysome sample) were pooled and treated equally for the remaining purification. The pooled samples were incubated with 100 μl of pre-equilibrated magnetic IgG-coupled Dynabeads M-270 Epoxy (Life Technologies) for 1 hour at 4°C. Beads were washed with 3 × 1 ml wash buffer [20 mM HEPES pH 7.4, 100 mM KOAc, 10 mM Mg(OAc)2, 1 mM DTT, 10 μg/ml tigecycline]. The Ccr4-Not:ribosome complexes were eluted from IgG beads by incubation with 70 U AcTEV protease (Thermo Fisher) in 50 μl of elution buffer [20 mM HEPES pH 7.4, 100 mM KOAc, 10 mM Mg(OAc)2, 1 mM DTT, 10 μg/ml tigecycline] for 1.5 hours at 4°C. For the cross-linked sample, chemical cross-linking was performed using 0.5 mM BS3 (Thermo Fisher) for 30 min on ice before the reaction was quenched with 20 mM Tris. All samples were kept on ice until cryo-EM grid preparation or were frozen in liquid nitrogen and stored at −80°C. For Northern blotting in the presence of CHX, the purification was performed following the same protocol, but instead of tigecycline, 100 μg/ml CHX were used in the lysis buffer and 10 μg/ml CHX were used during sucrose density centrifugation and affinity purification.
The sample leading to the eIF5A-bound (AA/PP/–) ribosome was prepared by culturing the Not4-FTPA strain in 10 l YPD medium supplemented with 5 μg/ml ampicillin and 10 μg/ml tetracycline and harvesting by centrifugation at OD600 of 0.9. The pellet was washed in water and lysis buffer [20 mM HEPES pH 7.4, 100 mM KOAc, 10 mM Mg (OAc)2, 1 mM DTT, 0.5 mM PMSF, 100 μg/ml CHX, protease inhibitor cocktail tablet (Roche)] and frozen in liquid nitrogen. Lysis was performed by grinding the frozen cell pellet in a Freezer Mill (6970 EFM). The ground powder was thawed in 15 ml of lysis buffer, and the lysate was cleared by centrifugation at 12,000g for 15 min at 4°C. Cleared lysate was incubated with 100 μl of pre-equilibrated magnetic IgG-coupled Dynabeads M-270 Epoxy (Life Technologies) for 1 hour at 4°C. Beads were washed with 3 × 2 ml wash buffer [20 mM HEPES pH 7.4, 100 mM KOAc, 10 mM Mg(OAc)2, 1 mM DTT, 10 μg/ml CHX]. Elution was performed by incubation with 200 U AcTEV protease (Thermo Fisher) in 300 μl of elution buffer [20 mM HEPES pH 7.4, 100 mM KOAc, 10 mM Mg(OAc)2, 1 mM DTT] for 1.5 hours at 4°C. The eluate was layered on top of a sucrose density gradient [10 to 50% sucrose in 20 mM HEPES pH 7.4, 100 mM KOAc, 10 mM Mg(OAc)2, 1 mM DTT, 10 μg/ml CHX in 14-mm–by–95-mm polyallomer tubes, SW40, Beckman Coulter] and centrifuged at 192,072g for 2.5 hours at 4°C. The gradient was subsequently fractionated from top to bottom using a Gradient Master (BioComp). The 80S fraction was pelleted for 1 hour at 436,000g at 4°C (TLA-100, Beckman Coulter); resuspended in 20 mM HEPES pH 7.4, 100 mM KOAc, 10 mM Mg(OAc)2, 1 mM DTT, and 10 μg/ml CHX; and kept on ice until cryo-EM grid preparation.
Electron microscopy and image processing
For all samples, 0.05% β-octylglucoside was added shortly before grid preparation. The final concentration of the ribosomal samples was 4 to 5 OD260 units per ml. Before sample application, grids were glow-discharged for 20 s at 2.2 × 10−1 torr using a PDC-32G plasma cleaner (Harrick). Cryo-EM grids were prepared by application of 3.5 μl of the sample containing purified Ccr4-Not:ribosome complexes onto R3/3 copper grids with 3-nm continuous carbon support (Quantifoil) and vitrification in liquid ethane using a Vitrobot Mark IV (Thermo Fisher) (45 s of wait time, 2.5 s of blotting time, blot force 0, 95% humidity, 4°C, Whatman 595 blotting paper). Data collection was performed on a Titan Krios TEM (Thermo Fisher) equipped with a Falcon II direct electron detector. Data were collected at 300 kV with a total dose of 28 e−/A2 fractionated over 10 frames with a pixel size of 1.084 Å per pixel and a target defocus range of −1.3 to −2.8 mm using software EPU (Thermo Fisher). The raw movie frames were aligned using MotionCor2 (31) and contrast transfer function (CTF) estimation was performed using Gctf (32). See table S1 for cryo-EM data, refinement statistics, and validation statistics.
