

Abstract
Focal adhesion kinase (FAK) activation plays a crucial role in vascular diseases. In endothelial cells, FAK activation is involved in the activation of pro-inflammatory signaling and the progression of atherosclerosis. Disturbed flow (D-flow) induces endothelial activation and senescence, but the exact role of FAK in D-flow-induced endothelial activation and senescence remains unclear. The objective of this study is to investigate the role of FAK SUMOylation in D-flow-induced endothelial activation and senescence. The results showed that D-flow induced reactive oxygen species (ROS) production via NADPH oxidase activation and activated a redox-sensitive kinase p90RSK, leading to FAK activation by upregulating FAK K152 SUMOylation and the subsequent Vav2 phosphorylation, which in turn formed a positive feedback loop by upregulating ROS production. This feedback loop played a crucial role in regulating endothelial activation and senescence. D-flow-induced endothelial activation and senescence were significantly inhibited by mutating a FAK SUMOylation site lysine152 to arginine. Collectively, we concluded that FAK K152 SUMOylation plays a key role in D-flow-induced endothelial activation and senescence by forming a positive feedback loop through ROS production
Keywords: Disturbed Flow, Reactive Oxygen Species, p90RSK, FAK, SUMOylation
Introduction:
Endothelial cell activation, characterized by inflammation (upregulated expression of chemokines, adhesion molecules, and proteins involved in cell-cell interactions), apoptosis, and senescence(1), is a hallmark of endothelial cell dysfunction. Endothelial cell dysfunction is an initial step toward the pathogenesis of atherosclerosis and cardiovascular disease (CVD), which is characterized by disrupted vascular tone (imbalanced vasodilation and vasoconstriction), chronic inflammatory reactions within the blood vessel wall, and elevated reactive oxygen species (ROS) production(9). By regulating both physiological (redox) and pathological (oxidative stress) signaling pathways(20,49), ROS exert crucial functions in human health and disease, including atherosclerosis(68). Cytoplasmic ROS are mainly generated by nicotinamide adenine dinucleotide phosphate (NADPH) oxidases (NOX)s(7). Particularly, the upregulation of NOX2, one of the major sources of cytoplasmic ROS, is associated with the inflammation and senescence of endothelial cells(19,52,51) and atherosclerosis(21). However, how ROS trigger chronic inflammatory state of atherosclerosis is not well defined.
In the vessels, endothelial cells are exposed to shear stress, generated by blood flow (16). Laminar flow maintains vascular homeostasis(27,59) by maintaining the expression of nitric oxide synthase (eNOS) and production of nitric oxide (NO), suppressing the expression of inflammatory chemokines and adhesion molecules and promoting the expression of anti-inflammatory and anti-atherogenic genes(60). By contrast, disturbed flow (D-flow) disrupts vascular homeostasis(27,61) by escalating ROS generation(1). Atherosclerosis tends to develop in areas of the vessels where endothelial cells experience D-flow(40,45,62). At D-flow regions, ROS production by endothelial cells is elevated. As such, it is critical to understand how the elevated ROS production is chronically maintained in the area of D-flow in the blood vessel. Previously, we have reported that, D-flow triggers endothelial cell inflammation and atherogenesis by promoting SUMOylation of certain proteins. SUMOylation is a dynamic and reversible form of post-translational modification (PTM) of proteins and is known to alter their functions. For example, SUMOylation is involved in activating nuclear factor-κB (NFκB) and also altering the functional roles of p53, KLF, and ERK5, thereby leading to activation of pro-inflammatory signaling which plays crucial roles in the progression of atherosclerosis. However, the involvement of SUMOylation in D-flow-induced ROS production in endothelial cells remains elusive.
The non-receptor protein tyrosine kinase focal adhesion kinase (FAK) is a scaffold protein that binds to itself and partners through its four-point-one, ezrin, radixin, moesin (FERM) domain(22). Ordinarily, FAK is located in the cytoplasm, and is expressed in most tissues and controls signaling between the plasma membrane and the nucleus(36,46). FAK regulates fundamental cellular processes in the integrin-induced cell adhesion, migration, proliferation, and survival(32,47,50,53,54). Endothelial FAK activation plays a crucial role in regulating endothelial cell inflammation and subsequent development and progression of atherosclerosis(65). Data obtained from a two-hybrid screen assay demonstrate that the N-terminus of FAK is able to bind SUMO E3 ligase of protein inhibitor of activated STAT1 (PIAS1), leading to FAK SUMOylation on the Lys 152 residue (K152) in the FERM domain, which is conserved among different species(36,46). Neither autophosphorylation nor catalytic activity is required for FAK SUMOylation. In contrast, FAK SUMOylation is required for FAK autophosphorylation(36) and increases FAK’s catalytic activity(36,48).
In this study, we investigated the role of FAK SUMOylation in D-flow-induced ROS production and subsequent endothelial cell inflammation and senescence. We found that FAK K152 SUMOylation is not only crucial for instigating endothelial cell inflammation, apoptosis and senescence but also for forming a positive feedback loop in which activation of Vav2 induces an increase of ROS production that feeds back to inflammation.
Results:
D-flow-induced FAK K152 SUMOylation promotes endothelial cell activation
SUMOylation involves the activity of deSUMOylation enzyme sentrin-specific proteases (SENPs), which deconjugate Small Ubiquitin-like Modifier (SUMO) proteins from the protein substrates. Previously, we have demonstrated that SENP2 expression inhibits ERK5 and p53 SUMOylation as well as the consequent endothelial inflammation and apoptosis, respectively. Accordingly, in this study, we transduced endothelial cells with the adenovirus expressing SENP2 (Ad-SENP2) to study the effect of D-flow on FAK SUMOylation (35). To confirm that the Ad-SENP2 transduced into cells is properly expressed, the control cells were transduced with the adenovirus expressing Lacz (Ad-Lacz) (Fig. 1A). The transduced cells were then exposed to D-flow, and total cell lysates were subjected to FAK SUMOylation detection. In the control cells, we noted an increase of FAK SUMOylation after D-flow (Fig.1A, B). However, this increase was completely abolished in the cells overexpressing SENP2 (Fig.1A,B). As the increase of FAK Y397 autophosphorylation represents FAK kinase activation(36), we examined the role of SUMOylation on this FAK phosphorylation. We found FAK Y397 phosphorylation by D-flow in the of control cells, but this phosphorylation was completely inhibited in cells overexpressing SENP2 (Fig. 1A,B). Among the six known isoforms of SENPs, SENP1 and 2 primarily localize in the nucleus like SENP 3,5,6, and 7, but they also present in the extranuclear compartments and the cytoplasm. SENP1 and 2 play a role in activating NF-kB, the master regulator of endothelial inflammation. Because SENP1 shares about 57% sequence identity with SENP2 within the catalytic domain, we also determined the role of SENP1 in D-flow-triggered FAK SUMOylation. By using each specific small interfering RNA (siRNA), we effectively knocked down (Fig. 1E) of each individual endogenous SENP 1 or 2 protein in endothelial cells, without altering the expression of SENP3 and 7 (Fig. 1E). In the cells treated with the control siRNA, we detected FAK SUMOylation only under D-flow condition, but not basal line condition. However, in the cells treated with SENP1 or 2 siRNA, FAK SUMOylation was readily detected even in basal line condition. This data indicates that similar to SENP2, SENP1 repress FAK SUMOylation. An increase of FAK SUMOylation is significantly higher in the cells treated with SENP2 siRNA, compared to that of SENP1 siRNA, suggesting that SENP2 inhibit FAK SUMOylation in a more efficient manner (Fig. 1C–D).
Figure 1: D-flow increases FAK K152 SUMOylation in endothelial cells.
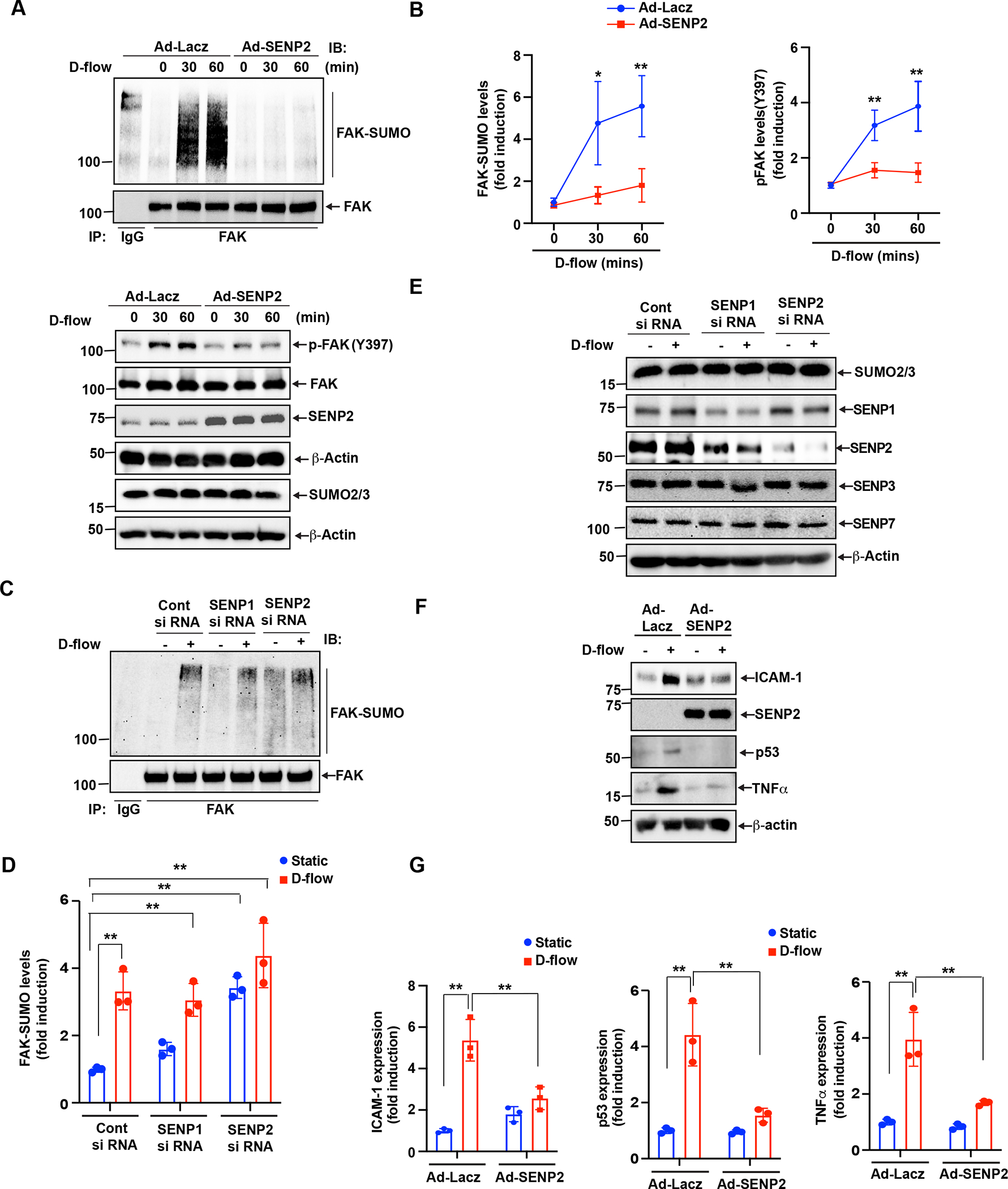
(A-B) HUVECs transduced with Ad-LacZ or -SENP2 were exposed to D-flow for different times as indicated. The level (intensity of region of interest) of SUMOylated FAK was detected in the resulted lysates. Briefly, the lysates were subjected to immunoprecipitation assay using anti-rabbit FAK (or immunoglobulin G as a control). The resulted proteins were then separated by electrophoresis and transferred onto PVDF membranes. Then, level of SUMOylated FAK was detected by immunoblotting with an anti-SUMO2/3. Finally, the same membrane was subjected to IB with anti-FAK as the input control. Shown are representative SUMOylated FAK and input FAK immunoblots (A-uper). Expression of SUMO2/3 was assessed by western blotting(A-lower). Y397 phosphorylated and total FAK were detected in the resulted lysates by IB with specific antibodies as indicated. Blot was reprobed for SENP2 and β-actin (loading control) to show normalization (A-lower). Intensity of the SUMOylated, and phosphorylated FAK region was quantified and represent mean±SD (n=3), **P<0.01 (B). (C-D) HUVECs were transfected with SENP1 or SENP2 or Control siRNA and exposed with and without D-flow. Equal amounts of protein from control and each treatment were immunoprecipitated with anti-FAK antibodies, followed by immunoblotting with the anti-SUMO2/3 antibody. Graph shows densitometric quantification of FAK SUMOylation, which was normalized by total FAK levels. Data represent mean ± SD, n = 3, **P < 0.01 (D). (E) Same Cell extracts were also analyzed by Western blotting for SENP1, SENP2, SENP3, and SENP7 expression levels using their specific antibodies and normalized to β-actin to show the effects of siRNAs on their target and off-target molecules. (F-G) ECs transduced with Ad-LacZ or Ad-SENP2 were exposed to DF for 24 hr. Expression of ICAM1, SENP2, TNF-α, and p53 analyzed by westernbltting and normalized to β-actin (C). Intensity of ICAM1, TNF-α and p53 was quantified and represent mean±SD (n=3) *P<0.05, **P<0.01 (G). For protein simple WES system, protein bands are shown as pseudoblots (A).
