

Abstract
Alzheimer’s disease (AD) is a pervasive, relentlessly progressive neurodegenerative disorder that includes both hereditary and sporadic forms linked by common underlying neuropathologic changes and neuropsychological manifestations. While a clinical diagnosis is often made on the basis of initial memory dysfunction that progresses to involve multiple cognitive domains, definitive diagnosis requires autopsy examination of the brain to identify amyloid plaques and neurofibrillary degeneration. Over the past 100 years, there has been remarkable progress in our understanding of the underlying pathophysiologic processes, pathologic changes, and clinical phenotypes of AD, largely because genetic pathways that include but expand beyond amyloid processing have been uncovered. This review discusses the current state of understanding of the genetics of AD with a focus on how these advances are both shaping our understanding of the disease and informing novel avenues and approaches for development of potential therapeutic targets.
Keywords: Alzheimer’s disease, neurodegeneration, amyloid, tau, genetics, aging
INTRODUCTION
Since the first published description of “a peculiar severe disease process of the cerebral cortex” by Dr. Alois Alzheimer in 1907 (1), Alzheimer’s disease (AD) has become recognized as a heterogeneous and polygenic group of both hereditary and sporadic neurodegenerative disorders with common underlying neuropathologic changes. In his landmark paper, Alzheimer described an individual who, at 51 years of age, presented with a history of paranoia, progressive sleep and memory disturbance, aggression, and confusion. Upon her death less than 5 years later, neuropathologic review of her brain revealed the plaques and tangles that are now considered pathognomonic for the disease (2). Today, although several variants of AD have been reported, the most common clinical presentation is similar to Alzheimer’s original case (albeit with later age of onset), and a clinical diagnosis is often based on initial memory dysfunction, which later progresses to involve multiple cognitive domains. As originally described, a neuropathologic diagnosis of AD requires the presence of both amyloid plaques and neurofibrillary degeneration (prominently including tangles), which can now be readily identified, with the aid of immunohistochemical stains, as comprising amyloid beta (Aβ) and pathologic tau, respectively (3).
Although Alzheimer’s initial findings provided the foundation for the current clinical and neuropathologic diagnostic criteria for AD, the conceptualization and characterization of this disease have changed and evolved over time, driven most recently by advances in our understanding of the underlying genetics. The amyloid plaques and neurofibrillary tangles (NFTs) Alzheimer described had previously been reported in the brains of elderly subjects both with and without dementia, and some researchers considered them a part of the aging process (4). What set Alzheimer’s case apart was that it occurred in a relatively young individual, and therefore AD was defined as a separate disease from what was observed in older people. In the second half of the twentieth century, however, AD became redefined as a single neurodegenerative process that could affect more commonly older but also younger adults—a clinically heterogeneous disease, united by a common underlying set of pathologic changes in the brain. AD is now commonly divided into late-onset AD (LOAD) cases, which have some identified genetic risk factors but often occur sporadically and are by far the most common, and early-onset AD (EOAD) cases, which usually include disease-causing autosomal dominant mutations in genes involved in the amyloid processing pathway.
More than 100 years after the first report, AD has been recognized as a public health crisis, affecting at least 40 million people worldwide. While the field still struggles with defining the etiology, diagnosis, and treatment of AD, progress has been made in our understanding of the role of genetics both in LOAD and in sporadic and familial EOAD forms. The general consensus is that this complex, multifactorial disease will require a precision medicine approach that demands a highly sophisticated understanding of its genetic underpinnings.
GENETIC CAUSE
In 1991, Goate et al. (5) used genetic markers to map a locus segregating with familial AD to chromosome 21. Recombinants between this locus and the APP (amyloid precursor protein) gene had been reported, but the authors found that this locus segregated with the families that had EOAD and not LOAD. They sequenced exon 17 of the APP gene and found a missense mutation that causes a valine-to-isoleucine change at amino acid 717 (p.Val717Ile). As it was initially discovered in an English family, the mutation is known as the London mutation. After this initial discovery, both Chartier-Harlin et al. (6) and Murrell et al. (7) found different mutations (V717G and V717F, respectively) at the same amino acid in other families with AD. Although the mutation identified by Murrell et al. became known as the Indiana mutation, it was in fact in an ethnically Romanian kindred. In 1993, Mullan et al. (8) found a double mutation located in the N terminus of APP (K670N/M671L) in two large Swedish families with EOAD (the Swedish mutation). Many additional mutations in APP have since been discovered, guiding research to focus on APP processing pathways (9).
The occurrence of EOAD in multiple families could not be explained by the known APP mutations. In 1995, Sherrington et al. (10) identified the first non-APP mutations in the PSEN1 (presenilin 1) gene in multiple families of different ethnicities. These included PSEN1 C410Y (Ashkenazi Jewish), H163R (American and French Canadian), M146L (Italian), L286V (German), and A246E (Anglo-Saxon-Celt). Shortly after this initial discovery, Campion et al. (11) identified six novel PSEN1 mutations in eight additional families, all within highly conserved regions of the gene. Many additional PSEN1 mutations have since been identified (12).
The next major genetic discovery in AD was the identification of PSEN2 (presenilin 2), a gene homologous to PSEN1 on chromosome 1 (13, 14). Rogaev et al. (14) reported two different mutations, M239V and N141I, that segregated with familial AD in an Italian pedigree and a Volga German pedigree, respectively, while Levy-Lahad et al. (13) also noted the N141I mutation in a Volga German kindred. With the technology for sequencing improving, Finckh et al. (15) sequenced individuals with EOAD and found additional mutations in both PSEN1 (F105L) and PSEN2 (T122P and M239I). Known mutations in these three genes, APP, PSEN1, and PSEN2, account for approximately 1% of autosomal dominantly inherited AD.
GENETIC RISK
The heritability of LOAD has been estimated to involve more than half to three-quarters of affected individuals. However, in contrast to EOAD, no causative genes have been identified for LOAD, but several risk genes have been described.
APOE
Using an affected-member-pedigree linkage analysis, Pericak-Vance et al. (16) found that the patients’ genomic markers did not show linkage with the previously identified chromosome 21 locus but instead with chromosome 19 (HAS 19). They then applied a standard likelihood (logarithm of the odds, or LOD, score) analysis and discovered the first significant linkage association for LOAD on chromosome 19. At the same time, Namba et al. (17) unexpectedly found that apolipoprotein E localized to NFTs and Aβ deposits in brain, in both senile plaques and cerebral vessels, in patients with AD. Apolipoprotein E is encoded by APOE on chromosome 19, and humans are unlike all other mammals in having three prevalent alleles (ε2, ε3, and ε4). In 1993, Strittmatter et al. (18) used an in vitro assay and showed that apoE binds to Aβ with high avidity and that APOE ε4 was found at a higher frequency among LOAD patients compared with unrelated, age-matched controls. Corder et al. (19) subsequently assessed 42 LOAD families and identified a gene dosage effect such that carriers of a single APOE ε4 allele had a hazard ratio of 2.84, while those who were homozygous for APOE ε4 (APOE ε4/ε4 genotype) had a hazard ratio of 8.07. Although additional studies have shown that the frequencies of APOE genotypes vary across different ethnic populations, the ε4 allele has repeatedly been associated with an increased risk of AD. Interestingly, a carrier of the PSEN1 (E280A) mutation, from a large Colombian EOAD cohort, also has two copies of the rare APOE3 Christchurch (R136S) mutation, and she did not develop cognitive impairments until her seventies, nearly 3 decades after the typical age of onset (20). This finding has piqued interest in the possible protective benefits of this mutation.
SORL1
After the discovery of the gene dosage–dependent risk of APOE ε4 with LOAD, other genes in the endocytic and cellular recycling pathways were investigated. Using a DNA microarray screen, Scherzer et al. (21) identified six genes that were differentially expressed in individuals with AD. One of these genes was SORL1, which encodes a neuronal apoE receptor. Rogaeva et al. (22) used previously identified single-nucleotide polymorphisms (SNPs) in these pathways and found that six SNPs in two different regions of the SORL1 gene were significantly associated with AD. Lee et al. (23) corroborated the intronic variants within SORL1 as being associated with familial and sporadic AD, and these findings were replicated in the TGen (21) data set and in an urban, multiethnic community (24). These variants are intronic and thought to be in regulatory sequences, which may affect the physiological role of SORL1 in the processing of APP (22).
MAPT
NFTs have been found in multiple neurodegenerative diseases, and MAPT (microtubule-associated protein tau) was initially investigated in association with frontotemporal dementia, leading to the discovery of multiple missense mutations, insertions, deletions, and splice-site mutations thought to be causative for some forms of the disease (25). Using the A169 polymorphism in exon 9 and the (CA)n-repeat polymorphism in intron 9, Roks et al. (26) performed an association study and found no mutations causally associated with EOAD. When initially looking for an association between MAPT and progressive supranuclear palsy (PSP), Baker et al. (27) sequenced the MAPT gene and identified a series of SNPs that were in complete disequilibrium with one another. These SNPs revealed two extended haplotypes (H1 and H2) that cover the entire MAPT gene. Further investigation revealed that the H1 haplotype is overrepresented in Caucasian patients with tauopathies, but subsequent studies of the relationship between MAPT haplotype and AD have been discordant, with some demonstrating an association between the H1 haplotype and AD and others failing to confirm these findings (28). Sun & Jia (29) evaluated only the H1 haplotype in a Chinese Han population, since the H2 haplotype is almost exclusively in Caucasian populations, and identified a SNP within the promoter of MAPT. The 347C allele was overrepresented in sporadic AD Chinese Han patients compared with healthy controls, and when evaluated in cell lines the SNP (347C/C versus 347 G/G) significantly increased transcriptional activity, thereby upregulating gene expression.
TREM2
In the early 2000s, researchers discovered that mutations in TREM2 could cause polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy (PLOSL), also known as Nasu–Hakola disease (30). One of the symptoms of PLOSL is presenile dementia with neurological abnormalities. TREM2 was not considered a candidate gene for dementia, however, until a Lebanese family with early-onset dementia but without PLOSL was found to have mutations in TREM2 (31). The implication of TREM2 in early-onset dementia led Guerreiro et al. (32) to study whole-exome/genome sequencing of 281 AD patients and 504 healthy controls. They found a disproportionate number of variants in exon 2 of TREM2 in the AD patients compared with the healthy controls, and, in a heterozygous state, these variants increased the risk of developing AD (32). Thus far, 46 genetic variants identified in TREM2 have been studied in association with LOAD (33). Different variants associate with LOAD in different populations, but within each group the variants lead to approximately a two- to four-fold-increased risk of developing LOAD (33). TREM2 variants have also been associated with frontotemporal dementia, amyotrophic lateral sclerosis, and Parkinson’s disease, suggesting that altered TREM2 function may nonspecifically increase risk for neurodegeneration, perhaps through microglial dysfunction (34).
Additional Risk Loci from Genome-Wide Association Studies
A more recent strategy for identifying AD genetic risk has been through genome-wide association studies (GWASs). Through 1,395 GWASs and 320 meta-analyses, a total of 695 genes have been identified as affecting the risk of LOAD (35). The top 10 genes most strongly associated with the risk of LOAD identified via GWASs are APOE, BIN1, CLU, ABCA7, CR1, PICALM, MS4A6A, CD33, MS4AE, and CD2AP. Several of these studies were done by combining multiple GWAS data sets with the idea that a common disease might present with a common variant, thus requiring large numbers of samples. When trying to confirm CLU, CR1, and PICALM as risk loci for LOAD, Jun et al. (36) investigated the SNPs within each gene in different populations (Caucasian, African American, Arab, and Caribbean Hispanic) as well as in a combined data set. In the Caucasian population, the SNP rs11136000 in CLU had an odds ratio of 0.91, the SNP rs3818361 in CR1 had an odds ratio of 1.14, and the SNP rs3851179 in PICALM had an odds ratio of 0.89. None of these SNPs were significantly associated with LOAD in any of the other ethnic groups analyzed either together or separately. Interestingly, when these SNPs were evaluated collectively with APOE genotypes, the authors found that the association with CLU was evident only in patients without the APOE ε4 allele, whereas the association with PICALM was evident only in patients with the APOE ε4 allele.