For the tigecycline monosome dataset, 9945 micrographs were used for automated particle picking with Gautomatch, resulting in 557,059 initial particles, of which 359,890 were selected for further processing upon 2D classification in RELION-2.1 (33). After an initial round of 3D refinement, 3D classification with a mask on the ribosomal intersubunit space was performed. The dataset featured two major classes: a ribosome lacking tRNAs but containing eIF5A (~31% of the data) and a ribosome containing P-site tRNA and additional density in the E-site, which was later identified as the NTD of Not5 (~48% of the data). The 176,111 particles belonging to the Not5-containing class were refined to a final resolution of 2.8 Å using 3D refinement, CTF refinement, and postprocessing in RELION-3.0 (34). The final volume was filtered according to local resolution using RELION-3.0. A classification scheme is provided in fig. S4A.
For the polysome dataset, 9611 micrographs were used for automated particle picking with Gautomatch, resulting in 828,516 initial particles, of which 435,002 were selected for further processing upon 2D classification in RELION-3.0. After an initial round of 3D refinement, 3D classification was performed with a mask on the ribosomal intersubunit space, yielding three major classes: a ribosome lacking tRNAs but containing eIF5A (~31% of the data), an AP/EE hybrid (~39% of the data), and a ribosome containing P-site tRNA and Not5 (~21% of the data). The Not5-containing class was further classified using a mask on Not5 and, finally, without a mask to further enrich particles containing Not5 and to remove bad particles. The final class containing P-site tRNA, Not5, and density for a nascent chain was refined to a resolution of 3.2 Å using 3D refinement, CTF refinement, and postprocessing in RELION-3.0. The final volume was filtered according to local resolution using RELION-3.0. A classification scheme is provided in fig. S4B.
For the CHX monosome dataset, 12,697 micrographs were used for automated particle picking, with Gautomatch resulting in 1,378,230 initial particles, of which 1,194,480 were selected for further processing upon 2D classification in RELION-3.0. After an initial round of 3D refinement and 3D classification, bad particles and empty ribosomes were discarded, resulting in 274,817 particles (100%) featuring A- and/or P-site tRNA, which were used for extensive 3D classification. This resulted in a class of (AA/PP/–) ribosomes containing eIF5A (~19%), a class of (–/PP/–) ribosomes containing Not5 (~18%), a class of ribosomes containing P-site tRNA and eRF1 in the A-site (~19%), a class of (AA/PP/–) ribosomes in a nonrolled state containing Not5 (~11%), a class of (AA/PP/–) ribosomes (~3%), a class of (–/PP/–) ribosomes (~16%), and a class of empty ribosomes bound to a hibernation factor (~14%). The 50,989 particles belonging to the eIF5A containing (AA/PP/–) class were refined to a final resolution of 3.1 Å using 3D refinement, CTF refinement, and postprocessing in RELION-3.0. The final volume was filtered according to local resolution using RELION-3.0. A classification scheme is provided in fig. S4C.
For the cross-linked monosome dataset, 10,654 micrographs were used for automated particle picking with Gautomatch, resulting in 589,191 initial particles, of which 337,215 were selected for further processing upon 2D classification in RELION-3.0. After an initial round of 3D refinement, 3D classification was performed using a mask comprising the ribosomal P- and E-sites, yielding a major class containing P-site tRNA and Not5 (~37% of the data). The class was further subclassified without a mask to remove low-quality particles. The final volume was refined to 2.9 Å using 3D refinement, CTF refinement, and postprocessing in RELION-3.0. A classification scheme is provided in fig. S4D.
Model building and refinement
A full model was built for the Not5-bound (–/PP/–) ribosome. The model was based on the crystal structure of the yeast 80S ribosome (PDB ID 4V88). Ribosomal proteins and RNA were modeled and refined using COOT (35) and PHENIX (36). A second copy of the ribosomal protein eL41 was identified adjacent to helix 54 and was then modeled and refined. Initiator-tRNA was modeled on the basis of the crystal structure of yeast tRNAiMet (PDB ID 1YFG) and subsequently refined. For identification of the additional density in the ribosomal E-site, we placed three helices of poly-Ala into the helical density and assigned individual, highly resolved side chains close to the D-loop of the tRNA as one specific amino acid (e.g., W), as belonging to a group of possible amino acids (e.g., [FYH]), or as any amino acid (X). The resulting pattern [VIL]-[NQ]-X-[FYH]-R-X(2)-[VIL]-[KRQ]-X-W was compared to the UniProt databank (restricted to S. cerevisiae) using the motif search function on prosite.expasy.org, yielding two hits: residues 56 to 66 of the Not5 subunit of Ccr4-Not and residues 834 to 844 of the SEH-associated protein 4. The SEH-associated protein could be excluded because the pattern had to be located within the first 100 residues of the unknown protein (N terminus clearly identifiable). Because the N termini of Not3 and Not5 are highly similar, we compared the density of distinctive residues such as I27 (Not5) versus Y27 (Not3), H59 (Not5) versus L58 (Not3), or L67 (Not5) versus Q66 (Not3). The density could be unambiguously assigned to Not5. Not52−113 was de novo modeled and refined.