We also examined the expression of inflammatory reponse genes including intercellular adhesion molecule 1 (ICAM-1) and tumor necrosis factor-α (TNF-α) as well as the cellular senescence marker p53. We found that SENP2 overexpression inhibits D-flow-induced the expression of both inflamatory and senescence markers (Fig.1F,G). These results suggest that FAK SUMOylation plays a role in D-flow-induced FAK activation and the subsequent endothelial inflammation.
To further demonstrate the involvement of FAK SUMOylation in D-flow-induced endothelial cell inflammation, we generated a FAK SUMOylation deficient mutant construct by substituting lysine152 to arginine (FAK K152R). We transiently overexpressed FAK K152R mutant or FAK wild type (WT) in endothelial cells prior to exposing them to D-flow and found that FAK SUMOylation triggered by D-flow in cells overexpressing the FAK WT (Fig. 2A, B) was completed abolished in cells overexpressing the FAK K152R (Fig. 2A, B). The increased FAK K152 SUMOylation induced by D-flow was also associated with the increase in FAK Y397 phosphorylation (Fig. 1A,B; Fig.2A, B). FAK activation in endothelial cells has a crucial role in D-flow-mediated the activation of transcription factor NF-κB. Whereas NF-κB activation induced by TNF-α or hydrogen peroxide (H2O2) is independent of FAK catalytic activity, D-flow-induced NF-κB depends on FAK’s catalytic activity(15,49). To study if FAK K152 SUMOylation plays a role in NF-κB activation by D-flow, we co-transfected endothelial cells with the FAK WT or the FAK K152R mutant and the dual luciferase NF-kB activity reporter gene that contains five NF-kB binding sites as an enhancer. As shown in Fig. 2C, while D-flow activated NF-kB in cells transfected with the FAK WT, it failed to activate NF-kB in cells expressing the FAK K152R mutant. We also found that D-flow-induced ICAM-1, TNF-α, and p53 expression was inhibited in the FAK K152R mutant expressing cells (Fig. 2A,B). These results suggest that FAK SUMOylation mediates endothelial cell inflammation and senescence due to D-flow.
Figure 2: D-flow-induced FAK K152 SUMOylation promotes endothelial cell inflammation and senescence.
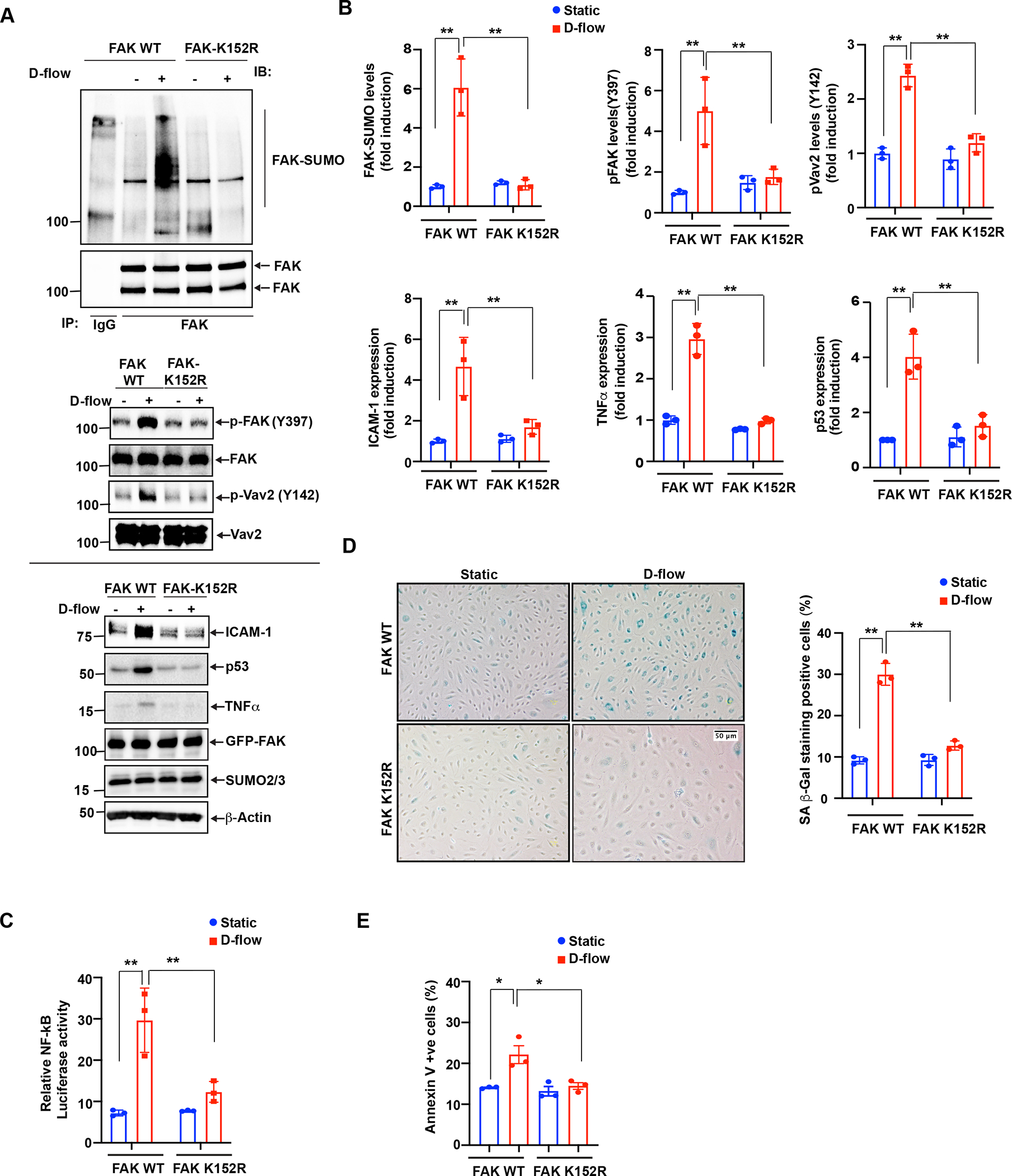
(A) HUVECs were transfected with FAK-WT or FAK-K152R plasmids for 36 hours and exposed to D-flow for 30 min or 24 hrs (for ICAM-1, TNF-α, and p53 expression), and cell extracts were prepared. To detect FAK SUMOylation, immunoprecipitation with anti-FAK was performed, followed by immunoblotting with Sumo2/3 antibody. Cell lysates were also immunoblotted for phosphorylation of FAK (p-FAK) and Vav2 (p-Vav2). The blots were sequentially reprobed with anti-FAK, anti-Vav2, anti-Sumo2/3 and β-actin for normalization and loading control. Shown is a representative set of data from three biological replicates. (B) Graphs show densitometric quantification of SUMOylated and phosphorylated FAK, phosphorylated Vav2 and ICAM-1, TNF-α, and p53 expression levels. Fold increases are shown after normalization using the total FAK and Vav2 and β-actin band intensity. Data represent mean ± SD, n = 3, **P < 0.01. (C) HUVECs were transfected with a mixture of either FAK-WT or FAK-K152R plasmid and the dual luciferase NF-kB activity reporter gene and exposed to D-flow for 24 hr. NF-kB activity in cell lysates was measured using the dual-luciferase reporter assay system. Relative NF-κB activity was determined by normalizing firefly luciferase activity to Renilla luciferase activity. Mean±SD (n=3) **P<0.01. (D) HUVECs transfected with FAK-WT or FAK-K152R were exposed to D-flow for 24 hr, and cell apoptosis was determined by annexin V staining as described in Materials and Methods. The graph shows the percent of cells in culture with annexin V staining. Mean±SD (n=3) *P<0.01. (E) HUVECs transfected as described in D were exposed to D-flow for 24 hr. Cells were stained for senescence-associated β-galactosidase (SA-βgal). The graph shows the percentage of SA-B gal positive cells. Mean±SD (n=3) **P<0.01.
Previously, we reported that D-flow can also induce endothelial cell senescence(37, 38). Here, we found that the FAK K152R mutation significantly decreased apoptosis and the number of D-flow-induced beta-galactosidase (SA- β gal) (Fig. 2D,E). SA- β gal and annexin V are commonly accepted biomarkers for identifying cellular senescence(17,63). Taken together, these data show that FAK SUMOylation plays a key role in inducing inflammation and senescence in endothelial cells exposed to D-flow.
D-flow-induced p90RSK activation upregulates FAK SUMOylation, Y397 phosphorylation, and subsequent VAV2 phosphorylation.
Previously, we have demonstrated the important role of the activated p90RSK pathway(22) in endothelial cell inflammation(34,40,59). p90RSK can be directly phosphorylated by ERK1/2 at the T359/S363 residues located within the carboxyl terminus domain, which in turn promoting p90RSK S380 autophosphorylation (39). An increase of p90RSK S380 autophosphorylation is required for the full activation of p90RSK kinase and therefore can be represented as p90RSK kinase activation (5). In this study, to determine the role of ERK1/2-independent p90RSK kinase activation on D-flow-mediated FAK SUMOylation, we measured the level of p90RSK S380 phosphorylation. We found that D-flow-induced FAK SUMOylation activation, FAK (Y397) phosphorylation, p90RSK kinase activity, and Vav2 (Y142) phosphorylation. However, this effect was completely inhibited by the p90RSK specific inhibitor, FMK-MEA(40) (Fig. 3A–D). To confirm these findings, we suppressed the activation of p90RSK kinase activity through siRNA-mediated depletion approach. We confirmed that p90RSK depletion substantially attenuated D-flow-mediated FAK SUMOylation (Fig.3 E–G).
Figure 3: p90RSK activation regulates both SUMOylation and Y397 phosphorylation of FAK and the subsequent VAV2 phosphorylation.