Using 94,437 individuals diagnosed with LOAD, Kunkle et al. (37) confirmed many previously identified genome-wide significant loci (CR1, BIN1, INPP5D, HLA-DRB1, TREM2, CD2AP, NYAP1, EPHA1, PTK2B, CLU, SPI1, MS4A2, PICALM, SORL1, FERMT2, SLC24A4, ABCA7, APOE, and CASS4). These genes may have pleiotropic associations with traits that are relevant to AD. To investigate this hypothesis further, Kunkle et al. (37) looked at the genetic architecture of LOAD in comparison to 792 other human diseases, traits, and behaviors. They corroborated Marioni et al.’s (38) findings that the genetic architecture of LOAD is more highly positively correlated with a maternal family history of AD than for paternal AD (genetic correlation = 0.91 and 0.66, respectively). When examining the genetic effect attributable to these candidate genes, Naj et al. (39) found that the most highly associated SNPs at each locus (other than APOE) had population-attributable fractions between only 2.72% and 5.97%. However, these estimates may vary widely among studies and populations.
To understand how all of these genetic risk loci might contribute to AD, Kunkle et al. (37) performed a pathway analysis for common and rare variants found in patients with LOAD. Most of the genes fell into the gene ontology pathway of activation of immune response; the most significant common variant pathway identified was protein–lipid complex assembly, while the most significant rare variant pathway was tau protein binding.
INSIGHTS INTO ALZHEIMER’S DISEASE
The neuropathologic and neuropsychologic changes of AD have been well characterized and are similar regardless of the genetic underpinnings. However, an increased understanding of AD genetics has aided in the elucidation of subtle differences associated with the various causative and risk genes, which in turn has helped shape the development of animal models and may ultimately lead to advances in the diagnosis, treatment, and prevention of AD.
Neuropathology and Pathophysiology
Postmortem neuropathologic examination remains the gold standard for a definitive diagnosis of AD. There are several nonspecific, but likely very important, changes that occur in the brain of an individual with AD that implicate a progressive neurodegenerative process, including synapse loss, neurodegeneration, and glial reaction. However, the pathologic diagnosis requires the presence of two disease-defining features: extracellular Aβ plaques and intraneuronal NFTs of hyperphosphorylated tau (Figure 1). While neurofibrillary degeneration is characteristic of many neurodegenerative diseases, its co-occurrence with Aβ plaques is what distinguishes AD. Cerebral cortical Aβ accumulation situates a person on a continuum of AD neuropathologic change (ADNC). The apparent spread of neurofibrillary degeneration out of perihippocampal structures to involve subregions of the hippocampus, and ultimately the cerebral cortex, is most closely associated with cognitive impairment and a clinical diagnosis of dementia (40). Note that, as expected for a chronic disease, mild pathologic changes of AD are commonly observed in individuals without significant cognitive impairment, while individuals with advanced pathologic changes of AD are usually diagnosed with dementia (3). However, exceptions to this simple clinicopathologic correlation are well known. Indeed, an area of intense research interest is apparently resilient individuals who have extensive AD pathologic changes without cognitive impairment (41).
Figure 1.
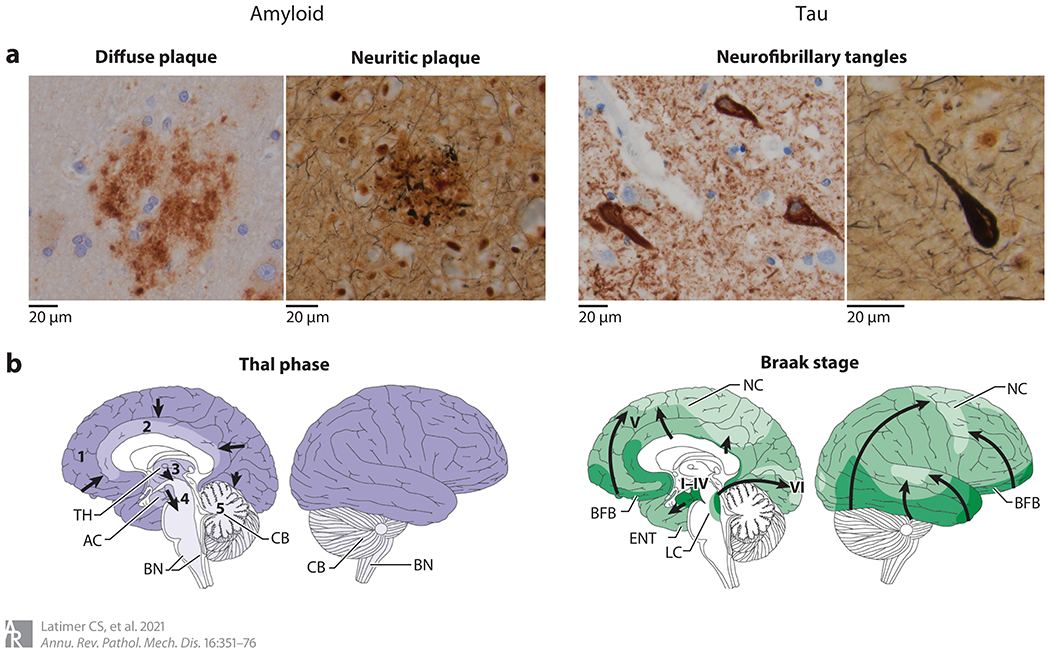
Neuropathologic changes of Alzheimer’s disease. (a) The hallmark features of Alzheimer’s disease neuropathologic change include extracellular amyloid beta (Aβ) plaques and intraneuronal neurofibrillary tangles of hyperphosphorylated tau. Aβ plaques include diffuse forms, highlighted by an anti-Aβ immunostain (left), and neuritic forms, characterized by the inclusion of dystrophic neurites containing hyperphosphorylated tau and highlighted by Bielschowsky silver stain (right). Neurofibrillary tangles can be identified with immunostains to hyperphosphorylated tau (left) as well as silver stains (right). Both amyloid and tau pathology appear to progress throughout the brain in stereotypical patterns. (b) Amyloid progression (purple), as described by Thal phases 1–5 and denoted by arrows, begins in the neocortex (phase 1), followed by limbic regions (phase 2), deep gray nuclei (phase 3), brainstem (phase 4), and, finally, the cerebellum (phase 5). Tau pathology (green), as described by Braak stages I–VI and denoted by arrows, begins in the mesial temporal structures (stages I–IV) before involving neocortex (stage V) and, finally, primary motor and sensory regions (stage VI). Abbreviations: AC, allocortex; BFB, basal forebrain; BN, brainstem nuclei; CB, cerebellum; ENT, entorhinal cortex; LC, locus ceruleus; NC, neocortex; TH, thalamus. Panel b adapted with permission from Reference 42; copyright 2015 Springer Nature.
Amyloid Beta Pathologic Change
A histologic diagnosis of AD hinges on the presence of extracellular Aβ, which can be found throughout the brain in several different forms of plaque, generally described as diffuse or neuritic (43). Neuritic plaques are more specific to ADNC, and in addition to accumulations of Aβ they contain dystrophic neurites, distorted by accumulations of intracellular hyperphosphorylated tau protein.
The neuropathologic assessment of amyloid pathology includes both the distribution of Aβ accumulation throughout the brain and the density of cerebral neuritic plaques (3, 44). Although neuritic plaques can be identified in cognitively normal subjects, studies implementing this semiquantitative technique have shown that neuritic plaque density positively correlates with risk of dementia (40). In 2002, Thal et al. (45) described the brain regions involved by amyloid plaques, dividing amyloid progression into five phases and showing that the higher phases are associated with an increased likelihood of dementia. In addition to parenchymal amyloid deposits, Aβ can be localized to blood vessels. Cerebral amyloid angiopathy (CAA) is observed to varying degrees in the setting of AD (and occasionally in the absence of plaques and tangles) and tends to involve the small arteries and arterioles in the meninges as well as in cortical gray matter. Because the normal constituents of the vessel wall are replaced by Aβ protein, the integrity of the wall is compromised, leading to a tendency, particularly in older adults, toward spontaneous hemorrhage.
Amyloid Pathophysiology
Amyloid plaques are derived from the sequential endoproteolytic cleavage of a transmembrane glycoprotein, APP. The gene encoding this protein is located on chromosome 21, and mutations in it are causative for AD (see the section titled Genetic Cause, above). APP processing can proceed along either a nonamyloidogenic pathway, driven by ⍺-secretases, or an amyloidogenic pathway, involving β-secretase activity. γ-Secretases, a complex of four individual proteins, including PSEN1 and PSEN2, further cleave the protein products generated by either α- or β-secretases. In the amyloidogenic pathway, following β-secretase cleavage, γ-secretase activity yields peptide fragments of varying lengths, but mainly those comprising 40 or 42 amino acids. Aβ42, the predominant Aβ peptide of neuritic plaques, is highly amyloidogenic and can form neurotoxic oligomeric species. In the nonamyloidogenic pathway, cleavage by α-secretases occurs near the middle of the Aβ region of APP, thereby precluding generation of Aβ; instead, the N terminus of the peptide is generated by subsequent γ-secretase activity within the transmembrane domain (46).
The amyloid cascade hypothesis postulates that the generation of Aβ peptides is the causative event in AD and that all other changes are a consequence of this primary process (47). This theory has been shaped by our combined understanding of the APP processing pathways, the neuropathologic features, and the genetics of AD. As described, the known disease-causing alleles in AD are related to APP processing (APP, PSEN1, and PSEN2), and experimental models have shown that they cause increased production of neurotoxic Aβ species and Aβ plaque formation. However, despite the concordance of genetics and Aβ deposition, the amyloid cascade hypothesis has failed to lead to disease-modifying therapies. Furthermore, Aβ plaques do not seem sufficient to cause clinical dementia, and the link between Aβ generation and neurofibrillary degeneration, the other neuropathologic hallmark of AD, remains enigmatic.
Pathologic Changes in Tau
Intracellular NFTs comprise extensively posttranslationally modified and, famously, hyperphosphorylated aggregates of tau. NFTs can be recognized on hematoxylin and eosin–stained sections as faintly basophilic, fibrillary structures within the neuronal cell body. These are best seen in the large pyramidal neurons of the hippocampus, where they often have a flame shape. Tangles are also identified in the cerebral cortex, with a preference for the larger neurons of layers III and V. Silver-based histochemical stains can be used to aid in the identification of tangles, as can antibodies to phosphorylated tau protein. These staining techniques also highlight the neuritic plaque–associated dystrophic neurites and background neuropil threads. The threads are nerve cell processes that have become distorted secondary to their accumulation of hyperphosphorylated tau. Finally, although not a diagnostic feature, glial tau is often present in AD and is found in subpial, subependymal, and perivascular locations (48). White matter is relatively devoid of pathologic tau.
The distribution of NFTs has been well described by the neuroanatomists Braak and Braak and their colleagues (49, 50), using both silver staining and immunohistochemical methods. In their method, the early stages of NFT formation (stages I–IV) are relatively confined to the medial temporal cortex. Neocortical NFTs denote stage V, while primary motor and sensory cortex are involved only late in the disease (stage VI). Although not all cases progress exactly in this manner, the Braak staging system provides a useful construct with which to determine the extent of pathologic tau throughout the brain. Use of this approach has demonstrated that the extent of NFT pathology correlates well with the clinical presentation, such that higher Braak stages are associated with an increased risk of dementia (40). Several studies have also used various quantitative and semiquantitative techniques to investigate the degree of pathologic tau burden, with the general finding that pathologic tau burden also correlates with clinical symptoms (41, 51).
While pathologic tau is considered characteristic of AD, features including NFTs, threads, and glial tau are not specific and are observed in many neurodegenerative diseases, most notably some forms of frontotemporal lobar degeneration (FTLD-tau), including PSP and corticobasal degeneration (CBD), as well as chronic traumatic encephalopathy (52). These other tauopathies have patterns of pathologic tau accumulation that differ from that described in AD, with glial and white matter tau often more prominent than in AD, although there is overlap. Also, like diffuse plaques, NFTs and some glial tau pathology in a restricted distribution can be considered age-related processes that are not necessarily associated with cognitive decline (53, 54).
Tau Pathophysiology
MAPT is encoded by a gene on chromosome 17 and aids in the stabilization of microtubules within neuronal processes. It is expressed in six isoforms produced by alternative splicing. The isoforms differ from one another on the basis of the number and location of microtubule binding domains, with three isoforms having three (3R tau) and three having four (4R tau). In the setting of neurodegenerative disease with pathologic tau, the tau protein becomes abnormally phosphorylated, either prompted by or a consequence of its disengagement from microtubules, and is then aggregated into paired helical filaments, ultimately forming NFTs. In AD, both 3R and 4R tau are abnormally phosphorylated, but in some of the primary tauopathies, pathological tau is restricted to either 3R tau (Pick’s disease) or 4R tau (CBD and PSP) (55).