The model of the eIF5A-bound (AA/PP/–) ribosome was derived from the Not5-bound ribosome. The model of eIF5A and the A-site tRNA were derived from a previously solved cryo-EM structure (PDB ID 5GAK). The models were refined to fit the (AA/PP/–) density map using COOT and PHENIX.
Cryo-EM densities and molecular models were visualized using ChimeraX (37).
Probe generation for tRNA Northern blotting
Specific digoxigenin (DIG)–labeled antisense probes for tRNA Northern blotting were generated by amplifying an unrelated DNA sequence of 230 nucleotides (nt) from a pcDNA5/FRT/TO-derived plasmid by PCR in the presence of 1 mM DIG-dUTP. The reverse primer used in the PCR reaction had a 3′ overhang of 30 nt, which were the reverse complement to the initial 30 bases at the 5′ end of the respective tRNAs.
Hybridizing sequence for tRNAiMet: 5′-CCCTGCGCGCTTCCACTGCGCCACGGCGCT-3′
Hybridizing sequence for tRNAAla: 5′-GGAGCGCGCTACCGACTACGCCACACGCCC-3′
tRNA Northern blotting
The samples for tRNA Northern blotting were prepared as described above. In essence, cells were lyzed in the presence of no antibiotic, 100 μg/ml tigecycline, or 100 μg/ml CHX. For sucrose density gradient centrifugation, the concentrations of antibiotics were reduced to 10 μg/ml. After fractionation, samples of the total 80S fractions of all preparations were taken, and the remaining 80S fractions were subjected to affinity purification as described above. Upon elution of the complexes through tag cleavage using AcTEV protease (Thermo Fisher), the ribosome concentrations were measured according to A254.
The Northern blots were performed according to Roche DIG Northern Starter Kit protocol. Equal amounts of ribosomes (~160 pmol) of each sample and 100 ng of yeast tRNA mix as control were separated on NOVEX TBE-Urea Gels 10% (Invitrogen), transferred to Nylon Hybond N+ membranes (Amersham), and probed against tRNAs with DIG-labeled antisense probes. Chemoluminescence was detected on an Amersham 600 imager (GE Healthcare).
Preparation of ribosome protected mRNA fragments for selective ribosome profiling (Not4- and L7-lped-based approach)
The yeast strains with genomically tagged Not4 (Not4-FTPA) or uL30 (uL30-TAP) were cultured in YPD medium and harvested by centrifugation at OD600 of 0.5 to 0.6. The harvested cell pellet was frozen in liquid nitrogen and then ground in liquid nitrogen using a mortar. The cell powder was resuspended with the no-drag lysis buffer (50 mM Tris pH7.5, 100 mM NaCl, 10 mM MgCl2, 0.01% NP-40, 1 mM DTT) to prepare the whole-cell lysate. The whole cell lysate was centrifuged at 39,000g for 30 min at 4°C, and the supernatant fraction was used for the purification step. The supernatant fraction was incubated with magnetic IgG-coupled Dynabeads M-270 Epoxy (Life Technologies) for 1 hour at 4°C, and then beads were washed with lysis buffer five times. Elution was performed by incubation with homemade His-TEV protease for 2 hours at 4°C. The elution containing 10 mg of total RNA was treated with 12.5 units of RNase I (Epicentre) at 23°C for 45 min. The ribosome protected mRNA fragments were extracted by TRIzole regent (Thermo Fisher Scientific) and used for the library preparation.
Preparation of ribosome protected mRNA fragments for monosome-enriched selective ribosome profiling
The yeast strain expressing genomically tagged Not4 (Not4-FTPA) was cultured in YPD medium until mid-log phase (OD600 = 0.5 to 0.6), and then the cell pellet was rapidly harvested by centrifugation. The harvested cell pellet was immediately frozen in liquid nitrogen and then ground in liquid nitrogen using a mortar. The cell powder was resuspended with lysis buffer containing 100 μg/ml tigecycline. The whole-cell lysate was centrifuged at 39,000g for 30 min at 4°C, and the supernatant fraction was used for the following step. For total ribo-seq (input), the supernatant fraction containing 10 μg of total RNA was treated with 12.5 units of RNase I (Epicentre) at 23°C for 45 min, and then ribosome fraction was sedimented through 1 M sucrose cushion. The ribosome protected mRNA fragments were extracted by TRIzole regent (Thermo Fisher Scientific) and used for the library preparation. For Not4-IPed ribo-seq, the supernatant fraction was incubated with magnetic IgG-coupled Dynabeads M-270 Epoxy (Life Technologies) for 3 hours at 4°C to ensure binding of Not4 to IgG and to partially digest mRNAs by endogenous nuclease. The Not4-bound IgG beads were washed with lysis buffer containing 100 μg/ml tigecycline. Elution was performed by incubation with homemade His-TEV protease for 2 hours at 4°C. The elution containing 10 mg of total RNA was treated with 12.5 units of RNase I (Epicentre) at 23°C for 45 min. The ribosome-protected mRNA fragments were extracted by TRIzole regent (Thermo Fisher Scientific) and used for the library preparation.