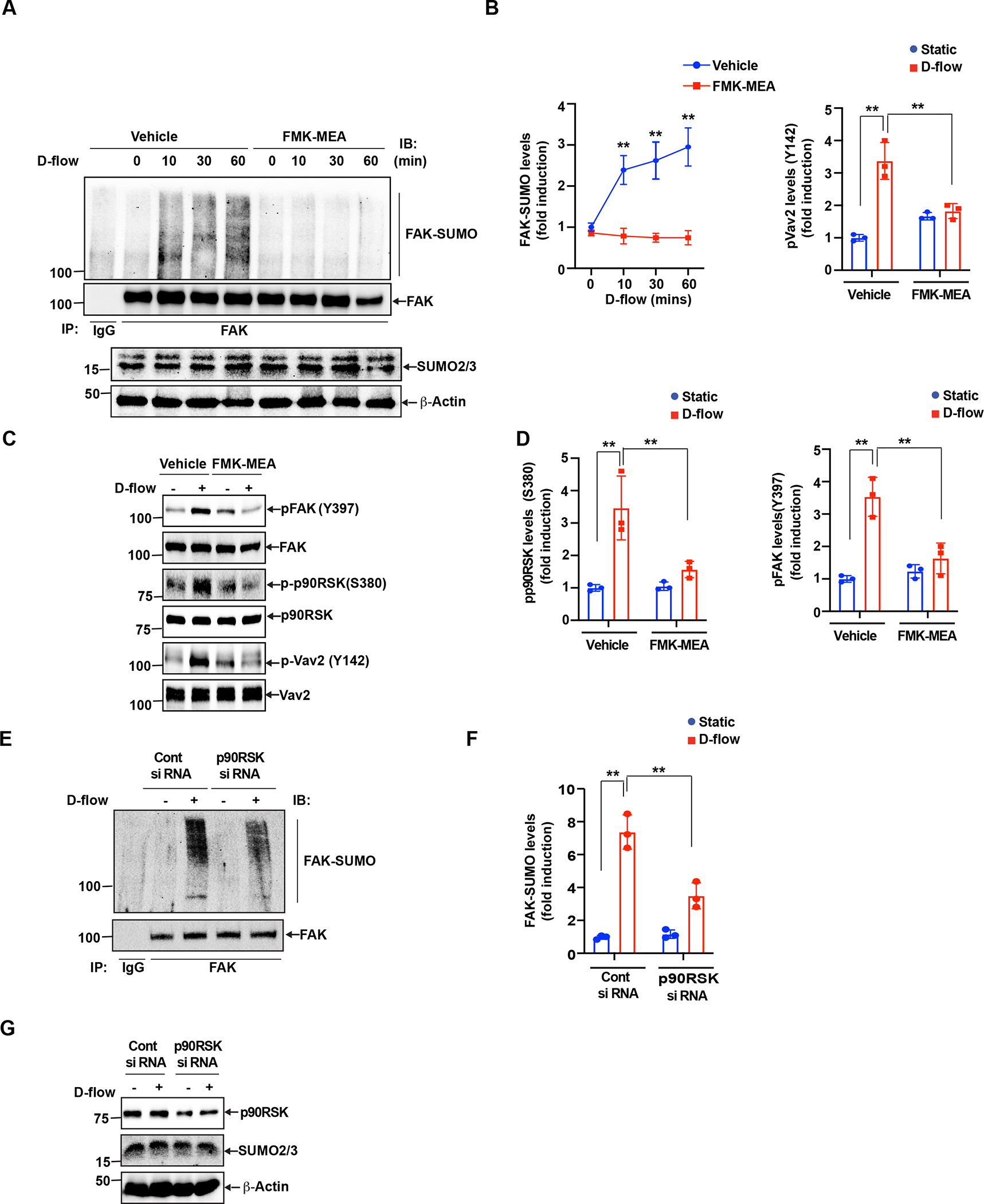
(A-B) A. (A) HUVECs were pre-treated with and without FMK-MEA (10 μM) for 1 hr and exposed to D-flow for 0 – 60 min. Cell lysates were immunoprecipitated with anti-FAK or control IgG and immunoblotted with anti-SUMO2/3 (top panel). Total cell lysates were immunoblotted with anti-FAK and anti-SUMO2/3 to show the presence of these proteins (lower panels). The blots are representative data from 3 independent experiments. (B) The panel shows densitometric quantification of FAK SUMOylation, which was normalized by the total FAK protein levels. Data represent mean ± SD. n = 3, **P < 0.01. (C) HUVECs were pretreated for 1 hr with FMK-MEA and exposed to D-flow for 30 min. Cell extracts were immunoblotted with anti-phospho-FAK, anti-phospho-p90RSK, anti-phospho-Vav2, and the blot was sequentially reprobed for the total p90RSK, FAK, and Vav2 levels to be used to normalize phosphorylation levels of each protein. The blots are representative data from 3 independent experiments. (D) Densitometric quantification of phosphorylation of the 3 proteins shown in C. Data represent mean ± SD. n = 3, **P < 0.01. (E-G) (E) Cells were transfected with p90RSK or control siRNA (100 nm) and then exposed to D-flow for 60 min. Equal amount of protein from control and each treatment were analyzed for FAK SUMOylation by immunoprecipitation using anti-FAK, and the immunocomplexes were analyzed by immune blotting with the anti-SUMO2/3 antibody. Quantitative densitometry data showing the effects control and p90RSK on the FAK SUMOylation on the D-flow response. Data represent mean ± SD, n = 3, **P < 0.01 (F). (G) The protein extracts were also analyzed for p90RSK levels to show the effects of the SiRNA on their target and off-target molecules.
FAK can activate the small RhoGTPase Rac1(13), an important component of the signaling machinery activated by the cell-extracellular adhesion molecules(10,18). Vav1 expression is normally restricted to hematopoietic cells, while Vav2 and Vav3 are more widely expressed (64, 55, 4). Vav2 is phosphorylated on at least three tyrosines (Y142, Y159 and Y172) in the acidic domain, and this phosphorylation results in an increase in Vav2 GEF activity (64, 42). Since we found the clear up-regulation of phospholylation of Y142 Vav2 after d-flow, to detect the Vav2 GEF activity, we detected Vav2 Y142 phosphorylation. Vav2 is a member of the Dbl protein family and is a Rac1 GEF (65). Accordingly, Vav2 activation also activates Rac1(25). We found that not only the FAK K152R SUMOylation deficient mutant (Fig.2A, B) but also FMK-MEA significantly inhibited D-flow-induced Vav2 phosphorylation (Fig. 3C,D). Based on previously published data(13) and those from this study, we hypothesize that D-flow triggers endothelial activation, senescence, and apoptosis through activating Vav2, which requires Vav2 Y142 phosphorylation(30). To elucidate this hypothesis, we treated endothelial cells with a specific Vav2 siRNA prior to D-flow exposure, and verified that the Vav2 siRNA effectively knocked down of endogenous Vav2 protein (Fig. 4C). When the cells treated with the control siRNA were exposure to D-flow, FAK SUMOylation was significantly increased. However, this increase was effectively suppressed by the Vav2 siRNA treatment (Fig. 4A, B). Next, we investigated the role of Vav2 in D-flow-induced NF-κB activation and subsequent inflammatory gene expression. Depletion of Vav2 significantly inhibited D-flow-induced NF-κB activation (Fig. 4C). We also found that D-flow induced ICAM-1, TNF-α, and p53 expression was significantly decreased by the depletion of Vav2 (Fig. 4 D,E). These results indicate a key role for Vav2 in EC activation, especially D-flow–induced EC activation. After D-flow exposure, we also measured telomere shortening for endothelial senescence, and the percentage of Annexin V positive cells for endothelial apoptosis (Fig.4 F,G). While all these parameters were upregulated by D-flow exposure, siRNA-induced Vav2 depletion significantly abolished these upregulations. Taken together, these results strongly supported the notion that Vav2 is involved in the regulation of D-flow-induced endothelial activation, senescence, and apoptosis.
Figure 4: The role played by Vav2 phosphorylation in endothelial cell activation and senescence.
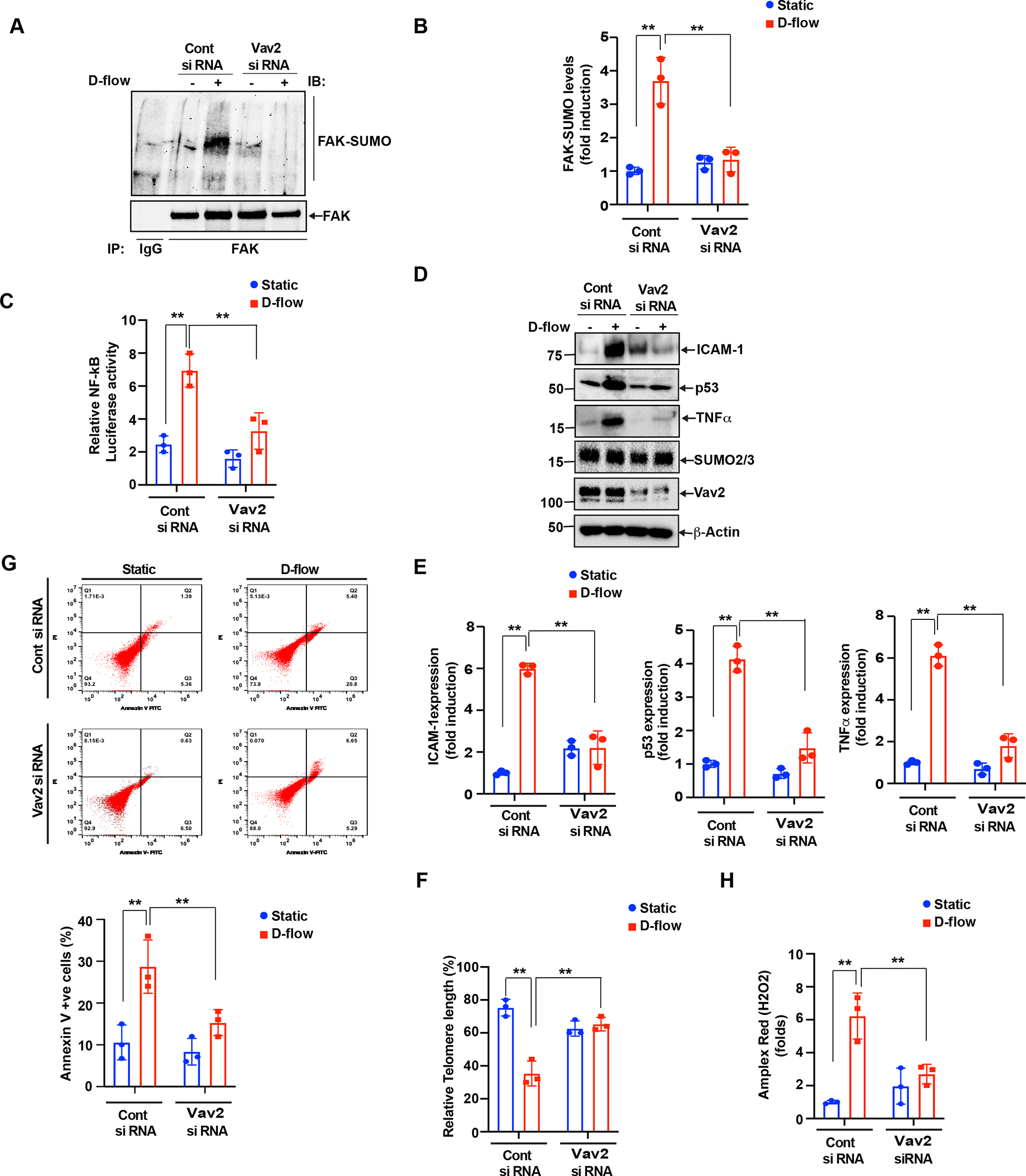
(A-B) (A). HUVECs were transfected with Control SiRNA or Vav2 siRNA (100 nm) and then exposed to D-flow 60 min and cell extracts were prepared. Equal amount of protein from control and each treatment were immunoprecipitated with anti-FAK antibody, and the immunocomplexes were analyzed by Western blotting for the SUMO2/3 antibody and normalized to FAK. The bar graphs represent densitometry quantitative analysis of three independent experiments. The values are presented as Mean ± SD n = 3, **P < 0.01 (B). (C) NF-kB activity was measured as we described in Figure 2C and Materials and Methods. The graph shows Mean±SD (n=3). **P<0.01. (D-E) (D) All of the conditions were the same as for A, except that after SiRNA transfection, the cells were exposed with D-flow or static conditions for 24 hours, and cell extracts were prepared. Equal amounts of protein from control and each treatment were analyzed by Western blotting for ICAM-1, TNF-α, p53, Vav2, and SUMO2/3 expression levels using their specific antibodies and normalized to β-actin. The bar graph represents densitometry Quantitative analysis of three independent experiments. Data represent mean ± SD values of three experiments n=3, **P < 0.01 (E). HUVECs pre-treated with either the control or Vav2 siRNA were exposed to D-flow for 24 hr. (F) Percentages of annexin V–positive cells determined by flow cytometry. Shown is a representative data from three biological replications. Mean±SD (n=3) **P<0.01. (G) The TL length was assayed using the TL PNA kit/FITC for flow cytometry. The mean fluorescence intensity was used to estimate the relative TL length. Data were analyzed using FlowJo software and results plotted as mean±SD (n=3). **P<0.01. (H) ROS levels in cells transfected with control siRNA or Vav2 siRNA and treated with or without D-flow for 2 hr. Data represent mean ± SD. n = 3, **P<0.01.
