

SUMMARY
Foxp3+ T regulatory (Treg) cells promote immunological tumor tolerance, but how their immune-suppressive function is regulated in the tumor microenvironment (TME) remains unknown. Here, we used intravital microscopy to characterize the cellular interactions that provide tumor-infiltrating Treg cells with critical activation signals. We found that the polyclonal Treg cell repertoire is pre-enriched to recognize antigens presented by tumor-associated conventional dendritic cells (cDCs). Unstable cDC contacts sufficed to sustain Treg cell function, whereas T helper cells were activated during stable interactions. Contact instability resulted from CTLA-4-dependent downregulation of co-stimulatory B7-family proteins on cDCs, mediated by Treg cells themselves. CTLA-4-blockade triggered CD28-dependent Treg cell hyper-proliferation in the TME, and concomitant Treg cell inactivation was required to achieve tumor rejection. Therefore, Treg cells self-regulate through a CTLA-4- and CD28-dependent feedback loop that adjusts their population size to the amount of local co-stimulation. Its disruption through CTLA-4-blockade may off-set therapeutic benefits in cancer patients.
Graphical Abstract
In brief
Intravital imaging studies of the tumor microenvironment provide insights into how Treg cells interact with dendritic cells and modulate their numbers through a CTLA-4- and CD28-dependent feedback loop. Disruption of this feedback loop by CTLA-4 blockade may thwart the efficacy of immune checkpoint therapy.
INTRODUCTION
Checkpoint blockade immunotherapy (CBI) enhances anti-tumor immune responses by inhibiting the interaction of molecules that restrain the activation and effector function of T cells, such as cytotoxic T lymphocyte-associated protein 4 (CTLA-4) and PD-1, with their ligands. This has allowed for long-term progression-free survival of a subset of cancer patients with otherwise dismal prognoses (Borghaei et al., 2015; Ferris et al., 2016; Motzer et al., 2018; Wolchok et al., 2017). Immune checkpoints are, however, only one component of a multi-layered system of regulatory mechanisms that synergize in the tumor microenvironment (TME) to antagonize immune-mediated elimination of malignant cells (Binnewies et al., 2018). Extending the benefits of CBI to more patients will likely require concurrent targeting of additional immune-regulatory mechanisms (Sanmamed and Chen, 2018). The activities of T regulatory (Treg) cells constitute another layer of immune regulation and include modulation of antigen presenting cell (APC) function, secretion of inhibitory cytokines, and consumption of T cell survival factors (Vignali et al., 2008). Consequently, approaches to target Treg cells are an important element of efforts to improve cancer therapy (Sakaguchi et al., 2020; Wing et al., 2019).
The mechanisms that prevent tumor eradication in cancer patients have evolved in order to maintain immune homeostasis in healthy individuals by containing self-reactive responses and to regulate the magnitude and duration of anti-pathogen responses to prevent exaggerated, pathogenic immune activation. However, these tolerance mechanisms need to be calibrated in order to avoid over-regulation of appropriate immune responses, which would impede pathogen elimination. The dependence of Treg cells on interleukin (IL)-2 produced by activated effector T cells, which ties Treg cell activation and expansion to the magnitude of the effector response, is one mechanism through which this control is achieved in secondary lymphoid organs (SLOs) (Liu et al., 2015; O’Gorman et al., 2009). However, how Treg cell function is regulated at effector sites, and especially in the TME, is not known.
The ability of Treg cells to establish tumor tolerance depends on their infiltration and local cognate antigen encounter in the TME (Bauer et al., 2014). This indicates that their function needs to be continually sustained by T cell antigen receptor (TCR) signals or by other signals that they receive during antigen-dependent cellular interactions. Accordingly, Treg cells isolated from human tumors express genes that reflect cellular activation (Plitas et al., 2016; De Simone et al., 2016; Zheng et al., 2017) and whose expression by Treg cells depends on TCR recognition in mice (Levine et al., 2014; Vahl et al., 2014). However, whether Treg cells recognize their cognate antigens indiscriminately on a wide range of tumor-associated APC, or through interactions with a specific subset, has not been examined.
Here, we used multiphoton intravital microscopy (MP-IVM) and a fluorescent reporter of nuclear factor of activated T cell (NFAT) activation to directly visualize how Treg cells receive TCR signals in the TME. We found that the preferentially self-reactive polyclonal Treg cell pool is pre-enriched for recognition of antigen in the TME. We also observed that unlike conventional CD4+ T helper (Th) cells, which were activated during stable APC contacts, tumor-infiltrating Treg cells were sustained through transient TCR signals that they received during unstable, short-lived interactions with conventional dendritic cells (cDCs), but not tumor-associated macrophages (TAMs). This contact instability was, however, not cell-intrinsic, but resulted from Treg cell-mediated and CTLA-4-dependent downregulation of CD80 and CD86 proteins on cDCs, which limited Treg cell co-stimulation via CD28 and controlled their proliferation within tumors. Disruption of this negative feedback loop through CTLA-4-targeted CBI stabilized Treg cell-cDC contacts in the TME, boosted Treg cell proliferation, and induced their local expansion in excess of that of effector T cells. Inactivating NFAT signaling in Treg cells during CTLA-4-targeted CBI unleashed its therapeutic anti-tumor efficacy, indicating that tumor-associated Treg cells continue to promote tumor tolerance even when their CTLA-4-dependent immune-suppressive function is disrupted.
RESULTS
The polyclonal TCR repertoire of Treg cells is enriched for tumor reactivity
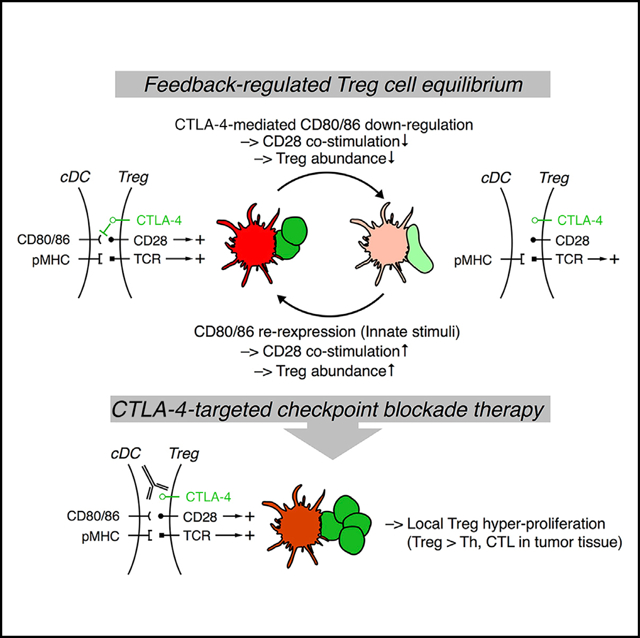
To examine the single cell-dynamics of Treg cell activation in tumor tissue by in vivo imaging, we selected a fusion of the NFAT1 regulatory domain and GFP (“NFAT-GFP”) as a fluorescent reporter of productive TCR engagement (Aramburu et al., 1998). NFAT-GFP accumulates in the nucleus of CD8+ T cells within 1–3 min after they form cognate APC contacts and redistributes to the cytoplasm with a half-life of 20 min upon contact cessation (Marangoni et al., 2013). We activated and transduced purified CD4+ Foxp3+ Treg cells from the SLOs of C57BL/6 mice with a bicistronic retroviral vector expressing NFAT-GFP and the fluorescent histone fusion protein H2B-RFP as a genetically encoded nuclear marker (Figure S1A). As a reference population, we used CD4+ Foxp3− Th cells (Figure S1B). Six days later, Treg, but not Th cells, expressed Foxp3 (Figure S1C) and both coordinately expressed NFAT-GFP and H2B-RFP (Figure S1D). These cells were then separately intravenously (i.v.) injected into sublethally irradiated CD11cmCherry reporter mice.
Six weeks later, mice were implanted subcutaneously (s.c.) in the back with MC38 colon carcinoma expressing a blue fluorescent histone H2B-Cerulean fusion protein (hereafter “MC38 H2B-Cer”), a model in which Treg cells strongly restrain anti-tumor immunity (Arce Vargas et al., 2017; Imai et al., 2007; Mahne et al., 2017). Ex vivo TCR-activation induces loss of the SLO homing receptor CCR7 and gain of trafficking receptors for migration into inflamed tissues, enabling Treg, and likely also Th cells, to directly infiltrate tumor tissue, bypassing the requirement for antigenic priming in tdLNs (Wang et al., 2012). Accordingly, 6 days after tumor implantation, when we installed dorsal skinfold chambers (DSFCs) around tumors to allow for MP-IVM analysis (Figure 1A), adoptively transferred Foxp3+ Treg cells and Foxp3− Th cells had accumulated in tumors (Figure 1B).
Figure 1. The polyclonal TCR repertoire of Treg cells is enriched for tumor reactivity.
(A) Experimental protocol.
(B) Phenotype of adoptively transferred T cells in the TME.
(C and D) MP-IVM micrographs (top panels) and individual cell images (middle panels) depicting the nucleo-cytoplasmic distribution of NFAT-GFP in an exemplary Treg (C) and Th cell (D) in the TME. Bottom panels show false-color representations of the NFAT signaling index (SI) (see Figures S1F–S1L for details). Numbers indicate min:s. Migratory tracks indicate 1-min intervals. Scale bars, 20 μm.
(E–G) 3D-instantaneous migratory velocities (E), arrest coefficient (F), and instantaneous NFAT SI frequency distribution (G) of Treg (n = 1,088 from 25 cells) and Th cells (n = 1,213 from 31 cells) from 3 individual movies each recorded in 2 independent experiments. Numbers in (G) refer to percentages of NFAT SI >0.5.
(H) Fraction of time in which NFAT SI >0.5 in individual cell tracks. Values above graphs refer to frequencies of cells in which NFAT >0.5 for greater than 20% of time.
(I) Instantaneous cell velocities with NFAT inactive (NFAT SI ≤0.5) or active (NFAT SI >0.5).
(J) Instantaneous NFAT SI as a function of distance to nearest APC. Numbers in grid indicate quadrant frequencies. Light blue symbols indicate instances of NFAT activation following a state of inactivity. p values calculated by Mann-Whitney U test.
Four and 5 days later, Treg cells were found both in the tumor parenchyma, identified by the blue fluorescence of tumor nuclei, as well as in the surrounding stroma. Treg cells in close vicinity or direct contact with CD11c-mCherry+ APCs frequently reduced their motility and redistributed NFAT-GFP from their cytoplasm to the nucleus (Figure 1C; Video S1). However, NFAT-GFP nuclear accumulation was not accompanied by sustained migratory arrest but occurred during low speed-scanning of APC surfaces (Video S2). Treg cells also transiently maintained NFAT activity while moving away from APCs, likely reflecting delayed nuclear export of NFAT1 upon cessation of a TCR stimulus (Figure S1E), as previously reported for CD8+ effector T cells (Marangoni et al., 2013). In contrast to Tregs, nearly all Th cells migrated at higher speed, did not arrest, and did not accumulate NFAT-GFP in the nucleus (Figures 1D–1F; Video S1). This indicated that most Th cells did not recognize antigen, in line with the prediction that transferred Th and Treg cells would predominantly be recruited to the TME based on homing receptor expression induced during polyclonal ex vivo activation.
To quantify the rate of T cell activation, we devised an automated algorithm to measure 3D central accumulation of NFAT-GFP in T cells, which was validated against manual as well as automated measurements of nuclear accumulation through independent methods (Figures S1F–S1L). This algorithm produced an instantaneous NFAT signaling index (NFAT SI), ranging from 0 to 1, and we defined 0.5 as the threshold above which we considered NFAT to be activated. By this measure, ~40% of all observed Treg cells (compared to 6% of Th cells) were activated, both based on instantaneous NFAT SI as well as on the frequency of individual cells with an NFAT SI >0.5 during more than 20% of the time they were observed (Figures 1G and 1H). Although Treg and especially rare Th cell activation events were associated with lower migratory velocity (Figure 1I), suggesting that they occurred during APC interactions, Treg cells also maintained NFAT activity while moving away from APCs. However, the onset of NFAT activity always occurred in direct contact with an APC (Figure 1J). Considering the likely antigen-independent recruitment of ex vivo activated Treg and Th cells to tumors, these observations suggest that the polyclonal Treg cell repertoire is pre-enriched for specificity for antigens presented in the TME, whereas only rare cells among the Th cell repertoire recognize such antigens. Second, although tumor-reactive Treg cells only partially stabilize their interactions with APCs in tumor tissue, these unstable interactions trigger productive TCR signals that activate the NFAT pathway.
Conventional DC activate Treg cells locally within the TME
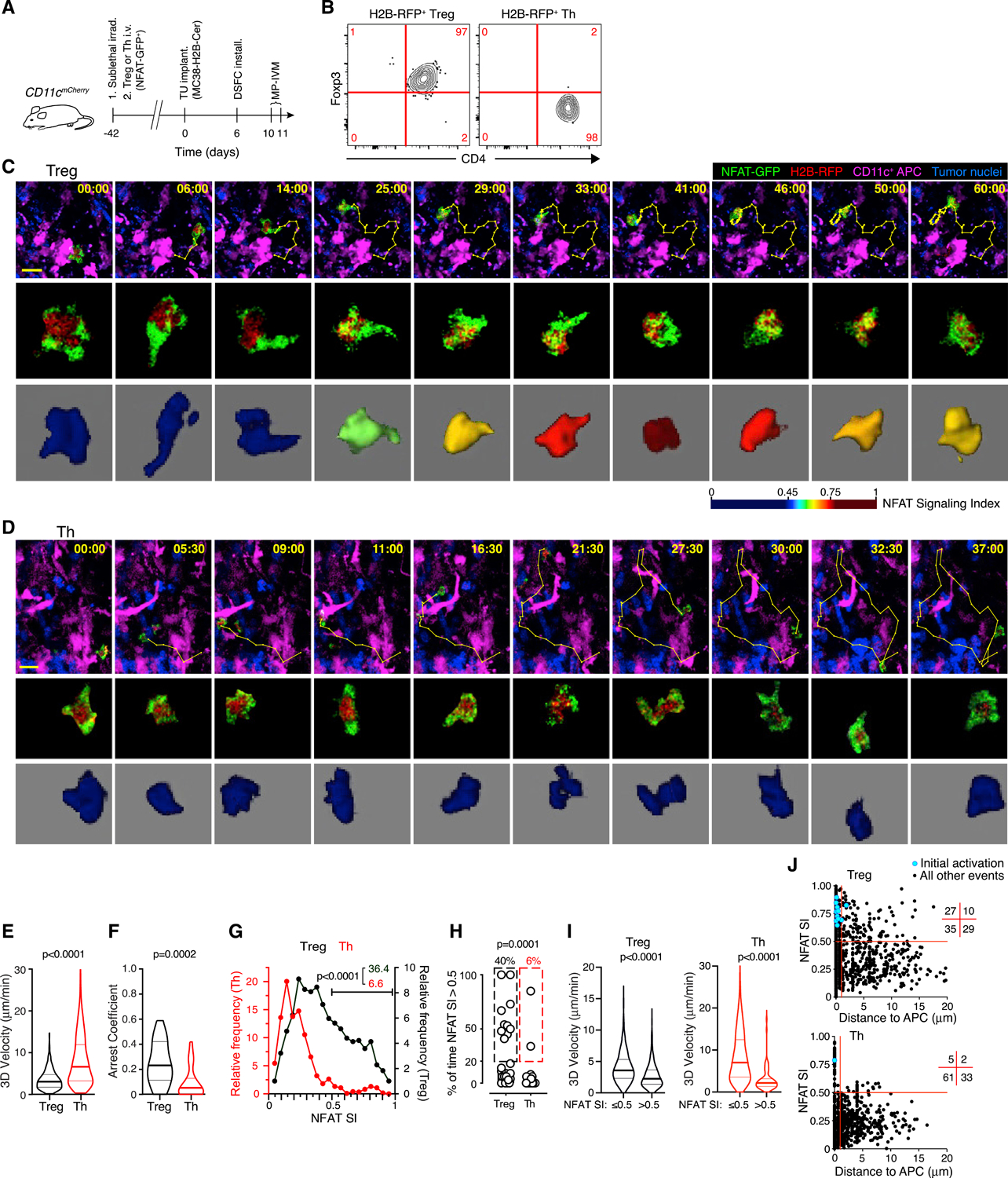
Although NFAT nuclear accumulation in Treg cells was always initiated in contact with a CD11c-mCherry+ APC, many APC contacts did not trigger NFAT-GFP activity. This raised the possibility that only a subset of CD11c+ APCs interact with Treg cells in a productive manner. Indeed, among all mCherry+ cells in tumor tissue, the majority were CD11c+ CD64+ F4/80+ TAM, as described (Franklin et al., 2014), and only a small fraction were cDCs. Among those, approximately half were CD172a+ cDC2 and fewer were XCR1+ cDC1 (Figures S2A and S2B). Because cDCs help recruit and activate tumor-infiltrating cytotoxic T lymphocytes (CTL) (Broz et al., 2014; Spranger et al., 2017), we hypothesized that they may also activate Treg cells. We therefore generated zDCDTR → B6 irradiation bone marrow chimeras (BMCs), where cDCs can be acutely ablated through diphtheria toxin (DT) treatment (Meredith et al., 2012). DT-treatment reduced the frequencies of XCR1-expressing cDC1 and CD172a-expressing cDC2 in the MC38 TME by 90% and 70%, respectively, relative to either vehicle-treated zDCDTR → B6 or DT-treated B6 → B6 control BMCs, while TAMs were not affected (Figures S2C and S2D). We transferred NFAT-GFP-expressing Treg cells into BMCs 3 weeks after bone marrow transfer, when the T cell compartment was not yet reconstituted, in order to enhance their engraftment. Another 3 weeks later, we implanted MC38 H2B-Cer tumors, installed DSFC, and treated animals either with DT or vehicle (Figure 2A). MP-IVM analysis of tumor-infiltrating Treg cells in vehicle-treated animals revealed a similar pattern of restrained cell motility and frequent NFAT nuclear accumulation as described above. In contrast, Treg cell migration was unconstrained and the frequency of NFAT activation events was reduced from about one third to 6%–7% in DT-treated mice (Figures 2B–2D; Video S3). Thus, local TCR-dependent Treg cell activation within the TME predominantly relies on antigen presentation by cDC.
Figure 2. cDCs activate Treg cells locally within the TME.
