

Abstract
Objective:
Our objective is to document progress in developing personalized therapy with fluoropyrimidine drugs (FPs) to improve outcomes for cancer patients and to identify areas requiring further investigation.
Background:
FPs including 5-fluorouracil (5-FU), are among the most widely used drugs for treating colorectal cancer (CRC) and other gastrointestinal (GI) malignancies. While FPs confer a survival benefit for CRC patients, serious systemic toxicities, including neutropenia, occur in ~30% of patients with lethality in 0.5–1% of patients. While serious systemic toxicities may occur in any patient, patients with polymorphisms in DPYD, which encodes the rate-limiting enzyme for pyrimidine degradation are at very high risk. Other genetic factors affecting risk for 5-FU toxicity, including miR-27a, are under investigation.
Methods:
Literature used to inform the text of this article was selected from PubMed.gov from the National Library of Medicine while regulatory documents were identified via Google search.
Conclusions:
Clinical studies to date have validated four DPYD polymorphisms (DPYD*2A, DPYD*13, c.2846A>T, HapB3) associated with serious toxicities in patients treated with 5-FU. Genetic screening for these is being implemented in the Netherlands and the UK and has been shown to be a cost-effective way to improve outcomes. Factors other than DPYD polymorphisms (e.g., miR-27a, TYMS, ENOSF1, p53) also affect 5-FU toxicity. Functional testing for deficient pyrimidine catabolism {defined as [U] >16 ng/mL or [UH2]:[U] <10} is being implemented in France and has demonstrated utility in identifying patients with elevated risk for 5-FU toxicity. Therapeutic drug monitoring (TDM) from plasma levels of 5-FU during first cycle treatment also is being used to improve outcomes and pharmacokinetic-based dosing is being used to increase the percent of patients within optimal area under the curve (AUC) (18–28 mg*h/L) values. Patients maintained in the optimal AUC range experienced significantly reduced systemic toxicities. As understanding the genetic basis for increased risk of 5-FU toxicity becomes more refined, the development of functional-based methods to optimize treatment is likely to become more widespread.
Keywords: Fluoropyrimidine (FP), colorectal cancer (CRC), dihydropyrimidine dehydrogenase (DPD), therapeutic drug monitoring (TDM)
Introduction
Fluoropyrimidine drugs (FPs) have been used for more than 50 years (1–3) and an estimated 2 million cancer patients are treated with FP drugs annually (4–6). In particular, FPs remain among the most effective drugs for treating GI malignancies, including colorectal cancer (CRC) (1.8 million), gastric (1 million), and pancreatic cancer (n=460,000) (7). Personalized medicine is advancing FP use in two ways: (I) Identifying the patient population for whom FPs constitute the preferred chemotherapeutic option and treating these patients with optimized regimens that include FPs (8); and (II) Identifying patient population for whom standard FP dosing poses a known pre-determined risk of serious toxicities, or even death, and either modifying FP dose or treating these patients with alternative drugs. The implementation of a personalized medicine approach to FP treatment has already contributed to a decline in mortality due to CRC, and these approaches are likely to become more widespread and applicable to treatment of other malignancies. This article will review tumor characteristics such as deficiencies in DNA mismatch repair and KRAS mutation status that are used for stratifying CRC patients, and determining if FP chemotherapy is a preferred option. The occurrence and origin of serious toxicities due to 5-FU treatment is also reviewed as are current measures to identify patients at high-risk for 5-FU toxicity thru pre-treatment screening for DPYD polymorphisms, reduced DPD activity, and other genetic factors. Approaches to personalize and optimize treatment thru TDM are also reviewed.
The author presents the following article in accordance with the Narrative Review Reporting Checklist (available at https://dx.doi.org/10.21037/pcm-21-17).
Methods
Literature used to inform the text of this article was selected from PubMed.gov from the National Library of Medicine. Full length manuscripts and Communications published in the English language between 1997 and 2021 were selected. Regulatory documents were identified via Google search.
Patient populations for whom FP-based regimens are a preferred option
The applicability of FP chemotherapy for treating CRC has evolved over its five decades of use, particularly in Western countries, and many findings are impacting treatment of other GI malignancies, worldwide. For patients with early stage CRC that is localized (stage I/II), or with regional lymph node involvement (stage III), surgery with curative intent is the preferred option, and adjuvant chemotherapy may be administered to reduce risk for disease recurrence (9). For patients with stage I colon cancer, surgery alone results in 95% 5-year disease-free survival (DFS) (10), and adjuvant chemotherapy is not administered. In stage II disease 5-year survival is 82–88%, and use of adjuvant chemotherapy remains controversial (11). An intergroup analysis reported a statistically significant improvement in 5-year DFS for stage II patients treated with chemotherapy, but improvement in overall survival was not statically significant (12). Follow-up analysis of long-term outcomes demonstrated 5-FU chemotherapy was associated with a 5% absolute improvement in survival at 8 years (13). For patients with high-risk stage II disease (e.g., T4 stage, poorly-differentiated tumors), the benefits of FP chemotherapy for risk reduction are considered by many oncologists to outweigh potential harmful effects due to treatment (14), and adjuvant chemotherapy, frequently FP doublet (5-FU/LV) may be administered (15). However, risks associated with FP-based treatment are real and potentially lethal, and for patients with stage II disease the potential risk-reducing benefits of FP regimens are offset by the potential detrimental effects of treatment (16).
For patients with stage III colon cancer the survival benefit of FP-based chemotherapy regimens outweighs potential risks for serious toxicities and the optimization of FP-based chemotherapy has evolved since initial report of therapeutic benefit in 1988 (17). Infusion dosing of 5-FU/LV (de Gramont schedule) was shown to cause reduced systemic toxicities relative to the Mayo and Roswell Park regimens that used bolus injection, although all schedules displayed similar survival benefit (9). 5-FU triplet therapy (e.g., 5-FU/LV/oxaliplatin—FOLFOX) chemotherapy was demonstrated to improve outcomes for patients with stage III CRC in the MOASIC trial and it is the current standard of care for stage III CRC patients. The exception remains tumors with deficient DNA mismatch repair (dMMR) for which FP-based regimens are ineffective and potentially harmful. Since FP chemotherapy is generally not effective in patients with tumors deficient in MMR (18), the MMR status is generally determined by immunohistochemistry (IHC) and/or by PCR-assessment of increased microsatellite instability (MSI-H) (19). Deficiencies in MMR occur either as the result of germ-line mutation conferring increased risk for colon cancer (e.g., Lynch syndrome; ~15%), or from epigenetic silencing of MMR genes (~85%). The mechanistic basis for lack of efficacy of FPs in dMMR CRC is not clearly understood, however MSI is also associated with elevated thymidylate synthase (TS) (20,21), which is the molecular target of FP chemotherapy (2).