Comorbid Neuropathologic Changes
In addition to Aβ plaques and neurofibrillary degeneration, several other pathologic changes are often comorbid with AD, including Lewy body disease (LBD), TDP-43 proteinopathy, hippocampal sclerosis (HS), and vascular brain injury (VBI). While each of these other diseases can cause cognitive decline on its own, the severity is usually worse when combined with AD, although it is unclear whether this is due to synergistic or additive effects.
Genetic Associations with Neuropathologic Features
There are subtle differences between the pathologic features of familial AD and those of sporadic cases (56). For example, in cases due to causative mutations in APP, PSEN1, and PSEN2, there is a greater burden of neocortical senile plaques and a higher ratio of Aβ42 to Aβ40 compared with cases of sporadic AD (57). This is perhaps not surprising, as these genes are involved in the processing of amyloid. CAA is also a more frequent finding in familial AD than in sporadic cases and is particularly severe in the setting of the APP mutations p.Asp694Asn and p.Ala713Thr. The p.Glu693Gln, or Dutch, mutation in APP is also associated with severe CAA but lacks significant parenchymal Aβ deposition. In stark contrast to Aβ deposition, there has been no well-characterized difference in pathologic tau burden between cases of sporadic AD and cases arising from causative mutations. CAA is also observed in sporadic AD, but to a lesser extent than in the familial forms.
With respect to comorbid pathologic changes, LBD, particularly brainstem and limbic Lewy bodies, is observed in AD with APP, PSEN1, or PSEN2 mutations more commonly than expected by chance (58, 59). VBI and TDP-43 copathology are rare in the setting of autosomal dominant AD mutations, in stark contrast to LOAD (60–62).
Neuropathologic Features of Alzheimer’s Disease Risk Genes
In the hope of identifying genetic associations with specific pathologic features, Beecham et al. (63) performed a GWAS on 3,887 AD patients and 1,027 healthy controls that used a clinicopathologic definition of AD, reducing the potential confounding effects of cases of dementia not due to ADNC. Having detailed neuropathologic data also enabled secondary analyses to identify genes associated with specific pathologies, including NFTs, neuritic plaques, LBD, CAA, HS, and VBI. This study confirmed 13 of the 21 previously identified gene associations with AD (APOE, CR1, BIN1, CLU, MS4A6A, PICALM, ABCA7, CD33, PTK2B, SORL1, MEF2C, 2CWPW1, and CASS4), 9 of which were strengthened. Subsequent analyses revealed that APOE was strongly associated with the pathologic features of AD, specifically NFTs, neuritic plaques, and CAA, as well as LBD; however, it was not associated with VBI or HS. The coincident neuropathologic features of AD showed only nominal associations with other known AD loci. Overall, finding additional LOAD risk alleles will require larger studies, with increased genotyping for candidate genes, to test for the effects of common and rare variants. At present, APOE is still the most highly associated and replicated risk locus associated with LOAD.
NEUROPSYCHOLOGY
In sporadic AD, anterograde amnesia is the hallmark neuropsychological feature and usually the primary presenting concern. Patients with AD frequently report an insidious onset and slowly progressive difficulty learning new information as a result of impaired encoding, retrieval, and, most strikingly, storage of new information. Semantic networks are generally disrupted early in the disease, commonly leading to impaired verbal recall. As the disease progresses, word-finding difficulty, impaired executive function (including working memory, abstract reasoning, problem solving, and judgment), and visuospatial impairment are often affected. However, simple attention, syntactic processing, long-term recall, and procedural memory generally remain relatively intact until later stages of the disease. The following subsections describe neuropsychological features of AD associated with known causal and risk genes (summarized in Figure 2).
Figure 2.
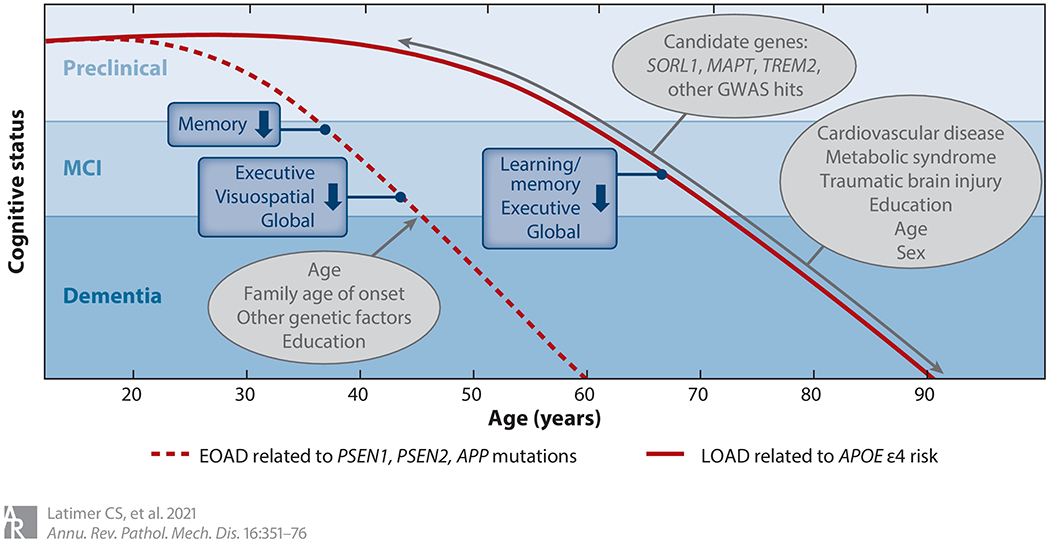
Genetic influence on cognition in EOAD and LOAD. Age of onset and disease duration represent the average for autosomal dominant EOAD or LOAD related to the presence of at least one APOE ε4 allele. Actual age of onset varies according to mutation type, age of onset in the family, and other disease-related and nonrelated factors. The gray ovals represent factors mediating cognitive decline. Abbreviations: APOE, gene encoding human apolipoprotein E; EOAD, early-onset Alzheimer’s disease; GWAS, genome-wide association study; LOAD, late-onset Alzheimer’s disease; MCI, mild cognitive impairment.
Autosomal Dominant Alzheimer’s Disease (APP, PSEN1, and PSEN2 Mutations)
With the exception of earlier dementia onset and accelerated cognitive decline, the cognitive profile most commonly described in autosomal dominant AD is predominantly amnestic and effectively indistinguishable from sporadic AD (64, 65). Although atypical cognitive presentations (e.g., visuospatial, executive, or language-predominant impairment) are reported, particularly among patients with PSEN1 mutations, a recent evaluation from the DIAN (Dominantly Inherited Alzheimer Network) observational cohort suggests that the rate of uncommon cognitive presentations in mutation carriers is either consistent with or lower than that reported in sporadic AD (66).
During preclinical stages, consistently poorer performance on episodic recall, executive, and visuospatial functions is reported in otherwise asymptomatic mutation carriers (64). The estimated onset of cognitive decline among mutation carriers ranges from 5 to 10 years prior to expected age of dementia onset, beginning with a decline in episodic verbal recall followed by other cognitive functions, including executive skills, processing speed, visuospatial abilities, and performance on global cognitive screening measures (67). The length of time between initial cognitive symptoms and dementia diagnosis may be mediated by factors such as age and education (64).
APOE
The APOE ε4 allele confers the greatest known genetic risk for LOAD; thus, the relationship between the APOE gene and cognition in AD has been studied extensively. Here, we summarize current knowledge relating APOE to cognition in AD and across the life span.
APOE and Alzheimer’s disease cognitive phenotype.
The presence of an APOE ε4 allele is associated most prominently with more severe learning and memory impairment in AD, while non–ε4 carriers with AD tend to have a greater diversity of cognitive deficits (68). These memory-specific deficits among ε4 carriers are associated with greater hippocampal atrophy in a dose-specific manner (69). In ADNI (Alzheimer’s Disease Neuroimaging Initiative), poorer memory performance by ε4 carriers was associated with greater medial temporal lobe atrophy; conversely, non–ε4 carriers had lower executive, verbal, and working memory scores in combination with greater frontoparietal atrophy (70).
Initial findings related to the association between the presence of an ε4 allele and AD progression and course were mixed (71, 72). Currently, there is a general consensus that while the APOE ε4 effect on progression of cognitive decline may be important in the early stages of the disease and in younger patients (73), it does not appear to substantially modify cognitive decline as the disease progresses (74). The presence of an APOE ε2 allele may be associated with slower cognitive decline in AD (75); however, given that the ε2 allele confers a lower overall risk for AD and represents a much smaller proportion of the overall population, sample sizes are small and conclusions are limited.
APOE across the life span.
Initial reports examining APOE allele effects throughout the life span supported antagonistic pleiotropy, such that the presence of an ε4 allele confers cognitive advantages early in life but disadvantages later in life (76). However, a recent meta-analysis showed that, across studies, young ε4 carriers did not statistically differ from noncarriers in any cognitive domain (77). Notably, socioeconomic factors such as education and health-related factors such as traumatic brain injury (TBI) may modify the effects of ε4 on cognition in younger ages (78, 79). With regard to middle age, although the ε4 allele is associated with increased ADNC (80), APOE-associated effects on cognition are unclear, with positive, negative, and null effects reported (81). Overall, in nondemented populations, APOE effects on cognition appear strongest in older adults, in which possession of an ε4 allele has been associated with impairments in learning and memory, working memory, executive function, and global cognition. Unlike in AD dementia, nondemented ε4 carriers demonstrate faster progression of cognitive symptoms, most prominently in memory, than noncarriers among initially nondemented adults (82). Non–ε4 carriers are also more likely to benefit from cognitive training (83) and are more likely to revert from mild cognitive impairment (MCI) to cognitively unimpaired (84). A dose effect of ε4 has also been reported, with more severe impairment and faster progression generally noted among ε4 homozygotes (85, 86). Alternatively, possession of an ε2 allele is associated with slower decline in episodic memory and executive function in nondemented older adults (82). Despite these reports, the conclusion that APOE is associated with cognitive performance in older adults independent from AD diagnosis is not necessarily justified. Indeed, not all studies support an association between the ε4 allele and cognitive performance and progression in this group, perhaps owing to factors such as sample size or inclusion criteria (87).
APOE interactions.
In addition to potentially independent effects on AD and cognition, APOE appears to interact with many other factors to affect cognitive performance. Interactions with age, sex, and education may all modulate the association between APOE and cognition. For example, cognitively normal female ε4 carriers show steeper declines in hippocampal volumes (88), and older adult female ε4 carriers have faster rates of decline in verbal recall (89). Education has been identified as a potential protective factor against the negative impact of APOE ε4 throughout the life span (90, 91).
The APOE ε4 allele may also compound the effects of other AD risk factors. Worse memory performance has been reported in patients with preclinical AD who have both an ε4 allele and a high Aβ burden (92). The ε4 allele also interacts with cerebrovascular disease to impede Aβ clearance, and it may have independent effects on the development of cerebrovascular disease and related cognitive functions (93). Similarly, the ε4 allele may predispose people to type 2 diabetes and metabolic syndrome, and ε4 interactions with diabetic status have been described with regard to cognitive functions including global cognition, episodic memory, and executive functions (94). Additionally, the negative cognitive impact of TBI may be more severe for combat veterans who are ε4 positive, particularly in the areas of memory and processing speed (95). Importantly, APOE may also interact with other genes (e.g., COMT, BDNF, TOMM40) to influence cognition (96).
SORL1
Despite the reported association between SORL1 variants and both familial and sporadic AD, the relationship between this gene and specific cognitive functions among nondemented adults is unresolved (97, 98); conflicting results in some studies may be related to disparities in study design, sample size, and possibly region, ethnicity, or sex. Among 780 nondemented Chinese participants, carriers of the SORL1 rs1699102 T allele demonstrated poorer episodic (verbal and nonverbal) recall and processing speed than noncarriers (99). Conversely, SORL1 rs1784933 genotypes were not associated with any cognitive measures administered to a sample of Chinese participants who were separated into AD, MCI, and cognitively unimpaired groups. In this study, however, the cognitively unimpaired participants were administered only the Mini Mental State Exam, which is considered an insensitive instrument among nondemented adults (100). In a very large, predominantly Caucasian sample that included participants from the Rotterdam Study and the Erasmus Rucphen Family Study, there was no association between AD or specific cognitive functions and SORL1 SNPs, and a meta-analysis of studies conducted up to that point confirmed this finding, once a study that enrolled Chinese participants was excluded (101).