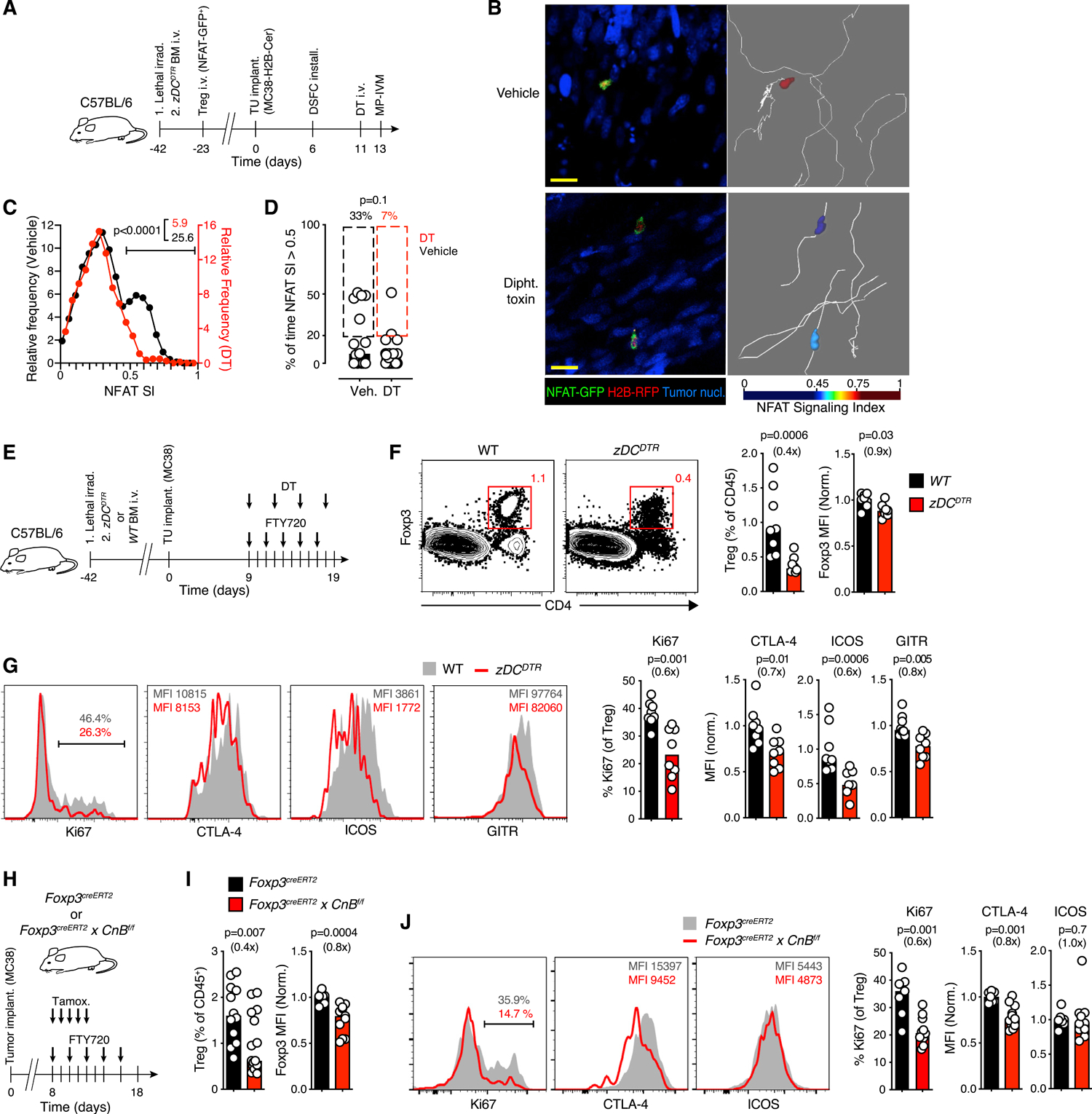
(A) Experimental protocol.
(B) MP-IVM micrographs of Treg cells in MC38 tumors in zDCDTR bone marrow chimeras. Images on right show representative migratory tracks and false-color-coded NFAT SI. Scale bar, 50 μm.
(C) Distribution of instantaneous Treg cell NFAT SI in the TME in presence (black, n = 933 from 15 cells) or following ablation (red, n = 824 from 28 cells) of cDC. Gate frequencies refer to NFAT SI >0.5.
(D) The fraction of time in which NFAT SI >0.5 in individual Treg cells. Percentages above graphs refer to frequencies of cells in which NFAT >0.5 for greater than 20% of time. Data in (C) and (D) are pooled from 4 recordings per condition.
(E) Experimental protocol.
(F and G) Frequency (gated on CD45+ cells) (F) and activation markers (G) on Treg cells in the TME following ablation of cDC. n = 8 mice per group pooled from two independent experiments.
(H) Experimental protocol.
(I and J) Frequency of Treg cells in the TME and expression of Foxp3 (I), and of activation markers (J) after CnB depletion. For Treg cell quantification, n = 12–16 in 3 independent experiments. For marker expression analysis, n = 7–11 from two separate experiments. Bars depict medians, symbols individual mice. MFIs normalized by the mean of control groups. p values calculated by Mann-Whitney U test. Fold change as compared to control groups in parentheses.
See also Figures S2 and S3 and Video S3.
To test the functional relevance of Treg cell interactions with cDCs specifically in tumor tissue, we ablated cDCs while at the same time blocking lymphocyte egress from SLOs using the functional S1P-receptor antagonist FTY720 (Figure 2E) to prevent further recruitment of Treg cells from tdLNs to tumors, as described (Di Pilato et al., 2019). Treatment with FTY720 alone increased the frequency of Treg cells in tdLN, as expected, accompanied by a decrease in tumor tissue. Treg cells in the TME also decreased their expression of Foxp3 and of markers of effector Treg (eTreg) cell differentiation (Figures S2E and S2F), revealing that the tumor-infiltrating Treg cell pool is continuously replenished from SLO. However, upon concomitant ablation of cDC, tumor-associated Treg cells became even less abundant and expressed even lower levels of Foxp3 (Figure 2F), proliferated less, and further reduced their expression of activation markers, such as CTLA-4, ICOS and GITR (Figure 2G). cDC-depletion also decreased the abundance of tumor-associated Th cells, albeit less profoundly than for Treg cells (Figure S2G). Similar changes were observed in tdLN (Figures S2G–S2I), in line with prior studies on CD11c+ DC depletion (Darrasse-Jèze et al., 2009). Thus, despite low frequency, cDCs play a non-redundant role in local Treg cell activation to maintain their population size in the TME.
To examine the relevance of the NFAT pathway for the maintenance of intratumoral Treg cells, we s.c. implanted MC38 tumors in the flanks of Foxp3creERT2 × CnBf/f mice. This allowed for tamoxifen-inducible deletion of the regulatory B subunit of the phosphatase calcineurin (CnB), which is required for TCR-induced NFAT activation (Marangoni et al., 2018). Following CnB deletion in Treg cells and concurrent FTY720 treatment (Figures 2H and S3A), Treg cells in tumors declined in frequency, expressed less Foxp3, proliferated less, and expressed less CTLA-4, while their ICOS levels remained unchanged (Figures 2I and 2J). CnB deletion without concurrent FTY720 treatment also diminished Treg cell proliferation and expression of markers of eTreg cell differentiation, but the decrease in Treg cell frequency did not reach statistical significance, presumably due to continued influx of eTreg cells generated in SLOs already prior to CnB deletion (Figures S3B and S3C). Thus, interruption of the CnB-NFAT signaling axis had similar effects on tumor-infiltrating Treg cells as removal of cDC, suggesting a critical role of this pathway among the signaling activities triggered by their interactions with cDC.
Treg cells destabilize intratumoral APC contacts with Th cells
Considering that the TCR repertoires of tumor-infiltrating Th and Treg cells are largely distinct (Ahmadzadeh et al., 2019; Malchow et al., 2013; Plitas et al., 2016), the lower stability of Treg cell-compared to Th cell-APC contacts may result from potentially lower affinity of thymic Treg cell-expressed TCRs for self-antigen compared to the affinity of Th cell-expressed TCRs for foreign antigen (Josefowicz et al., 2012). We therefore compared the behavior of Treg and Th cells expressing the same TCR. CT26 is a colon carcinoma model on the BALB/c background where, similarly to the MC38 model, Treg cells restrain anti-tumor immunity (Arce Vargas et al., 2017; Golgher et al., 2002). When CT26 is engineered to express influenza hemagglutinin (HA), adoptive transfer of HA-specific Treg cells further enhances tumor tolerance and prevents rejection of CT26HA tumors by HA-specific CTL (Bauer et al., 2014; Chen et al., 2005; Mempel et al., 2006). We purified naive, HA-specific CD4+ Th cells from HA-TCR transgenic mice (Kirberg et al., 1994) and HA-specific Foxp3+ Treg cells from HA-TCR mice that co-express HA as a self-antigen (Bauer et al., 2014; Klein et al., 2003; Marangoni et al., 2013). Both were activated in vitro, transduced to co-express NFAT-GFP and H2B-RFP, and injected into sublethally irradiated hosts generated by crossing CD11cmCherry (C57BL/6 background) to BALB/c mice. CD11cmCherry × BALB/c (F1) hosts were used to visualize mCherry+ APCs in mice where HA is presented to HA-TCR transgenic T cells on I-Ed (Bauer et al., 2014). Six weeks later, mice were implanted with CT26HA expressing H2B-Cerulean (CT26HA H2B-Cer), and DSFCs were installed to permit MP-IVM analysis (Figure 3A).
Figure 3. Treg cells destabilize intratumoral APC contacts with Th cells.
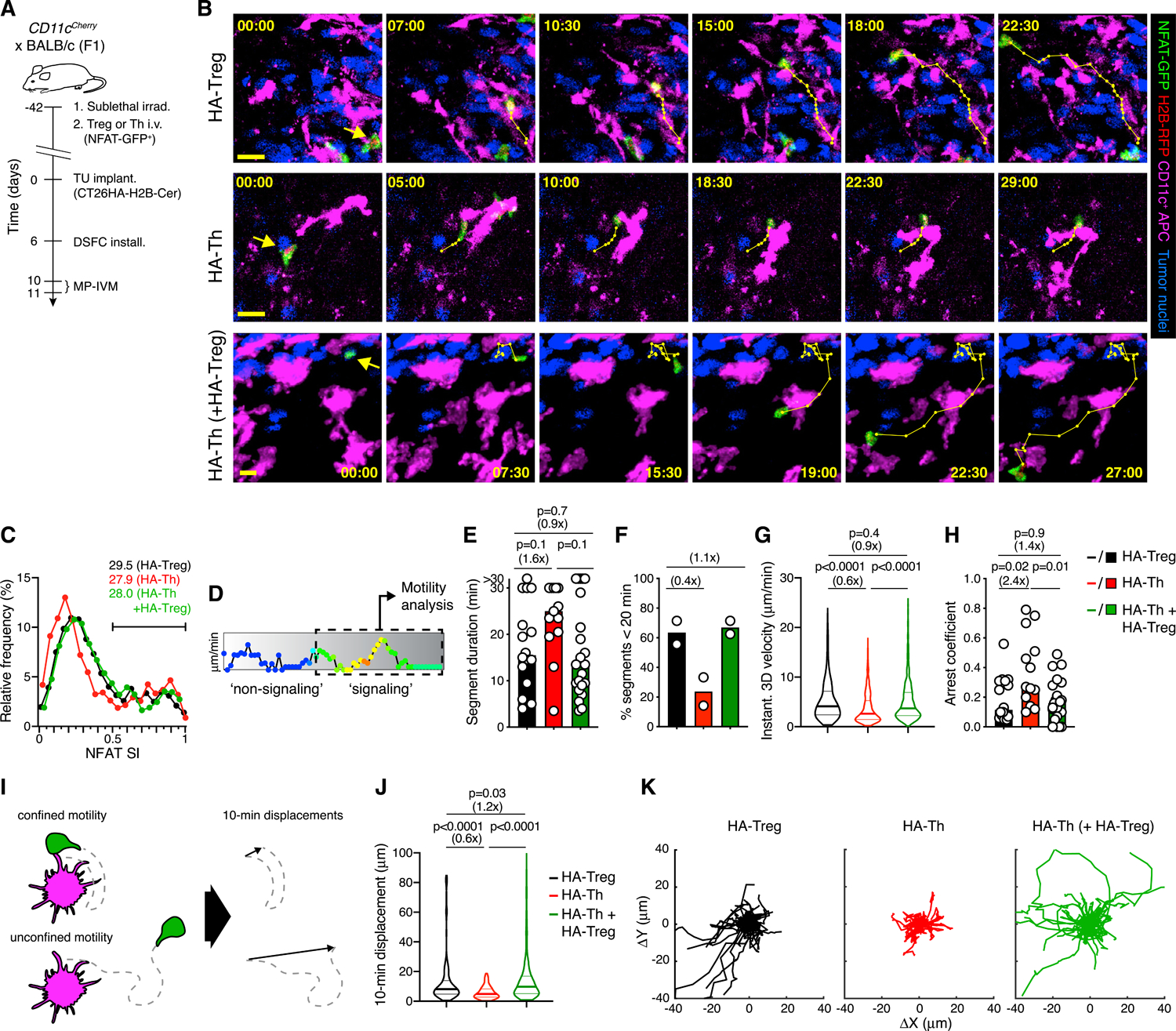
(A) Experimental protocol.
(B) MP-IVM micrographs illustrating cell motility during NFAT activation in representative HA-Treg (top), HA-Th (middle), and HA-Th cells in presence of non-visualized HA-Treg cells (bottom), in the CT26HA TME. Numbers indicate min:s. Migratory tracks indicate 1-min intervals. Scale bars, 20 μm.
(C) Distribution of instantaneous NFAT SI of HA-Treg (n = 2,259), HA-Th (n = 2,177), and HA-Th + HA-Treg (n = 3,322). Numbers refer to fraction of NFAT SI >0.5.
(D) Gating on signaling segments within tracks for motility analysis shown in (E)–(K).
(E and F) Duration of signaling segments (E) and percentage of segments ≤20 min (F). n = 16 (HA-Treg), 13 (HA-Th), and 22 segments (HA-Th + HA-Treg) recorded in 2 independent experiments.
(G) Instantaneous 3D velocities in signaling segments. n = 582 (HA-Treg), 608 (HA-Th), and 860 (HA-Th + HA-Treg).
(H) Arrest coefficients in signaling segments. n = 16 (HA-Treg), 13 (HA-Th), and 22 (HA-Th + HA-Treg).
(I) Schematic depiction of the 10-min displacement metric.
(J) 10-min displacement indices. n = 305 (HA-Treg), 372 (HA-Th), and 482 (HA-Th + HA-Treg).
(K) Migratory tracks of non-overlapping 10-min signaling segments with normalized starting coordinates. n = 36 (HA-Treg), 34 (HA-Th), and 52 (HA-Th in presence of HA-Treg). Data in (C)–(K) were collected from 6 (HA-Treg), 6 (HA-Th), and 5 (HA-Th + HA-Treg) independent recordings from two independent experiments. Bars represent medians and white symbols individual segments. Values in parenthesis above graphs indicate fold change of medians. p values calculated by Mann-Whitney U test.
HA-specific Treg cells behaved similarly in a TME where HA antigen was abundantly expressed as polyclonal Treg cells did in MC38 tumors: NFAT-GFP rapidly accumulated in their nucleus upon encounter with CD11c-mCherry+ APCs, was retained during partially stabilized contacts, and slowly relocated to the cytoplasm when Treg cells dissociated and migrated away from APCs (Figure 3B; Video S4). In contrast, HA-specific Th cells accumulated NFAT-GFP in their nucleus mostly during fully stabilized APC contacts and sustained these contacts for longer periods of time than HA-Treg cells. However, the overall rate of NFAT activation was similar between HA-Treg and HA-Th cells, indicating that their exposure and sensitivity toward their shared cognate antigen was comparable (Figure 3C). To quantify these observations, we divided each cell track into NFAT signaling and non-signaling segments and analyzed these separately (Figures 3D and S4A). Although the behavior of non-signaling HA-Treg and HA-Th cells was similar (Figures S4B–S4G), HA-Treg cell signaling segments tended to be of shorter duration (Figures 3E and 3F), and signaling HA-Treg cells migrated faster and were less arrested than HA-Th cells (Figures 3G and 3H). Treg cells also displaced twice as far as Th cells while signaling, as reflected by the 10-min displacement index, indicating that they traveled further without turning, whereas Th cells made more frequent sharp-angled turns and therefore remain confined to a small tissue region (Figures 3I–3K). Treg and Th cell displacement was however similar while NFAT was inactive (Figures S4F and S4G). The differences in how Treg and Th cells interact with APCs presenting their cognate antigen are thus not driven by intrinsic differences in TCR affinity.
Treg cells can destabilize Th cell interactions with DC in lymph nodes in autoimmune settings (Tadokoro et al., 2006; Tang et al., 2006), potentially by altering the way that these DC present antigen. We therefore hypothesized that Treg cells also alter the way tumor-associated APCs present antigen and thereby reduce the stability of their own APC interactions, as well as those of effector T cells. To test this hypothesis, we injected a third group of mice not only with NFAT-GFP-expressing HA-Th, but also with non-fluorescent HA-Treg cells. Even when less numerous than endogenous Treg cells, adoptively transferred HA-Treg enhance immune-regulation in the CT26HA TME, likely due to the abundance of their cognate antigen (Bauer et al., 2014; Marangoni et al., 2013). Indeed, in the presence of HA-Treg, HA-Th cells activated NFAT-GFP at the same rate as in their absence, but showed signs of reduced APC contact stability and behaved similarly to HA-Treg cells based on all measures of motility, including during phases of NFAT activity (Figures 3B–3K and S4B–S4G). These observations indicate that differences in how Treg and Th cells interact with APCs in tumor tissue are predominantly driven by changes that Treg cells introduce to the TME. These changes likely include altered APC function, resulting in destabilization of APC contacts not only with Th, but also with Treg cells.
CTLA-4 blockade increases the density of co-stimulatory ligands on tumor-associated cDCs
How could Treg cells impair the stability of Th cell as well as their own interactions with tumor-associated APCs? Because Treg cells can use CTLA-4 to reduce the amount of CD80 and CD86 on APCs in SLOs, including through CTLA-4-mediated trans-endocytosis (Ovcinnikovs et al., 2019; Qureshi et al., 2011; Wing et al., 2008), and considering the high expression of CTLA-4 on tumor-infiltrating Treg cells (Simpson et al., 2013), we hypothesized that they use CTLA-4 to negatively regulate the stability of T cell-APC interactions in tumor tissue by reducing APC co-stimulatory capacity. To block CTLA-4 function and examine the short-term effects on CD80 and CD86 expression in MC38 tumors, we used the CTLA-4 antibody UC10–4F10–11 (henceforth 4F10) (Walunas et al., 1994). Pretreatment of splenocytes with 4F10 blocked binding of CD80- and CD86-Fc chimeras to Treg cells at a similar ED50 as it blocked its own binding (Figure 4A). Therefore, and because a single i.v. injection of 4F10 reduced ex vivo monoclonal antibody (mAb)-staining of CTLA-4 on tumor-infiltrating Treg cells by 80% (Figure 4B), we estimated that it blocked CD80/CD86-binding to a similar degree in vivo. Of note, 4F10 injection did not reduce Treg cell frequency in tumor tissue, indicating that it does not cause their Fc receptor-dependent depletion as seen with clone 9H10 αCTLA-4 mAb in mice (Figure 4C) (Simpson et al., 2013) and with ipilimumab in a minority of patients with specific FcγRIIIA polymorphisms (Arce Vargas et al., 2018; Romano et al., 2015). Thus, our mouse model replicates the most common situation in humans where αCTLA-4 mAbs ipilimumab or tremelimumab do not deplete tumor-associated Treg cells (Sharma et al., 2019).