NADPH oxidase 2 regulates the D-flow-induced endothelial cell activation
The activated Rac1 signaling can regulate ROS production(3,14). The NADPH oxidase isoform 2 (NOX2)(11), one of the major enzymes that promote ROS production, is modulated by Rac1(3) and Vav2(68). NOX2 mediates endothelial cell activation and senescence(29,52,53) and plays a pro-atherosclerotic role(19). As NOX2 is a major generator of oxygen radicals(26,56), Vav2 activation mediated by FAK SUMOylation might be involved in D-flow-triggered ROS production. Indeed, we found that depletion of Vav2 significantly inhibited D-flow-triggered ROS production (Fig. 4H). From these results, we propose that NOX2 activation induced by Vav2 plays a crucial role in D-flow-induced ROS production and also in inducing the D-flow-dependent endothelial cell activation and senescence. To further test the validity of the proposed line of molecular events, first we used the Amplex-Red-based assay to measure the formation of H2O2 in cells. In endothelial cells exposed to D-flow, we observed a significant increase of H2O2 formation in a time-dependent fashion (Fig. 5A). However, the increase of H2O2 formation stimulated by D-flow exposure was markly decreased when endothelial cells were pre-treated with di phenyleneiodonium (DPI), a NOX/NADPH Oxidase inhibitor(44), or manganese(III) tetrakis(1-methyl-4-pyridyl)porphyrin (MnTMPyP) (41), or polyethylene glycol (PEG)-catalase (6) (Fig.5B,C). Similarly, siRNA-mediated NOX2 depletion also significantly suppressed the formation of H2O2 triggered by D-flow (Fig. 5D). We also knocked down NOX2 using siRNA, exposed the cells to D-flow, and assayed for further downstream events related to endothelial cell activation and senescence. Whereas D-flow increases NF-kB activation (Fig. 5E), telomere shortening (Fig. 5F), and the percentage of Annexin V (Fig. 5G) positive cells in the control endothelial cells, these increases were blunted in the NOX2 depleted cells (Fig.5 E–G). All together our study shows that Vav2-induced NOX2 activation is required for the DF-triggered ROS production and endothelial cell activation and senescence.
Figure 5: NADPH oxidase 2 activation in DF-induced endothelia cell activation and senescence.
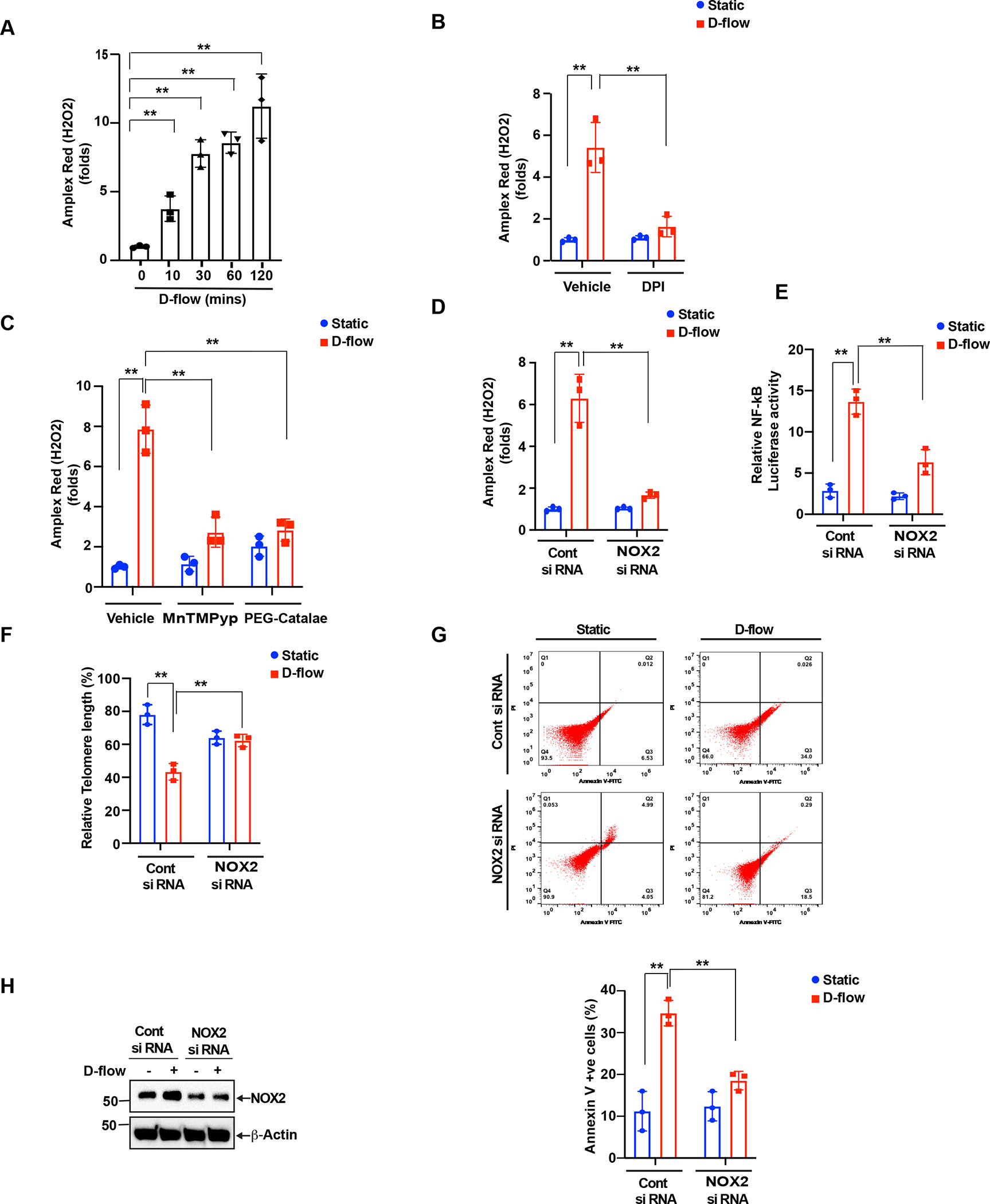
Modulation of cellular hydrogen peroxide production. (A). HUVECs cells were exposed with D-flow, and H2O2 levels determined by Amplex Red assay. Panel A shows the time course of H2O2 production following treatment with D-flow. (B). H2O2 production in HUVECs that were pre-treated with or without DPI (10 μM) for 1 hr and exposed or not exposed to D-flow for 2 hr. Data are presented as mean ± SD (n=3). **P<0.01. In panel C shows cells were pre-treated with PEG-catalase (100 U/ml) or MnTMPyP (10 μM) for 1hr and then exposed to D-flow or static conditions for 2hrs and measured H2O2 levels by using Amplex Red assay. (D). Cells were transfected with siRNA (100 nm) to knock down NOX2 and then exposed to D-flow and static conditions for 2 hrs. H2O2 levels were measured with Amplex Red assay. Data are presented as mean ± SD (n=3). **P<0.01. (E) NOX2 depletion inhibits NF-κB activation. HUVECs were transfected with control or NOX2 targeted siRNA and then treated with or without D-flow for 24 hr. NF-κB activation was determined by the dual luciferase assay. Data represent mean ± SD (n = 3). **P< 0.01. (F and G) HUVECs prepared as described in C were stained with FITC annexin V and the percent of stained cells is shown (F). Telomere length was determined by using the TL PNA kit (G). Data represent mean ± SD (n = 3). **P < 0.01. (H). Equal amounts of protein from the same cell extracts from each condition were also analyzed by Western blotting for NOX2 levels to show the efficacy of the siRNA on its target molecule level.
Vav2-induced ROS production feeds back to endothelial cell activation
Our data as well as published reports have indicated that both p90RSK and FAK are upstream of VAV2 phosphorylation (Fig. 1–5)(14,21,26,55). Since Vav2 has a role in D-Flow-induced ROS production which then instigates endothelial cell activation and senescence, we initially thought that the inhibition of ROS production would not show any effects on the activation of p90RSK-FAK SUMOylation-Vav2 signaling cascade. Surprisingly, we observed that both siRNA-mediated NOX2 depletion and pharmacological inhibition of NADPH oxidase attenuated D-flow-dependent FAK SUMOylation (Fig.6 A–G). Furthermore, DPI pre-treatment inhibited the elevation of not only p90RSK kinase activity but also FAK phosphorylation on Y397 as well as Vav2 phosphorylation on Y142, all of which are induced by DF (Fig. 6H,I). These data show that ROS production regulates both upstream and downstream of p90RSK-FAK-Vav2 signaling. To examine the effects of siRNA-induced NOX2 depletion and DPI pre-treatment on D-flow-mediated the elevated expression of ICAM-1, TNF-α, and p53, we noted that the increase of these inflammatory and senscence molecules was significantly inhibited (Fig.6 J–K). This results indicate a key role for ROS production in D-flow induced endothelial activation, senescence, and apoptosis. This observation can only be explained by the presence of a positive feedback loop. The establishment of this feedback loop instigates endothelial cell activation and senescence by D-flow (Fig.7).
Figure 6: Vav2 role in Nox2 dependent ROS production mediated p90RSK-FAK positive inflammatory feedback loop.