These differences may be further influenced by sex as a potential mediator and possible confounder in earlier studies. Reynolds et al. (102) reported associations between multiple SORL1 SNPs and cognitive trajectories across spatial memory, episodic memory, and verbal cognitive domains. These associations differed according to sex: Males who were homozygous for the rs2070045 G allele showed initially protective effects across these cognitive domains, while female carriers of this allele showed worse performance overall. Conversely, another study showed that for the same SNP, males who were homozygous for the G and T alleles performed worse on executive function measures than male heterozygotes, while female T allele carriers exhibited worse performance than female homozygous G carriers (103). Yet another study found that initially nondemented males with the rs11218343 risk allele were more susceptible than females to global cognitive decline (104). Thus, the relationship between this gene and factors such as sex and ethnicity need to be further elucidated to provide greater understanding. In addition, an understanding of the role of SORL1 across the life span is currently lacking, with only one small study suggesting no association between SORL1 polymorphisms and cognition during middle age (105).
MAPT
The MAPT H1 haplotype has been associated with MCI and more rapid progression to dementia (106, 107); however, reports vary, and the independent impact of MAPT genotype on cognition in AD remains unclear. Asymptomatic carriers of MAPT or two other FTLD-associated mutations (GRN, C9orf72) show worse executive function, language, and visuospatial construction up to 5 years prior to expected symptom onset (108). Studies of MAPT in other diseases produce less consistent results, however. For example, carriers of the H1c subhaplotype may perform better on tasks of general cognitive function, executive function, and attention among those with PSP (109), and Mata et al. (110) found no association with cognition in Parkinson’s disease. Additional research into the role of the MAPT gene and cognition specific to AD is needed.
TREM2
To date, very little information is available regarding specific cognitive functions and TREM2 variants. Data collected for the Wisconsin Registry for Alzheimer’s Prevention suggested a possible association between TREM2 R47H carrier status and cognitive speed and flexibility, which is not surprising given the association between TREM2 variants and non-AD dementias that are likely to produce impairments in attention and executive function (111). However, these associations failed to meet statistical significance; thus, more research pertaining to the relationship between cognition and TREM2 variants is needed.
Genome-Wide Association Studies (Additional Candidate Genes)
Additional candidate AD risk genes identified through GWAS have been evaluated with regard to cognitive performance and decline in nondemented groups. Several, including loci on BIN1, CLU, CR1, CELF1, and ABCA7, were associated with baseline and/or subsequent decline in episodic learning or memory (112–114). Loci on genes associated with language (BIN1, CLU, CR1, MS4A4E) and with executive function/working memory (ABCA7, CLU, EPHA1, SORL1, INPP5D, FERMT2) have also been reported (112, 113). Further studies are needed to determine which of these genes are predominantly associated with decline across specific cognitive domains.
ANIMAL MODELS
As our understanding of how the underlying genetics of AD influence the pathologic features and clinical presentation has evolved, so has our approach to developing animal models with which to probe the mechanistic underpinnings of AD. The various models can be considered with respect to the methods used in their development, ranging from those that are naturally occurring to those that are induced, either with chemicals or with genetic engineering (Figure 3).
Figure 3.
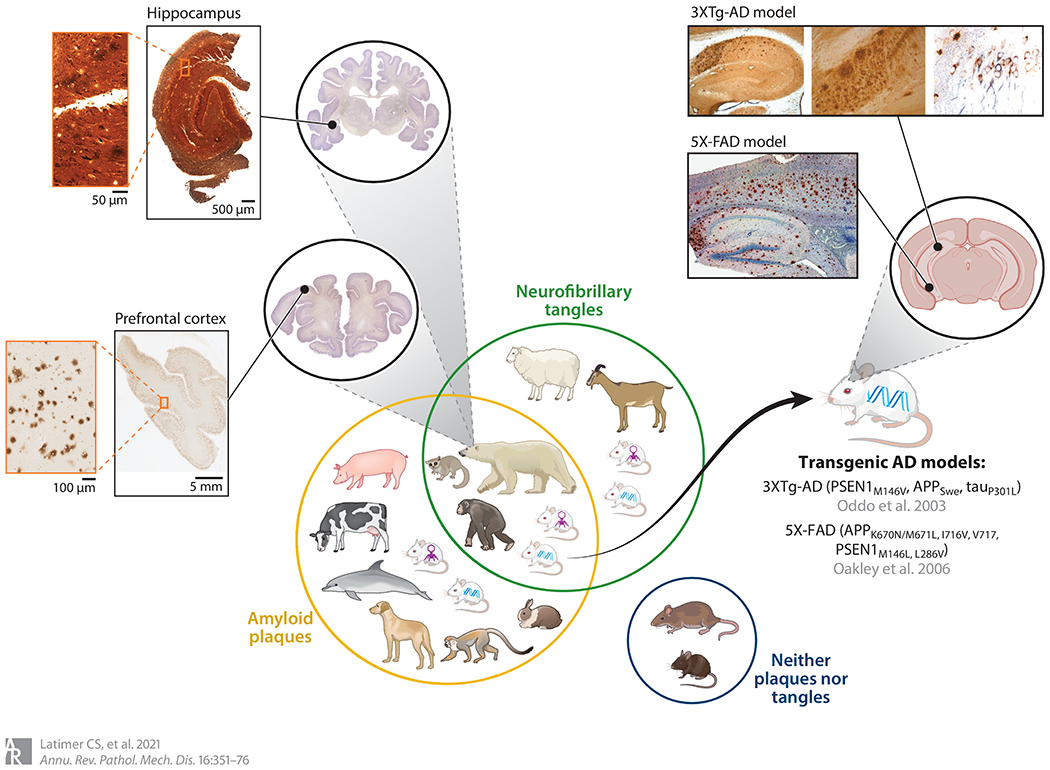
A Venn diagram of animal models of Alzheimer’s disease (AD). The yellow circle encompasses animals that develop amyloid beta (Aβ) plaques; the green circle encompasses animals that develop neurofibrillary tangles. Animals that, like humans, can develop both are shown in the overlap of the green and yellow circles. Rats and mice do not naturally develop either plaques or tangles, and thus are in their own circle. Adeno-associated vector (AAV) mice (symbolized by purple virus) and transgenic mice (symbolized by blue DNA strand) can develop both plaques and tangles in the same brain regions as in human AD brain, including hippocampus. The hippocampal pathology seen in these models is illustrated in the zoomed-in images of mouse brain sections. Images of the hippocampus from the 3XTg-AD model show that the transgenic-induced neurofibrillary tangles and Aβ plaques, as in human brain, are highlighted by silver stain (left, center), and neurofibrillary tangles are also highlighted by immunostains to hyperphosphorylated tau (right) (image adapted with permission from Reference 115; copyright 2003 Cell Press). The amyloid plaques in these models can also be highlighted by an anti-Aβ immunostain, shown in the zoomed-in images of mouse hippocampal section from the 5X-FAD model (image adapted from Reference 116; copyright 2006 Society for Neuroscience). Similarly, the zoomed-in images of the polar bear brain show that the naturally occurring tangles, highlighted in the hippocampus with a silver stain (top), and plaques in prefrontal cortex, highlighted by an anti-Aβ immunostain (bottom), found in aged polar bears replicate the pattern of pathology seen in human AD brain (polar bear brain images reproduced with permission from http://brainmuseum.org; specimens used for this publication are from the Department of Health Affairs, Neuroanatomical Collections Division, of the National Museum of Health and Medicine, the University of Wisconsin, and the Michigan State Comparative Mammalian Brain Collections, supported by the US National Science Foundation).
Naturally Occurring Models
Animal models of human diseases possess inestimable value as a source of understanding causation, pathogenesis, and therapeutic responsiveness. Approximately 70 years after the first diagnosis of AD, the quest for an animal model of the disease began, and in 1970 neuritic plaques were discovered in the cortex of domesticated dogs (117). Neuritic plaques were also identified in the cortex of aged rhesus monkeys (118). Gradually, investigators added squirrel monkeys, orangutans, polar bears, black bears, rabbits, cows, pigs, and guinea pigs to the list of animals whose brains contain Aβ plaques (117). Mice and rats do not develop Aβ plaques, even though Johnstone et al. (119) found that the sequence of mouse and rat cDNA was approximately 90% homologous to that of humans. The other major component of ADNC, neurofibrillary degeneration, seems quite rare in other species but has been found in aged bears, sheep, and mouse lemurs (120–122).
Animals have also been used to model the cognitive changes of AD. Ruehl et al. (123) found that dogs not only experience AD-like neuropathologic change within the same brain regions as humans but also develop cognitive impairment, making elderly dogs a spontaneously occurring model (124). Behavioral impairments in aged monkeys also resemble those in aged humans and positively correlate with number of neuritic plaques (125). Latimer et al. (126) investigated sporadic age-associated brain Aβ deposition and AD disease pathologic changes in African green monkeys. Aβ plaques were found in the cortex in all of the aged vervets, equating to Thal phase 1 in humans. The vervets did not present with true tau tangle pathology; instead, the immunoreactive lesions were localized to small cells with granular cytoplasmic immunoreactivity. With advancing age, the spontaneous development of these pathologic changes in the monkeys were remarkably similar to those observed in human patients with AD (126).
Transgenic Models
Numerous transgenic mouse models have been developed with the hope of better replicating the symptoms and pathologic changes of AD (127). Using a vector to insert the human APP gene with the V717F (Indiana) mutation, Games et al. (128) produced the first mouse model that developed Aβ aggregates in cortex and hippocampus with age. These mice also demonstrated cognitive impairments and behavioral abnormalities. Hsiao et al. (129) made another transgenic mouse that overexpresses APP by inserting the Swedish double mutation of APP695 (K670N and M671L). This mouse develops plaque-like deposits of Aβ40 and Aβ42/43 in the frontal, temporal, and entorhinal cortices; hippocampus; presubiculum; subiculum; and cerebellum. Sturchler-Pierrat et al. (130) made one of the first transgenic APP models that had multiple familial AD mutations; this was also the first model to display neuron loss. Several other models using APP mutations to drive expression of human APP followed, including the TgCRND8 mouse (131) and the double-mutant (Swedish and Indiana) APP mouse (132), each demonstrating similar pathologic and behavioral features. These models were soon followed by others that involved the PSEN1 and PSEN2 genes. The first PSEN1 null mice presented with skeletal and cerebral abnormalities and died shortly after birth (133). In contrast, transgenic mice overexpressing human PSEN1 have relatively few distinctive phenotypes and no plaque-like accumulations (127). PSEN2 knockout mice have a far less severe phenotype than PSEN1 knockouts but also do not develop plaques (134). In 1998, the first multiple transgenic AD mouse model was created by combining the APP Swedish double mutation and the PSEN1 (M146L) mutation (the APP+PS1 mouse). In these mice, Aβ40 and Aβ42/43 levels are up to fivefold higher than in the singly transgenic APP Swedish double-mutant mice (135).
The next AD gene pursued for transgenic models was BACE1 (β-secretase 1), and the first knockout mouse was found to be outwardly healthy with no apparent differences from control mice (136). Overexpression of human BACE1, resulting in levels of BACE1 protein that were 4- to 10-fold higher than those of endogenous mouse BACE, generated a model that showed subtle behavioral changes (137). BACE1 knockout mice were also crossed with mice harboring the transgenic APP Swedish double mutation to create another multitransgenic mouse. However, these BACE1 knockout/APP transgenic mice did not develop Aβ plaques or have β-site amyloid cleavage products (127). Along with β-secretase, APP is cleaved by ⍺-secretase to produce Aβ fragments. To investigate how altering this gene would affect AD pathologic features, Moechars et al. (138) generated a transgenic mouse that expressed a double-mutant form of human APP and a disturbed ⍺-secretase cleavage site. These mice had increased expression of APP in their brains and more β-site cleaved amyloid peptides, along with reactive gliosis in the amygdala, cortex, and hippocampus and abnormal neurons in the dentate gyrus.