Figure 4. CTLA-4 blockade increases the density of co-stimulatory ligands on tumor-associated cDC.
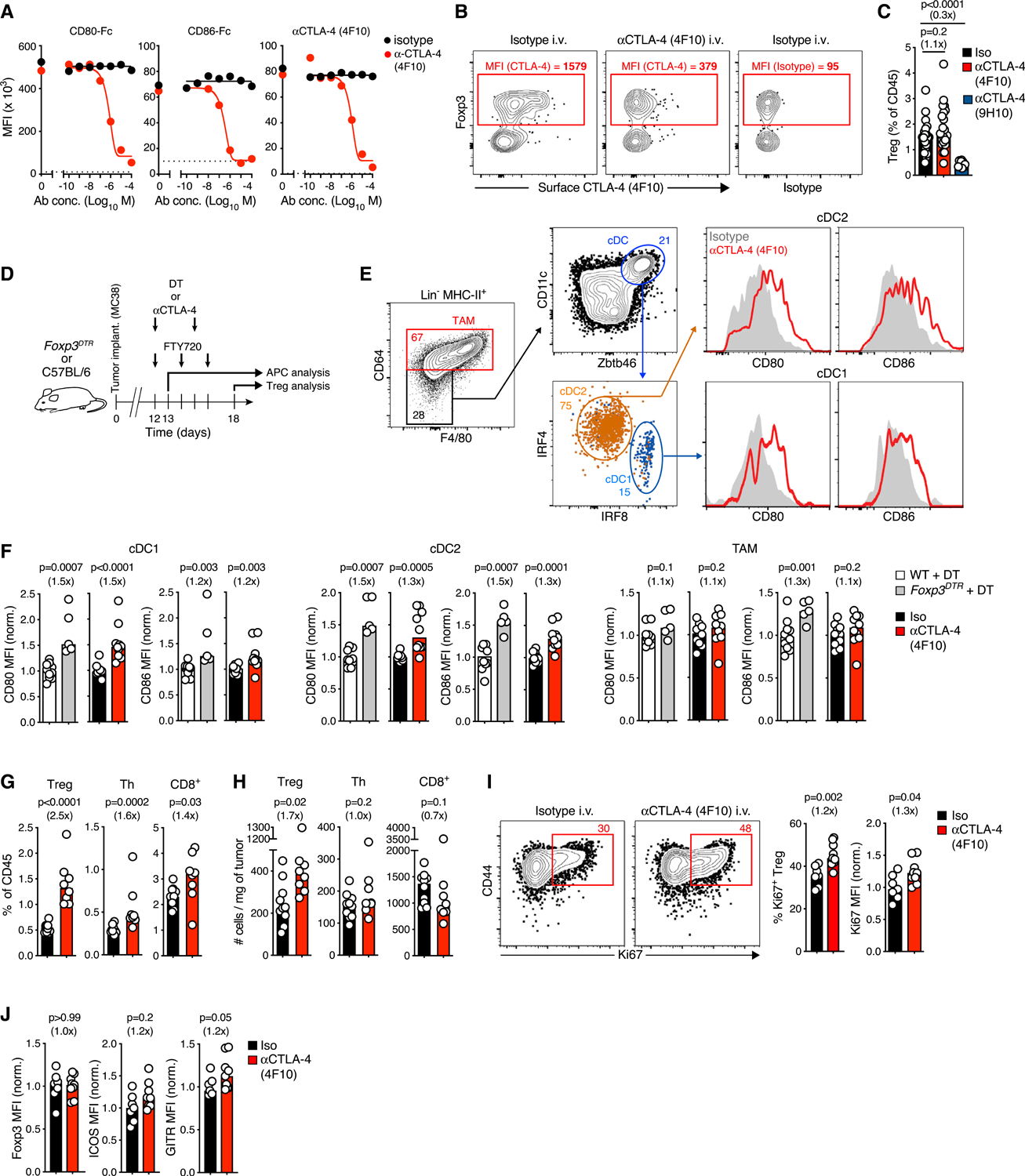
(A) Fixed/permeabilized splenic Treg cells were treated with αCD28 mAb 37.51 to prevent binding of CD80/CD86 to CD28, αCTLA-4 mAb 4F10 added at indicated concentrations, and binding of CD80-hIgG1Fc, CD86-hIgG1Fc, or 4F10 to unbound CTLA-4 was determined. Dotted lines represent background staining (secondary Ab without CD80/CD86-hIgG1Fc or isotype control mAb).
(B) Tumor-bearing mice received 750 μg of 4F10 or isotype control i.v. and 12 h later, unbound cell surface CTLA-4 on tumor-infiltrating T cells was stained ex vivo with fluorescent 4F10. The difference in residual ex vivo binding of fluorescent αCTLA-4 (after substraction of isotype staining MFI) was used to estimate functional in vivo CTLA-4 blockade.
(C) Percentage of Treg cells in tumor tissue 24 h after injection of αCTLA-4 clone 4F10 (n = 21), clone 9H10 (n = 9), or isotype control (n = 19) mAb (pooled from five independent experiments).
(D) Experimental protocol.
(E) Gating strategy to quantify expression of CD80/CD86 on cDC1, cDC2, and TAMs in tumor tissue. Numbers indicate gate frequencies.
(F) CD80/CD86 on cDC1, cDC2, and TAMs 24 h after CTLA-4 blockade or ablation of Treg cells. n = 10 (αCTLA-4 clone 4F10), n = 10 (isotype), n = 5 (DT treatment in Foxp3DTR), and n = 10 (DT treatment in WT) (pooled from two separate experiments).
(G and H) Frequency (G) and numbers (H) of the indicated cell types in tumors after treatment with αCTLA-4 mAb clone 4F10 (n = 8) or isotype-matched control mAb (n = 10) (pooled from two independent experiments).
(I and J) Expression of proliferation (I) and activation markers (J) in tumor-associated Treg cells after CTLA-4 blockade. n = 9 (αCTLA-4) and n = 7 (isotype), pooled from two separate experiments. Bar graphs indicate medians, each symbol an individual animal. MFI are normalized to means of the control group, values in parenthesis indicate fold change relative to medians of the control group, and p values are calculated by Mann-Whitney U test.
See also Figure S5.
24 h after CTLA-4 blockade, IRF8-expressing cDC1 in tumor tissue had increased their expression of CD80 and CD86 to a similar extent as observed 1 day following DT-treatment-induced ablation of Treg cells in Foxp3DTR mice, indicating that CTLA-4 is chiefly responsible for Treg cell-mediated regulation of the CD80/CD86 cell surface pool in these APCs (Figures 4D–4F). This increase was slightly less pronounced in IRF4-expressing cDC2 following CTLA-4 blockade relative to ablation of Treg cells, suggesting the existence of additional, but less effective mechanisms by which Treg cells control co-stimulatory capacity in this DC subset. No increase was detectable on TAMs. At the same time, short-term CTLA-4 blockade did not affect expression of markers of general DC activation, such as CD40, MHC-I, and MHC-II (Figure S5A). Moreover, administration of αPD-1 mAbs, a different form of CBI, caused little or no upregulation of CD80 and CD86 within 24 h (Figure S5A). Thus, αCTLA-4 likely increases CD80 and CD86 expression through a specific mechanism and not through a general inflammatory response that enhances DC activation. An increase of CD80/CD86 expression, but not of CD40, MHC-I, and MHC-II, was also observed in tdLN following CTLA-4 blockade (Figures S5B–S5D). There, CD80/CD86 modulation was more pronounced on migratory compared to resident cDC, as previously observed in immune homeostasis in Ctla4−/− mice (Ovcinnikovs et al., 2019).
To examine how enhanced co-stimulation affected Treg cells in the TMEs, we extended CTLA-4 blockade through a second injection of 4F10 while interrupting recruitment of T cells from tdLN through FTY720 treatment (Figure 4D). Within 6 days, the abundance of Treg cells in tumor tissue more than doubled compared to control animals, paralleled by a rise in their proliferation rate (defined as the fraction of cells expressing Ki67), while expression of Foxp3, ICOS, and GITR did not change (Figures 4G–4J). Th and CD8+ T cell frequencies also increased in the TME after CTLA-4 blockade, yet not as strongly, and their absolute numbers did not (Figures 4G and 4H), even though more Th cells proliferated (Figure S5E). Again, similar changes were observed in the tdLN where the proportion of CD44hi CD62L− eTreg among total Treg cells and their proliferation rate, as well as expression of ICOS, but not of GITR and Foxp3, increased (Figures S5F–S5L). Hence, CTLA-4 restricts the expression CD80 and CD86 on tumor-infiltrating cDCs and limits local Treg cell proliferation.
Treg cells self-regulate their intratumoral population size via CTLA-4 in trans
A variety of mechanisms are conceivable through which CTLA-4 could regulate the size of the tumor-associated Treg cell pool. It may act in cis (e.g., by transducing inhibitory signals) or by competing with CD28 for CD80/86-binding at the immunological synapse (Egen and Allison, 2002; Pentcheva-Hoang et al., 2004). Alternatively, CTLA-4 may act in trans, for instance by restricting effector T cell activation and their secretion of pro-inflammatory cytokines that would otherwise enhance Treg activation or increase CD80/CD86 expression on APCs to enhance Treg cell co-stimulation (for review, see Walker and Sansom, 2015). To determine whether CTLA-4 controls tumor-associated Treg cell proliferation in cis or in trans, we generated mixed irradiation BMCs from CD45.2 Foxp3creERT2 × Ctla4f/f and CD45.1 wild-type (WT) donor bone marrow transplanted into CD45.1 WT hosts (Foxp3creERT2 × Ctla4f/f BMCs), so that a fraction of the Treg cell population could acutely be rendered CTLA-4-deficient through tamoxifen treatment. If CTLA-4 acts in cis, CTLA-4-deficient cells should increase their proliferation despite the presence of CTLA-4-sufficient Treg cells in the same animal. If CTLA-4 acts in trans, the larger CTLA-4-sufficient Treg cell population should prevent hyper-proliferation of CTLA-4-deficient cells. When we implanted these BMCs with MC38 tumors, treated them with tamoxifen to delete CTLA-4 in a fraction of Treg cells, and then with FTY720 to block further T cell recruitment from tdLN (Figures 5A and 5B), loss of CTLA-4 did not increase the proliferative rate or abundance of Treg cells, neither in the TME (Figures 5C and 5D) nor in the tdLN (Figures S6A–S6D), compared to control BMCs. Moreover, Treg cells expressed the same levels of Foxp3, ICOS, and GITR at both sites, irrespective of whether they expressed CTLA-4 or not (Figures S6E and S6F). Thus, CTLA-4 regulates the proliferative rate, abundance, and activation state of tumor-reactive Treg cells not in cis, but in trans.
Figure 5. Treg cells self-regulate their intratumoral population size via CTLA-4 in trans.
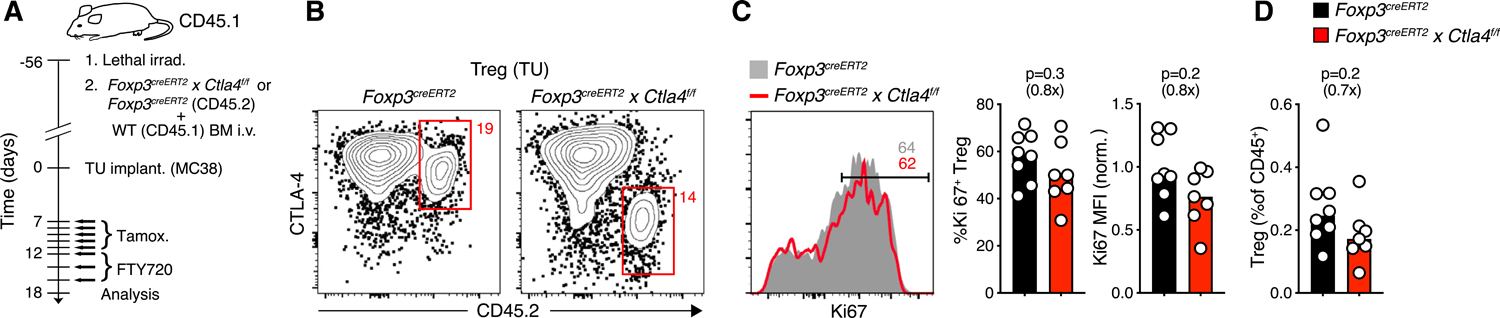
(A) Experimental protocol.
(B) CTLA-4 expression in CD45.2+ Ctla4f/f or control Treg cells in the TME following tamoxifen treatment.
(C and D) Expression of Ki67 (C) and frequency (D) of CTLA-4-sufficient (n = 8) or CTLA-4-deficient (n = 7) CD45.2+ Treg cells in the TME (pooled from two experiments). Fold change as compared to control groups indicated in parentheses. MFI was normalized by the mean of control groups. p values calculated by Mann-Whitney U test.
See also Figure S6.
IL-2 supports Treg cell maintenance in tumors but does not drive their hyper-proliferative response to CTLA-4 blockade
Among effector T cell-produced factors that could enhance Treg cell activity, IL-2 is the best characterized for its key role in peripheral Treg cell homeostasis (D’Cruz and Klein, 2005; Fontenot et al., 2005). Upon ex vivo re-stimulation, both Th and CD8+ T cells produced IL-2 in the MC38 TME, whereas Treg cells did not, as expected (Figures S6G and S6H). In tdLN, Th cells were the major producers of IL-2, while little cytokine was produced by CD8+ cells. However, the frequency and numbers of IL-2-producing cells did not increase following 6 days of αCTLA-4 treatment, neither in the TME nor in tdLN. To also assess the expression of IL-2 in situ, we repeated this experiment in Il2GFP reporter mice, in which GFP expression provides a record of recent Il2-transcription without a requirement for ex vivo re-stimulation (DiToro et al., 2018). The results mirrored those on IL-2 protein expression (Figures S6I–S6K). There was also no change in the expression of tumor necrosis factor (TNF), interferon (IFN)-γ, or granzyme B by effector T cells, with the exception of a moderate increase in IFN-γ produced by Th cells in tdLN upon ex vivo re-stimulation (Figures S7A and S7B).
To also functionally test a potential role for IL-2 in αCTLA-4 treatment-induced effects on tumor-associated Treg cells, we injected mice with mAbs to block its interaction with both the IL-2Rα and IL-2Rβ receptor chains, as described (Boyman et al., 2006) (Figure 6A). Indeed, this caused a reduction of Treg cell abundance and proliferative rate by ~50% in the TME and tdLN (Figures 6B and S7C), indicating that IL-2 maintains Treg cell populations not only in SLOs, as described (Liu et al., 2015; O’Gorman et al., 2009; Smigiel et al., 2014), but also at immune effector sites, such as the TME. However, when we blocked CTLA-4 in addition to IL-2, the increase in Treg cell proliferative rate and abundance was of the same or greater magnitude as in the absence of IL-2 blockade, albeit starting from a lower baseline (Figures 6B and S7C). Hence, although IL-2 contributes to the maintenance of intratumoral Treg cells, it does not facilitate their hyper-proliferative response and expansion following αCTLA-4 treatment.
Figure 6. CTLA-4 blockade expands tumor-associated Treg cells through enhanced CD28 co-stimulation.
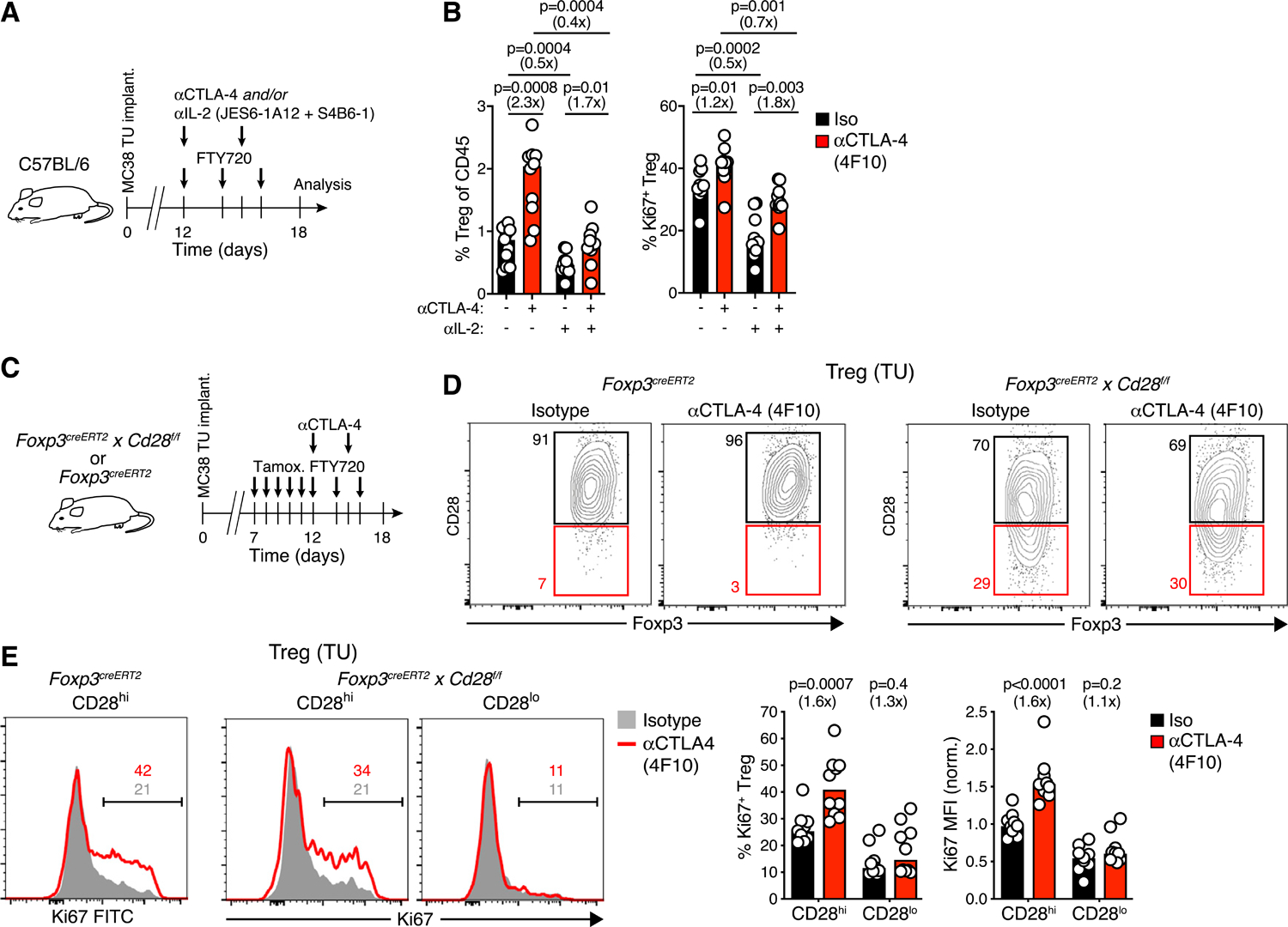
(A) Experimental protocol.