Treatment of metastatic CRC (mCRC) also continues to rely heavily on FP-based chemotherapy regimens (8). However, the use of FPs for mCRC is evolving with refined implementation based on biomarker-based stratification to deliver optimal treatment. For mCRC, FP doublet chemotherapy is widely used in combination with targeted therapies directed at EGFR, such as the monoclonal antibody Cetuximab. However, tumors that display activating mutations in KRAS or BRAF negate the efficacy of agents targeting EGFR, and in these cases FP-based chemotherapy (doublet or triplet) is frequently used in combination with anti-VEGF therapy (e.g., Bevacizumab). For highly aggressive mCRC, as occurs with BRAF mutations, four component FP-based regimens FOLFOXIRI and FOLFIRINOX are used (22). FOLFIRINOX was also recently shown to be a preferred option for adjuvant chemotherapy of pancreatic cancer (23), and to be an effective option in patients with advanced gastroesophageal adenocarcinoma (24). However, its use was associated with higher incidence of toxic effects (23), demonstrating the need to better understand and manage toxicities related to FP use.
Frequency and types of serious toxicities
Nearly all cytotoxic chemotherapy drugs cause serious systemic toxicities and the side effect profiles for 5-FU and other FPs are considered relatively manageable in most instances. Diarrhea and nausea are common side effects that generally present within 7–10 days after initial treatment and are most often manageable. Dermatological toxicities, such as hand-foot syndrome, are common and generally occur later in treatment (25). Risk for severe toxicities is dependent on the FP and schedule used as well as on the genetic features of individual patients. The Mayo schedule is widely used for 5-FU administration and the primary toxicities associated with its use include neutropenia (69%), mucositis (58%), and leukopenia (46%) (26). Female patients have a higher incidence of severe toxicity and death than male patients and the incidence of severe toxicities is more frequent in elderly patients (27). Cardiotoxicity, while relatively rare, is a potentially lethal occurrence with high risk for long-term morbidity and mortality (28), and its risk with 5-FU is second only to anthracyclines (29). 5-FU crosses the blood-brain barrier and its use has been associated with cognitive impairment (30).
Patient population susceptible to serious toxicities from FPs
FPs are central to clinical management of GI-malignancies (3) (e.g., colorectal, gastric, pancreatic) and are widely used to treat head-and-neck cancer (31), basal cell carcinoma (32), and other malignancies. While their widespread use attests to FPs clinical activity, serious treatment-related toxicities occur in a sizable minority of patients (33). For example, approximately 30% of CRC patients treated with 5-FU or capecitabine experienced severe (≥ grade 3) toxicities, with 10–20% requiring hospitalization for toxicity management (34). Toxicities ultimately cause death in 0.5–1% of patients. In the U.S. alone, the number of lethalities associated with 5-FU treatment exceeds 1,300 per year (35). Further, the cost of managing toxicities that result from 5-FU treatment is substantial, with one study finding that the expected cost of hospitalization related to 5-FU toxicity was $3,674 per patient, approximately $2,700 higher among 5-FU patients than patients not treated with 5-FU (36).
The occurrence of serious toxicities following 5-FU treatment is much more frequent in patients that are deficient in the catabolism of 5-FU (Figure 1). While the anti-cancer activities of FPs result primarily from conversion to FdUMP and inhibition of TS (2,37), up to 90% of 5-FU is degraded (38). The first and rate-limiting step in 5-FU degradation (39,40) is catalyzed by dihydropyrimidine dehydrogenase (DPD). Deficiencies in DPD activity occur in an estimated 3–8% of patients (34,41), and certain populations such as individuals of African descent, are at especially high risk to drug-induced toxicities due to variability in drug metabolism (42) while in Asian populations a distinct spectrum of polymorphisms affecting DPD activity predominates (43). Patients deficient in DPD activity are at very high risk for serious 5-FU-related toxicities.
Figure 1.
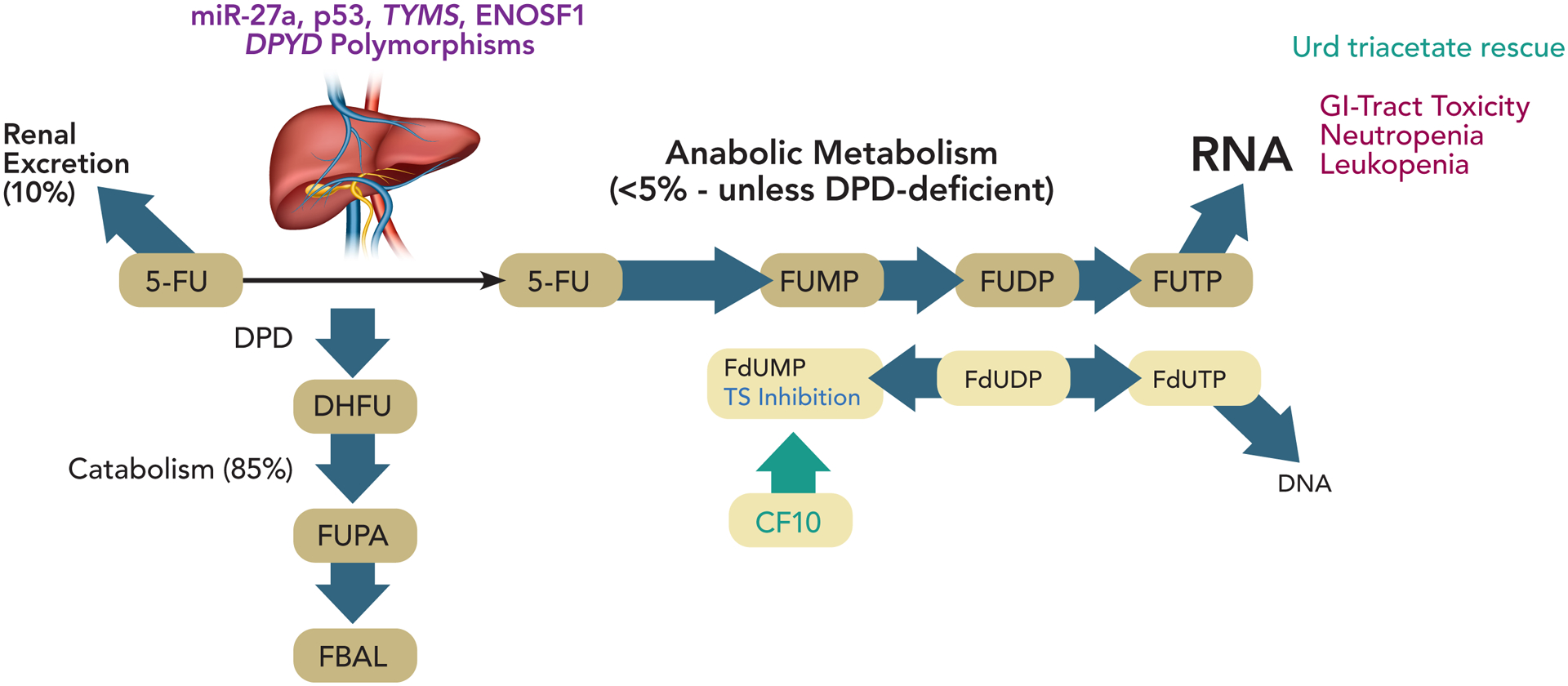
Depiction of fluoropyrimidine metabolism and genetic factors influencing fluoropyrimidine toxicity. In most cancer patients >80% of 5-FU is degraded in the liver. However, for patients with certain DPYD polymorphisms reduced catabolism is accompanied by increased anabolic metabolism that perturbs RNA-mediated processes, particularly in the GI-tract and bone marrow, causing serious systemic toxicities. Several factors other than DPYD polymorphisms affect 5-FU toxicity including polymorphisms in miR-27a, polymorphisms in TYMS, an enzymatic target of 5-FU and levels of ENOSF1, a regulator of TYMS. p53 also regulates DPYD expression. The fluoropyrimidine polymer CF10 is directly converted to FdUMP and may circumvent issues related to genetic factors affecting 5-FU toxicity.