Library preparation for ribosome profiling
Library preparation was performed according to the method previously described with the following modifications (38). As linker DNA, 5′-(Phos)NNNNNIIIIITGATCGGAAGAGCACACGTCTGAA(ddC)-3′ [where (Phos) indicates 5′ phosphorylation and (ddC) indicates a terminal 2′,3′-dideoxycytidine] was used. The Ns and Is indicate a random barcode for eliminating PCR duplication and multiplexing barcode, respectively. The linkers were preadenylated with a 5′ DNA Adenylation kit (NEB) and then used for the ligation reaction. Unreacted linkers were digested by 5′ deadenylase (NEB) and RecJ exonuclease (epicentre) at 30°C for 45 min. An oligo 5′-(Phos)NNAGATCGGAAGAGCGTCGTGTAGGGAAAGAG(iSp18)GTGACTGGAGTTCAGACGTGTGCTC-3′ [where (Phos) indicated 5′ phosphorylation and Ns indicate a random barcode] was used for reverse transcription. PCR was performed with oligos 5′-AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTC-3′ and 5′-CAAGCAGAAGACGGCATACGAGATJJJJJJGTGACTGGAGTTCAGACGTGTG-3′ [where Js indicate the reverse complement of the index sequence discovered during Illumina sequencing]. The libraries were sequenced on a HiSeq 4000 (Illumina).
Data analysis for selective ribosome profiling
Sequencing reads were demultiplexed and stripped of 3′ linker sequence using FASTX-toolkit v0.0.14. The unique molecular identifiers (UMIs), which can serve to remove PCR duplications generated during library preparation, were extracted by UMI-tools v1.0.1 (39). The datasets after these steps are deposited at Gene Expression Omnibus (GEO): GSE131408 and GSE144250. The reads were first filtered by mapping to Bowtie Index composed of noncoding RNA genes using Bowtie2 v2.2.5. Reads were then mapped to the genome using Tophat v2.1.1. Only uniquely mapping reads from the final genomic alignment were used for subsequent analysis. We estimated the position of the A-site from the 5′ end of the reads at the initiation codon on the basis of the length of each footprint using plastids v0.4.7 (40). The mapped read counts were calculated by plastids v0.4.7.
For the analysis of ribosome pause score (Fig. 2F), all footprints containing short form (21 to 22 nt for Not4-IPed and 21 to 23 nt for L7-IPed ribo-seq) and classical form (28 to 30 nt for Not4-IPed and 29 to 31 nt for L7-IPed ribo-seq) were used. The offsets were 17 for 23-nt reads; 16 for 22-, 29-, 30-, and 31-nt reads; and 15 for 21- and 28-nt reads. As previously published (41), we basically took the footprints accumulation over the average of the footprint density in the given ORFs. Analyses were restricted to the mRNAs with 0.5 footprints per codon and more. The averaged pause scores on given codons were computed by R v3.3.2.
For calculating Not4-enrichment (Fig. 5), we employed the monosome-enriched ribosome profiling dataset. All footprints containing short form (21 to 24 nt for input and 20 to 23 nt for Not4-IPed ribo-seq) and classical form (29 to 33 nt for input and 28 to 30 nt for Not4-IPed ribo-seq) were used for analysis. The offsets used in this analysis were 17 for 33-nt reads; 16 for 21- to 24- and 29- to 32-nt reads; and 15 for 20- and 28-nt reads. Transcript-level Not4-enrichment analysis was computed by R package “DESeq” (42). Transcripts that mapped less than 25 reads were omitted from transcript-level enrichment analysis. Codon stabilization coefficient (CSC, fraction of optimality (codon optimality), and tAI were referred for (4, 43, 44). For meta-gene assay of Not4-enrichment at individual codons within the ORF, we first calculated reads per million (RPM) for each codon from overall ORFs (n = 6696 genes), excluding initiation and termination codons, and then mean RPM at each codon (n = 61 amino acidcoding codons) from Not4-IPed ribo-seq dataset were divided by input one.
Analytical sucrose density gradient centrifugation
Yeast cells were grown exponentially at 30°C, treated with 0.1 mg/ml of CHX for 5 min before harvesting, and then harvested by centrifugation. The harvested cell pellet was frozen and ground in liquid nitrogen using a mortar. The cell powder was resuspended with lysis buffer [20 mM HEPES-KOH, pH 7.4, 100 mM potassium acetate, 2 mM magnesium acetate, 0.5 mM dithiothreitol, 1 mM phenylmethyl-sulfonyl fluoride, 1 tablet/10 ml Complete mini EDTA-free (11836170001, Roche)] to prepare the lysate. The lysate (the equivalent of 50 A260 units) was layered on top of the 10 to 50% sucrose gradients and then centrifuged at 150,000g in a P28S rotor (Hitachi Koki, Japan) for 2.5 hours at 4°C. The polysome profiles were generated by continuous absorbance measurement at 254 nm using a single-path UV-1 optical unit (ATTO Biomini UV-monitor) connected to a chart recorder (ATTO digital mini-recorder). Proteins in each fraction were separated by 10% Nu-PAGE and transferred to PVDF membranes (Millipore; IPVH00010). After blocking with 5% skim milk, the blots were incubated with the Anti-HA-Peroxidase (12013819001, Roche), and detected by Image-Quant LAS4000 (GE Healthcare).