(B) Frequency of tumor-associated Treg and of Ki67+ Treg cells after blockade of CTLA-4 and/or IL-2. n = 10–11 per group in two independent experiments.
(C) Experimental protocol.
(D) Tamoxifen-induced downregulation of CD28 on Treg cells in the TME of Foxp3creERT2 control (left) or Foxp3creERT2 × CD28f/f (right) mice treated with αCTLA-4 (4F10) or isotype mAbs. Gates defining CD28lo cells are based on CD28 expression in Foxp3creERT2 control animals.
(E) Ki67 expression in CD28hi and CD28lo Treg cells in Foxp3creERT2 control (left, no CD28lo cells detected) or Foxp3creERT2 × CD28f/f mice (right) following αCTLA-4 or isotype mAb treatment. Numbers depict cell percentages. Bars represent medians, each symbol represents an animal. Fold change as compared to control groups indicated in parentheses. MFI normalized to the mean of controls; p values calculated by Mann-Whitney U test.
See also Figure S7.
Treg cells calibrate their local proliferation by tuning the amount of CD28 co-stimulation they receive
Thymic Treg cells require CD28 for development and peripheral homeostasis (Salomon et al., 2000; Zhang et al., 2013) and to sustain their function in the TME (Marangoni et al., 2018). We therefore hypothesized that Treg cells limit their own proliferative expansion at effector sites by using CTLA-4 to regulate the density of the CD28 ligands CD80/CD86 on APCs. This would create a rheostat mechanism through which Treg cells constantly adjust their population size by expanding in response to inflammation-induced increases of CD80 and CD86 expression, and by contracting based on the extent to which they then deplete these CD28 ligands from APC. In the context of infections, such a mechanism may help to avoid inappropriate over-regulation of immune responses by Treg cells. We therefore examined how CD28-deficient Treg cells respond to CTLA-4 blockade in the TME by implanting MC38 tumors into Foxp3creERT2 × Cd28f/f mice, which we treated with tamoxifen to delete CD28 in tumor-infiltrating Treg cells (Figures 6C and 6D). Those cells that downregulated CD28 protein below the levels expressed by control Treg cells in Foxp3creERT2 mice (CD28lo) proliferated at a reduced rate compared to their CD28hi counterparts already at baseline, demonstrating a requirement for CD28 to sustain Treg cell proliferation in the TME. More importantly, however, CD28lo Treg cells showed no proliferative response to CTLA-4 blockade, in contrast to CD28hi Treg cells, both in the TME and in tdLN (Figures 6E, S7D, and S7E). αCTLA-4 treatment also did not affect CD28lo Treg cell expression of Foxp3, ICOS, and GITR in the TME and in tdLN, with the exception of a trend toward lower expression of ICOS in tdLN (Figures S7F and S7G). Hence, CTLA-4-mediated control of CD80/CD86 on APC allows Treg cells to calibrate their own activity in the TME by regulating CD28 co-stimulation they themselves receive. CTLA-4-blocking antibody therapy interrupts this mechanism of negative feedback control.
Disruption of the CTLA-4/CD28 rheostat stabilizes Treg cell-APC interactions
Treg cells migrating on lipid bilayers seeded with TCR ligands decrease their speed in response to increasing quantities of CD80 (Thauland et al., 2014). We therefore hypothesized that the instability of Treg cell-cDC interactions observed in the TME resulted from reduced co-stimulation following CTLA-4-mediated depletion of CD80 and CD86 on cDC. To test this, we implanted MC38 H2B-Cer tumors into Foxp3GFP × CD11cmCherry (F1) reporter mice to visualize Treg cell-APC interactions in the TME by MP-IVM (Figure 7A) and used the 3D-colocalization of red (mCherry) and green (GFP) fluorescence that occurred during these interactions for an automated measure of cell-cell contact (Figure 7B). Before and following injection of isotype control mAb, individual APCs interacted with Treg cells for on average 10 min per 1 h of observation time, although some individual APCs cumulatively interacted for more than 60 min per hour through simultaneous contacts with multiple Treg cells, likely reflecting cognate Treg-cDC interactions at baseline. In contrast, cumulative Treg cell contact time per APC increased within hours of 4F10 injection and doubled by 18–20 h, with individual APCs reaching several hours of cumulative contact time per hour (Figures 7C–7E; Video S5). We also quantified the number of contacts per APC (Figures 7F and S7H) and contact duration (Figures 7G and S7I). When considering all APCs (that mostly comprise TAM), the increases in contact frequency or duration at 18 h post αCTLA-4 treatment start did not reach statistical significance (Figures 7F and 7G). However, although the landscape of CD11c-mCherry+ APCs in the TME changed considerably over time, we could, in several cases, identify the likely same APCs or APC clusters before and after either αCTLA-4 or isotype treatment, based on their morphology and position in the TME (Figure S7J). Analysis of Treg cells surrounding these specific APCs revealed a decrease in velocity over time, suggesting stabilization of Treg cell contacts following αCTLA-4 but not isotype antibody treatment (Figure 7H). Hence, CTLA-4-targeted CBI stabilizes Treg cell-APC contacts in the TME, which is followed by local Treg cell expansion.
Figure 7. Treg cells limit the therapeutic efficacy of CBI even when their CTLA-4-dependent function is disabled.
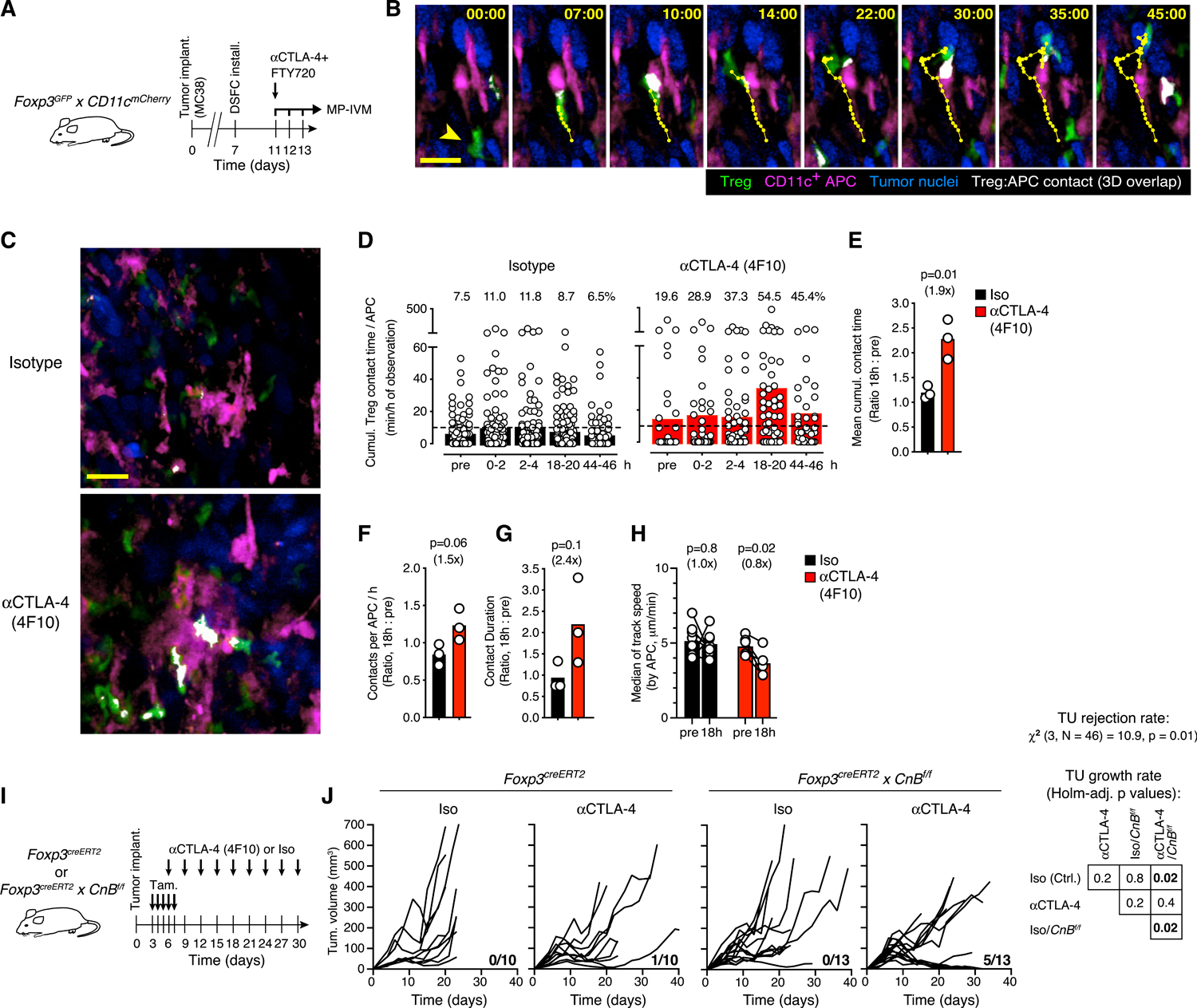
(A) Experimental protocol.
(B) MP-IVM micrographs illustrating the dynamic physical contact zone (white) between a Treg cell (green, arrowhead in first frame) and a CD11c+ APC (magenta) in the TME, as determined by their overlapping fluorescent signals. Numbers indicate min:s. Migratory tracks indicate 1-min intervals. Scale bar, 20 μm.
(C) MP-IVM micrographs recorded 18 h after αCTLA-4 4F10 or isotype control mAb treatment. Note increased Treg cell-APC contacts (white) upon CTLA-4 blockade. Scale bar, 50 μm.
(D) Cumulative Treg cell contact time per hour for individual APCs in the same region of interest of the tumor at various time points. Symbols represent individual APCs and bars means. Numbers above graphs indicate percentages of APCs interacting with Treg cells for >10 min (dashed line) per hour.
(E–G) Ratios of mean cumulative Treg cell contact time per APC (E) mean number of Treg cell contacts per APC (F) and median Treg cell-APC contact duration (G) between 18 h after and before injection of either αCTLA-4 or isotype mAbs. Bars depict means, symbols individual experiments. p value calculated by Student’s t test.
(H) Median track speed of Treg cells within a 20 μm radius of selected APCs identifiable before and 18 h after αCTLA-4 (n = 6) or isotype mAbs (n = 8) administration. Bars represent means, paired symbols individual areas. p values calculated by ratio paired Student’s t test. In (E) to (H), numbers in parentheses represent the fold change from the control group.
(I) Experimental protocol.
(J) Growth curves of MC38 tumors upon 4F10-mediated blockade of CTLA-4 and/or Treg cell-specific deletion of CnB. n = 10–13 per group. The fraction of mice that rejected tumors is indicated in each graph (pooled from three independent experiments). The rates of tumor rejection were compared using χ2 test. Tumor growth rates were compared using type II ANOVA followed by Holm-corrected pairwise comparisons. Table shows Holm-adjusted p values.
Treg cells limit the therapeutic efficacy of CBI even when their CTLA-4-dependent function is disabled
Treg cells utilize CTLA-4 for a core mechanism of immune suppression in mice and humans (Schubert et al., 2014; Wing et al., 2008). Thus, following antibody blockade of CTLA-4, Treg cells may have lost their ability to restrict the anti-tumor immune response, in which case their proliferative expansion would be inconsequential. However, Treg cells command over a variety of alternative suppressive mechanisms (Vignali et al., 2008). To test whether the residual TCR-dependent immunosuppressive functions of hyper-proliferating Treg cells restrict the therapeutic anti-tumor efficacy of CTLA-4 blockade, we disabled Treg cell activation by deleting CnB through tamoxifen treatment of MC38 tumor-bearing Foxp3creERT2 × CnBf/f mice (Figure 7I). αCTLA-4 monotherapy using the non-depleting 4F10 antibody resulted in a minor delay of MC38 tumor progression, as previously observed (Allard et al., 2013; Kocak et al., 2006; Selby et al., 2013). Induced deletion of CnB from Treg cells also caused transient growth stagnation but no rejection of MC38 tumors, as reported (Marangoni et al., 2018). However, the combination of 4F10 treatment and Treg cell inactivation caused elimination of MC38 tumors in more than a third of animals (Figure 7J). Thus, Treg cells that expand in the TME but are deprived of a key mechanism of suppression upon blockade of CTLA-4 continue to oppose the full therapeutic efficacy of CBI.
DISCUSSION
It has been shown that Treg cells need to encounter their cognate antigen locally in the TME in order to promote tumor tolerance (Bauer et al., 2014), but the nature of the antigens they recognize and the identity of the APCs that present these are not well defined. Treg cells infiltrating mouse prostate tumors are enriched for specificity toward a prostate tissue self-antigen also expressed in the thymus in an AIRE-dependent manner, suggesting that tumor-infiltrating Treg cells are preferentially of thymic origin (Leonard et al., 2017; Malchow et al., 2013). This is further supported by the lack of overlap between the TCR repertoires of Th and Treg cells in human tumors, which indicates that few intratumoral Treg cells derive from peripheral conversion of conventional CD4+ T cells (Plitas et al., 2016). Here, we observed that at least one-third of Treg cells from SLOs of healthy mice, when injected into tumor-bearing mice without prior enrichment for tumor reactivity, receive productive TCR signals in the TME, in contrast to very rare Th cells from the same source. This indicates that non-mutational self-antigens are presented in the TME to activate a significant fraction of the thymic Treg cell repertoire. Those self-antigens, which may be tissue-specific or not, could be expressed by cancer cells (as modeled in this study by HA-specific Treg cells infiltrating HA-expressing CT26 tumors) or by cells of the tumor stroma and conceivably be presented to Treg cells by any MHC II-expressing phagocyte in the TME, including TAM. However, we found here that primarily, if not exclusively, rare cDCs activate intratumoral Treg cells. The fact that TCR signaling was also observed in only one-third of clonal HA-specific Treg cells in CT26HA tumors, despite the abundant expression of HA by cancer cells in this model, may reflect limited uptake and presentation of cancer cell-expressed antigens by cDCs in the tumor stroma, or the general scarcity of cDCs.
Among cDCs, cDC1 are specialized for cross-presentation of exogenous antigens to CD8+ T cells, (Broz et al., 2014; Roberts et al., 2016; Salmon et al., 2016), whereas cDC2 are particularly effective at antigen presentation to CD4+ T cells (Binnewies et al., 2019; Gao et al., 2013; Krishnaswamy et al., 2017). In lack of an experimental approach to selectively ablate cDC2, we could not determine their specific role in antigen presentation to Treg cells. However, elimination of cDC1 in Xcr1DTR mice (Yamazaki et al., 2013) did not affect Treg cell abundance or activation state in the TME (data not shown), which aligns with the observation that cDC1 are not critical for thymic Treg cell development (Leventhal et al., 2016). Therefore, intratumoral Treg cell activation may be mediated either selectively by cDC2 or, redundantly, by both cDC1 and cDC2. Because efficient downregulation of CD80/CD86 on APCs via CTLA-4 requires T cell antigen recognition in vivo (Ovcinnikovs et al., 2019), our observation that intratumoral Treg cells regulate CD80/CD86 density on cDC1 and cDC2 indicates that they engage with both cDC subsets in cognate interactions, during which they may also receive activating TCR signals to sustain their function.
The frequency and dynamics of T cell-APC interactions and of the resulting TCR-dependent signaling activities determine the functional outcome of effector T cell activation. Stable and sustained DC contacts culminate in T cell immunity whereas unstable and short-lived contacts lead to T cell tolerance (Fife et al., 2009; Marangoni et al., 2013; Tadokoro et al., 2006; Tang et al., 2006). A proposed mechanism is that repetitive short-lived interactions suffice to sustain the function of NFAT transcription factors due to their rapid activation and slow de-activation kinetics (Marangoni et al., 2013), whereas stable contacts are required for the function of other nuclear factors, such as AP-1, which cooperates with NFAT in the induction of T cell effector programs. Isolated NFAT activation, on the other hand, induces T cell anergy and dysfunction programs (Heissmeyer et al., 2004; Macián et al., 2002; Martinez et al., 2015). We found here that Treg cells deviate from this pattern in that unstable cDC contacts and intermittent NFAT activation sustained their proliferation and suppressive effector state in the TME. Although we have not tested what additional nuclear factors are activated during short-lived Treg cell-APC contacts, it is interesting to note that NFAT can also interact with Foxp3 to induce expression of CD25 and CTLA-4, and thereby equip Treg cells with suppressive function (Wu et al., 2006). During thymic Treg cell development, TCR-driven activation of AP-1, likely in cooperation with NFAT, facilitates chromatin remodeling at Treg cell-specific enhancers and prepares these sites for Foxp3 binding (Samstein et al., 2012). Constitutively nuclear Foxp3 may therefore replace the function fulfilled by AP-1 in effector T cells, at least on some genes, and thereby relieve Treg cells from the requirement for stable cDC contacts and enable them to utilize transient NFAT signals to sustain their immunosuppressive gene expression program.
Several studies have identified differences in the way Treg and Th cells transduce TCR signals (Gavin et al., 2002; Hickman et al., 2006; Yan et al., 2015). However, these do not seem to explain the differences in intratumoral cDC contact stability reported here. Instead, we found that differences in how Treg cells and Th cells, even those expressing the same TCR, interact with intratumoral APCs were cell-extrinsic and resulted from the ability of Treg cells to alter the antigen-presenting function of APC, reminiscent of the capacity to also destabilize CTL contacts with intratumoral APCs (Marangoni et al., 2013).
In SLOs, IL-2 secreted during incipient autoreactive Th cell responses induces localized Treg cell activity that rapidly quells these responses (Liu et al., 2015). However, prior studies did not reveal a role for IL-2 in the maintenance of CD44hi eTreg cells, neither in SLOs nor in non-lymphoid tissues (Smigiel et al., 2014). It was therefore unexpected that IL-2 helped sustain the size of the tumor-infiltrating Treg cell pool composed exclusively of eTreg cells. eTreg cells have poor access to IL-2 produced in the T cell areas of SLOs due to their lack of CCR7 expression (Smigiel et al., 2014) but may have access in the TME by co-clustering with IL-2-secreting effector T cells around the same, rare cDCs.