Although deficiency in DPD activity in most individuals is considered to result from polymorphisms in DPYD affecting enzyme expression and function, other causes, such as variability in miR-27a regulation of DPYD have been identified (44). miR-27a and miR-27b bind a conserved recognition site on DPYD repressing its expression and mouse liver DPD activity was inversely correlated with miR-27a expression. Further a variant of miR-27a was associated with reduced DPD activity in healthy volunteers (44), and miR-27a variants improve predictive values of DPYD variants for 5-FU toxicity. Intriguingly, p53 also modulates pyrimidine catabolism by repressing DPYD expression (45). Polymorphisms in TYMS, which encodes thymidylate synthase (TS), the principal target of 5-FU are also implicated in 5-FU toxicity and combined DPYD and TYMS genotyping could identify a majority of patients at elevated risk for adverse events (46). An isoform of Enolase Superfamily member 1 (ENOSF1) regulates TYMS and is implicated in capecitabine toxicity (47).
DPYD polymorphisms and increased risk for 5-FU toxicity
DPYD polymorphisms with clinical impact
Deficiencies in DPD activity is causative of increased risk for severe toxicity following FP treatment, with 60–80% of DPD-deficient individuals experiencing dose-limiting and potentially life-threatening toxicities, while the corresponding incidence in DPD-proficient patients is 10–20% (48). Although >160 SNPs in DPYD have been reported (49) as outlined by the Clinical Pharmacogenetics Implementation Consortium (CPIC) guidelines (6), relatively few are both highly prevalent and have a substantial effect on DPD activity to justify pre-emptive screening. The potential risk for serious toxicities related to the most prevalent DPYD polymorphisms associated with reduced or total dysfunction of DPD activity has stimulated national healthcare programs to implement screening programs to reduce serious toxicities in patients treated with 5-FU, capecitabine, or tegafur. The Dutch Pharmacogenetics Working Group (DPWG) identified four DPYD variants for which sufficient evidence for reduced/complete DPD dysfunction (50) and the UK chemotherapy board (51) also considers these same four to have serious clinical impact affecting FP toxicity and since 2020 recommends pre-treatment pharmacogenomic screening for these (Table 1): DPYD*2A (rs3918290), DPYD*13 (rs55886062), c.2846A>T (rs67376798) and c.1236G>A (DPYD-Hap-B3; rs56038477). The clinical relevance of these four SNPs for increased risk of serious toxicities resulting from FP treatment was established by meta-analysis of multiple clinical studies (53). The c.2846A>T and Hap-B3 variants are associated with reduced functionality with modified initial FP treatment regimens recommended (e.g., 50% dose reduction) while the *2A and *13 variants are considered to encode a fully dysfunctional enzyme precluding FP use (50). Case reports document fatal 5-FU toxicity in patients with specific SNPs (54). Neutropenia is the most common toxicity associated with DPD deficiency with one study finding stage IV neutropenia occurring in 55% of patients with decreased DPD activity while the rate in patients with normal DPD activity was only 13% (55) and similar differential toxicity was found in other studies (56). Each of the four DPYD SNPs considered to have serious clinical impact by the DPWG and UK chemotherapy board was associated with a significant increased risk for severe neutropenia. Patients with these SNPs display both increased incidence and more rapid onset of serious neutropenia (57).
Table 1.
DPYD Polymorphisms with Established Prognostic Value for 5-FU Toxicity
| DPYD variant | NCBI SNP reference | Nucleotide change | Protein effect | RRa |
|---|---|---|---|---|
| DPYD*2A | rs3918290 | 1905+1G>A | Exon Skipping | 2.87 |
| DPYD*13 | rs55886062 | 1679 T>G | Ile 560 Ser | 4.3 |
| c.2846A>T | rs67376798 | 2846 A>T | Asp 949 Val | 3.11 |
| HapB3 | s56038477 | 1236 G>A | Glu 412 Glu, in haplotype B3 | 1.72 |
relative risk for overall grade ≥3 fluoropyrimidine-related toxicity compared with non-carriers of this variant (52). CPIC, Clinical Pharmacogenetics Implementation Consortium; HapB3, haplotype B3; RR, relative risk; NCBI, National Center for Biotechnology Information.
Importantly, pre-treatment screening for DPD-deficiency based on screening for relatively prevalent DPYD polymorphisms significantly improves outcomes when used in conjunction with personalized dose management. The recommendations adopted in the UK parallel results from clinical studies in the Netherlands (58) and other countries that demonstrated prospective screening for clinically-relevant DPYD variants was feasible, and that DPYD genotype to guide dose improved outcomes. Patient management to reduce risk of toxicities in DPD-deficient individuals includes dose reduction in first-cycle dose of 5-FU or capecitabine followed by dose titration guided by toxicities in subsequent cycles. Collectively these studies established that dose adjustments based on prospective DPYD genotyping decreased toxicity while maintaining efficacy (52). Importantly, the data also support that prospective DPYD screening not only decreases the incidence of serious toxicities, but reduces overall cost of treatment by an amount that is greater than the cost of screening (59).