The first transgenic model to develop both amyloid plaques and NFTs was the 3XTg-AD mouse, which contain the PSEN1M146V, APPSwe, and tauP301L transgenes (115). These mice progressively develop plaques followed by tangles in AD-relevant brain regions. Cognitively, the mice begin to show alterations in retention at 6 months of age (139). When tested with the Morris water maze, 6-month-old 3XTg-AD mice required 6 days of training to reach an escape latency of less than 20 s, whereas at 2 months of age they required only 3 days of training. Oddo et al. (140) tried to improve the cognitive function of 3XTg-AD mice by actively or passively immunizing them against Aβ42. Upon immunization against Aβ42, aged 3XTg-AD mice were exposed to two behavioral tests; the immunized mice showed significant improvements in behavioral dysfunction compared with the unimmunized controls. These studies encapsulate both the hope that model systems will help uncover disease modifying therapies for AD and the challenging reality of translating those efforts to humans, as the treatments have ultimately failed in human trials due to unacceptable toxicity.
Investigators have developed several transgenic mouse models with mutations that cause plaques to develop gradually across much of the normal mouse life span, but in 2006, Oakley et al. (116) generated a mouse model that coexpresses five causative AD mutations, resulting in very high cerebral Aβ42 deposition starting at age 1.5 months. This model, referred to as 5XFAD, was designed using transgenic human APP with three mutations (K670N/M671L, I716V, V717I) and human PSEN1 with two mutations (M146L and L286V). Behavioral studies show that at 2 months of age the 5XFAD mice had similar spontaneous alternation in the Y maze as age-matched nontransgenic littermates, but by 4 to 5 months of age, the 5XFAD mice had become significantly impaired, demonstrating that these mice develop memory deficits with age.
The 5XFAD mouse model has allowed researchers to investigate different aspects of known AD pathology in a model that mimics some of pathology observed in humans. Ohno et al. (141) were able to reverse the pathologic changes in the 5XFAD mouse brains by crossing them with a BACE1−/− mouse. An immunoblot analysis showed that BACE1 levels were significantly increased in 5XFAD mice compared with the 5XFAD/BACE1−/− mice, which did not have any BACE1 protein. Immunostaining for Aβ42 showed that while the 5XFAD mice had a large quantity of Aβ deposition in the cerebral cortex and subiculum at 18 months of age, this was completely abolished in all brain regions of the 5XFAD/BACE1−/− mice, making them indistinguishable from wild-type control mice. To investigate how the APOE genotype affects Aβ plaques, Youmans et al. (142) crossed 5XFAD mice with mice containing human APOE ɛ2, ɛ3, or ɛ4, but immunostaining for Aβ showed that the overall regional pattern of accumulation was similar, with no changes due to APOE genotype. Overall, manipulation of genes known to cause familial AD, along with prominent risk genes like APOE, has created numerous transgenic models, each of which provides insight into the molecular mechanisms underlying the pathological and clinical progression of AD (127, 143). These efforts are continuing with incorporation of more AD risk variants in increasingly complex combinations in an attempt to improve on existing AD models.
Adeno-Associated Virus Models
The addition of adeno-associated virus (AAV) models has enabled investigations of combinational testing of several genes or variants in parallel. AAVs infect cells via a universal cellular receptor, and their molecular interactions differ in a serotype-dependent manner. Rosenberg et al. (144) found that AAVrh.10 was the best serotype for widespread central nervous system expression of APOE2 in African green monkeys, while Klein et al. (145) found that serotypes 9 and 10 worked best when inducing robust loss of tyrosine hydroxylase immunoreactive cells in the substantia nigra of rats.
AAVs have been used to induce AD-like symptoms or pathologic changes, as well as to evaluate potential interventions. Klein et al. (145) injected rats with AAV tau vectors to induce dopaminergic cell loss and found that, when injecting different serotypes of AAV tau into the substantia nigra, they could model graded disease states with respect to early expression levels and severity of striatal dopamine loss. Transgenic AD mouse lines have also been injected with various AAVs in an attempt to reproduce different aspects of AD. For example, in order to induce tau pathology, Ubhi et al. (146) bilaterally injected transgenic APP mice with an AAV that had a mutated tau cassette (AAV2-mut Tau) in the hippocampus. Within 3 months of the AAV2-mut Tau injection, the mice developed localized increased accumulation of tau and associated neurodegeneration. Chu et al. (147) used an Tg2576 AD mouse model carrying the Swedish double mutation and bilaterally injected AAV2/1 containing a 5-lipoxygenase cassette into the third ventricle. These mice are an AD-like amyloidosis model, and following the AAV injection they developed worsening behavioral deficits along with a significant increase in Aβ. Drummond et al. (148) injected AAV2 with various cassettes (Aβ mutant or mutant APP) into the hippocampus of mice; they found no accumulation of Aβ but instead saw increased microgliosis and altered permeability of the blood–brain barrier. Numerous other models of AD with concomitant AAV injection showing similar pathologic changes have been reported (149, 150).
In addition to using AAVs to induce disease, investigators have extensively evaluated them as a treatment strategy (gene therapy) in multiple diseases, including AD. Zhao et al. (151) gave intrahippocampal injections of an AAV with overexpression of APOE2 to two AD mouse models, APOE knockout mice and a mouse model of brain amyloidosis (PDAPP mice) (128) with the Indiana mutation, and found that soluble and insoluble Aβ in their brain, as well as the overall Aβ burden, was reduced. Wang et al. (152) used an AAV with a neurotrophin receptor to reduce brain Aβ burden in APP/PSEN1 transgenic mice. Through intramuscular delivery of this AAV, they significantly improved the behavioral phenotype of the mice, reduced the brain Aβ burden, and attenuated tau hyperphosphorylation and neuroinflammation. Yang et al. (153) also decreased soluble Aβ levels in APP/PSEN1 mice by using AAV5 with interleukin (IL)-17A. Overexpression of IL-17A improved glucose metabolism, decreased soluble Aβ levels in the hippocampus and cerebrospinal fluid, and reduced CAA. When He et al. (154) injected APP/PSEN1 transgenic mice with an AAV that contained a Cdk5 inhibitory peptide cassette, brain atrophy and memory loss were prevented. These models as well as others demonstrate the therapeutic potential of AAVs (149, 150).
CONCLUSIONS
Since Alois Alzheimer’s first description of AD more than 100 years ago, there has been substantial progress in our understanding of the underlying pathophysiologic processes, pathologic changes, and clinical phenotypes of this pervasive and devastating neurodegenerative disease. Much of this advancement has been fueled and enhanced by the elucidation of the relevant genetic pathways (Figure 4). Importantly, although amyloid processing is still recognized as a major component of the disease, this research has exposed a broader network of the relevant pathways involved in disease initiation and progression that may represent novel avenues for therapeutic target development.
Figure 4.
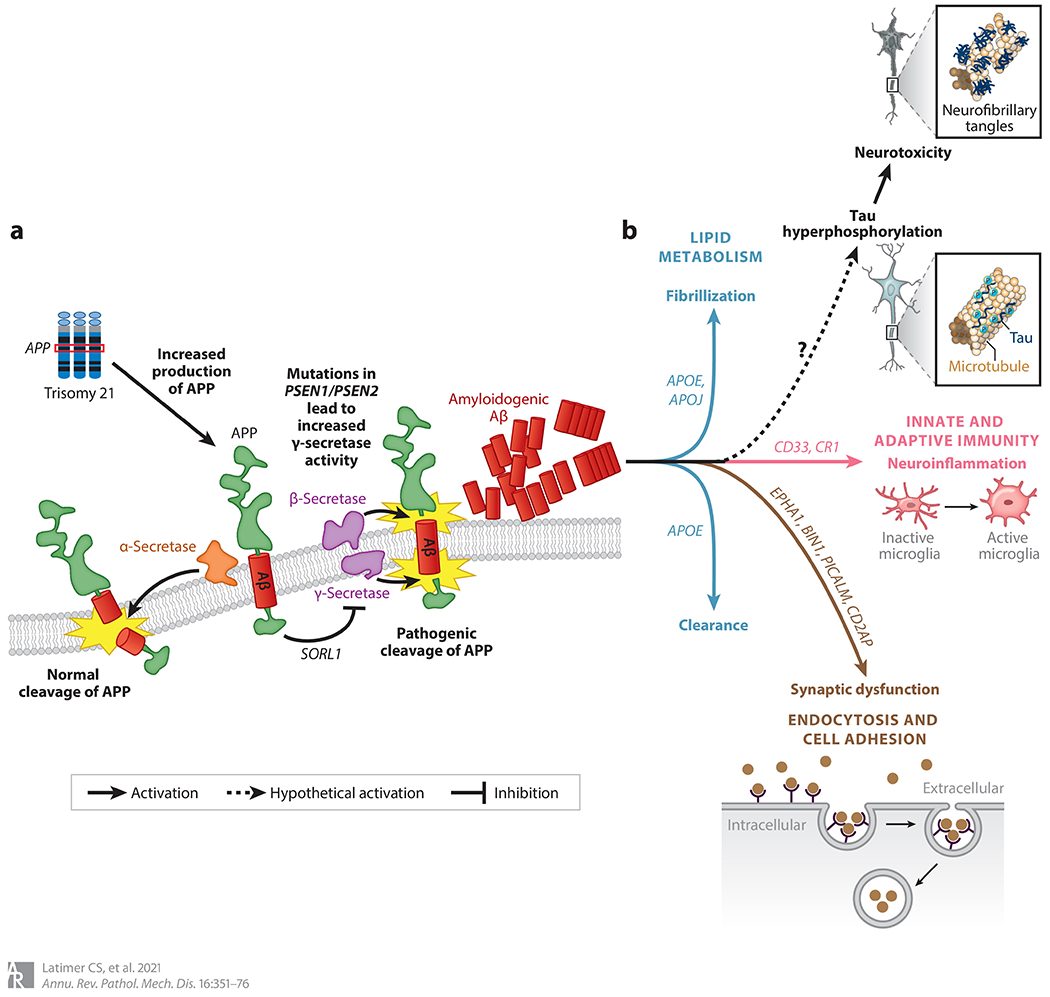
Genetics and pathophysiology of Alzheimer’s disease (AD). (a) Amyloid processing and misprocessing. Amyloid precursor protein (APP) (green) is a transmembrane protein encoded by the APP gene; it is located on chromosome 21 and is therefore overexpressed in trisomy 21. Once at the plasma membrane, APP can be either physiologically cleaved by α-secretase (orange) or pathologically cleaved by β- and γ-secretases (purple) to generate amyloidogenic amyloid beta (Aβ) (red). This process is also influenced by the activity of SORL1, which sequesters APP in the Golgi network. The importance of these processing steps was initially revealed by genetic studies of AD, and they are still recognized as a major component of the disease (panel adapted with permission from Reference 155 copyright 2008 Canadian Medical Association). (b) An expanded network of AD-relevant genes that are implicated in disease initiation and progression and fall into the broad categories of lipid metabolism (blue line), innate and adaptive immunity (pink line), and cell adhesion and endocytosis (brown line). Although the genetics underlying the tau pathology of AD remain somewhat elusive, there appears to be a link between pathologic amyloid protein and tau pathology that is as yet poorly understood (dashed line with question mark).
Our increasing ability to interrogate the genome is a direct result of advances in our technological tools and analytical methods over the past few decades. We can now evaluate thousands of genes through GWASs, leading to an increase in the discovery of risk genes for AD that alone may not impart a substantial risk but may highlight possible molecular mechanisms underlying AD pathophysiology. For example, there are currently approximately 25 known risk genes in addition to APOE. These genes fall into broad categories, and while the exact role for each individual gene in the pathogenesis of AD remains to be determined, the broad categories point to mechanistic themes that include lipid metabolism, innate and adaptive immunity, cell adhesion, and endocytosis.
With the multitude of pathophysiologic pathways implicated by genetic risk thus far, there are already diverse potential therapeutic targets to pursue. As we continue to learn more about the underlying genetics of AD, we should be able to better predict an individual’s risk for developing AD, their likely AD phenotype, and how their disease will progress. Ultimately, as genetics have helped shape clinical management for other diseases, particularly cancer, the genetics of AD will hopefully inform a precision medicine approach wherein preventive and therapeutic strategies are tailored to the individual to maximize clinical impact.
ACKNOWLEDGMENTS
The writing of this review was supported by grants from the National Institutes of Health: AG047366, NS062684, AG57707, and AG053983 (to T.M.); AG005136 (to C.D.K.); and AG065426 (to C.S.L.). We thank Dr. Kathleen Montine for her editorial assistance.