Treg cell hyper-proliferation following CTLA-4 blockade did, however, not result from enhanced exposure to IL-2, but was entirely dependent on co-stimulation via CD28 and therefore likely driven by the rapidly ensuing increase of CD80 and CD86 expression on APC. Therefore, our study reveals a second, distinct mechanism by which Treg cells self-regulate by tying their proliferative expansion to a key outcome of their suppressive activity. Unlike regulation by IL-2 availability, however, this mechanism is independent of the presence of activated effector cells and may calibrate Treg cell activity also at those effector sites where IL-2-secreting cells are sparse.
Even though Treg cells use CTLA-4 to inhibit anti-tumor immunity (Wing et al., 2008), they continued to obstruct tumor elimination following CTLA-4 blockade, pointing to their deployment of alternative mechanisms of suppression. This is not without precedent, because Treg cells upregulate IL-10 and continue to protect mice from experimental autoimmune encephalomyelitis upon deletion of CTLA-4 (Paterson et al., 2015). We do not know whether expansion of Treg cells deprived of their capacity to deplete co-stimulatory ligands on cDCs produces a net increase in immunosuppression in the TME. The residual suppressive function of expanded Treg cells is, however, functionally relevant in our preclinical tumor model and may potentially also offset the efficacy of αCTLA-4 CBI in cancer patients. Treg cell depletion or functional inactivation in the TME could therefore synergize with CTLA-4 blockade. In mouse models, the efficacy of αCTLA-4 CBI correlates with the degree of intratumoral Treg cell depletion mediated by binding of the therapeutic antibody to Fc-receptors on local phagocytes (Bulliard et al., 2013; Marabelle et al., 2013; Selby et al., 2013; Simpson et al., 2013). The contribution of Treg cell depletion to the efficacy of clinical αCTLA-4 therapy is controversial and may vary with the size and composition of the immune infiltrate, the therapeutic antibody isotype and glycosylation, and patient Fc-receptor polymorphisms that determine therapeutic antibody binding affinity (Arce-Vargas et al., 2018; Ferrara et al., 2019; Romano et al., 2015; Sharma et al., 2019). Consequently, ipilimumab is now being engineered to enhance Fc-receptor binding and intratumoral Treg cell depletion. This strategy has proven effective in Fc-receptor humanized mice (Korman et al., 2017) but also produced greater toxicity in monkeys, presumably due to extratumoral Treg cell depleting activity (Price et al., 2018). Another limitation of highly effective depletion of CTLA-4 expressing cells is the inadvertent removal of anti-tumor CTL that also upregulate this molecule on the cell membrane, especially once they are relieved from Treg cell-mediated suppression (Ha et al., 2019). In mouse models, this can be avoided by delaying concurrent treatments designed to activate CTL, such as tumor vaccination, relative to αCTLA-4 CBI, however, this strategy may be impractical in human therapy given the longer half-life of ipilimumab and its derivatives compared to antibodies used in mice. Strategies for more selective depletion of intratumoral Treg cell using mAbs targeting molecules other than CTLA-4, such as CCR4, CCR8, and OX40 (Kamada et al., 2019; Villarreal et al., 2018), may therefore be preferable for combination with ipilimumab.
A precise mechanistic understanding of cancer immunotherapies is the basis for their continuous improvement. Here, we report that, in a preclinical tumor model, Treg cells act as a biological rheostat for the expression of the co-stimulatory proteins CD80 and CD86 on cDC, which are induced by innate stimuli. Treg cells achieve this by using their dependence on CD28 co-stimulatory signals to continually adjust their population size to the extent to which they have depleted the CD28 ligands CD80 and CD86. The resulting equilibrium state is interrupted by therapeutic CTLA-4 blockade, which produces an intratumoral pool of hyper-proliferative Treg cells that are deprived of their use of CTLA-4, but maintain immunosuppressive function and continue to antagonize immune-mediated tumor rejection.
Limitations of the study
Our observation that Treg cells self-regulate in trans is not compatible with models of CTLA-4 inhibitory signaling or cell-intrinsic competition with CD28 for binding to B7 proteins at the immunological synapse but support models in which Treg cells use CTLA-4 to alter APC function. We propose that the rapid and selective downregulation of CD80 and CD86 on cDCs is most compatible with CTLA-4-mediated trans-endocytosis (Qureshi et al., 2011). However, other mechanisms, such as cellular competition for access to B7 proteins on APC, reverse signaling via CD80 (Kowalczyk et al., 2014), or “masking” of B7 proteins on all APCs by soluble splice variants of CTLA-4 (Magistrelli et al., 1999), may also contribute to Treg cell self-regulation. The role of trans-endocytosis in Treg cell function in the TME and other immune effector sites thus needs to be further defined.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Thorsten Mempel (tmempel@mgh.harvard.edu).
Materials availability
The MW-NFAT-GFP-IRES-H2B-RFP retroviral construct is available upon request.
Data and code availability
Imaris scripts to calculate NFAT SI using the 3D central accumulation algorithm, as well as the MATLAB cell motility analysis scripts are available upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Ppp3r1f/f (here referred to as CnBf/f) (Neilson et al., 2004), Foxp3cre (Rubtsov et al., 2008), Foxp3creERT2 (Rubtsov et al., 2010), Foxp3GFP (Lin et al., 2007), Foxp3DTR (Kim et al., 2007), zDCDTR (Zbtb46DTR) (Meredith et al., 2012), C57BL/6, C57BL/6 CD45.1, and BALB/c mice were from Jackson laboratories. Cd28f/f (Zhang et al., 2013), Cd11cmCherry (Khanna et al., 2010), Ctla4f/f (Paterson et al., 2015), Il2GFP (DiToro et al., 2018), and pgk-HA × TCR-HA transgenic mice as a source of HA-specific Treg cells (Klein et al., 2003) were directly obtained from the investigators who had generated them. Mice were enrolled in experiments at 6–12 weeks of age. Per NIH guidelines, both male and female mice were used for experiments whenever possible, and results pooled when comparable. In all cases, mice were bred, housed, enrolled in experiments and euthanized according to protocols approved by the Institutional Animal Care and Use Committee (IACUC) of the Massachusetts General Hospital and of the University of California - Irvine.
Cells
Platinum-E cells were purchased from Cell Biolabs (Cat# RV101) and grown in DMEM 1640 supplemented with 10% fetal calf serum (Atlanta Biologicals), 1 μg/ml puromycin and 10 μg/ml blasticidin (both from Invivogen) under 37°C/5% CO2 conditions. Platinum-E cells are derived from the female human 293T cell line. This cell line has not been authenticated.
MC38 colon carcinoma cells were purchased from Kerafast (Cat# ENH204-FP) and grown in DMEM 1640 supplemented with 10% fetal calf serum (Atlanta Biologicals) under 37°C/5% CO2 conditions. MC38 cells are derived from female mice (Corbett et al., 1975). This cell line has not been authenticated.
CT26 colon carcinoma cells were purchased from ATCC (Cat# CRL-2638) and grown in DMEM 1640 supplemented with 10% fetal calf serum (Atlanta Biologicals) under 37°C/5% CO2 conditions. CT26 cells are derived from female mice (Castle et al., 2014). This cell line has not been authenticated.
All cell lines were routinely tested for mycoplasma contamination and found to be negative.
Viral constructs
The retroviral construct MW NFAT-GFP-IRES-H2B-RFP was cloned as follows. As a retroviral vector backbone, we selected an MSCV-based plasmid further engineered to contain a multiple cloning site (MCS), an internal ribosomal entry site (IRES), the reporter gene ΔLNGFR and a WPRE element to stabilize the transgenic RNA (MiNW, a kind gift of Dr. Roberto Bonasio). The ΔLNGFR gene was substituted with the tdTomato gene using standard cloning techniques to obtain the MiTW plasmid. The NFAT-GFP ORF (Aramburu et al., 1998) (ADDGENE 11107) was amplified by high-fidelity PCR (PfuUltra DNA Polymerase, Agilent) and cloned within the BglII – EcoRI sites in the MCS of MiTW using the following primers: 5′-GCTAGATCTCACCATGGACGTCCC-3′ and 5′-GCTCAATTGTTACTTGTACAGCTCGTCCA-3′. To substitute tdTomato with H2B-RFP, we first inserted unique MfeI and PacI sites by site-specific mutagenesis (QuikChange lightning site-directed mutagenesis kit from Agilent Technologies) to the 5′ and the 3′ of tdTomato, respectively, using the following primers: for Mfe I 5′-GGCCACAACCCATGTCAATTGCCATGGTGAGCAAGG-3′ and 5′-CCTTGCTCACCATGGCAATTGACATGGGTTGTGGCC-3′ and for PacI 5′-CGAGCTGTACAAGTAATTAATTAAGGATCCTAATCAACCTCTGG-3′ and 5′-CCAGAGGTTGATTAGGATCCTTAATTAATTACTTGTACAGCTCG-3′. We finally amplified H2B-RFP from pHIV-ef-1α-H2BmRFP1 (Welm et al., 2008) (ADDGENE 18982) by PCR using primers 5′-GCTCAATTGCACCATGGCCACAACCATGTCAC-3′ and 5′-GCCGTTAATTAATTAGGCGCCGGTGGAGT-3′ and cloned it between the MfeI-PacI sites to generate the MW NFAT-GFP-IRES-H2B-RFP vector.
All constructs were verified by Sanger sequencing, propagated in XL-10 Gold Ultracompetent Cells and grown at 37°C in a shaking incubator for 18 hours. Plasmids were purified using a commercial kit (QIAGEN) and DNA content quantified by spectrophotometry. Plasmid stocks were stored at −80°C.
METHOD DETAILS
Generation of packaging cell lines and preparation of viral stocks
To generate stable packaging cell lines, we adopted a two-step strategy. In the first step, we generated VSV-g pseudotyped retroviral vectors to infect human cells (“shuttling” vectors). In the second step, we used the shuttling vectors to stably transduce packaging cells.
Shuttling vectors were generated by transient transfection of Platinum-E cells, which express retroviral gag-pol and mouse ecotropic env, with the retroviral plasmid MW NFAT-GFP-IRES-H2B-RFP and the VSV-g containing plasmid pMD2.g (ADDGENE 12259) using Lipofectamine 2000 (Invitrogen). Cell culture medium was changed after 16 h to eliminate Lipofectamine, and supernatant containing the shuttling vectors was harvested every 24h thereafter. Shuttling vectors were then applied onto fresh Platinum-E cells, which were subsequently spin-fected (1000 × g for 90 min at 32°C) to facilitate infection. Transduction was performed twice. Transduced GFP+ RFP+ Platinum-E cells were purified by FACS and used as a continuous and stable source of MW NFAT-GFP-IRES-H2B-RFP retroviral vectors.
In vitro transduction of Treg and Th cells with retroviral vectors
Polyclonal or HA-specific Treg cells were enriched from spleen and lymph nodes of C57BL/6 or pgk-HA × HA-TCR mice using the CD4+ CD25+ Regulatory T Cell Isolation Kit from Miltenyi. To ensure that no contaminating Th cells were present, we further FACS-purified CD4+ CD25hi CD62L+ cells. An analogous strategy was used to purify polyclonal or HA-specific Th: we used the CD4+ CD25+ Regulatory T Cell Isolation Kit to enrich CD4+CD25− Th, which were further purified by FACS to obtain a CD4+ CD25− CD62L+ population.
Treg and Th cells were resuspended at a concentration of 106/ml in DMEM with 10% FCS (Atlanta Biologicals), glucose (4.5 g/L), 1 mM GlutaMax, 5 mM HEPES, 100 μM non-essential amino acids, 0.5 mM sodium pyruvate, 55 μM β-mercaptoethanol and 400 ng/ml recombinant IL-2 (R&D). To activate the cells, αCD3/CD28 mAb-coated beads (Invitrogen) were added at a 4 to 1 bead to cell ratio for two days (Tang et al., 2004). On the third day, we substituted the cell medium with MW NFAT-GFP-IRES-H2B-RFP supernatant supplemented with 400 ng/ml IL-2, spin-fected the cells at 1000 × g for 90 min at 32°C, and finally substituted retroviral supernatant with fresh medium containing 400 ng/ml IL-2. The spin-fection procedure was repeated on day 4, followed by removal of the αCD3/CD28-coated beads using a cell magnet (Invitrogen). Cell culture was continued in presence of 400 ng/ml IL-2 to day 6, adjusting the cell concentration to 106/ml. Before use in experiments, each cell preparation was quality controlled for Foxp3 expression (> 90% for Treg, < 5% for Th cells) and NFAT-GFP / H2B-RFP transduction rate (> 30%).
Preparation of mice for MP-IVM studies
We intravenously injected 0.5–1 × 106 NFAT-GFP and H2B-RFP co-transduced polyclonal Treg or Th cells into CD11cmCherry mice following their sublethal irradiation (450 rad) in order to achieve partial lymphodepletion and facilitate the engraftment of adoptively transferred cells, as previously described (Wright et al., 2009). Mice were allowed to recover for 6 weeks before being injected with 0.5 × 106 MC38 cells engineered to express H2B-Cerulean (MC38 H2B-Cer) in the center of the back, approximately 1 cm to the right of the midline. An additional 106 MC38 H2B-Cer cells were injected in the homolateral scruff of the neck, to ensure adequate antigen drainage to the regional LN. Six days after tumor injection, we surgically implanted a dorsal skinfold chamber (DSFC) in a way that the tumor injected in the back was centered in the optical window of the DSFC. Analgesia was achieved by injecting 5 mg/kg carprofen s.c. pre-operatively and every 24 h thereafter until the end of the experiment, and surgical anesthesia was achieved through isoflurane inhalation. MP-IVM analysis was performed at multiple time points. An identical procedure, except for the use of (CD11cmCherry × BALB/c) F1 mice as tumor hosts and injection of CT26HA H2B-Cer tumors, was employed to image HA-specific Treg and Th cells.
To study polyclonal Treg cell activation in mice devoid of cDC, we lethally irradiated (950 rad) B6 mice and transplanted them with 10 × 106 bone marrow cells from zDCDTR mice. Twenty days later, a time point at which the T cell compartment is still incompletely reconstituted, we adoptively transferred 0.5 – 1 × 106 NFAT-GFP and H2B-RFP co-transduced polyclonal Treg cells. Six weeks after irradiation, mice were implanted with MC38 H2B-Cer tumors in DSFC for visualization by MP-IVM. To deplete cDC, mice received 20 μg/kg diphtheria toxin i.p. We compared either DT- with vehicle-treated zDCDTR − > B6 BMCs or DT-treated zDCDTR − > B6 with DT-treated B6 − > B6 BMCs.
Treg cell interactions with CD11c+ tumor-associated APCs before and after CTLA-4 blockade (through i.v. injection of 750 μg 4F10 mAb) were visualized directly in Foxp3GFP × CD11cmCherry mice with DSFC-implanted MC38 H2B-Cer tumors.
MP-IVM recordings
Mice were anaesthetized with inhaled isoflurane and the DSFC was secured on a custom-built stage for imaging on an Olympus MPE-RS multiphoton microscope. Imaging depth was typically 30–120 μm below the DSFC glass. For multiphoton excitation and second harmonic generation, a MaiTai DeepSee HP and a co-axially aligned Insight X3 Ti:sapphire laser (Newport/Spectra-Physics) were tuned to 850 and 985 nm for optimized excitation of the fluorescent probes used. For four-dimensional recordings of cell migration, stacks of 11 optical sections (512 × 512 pixels) with 4 μm z-spacing were acquired every 30 or 60 s to provide imaging volumes of 40 μm in depth. Emitted light and second harmonic signals were detected through 455/50 nm, 525/50 nm, 590/50 nm, and 665/65 band-pass filters with non-descanned detectors. Datasets were transformed in Imaris 8.4 (Bitplane) to generate maximum intensity projections (MIP) and exported as QuickTime movies.
Calculation of NFAT Signaling Index (SI) through the 3D central accumulation algorithm
To estimate the fraction of nuclear among total cellular NFAT-GFP, we devised a new algorithm called “3D central accumulation” and compared its performance to the “2D radial fluorescence intensity” algorithm (Marangoni et al., 2013), using an existing dataset of four recordings of HA-specific NFAT-GFP and H2B-RFP co-transduced CD8+ cytotoxic T lymphocytes (CTL) undergoing TCR activation in HA-expressing tumors, and one recording of HA-specific NFAT-GFP and H2B-RFP co-transduced CD8+ T cells interacting with HA peptide-loaded B cells in a tdLN (Marangoni et al., 2013). The 3D central accumulation algorithm calculates a spherical pseudo-nucleus around the center of mass of each 3D-cell object constructed based on a cell’s NFAT-GFP signal. The volume of the pseudo-nucleus was set to 30% of the average Treg cell body and 20% of the average Th cell body to approximate the actual average volumes of nuclei in each cell type, derived from measurements of authentic H2B-RFP nuclear signals. The NFAT SI were then calculated as the fraction of nuclear (contained within the pseudo-nucleus) to total cellular NFAT GFP signal, normalized across datasets to range from 0 to 1. This strategy bears many predicted advantages over the legacy 2D radial fluorescence intensity method, in that it considers the 3D shape of the cell and is robust against errors due to faint H2B-RFP signals and the interference of autofluorescence or the mCherry signals from APC, which may render the correct delineation of the true nucleus unreliable (Figure S1F). Comparison of the NFAT SI determined by the 2D and the 3D algorithms using a threshold of 0.5 to define non-signaling (SI < 0.5) or signaling (SI ≥ 0.5) resulted in 85% of cells being concordantly classified (Figure S1G). However, discordant determination also occurred. To compare the performance of the 2D and the 3D algorithms as binary classifiers, we compared the measurements obtained by the 2D and 3D automated algorithms with our visual classification as “signaling” or “not signaling.” A table for each algorithm, reporting the number of true positive (TP), false positive (FP), true negative (TN) and false negative (FN) calls for each of the five recordings is shown in Figure S1H. Based on these numbers, we calculated for each method the accuracy (the percentage of correctly classified instances, (TP+TN) / (TP+TN+FP+FN)), specificity (the ability to correctly classify non-signaling cells as such, TN/(TN+FP)) and sensitivity (the ability to correctly classify signaling cells as such, TP/(TP+FN)). We found that the 3D method was more accurate than the 2D method, essentially because of a rise in specificity, while sensitivity was comparable at values ~90% (Figure S1I). Accordingly, the alpha error was lower for the 3D than for the 2D method, meaning that a positive call by the algorithm has a higher probability to be a signaling event. On the other hand, the beta error that is strictly connected to sensitivity was comparable between the two methods, and both miss ~10% of signaling events (Figure S1I).