In the sucrose density gradient centrifugation analysis of fig. S5H, yeast cells were grown exponentially at 30°C and then harvested by centrifugation without CHX treatment. The lysate (the equivalent of 50 A260 units) were layered on top of the 10 to 50% sucrose gradients and then centrifuged at 270,700g in a SW41Ti rotor (Beckman Coulter) for 80 min at 4°C. Other experimental handling was similar to that described above.
Transcriptional shutoff and RNA Northern blot analysis
For the GAL1 UAS transcriptional shutoff analysis, cells expressing the appropriate plasmids were grown at 24°C in synthetic media with 2% galactose/1% sucrose to allow for expression of the reporter mRNA. Cells were shifted to synthetic media without sugar at an OD600 = 0.4, and then transcription was repressed by adding glucose to a final concentration of 4%. Cells were collected at the time points indicated in the figures. For the CHX-treated experiments, different concentrations of CHX (0, 0.2, 50, or 100 mg/ml) were added to cells (OD600 = 0.4) for 40 min, followed by the transcriptional shutoff analysis.
Total RNA was extracted by phenol/chloroform and precipitated with 95% EtOH overnight. RNA (30 μg) was separated on 1.4% agarose-formaldehyde gels at 100 V for 1.5 hours, transferred to nylon membranes, and probed with 32P-labeled antisense oligonucleotides to detect poly (G) (oJC168), HIS3 (oJC2564), or SCR1 (oJC306) (table S3). Blots were exposed to PhosphorImager screens, scanned by Typhon 9400, and quantified by ImageQuant.
Half-life measurement and RNA Northern blot for HIS3 mRNA stability analysis
For the HIS3 mRNA stability analysis, cells expressing the respective plasmids were grown at 30°C in synthetic media with 2% galactose after preculture in synthetic media with 2% raffinose to allow for expression of the reporter mRNA. At an OD600 = 0.5, galactose-containing media was removed and then transcription was repressed by adding glucose to a final concentration of 2%. Cells were collected at the time points indicated in the figures. See Fig. 5E for HIS3 reporter mRNA half-life measurements and fig. S6 for Northern blots of HIS3 reporter mRNA.
Total RNA was extracted by phenol/chloroform and refined with ethanol precipitation. RNA (500 or 1000 ng) was mixed with 27 μl of glyoxal mix [600 μl of DMSO, 200 μl of deionized 40% glyoxal, 120 μl of 10x MOPS buffer (200 mM MOPS, 50 mM NaOAc, 10 mM EDTA, pH 7.0), 62.5 μl of 80% glycerol, and 17.5 μl of DEPC-treated water in 1 ml] and 3 μl of RNA loading buffer (50% glycerol, 10 mM EDTA pH 8.0, 0.05% bromophenol blue, 0.05% xylene cyanol) and separated on 1.2% agarose-MOPS gels at 200 V for 40 min, the transferred to Hybond-N+ membrane (GE healthcare) with 20xSSC (3M NaCl, 300 mM Trisodium citrate dihydrate) for 20 hours using a capillary system. After cross-linking on a membrane by CL-1000 ultraviolet cross-linker (UVP) at 120 mJ/cm2, the membrane was incubated with DIG Easy Hyb Granules (Roche) for 1 hour in a hybridization oven at 50°C. 5′ end DIG-labeled oligonucleotide for the FLAG-Tag sequence (5′-CTTGTCGTCGTCGTCCTTGTAGTC-3′) was added and incubated for more than 18 hours, followed by an additional wash with wash buffer I (2.0x SSC, 0.1% SDS) and wash buffer II (0.1x SSC, 0.1% SDS) for 15 min at 50°C. The membrane was then incubated with 1x maleic acid buffer (100 mM maleic acid, 150 mM NaCl, pH 7.0, adjusted by NaOH) containing Blocking Reagent (Roche) for 60 min at room temperature. Anti-Digoxigenin-AP, Fab fragments (Roche), was added to Blocking Reagent and further incubated for 1 hour. For washing, membrane was incubated with wash buffer III (1x maleic acid buffer, 0.3% tween 20) for 10 min, three times, and equilibrated by equilibration buffer (100 mM Tris-HCl, 100 mM NaCl, pH 9.5). For detection, membrane was reacted with CDP-star (Roche) for 10 min, and chemiluminescence was detected by LAS-4000 (GE healthcare). Relative RNA levels were determined using Multi Gauge v3.0 (Fujifilm, Japan) by comparison to a standard curve using a series of dilutions of samples from time point 0 hours (just after transcription shutoff). The displayed values represent the averages of biological triplicates (N = 3) and their standard deviations.