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Alzheimer A 1907. Über eine eigenartige Erkrankung der Hirnrinde. Allg. Z. Psychiatr. Psych.-Gerichtl. Med 64:146–48 [Google Scholar]
- 2.Maurer K, Volk S, Gerbaldo H. 1997. Auguste D and Alzheimer’s disease. Lancet 349:1546–49 [DOI] [PubMed] [Google Scholar]
- 3.Montine TJ, Phelps CH, Beach TG, Bigio EH, Cairns NJ, et al. 2012. National Institute on Aging–Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta Neuropathol. 123:1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roth M 1955. The natural history of mental disorder in old age. J. Ment. Sci 101:281–301 [DOI] [PubMed] [Google Scholar]
- 5.Goate A, Chartier-Harlin MC, Mullan M, Brown J, Crawford F, et al. 1991. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature 349:704–6 [DOI] [PubMed] [Google Scholar]
- 6.Chartier-Harlin MC, Crawford F, Houlden H, Warren A, Hughes D, et al. 1991. Early-onset Alzheimer’s disease caused by mutations at codon 717 of the beta-amyloid precursor protein gene. Nature 353:844–46 [DOI] [PubMed] [Google Scholar]
- 7.Murrell J, Farlow M, Ghetti B, Benson MD. 1991. A mutation in the amyloid precursor protein associated with hereditary Alzheimer’s disease. Science 254:97–99 [DOI] [PubMed] [Google Scholar]
- 8.Mullan M, Tsuji S, Miki T, Katsuya T, Naruse S, et al. 1993. Clinical comparison of Alzheimer’s disease in pedigrees with the codon 717 Val→Ile mutation in the amyloid precursor protein gene. Neurobiol. Aging 14:407–19 [DOI] [PubMed] [Google Scholar]
- 9.Hardy J 2017. The discovery of Alzheimer-causing mutations in the APP gene and the formulation of the “amyloid cascade hypothesis.” FEBS J. 284:1040–44 [DOI] [PubMed] [Google Scholar]
- 10.Sherrington R, Rogaev EI, Liang Y, Rogaeva EA, Levesque G, et al. 1995. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature 375:754–60 [DOI] [PubMed] [Google Scholar]
- 11.Campion D, Flaman JM, Brice A, Hannequin D, Dubois B, et al. 1995. Mutations of the presenilin I gene in families with early-onset Alzheimer’s disease. Hum. Mol. Genet 4:2373–77 [DOI] [PubMed] [Google Scholar]
- 12.Cacace R, Sleegers K, Van Broeckhoven C. 2016. Molecular genetics of early-onset Alzheimer’s disease revisited. Alzheimer’s Dement. 12:733–48 [DOI] [PubMed] [Google Scholar]
- 13.Levy-Lahad E, Wasco W, Poorkaj P, Romano DM, Oshima J, et al. 1995. Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science 269:973–77 [DOI] [PubMed] [Google Scholar]
- 14.Rogaev EI, Sherrington R, Rogaeva EA, Levesque G, Ikeda M, et al. 1995. Familial Alzheimer’s disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer’s disease type 3 gene. Nature 376:775–78 [DOI] [PubMed] [Google Scholar]
- 15.Finckh U, Müller-Thomsen T, Mann U, Eggers C, Marksteiner J, et al. 2000. High prevalence of pathogenic mutations in patients with early-onset dementia detected by sequence analyses of four different genes. Am. J. Hum. Genet 66:110–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pericak-Vance MA, Bebout JL, Gaskell PC Jr., Yamaoka LH, Hung WY, et al. 1991. Linkage studies in familial Alzheimer disease: evidence for chromosome 19 linkage. Am. J. Hum. Genet 48:1034–50 [PMC free article] [PubMed] [Google Scholar]
- 17.Namba Y, Tomonaga M, Kawasaki H, Otomo E, Ikeda K. 1991. Apolipoprotein E immunoreactivity in cerebral amyloid deposits and neurofibrillary tangles in Alzheimer’s disease and kuru plaque amyloid in Creutzfeldt-Jakob disease. Brain Res. 541:163–66 [DOI] [PubMed] [Google Scholar]
- 18.Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, Enghild J, et al. 1993. Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. PNAS 90:1977–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, et al. 1993. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261:921–23 [DOI] [PubMed] [Google Scholar]
- 20.Arboleda-Velasquez JF, Lopera F, O’Hare M, Delgado-Tirado S, Marino C, et al. 2019. Resistance to autosomal dominant Alzheimer’s disease in an APOE3 Christchurch homozygote: a case report. Nat. Med 25:1680–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Scherzer CR, Offe K, Gearing M, Rees HD, Fang G, et al. 2004. Loss of apolipoprotein E receptor LR11 in Alzheimer disease. Arch. Neurol 61:1200–5 [DOI] [PubMed] [Google Scholar]
- 22.Rogaeva E, Meng Y, Lee JH, Gu Y, Kawarai T, et al. 2007. The neuronal sortilin-related receptor SORL1 is genetically associated with Alzheimer disease. Nat. Genet 39:168–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee JH, Cheng R, Honig LS, Vonsattel JP, Clark L, Mayeux R. 2008. Association between genetic variants in SORL1 and autopsy-confirmed Alzheimer disease. Neurology 70:887–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee JH, Cheng R, Schupf N, Manly J, Lantigua R, et al. 2007. The association between genetic variants in SORL1 and Alzheimer disease in an urban, multiethnic, community-based cohort. Arch. Neurol 64:501–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Strang KH, Golde TE, Giasson BI. 2019. MAPT mutations, tauopathy, and mechanisms of neurodegeneration. Lab. Investig 99:912–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roks G, Dermaut B, Heutink P, Julliams A, Backhovens H, et al. 1999. Mutation screening of the tau gene in patients with early-onset Alzheimer’s disease. Neurosci. Lett 277:137–39 [DOI] [PubMed] [Google Scholar]
- 27.Baker M, Litvan I, Houlden H, Adamson J, Dickson D, et al. 1999. Association of an extended haplotype in the tau gene with progressive supranuclear palsy. Hum. Mol. Genet 8:711–15 [DOI] [PubMed] [Google Scholar]
- 28.Kalinderi K, Fidani L, Bostantjopoulou S. 2009. From 1997 to 2007: a decade journey through the H1 haplotype on 17q21 chromosome. Parkinsonism Relat. Disord 15:2–5 [DOI] [PubMed] [Google Scholar]
- 29.Sun W, Jia J. 2009. The +347 C promoter allele up-regulates MAPT expression and is associated with Alzheimer’s disease among the Chinese Han. Neurosci. Lett 450:340–43 [DOI] [PubMed] [Google Scholar]
- 30.Colonna M 2003. TREMs in the immune system and beyond. Nat. Rev. Immunol 3:445–53 [DOI] [PubMed] [Google Scholar]
- 31.Chouery E, Delague V, Bergougnoux A, Koussa S, Serre JL, Megarbane A. 2008. Mutations in TREM2 lead to pure early-onset dementia without bone cysts. Hum. Mutat 29:E194–204 [DOI] [PubMed] [Google Scholar]
- 32.Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, et al. 2013. TREM2 variants in Alzheimer’s disease. N. Engl. J. Med 368:117–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Carmona S, Zahs K, Wu E, Dakin K, Bras J, Guerreiro R. 2018. The role of TREM2 in Alzheimer’s disease and other neurodegenerative disorders. Lancet Neurol. 17:721–30 [DOI] [PubMed] [Google Scholar]
- 34.Colonna M, Wang Y. 2016. TREM2 variants: new keys to decipher Alzheimer disease pathogenesis. Nat. Rev. Neurosci 17:201–7 [DOI] [PubMed] [Google Scholar]
- 35.Bertram L, McQueen MB, Mullin K, Blacker D, Tanzi RE. 2007. Systematic meta-analyses of Alzheimer disease genetic association studies: the AlzGene database. Nat. Genet 39:17–23 [DOI] [PubMed] [Google Scholar]
- 36.Jun G, Naj AC, Beecham GW, Wang LS, Buros J, et al. 2010. Meta-analysis confirms CR1, CLU, and PICALM as Alzheimer disease risk loci and reveals interactions with APOE genotypes. Arch. Neurol 67:1473–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kunkle BW, Grenier-Boley B, Sims R, Bis JC, Damotte V, et al. 2019. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat. Genet 51:414–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Marioni RE, Harris SE, Zhang Q, McRae AF, Hagenaars SP, et al. 2018. GWAS on family history of Alzheimer’s disease. Transl. Psychiatry 8:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Naj AC, Jun G, Beecham GW, Wang LS, Vardarajan BN, et al. 2011. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat. Genet 43:436–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nelson PT, Alafuzoff I, Bigio EH, Bouras C, Braak H, et al. 2012. Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J. Neuropathol. Exp. Neurol 71:362–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Latimer CS, Burke BT, Liachko NF, Currey HN, Kilgore MD, et al. 2019. Resistance and resilience to Alzheimer’s disease pathology are associated with reduced cortical pTau and absence of limbic-predominant age-related TDP-43 encephalopathy in a community-based cohort. Acta Neuropathol. Commun 7:91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brettschneider J, Del Tredici K, Lee VM-Y, Trojanowski JQ. 2015. Spreading of pathology in neurodegenerative diseases: a focus on human studies. Nat. Rev. Neurosci 16:109–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tomlinson BE, Blessed G, Roth M. 1968. Observations on the brains of non-demented old people. J. Neurol. Sci 7:331–56 [DOI] [PubMed] [Google Scholar]
- 44.Jack CR Jr., Bennett DA, Blennow K, Carrillo MC, Dunn B, et al. 2018. NIA-AA Research Framework: toward a biological definition of Alzheimer’s disease. Alzheimer’s Dement. 14:535–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thal DR, Rub U, Orantes M, Braak H. 2002. Phases of Aβ-deposition in the human brain and its relevance for the development of AD. Neurology 58:1791–800 [DOI] [PubMed] [Google Scholar]
- 46.Zhang YW, Thompson R, Zhang H, Xu H. 2011. APP processing in Alzheimer’s disease. Mol. Brain 4:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hardy JA, Higgins GA. 1992. Alzheimer’s disease: the amyloid cascade hypothesis. Science 256:184–85 [DOI] [PubMed] [Google Scholar]
- 48.Nolan A, Resende EDF, Peterson C, Neylan K, Spina S, et al. 2019. Astrocytic tau deposition is frequent in typical and atypical Alzheimer disease presentations. J. Neuropathol. Exp. Neurol 78:1112–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K. 2006. Staging of Alzheimer disease–associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 112:389–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Braak H, Braak E. 1991. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 82:239–59 [DOI] [PubMed] [Google Scholar]
- 51.Murray ME, Graff-Radford NR, Ross OA, Petersen RC, Duara R, Dickson DW. 2011. Neuropathologically defined subtypes of Alzheimer’s disease with distinct clinical characteristics: a retrospective study. Lancet Neurol. 10:785–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kovacs GG. 2015. Neuropathology of tauopathies: principles and practice. Neuropathol. Appl. Neurobiol 41:3–23 [DOI] [PubMed] [Google Scholar]
- 53.Crary JF, Trojanowski JQ, Schneider JA, Abisambra JF, Abner EL, et al. 2014. Primary age-related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol. 128:755–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kovacs GG, Ferrer I, Grinberg LT, Alafuzoff I, Attems J, et al. 2016. Aging-related tau astrogliopathy (ARTAG): harmonized evaluation strategy. Acta Neuropathol. 131:87–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Buee L, Delacourte A. 1999. Comparative biochemistry of tau in progressive supranuclear palsy, corticobasal degeneration, FTDP-17 and Pick’s disease. Brain Pathol. 9:681–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schellenberg GD, Montine TJ. 2012. The genetics and neuropathology of Alzheimer’s disease. Acta Neuropathol. 124:305–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wolfe MS. 2007. When loss is gain: Reduced presenilin proteolytic function leads to increased Aβ42/Aβ40. EMBO Rep. 8:136–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Halliday G, Brooks W, Arthur H, Creasey H, Broe GA. 1997. Further evidence for an association between a mutation in the APP gene and Lewy body formation. Neurosci. Lett 227:49–52 [DOI] [PubMed] [Google Scholar]