Functional implications of DPYD polymorphisms
The effects of SNPs on DPD protein sequence and function have been validated in many cases. For example, the *2A (rs3918290) locus alters a splice recognition sequence leading to a 165 base pair deletion and expression of an inactive DPD protein (60). DPYD*13 (rs55886062) and rs67376798 result in amino acid changes decreasing DPD activity. Other SNPs that are more prevalent, including DPYD*9A (~40% heterozygous, 10% homozygous) were implicated in affecting FP toxicity, but are not presently considered important for pre-treatment screening. A preliminary study indicated *9A was associated with increased risk for serious toxicities, however a later study with larger sample did not reproduce the earlier trend (61). While occurrence of multiple DPYD variants in a single individual is rare, it can have serious consequences. For example, a recent study found 4 of 7 patients carrying multiple DPYD variants experienced severe toxicity after receiving FP therapy (62). The DPWG considered how combinations of DPYD variants impacted overall enzyme activity as indicated by a gene activity score (50). It should be noted that the occurrence of multiple variants (~0.2%) is more common than the DPYD*13 variant (0.1%) that is widely included in DPYD genetic screens and this possibility must be considered (62). Further, it is important to note that the prevalence of DPYD SNPs associated with DPD deficiency and severe FP toxicity varies considerably in different patient populations. For example, the HapB3 SNP (rs75017182) is approximately 10-fold more prevalent in Europeans compared to either African or East Asian populations while other SNPs are more prevalent in these populations (48).
Financial implications of DPD testing
The occurrence of serious toxicities related to 5-FU treatment of patients with low DPD activity has serious economic consequences. For example, Murphy et al., calculated the cost of toxicity-related hospitalizations in patients with DPYD polymorphisms associated with increased toxicity risk was 10-fold higher than the cost of prospective testing for all patients (63). Martens et al noted an increase in pre-treatment DPD testing from 1% in Q2 of 2017 to 87% in Q4-2018. The availability of clear protocols and evidence of their utility were found to be key factors in increasing testing in the Amsterdam University Medical Centers (64).
Functional testing of DPD activity
Since only 39–61% of patients with serious 5-FU toxicities display polymorphisms associated with reduced DPD activity (41,65) and <20% of patients that experience serious FP-related toxicities are identified by genotyping DPYD (34) other factors must contribute to 5-FU toxicity. The need to more generally identify patients with high-risk has resulted in efforts to quantify patient-specific DPD activity prior to initiating 5-FU treatment to identify patients at elevated risk for serious toxicities. While polymorphisms in DPYD are a principal contributor to serious 5-FU toxicities, they are not the only cause. For example miR-27 regulation of DPYD is another potential causes of reduced DPD activity (Figure 1). Also p53 repressively regulates DPYD expression (45). Further, there are additional contributing factors including polymorphisms in TYMS and variants in ENOSF1 affecting 5-FU toxicity. Several functional endpoints related to DPD activity and pyrimidine metabolism have been developed and tested clinically including: (I) DPD activity in PBMCs; (II) plasma uracil concentrations; (III) 2-13C breath test; (IV) Uracil test dose (Figure 2).
Figure 2.
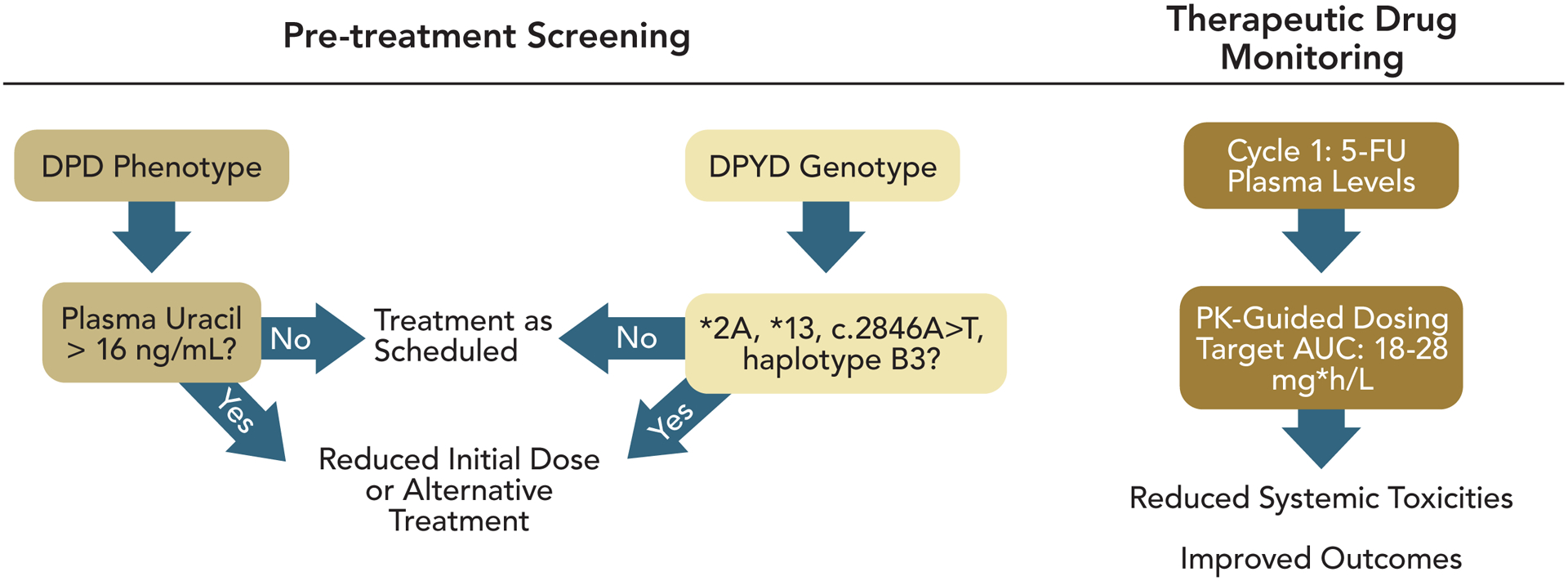
Depiction of strategies to reduce the occurrence of 5-FU-related toxicities. Pre-treatment screening is used to identify patients at elevated risk for 5-FU-related toxicities due to either polymorphisms in the DPYD gene associated with reduced DPD functional activity or measuring plasma Uracil values from which deficiencies in pyrimidine metabolism can be inferred. Initial 5-FU dosing may be reduced based on pre-treatment screening results. TDM permits 5-FU dose modulation based on plasma 5-FU levels during cycle 1 treatment such that subsequent cycles are more likely to be in the optimal range.
Pallet et al. (66) assessed characteristics of 3,680 patients that had completed testing to establish their DPD phenotypes and DPYD genotypes. DPD functional activity was assessed both from uracil plasma concentrations ([U]), and the ratio of dihydrouracil to uracil ([UH2]:[U]). Reduced DPD activity is defined as [U] >16 ng/mL or [UH2]:[U] <10. DPYD SNPs associated with reduced DPD activity were detected in 4.5% of patients; however, their detection had only moderate predictive value in identifying patients with functional DPD deficiencies. The authors concluded that a clinical trial comparing toxicity rates after dose adjustments based on genotype vs phenotype testing for DPD deficiency was needed to identify the best strategy for mitigating risk of 5-FU treatment. In another study, plasma uracil concentrations were found to be superior to detection of gene polymorphisms for predicting which patients will experience serious 5-FU-related toxicities. Meulendijks et al. (67), conducted testing in 550 patients and elevated plasma uracil concentration was significantly associated with global severe toxicity (P=0.0009), severe gastrointestinal (GI) toxicity (P<0.0001), toxicity-related hospitalization (P<0.0001), and fatal treatment-related toxicity (P=0.001). None of the DPYD or TYMS variants were significantly associated with severe toxicities in this study.