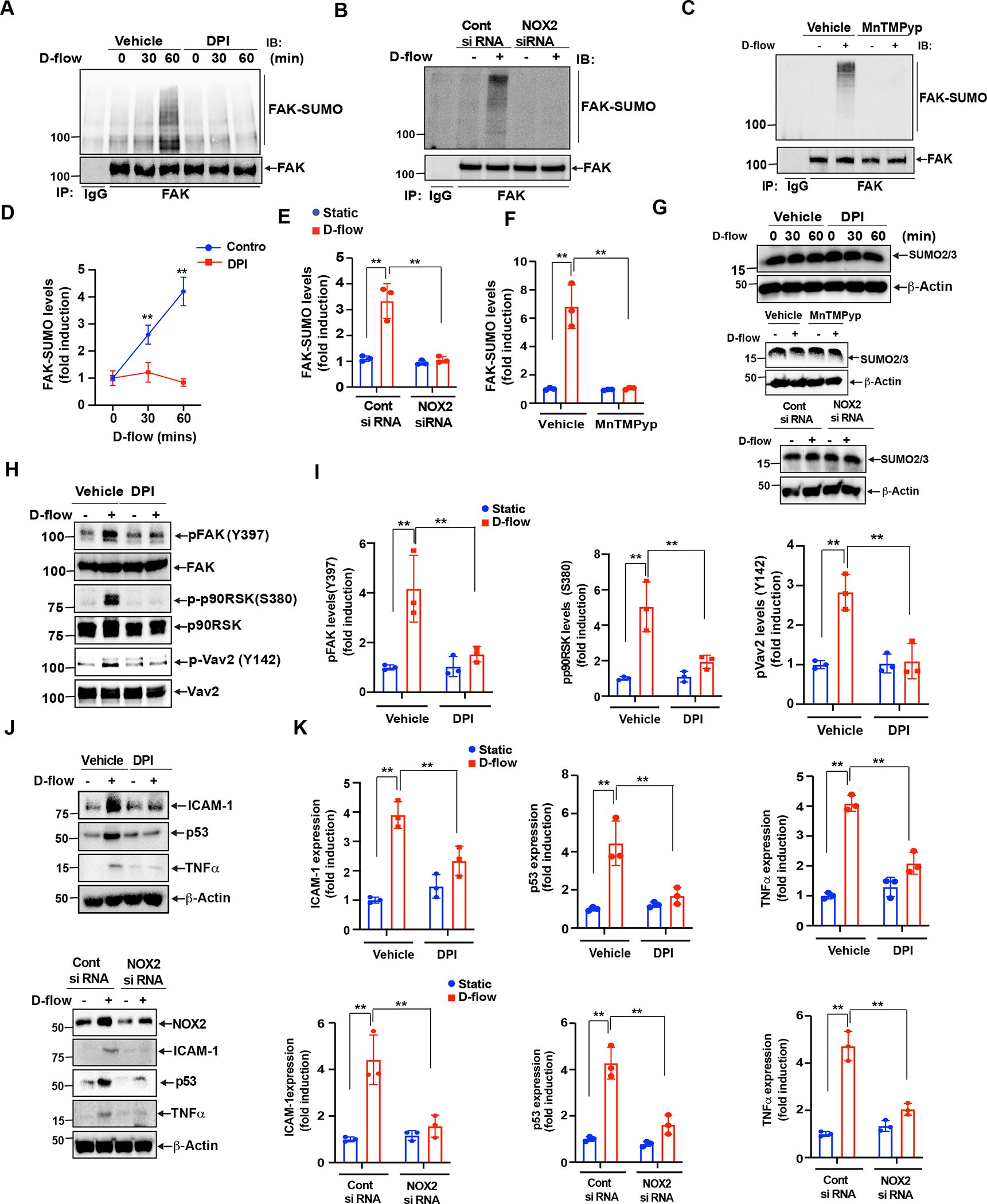
(A, B). HUVECs were pretreated with either DMSO (vehicle) or DPI (10 μM) for 1 hr followed by D-flow stimulation for the indicated times. FAK SUMOylation was detected by immunoprecipitation with anti-FAK or IgG as a control followed by Western blotting with anti-SUMO2/3. The blot was sequentially reprobed for FAK for normalization. The blots are representative data from 3 independent experiments. Quantification of FAK SUMOylation levels is shown after normalization by the total protein levels. Data represent mean ± SD (n = 3–4). **P<0.01 (B). (C-G) Cells were pretreated with 10 μM MnTMPyp for 1hr or transfected with siRNA to knock down NOX2 (100 nm). After that cells were exposed with D-flow for 60 min and cell extracts were prepared. An equal amount of protein from control and each treatment was immunoprecipitated with anti-FAK or anti IgG and the immunocomplexes were immunoblotted with anti-SUMO2/3 antibody. The blots were sequentially reprobed with anti-FAK antibody to show normalization, lane loading control. (G). Equal amounts of protein from the same cell extracts were also analyzed by Western blotting for SUMO2/3, to show the expression of SUMO2/3 and normalized to β-actin. (E-F) shows densitometric quantification of FAK SUMOylation, which was normalized by total FAK protein levels. Data represent mean ± SD, n = 3, **P < 0.01. (H, I) (H). HUVECs were pretreated with either vehicle or DPI for 1 h followed by D-flow stimulation for 1 hour and cell extracts were analyzed by Western blotting for phospho-FAK, phospho-p90RSK and phospho-Vav2 using phosphorylation-specific antibodies against these proteins. The blots were reprobed with anti-FAK, anti-p90RSK and anti-Vav2 to show the total protein expression and to also normalize phosphorylation levels. The blots are representative data from 3 independent experiments. Quantification by densitometry of phosphorylation levels of FAK, p90RSK and Vav2. Data represent mean ± SD (n = 3). **P < 0.01. (I). (J, K) (J) HUVECs were pretreated with DPI for 1 hr or transfected with siRNA to knock down Nox2 and then stimulated by D-flow or no flow for 24 hr. ICAM-1, TNF-α, and p53 expression detected by Western blotting is shown. Blots were reprobed with anti-β-actin antibody to show loading controls. was used as loading control. The blots are representative data from 3 independent experiments. (K) Densitometry was used to quantify ICAM-1, TNF-α, and p53 expression levels. Data represent mean ± SD(n = 3). **P < 0.01.
Figure 7.
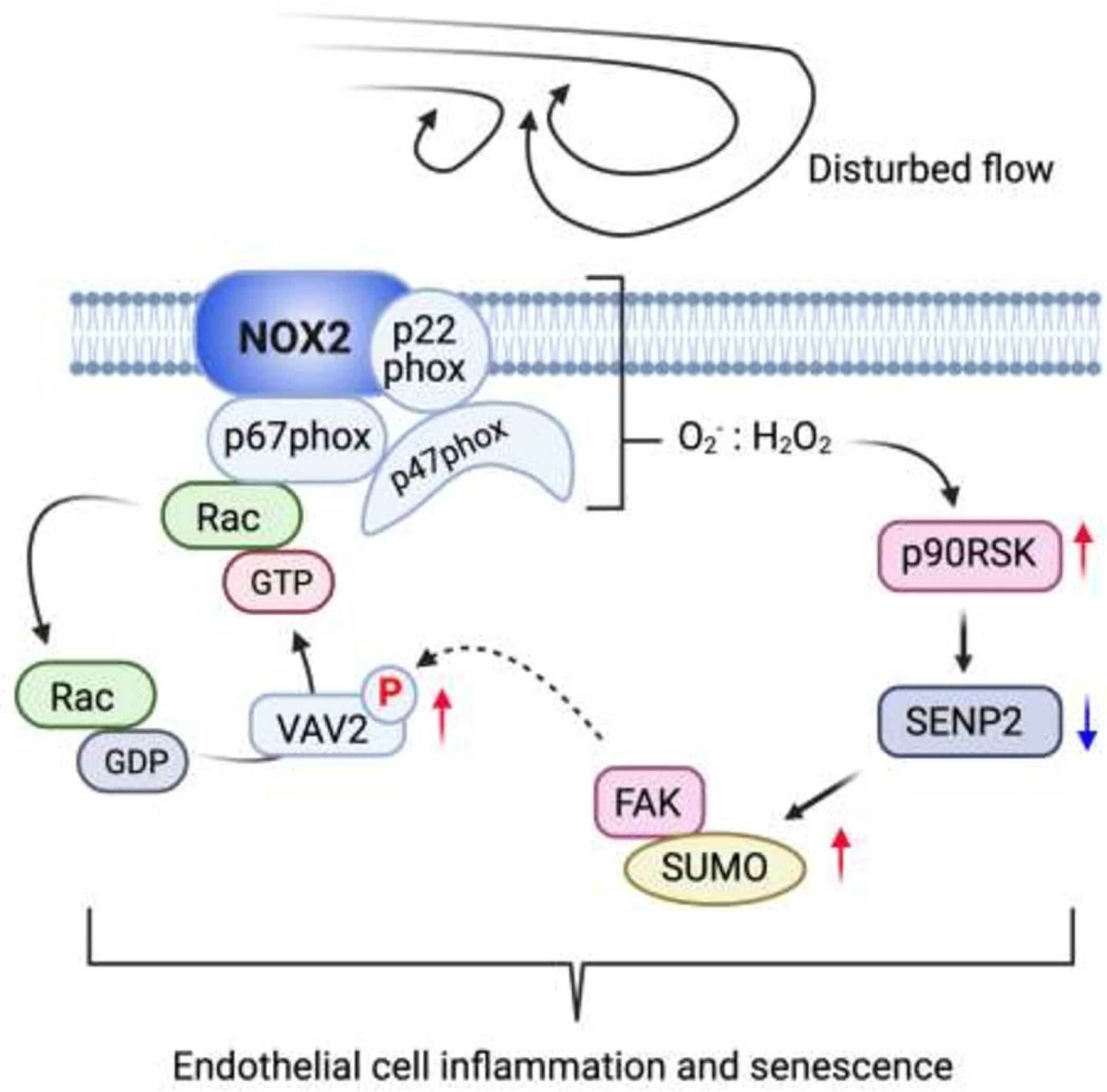
Summary graphic illustration.
Discussion
In the present study, we have made several new findings: (1) FAK K152 SUMOylation is vital for the induction of endothelial cell activation, apoptosis, and senescence by D-flow; (2) Endothelial cell activation by D-flow is mediated by FAK K152 SUMOylation and VAV2-regulated NOX2 activation; and (3) via ROS-induced p90RSK activation, FAK K152 SUMOylation not only activates pro-inflammatory downstream events but also feeds back on to the inflammatory response by intensifying the VAV2-regulated NOX2 signaling and thereby inducing more ROS production. These findings suggest that FAK K152 SUMOylation is one of the major but unexplored PTMs underlying the D-flow-induced sustained ROS production and chronic endothelial cell activation. Although the sustained ROS production-mediated chronic endothelial cell activation is vital for the progression of atherosclerosis, its underlying mechanism remains unclear. Our findings suggest that FAK activation in endothelial cells can be an attractive therapeutic target for atherosclerosis and CVD and highlight the importance of SUMOylation-dependent signaling in D-flow-induced atherosclerosis. The interplay between FAK and p90RSK has been suggested. For example, Yurdagul et al. have reported that oxLDL increased Ikb kinase activation via FAK-dependent p90RSK activation(67). In this study, we found that p90RSK-FAK signaling is a part of a positive feedback loop in which ROS completes the loop formation. In addition, the inter-activation between p90RSK and FAK plays a crucial role for chronic endothelial cell activation.
Elevated ROS production can regulate SUMOylation(57,66). In the present study, we found that increased FAK K152 SUMOylation leads to more production of ROS regulated by NOX2. We show that NOX2 induces sustained ROS generation through a FAK-p90RSK-VAV2-dependent mechanism. Treatment of endothelial cells with the NOX inhibitor reduces ROS production triggered by D-flow. This is associated with the reduction of D-flow-induced FAK K152 SUMOylation and FAK-p90RSK-Vav2 activation. Consistent with this, both siRNA of NOX2 and Vav2 dramatically decrease D-flow-mediated endothelial cell activation. Together, these results suggest that FAK SUMOylation can be initiated by D-flow-induced ROS production, but at the same time FAK-SUMOylation can also induce ROS production by activating the FAK-p90RSK-Vav2 module. Therefore, ROS-induced FAK SUMOylation forms a positive feedback loop, which accelerates D-flow-induced ROS production and endothelial cell activation. Lin et al.(43) have suggested the role of p90RSK in upregulating ROS production. They generated FSP-1-specific p90RSK transgenic mice (RSK-Tg), which overexpressed p90RSK specifically in fibroblasts, and found a significant increase in fibroblast numbers and tubular epithelial damage after obstructive injury in the kidney. They also found hyperproduction of ROS by RSK-Tg fibroblasts, which caused epithelial apoptosis by activating b-catenin and FOXO1 activity. These data also suggest the significant role of p90RSK-mediated ROS production not only in D-flow-induced endothelial cell activation and atherosclerosis, but also in obstructive chronic kidney disease. Therefore, to determine the contribution of FAK-SUMOylation in regulating p90RSK-mediated ROS production would be critical for devising therapeutic interventions against D-flow-induced vascular dysfunction, atherosclerosis and obstructive chronic kidney disease. It is worth noting that mitochondrial ROS may also contribute to this positive feedback loop. Recently, Fukai T et al.(24) have reviewed the crosstalk between NADPH oxidase and mitochondria-induced ROS, namely “ROS-induced ROS release”. The authors suggested that “ROS-induced ROS release” might generate another positive feedback loop and maintain persistent ROS production with sustained signaling, as we reported in this manuscript. It is possible that the positive feedback loop formed by FAK SUMOylation and “ROS-induced ROS release” triggered by both NADPH oxidase and mitochondrial play a key role in regulating long lasting effects of D-flow in concert.