Finally, we sought to further compare the ability of the 3D and the 2D algorithms to produce precise continuous NFAT SI measurements, using an unbiased reference method. This reference method uses the H2B-RFP signal of selected cells with clearly visualized nuclei that do not overlap with autofluorescence or CD11cmCherry signals and calculates the NFAT SI based on the ratio of NFAT-GFP signal within this “true” nucleus and NFAT-GFP signal in the entire 3D-cell object (Figure S1J). Unfortunately, this method is not broadly applicable as the nucleus is often difficult to detect because of faint H2B-RFP signals or interference from mCherry+ APC (see also Figure S1F). To compare the performance of the 2D or the 3D algorithms to this reference method, we calculated for each data point the absolute value of the difference between the SI calculated with either the 2D or 3D algorithm and the SI calculated with the reference method. This absolute difference is high when data points are misclassified, and low when data points are correctly classified (Figure S1K). The 3D method generated significantly fewer data points with high absolute differences (Figure S1L). Thus, the 3D central accumulation algorithm tends to make fewer errors than the 2D method both as a binary and as a continuous classifier. It is also more robust in comparison to an automated NFAT SI calculation method that uses actual nuclear measurements, but which is applicable to only a small subset of cells. For these reasons, we chose the 3D central accumulation algorithm to generate the NFAT SI.
Calculation of cell motility parameters
To correlate migratory dynamics with NFAT signaling activity, tracks were divided into signaling and non-signaling segments, as follows. The series of instantaneous NFAT SI of each track was smoothed using a 10-minute moving average. A signaling segment was identified when the averaged NFAT SI remained above a threshold of 0.5 for a minimum of 3 minutes, however tolerating values under the threshold for a maximum of 1 minute. Each part of a track longer than 3 minutes and not identified as a signaling segment was identified as a non-signaling segment. If a track contained more than 3 consecutive time points during which no measurement was possible, e.g., due to the transient departure of most of the cell body from the field of view, the track was split in two. When the moving average window included time-points lacking a measurement, these were excluded from the average NFAT SI calculation.
Dynamic parameters of tracks or segments (% of time with or duration of active NFAT, 3D velocity, arrest coefficient, 10-minute displacement) were analyzed in MATLAB (Mathworks), using non-smoothed data. Percentage of time with active NFAT was calculated using a NFAT SI threshold of 0.5. Arrest coefficient was defined as the fraction of time in a track or segment that a cell was migrating at a velocity below 2 μm/min.
Estimation of NFAT deactivation kinetics in Treg and Th cells
CD4+ T cells were purified from splenocytes of Foxp3GFP mice by immunomagnetic selection and were stimulated with 1 μg/ml ionomycin at 37°C for 10 minutes to induce maximal activation of NFAT. NFAT activation was then blocked with 1 μM cyclosporine A (CsA) for various amounts of time. Five minutes before each time point, cells were allowed to adhere to poly-L-lysine-coated 12 mm circular coverglasses, and then fixed with 4% formalin for 15 min. Cells were permeabilized with PBS 5% BSA 0.3% Triton for 60 min and reacted with αNFAT1 D43B1 antibody (Cell Signaling Technologies) at 1:50 dilution overnight at 4°C. NFAT1 reactivity was revealed with 4 μg/ml αrabbit IgG Fab Alexa Fluor 633 for 1 hour at room temperature. Nuclei were counterstained with 100 nM DAPI for 5 min at room temperature. Slides were mounted and analyzed using a Zeiss LSM780 confocal microscope.
CTLA-4 blockade in vitro
To determine whether αCTLA-4 clone 4F10 blocks CTLA-4 binding to CD80 and CD86, we permeabilized splenocytes and treated them with graded doses of 4F10 or an isotype control mAb for 60 min at 4°C. All subsequent incubations lasted 30 min at 4°C. Cells were washed, and the amount of residual functionally available CTLA-4 was determined by incubation with 2 μg/ml of recombinant CD80-hIgG1 Fc or CD80-hIgG1 Fc (Biolegend), revealed by subsequent incubation first with 1.25 μg/ml of PE-conjugated goat αhu-man IgG (Southern Biotech) and then with 5 μg/ml PE-conjugated αgoat IgG Abs (Invitrogen). To prevent binding of CD80-hIgG1 Fc or CD80-hIgG1 Fc to CD28 on splenocytes, we blocked CD28 with 10 μg/ml of αCD28 clone 37.51 (Biolegend) during incubation with titrated doses of 4F10. Alternatively to CD80- and CD86-Fc, residual non-blocked CTLA-4 was revealed by incubation with 4 μg/ml APC-conjugated 4F10 mAb (Millipore) under conditions at which displacement of the originally applied blocking antibody is negligible (data not shown). Further staining for CD4 and Foxp3 through conventional protocols identified Treg and Th cells.
CTLA-4, PD-1, and IL-2 blockade in vivo
Twelve days after the injection of 106 MC38 cells, CTLA-4 was blocked by i.v. injection of 750 μg 4F10 mAb (BioXCell). Injections were repeated every 3 days. As control Ab, we used Armenian Hamster IgG (BioXCell) at an identical dosing schedule. Concomitant to CTLA-4 blockade, 1 mg/kg FTY720 was injected i.p. every other day to block lymphocyte egress from tdLN and other SLO, thus allowing us to study immunological effects of CTLA-4 blockade selectively on cells within the tumor environment. CTLA-4 treatment was continued for 6 days, which, based on preliminary experiments, was the earliest time point at which tumor-associated Treg cell expansion was evident. Treg cell depletion activity by 4F10 mAbs was compared to the αCTLA-4 clone 9H10 (Simpson et al., 2013) administered at an identical dosing schedule. For experiments to evaluate the role of IL-2 in tumor-associated Treg cell expansion, IL-2 was blocked concomitantly to αCTLA-4 and FTY720 administration by i.v. injection of 750 μg of JES6–1A12 (blocking the interaction of IL-2 with IL2Rβ) and 750 μg of S4B6–1 (blocking the interaction of IL-2 with IL2Rα) (Boyman et al., 2006). To block PD-1, we administered 200 μg of αPD-1 mAb clone 29F.1A12 i.p. every other day. This clone facilitates rejection of immunogenic tumors (Di Pilato et al., 2019). All blocking mAbs were from BioXCell. Rat IgG (Sigma) was injected as control for αIL-2 and αPD-1 mAb treatments.
Generation and use of bone marrow chimeras for flow cytometry studies
For analysis of Treg cell activation in absence of cDC, we injected 10 × 106 bone marrow cells from zDCDTR (Zbtb46DTR) or control mice into lethally irradiated (950 rad) B6 recipients. Upon hematopoietic reconstitution, bone marrow chimeras (BMCs) were s.c. injected with 106 MC38 cells. When tumors were established, mice received diphtheria toxin (20 μg/kg on the first day, then 4 μg/kg every third day) concomitantly with 1 mg/kg FTY720 every other day.
To assess whether CTLA-4 controls Treg cell expansion in cis or in trans, we generated radiation BMCs by injecting a mixture (10 × 106 total cells) of CD45.2 Ctla4f/f × Foxp3creERT2 and CD45.1 B6 bone marrow into lethally irradiated (950 rad) CD45.1 mice. Control BMCs were generated by injecting a mixture of CD45.2 Foxp3creERT2 and CD45.1 B6 bone marrow into lethally irradiated CD45.1 mice. The mixture of CD45.2+ and CD45.1+ bone marrow was titrated to produce an approximately 1:5 ratio within the Treg cell compartment upon reconstitution, in order to maximize the chance of observing trans effects due to the abundance of CD45.1+ WT Treg cells, while not changing the odds of observing cis effects intrinsic on the CD45.2+ population. Upon hematopoietic reconstitution, mice received 106 MC38 cells s.c.. Tamoxifen was dissolved in 10% ethanol + 90% olive oil and injected i.p. at a dose of 1 mg/day for 5 consecutive days. FTY720 was administered i.p. at 1 mg/kg every other day starting on the day after the termination of tamoxifen treatment.
Flow Cytometry
Single cell suspensions were prepared by mechanical dissociation of tdLN and by digestion of finely minced tumor tissue for 30 min at 37°C using DMEM 10% FCS supplemented with 1.5 mg/ml Collagenase IV (Worthington) and 50 U/ml DNase I (Roche).
Dead cells were stained through exposure to Zombie Red (1:400) or Zombie Yellow (1:200), diluted in PBS, for 15 min at 4°C. Cells were subsequently treated with Fc-Block for 10 min at room temperature to decrease nonspecific Ab binding. mAb staining was carried out at 4°C for 30 min.
The staining panel to enumerate T cells and analyze Treg cell activation included Zombie Red and αCD45, αCD8, αCD4, αFoxp3, αCD44, αCD62L and mAbs against various activation markers including Ki67, ICOS, GITR, and CTLA-4. Absolute cell counts were obtained either by combining a separate cell concentration determination from a Neubauer Haemocytometer with flow cytometric population data or by adding an internal bead counting standard (Life Technologies) to the flow cytometric sample and expressed either as cells/organ (tdLNs) or cells/gram of tissue (TU).
To measure cytokine production by T cells, 2 × 106 live cells from tdLN or tumors were stimulated with PMA (50 ng/ml) and ionomycin (1 μg/ml) in the presence of Brefeldin A (5 μg/ml) for 4 h at 37°C. Cells were then stained using Zombie Yellow and αCD45, αCD8, αCD4, αFoxp3, αCD44, αIFNγ, αTNF and αIL-2 mAbs. Re-stimulated IL-2 production was determined by subtracting background production (culture for 4h with Brefeldin A but without stimulation) from the production during 4h of PMA/ionomycin/Brefeldin A treatment. Measurement of GFP as a reporter of transcriptional activity of the Il2 locus was performed directly ex vivo in tumor-bearing Il2GFP mice treated with αCTLA-4 (4F10) or isotype control Abs. Since the fluorescent GFP signal is lost upon cell permeabilization required for Foxp3 staining, we validated the combination of surface markers CD4, CD25, and 4–1BB to identify cells highly enriched for Foxp3+ expression (Figure S6I) and used these markers to identify Treg cells in absence of Foxp3 staining in samples from Il2GFP mice.
We used two panels to analyze APC. For analysis of APC in CD11cmCherry mice, we relied solely on cell surface markers to avoid loss of soluble mCherry protein from the cytoplasm during permeabilization of cells for intracellular staining. This panel included Zombie Yellow, a lineage cocktail of mAbs against TCRβ, CD19 and Gr-1, as well as αCD45, αCD64, αF4/80, αCD40, αMHC-I Kb, αMHC-II, αCD26, αCD11c, αCD103, αCD11b, αXCR1, αCD172a (SIRPα), αCD80, and αCD86 mAbs. The panel to analyze transcription factors within APC included: Zombie Yellow, a lineage cocktail against TCRβ, CD19 and Gr-1, as well as αCD45, αCD64, αF4/80, αMHC-II, αZbtb46, αCD11c, αIRF4, and αIRF8 mAbs.
To quantify the expression of surface CTLA-4 on tumor-associated Treg and Th cells, we stained cells with 4 μg/ml αCTLA-4 4F10 mAb or isotype controls, conjugated to APC. The signal from the APC-conjugated antibodies was then amplified by incubation with 5 μg/ml biotinylated αAPC antibodies (Biolegend), followed by 2 μg/ml APC-conjugated streptavidin (Biolegend). After CTLA-4 surface staining, cells were stained for CD4 and Foxp3 to identify Treg and Th cells.
PCR
Deletion of the floxed CnB alleles upon tamoxifen treatment of Foxp3creERT2 × CnBf/f mice was assessed in Treg cells from tdLN and tumors. We took advantage of the expression of GFP from the Foxp3creERT2 (Foxp3GFP-creERT2) allele to FACS-purify CD4+GFP+ Treg cells by double-sorting to achieve > 98% purity. DNA was extracted immediately using the PicoPure DNA extraction kit (Arcturus) and the rearrangement of the CnB locus was analyzed by PCR.
Tumor growth after CTLA-4 blockade and CnB deletion in Treg cells
One million MC38 cells were injected in both flanks of Foxp3creERT2 × CnBf/f or Foxp3creERT2 control mice. Because the MC38 cell line originated in female mice (Corbett et al., 1975), we used female mice as tumor recipients in these experiments. Three days later, CnB was deleted in Treg cells by i.p. injections of 1 mg Tamoxifen for 5 consecutive days. CTLA-4 blockade (through 4F10 mAb) was initiated six days after tumor injection, so that deletion of CnB and blockade of CTLA-4 occurred approximately at the same time. 4F10 (750 μg) was injected i.v. every 3 days.
QUANTIFICATION AND STATISTICAL ANALYSIS
Numbers of individual cells, recordings, and animals analyzed are indicated in the figure legends. Student’s t test and Mann-Whitney U test were used for comparisons of datasets with normal and non-normal distribution, respectively, using Prism (GraphPad). For multiple comparisons, we used type II ANOVA with Holm correction. For categorical variables, we used the χ2 test. Statistical significance was considered reached for p values < 0.05.
Supplementary Material
Figure S1. An NFAT-GFP reporter system to study Treg cell activation, related to Figure 1 (A), Scheme of the retroviral vector MW NFAT-GFP-IRES-H2B-RFP for coordinate expression of NFAT-GFP and H2B-RFP. (B), Protocol to transduce polyclonal Treg and Th cells with MW NFAT-GFP-IRES-H2B-RFP. C-D, Expression of Foxp3 (C) and of NFAT-GFP and H2B-RFP (D) in Treg and Th cells at the end of the 6-day culture and transduction protocol. (E), Comparable NFAT deactivation kinetics in Treg and Th cells. Purified splenic CD4+ T cells from Foxp3GFP mice were stimulated with ionomycin, which caused accumulation of NFAT in the nucleus. Cells were then exposed to cyclosporine A (CsA), which blocks NFAT activation through inhibition of calcineurin, fixed at various time points thereafter, and stained with αNFAT1 mAb and DAPI. Kinetics of NFAT relocation to the cytoplasm were similar in GFP+ Treg (arrows) and GFP− Th cells. Scale bar = 10 μm. (F), Illustration of a previously published algorithm for quantification of NFAT activation from 2-dimensional (2D) images of T cells in vivo (left), its limitations (center), and a 3D-algorithm newly developed for this study (right, see also Method details). G-I, Performance of NFAT SI generated by each algorithm as a binary classifier of cells as “signaling” (NFAT SI ≥ 0.5) or “non-signaling” (NFAT SI < 0.5). Correlation of NFAT SI computed by each algorithm in one representative movie out of five (G), confusion matrices of all five movies analyzed (H), and quantification of accuracy, specificity, sensitivity, alpha-, and beta-errors (I). n = 1210 cells from 5 recording of HA-specific NFAT-GFP and H2B-RFP co-expressing CTL in tumor tissue or tdLN of CT26HA tumor-implanted BALB/c mice (dataset from Marangoni et al., 2013). Bars in (I) represent the mean value and symbols individual movies. p value calculated by paired Student t test. (J), Approach to validate each algorithm against a “ground truth” reference method. K-L, Correlation of instantaneous NFAT SI generated by each algorithm with the reference method (K) and distribution of median deviation of individual data points (L). In (K), data points highlighted by squares represent an instance where the 3D Central accumulation algorithm provided an accurate NFAT SI, while the 2D radial profile method provided a much higher, incorrect NFAT SI for the same cell, relative to the reference method. The deviation of the calculated NFAT SI from the reference value for that data point is indicated at the top of each plot. p value in (L) calculated by Kolmogorov-Smirnov test. See STAR Methods for additional details.
Figure S2. Effects of cDC ablation on Treg cell activation, related to Figure 2 A-B, Identification of mCherry-expressing cells in CD11cmCherry mice. The gating scheme and population frequencies are reported in A, and the percentage of each population among mCherry-expressing tumor-associated cells is shown in B. C-D, Selective ablation of cDC in zDCDTR and control bone marrow chimeras treated with DT. Gating scheme and gate frequencies are shown in (C), quantification of cDC1, cDC2, and TAM in (D). n = 4–5 per group from two experiments. E-F, Treg cell frequencies and expression of various activation markers in tumors (E) and tdLN (F) following administration of FTY720. n = 7–13 per group from two independent experiments. G-I, Effects of cDC ablation in zDCDTR BMCs on the frequency of Th and CD8+ T cells in the TME and tdLN (G), expression of activation markers in Treg cells in tdLN (H), frequency of CD44hi CD62L− eTreg cells and their expression of Ki67 in tdLN (I). Data in (G-I) (n = 8 mice per group) are pooled from two independent experiments. MFI were normalized to the mean of the control groups. Bars represent medians, each symbol represents an individual animal. Numbers in parentheses represent fold change. p values were calculated by Mann-Whitney U test.
Figure S3. Effects of CnB deletion on Treg cell activation, related to Figure 2 (A), PCR analysis of CnB deletion in Treg cells in tdLN (lanes 5/6) and tumors (lanes 7/8) of tamoxifen treated Foxp3creERT2 or Foxp3creERT2 × CnBf/f mice. Foxp3cre and Foxp3cre × CnBf/f mice, in which CnB is constitutively deleted in Treg cells were used as controls (lanes 3/4). The ~3000-bp bands correspond to the non-rearranged CnBf/f or the WT CnB loci, whereas the 200-bp band corresponds to the rearranged CnBf/f locus, indicating deletion of CnB. Vertical lines denote non-adjacent lanes, stitched together for display purposes. B-C, Treg cell frequencies and expression of various activation markers in tumors (B) and tdLN (C) following tamoxifen-induced deletion of CnB without concurrent FTY720 treatment. n = 11–14 per group from two independent experiments. MFI were normalized to the mean of the control groups. Bars represent medians, each symbol represents an individual mouse. Numbers in parentheses represent fold change. p values were calculated by Mann-Whitney U test.
Figure S4. Analysis of non-signaling segments of migratory tracks of HA-Treg, HA-Th, and HA-Th cells in the presence of non-visualized HA-Treg cells, related to Figure 3 (A), Gating on non-signaling segments for motility analysis shown in (B-G). B-C, Duration of non-signaling segments (B) and percentage of segments with a duration of 20 minutes or less (C). n = 42 (HA-Treg), 45 (HA-Th) and 67 segments (HA-Th in the presence of HA-Treg cells) recorded in 2 independent experiments. (D), Instantaneous 3D velocities in non-signaling segments. n = 1409 (HA-Treg), 1520 (HA-Th) and 2408 (HA-Th in the presence of HA-Treg cells). (E), Arrest coefficients of non-signaling segments. n = 42 (HA-Treg), n = 45 (HA-Th) and n = 67 (HA-Th in presence of HA-Treg cells). (F), 10-min displacements in non-signaling segments. n = 700 (HA-Treg), n = 711 (HA-Th), and n = 1230 (HA-Th in presence of HA-Treg cells). (G), Migratory tracks of non-overlapping 10-minute non-signaling segments with normalized starting coordinates. n = 89 (HA-Treg), 92 (HA-Th) and 155 (HA-Th in the presence of HA-Treg cells). Data were collected in 6 (HA-Treg), 6 (HA-Th), and 5 (HA-Th in the presence of HA-Treg cells) movies, recorded in two independent experiments. Bars in (B, C, E) depict medians, and symbols represent individual non-signaling segments. p values calculated by Mann-Whitney U test. Fold change relative to the HA-Treg cell group median is shown in parenthesis.