Several efforts are ongoing to make widespread testing of uracil and dihydrouracil prior to FP treatment more accurate and more economical. To automate testing of uracil and dihydrouracil in plasma, Robin et al. developed a LC-MS method with an automated sample preparation system that may enable widespread implementation of pre-treatment testing for DPD functional activity (68). Uracil and dihydrouracil concentrations in saliva may provide a more convenient and less invasive source of biological fluids to identify patients at high risk for 5-FU toxicity. Both fresh saliva and dried saliva spots were shown to display prognostic value (69). Another approach is use of a breath test to detect uracil degradation. However, this approach, while rapid and reliable, requires use of either radioactive isotopes or mass spectrometry (70), and thus may have less potential for widespread implementation. The French cancer institute and French health authority recommend measurement of uracil blood level and implementation of alternative regimens for cancer patients with elevated levels (71).
Interventions to reduce FP toxicity
Since severe toxicity in response to 5-FU treatment can occur in any patient, regardless of DPYD status or DPD activity, pre-treatment screening for these will reduce but not eliminate the occurrence of severe toxic responses. Further, these pre-treatment screening approaches will not enable dose modification for optimal response in the majority of patients that do not display serious DPD deficiencies. For these reasons, approaches to TDM are being implemented to adjust 5-FU dosing for optimal therapeutic response (72). 5-FU is now primarily dosed as an infusion over 46 h and this is used in combination chemotherapy regimens such as FOLFOX and FOLFIRI. Infusion dosing has generally replaced bolus administration and has an improved safety profile although severe toxicities still occur, largely based on large inter-patient variability in 5-FU plasma levels.
As with other cytotoxic agents, 5-FU dose is calculated based on patient body surface area (BSA), however BSA-based dosing does not account for inter-patient variability in plasma clearance which ranged from 0.49 to 4.93 L/h/m2 in one study (73). Further, this inter-patient variability in plasma clearance correlates with therapeutic response with elevated clearance associated with poor outcomes (74). Multiple studies have now demonstrated that adjusting 5-FU dose based on plasma levels is feasible and can improve outcomes [reviewed in ref (72)]. For example, a recent study showed that implementing an adjusted, PK-guided dose of 5-FU in cycle 2 based 5-FU plasma levels measured during cycle 1 resulted in increasing the percent of patients within optimal area under the curve (AUC; 18–28 mg*h/L) values from 49.3% to 66.9%. Patients maintained in the optimal AUC range experienced significantly reduced systemic toxicities. For example relative risk for neutropenia for overexposed patients based on AUC was 3.05 (1.55–6.01; P=0.004), while patients with at least one AUC below the target during the first 3 cycles displayed significantly reduced progression-free survival (75).
The serious systemic toxicities such as neutropenia that occur in patients deficient in 5-FU catabolism result from RNA-mediated effects of the drug and not on-target effects related to FdUMP, the metabolite primarily responsible for anti-tumor activity. Among anabolic metabolites, ribonucleotides (e.g., FUTP) are produced at much higher levels than the corresponding deoxyribonucleotides by ~10:1 ratio (76). Under conditions of reduced catabolism, such as in patients with DPYD polymorphisms, increased misincorporation of FUTP into RNA causes extensive damage to leukocytes and cells of the GI-tract by an unspecified process. The serious toxicities associated with 5-FU overtreatment can be reversed by treatment with uridine triacetate (77), which increases UTP pools and competes with FUTP for RNA incorporation. Both leukopenia (78) and 5-FU-induced GI-tract toxicity (79) can be reversed with Uridine treatment. An alternative approach to reducing 5-FU’s RNA-mediated effects and this its systemic toxicities while increasing levels of FdUMP responsible for anti-tumor activity is use of fluoropyrimidine polymers (80,81). The Gmeiner lab has pioneered testing of FP polymers (e.g., CF10) in preclinical models (82–84), and demonstrated these result in sustained high plasma levels of FdU and FdUMP while not causing serious systemic toxicities that occur with 5-FU treatment, in part due to reduced dependence on DPD-mediated degradation (80).
Conclusions
FPs remain the backbone for combination chemotherapy regimens used to treat CRC and are among the most important drugs for treatment of pancreatic cancer and other GI-malignancies. While FP use confers a survival benefit, a significant portion of patients experience serious toxicities that are expensive to manage and may be lethal. Risk/benefit analysis limits 5-FU use in some patient populations that could benefit from treatment if toxicities were not a concern. While serious toxicities may occur in any patient, patients with DPYD polymorphisms reducing pyrimidine catabolism are known to be at elevated risk. Genetic screening is being implemented in The Netherlands and UK and other locations to identify these high-risk patients and pre-emptively adjust FP dose. Outcomes and cost-analysis indicates genetic-based screening is effective. A more general approach involving functional screening for patients with atypical pyrimidine catabolism is also being implemented in France and other locations and may permit improved dosing for a wider spectrum of patients than those with specific DPYD polymorphisms. TDM utilizing PK-guided dosing to personalize treatment is demonstrated to increase the percent of patients that achieve an optimal dose range and this approach is both reducing the incidence of serious systemic toxicities and improving outcomes. Since the most frequent serious systemic toxicities of 5-FU appear to be unrelated to the on-target effects of the primary anti-tumor metabolite, FdUMP, next generation FPs, such as FP polymers may reduce the need for identifying patients who are at high risk for toxicities with treatment with the current FP drugs.
Acknowledgments
Funding:
This work was supported by National Institutes of Health (grant numbers CA218933, CA012197).
Footnotes
Reporting Checklist: The author has completed the Narrative Review Reporting Checklist. Available at https://dx.doi.org/10.21037/pcm-21-17
Conflicts of Interest: The author has completed the ICMJE uniform disclosure form (available at https://dx.doi.org/10.21037/pcm-21-17). WHG receives grants from National Institutes of Health (grant numbers CA218933, CA012197) and has patent for PCT/US2019/033902 pending. WHG serves as an unpaid editorial board member of Precision Cancer Medicine from Aug 2018 to Jul 2022. The author has no other conflicts of interest to declare.