It is not clear why two steps are necessary to fully activate FAK’s catalytic function. One possibility is that FAK SUMOylation-mediated FAK activation may not be able to activate all of the FAK’s substrates so that only a limited number of them can be activated by SUMOylation. It is also not clear how FAK SUMOylation can promote FAK kinase activity. One possibility is that SUMOylated FAK may inhibit FAK phosphatase such as protein-tyrosine phosphatase D1 (PTPD1)(12) from binding, thereby maintaining its activated state which is necessary for chronic endothelial cell activation. Another possible scenario might be for SUMOylated FAK to bind an upstream kinase such as Src(31) to keep it activated. Further investigation is necessary to determine the exact mechanism for the sustained FAK activity and FAK SUMOylation.
Lastly, we found that the deSUMOylation enzyme SENP2 is essential for FAK SUMOylation(36,46). SENP2 controls protein SUMOylation(35) by removing SUMO from the SUMOylated substrate(29). SUMOylation has been shown to regulate a wide range of cellular functions, many of which are associated with activities in the nucleus. Consisting of multiple nuclear localization signals (NLS) and nuclear export signals (NES), SENP2 can be localized in the nucleus but can also shuttle between the nucleus and the cytoplasm(33). SENP2 shuttling between the nucleus and the cytoplasm is modulated by its PTMs(2,29). Previously, we have reported that the depletion of SENP2 in endothelial cells leads to their apoptosis and inflammation by enhancing the SUMOylation of p53 and ERK5, respectively(29). We further showed that, by phosphorylating SENP2 on the Threonine 368 residue (T368), D-flow triggers SENP2 nuclear export, thereby reducing the overall deSUMOylation activity in the nucleus. Consequently, SUMOylation of p53 and ERK5(26) is enhanced in the nucleus, increasing endothelial cell apoptosis, inflammation, and atherosclerosis (2,28,29,27). As FAK is mainly localized in the cytoplasm(69), SENP2 nuclear export induced by D-flow should decrease FAK SUMOylation. However, in this study, we found that D-flow-induced FAK SUMOylation was inhibited in endothelial cells over expressing SENP2(2). Therefore, overexpress SENP2 inhibits FAK K152 SUMOylation after D-flow might not be due to SENP2 nuclear export induced by SENP2 T368 phosphorylation. Mass spectrometry (MS)-based phospho-proteomics identified that SENP2 can be phosphorylated on Serine 32 (RRSDS), Serine 333 (LGSGS), and Serine 344 (KVSII) residues(8,68). Among these residues, Serine 344 (S344) is positioned proximal to SENP2’s catalytic domain(23) and therefore may alter its deSUMOylation activity, and deactivation of SENP2 may play a role of D-flow-induced FAK SUMOylation. We will explore these possibilities in the next study.
Overall, the major finding in this study is that FAK K152 SUMOylation forms a positive feedback loop, which induces the persistent activation of FAK and H2O2 production. This persistent activation of positive feedback loop is the key leads to endothelial activation and the subsequent atherosclerotic plaque formation. To corroborate this finding, we could have generated the FAK K152R SUMOylation site mutant knock-in (KI) mice and detect the formation of atherosclerosis in the resulted KI mice. Because FAK SUMOylation deficient K152R mutant decreases FAK kinase activation and we did not see any functional difference between FAK K152R and the inhibition of FAK kinase activity, presumably, the phenotype of FAK K152R and FAK kinase inhibitor on atherosclerosis would be similar. Indeed, Yamaura T et al. (65) have recently demonstrated therapeutic properties of FAK inhibitor using a mouse model of atherosclerosis. Furthermore, it is very difficult to prove the existence of the positive feedback loop mouse models. As such, the additional FAK K152R KI mouse model may not provide us significant knowledge than those obtained from our in vitro studies.
Innovation
The interplay between ROS and chronic inflammatory disease such as atherosclerosis, diabetes, and hypertension has been suggested. However, the mechanism how the ROS production can chronically maintain pro-inflammatory status is still not completely uncovered. The current study demonstrates for the first time that p90RSK activation-mediated FAK K152 SUMOylation plays a crucial role for VAV2-regulated NOX2 activation and subsequent ROS production, which forms a positive feedback loop by activating p90RSK and triggers inflammation and senescence. Our work illustrates a novel role of p90RSK-mediated FAK SUMOylation in ROS production, providing a potential therapeutic target for the treatment of chromic pro-inflammatory disease.
Summary Graphic Illustration
Disturbed flow increased FAK K152 SUMOylation regulating p90RSK and SENP2 function. FAK K152 SUMOylation upregulates FAK kinase activity and increases Vav2 tyrosine phosphorylation and subsequently upregulates NOX2 activation. Lastly, this Vav2-induced ROS production via NOX2 activation feeds back to endothelial cells activation by activating redox-sensitive kinase of p90RSK. This positive feedback loop consisting with p90RSK, SENP2, FAK, and Vav2-NOX2 maintains ROS production and promotes endothelial inflammation and senescence.
Materials and Methods
Antibodies and reagents: Antibodies and reagents:
The following Antibodies and chemicals were acquired: Anti-phopsho-FAK Y397 (3283), anti-FAK (3285), anti-phospho-p90RSK S380 (9341), anti-Vav2 (2848), anti-SENP1 (11929), anti-p53 (2527), anti-SENP3 (5591) and anti-ICAM1 (4915) were purchased from Cell Signaling Technology (Beverly, MA). Anti-Nox2 (NBP2–67680), and anti-TNFa (NBP1–19532) was purchased from Novus biologicals. (Centennial, CO). Anti- SENP2 (109590), anti-SENP7 (ab186963) purchased from abcam (Waltham, MA). In addition, anti-p90RSK (MAB 2056) was from R&D Systems (Minneapolis, MN), anti-β-actin (A3854), Catalase-polyethylene glycol (C4963) from Sigma-Aldrich (St. Louis, MO), anti- phospho-Vav2 (PA5–105187) from Thermo Fisher Scientific (Waltham, MA) and anti-SUMO2/3 (AP1224a) was from Abcepta, Inc. (San Diego, CA). Protease inhibitor cocktail (P8340), PMSF (36978), NEM (E3876) and Diphenyleneiodonium chloride (D2926) were purchased from Sigma-Aldrich (St. Louis, MO). The TL PNA kit/FITC flow cytometry kit (K5327) and Quick ChangeII Site-Directed Mutagenesis Kit (200523) were purchased from Agilent Technology (Santa Clara, CA). The dual luciferase assay kit (E1910) was from Promega Life Sciences (Fitchburg, WI). Amplex® Red assay kit (A22188) and Lipofectamine 2000 transfection reagent (11668027) were from Thermo Fisher Scientific (Waltham, MA).The manganese(III) tetrakis(1-methyl-4-pyridyl)porphyrin (MnTMPyP) (475872) purchased from EMD Millipore (Burlington, MA)
siRNA:
Short interfering RNA (siRNA-SMARTpool) targeting human Nox2 siRNA (L-011021–00-0010), human Vav2 siRNA (L-005199–00-0010) and, human p90RSK siRNA (L-003025–00-0005) was purchased from Dharmacon RNAi technologies. SENP1 (WD05005417), SENP2 (WD08342101) and, control non-targeting (WD08342103) siRNA was purchased from Sigma-Aldrich.
Generation of plasmids and adenoviruses:
Plasmid containing the FAK wild type (pCMV-GFP-FAK-WT) was obtained from Addgene (#50515). The pCMV-GFP-FAKK152R mutant was generated by site-directed mutagenesis of pCMV-GFP-FAK-WT using the Quik Change site-directed mutagenesis kit (Agilent Technologies, Santa Clara, CA), following the manufacturer’s instructions. The Flag-tagged adenovirus expressing SENP2-WT was generated by cloning the corresponding insert from pCMV-Flag-SENP2-WT into the pENTR1A vector (Life Technologies, Carlsbad, CA) at sites recognized by the restriction enzymes Kpn1 and EcoRV, and then a recombinase reaction was performed to get a pDEST based vector (Invitrogen Gateway LR Clonase II Enzyme mix, #11791100, Thermo Fisher, Waltham, MA) following the manufacturer’s instructions. An adenovirus containing β-galactosidase (Ad-LacZ) was used as a control (24). All constructs were verified by DNA sequencing by using vector specific primers.
Human endothelial Cells:
Human umbilical vein endothelial cells (HUVECs) were isolated from collagenase-digested umbilical cord veins (59) and collected in M200 medium supplemented with LSGS (Cascade Biologics, Inc., Portland, OR) and 2% FBS (Atlanta Biologicals, Inc., Lawrenceville, GA). HUVECs were cultured in petri dishes coated with 0.2% gelatin type A (#901771; MP Biomedicals, Santa Ana, CA), in Endothelial Cell Medium (ECM, #1001, ScienCell, Carlsbad, CA). The media was supplemented with 5% (v/v) FBS (#0025, ScienCell), 5 mL of Endothelial Cell Growth Supplement (ECGS, #1052, ScienCell, Carlsbard, CA) and 5 mL of penicillin/streptomycin (P/S, #0503, ScienCell, Carlsbard, CA). All cells were maintained in culture at 37°C in a 5% CO2 humidified atmosphere. HUVECs were used for experiments between 3 and 6 passages.
Inhibitor treatment:
p90RSK specific inhibitor, Fluoromethyl Ketone-MEA (FMK-MEA), and NADPH oxidase inhibitors, diphenyleneiodonium chloride (DPI), Manganese(III) tetrakis(1-methyl-4-pyridyl)porphyrin (MnTMPyP), or polyethylene glycol (PEG)-catalase were dissolved in DMSO. Cells were pre-treated with specified concentrations of inhibitors or the vehicle control for 1 hr prior to disturbed flow (D-flow) exposure.
Disturbed-flow (D-flow) studies:
To study the effect of DF on HUVECs in vitro, we used a cone-and-plate apparatus as we reported previously(29) HUVECs were cultured in 10 cm dishes to confluence in ECM medium. Confluent monolayers of HUVECs in 10 cm Petri dishes were exposed to DF using a grooved cone as we described previously (29) The DF system was kept at 37°C in a 5% CO2 humidified atmosphere.
Transient expression of proteins:
HUVECs were transfected with siRNA at a final concentration of 100 nM or plasmid DNAs using Lipofectamine 2000 transfection reagent, according to the manufacturer’s instructions. After transfection, cells were allowed to recover in the complete medium for 24–48 h. For adenovirus transduction, we used 20 multiplicities of infection for each adenovirus
Western blotting analysis:
HUVECs were washed twice with cold PBS, and whole cell lysates were prepared in RIPA buffer (50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1 mM EDTA, 1% Nonidet P40, 0.1% SDS, 1 mM dithiothreitol, 1:200-diluted protease inhibitor cocktail (P8340, Sigma-Aldrich, St. Louis, MO), and 1 mM PMSF). Total lysates were resolved by SDS-PAGE and electrotransferred onto a Hybond enhanced chemiluminescence PVDF membrane, which was then incubated with antibodies against each of the proteins to be detected in the lysate. Bound antibodies were visualized using enhanced chemiluminescence detection reagents (Amersham Pharmacia Biotech, Buckinghamshire, UK) according to the manufacturer’s instructions and quantified by densitometry using ImageJ software. B-actin was used as the loading control and was always probed together with the specific protein of interest. The immunoblotted band intensities of the proteins of interest were standardized against the intensity of the anti-B actin band. In most cases, anti-B-actin immunoblots were not shown in each figure to save space.