Polyribosome analysis and Western blotting
CHX (100 μg/ml) was added when cells reach to OD600 = 0.4 before harvesting. For detecting Dhh1p by Western blotting, cells were cross-linked at a final concentration of 0.25% formaldehyde for 5 min, then treated with 125 mM glycine for 10 min to quench cross-linking before adding CHX. Cells were then lysed into lysis buffer (10 mM Tris pH 7.4, 100 mM NaCl, 30 mM MgCl2, 1 mM DTT, 100 μg/ml CHX) by vortexing with glass beads, and cleared using the hot needle puncture method. 1% Triton X-100 was added into the supernatant. 7.5 OD260 units were loaded on 15 to 45% (w/w) sucrose gradients prepared on a Biocomp Gradient Master in gradient buffer (50 mM Tris-acetate pH 7.0, 50 mM NH4Cl, 12 mM MgCl2, 1 mM DTT) and centrifuged in a SW-41Ti rotor at 41,000 revolutions per minute for 2 hours and 26 min (to detect Dhh1p signal, the centrifugation step is optimized to 1 hour 13 min for all groups in fig. S5I (45) at 4°C]. Gradients were fractionated by using a Brandel Fractionation System and an ISCO UA-6 ultraviolet detector. Fractions were precipitated at −20°C with 10% TCA (final concentration) overnight. Pellets were washed with 80% acetone, resuspended in 50 ml of SDS-PAGE loading buffer, and boiled at 95°C for 5 min, then separated by 10% SDS polyacrylamide gels, followed by Western blotting with primary antibodies (anti-HA [BioLegend, PRB-101C], anti-Rpl4 [Proteintech, 11302–1-AP], anti-Dhh1p at 4°C overnight and incubated with secondary antibodies (goat-anti-Mouse [Santa Cruz sc-2005] and goat-anti-Rabbit [Pierce 31460]) at room temperature for 1 hour. Signal was detected by chemiluminescence using Blue Ultra Autorad film.
Endogenous mRNA Northern blot for EDC1 mRNAs
For the endogenous mRNA Northern blot, the indicated cells were grown at 30°C in synthetic media with 2% glucose. Cells were harvested at an OD600 = 0.7. Total RNA was extracted by phenol/chloroform and ethanol precipitation. RNA (5 μg) was separated on 1.2% agarose-formaldehyde gels at 200 V for 60 min, and then transferred to nylon membranes. After the RNA-transfer step, experimental handling was similar to that described for the HIS3 half-life measurements. Relative RNA levels were determined using Multi Gauge v3.0 (Fujifilm, Japan). The displayed values represent the averages of biological triplicates (N = 3) and their standard deviations.
DIG-labeled EDC1 probes were prepared by PCR-based nucleic acid labeling using PCR DIG Probe Synthesis kit (Roche, NJ, USA) according to the procedure specified by the manufacturer and primers 5′- ATGTCGACGGATACCATGTATTTCAACAG-3′ and 5′- TTAGCCTCCCTTGGACCATTAGTG-3′.
Supplementary Material
ACKNOWLEDGMENTS
Computations were partially performed on the NIG supercomputer at ROIS National Institute of Genetics. We thank H. Sieber, C. Ungewickell, S. Rieder, L. Kater, and J. Schneider for technical support. This work used the Vincent J. Coates Genomics Sequencing Laboratory at UC Berkeley, supported by NIH S10 Instrumentation Grant OD018174. We also thank T. Fröhlich and LAFUGA for mass spectrometry analysis.
Funding:
This study was supported by a Ph.D. fellowship by Boehringer Ingelheim Fonds to R.Bu., a Deutsche Forschungsgemeinschaft grant (BE1814/15–1) to J.Ch., Grants-in-Aid for Scientific Research (KAKENHI) from the Japan Society for the Promotion of Science (26116003 and 18H03977 to T.I.; 19K06481 to Y.Matsuo), Research Grants in the Natural Sciences from the Takeda Foundation (to T.I.), Research Grants in the Medical Sciences from Kato Memorial Bioscience Foundation (to Y.Matsuo), and NIH grants (GM118018 and GM115086 to J.Co.).
Footnotes
Competing interests: The authors declare no competing interests.
Data and materials availability: The sequencing data for ribosome profiling experiments have been deposited in the National Center for Biotechnology Information′s Gene Expression Omnibus (GEO) and are accessible through GEO series accession numbers GSE131408 and GSE144250. Cryo-EM maps and molecular models have been deposited with in the Electron Microscopy Data Bank with accession codes EMD-10431 (Not5–80S) and EMD-10537 (eIF5A-80S) and in the Protein Data Bank (PDB) with IDs 6TB3 (Not5–80S) and 6TNU (eIF5A-80S).
SUPPLEMENTARY MATERIALS
MDAR Reproducibility Checklist
View/request a protocol for this paper from Bio-protocol.