Ethical Statement: The author is accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
References
- 1.Wilson PM, Danenberg PV, Johnston PG, et al. Standing the test of time: targeting thymidylate biosynthesis in cancer therapy. Nat Rev Clin Oncol 2014;11:282–98. [DOI] [PubMed] [Google Scholar]
- 2.Gmeiner WH. Chemistry of Fluorinated Pyrimidines in the Era of Personalized Medicine. Molecules 2020;25:3438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gmeiner WH. Fluoropyrimidine Modulation of the Anti-Tumor Immune Response-Prospects for Improved Colorectal Cancer Treatment. Cancers (Basel) 2020;12:1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Deac AL, Burz CC, Bocşe HF, et al. A review on the importance of genotyping and phenotyping in fluoropyrimidine treatment. Med Pharm Rep 2020;93:223–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ly RC, Schmidt RE, Kiel PJ, et al. Severe Capecitabine Toxicity Associated With a Rare DPYD Variant Identified Through Whole-Genome Sequencing. JCO Precis Oncol 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Amstutz U, Henricks LM, Offer SM, et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) Guideline for Dihydropyrimidine Dehydrogenase Genotype and Fluoropyrimidine Dosing: 2017 Update. Clin Pharmacol Ther 2018;103:210–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Worldwide Cancer Data. Washington, DC: World Cancer Research Fund American Institute for Cancer Research, 2018. [Google Scholar]
- 8.Gmeiner WH. Recent Advances in Our Knowledge of mCRC Tumor Biology and Genetics: A Focus on Targeted Therapy Development. Onco Targets Ther 2021;14:2121–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chakrabarti S, Peterson CY, Sriram D, et al. Early stage colon cancer: Current treatment standards, evolving paradigms, and future directions. World J Gastrointest Oncol 2020;12:808–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Osterman E, Glimelius B. Recurrence Risk After Up-to-Date Colon Cancer Staging, Surgery, and Pathology: Analysis of the Entire Swedish Population. Dis Colon Rectum 2018;61:1016–25. [DOI] [PubMed] [Google Scholar]
- 11.Böckelman C, Engelmann BE, Kaprio T, et al. Risk of recurrence in patients with colon cancer stage II and III: a systematic review and meta-analysis of recent literature. Acta Oncol 2015;54:5–16. [DOI] [PubMed] [Google Scholar]
- 12.Gill S, Loprinzi CL, Sargent DJ, et al. Pooled analysis of fluorouracil-based adjuvant therapy for stage II and III colon cancer: who benefits and by how much? J Clin Oncol 2004;22:1797–806. [DOI] [PubMed] [Google Scholar]
- 13.Sargent D, Sobrero A, Grothey A, et al. Evidence for cure by adjuvant therapy in colon cancer: observations based on individual patient data from 20,898 patients on 18 randomized trials. J Clin Oncol 2009;27:872–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jee SH, Moon SM, Shin US, et al. Effectiveness of Adjuvant Chemotherapy with 5-FU/Leucovorin and Prognosis in Stage II Colon Cancer. J Korean Soc Coloproctol 2011;27:322–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moran RG, Keyomarsi K. Biochemical rationale for the synergism of 5-fluorouracil and folinic acid. NCI Monogr 1987;(5):159–63. [PubMed] [Google Scholar]
- 16.Varghese A Chemotherapy for Stage II Colon Cancer. Clin Colon Rectal Surg 2015;28:256–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wolmark N, Fisher B, Rockette H, et al. Postoperative adjuvant chemotherapy or BCG for colon cancer: results from NSABP protocol C-01. J Natl Cancer Inst 1988;80:30–6. [DOI] [PubMed] [Google Scholar]
- 18.Sargent DJ, Marsoni S, Monges G, et al. Defective mismatch repair as a predictive marker for lack of efficacy of fluorouracil-based adjuvant therapy in colon cancer. J Clin Oncol 2010;28:3219–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kawakami H, Zaanan A, Sinicrope FA. Microsatellite instability testing and its role in the management of colorectal cancer. Curr Treat Options Oncol 2015;16:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Öhrling K, Karlberg M, Edler D, et al. A combined analysis of mismatch repair status and thymidylate synthase expression in stage II and III colon cancer. Clin Colorectal Cancer 2013;12:128–35. [DOI] [PubMed] [Google Scholar]
- 21.Gatalica Z, Vranic S, Xiu J, et al. High microsatellite instability (MSI-H) colorectal carcinoma: a brief review of predictive biomarkers in the era of personalized medicine. Fam Cancer 2016;15:405–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Adenis A, Mazard T, Fraisse J, et al. FOLFIRINOX-R study design: a phase I/II trial of FOLFIRINOX plus regorafenib as first line therapy in patients with unresectable RAS-mutated metastatic colorectal cancer. BMC Cancer 2021;21:564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Conroy T, Hammel P, Hebbar M, et al. FOLFIRINOX or Gemcitabine as Adjuvant Therapy for Pancreatic Cancer. N Engl J Med 2018;379:2395–406. [DOI] [PubMed] [Google Scholar]
- 24.Park H, Jin RU, Wang-Gillam A, et al. FOLFIRINOX for the Treatment of Advanced Gastroesophageal Cancers: A Phase 2 Nonrandomized Clinical Trial. JAMA Oncol 2020;6:1231–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kwakman JJM, Elshot YS, Punt CJA, et al. Management of cytotoxic chemotherapy-induced hand-foot syndrome. Oncol Rev 2020;14:442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Garg MB, Lincz LF, Adler K, et al. Predicting 5-fluorouracil toxicity in colorectal cancer patients from peripheral blood cell telomere length: a multivariate analysis. Br J Cancer 2012;107:1525–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tsalic M, Bar-Sela G, Beny A, et al. Severe toxicity related to the 5-fluorouracil/leucovorin combination (the Mayo Clinic regimen): a prospective study in colorectal cancer patients. Am J Clin Oncol 2003;26:103–6. [DOI] [PubMed] [Google Scholar]
- 28.Yuan C, Parekh H, Allegra C, et al. 5-FU induced cardiotoxicity: case series and review of the literature. Cardiooncology 2019;5:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sara JD, Kaur J, Khodadadi R, et al. 5-fluorouracil and cardiotoxicity: a review. Ther Adv Med Oncol 2018;10:1758835918780140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lyons L, ElBeltagy M, Bennett G, et al. Fluoxetine counteracts the cognitive and cellular effects of 5-fluorouracil in the rat hippocampus by a mechanism of prevention rather than recovery. PLoS One 2012;7:e30010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kumagai A, Iijima S, Nomiya T, et al. A pilot study of the clinical evidence for the methodology for prevention of oral mucositis during cancer chemotherapy by measuring salivary excretion of 5-fluorouracil. BDJ Open 2018;4:17041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ikediobi O Pharmacogenomics of Fluoropyrimidines-A Personalized Approach for Treating and Preventing Keratinocyte Carcinomas. JAMA Dermatol 2021;157:357–8. [DOI] [PubMed] [Google Scholar]