Figure S5. Increase of co-stimulatory ligands on APC and Treg cell hyper-proliferation in tdLN of mice treated with αCTLA-4 mAb, related to Figure 4 (A), Expression of CD80 and 86 as well as various markers of general DC activation on tumor-associated cDC1 and cDC2, 24 hours after administration of αCTLA-4 (clone 4F10), αPD-1, or an isotype control mAb. n = 8–9 per group from two independent experiments. (B), Gating strategy to quantify CD80 and CD86 expression within cDC1 and cDC2 subsets among resident DC (rDC) and migratory DC (mDC) in tdLN. Numbers indicate gate frequencies. (C), Normalized MFI of CD80 and CD86 on indicated subsets 24 h after either ablation of all Treg cells (gray versus white bars) or CTLA-4 blockade (red versus black bars). n = 10 (DT treatment in WT mice), n = 5 (DT treatment in Foxp3DTR mice), n = 10 (isotype control), n = 10 (αCTLA-4 4F10), pooled from two separate experiments. (D), Normalized MFI of CD40, MHC-I and MHC-II on the indicated DC subsets 24 h after CTLA-4 blockade or treatment with isotype control Abs. n = 8–9 per group from two independent experiments. (E), Ki67 expression by Th and CD8+T cells in tumors and tdLN. Data represent 7–10 mice per group from two independent experiments. F-G, Frequency (F) and absolute numbers (G) of indicated T cell types in tdLN 6 days after treatment with αCTLA-4 mAb clone 4F10 (n = 8) or isotype-matched control mAb (n = 10), and FTY720. Data are pooled from two independent experiments. H-L, Frequencies of eTreg cells (H), their expression of Ki67 (I), and of various activation markers (J-L) in tdLN 6 days after treatment with αCTLA-4 mAb or isotype-matched control mAb. Data represent n = 10 per group pooled from two independent experiments. Numbers within gates represent cell percentages. Bars indicate medians, each symbol represents an individual animal. p values were calculated by Mann-Whitney U test. Fold change relative to the median of the control group is shown in parentheses. MFI were normalized to mean values in control groups.
Figure S6. CTLA-4 does not regulate Treg cells in cis in tdLN, related to Figure 5 (A), Experimental protocol. (B), Tamoxifen-induced loss of CTLA-4 expression in CD45.2+ Treg cells in tdLN. C-D, Expression of Ki67 in CTLA-4-sufficient or -deficient CD45.2+ Treg cells (C) and their frequency (D) in tdLN. In (B-C) numbers represent cell percentages within gates. Ten mice per group were analyzed in two experiments. E-F, Expression of activation markers in CTLA-4-sufficient or -deficient Treg cells within tumors (E) or tdLN (F). n = 7–8 (E) and 10 per group (F). G-H, Production of IL-2 by Treg, Th, and CD8+ T cells from tumors and tdLN upon ex vivo re-stimulation with PMA and ionomycin, following 6 days of αCTLA-4 or isotype mAb treatment, as described for Figure 4D. Background IL-2 production (4h culture in absence of PMA/ionomycin) was subtracted from IL-2 production after 4h stimulation with PMA/ionomycin for each individual mouse to determine the frequencies of IL-secreting cells (G). Based on these and absolute cell counts, the absolute numbers of IL-2 producing cells were calculated (H). n = 14–16 per group in two separate experiments. (I), Gating strategy to identify Treg cells in tumors and tdLN using surface markers. J-K, In situ transcriptional activity of the Il2 gene in Treg, Th, and CD8+ T cells from tumors and tdLN in Il2GFP mice after 6 days of αCTLA-4 treatment, as described for Figure 4D. The gating strategy in (I) was used to avoid loss of GFP signal during cell fixation/permeabilization for Foxp3 staining. n = 3 mice per group. Numbers indicate gate frequencies. Bars show median values, each symbol represents an individual animal. MFIs were normalized to the mean values in control groups. p values calculated by Mann-Whitney U test. Fold change compared to the median of the control group in parentheses.
Figure S7. Teff cell functions are not increased after CTLA-4 blockade whereas Treg cell expansion depends on CD28, related to Figures 6 and 7 A-B, Expression of granzyme B, and of the effector cytokines IFNγ and TNF in PMA/ionomycin re-stimulated Th and CD8+ T cells in the TME (A) and in tdLN (B) following 6 days of CTLA-4 blockade. n = 7–9 per group from two independent experiments. (C), Frequency of Treg cells among live hematopoietic cells and of Ki67+ eTreg cells in tdLN after blockade of CTLA-4 and/or IL-2. n = 10–11 per group in two separate experiments. (D), Experimental protocol. (E), Frequency and expression of Ki67 in CD28hi and CD28lo eTreg cells in tdLN after CTLA-4 blockade. F-G, Expression of the indicated activation markers on CD28hi and CD28lo Treg cells in the TME (F) and in eTreg cells in tdLN (G) after CTLA-4 blockade. Data represent 9–10 mice per group pooled from two independent experiments. Bars show medians, each symbol represents an individual animal. Numbers represent cell frequencies within gates. MFIs were normalized to the mean values in control groups. p values calculated by Mann-Whitney U test. Fold change as compared to the median of the control group in parentheses. H-I, Number of Treg cells contacting each APC per hour (H) and duration of individual contacts (I) at various time points in the same regions of interests of the TME. In (H), symbols represent individual APC and bars show means. In (I), symbols represent individual Treg:APC contacts and bars show medians. (J), Example of an APC (oval) located at the tumor/stroma border (dashed line), identified both before and following 18h of αCTLA-4 treatment. Scale bar = 50μm.
KEY RESOURCES TABLE
| REAGENT or RESOURCESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-4-1BB PE (clone: 17B5) | Biolegend | Cat# 106105; RRID:AB_2205693 |
| Anti-APC Biotin (clone: APC003) | BioLegend | Cat# 408003; RRID:AB_345359 |
| Armenian Hamster IgG APC (clone: eBio299Arm) | eBioscience | Cat# 17-4888-81; RRID:AB_470192 |
| Armenian Hamster IgG (lot: 708418O10) | BioXCell | Cat# BE0091; RRID:AB_1107773 |
| Anti-CD103 Per-CP-eFluor710 (clone: 2E7) | eBioscience | Cat#46-1031-80; RRID:AB_2573703 |
| Anti-CD11b BUV737 (clone: M1/70) | BD Horizon | Cat# 564443; RRID:AB_2738811 |
| Anti-CD11c Alexa700 (clone: HL3) | BD PharMingen | Cat# 560583; RRID:AB_1727421 |
| Anti-CD16-32 TruStain FcX (clone: 93) | BioLegend | Cat# 101320; RRID:AB_1574975 |
| Anti-CD172a SIRPα FITC (clone: P84) | BioLegend | Cat# 144005; RRID:AB_11204432 |
| Anti-CD19 PE-Cy5 (clone: 6D5) | BioLegend | Cat# 115509; RRID:AB_313644 |
| Anti-CD25 BV605 (clone: PC61) | BioLegend | Cat# 102035; RRID:AB_313644 |
| Anti-CD25 PE-CY7 (clone: PC61) | Biolegend | Cat# 102010; RRID:AB_312859 |
| Anti-CD26 BV711 (clone: H194-112) | BD Biosciences | Cat# 740678; RRID:AB_2740365 |
| Anti-CD26 PE (clone: H194-112) | BioLegend | Cat# 137803; RRID:AB_2093729 |
| Anti-CD28 (clone: 37.51) | BioLegend | Cat# 102101; RRID:AB_312866 |
| Anti-CD28 BV421 (clone: 37.51) | BioLegend | Cat# 102127; RRID:AB_2650628 |
| Anti-CD4 BUV563 (clone: GK1.5) | BD Horizon | Cat# 565709; RRID:AB_2739335 |
| Anti-CD4 BV510 (clone: RM4-5) | BioLegend | Cat# 100559; RRID:AB_2562608 |
| Anti-CD4 BV605 (clone: RM4-5) | BioLegend | Cat# 100547; RRID:AB_11125962 |
| Anti-CD40 PE-CY7 (clone: 3/23) | BioLegend | Cat# 124621; RRID:AB_10933422 |
| Anti-CD44 Alexa700 (clone: IM7) | BioLegend | Cat# 103026; RRID:AB_493713 |
| Anti-CD44 BV421 (clone: IM7) | BioLegend | Cat# 103039; RRID:AB_10895752 |
| Anti-CD45 BUV395 (clone: 30-F11) | BD Horizon | Cat# 564279; RRID:AB_2651134 |
| Anti-CD45 APC-Cy7 (clone: 30-F11) | Biolegend | Cat# 103115; RRID:AB_312980 |
| Anti-CD45.1 BV421 (clone: A20) | BioLegend | Cat# 110731; RRID:AB_10896425 |
| Anti-CD45.1 PerCP-Cy5.5 (clone: A20) | BioLegend | Cat# 110727; RRID:AB_893348 |
| Anti-CD45.2 APC (clone: 104) | BioLegend | Cat# 109813; RRID:AB_389210 |
| Anti-CD45.2 APC-Cy7 (clone: 104) | BioLegend | Cat# 109824; RRID:AB_830789 |
| Anti-CD62L BUV737 (clone: MEL-14) | BD Horizon | Cat# 565213; RRID:AB_2721774 |
| Anti-CD62L BV650 (clone: MEL-14) | Biolegend | Cat# 104453; RRID:AB_2800559 |
| Anti-CD64 PE/Dazzle 594 (clone: X54-5/7.1) | BioLegend | Cat# 139319; RRID:AB_2566558 |
| Anti-CD64 APC (clone: X54-5/7.1) | BioLegend | Cat# 139305; RRID:AB_11219205 |
| Anti-CD80 Alexa488 (clone: 16-10A1) | BioLegend | Cat# 104716; RRID:AB_492822 |
| Anti-CD86 BV421 (clone: GL-1) | BioLegend | Cat# 105032; RRID:AB_2650895 |
| Anti-CD86 BV785 (clone: GL-1) | BioLegend | Cat# 105043; RRID:AB_2566722 |
| Anti-CD8a APC-Cy7 (clone: 53-6.7) | BioLegend | Cat# 100714; RRID:AB_312753 |
| Anti-CD8a BV650 (clone: 53-6.7) | BioLegend | Cat# 100741; RRID:AB_11124344 |
| Anti-CD8a BV785 (clone: 53-6.7) | BioLegend | Cat# 100749; RRID:AB_11218801 |
| Anti-CTLA-4 (clone: UC10-4F10-11) (lot: 619017J1) | BioXCell | Cat# BP0032; RRID:AB_1107598 |
| Anti-CTLA-4 (clone: 9H10) | BioXCell | Cat# BE0131; RRID:AB_10950184 |
| Anti-CTLA-4 APC (clone: UC10-4F10-11) | Millipore | Cat# MABF389; RRID:AB_2892076 |
| F(ab’)2 Donkey anti-goat IgG (H+L) PE | Invitrogen | Cat# 31860; RRID:AB_429714 |
| F(ab’)2 Goat anti-rabbit IgG (H+L) Alexa633 | Invitrogen | Cat#A-21072; RRID:AB_2535733 |
| Anti-F4/80 APC Fire 750 (clone: BM8) | BioLegend | Cat# 123151; RRID:AB_2616724 |
| Anti-F4/80 Pacific Blue (clone: BM8) | BioLegend | Cat# 123124; RRID:AB_893475 |
| Anti-Foxp3 Alexa700 (clone: FJK-16 s) | eBioscience | Cat# 56-5773-82; RRID:AB_1210557 |
| Anti-Foxp3 APC (clone: FJK-16 s) | eBioscience | Cat# 17-5773-82; RRID:AB_469457 |
| Anti-Foxp3 PE (clone: FJK-16 s) | eBioscience | Cat# 12-5773-82; RRID:AB_465936 |
| Anti-GITR PE-Cy5 (clone: YGITR765) | BioLegend | Cat# 120220 |
| Goat anti-human IgG-Fc PE | SouthernBiotech | Cat# 2014-09; RRID:AB_2795582 |
| Anti-Granzyme B PE-Dazzle (clone: GB11) | BD Horizon | Cat# 563388; RRID:AB_2738174 |
| Anti-H-2Kb Alexa647 (clone: AF6-88.5) | Biolegend | Cat# 116511; RRID:AB_492918 |
| Anti-I-A/I-E (MHC-II) BV605 (clone: M5-114.15.2) | BioLegend | Cat# 107639; RRID:AB_2565894 |
| Anti-ICOS BV785 (clone: C398.4A) | BioLegend | Cat# 313533; RRID:AB_2629728 |
| Anti-IL-2 (clone: JES6-1A12) | BioXCell | Cat# BE0043; RRID:AB_1107702 |
| Anti-IL-2 (clone: S4B6-1) | BioXCell | Cat# BE0043-1; RRID:AB_1107705 |
| Anti-IL-2 PE (clone: JES6-5H4) | Biolegend | Cat# 503807; RRID:AB_315301 |
| Anti-IL-2 APC (clone: JES6-5H4) | Biolegend | Cat# 503809; RRID:AB_315303 |
| Anti-IFNg PE-CY7 (clone: XMG1.2) | Biolegend | Cat# 505825; RRID:AB_1595591 |
| Anti-IRF4 PE-Cy7 (clone: 3E4) | eBioscience | Cat# 25-9858-82; RRID:AB_2573558 |
| Anti-IRF8 APC (clone: V3GYWCH) | eBioscience | Cat# 17-9852-80; RRID:AB_2573317 |
| Anti-Ki67 FITC (clone: B56) | BD PharMingen | Cat# 556026; RRID:AB_396302 |
| Anti-Ly-6G/Ly-6C (Gr1) PE-Cy5 (clone: RB6-8C6) | BioLegend | Cat# 108409; RRID:AB_313374 |
| Anti-NK1.1 PE-Cy5 (clone: PK136) | Biolegend | Cat# 108715; RRID:AB_493591 |
| Anti-NFAT1 (clone: D43B1) | Cell Signaling | Cat#5861T; RRID:AB_10834808 |
| Anti-PD-1 (clone: 29F.1A12) | BioXCell | Cat# BE0273; RRID:AB_2687796 |
| Anti-PD-1 Per-CP-eFluor710 (clone: RMP1-30) | eBioscience | Cat# 46-9981-80; RRID:AB_11149347 |
| Rat IgG | Sigma | Cat# I4131; RRID:AB_1163627 |
| Rat IgG1,κ FITC (clone: RTK2071) | Biolegend | Cat# 400405, RRID:AB_326511 |
| Rat IgG1,κ PE-CY7 (clone: RTK2071) | Biolegend | Cat# 400415; RRID:AB_326521 |
| Rat IgG2b,κ APC (clone: RTK4530) | Biolegend | Cat# 400611; RRID:AB_326555 |
| Streptavidin APC | BioLegend | Cat# 405207 |
| Anti-TCR β chain PE-Cy5 (clone: H57-597) | BioLegend | Cat# 109209; RRID:AB_313432 |
| Anti-TNF FITC (clone: MP6-XT22) | Biolegend | Cat# 506303; RRID:AB_315424 |
| Anti-XCR1 BV650 (clone: ZET) | BioLegend | Cat# 148220; RRID:AB_2566410 |
| Anti-Zbtb46 PE (clone: U4-1374) | BD PharMingen | Cat# 565832; RRID:AB_2739372 |
| Chemicals, peptides, and recombinant proteins | ||
| CD80-Fc chimera | BioLegend | Cat# 555404 |
| CD86-Fc chimera | BioLegend | Cat#771704 |
| FTY720 | Cayman | Cat# 10006292 |
| Diphtheria Toxin | Calbiochem | Cat# 32232619/07/ |
| Tamoxifen | Sigma | Cat# T5648 |
| Brefeldin A | Biolegend | Cat # 420601 |
| Phorbol myristate-acetate | Sigma | Cat#P1585 |
| Ionomycin | Sigma | Cat# I0634 |
| IL-2 (cys160Ser) | R&D | Cat# 1150-ML |
| Critical commercial assays | ||
| CD4+ CD25+ Regulatory T Cell Isolation Kit | Miltenyi | Cat# 130-091-041 |
| Experimental models: cell lines | ||
| Mouse: MC38 cells | Kerafast | Cat# ENH204-FP |
| Mouse: CT26 cells | ATCC | Cat# CRL-2638 |
| Human: Platinum-E cells | Cell Biolabs | Cat#RV101 |
| Experimental models: organisms/strains | ||
| Mouse: B6(Cg)-Zbtb46tm1(HBEGF)Mnz/J | Jackson Laboratories | JAX: 019506 |
| Mouse: CD11cmCherry | Kamal Khanna | Khanna et al., 2010 |
| Mouse: B6;129S-Ppp3r1tm2Grc/J (CnBf/f) | Jackson Laboratories | JAX: 017692 |
| Mouse: C57BL/6-Cd28tm1Ltu/J (CD28f/f) | Laurence Turka | JAX: 024282 |
| Mouse: B6.SJL-PtprcaPepcb/BoyJ (B6 CD45.1) | Jackson Laboratories | JAX: 002014 |
| Mouse: C57BL/6 CTLA-4 floxed | Arlene Sharpe | Paterson et al., 2015 |
| Mouse: Foxp3tm9(EGFP/cre/ERT2)Ayr/J (Foxp3creERT2) | Jackson Laboratories | JAX: 016961 |
| Mouse: BALB/c TCR-HA | Harald von Boehmer | Kirberg et al., 1994 |
| Mouse: BALB/c pgk-HA | Harald von Boehmer | Klein et al., 2003 |
| Mouse: B6.129(Cg)-Foxp3tm3(DTR/GFP)Ayr/J (Foxp3DTR) | Jackson Laboratories | JAX: 016958 |
| Mouse: B6.Cg-Foxp3tm2Tch/J (Foxp3GFP) | Jackson Laboratories | JAX: 006772 |
| Mouse: B6.129(Cg)-Foxp3tm4(YFP/icre)Ayr/J (Foxp3cre) | Jackson Laboratories | JAX: 016959 |
| Mouse: C57BL/6 IL-2GFP | Casey Weaver | DiToro et al., 2018 |
| Mouse: C57BL/6J | Jackson Laboratories | JAX: 000664 |
| Mouse: BALB/cJ | Jackson Laboratories | JAX: 000651 |
| Oligonucleotides | ||
| PCR primer: CnB FW: 5′-CAATGCAGTCCGCTGTAGTTC-3′ | Thorsten Mempel | Marangoni et al., 2018 |
| PCR primer: CnB REV: 5′-AGCCTCCACATACACAGATAC-3′ | Thorsten Mempel | Marangoni et al., 2018 |
| Recombinant DNA | ||
| Retroviral plasmid: MW NFAT-GFP-IRES-H2B-RFP | This paper | N/A |
| Plasmid: cmv-VSVg (pMD2.G) | Addgene | Plasmid# 12259; RRID:Addgene_12259 |
| Plasmid: HA-NFAT1(4-460)-GFP | Addgene | Plasmid# 11107; RRID:Addgene_11107 |
| Plasmid: pHIV-H2BmRFP | Addgene | Plasmid# 18982; RRID:Addgene_18982 |
| Software and algorithms | ||
| Imaris scripts for NFAT-SI calculation (3D central accumulation) | This paper | N/A |
| Imaris 8.4 | Bitplane | https://imaris.oxinst.com |
| ImageJ | Freeware/NIH | https://imagej.nih.gov/ij/ |
| Prism | GraphPad | https://www.graphpad.com |
| FlowJo | Treestar | https://www.flowjo.com/ |
| Other | ||
| Zombie Red Fixable Viability Dye | BioLegend | Cat#423109 |
| Zombie Yellow Fixable Viability Dye | BioLegend | Cat#423103 |
| Dynabeads Mouse T-Activator CD3/CD28 | Invitrogen | Cat#114.52D |
Highlights.