REFERENCES AND NOTES
- 1.Collart MA, The Ccr4-Not complex is a key regulator of eukaryotic gene expression. WIREs RNA 7, 438–454 (2016). doi: 10.1002/wrna.1332; pmid: 26821858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schäfer IB et al. , Molecular Basis for poly(A) RNP Architecture and Recognition by the Pan2-Pan3 Deadenylase. Cell 177,1619–1631.e21 (2019). doi: 10.1016/j.cell.2019.04.013; pmid: 31104843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Parker R, RNA degradation in Saccharomyces cerevisae. Genetics 191, 671–702 (2012). doi: 10.1534/genetics.111.137265; pmid: 22785621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Presnyak V et al. , Codon optimality is a major determinant of mRNA stability. Cell 160, 1111–1124 (2015). doi: 10.1016/j.cell.2015.02.029; pmid: 25768907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bazzini AA et al. , Codon identity regulates mRNA stability and translation efficiency during the maternal-to-zygotic transition. EMBO J. 35, 2087–2103 (2016). doi: 10.15252/embj.201694699; pmid: 27436874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu Q et al. , Translation affects mRNA stability in a codon-dependent manner in human cells. eLife 8, e45396 (2019). doi: 10.7554/eLife.45396; pmid: 31012849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Webster MW et al. , mRNA Deadenylation Is Coupled to Translation Rates by the Differential Activities of Ccr4-Not Nucleases. Mol. Cell 70, 1089–1100.e8 (2018). doi: 10.1016/j.molcel.2018.05.033; pmid: 29932902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Radhakrishnan A et al. , The DEAD-Box Protein Dhh1p Couples mRNA Decay and Translation by Monitoring Codon Optimality. Cell 167, 122–132.e9 (2016). doi: 10.1016/j.cell.2016.08.053; pmid: 27641505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tesina P et al. , Structure of the 80S ribosome-Xrn1 nuclease complex. Nat. Struct. Mol. Biol. 26, 275–280 (2019). doi: 10.1038/s41594-019-0202-5; pmid: 30911188 [DOI] [PubMed] [Google Scholar]
- 10.Dimitrova LN, Kuroha K, Tatematsu T, Inada T, Nascent peptide-dependent translation arrest leads to Not4p-mediated protein degradation by the proteasome. J. Biol. Chem. 284, 10343–10352 (2009). doi: 10.1074/jbc.M808840200; pmid: 19204001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Preissler S et al. , Not4-dependent translational repression is important for cellular protein homeostasis in yeast. EMBO J. 34, 1905–1924 (2015). doi: 10.15252/embj.201490194; pmid: 25971775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ikeuchi K et al. , Collided ribosomes form a unique structural interface to induce Hel2-driven quality control pathways. EMBO J. 38, e100276 (2019). doi: 10.15252/embj.2018100276; pmid: 30609991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Panasenko OO, Collart MA, Presence of Not5 and ubiquitinated Rps7A in polysome fractions depends upon the Not4 E3 ligase. Mol. Microbiol. 83, 640–653 (2012). doi: 10.1111/j.1365-2958.2011.07957.x; pmid: 22243599 [DOI] [PubMed] [Google Scholar]
- 14.Villanyi Z et al. , The Not5 subunit of the ccr4-not complex connects transcription and translation. PLOS Genet. 10, e1004569 (2014). doi: 10.1371/journal.pgen.1004569; pmid: 25340856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alhusaini N, Coller J, The deadenylase components Not2p, Not3p, and Not5p promote mRNA decapping. RNA 22, 709–721 (2016). doi: 10.1261/rna.054742.115; pmid: 26952104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Muhlrad D, Parker R, The yeast EDC1 mRNA undergoes deadenylation-independent decapping stimulated by Not2p, Not4p, and Not5p. EMBO J. 24, 1033–1045 (2005). doi: 10.1038/sj.emboj.7600560; pmid: 15706350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Basavappa R, Sigler PB, The 3 A crystal structure of yeast initiator tRNA: Functional implications in initiator/elongator discrimination. EMBO J. 10, 3105–3111 (1991). doi: 10.1002/j.1460-2075.1991.tb07864.x; pmid: 1915284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tate JG et al. , COSMIC: The Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 47, D941–D947 (2019). doi: 10.1093/nar/gky1015; pmid: 30371878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.De Keersmaecker K et al. , Exome sequencing identifies mutation in CNOT3 and ribosomal genes RPL5 and RPL10 in T-cell acute lymphoblastic leukemia. Nat. Genet. 45, 186–190 (2013). doi: 10.1038/ng.2508; pmid: 23263491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Budkevich TV et al. , Regulation of the mammalian elongation cycle by subunit rolling: A eukaryotic-specific ribosome rearrangement. Cell 158, 121–131 (2014). doi: 10.1016/j.cell.2014.04.044; pmid: 24995983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schmidt C et al. , Structure of the hypusinylated eukaryotic translation factor eIF-5A bound to the ribosome. Nucleic Acids Res. 44,1944–1951 (2016). doi: 10.1093/nar/gkv1517; pmid: 26715760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu CC, Zinshteyn B, Wehner KA, Green R, High-Resolution Ribosome Profiling Defines Discrete Ribosome Elongation States and Translational Regulation during Cellular Stress. Mol. Cell 73, 959–970.e5 (2019). doi: 10.1016/j.molcel.2018.12.009; pmid: 30686592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hanson