Immunoprecipitation (IP) assays:
Cell lysates were prepared in the cell lysis RIPA buffer (50 mM Tris-HCl [pH 7.4], 150 mM NaCl, 1 mM EDTA, 1% Nonidet P40, 0.1% SDS, 1 mM dithiothreitol, 1:200-diluted protease inhibitor cocktail [P8340, Sigma-Aldrich, St. Louis, MO], and 1 mM PMSF) on ice. Protein concentrations were determined by protein quantification assay kit (Pierce Biotechnology, Waltham, MA). Lysates (0.6 mg) were incubated with anti-FAK antibody or IgG overnight and then incubated with protein A/G beads for two hrs at room temperature. The immunoprecipitate was collected by centrifugation, washed three times with the cell lysis buffer, and subjected to the Western blotting assay.
NF-kB activity assay:
NF-κB activity was assayed using the PathDetect Signal Transduction Pathway trans-Reporting Systems (Stratagene, San Diego, CA) as we described previously (38).
Apoptosis assay:
HUVECs collected as cell suspension and incubated with ethylenediaminetetraacetic acid (10 mM in PBS). Apoptotic cells were determined using Annexin V-FITC Apoptosis Detection Kit (ab14085, Abcam, Cambridge, UK) according to the manufacturer’s instructions. Annexin V-positive cells were measured using a BD Accuri C6 Flow Cytometer (BD Biosciences, San Jose, CA), and data were analyzed using the FlowJo software program.
Senescence-associated β-galactosidase (SA-β-gal) staining:
HUVEC senescence was determined using Senescence β-Galactosidase Staining Kit (#9860, Cell Signaling Technology, Danvers, MA) per manufacturer’s instructions. Briefly, cells were washed once with PBS and fixed with 4% paraformaldehyde in PBS for 15 min at room temperature. The cells were washed twice with PBS and incubated in a sealed container with β-Galactosidase staining solution, pH 6.0 in darkness at 37 °C overnight. The stained cells were examined under a light microscope and the percentage of positive senescent cells were quantified.
ROS detection:
Intracellular H2O2 was measured with Amplex® Red assay kit (Molecular Probes, Life Technologies) according to manufacturer’s instructions. After the treatments indicated in the figure legends, trypsinized cells were suspended in Krebs-HEPES buffer (HBSS, in mM: NaCl 98.0, KCl 4.7, NaHCO3 25.0, MgSO4 1.2, 137 KH2PO4 1.2, CaCl2 2.5, D-glucose 11.1 and HEPES-Na 20.0) containing Amplex® Red reagent (10 mM) and horseradish peroxidase (0.1 U/ml). The fluorescence intensities were measured in a Synergy HTX Spectroflurometer (BioTek) with excitation at 550 nm and emission at 590 nm at 37°C.
Telomere length assay:
Telomere length was measured using the Telomere PNA kit with FITC ((#K5327, DAKO, Glostrup, Denmark) according to the manufacturer’s instructions. Briefly, cells were washed with PBS and equal number of cells were suspended in hybridization solution containing either fluorescein conjugated PNA telomere probe or in the hybridization solution without probe and heated to 82°C for 10 min. Mix gently and place the samples in the dark at room temperature overnight. Cells were then washed twice for 10 min each, using the washing solution provided with the kit at 40°C. Cells were resuspended in the 0.5 mL of DNA staining solution and incubated for 2 hr in the dark at 4°C. After washing cells, analyzed with a BD Accuri C6 flow cytometer using the FL-1 channel to detect the FITC signal and the FL-3 channel for DNA staining (Propidium Iodide). Human 1301 cells (T-cell leukemia, 01151619–1VL, Millipore Sigma, Burlington, MA) was used as a positive control. These control cells were cultured in RPMI 1640 medium containing 2 mM L-glutamine and 10% FBS. Relative telomere length (RTL) was calculated as the ratio between the fluorescence signal of each sample and that of the control cell after correcting the fluorescence value for the DNA index of the cells. The gained data was represented as RTL(38).
Statistical analysis:
Data are presented as the mean ± standard deviation (SD). Differences between two independent groups were determined by the Student t-test (two-tailed) or, when applicable, one-way ANOVA, followed by Bonferroni post hoc testing for multiple group comparisons, using Prism software (version 5.0; GraphPad Software, La Jolla, CA). When groups exhibited unequal variances, the Welch ANOVA was used to perform multiple group comparisons. P-values <0.01 were considered statistically significant.
Disturbed flow increased FAK K152 SUMOylation regulating p90RSK and SENP2 function.
FAK K152 SUMOylation upregulates FAK kinase activity and increases Vav2 tyrosine phosphorylation and subsequently upregulates NOX2 activation.
Vav2 -induced ROS production via NOX2 activation forms a positive feedback loop by activating redox-sensitive kinase of p90RSK, and consequently induces endothelial activation.
This positive feedback loop consisting with p90RSK, SENP2, FAK, and Vav2-NOX2 maintains ROS production and promotes endothelial inflammation and senescence.
Acknowledgments:
We thank Mrs. Carolyn Giancursio for her technical support. We thank Dr. Jun-ichi Abe for his critical reading of this manuscript. We thank Amy Ninetto, Scientific Editor, Research Medical Library, MD Anderson Cancer Center, for editing the manuscript. This work is supported by funding from the National Institutes of Health to NTL (HL-134740).
Abbreviations
- CVD
cardiovascular disease
- DF
disturbed flow
- DPI
diphenyleneiodonium chloride
- FAK
Focal adhesion kinase
- FMK-MEA
Fluoromethyl Ketone-MEA
- SENP2
sentrin-specific isopeptidase 2
- siRNA
small interfering RNA
- SUMO
Small Ubiquitin-related Modifier
- PTM
post-translational modification
- ICAM-1
inter-cellular adhesion
- ROS
reactive oxygen species
- TNF-α
Tumor necrosis factor α
Footnotes
Author disclosures: The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Abe J, Berk BC. Novel mechanisms of endothelial mechanotransduction. Arterioscler Thromb Vasc Biol 34: 2378–86, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abe JI, Ko KA, Kotla S, Wang Y, Paez-Mayorga J, Shin IJ, Imanishi M, Vu HT, Tao Y, Leiva- Juarez MM, Thomas TN, Medina JL, Won JH, Fujii Y, Giancursio CJ, McBeath E, Shin JH, Guzman L, Abe RJ, Taunton J, Mochizuki N, Faubion W, Cooke JP, Fujiwara K, Evans SE, Le NT. MAGI1 as a link between endothelial activation and ER stress drives atherosclerosis. JCI Insight 4, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Acevedo A, Gonzalez-Billault C. Crosstalk between Rac1-mediated actin regulation and ROS production. Free Radic Biol Med 116: 101–113, 2018. [DOI] [PubMed] [Google Scholar]
- 4.Aghazadeh B, Lowry WE, Huang XY and Rosen MK. Structural basis for relief of autoinhibition of the Dbl homology domain of proto-oncogene Vav by tyrosine phosphorylation. Cell. 2000;102:625–33 [DOI] [PubMed] [Google Scholar]
- 5.Anjum R and Blenis J. The RSK family of kinases: emerging roles in cellular signalling. Nat RevMol Cell Biol. 2008;9:747–58. [DOI] [PubMed] [Google Scholar]
- 6.Beckman JS, Minor RL Jr, White CW, Repine JE, Rosen GM, Freeman BA. Superoxide dismutase and catalase conjugated to polyethylene glycol increases endothelial enzyme activity and oxidant resistance. J Biol Chem. 1988. May 15;263(14):6884–92. [PubMed] [Google Scholar]
- 7.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev 87: 245–313, 2007. [DOI] [PubMed] [Google Scholar]
- 8.Bian Y, Song C, Cheng K, Dong M, Wang F, Huang J, Sun D, Wang L, Ye M, Zou H. An enzyme assisted RP-RPLC approach for in-depth analysis of human liver phosphoproteome. J Proteomics 96: 253–62, 2014. [DOI] [PubMed] [Google Scholar]
- 9.Boulanger CM. Endothelium. Arterioscler Thromb Vasc Biol 36: e26–31, 2016. [DOI] [PubMed] [Google Scholar]
- 10.Brakebusch C, Bouvard D, Stanchi F, Sakai T, Fassler R. Integrins in invasive growth. J Clin Invest 109: 999–1006, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Burtenshaw D, Hakimjavadi R, Redmond EM, Cahill PA. Nox, Reactive Oxygen Species and Regulation of Vascular Cell Fate. Antioxidants (Basel) 6, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carlucci A, Gedressi C, Lignitto L, Nezi L, Villa-Moruzzi E, Avvedimento EV, Gottesman M, Garbi C, Feliciello A. Protein-tyrosine phosphatase PTPD1 regulates focal adhesion kinase autophosphorylation and cell migration. J Biol Chem 283: 10919–29, 2008. [DOI] [PubMed] [Google Scholar]
- 13.Chen R, Kim O, Li M, Xiong X, Guan JL, Kung HJ, Chen H, Shimizu Y, Qiu Y. Regulation of the PH-domain-containing tyrosine kinase Etk by focal adhesion kinase through the FERM domain. Nat Cell Biol 3: 439–44, 2001. [DOI] [PubMed] [Google Scholar]
- 14.Chiarugi P, Pani G, Giannoni E, Taddei L, Colavitti R, Raugei G, Symons M, Borrello S, Galeotti T, Ramponi G. Reactive oxygen species as essential mediators of cell adhesion: the oxidative inhibition of a FAK tyrosine phosphatase is required for cell adhesion. J Cell Biol 161: 933–44, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chien S, Chiu JJ, Li YS. Focal adhesion kinase phosphorylation in flow-activation of endothelial NF-kappaB. Focus on “Focal adhesion kinase modulates activation of NF-kappaB by flow in endothelial cells”. Am J Physiol Cell Physiol 297: C800–1, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chiu JJ, Chien S. Effects of disturbed flow on vascular endothelium: pathophysiological basis and clinical perspectives. Physiol Rev 91:327–87, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.De Mera-Rodriguez JA, Alvarez-Hernan G, Ganan Y, Martin-Partido G, Rodriguez-Leon J, Francisco-Morcillo J. Is Senescence-Associated beta-Galactosidase a Reliable in vivo Marker of Cellular Senescence During Embryonic Development? Front Cell Dev Biol 9: 623175, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Del Pozo MA, Price LS, Alderson NB, Ren XD, Schwartz MA. Adhesion to the extracellular matrix regulates the coupling of the small GTPase Rac to its effector PAK. EMBO J 19: 2008–14, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fan LM, Cahill-Smith S, Geng L, Du J, Brooks G, Li JM. Aging-associated metabolic disorder induces Nox2 activation and oxidative damage of endothelial function. Free Radic Biol Med 108: 940–951, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Finkel T Signal transduction by reactive oxygen species. J Cell Biol 194: 7–15, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Forrester SJ, Kikuchi DS, Hernandes MS, Xu Q, Griendling KK. Reactive Oxygen Species in Metabolic and Inflammatory Signaling. Circ Res 122: 877–902, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Frame MC, Patel H, Serrels B, Lietha D, Eck MJ. The FERM domain: organizing the structure and function of FAK. Nat Rev Mol Cell Biol 11: 802–14, 2010. [DOI] [PubMed] [Google Scholar]
- 23.Frodin M, Gammeltoft S. Role and regulation of 90 kDa ribosomal S6 kinase (RSK) in signal transduction. Mol Cell Endocrinol 151: 65–77, 1999. [DOI] [PubMed] [Google Scholar]