- 59.Leverenz JB, Fishel MA, Peskind ER, Montine TJ, Nochlin D, et al. 2006. Lewy body pathology in familial Alzheimer disease: evidence for disease- and mutation-specific pathologic phenotype. Arch. Neurol 63:370–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jayadev S, Leverenz JB, Steinbart E, Stahl J, Klunk W, et al. 2010. Alzheimer’s disease phenotypes and genotypes associated with mutations in presenilin 2. Brain 133:1143–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nelson PT, Dickson DW, Trojanowski JQ, Jack CR, Boyle PA, et al. 2019. Limbic-predominant age-related TDP-43 encephalopathy (LATE): consensus working group report. Brain 142:1503–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sonnen JA, Larson EB, Crane PK, Haneuse S, Li G, et al. 2007. Pathological correlates of dementia in a longitudinal, population-based sample of aging. Ann. Neurol 62:406–13 [DOI] [PubMed] [Google Scholar]
- 63.Beecham GW, Hamilton K, Naj AC, Martin ER, Huentelman M, et al. 2014. Genome-wide association meta-analysis of neuropathologic features of Alzheimer’s disease and related dementias. PLOS Genet. 10:e1004606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ringman JM, Diaz-Olavarrieta C, Rodriguez Y, Chavez M, Fairbanks L, et al. 2005. Neuropsychological function in nondemented carriers of presenilin-1 mutations. Neurology 65:552–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Storandt M, Balota DA, Aschenbrenner AJ, Morris JC. 2014. Clinical and psychological characteristics of the initial cohort of the Dominantly Inherited Alzheimer Network (DIAN). Neuropsychology 28:19–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tang M, Ryman DC, McDade E, Jasielec MS, Buckles VD, et al. 2016. Neurological manifestations of autosomal dominant familial Alzheimer’s disease: a comparison of the published literature with the Dominantly Inherited Alzheimer Network observational study (DIAN-OBS). Lancet Neurol. 15:1317–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bateman RJ, Xiong C, Benzinger TL, Fagan AM, Goate A, et al. 2012. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N. Engl. J. Med 367:795–804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dickerson BC, Wolk DA (Alzheimer’s Dis. Neuroimag. Initiat.). 2011. Dysexecutive versus amnesic phenotypes of very mild Alzheimer’s disease are associated with distinct clinical, genetic and cortical thinning characteristics. J. Neurol. Neurosurg. Psychiatry 82:45–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Saeed U, Mirza SS, MacIntosh BJ, Herrmann N, Keith J, et al. 2018. APOE-ε4 associates with hippocampal volume, learning, and memory across the spectrum of Alzheimer’s disease and dementia with Lewy bodies. Alzheimer’s Dement. 14:1137–47 [DOI] [PubMed] [Google Scholar]
- 70.Wolk DA, Dickerson BC (Alzheimer’s Dis. Neuroimag. Initiat.). 2010. Apolipoprotein E (APOE) genotype has dissociable effects on memory and attentional-executive network function in Alzheimer’s disease. PNAS 107:10256–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hoyt BD, Massman PJ, Schatschneider C, Cooke N, Doody RS. 2005. Individual growth curve analysis of APOEε4-associated cognitive decline in Alzheimer disease. Arch. Neurol 62:454–59 [DOI] [PubMed] [Google Scholar]
- 72.Stern Y, Brandt J, Albert M, Jacobs DM, Liu X, et al. 1997. The absence of an apolipoprotein ε4 allele is associated with a more aggressive form of Alzheimer’s disease. Ann. Neurol 41:615–20 [DOI] [PubMed] [Google Scholar]
- 73.Cosentino S, Scarmeas N, Helzner E, Glymour MM, Brandt J, et al. 2008. APOEε4 allele predicts faster cognitive decline in mild Alzheimer disease. Neurology 70:1842–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Allan CL, Ebmeier KP. 2011. The influence of ApoE4 on clinical progression of dementia: a meta-analysis. Int. J. Geriatr. Psychiatry 26:520–26 [DOI] [PubMed] [Google Scholar]
- 75.Martins CA, Oulhaj A, de Jager CA, Williams JH. 2005. APOE alleles predict the rate of cognitive decline in Alzheimer disease: a nonlinear model. Neurology 65:1888–93 [DOI] [PubMed] [Google Scholar]
- 76.Han SD, Bondi MW. 2008. Revision of the apolipoprotein E compensatory mechanism recruitment hypothesis. Alzheimer’s Dement. 4:251–54 [DOI] [PubMed] [Google Scholar]
- 77.Weissberger GH, Nation DA, Nguyen CP, Bondi MW, Han SD. 2018. Meta-analysis of cognitive ability differences by apolipoprotein E genotype in young humans. Neurosci. Biobehav. Rev 94:49–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kassam I, Gagnon F, Cusimano MD. 2016. Association of the APOE-ε4 allele with outcome of traumatic brain injury in children and youth: a meta-analysis and meta-regression. J. Neurol. Neurosurg. Psychiatry 87:433–40 [DOI] [PubMed] [Google Scholar]
- 79.Li X, Hildebrandt A, Sommer W, Wilhelm O, Reuter M, et al. 2019. Cognitive performance in young APOE ε4 carriers: a latent variable approach for assessing the genotype–phenotype relationship. Behav. Genet 49:455–68 [DOI] [PubMed] [Google Scholar]
- 80.Bussy A, Snider BJ, Coble D, Xiong C, Fagan AM, et al. 2019. Effect of apolipoprotein E4 on clinical, neuroimaging, and biomarker measures in noncarrier participants in the Dominantly Inherited Alzheimer Network. Neurobiol. Aging 75:42–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Flory JD, Manuck SB, Ferrell RE, Ryan CM, Muldoon MF. 2000. Memory performance and the apolipoprotein E polymorphism in a community sample of middle-aged adults. Am. J. Med. Genet 96:707–11 [DOI] [PubMed] [Google Scholar]
- 82.O’Donoghue MC, Murphy SE, Zamboni G, Nobre AC, Mackay CE. 2018. APOE genotype and cognition in healthy individuals at risk of Alzheimer’s disease: a review. Cortex 104:103–23 [DOI] [PubMed] [Google Scholar]
- 83.López-Higes R, Rodríguez-Rojo IC, Prados JM, Montejo P, Del-Río D, et al. 2017. APOE ε4 modulation of training outcomes in several cognitive domains in a sample of cognitively intact older adults. J. Alzheimer’s Dis 58:1201–15 [DOI] [PubMed] [Google Scholar]
- 84.Kelly DA, Seidenberg M, Reiter K, Nielson KA, Woodard JL, et al. 2018. Differential 5-year brain atrophy rates in cognitively declining and stable APOE-ε4 elders. Neuropsychology 32:647–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Caselli RJ, Reiman EM, Osborne D, Hentz JG, Baxter LC, et al. 2004. Longitudinal changes in cognition and behavior in asymptomatic carriers of the APOE e4 allele. Neurology 62:1990–95 [DOI] [PubMed] [Google Scholar]
- 86.Yaffe K, Cauley J, Sands L, Browner W. 1997. Apolipoprotein E phenotype and cognitive decline in a prospective study of elderly community women. Arch. Neurol 54:1110–14 [DOI] [PubMed] [Google Scholar]
- 87.Lange KL, Bondi MW, Salmon DP, Galasko D, Delis DC, et al. 2002. Decline in verbal memory during preclinical Alzheimer’s disease: examination of the effect of APOE genotype. J. Int. Neuropsychol. Soc 8:943–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Shen S, Zhou W, Chen X, Zhang J (Alzheimer’s Dis. Neuroimag. Initiat.). 2019. Sex differences in the association of APOE ε4 genotype with longitudinal hippocampal atrophy in cognitively normal older people. Eur. J. Neurol 26:1362–69 [DOI] [PubMed] [Google Scholar]
- 89.Wang X, Zhou W, Ye T, Lin X, Zhang J (Alzheimer’s Dis. Neuroimag. Initiat.). 2019. Sex difference in the association of APOE4 with memory decline in mild cognitive impairment. J. Alzheimer’s Dis 69:1161–69 [DOI] [PubMed] [Google Scholar]
- 90.Ishioka YL, Gondo Y, Fuku N, Inagaki H, Masui Y, et al. 2016. Effects of the APOE ε4 allele and education on cognitive function in Japanese centenarians. Age 38:495–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wang HX, Gustafson DR, Kivipelto M, Pedersen NL, Skoog I, et al. 2012. Education halves the risk of dementia due to apolipoprotein ε4 allele: a collaborative study from the Swedish Brain Power Initiative. Neurobiol. Aging 33:1007.e1–7 [DOI] [PubMed] [Google Scholar]
- 92.Kantarci K, Lowe V, Przybelski SA, Weigand SD, Senjem ML, et al. 2012. APOE modifies the association between Aβ load and cognition in cognitively normal older adults. Neurology 78:232–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tai LM, Thomas R, Marottoli FM, Koster KP, Kanekiyo T, et al. 2016. The role of APOE in cerebrovascular dysfunction. Acta Neuropathol. 131:709–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zhen J, Lin T, Huang X, Zhang H, Dong S, et al. 2018. Association of ApoE genetic polymorphism and type 2 diabetes with cognition in non-demented aging Chinese adults: a community based cross-sectional study. Aging Dis. 9:346–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Merritt VC, Clark AL, Sorg SF, Evangelista ND, Werhane ML, et al. 2018. Apolipoprotein E (APOE) ε4 genotype is associated with reduced neuropsychological performance in military veterans with a history of mild traumatic brain injury. J. Clin. Exp. Neuropsychol 40:1050–61 [DOI] [PubMed] [Google Scholar]
- 96.Fan J, Tao W, Li X, Li H, Zhang J, et al. 2019. The contribution of genetic factors to cognitive impairment and dementia: apolipoprotein E gene, gene interactions, and polygenic risk. Int. J. Mol. Sci 20:1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Houlihan LM, Harris SE, Luciano M, Gow AJ, Starr JM, et al. 2009. Replication study of candidate genes for cognitive abilities: the Lothian Birth Cohort 1936. Genes Brain Behav. 8:238–47 [DOI] [PubMed] [Google Scholar]
- 98.Seshadri S, DeStefano AL, Au R, Massaro JM, Beiser AS, et al. 2007. Genetic correlates of brain aging on MRI and cognitive test measures: a genome-wide association and linkage analysis in the Framingham Study. BMC Med. Genet 8(Suppl. 1):S15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Li H, Lv C, Yang C, Wei D, Chen K, et al. 2017. SORL1 rs1699102 polymorphism modulates age-related cognitive decline and gray matter volume reduction in non-demented individuals. Eur. J. Neurol 24:187–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Chou CT, Liao YC, Lee WJ, Wang SJ, Fuh JL. 2016. SORL1 gene, plasma biomarkers, and the risk of Alzheimer’s disease for the Han Chinese population in Taiwan. Alzheimer’s Res. Ther 8:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Liu F, Ikram MA, Janssens AC, Schuur M, de Koning I, et al. 2009. A study of the SORL1 gene in Alzheimer’s disease and cognitive function. J. Alzheimer’s Dis 18:51–64 [DOI] [PubMed] [Google Scholar]
- 102.Reynolds CA, Zavala C, Gatz M, Vie L, Johansson B, et al. 2013. Sortilin receptor 1 predicts longitudinal cognitive change. Neurobiol. Aging 34:1710.e11–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Liang Y, Li H, Lv C, Shu N, Chen K, et al. 2015. Sex moderates the effects of the Sorl1 gene rs2070045 polymorphism on cognitive impairment and disruption of the cingulum integrity in healthy elderly. Neuropsychopharmacology 40:1519–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Nettiksimmons J, Tranah G, Evans DS, Yokoyama JS, Yaffe K. 2016. Gene-based aggregate SNP associations between candidate AD genes and cognitive decline. Age 38:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Korthauer LE, Awe E, Frahmand M, Driscoll I. 2018. Genetic risk for age-related cognitive impairment does not predict cognitive performance in middle age. J. Alzheimer’s Dis 64:459–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Di Maria E, Cammarata S, Parodi MI, Borghi R, Benussi L, et al. 2010. The H1 haplotype of the tau gene (MAPT) is associated with mild cognitive impairment. J. Alzheimer’s Dis 19:909–14 [DOI] [PubMed] [Google Scholar]