G, Alhusaini N, Morris N, Sweet T, Coller J, Translation elongation and mRNA stability are coupled through the ribosomal A-site. RNA 24,1377–1389 (2018). doi: 10.1261/rna.066787.118; pmid: 29997263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Petropoulos AD, Green R, Further in vitro exploration fails to support the allosteric three-site model. J. Biol. Chem. 287, 11642–11648 (2012). doi: 10.1074/jbc.C111.330068; pmid: 22378789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Suzuki T et al. , CNOT3 suppression promotes necroptosis by stabilizing mRNAs for cell death-inducing proteins. Sci. Rep. 5, 14779 (2015). doi: 10.1038/srep14779; pmid: 26437789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chan LY, Mugler CF, Heinrich S, Vallotton P, Weis K, Non-invasive measurement of mRNA decay reveals translation initiation as the major determinant of mRNA stability. eLife 7, e32536 (2018). doi: 10.7554/eLife.32536; pmid: 30192227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sohrabi-Jahromi S et al. , Transcriptome maps of general eukaryotic RNA degradation factors. eLife 8, e47040 (2019). doi: 10.7554/eLife.47040; pmid: 31135339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Panasenko OO et al. , Co-translational assembly of proteasome subunits in NOT1-containing assemblysomes. Nat. Struct. Mol. Biol. 26, 110–120 (2019). doi: 10.1038/s41594-018-0179-5; pmid: 30692646 [DOI] [PubMed] [Google Scholar]
- 29.Longtine MS et al. , Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14, 953–961 (1998). doi: 10.1002/(SICI)1097-0061(199807)14:10<953:AID-YEA293>3.0.C0;2-U; pmid: 9717241 [DOI] [PubMed] [Google Scholar]
- 30.Janke C et al. , A versatile toolbox for PCR-based tagging of yeast genes: New fluorescent proteins, more markers and promoter substitution cassettes. Yeast 21, 947–962 (2004). doi: 10.1002/yea.1142; pmid: 15334558 [DOI] [PubMed] [Google Scholar]
- 31.Zheng SQ et al. , MotionCor2: Anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017). doi: 10.1038/nmeth.4193; pmid: 28250466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang K, Gctf: Real-time CTF determination and correction. J. Struct. Biol. 193,1–12 (2016). doi: 10.1016/j.jsb.2015.11.003; pmid: 26592709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kimanius D, Forsberg BO, Scheres SH, Lindahl E, Accelerated cryo-EM structure determination with parallelisation using GPUs in RELION-2. eLife 5, e18722 (2016). doi: 10.7554/eLife.18722; pmid: 27845625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zivanov J et al. , New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife 7, e42166 (2018). doi: 10.7554/eLife.42166; pmid: 30412051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Emsley P, Cowtan K, Coot: Model-building tools for molecular graphics. Acta Crystallogr. D 60, 2126–2132 (2004). doi: 10.1107/S0907444904019158; pmid: 15572765 [DOI] [PubMed] [Google Scholar]
- 36.Afonine PV et al. , Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr D 68, 352–367 (2012). doi: 10.1107/S0907444912001308; pmid: 22505256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Goddard TD et al. , UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 27, 14–25 (2018). doi: 10.1002/pro.3235; pmid: 28710774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ingolia NT, Brar GA, Rouskin S, McGeachy AM, Weissman JS, The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome-protected mRNA fragments. Nat. Protoc. 7, 1534–1550 (2012). doi: 10.1038/nprot.2012.086; pmid: 22836135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Smith T, Heger A, Sudbery I, UMI-tools: Modeling sequencing errors in Unique Molecular Identifiers to improve quantification accuracy. Genome Res. 27, 491–499 (2017). doi: 10.1101/gr.209601.116; pmid: 28100584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dunn JG, Weissman JS, Plastid: Nucleotide-resolution analysis of next-generation sequencing and genomics data. BMC Genomics 17, 958 (2016). doi: 10.1186/s12864-016-3278-x; pmid: 27875984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Woolstenhulme CJ, Guydosh NR, Green R, Buskirk AR, High-precision analysis of translational pausing by ribosome profiling in bacteria lacking EFP. Cell Rep. 11, 13–21 (2015). doi: 10.1016/j.celrep.2015.03.014; pmid: 25843707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Anders S, Huber W, Differential expression analysis for sequence count data. Genome Biol. 11, R106 (2010). doi: 10.1186/gb-2010-11-10-r106; pmid: 20979621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pechmann S, Frydman J, Evolutionary conservation of codon optimality reveals hidden signatures of cotranslational folding. Nat. Struct Mol. Biol. 20, 237–243 (2013). doi: 10.1038/nsmb.2466; pmid: 23262490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Drummond DA, Raval A, Wilke CO, A single determinant dominates the rate of yeast protein evolution. Mol. Biol. Evol. 23, 327–337 (2006). doi: 10.1093/molbev/msj038; pmid: 16237209 [DOI] [PubMed] [Google Scholar]
- 45.Sweet T, Kovalak C, Coller J, The DEAD-box protein Dhh1 promotes decapping by slowing ribosome movement. PLOS Biol. 10, e1001342 (2012). doi: 10.1371/journal.pbio.1001342; pmid: 22719226 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.