- 33.Meta-Analysis Group In Cancer; Lévy E, Piedbois P, et al. Toxicity of fluorouracil in patients with advanced colorectal cancer: effect of administration schedule and prognostic factors. J Clin Oncol 1998;16:3537–41. [DOI] [PubMed] [Google Scholar]
- 34.Meulendijks D, Cats A, Beijnen JH, et al. Improving safety of fluoropyrimidine chemotherapy by individualizing treatment based on dihydropyrimidine dehydrogenase activity - Ready for clinical practice? Cancer Treat Rev 2016;50:23–34. [DOI] [PubMed] [Google Scholar]
- 35.El-Rayes B Managing Severe Toxicities Associated with 5-FU and Capecitabine: An Oncologists’s Perspective. Medscape Educ Oncol 2017. Available online: https://www.medscape.org/viewarticle/872909_sidebar2 [Google Scholar]
- 36.Delea TE, Vera-Llonch M, Edelsberg JS, et al. The incidence and cost of hospitalization for 5-FU toxicity among Medicare beneficiaries with metastatic colorectal cancer. Value Health 2002;5:35–43. [DOI] [PubMed] [Google Scholar]
- 37.Longley DB, Harkin DP, Johnston PG. 5-fluorouracil: mechanisms of action and clinical strategies. Nat Rev Cancer 2003;3:330–8. [DOI] [PubMed] [Google Scholar]
- 38.Johnson MR, Hageboutros A, Wang K, et al. Life-threatening toxicity in a dihydropyrimidine dehydrogenase-deficient patient after treatment with topical 5-fluorouracil. Clin Cancer Res 1999;5:2006–11. [PubMed] [Google Scholar]
- 39.Diasio RB. The role of dihydropyrimidine dehydrogenase (DPD) modulation in 5-FU pharmacology. Oncology (Williston Park) 1998;12:23–7. [PubMed] [Google Scholar]
- 40.Diasio RB, Johnson MR. Dihydropyrimidine dehydrogenase: its role in 5-fluorouracil clinical toxicity and tumor resistance. Clin Cancer Res 1999;5:2672–3. [PubMed] [Google Scholar]
- 41.Johnson MR, Diasio RB. Importance of dihydropyrimidine dehydrogenase (DPD) deficiency in patients exhibiting toxicity following treatment with 5-fluorouracil. Adv Enzyme Regul 2001;41:151–7. [DOI] [PubMed] [Google Scholar]
- 42.Mattison LK, Fourie J, Desmond RA, et al. Increased prevalence of dihydropyrimidine dehydrogenase deficiency in African-Americans compared with Caucasians. Clin Cancer Res 2006;12:5491–5. [DOI] [PubMed] [Google Scholar]
- 43.Leung HW, Chan AL. Association and prediction of severe 5-fluorouracil toxicity with dihydropyrimidine dehydrogenase gene polymorphisms: A meta-analysis. Biomed Rep 2015;3:879–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Offer SM, Butterfield GL, Jerde CR, et al. microRNAs miR-27a and miR-27b directly regulate liver dihydropyrimidine dehydrogenase expression through two conserved binding sites. Mol Cancer Ther 2014;13:742–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gokare P, Finnberg NK, Abbosh PH, et al. P53 represses pyrimidine catabolic gene dihydropyrimidine dehydrogenase (DPYD) expression in response to thymidylate synthase (TS) targeting. Sci Rep 2017;7:9711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Saif MW, Hachem H, Purvey S, et al. Pharmacogenetic Variants in the DPYD and TYMS Genes are Clinically Significant Predictors of Fluoropyrimidine Toxicity: Are We Ready for Use in our Clinical Practice. Arch Pharmacol Ther 2020;2:6–8. [PMC free article] [PubMed] [Google Scholar]
- 47.Palles C, Fotheringham S, Chegwidden L, et al. An Evaluation of the Diagnostic Accuracy of a Panel of Variants in DPYD and a Single Variant in ENOSF1 for Predicting Common Capecitabine Related Toxicities. Cancers (Basel) 2021;13:1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhou Y, Dagli Hernandez C, Lauschke VM. Population-scale predictions of DPD and TPMT phenotypes using a quantitative pharmacogene-specific ensemble classifier. Br J Cancer 2020;123:1782–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Toffoli G, Giodini L, Buonadonna A, et al. Clinical validity of a DPYD-based pharmacogenetic test to predict severe toxicity to fluoropyrimidines. Int J Cancer 2015;137:2971–80. [DOI] [PubMed] [Google Scholar]
- 50.Lunenburg CATC, van der Wouden CH, Nijenhuis M, et al. Dutch Pharmacogenetics Working Group (DPWG) guideline for the gene-drug interaction of DPYD and fluoropyrimidines. Eur J Hum Genet 2020;28:508–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Clinical Commissioning Urgent Policy Statement Pharmacogenomic testing for DPYD polymorphisms with fluoropyrimidine therapies [URN 1869] (200603P). Available online: https://www.england.nhs.uk/wp-content/uploads/2020/11/1869-dpyd-policy-statement.pdf
- 52.Lunenburg CATC, Henricks LM, Guchelaar HJ, et al. Prospective DPYD genotyping to reduce the risk of fluoropyrimidine-induced severe toxicity: Ready for prime time. Eur J Cancer 2016;54:40–8. [DOI] [PubMed] [Google Scholar]
- 53.Meulendijks D, Henricks LM, Sonke GS, et al. Clinical relevance of DPYD variants c.1679T>G, c.1236G>A/HapB3, and c.1601G>A as predictors of severe fluoropyrimidine-associated toxicity: a systematic review and meta-analysis of individual patient data. Lancet Oncol 2015;16:1639–50. [DOI] [PubMed] [Google Scholar]
- 54.Fidai SS, Sharma AE, Johnson DN, et al. Dihydropyrimidine dehydrogenase deficiency as a cause of fatal 5-Fluorouracil toxicity. Autops Case Rep 2018;8:e2018049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.van Kuilenburg AB, Haasjes J, Richel DJ, et al. Clinical implications of dihydropyrimidine dehydrogenase (DPD) deficiency in patients with severe 5-fluorouracil-associated toxicity: identification of new mutations in the DPD gene. Clin Cancer Res 2000;6:4705–12. [PubMed] [Google Scholar]
- 56.Morel A, Boisdron-Celle M, Fey L, et al. Clinical relevance of different dihydropyrimidine dehydrogenase gene single nucleotide polymorphisms on 5-fluorouracil tolerance. Mol Cancer Ther 2006;5:2895–904. [DOI] [PubMed] [Google Scholar]
- 57.Ruzzo A, Graziano F, Galli F, et al. Dihydropyrimidine dehydrogenase pharmacogenetics for predicting fluoropyrimidine-related toxicity in the randomised, phase III adjuvant TOSCA trial in high-risk colon cancer patients. Br J Cancer 2017;117:1269–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Henricks LM, Lunenburg CATC, de Man FM, et al. DPYD genotype-guided dose individualisation of fluoropyrimidine therapy in patients with cancer: a prospective safety analysis. Lancet Oncol 2018;19:1459–67. [DOI] [PubMed] [Google Scholar]