- 107.Samaranch L, Cervantes S, Barabash A, Alonso A, Cabranes JA, et al. 2010. The effect of MAPT H1 and APOE ε4 on transition from mild cognitive impairment to dementia. J. Alzheimer’s Dis 22:1065–71 [DOI] [PubMed] [Google Scholar]
- 108.Staffaroni AM, Bajorek L, Casaletto KB, Cobigo Y, Goh SM, et al. 2019. Assessment of executive function declines in presymptomatic and mildly symptomatic familial frontotemporal dementia: NIH-EXAMINER as a potential clinical trial endpoint. Alzheimer’s Dement. 16:11–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Gerstenecker A, Roberson ED, Schellenberg GD, Standaert DG, Shprecher DR, et al. 2017. Genetic influences on cognition in progressive supranuclear palsy. Mov. Disord 32:1764–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Mata IF, Leverenz JB, Weintraub D, Trojanowski JQ, Hurtig HI, et al. 2014. APOE, MAPT, and SNCA genes and cognitive performance in Parkinson disease. JAMA Neurol. 71:1405–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Engelman CD, Darst BF, Bilgel M, Vasiljevic E, Koscik RL, et al. 2018. The effect of rare variants in TREM2 and PLD3 on longitudinal cognitive function in the Wisconsin Registry for Alzheimer’s Prevention. Neurobiol. Aging 66:177.e1–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Andrews SJ, Das D, Anstey KJ, Easteal S. 2017. Late onset Alzheimer’s disease risk variants in cognitive decline: the PATH Through Life study. J. Alzheimer’s Dis 57:423–36 [DOI] [PubMed] [Google Scholar]
- 113.Cruz-Sanabria F, Bonilla-Vargas K, Estrada K, Mancera O, Vega E, et al. 2018. Analysis of cognitive performance and polymorphisms of SORL1, PVRL2, CR1, TOMM40, APOE, PICALM, GWAS_14q, CLU, and BIN1 in patients with mild cognitive impairment and cognitively healthy controls. Neurologia. 10.1016/j.nrl.2018.07.002 [DOI] [PubMed] [Google Scholar]
- 114.Franzmeier N, Rubinski A, Neitzel J, Ewers M (Alzheimer’s Dis. Neuroimag. Initiat.). 2019. The BIN1 rs744373 SNP is associated with increased tau-PET levels and impaired memory. Nat. Commun 10:1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, et al. 2003. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Aβ and synaptic dysfunction. Neuron 39:409–21 [DOI] [PubMed] [Google Scholar]
- 116.Oakley H, Cole SL, Logan S, Maus E, Shao P, et al. 2006. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J. Neurosci 26:10129–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Youssef SA, Capucchio MT, Rofina JE, Chambers JK, Uchida K, et al. 2016. Pathology of the aging brain in domestic and laboratory animals, and animal models of human neurodegenerative diseases. Vet. Pathol 53:327–48 [DOI] [PubMed] [Google Scholar]
- 118.Wisniewski HM, Ghetti B, Terry RD. 1973. Neuritic (senile) plaques and filamentous changes in aged rhesus monkeys. J. Neuropathol. Exp. Neurol 32:566–84 [DOI] [PubMed] [Google Scholar]
- 119.Johnstone EM, Chaney MO, Norris FH, Pascual R, Little SP. 1991. Conservation of the sequence of the Alzheimer’s disease amyloid peptide in dog, polar bear and five other mammals by cross-species polymerase chain reaction analysis. Mol. Brain Res 10:299–305 [DOI] [PubMed] [Google Scholar]
- 120.Bons N, Rieger F, Prudhomme D, Fisher A, Krause KH. 2006. Microcebus murinus: a useful primate model for human cerebral aging and Alzheimer’s disease? Genes Brain Behav. 5:120–30 [DOI] [PubMed] [Google Scholar]
- 121.Cork LC, Powers RE, Selkoe DJ, Davies P, Geyer JJ, Price DL. 1988. Neurofibrillary tangles and senile plaques in aged bears. J. Neuropathol. Exp. Neurol 47:629–41 [DOI] [PubMed] [Google Scholar]
- 122.Nelson PT, Greenberg SG, Saper CB. 1994. Neurofibrillary tangles in the cerebral cortex of sheep. Neurosci. Lett 170:187–90 [DOI] [PubMed] [Google Scholar]
- 123.Ruehl WW, Bruyette DS, DePaoli A, Cotman CW, Head E, et al. 1995. Canine cognitive dysfunction as a model for human age-related cognitive decline, dementia and Alzheimer’s disease: clinical presentation, cognitive testing, pathology and response to 1-deprenyl therapy. Prog. Brain Res 106:217–25 [DOI] [PubMed] [Google Scholar]
- 124.Sarasa M, Pesini P. 2009. Natural non-transgenic animal models for research in Alzheimer’s disease. Curr. Alzheimer’s Res 6:171–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Struble RG, Price DL Jr., Cork LC, Price DL. 1985. Senile plaques in cortex of aged normal monkeys. Brain Res. 361:267–75 [DOI] [PubMed] [Google Scholar]
- 126.Latimer CS, Shively CA, Keene CD, Jorgensen MJ, Andrews RN, et al. 2019. A nonhuman primate model of early Alzheimer’s disease pathologic change: implications for disease pathogenesis. Alzheimer’s Dement. 15:93–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Kobayashi DT, Chen KS. 2005. Behavioral phenotypes of amyloid-based genetically modified mouse models of Alzheimer’s disease. Genes Brain Behav. 4:173–96 [DOI] [PubMed] [Google Scholar]
- 128.Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, et al. 1995. Alzheimer-type neuropathology in transgenic mice overexpressing V717F beta-amyloid precursor protein. Nature 373:523–27 [DOI] [PubMed] [Google Scholar]
- 129.Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, et al. 1996. Correlative memory deficits, Aβ elevation, and amyloid plaques in transgenic mice. Science 274:99–102 [DOI] [PubMed] [Google Scholar]
- 130.Sturchler-Pierrat C, Abramowski D, Duke M, Wiederhold KH, Mistl C, et al. 1997. Two amyloid precursor protein transgenic mouse models with Alzheimer disease–like pathology. PNAS 94:13287–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Chishti MA, Yang DS, Janus C, Phinney AL, Horne P, et al. 2001. Early-onset amyloid deposition and cognitive deficits in transgenic mice expressing a double mutant form of amyloid precursor protein 695. J. Biol. Chem 276:21562–70 [DOI] [PubMed] [Google Scholar]
- 132.Hsia AY, Masliah E, McConlogue L, Yu GQ, Tatsuno G, et al. 1999. Plaque-independent disruption of neural circuits in Alzheimer’s disease mouse models. PNAS 96:3228–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Shen J, Bronson RT, Chen DF, Xia W, Selkoe DJ, Tonegawa S. 1997. Skeletal and CNS defects in presenilin-1-deficient mice. Cell 89:629–39 [DOI] [PubMed] [Google Scholar]
- 134.Herreman A, Hartmann D, Annaert W, Saftig P, Craessaerts K, et al. 1999. Presenilin 2 deficiency causes a mild pulmonary phenotype and no changes in amyloid precursor protein processing but enhances the embryonic lethal phenotype of presenilin 1 deficiency. PNAS 96:11872–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Holcomb L, Gordon MN, McGowan E, Yu X, Benkovic S, et al. 1998. Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat. Med 4:97–100 [DOI] [PubMed] [Google Scholar]
- 136.Luo Y, Bolon B, Kahn S, Bennett BD, Babu-Khan S, et al. 2001. Mice deficient in BACE1, the Alzheimer’s beta-secretase, have normal phenotype and abolished beta-amyloid generation. Nat. Neurosci 4:231–32 [DOI] [PubMed] [Google Scholar]
- 137.Harrison SM, Harper AJ, Hawkins J, Duddy G, Grau E, et al. 2003. BACE1 (beta-secretase) transgenic and knockout mice: identification of neurochemical deficits and behavioral changes. Mol. Cell Neurosci 24:646–55 [DOI] [PubMed] [Google Scholar]
- 138.Moechars D, Lorent K, De Strooper B, Dewachter I, Van Leuven F. 1996. Expression in brain of amyloid precursor protein mutated in the alpha-secretase site causes disturbed behavior, neuronal degeneration and premature death in transgenic mice. EMBO J. 15:1265–74 [PMC free article] [PubMed] [Google Scholar]
- 139.Billings LM, Oddo S, Green KN, McGaugh JL, LaFerla FM. 2005. Intraneuronal Aβ causes the onset of early Alzheimer’s disease–related cognitive deficits in transgenic mice. Neuron 45:675–88 [DOI] [PubMed] [Google Scholar]
- 140.Oddo S, Vasilevko V, Caccamo A, Kitazawa M, Cribbs DH, LaFerla FM. 2006. Reduction of soluble Aβ and tau, but not soluble Aβ alone, ameliorates cognitive decline in transgenic mice with plaques and tangles. J. Biol. Chem 281:39413–23 [DOI] [PubMed] [Google Scholar]
- 141.Ohno M, Cole SL, Yasvoina M, Zhao J, Citron M, et al. 2007. BACE1 gene deletion prevents neuron loss and memory deficits in 5XFAD APP/PS1 transgenic mice. Neurobiol. Dis 26:134–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Youmans KL, Tai LM, Nwabuisi-Heath E, Jungbauer L, Kanekiyo T, et al. 2012. APOE4-specific changes in Aβ accumulation in a new transgenic mouse model of Alzheimer disease. J. Biol. Chem 287:41774–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.LaFerla FM, Green KN. 2012. Animal models of Alzheimer disease. Cold Spring Harb. Perspect. Med 2:a006320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Rosenberg JB, Kaplitt MG, De BP, Chen A, Flagiello T, et al. 2018. AAVrh.10-mediated APOE2 central nervous system gene therapy for APOE4-associated Alzheimer’s disease. Hum. Gene Ther. Clin. Dev 29:24–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Klein RL, Dayton RD, Tatom JB, Henderson KM, Henning PP. 2008. AAV8, 9, Rh10, Rh43 vector gene transfer in the rat brain: effects of serotype, promoter and purification method. Mol. Ther 16: 89–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Ubhi K, Rockenstein E, Doppler E, Mante M, Adame A, et al. 2009. Neurofibrillary and neurodegenerative pathology in APP-transgenic mice injected with AAV2-mutant TAU: neuroprotective effects of Cerebrolysin. Acta Neuropathol. 117:699–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Chu J, Giannopoulos PF, Ceballos-Diaz C, Golde TE, Pratico D. 2012. Adeno-associated virus-mediated brain delivery of 5-lipoxygenase modulates the AD-like phenotype of APP mice. Mol. Neurodegener 7:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Drummond ES, Muhling J, Martins RN, Wijaya LK, Ehlert EM, Harvey AR. 2013. Pathology associated with AAV mediated expression of beta amyloid or C100 in adult mouse hippocampus and cerebellum. PLOS ONE 8:e59166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Combs B, Kneynsberg A, Kanaan NM. 2016. Gene therapy models of Alzheimer’s disease and other dementias. Methods Mol. Biol 1382:339–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Ittner LM, Klugmann M, Ke YD. 2019. Adeno-associated virus–based Alzheimer’s disease mouse models and potential new therapeutic avenues. Br. J. Pharmacol 176:3649–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Zhao L, Gottesdiener AJ, Parmar M, Li M, Kaminsky SM, et al. 2016. Intracerebral adeno-associated virus gene delivery of apolipoprotein E2 markedly reduces brain amyloid pathology in Alzheimer’s disease mouse models. Neurobiol. Aging 44:159–72 [DOI] [PubMed] [Google Scholar]
- 152.Wang QH, Wang YR, Zhang T, Jiao SS, Liu YH, et al. 2016. Intramuscular delivery of p75NTR ectodomain by an AAV vector attenuates cognitive deficits and Alzheimer’s disease-like pathologies in APP/PS1 transgenic mice. J. Neurochem 138:163–73 [DOI] [PubMed] [Google Scholar]
- 153.Yang J, Kou J, Lalonde R, Fukuchi KI. 2017. Intracranial IL-17A overexpression decreases cerebral amyloid angiopathy by upregulation of ABCA1 in an animal model of Alzheimer’s disease. Brain Behav. Immun 65:262–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.He Y, Pan S, Xu M, He R, Huang W, et al. 2017. Adeno-associated virus 9–mediated Cdk5 inhibitory peptide reverses pathologic changes and behavioral deficits in the Alzheimer’s disease mouse model. FASEB J. 31:3383–92 [DOI] [PubMed] [Google Scholar]
- 155.Patterson C, Feightner JW, Garcia A, Hsiung G-YR, MacKnight C, Sadovnick AD. 2008. Diagnosis and treatment of dementia. 1. Risk assessment and primary prevention of Alzheimer disease. CMAJ 178:548–56 [DOI] [PMC free article] [PubMed] [Google Scholar]