- 24.Fukai T and Ushio-Fukai M. Cross-Talk between NADPH Oxidase and Mitochondria: Role in ROS Signaling and Angiogenesis. Cells. 2020;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Garvin AJ, Walker AK, Densham RM, Chauhan AS, Stone HR, Mackay HL, Jamshad M, Starowicz K, Daza-Martin M, Ronson GE, Lanz AJ, Beesley JF, Morris JR. The deSUMOylase SENP2 coordinates homologous recombination and nonhomologous end joining by independent mechanisms. Genes Dev 33: 333–347, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gorlach A, Brandes RP, Nguyen K, Amidi M, Dehghani F, Busse R. A gp91phox containing NADPH oxidase selectively expressed in endothelial cells is a major source of oxygen radical generation in the arterial wall. Circ Res 87: 26–32, 2000. [DOI] [PubMed] [Google Scholar]
- 27.Heo KS, Le NT, Cushman HJ, Giancursio CJ, Chang E, Woo CH, Sullivan MA, Taunton J, Yeh ET, Fujiwara K, Abe J. Disturbed flow-activated p90RSK kinase accelerates atherosclerosis by inhibiting SENP2 function. J Clin Invest 125: 1299–310, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Heo KS, Berk BC, Abe J. Disturbed Flow-Induced Endothelial Proatherogenic Signaling Via Regulating Post-Translational Modifications and Epigenetic Events. Antioxid Redox Signal 25: 435–50, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Heo KS, Chang E, Le NT, Cushman H, Yeh ET, Fujiwara K, Abe J. De-SUMOylation enzyme of sentrin/SUMO-specific protease 2 regulates disturbed flow-induced SUMOylation of ERK5 and p53 that leads to endothelial dysfunction and atherosclerosis. Circ Res 112: 911–23, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hornstein I, Alcover A, Katzav S. Vav proteins, masters of the world of cytoskeleton organization. Cell Signal 16: 1–11, 2004. [DOI] [PubMed] [Google Scholar]
- 31.Huveneers S, Danen EH. Adhesion signaling - crosstalk between integrins, Src and Rho. J Cell Sci 122: 1059–69, 2009. [DOI] [PubMed] [Google Scholar]
- 32.Ilic D, Furuta Y, Kanazawa S, Takeda N, Sobue K, Nakatsuji N, Nomura S, Fujimoto J, Okada M, Yamamoto T. Reduced cell motility and enhanced focal adhesion contact formation in cells from FAK-deficient mice. Nature 377: 539–44, 1995. [DOI] [PubMed] [Google Scholar]
- 33.Itahana Y, Yeh ET, Zhang Y. Nucleocytoplasmic shuttling modulates activity and ubiquitination-dependent turnover of SUMO-specific protease 2. Mol Cell Biol 26: 4675–89, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Itoh S, Ding B, Shishido T, Lerner-Marmarosh N, Wang N, Maekawa N, Berk BC, Takeishi Y, Yan C, Blaxall BC, Abe J. Role of p90 ribosomal S6 kinase-mediated prorenin-converting enzyme in ischemic and diabetic myocardium. Circulation 113: 1787–98, 2006. [DOI] [PubMed] [Google Scholar]
- 35.Johnson ES. Protein modification by SUMO. Annu Rev Biochem 73: 355–82, 2004. [DOI] [PubMed] [Google Scholar]
- 36.Kadare G, Toutant M, Formstecher E, Corvol JC, Carnaud M, Boutterin MC, Girault JA. PIAS1 mediated sumoylation of focal adhesion kinase activates its autophosphorylation. J Biol Chem 278: 47434–40, 2003. [DOI] [PubMed] [Google Scholar]
- 37.Kotla S, Le NT, Vu HT, Ko KA, Gi YJ, Thomas TN, Giancursio C, Lusis AJ, Cooke JP, Fujiwara K, Abe JI. Endothelial senescence-associated secretory phenotype (SASP) is regulated by Makorin-1 ubiquitin E3 ligase. Metabolism 100: 153962, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kotla S, Vu HT, Ko KA, Wang Y, Imanishi M, Heo KS, Fujii Y, Thomas TN, Gi YJ, Mazhar H, Paez-Mayorga J, Shin JH, Tao Y, Giancursio CJ, Medina JL, Taunton J, Lusis AJ, Cooke JP, Fujiwara K, Le NT, Abe JI. Endothelial senescence is induced by phosphorylation and nuclear export of telomeric repeat binding factor 2-interacting protein. JCI Insight 4, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lara R, Seckl MJ and Pardo OE. The p90 RSK family members: common functions and isoform specificity. Cancer Res. 2013;73:5301–8 [DOI] [PubMed] [Google Scholar]
- 40.Le NT, Heo KS, Takei Y, Lee H, Woo CH, Chang E, McClain C, Hurley C, Wang X, Li F, Xu H, Morrell C, Sullivan MA, Cohen MS, Serafimova IM, Taunton J, Fujiwara K, Abe J. A Crucial Role for p90RSK-Mediated Reduction of ERK5 Transcriptional Activity in Endothelial Dysfunction and Atherosclerosis. Circulation 127: 486–99, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liang HL, Hilton G, Mortensen J, Regner K, Johnson CP, Nilakantan V. MnTMPyP, a cell-permeant SOD mimetic, reduces oxidative stress and apoptosis following renal ischemia-reperfusion. Am J Physiol Renal Physiol. 2009. February;296(2):F266–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lopez-Lago M, Lee H, Cruz C, Movilla N and Bustelo XR. Tyrosine phosphorylation mediates both activation and downmodulation of the biological activity of Vav. Molecular and cellular biology. 2000;20:1678–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lin L, Shi C, Sun Z, Le NT, Abe JI, Hu K. The Ser/Thr kinase p90RSK promotes kidney fibrosis by modulating fibroblast-epithelial crosstalk. J Biol Chem 294: 9901–9910, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Massart C, Giusti N, Beauwens R, Dumont JE, Miot F, Sande JV. Diphenyleneiodonium, an inhibitor of NOXes and DUOXes, is also an iodide-specific transporter. FEBS Open Bio 4: 55–9, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Minamino T, Miyauchi H, Yoshida T, Ishida Y, Yoshida H, Komuro I. Endothelial cell senescence in human atherosclerosis: role of telomere in endothelial dysfunction. Circulation 105: 1541–4, 2002. [DOI] [PubMed] [Google Scholar]
- 46.Mitra SK, Hanson DA, Schlaepfer DD. Focal adhesion kinase: in command and control of cell motility. Nat Rev Mol Cell Biol 6: 56–68, 2005. [DOI] [PubMed] [Google Scholar]
- 47.Mitra SK, Schlaepfer DD. Integrin-regulated FAK-Src signaling in normal and cancer cells. Curr Opin Cell Biol 18: 516–23, 2006. [DOI] [PubMed] [Google Scholar]
- 48.Naser R, Aldehaiman A, Diaz-Galicia E, Arold ST. Endogenous Control Mechanisms of FAK and PYK2 and Their Relevance to Cancer Development. Cancers (Basel) 10, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Palazzo AL, Evensen E, Huang YW, Cesano A, Nolan GP, Fantl WJ. Association of reactive oxygen species-mediated signal transduction with in vitro apoptosis sensitivity in chronic lymphocytic leukemia B cells. PLoS One 6: e24592, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Petzold T, Orr AW, Hahn C, Jhaveri KA, Parsons JT, Schwartz MA. Focal adhesion kinase modulates activation of NF-kappaB by flow in endothelial cells. Am J Physiol Cell Physiol 297: C814–22, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Richardson A, Malik RK, Hildebrand JD, Parsons JT. Inhibition of cell spreading by expression of the C-terminal domain of focal adhesion kinase (FAK) is rescued by coexpression of Src or catalytically inactive FAK: a role for paxillin tyrosine phosphorylation. Mol Cell Biol 17: 6906–14, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rojas M, Lemtalsi T, Toque HA, Xu Z, Fulton D, Caldwell RW, Caldwell RB. NOX2-Induced Activation of Arginase and Diabetes-Induced Retinal Endothelial Cell Senescence. Antioxidants (Basel) 6, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Salazar G NADPH Oxidases and Mitochondria in Vascular Senescence. Int J Mol Sci 19, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schlaepfer DD, Mitra SK. Multiple connections link FAK to cell motility and invasion. Curr Opin Genet Dev 14: 92–101, 2004. [DOI] [PubMed] [Google Scholar]
- 55.Schuebel KE, Bustelo XR, Nielsen DA, Song BJ, Barbacid M, Goldman D and Lee IJ. Isolation and characterization of murine vav2, a member of the vav family of proto-oncogenes. Oncogene. 1996;13:363–71. [PubMed] [Google Scholar]
- 56.Souza HP, Laurindo FR, Ziegelstein RC, Berlowitz CO, Zweier JL. Vascular NAD(P)H oxidase is distinct from the phagocytic enzyme and modulates vascular reactivity control. Am J Physiol Heart Circ Physiol 280: H658–67, 2001. [DOI] [PubMed] [Google Scholar]
- 57.Stankovic-Valentin N, Melchior F. Control of SUMO and Ubiquitin by ROS: Signaling and disease implications. Mol Aspects Med 63: 3–17, 2018. [DOI] [PubMed] [Google Scholar]
- 58.Takahashi M, Berk BC. Mitogen-activated protein kinase (ERK1/2) activation by shear stress and adhesion in endothelial cells. Essential role for a herbimycin-sensitive kinase. J Clin Invest. 98: 2623–31, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Takeishi Y, Huang Q, Abe J, Che W, Lee JD, Kawakatsu H, Hoit BD, Berk BC, Walsh RA. Activation of mitogen-activated protein kinases and p90 ribosomal S6 kinase in failing human hearts with dilated cardiomyopathy. Cardiovasc Res 53: 131–7., 2002. [DOI] [PubMed] [Google Scholar]
- 60.Traub O, Berk BC. Laminar shear stress: mechanisms by which endothelial cells transduce an atheroprotective force. Arterioscler Thromb Vasc Biol 18: 677–85, 1998. [DOI] [PubMed] [Google Scholar]
- 61.Urbich C, Stein M, Reisinger K, Kaufmann R, Dimmeler S, Gille J. Fluid shear stress-induced transcriptional activation of the vascular endothelial growth factor receptor-2 gene requires Sp1-dependent DNA binding. FEBS Lett 535: 87–93, 2003. [DOI] [PubMed] [Google Scholar]
- 62.Vanhoutte PM. Endothelial dysfunction: the first step toward coronary arteriosclerosis. Circ J 73: 595–601, 2009. [DOI] [PubMed] [Google Scholar]
- 63.Wang AS, Dreesen O. Biomarkers of Cellular Senescence and Skin Aging. Front Genet 9: 247, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wilsbacher JL, Moores SL and Brugge JS. An active form of Vav1 induces migration of mammary epithelial cells by stimulating secretion of an epidermal growth factor receptor ligand. Cell Commun Signal. 2006;4:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yamaura T, Kasaoka T, Iijima N, Kimura M, Hatakeyama S. Evaluation of therapeutic effects of FAK inhibition in murine models of atherosclerosis. BMC Res Notes 12: 200, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yang P, Hu S, Yang F, Guan XQ, Wang SQ, Zhu P, Xiong F, Zhang S, Xu J, Yu QL, Wang CY. Sumoylation modulates oxidative stress relevant to the viability and functionality of pancreatic beta cells. Am J Transl Res 6: 353–60, 2014. [PMC free article] [PubMed] [Google Scholar]
- 67.Yurdagul A Jr., Sulzmaier FJ, Chen XL, Pattillo CB, Schlaepfer DD, Orr AW. Oxidized LDL induces FAK-dependent RSK signaling to drive NF-kappaB activation and VCAM-1 expression. J Cell Sci 129: 1580–91, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zheng H, Kim J, Liew M, Yan JK, Herrera O, Bok JW, Kelleher NL, Keller NP, Wang Y. Redox metabolites signal polymicrobial biofilm development via the NapA oxidative stress cascade in Aspergillus. Curr Biol 25: 29–37, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhou J, Yi Q, Tang L. The roles of nuclear focal adhesion kinase (FAK) on Cancer: a focused review. J Exp Clin Cancer Res 38: 250, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]