- 59.Deenen MJ, Meulendijks D, Cats A, et al. Upfront Genotyping of DPYD*2A to Individualize Fluoropyrimidine Therapy: A Safety and Cost Analysis. J Clin Oncol 2016;34:227–34. [DOI] [PubMed] [Google Scholar]
- 60.Ezzeldin H, Diasio R. Dihydropyrimidine dehydrogenase deficiency, a pharmacogenetic syndrome associated with potentially life-threatening toxicity following 5-fluorouracil administration. Clin Colorectal Cancer 2004;4:181–9. [DOI] [PubMed] [Google Scholar]
- 61.Maharjan AS, McMillin GA, Patel GK, et al. The Prevalence of DPYD*9A(c.85T>C) Genotype and the Genotype-Phenotype Correlation in Patients with Gastrointestinal Malignancies Treated With Fluoropyrimidines: Updated Analysis. Clin Colorectal Cancer 2019;18:e280–6. [DOI] [PubMed] [Google Scholar]
- 62.Lunenburg CATC, Henricks LM, van Kuilenburg ABP, et al. Diagnostic and Therapeutic Strategies for Fluoropyrimidine Treatment of Patients Carrying Multiple DPYD Variants. Genes (Basel) 2018;9:585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Murphy C, Byrne S, Ahmed G, et al. Cost Implications of Reactive Versus Prospective Testing for Dihydropyrimidine Dehydrogenase Deficiency in Patients With Colorectal Cancer: A Single-Institution Experience. Dose Response 2018;16:1559325818803042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Martens FK, Huntjens DW, Rigter T, et al. DPD Testing Before Treatment With Fluoropyrimidines in the Amsterdam UMCs: An Evaluation of Current Pharmacogenetic Practice. Front Pharmacol 2019;10:1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Milano G, Etienne MC, Pierrefite V, et al. Dihydropyrimidine dehydrogenase deficiency and fluorouracil-related toxicity. Br J Cancer 1999;79:627–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pallet N, Hamdane S, Garinet S, et al. A comprehensive population-based study comparing the phenotype and genotype in a pretherapeutic screen of dihydropyrimidine dehydrogenase deficiency. Br J Cancer 2020;123:811–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Meulendijks D, Henricks LM, Jacobs BAW, et al. Pretreatment serum uracil concentration as a predictor of severe and fatal fluoropyrimidine-associated toxicity. Br J Cancer 2017;116:1415–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Robin T, Saint-Marcoux F, Toinon D, et al. Automatic quantification of uracil and dihydrouracil in plasma. J Chromatogr B Analyt Technol Biomed Life Sci 2020;1142:122038. [DOI] [PubMed] [Google Scholar]
- 69.Neto OV, Raymundo S, Franzoi MA, et al. DPD functional tests in plasma, fresh saliva and dried saliva samples as predictors of 5-fluorouracil exposure and occurrence of drug-related severe toxicity. Clin Biochem 2018;56:18–25. [DOI] [PubMed] [Google Scholar]
- 70.Cunha-Junior GF, De Marco L, Bastos-Rodrigues L, et al. (13)C-uracil breath test to predict 5-fluorouracil toxicity in gastrointestinal cancer patients. Cancer Chemother Pharmacol 2013;72:1273–82. [DOI] [PubMed] [Google Scholar]
- 71.Hodroj K, Barthelemy D, Lega JC, et al. Issues and limitations of available biomarkers for fluoropyrimidine-based chemotherapy toxicity, a narrative review of the literature. ESMO Open 2021;6:100125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lee JJ, Beumer JH, Chu E. Therapeutic drug monitoring of 5-fluorouracil. Cancer Chemother Pharmacol 2016;78:447–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gamelin E, Boisdron-Celle M, Guérin-Meyer V, et al. Correlation between uracil and dihydrouracil plasma ratio, fluorouracil (5-FU) pharmacokinetic parameters, and tolerance in patients with advanced colorectal cancer: A potential interest for predicting 5-FU toxicity and determining optimal 5-FU dosage. J Clin Oncol 1999;17:1105. [DOI] [PubMed] [Google Scholar]
- 74.Seitz JF, Cano JP, Rigault JP, et al. Chemotherapy of extensive digestive cancers with 5-fluorouracil: relation between the clinical response and plasma clearance of the drug. Gastroenterol Clin Biol 1983;7:374–80. [PubMed] [Google Scholar]
- 75.Morawska K, Goirand F, Marceau L, et al. 5-FU therapeutic drug monitoring as a valuable option to reduce toxicity in patients with gastrointestinal cancer. Oncotarget 2018;9:11559–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Noordhuis P, Holwerda U, Van der Wilt CL, et al. 5-Fluorouracil incorporation into RNA and DNA in relation to thymidylate synthase inhibition of human colorectal cancers. Ann Oncol 2004;15:1025–32. [DOI] [PubMed] [Google Scholar]
- 77.Saif MW, Diasio RB. Benefit of uridine triacetate (Vistogard) in rescuing severe 5-fluorouracil toxicity in patients with dihydropyrimidine dehydrogenase (DPYD) deficiency. Cancer Chemother Pharmacol 2016;78:151–6. [DOI] [PubMed] [Google Scholar]
- 78.van Groeningen CJ, Peters GJ, Leyva A, et al. Reversal of 5-fluorouracil-induced myelosuppression by prolonged administration of high-dose uridine. J Natl Cancer Inst 1989;81:157–62. [DOI] [PubMed] [Google Scholar]
- 79.Pritchard DM, Watson AJ, Potten CS, et al. Inhibition by uridine but not thymidine of p53-dependent intestinal apoptosis initiated by 5-fluorouracil: evidence for the involvement of RNA perturbation. Proc Natl Acad Sci U S A 1997;94:1795–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gmeiner WH, Dominijanni A, Haber AO, et al. Improved Antitumor Activity of the Fluoropyrimidine Polymer CF10 in Preclinical Colorectal Cancer Models through Distinct Mechanistic and Pharmacologic Properties. Mol Cancer Ther 2021;20:553–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Haber AO, Jain A, Mani C, et al. AraC-FdUMP10 Is a Next-Generation Fluoropyrimidine with Potent Antitumor Activity in PDAC and Synergy with PARG Inhibition. Mol Cancer Res 2021;19:565–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Pardee TS, Gomes E, Jennings-Gee J, et al. Unique dual targeting of thymidylate synthase and topoisomerase1 by FdUMP10 results in high efficacy against AML and low toxicity. Blood 2012;119:3561–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Pardee TS, Stadelman K, Jennings-Gee J, et al. The poison oligonucleotide F10 is highly effective against acute lymphoblastic leukemia while sparing normal hematopoietic cells. Oncotarget 2014;5:4170–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gmeiner WH, Lema-Tome C, Gibo D, et al. Selective anti-tumor activity of the novel fluoropyrimidine polymer F10 towards G48a orthotopic GBM tumors. J Neurooncol 2014;116:447–54. [DOI] [PMC free article] [PubMed] [Google Scholar]