The TCR repertoire of Treg cells is enriched for reactivity to antigens in the TME
Tumor Treg cells use CTLA-4 to destabilize their own interactions with dendritic cells
CTLA-4 blockade causes the CD28-mediated expansion of tumor-associated Treg cells
Following CTLA-4 blockade, Treg cells continue to promote tumor immune tolerance
ACKNOWLEDGMENTS
This work was supported by a Melanoma Research Foundation award to T.R.M., by NIH grants R01 AI123349 to T.R.M. and R21 AR072849 to F.M., and by an Elizabeth O’Brien/Charles King Trust fellowship to F.M. A.Z. was supported by a scholarship from the Shakhmardan Yessenov Foundation and by Nazarbayev University. M.C. was supported by a fellowship from the Italian Biochemistry Society (SIB). J.N.P. was supported by DFG research fellowship (PR 1652/1-1).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.cell.2021.05.027.
DECLARATION OF INTERESTS
T.R.M. is a founder and shareholder in Monopteros Therapeutics, Inc. This commercial relationship is unrelated to this study.
REFERENCES
- Ahmadzadeh M, Pasetto A, Jia L, Deniger DC, Stevanović S, Robbins PF, and Rosenberg SA (2019). Tumor-infiltrating human CD4+ regulatory T cells display a distinct TCR repertoire and exhibit tumor and neoantigen reactivity. Sci. Immunol 4, eaao4310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allard B, Pommey S, Smyth MJ, and Stagg J (2013). Targeting CD73 enhances the antitumor activity of anti-PD-1 and anti-CTLA-4 mAbs. Clin. Cancer Res 19, 5626–5635. [DOI] [PubMed] [Google Scholar]
- Aramburu J, Garcia-Cózar F, Raghavan A, Okamura H, Rao A, and Hogan PG (1998). Selective inhibition of NFAT activation by a peptide spanning the calcineurin targeting site of NFAT. Mol. Cell 1, 627–637. [DOI] [PubMed] [Google Scholar]
- Arce Vargas F, Furness AJS, Solomon I, Joshi K, Mekkaoui L, Lesko MH, Miranda Rota E, Dahan R, Georgiou A, Sledzinska A, et al. ; Melanoma TRACERx Consortium; Renal TRACERx Consortium; Lung TRACERx Consortium (2017). Fc-Optimized Anti-CD25 Depletes Tumor-Infiltrating Regulatory T Cells and Synergizes with PD-1 Blockade to Eradicate Established Tumors. Immunity 46, 577–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arce Vargas F, Furness AJS, Litchfield K, Joshi K, Rosenthal R, Ghorani E, Solomon I, Lesko MH, Ruef N, Roddie C, et al. ; TRACERx Melanoma; TRACERx Renal; TRACERx Lung consortia (2018). Fc Effector Function Contributes to the Activity of Human Anti-CTLA-4 Antibodies. Cancer Cell 33, 649–663.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer CA, Kim EY, Marangoni F, Carrizosa E, Claudio NM, and Mempel TR (2014). Dynamic Treg interactions with intratumoral APCs promote local CTL dysfunction. J. Clin. Invest 124, 2425–2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, Coussens LM, Gabrilovich DI, Ostrand-Rosenberg S, Hedrick CC, et al. (2018). Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med 24, 541–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binnewies M, Mujal AM, Pollack JL, Combes AJ, Hardison EA, Barry KC, Tsui J, Ruhland MK, Kersten K, Abushawish MA, et al. (2019). Unleashing Type-2 Dendritic Cells to Drive Protective Antitumor CD4+ T Cell Immunity. Cell 177, 556–571.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE, Chow LQ, Vokes EE, Felip E, Holgado E, et al. (2015). Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N. Engl. J. Med 373, 1627–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyman O, Kovar M, Rubinstein MP, Surh CD, and Sprent J (2006). Selective stimulation of T cell subsets with antibody-cytokine immune complexes. Science 311, 1924–1927. [DOI] [PubMed] [Google Scholar]
- Broz ML, Binnewies M, Boldajipour B, Nelson AE, Pollack JL, Erle DJ, Barczak A, Rosenblum MD, Daud A, Barber DL, et al. (2014). Dissecting the tumor myeloid compartment reveals rare activating antigen-presenting cells critical for T cell immunity. Cancer Cell 26, 638–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulliard Y, Jolicoeur R, Windman M, Rue SM, Ettenberg S, Knee DA, Wilson NS, Dranoff G, and Brogdon JL (2013). Activating Fc γ receptors contribute to the antitumor activities of immunoregulatory receptor-targeting antibodies. J. Exp. Med 210, 1685–1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castle JC, Loewer M, Boegel S, de Graaf J, Bender C, Tadmor AD, Boisguerin V, Bukur T, Sorn P, Paret C, et al. (2014). Immunomic, genomic and transcriptomic characterization of CT26 colorectal carcinoma. BMC Genomics 15, 190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M-L, Pittet MJ, Gorelik L, Flavell RA, Weissleder R, von Boehmer H, and Khazaie K (2005). Regulatory T cells suppress tumor-specific CD8 T cell cytotoxicity through TGF-beta signals in vivo. Proc. Natl. Acad. Sci. USA 102, 419–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbett TH, Griswold DP Jr., Roberts BJ, Peckham JC, and Schabel FM Jr. (1975). Tumor induction relationships in development of transplantable cancers of the colon in mice for chemotherapy assays, with a note on carcinogen structure. Cancer Res. 35, 2434–2439. [PubMed] [Google Scholar]
- D’Cruz LM, and Klein L (2005). Development and function of agonist-induced CD25+Foxp3+ regulatory T cells in the absence of interleukin 2 signaling. Nat. Immunol 6, 1152–1159. [DOI] [PubMed] [Google Scholar]
- Darrasse-Jèze G, Deroubaix S, Mouquet H, Victora GD, Eisenreich T, Yao KH, Masilamani RF, Dustin ML, Rudensky A, Liu K, and Nussenzweig MC (2009). Feedback control of regulatory T cell homeostasis by dendritic cells in vivo. J. Exp. Med 206, 1853–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Simone M, Arrigoni A, Rossetti G, Gruarin P, Ranzani V, Politano C, Bonnal RJP, Provasi E, Sarnicola ML, Panzeri I, et al. (2016). Transcriptional Landscape of Human Tissue Lymphocytes Unveils Uniqueness of Tumor-Infiltrating T Regulatory Cells. Immunity 45, 1135–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Pilato M, Kim EY, Cadilha BL, Prüßmann JN, Nasrallah MN, Seruggia D, Usmani SM, Misale S, Zappulli V, Carrizosa E, et al. (2019). Targeting the CBM complex causes Treg cells to prime tumours for immune checkpoint therapy. Nature 570, 112–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiToro D, Winstead CJ, Pham D, Witte S, Andargachew R, Singer JR, Wilson CG, Zindl CL, Luther RJ, Silberger DJ, et al. (2018). Differential IL-2 expression defines developmental fates of follicular versus nonfollicular helper T cells. Science 361, eaao2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egen JG, and Allison JP (2002). Cytotoxic T lymphocyte antigen-4 accumulation in the immunological synapse is regulated by TCR signal strength. Immunity 16, 23–35. [DOI] [PubMed] [Google Scholar]
- Ferrara R, Susini S, and Marabelle A (2019). Anti-CTLA-4 Immunotherapy Does Not Deplete FOXP3+ Regulatory T Cells (Tregs) in Human Cancers-Letter. Clin. Cancer Res 25, 3468. [DOI] [PubMed] [Google Scholar]
- Ferris RL, Blumenschein G Jr., Fayette J, Guigay J, Colevas AD, Licitra L, Harrington K, Kasper S, Vokes EE, Even C, et al. (2016). Nivolumab for recurrent squamous-cell carcinoma of the head and neck. N. Engl. J. Med 375, 1856–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fife BT, Pauken KE, Eagar TN, Obu T, Wu J, Tang Q, Azuma M, Krummel MF, and Bluestone JA (2009). Interactions between PD-1 and PD-L1 promote tolerance by blocking the TCR-induced stop signal. Nat. Immunol 10, 1185–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontenot JD, Rasmussen JP, Gavin MA, and Rudensky AY (2005). A function for interleukin 2 in Foxp3-expressing regulatory T cells. Nat. Immunol 6, 1142–1151. [DOI] [PubMed] [Google Scholar]
- Franklin RA, Liao W, Sarkar A, Kim MV, Bivona MR, Liu K, Pamer EG, and Li MO (2014). The cellular and molecular origin of tumor-associated macrophages. Science 344, 921–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Nish SA, Jiang R, Hou L, Licona-Limón P, Weinstein JS, Zhao H, and Medzhitov R (2013). Control of T helper 2 responses by transcription factor IRF4-dependent dendritic cells. Immunity 39, 722–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavin MA, Clarke SR, Negrou E, Gallegos A, and Rudensky A (2002). Homeostasis and anergy of CD4(+)CD25(+) suppressor T cells in vivo. Nat. Immunol 3, 33–41. [DOI] [PubMed] [Google Scholar]
- Golgher D, Jones E, Powrie F, Elliott T, and Gallimore A (2002). Depletion of CD25+ regulatory cells uncovers immune responses to shared murine tumor rejection antigens. Eur. J. Immunol 32, 3267–3275. [DOI] [PubMed] [Google Scholar]
- Ha D, Tanaka A, Kibayashi T, Tanemura A, Sugiyama D, Wing JB, Lim EL, Teng KWW, Adeegbe D, Newell EW, et al. (2019). Differential control of human Treg and effector T cells in tumor immunity by Fc-engineered anti-CTLA-4 antibody. Proc. Natl. Acad. Sci. USA 116, 609–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heissmeyer V, Macián F, Im SH, Varma R, Feske S, Venuprasad K, Gu H, Liu YC, Dustin ML, and Rao A (2004). Calcineurin imposes T cell unresponsiveness through targeted proteolysis of signaling proteins. Nat. Immunol 5, 255–265. [DOI] [PubMed] [Google Scholar]
- Hickman SP, Yang J, Thomas RM, Wells AD, and Turka LA (2006). Defective activation of protein kinase C and Ras-ERK pathways limits IL-2 production and proliferation by CD4+CD25+ regulatory T cells. J. Immunol 177, 2186–2194. [DOI] [PubMed] [Google Scholar]
- Imai H, Saio M, Nonaka K, Suwa T, Umemura N, Ouyang G-F, Nakagawa J, Tomita H, Osada S, Sugiyama Y, et al. (2007). Depletion of CD4+CD25+ regulatory T cells enhances interleukin-2-induced antitumor immunity in a mouse model of colon adenocarcinoma. Cancer Sci. 98, 416–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josefowicz SZ, Lu L-F, and Rudensky AY (2012). Regulatory T cells: mechanisms of differentiation and function. Annu. Rev. Immunol 30, 531–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamada T, Togashi Y, Tay C, Ha D, Sasaki A, Nakamura Y, Sato E, Fukuoka S, Tada Y, Tanaka A, et al. (2019). PD-1+ regulatory T cells amplified by PD-1 blockade promote hyperprogression of cancer. Proc. Natl. Acad. Sci. USA 116, 9999–10008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanna KM, Blair DA, Vella AT, McSorley SJ, Datta SK, and Lefrançois L (2010). T cell and APC dynamics in situ control the outcome of vaccination. J. Immunol 185, 239–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JM, Rasmussen JP, and Rudensky AY (2007). Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat. Immunol 8, 191–197. [DOI] [PubMed] [Google Scholar]
- Kirberg J, Baron A, Jakob S, Rolink A, Karjalainen K, and von Boehmer H (1994). Thymic selection of CD8+ single positive cells with a class II major histocompatibility complex-restricted receptor. J. Exp. Med 180, 25–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein L, Khazaie K, and von Boehmer H (2003). In vivo dynamics of antigen-specific regulatory T cells not predicted from behavior in vitro. Proc. Natl. Acad. Sci. USA 100, 8886–8891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kocak E, Lute K, Chang X, May KFJ Jr., Exten KR, Zhang H, Abdes-salam SF, Lehman AM, Jarjoura D, Zheng P, and Liu Y (2006). Combination therapy with anti-CTL antigen-4 and anti-4–1BB antibodies enhances cancer immunity and reduces autoimmunity. Cancer Res. 66, 7276–7284. [DOI] [PubMed] [Google Scholar]
- Korman AJ, Engelhardt J, Loffredo J, Valle J, Akter R, Vuyyuru R, Bezman N, So P, Graziano R, Tipton K, et al. (2017). Abstract SY09–01: Next-generation anti-CTLA-4 antibodies. Cancer Res. 77, SY09–01. [Google Scholar]
- Kowalczyk A, D’Souza CA, and Zhang L (2014). Cell-extrinsic CTLA4-mediated regulation of dendritic cell maturation depends on STAT3. Eur. J. Immunol 44, 1143–1155. [DOI] [PubMed] [Google Scholar]
- Krishnaswamy JK, Gowthaman U, Zhang B, Mattsson J, Szeponik L, Liu D, Wu R, White T, Calabro S, Xu L, et al. (2017). Migratory CD11b+ conventional dendritic cells induce T follicular helper cell-dependent antibody responses. Sci. Immunol 2, eaam9169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard JD, Gilmore DC, Dileepan T, Nawrocka WI, Chao JL, Schoenbach MH, Jenkins MK, Adams EJ, and Savage PA (2017). Identification of Natural Regulatory T Cell Epitopes Reveals Convergence on a Dominant Autoantigen. Immunity 47, 107–117.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leventhal DS, Gilmore DC, Berger JM, Nishi S, Lee V, Malchow S, Kline DE, Kline J, Vander Griend DJ, Huang H, et al. (2016). Dendritic Cells Coordinate the Development and Homeostasis of Organ-Specific Regulatory T Cells. Immunity 44, 847–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine AG, Arvey A, Jin W, and Rudensky AY (2014). Continuous requirement for the TCR in regulatory T cell function. Nat. Immunol 15, 1070–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W, Haribhai D, Relland LM, Truong N, Carlson MR, Williams CB, and Chatila TA (2007). Regulatory T cell development in the absence of functional Foxp3. Nat. Immunol 8, 359–368. [DOI] [PubMed] [Google Scholar]
- Liu Z, Gerner MY, Van Panhuys N, Levine AG, Rudensky AY, and Germain RN (2015). Immune homeostasis enforced by co-localized effector and regulatory T cells. Nature 528, 225–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macián F, García-Cózar F, Im S-H, Horton HF, Byrne MC, and Rao A (2002). Transcriptional mechanisms underlying lymphocyte tolerance. Cell 109, 719–731. [DOI] [PubMed] [Google Scholar]
- Magistrelli G, Jeannin P, Herbault N, Benoit De Coignac A, Gauchat JF, Bonnefoy JY, and Delneste Y (1999). A soluble form of CTLA-4 generated by alternative splicing is expressed by nonstimulated human T cells. Eur. J. Immunol 29, 3596–3602. [DOI] [PubMed] [Google Scholar]
- Mahne AE, Mauze S, Joyce-Shaikh B, Xia J, Bowman EP, Beebe AM, Cua DJ, and Jain R (2017). Dual Roles for Regulatory T-cell Depletion and Costimulatory Signaling in Agonistic GITR Targeting for Tumor Immunotherapy. Cancer Res. 77, 1108–1118. [DOI] [PubMed] [Google Scholar]
- Malchow S, Leventhal DS, Nishi S, Fischer BI, Shen L, Paner GP, Amit AS, Kang C, Geddes JE, Allison JP, et al. (2013). Aire-dependent thymic development of tumor-associated regulatory T cells. Science 339, 1219–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marabelle A, Kohrt H, Sagiv-Barfi I, Ajami B, Axtell RC, Zhou G, Rajapaksa R, Green MR, Torchia J, Brody J, et al. (2013). Depleting tumor-specific Tregs at a single site eradicates disseminated tumors. J. Clin. Invest 123, 2447–2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marangoni F, Murooka TT, Manzo T, Kim EY, Carrizosa E, Elpek NM, and Mempel TR (2013). The transcription factor NFAT exhibits signal memory during serial T cell interactions with antigen-presenting cells. Immunity 38, 237–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marangoni F, Zhang R, Mani V, Thelen M, Ali Akbar NJ, Warner RD, Äijö T, Zappulli V, Martinez GJ, Turka LA, and Mempel TR (2018). Tumor Tolerance-Promoting Function of Regulatory T Cells Is Optimized by CD28, but Strictly Dependent on Calcineurin. J. Immunol 200, 3647–3661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez GJ, Pereira RM,Äijö T, Kim EY, Marangoni F, Pipkin ME, Togher S, Heissmeyer V, Zhang YC, Crotty S, et al. (2015). The transcription factor NFAT promotes exhaustion of activated CD8+ T cells. Immunity 42, 265–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mempel TR, Pittet MJ, Khazaie K, Weninger W, Weissleder R, von Boehmer H, and von Andrian UH (2006). Regulatory T cells reversibly suppress cytotoxic T cell function independent of effector differentiation. Immunity 25, 129–141. [DOI] [PubMed] [Google Scholar]
- Meredith MM, Liu K, Darrasse-Jeze G, Kamphorst AO, Schreiber HA, Guermonprez P, Idoyaga J, Cheong C, Yao K-H, Niec RE, and Nussenzweig MC (2012). Expression of the zinc finger transcription factor zDC (Zbtb46, Btbd4) defines the classical dendritic cell lineage. J. Exp. Med 209, 1153–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motzer RJ, Tannir NM, McDermott DF, Arén Frontera O, Melichar B, Choueiri TK, Plimack ER, Barthélémy P, Porta C, George S, et al. ; CheckMate 214 Investigators (2018). Nivolumab plus Ipilimumab versus Sunitinib in advanced renal-cell carcinoma. N. Engl. J. Med 378, 1277–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neilson JR, Winslow MM, Hur EM, and Crabtree GR (2004). Calcineurin B1 is essential for positive but not negative selection during thymocyte development. Immunity 20, 255–266. [DOI] [PubMed] [Google Scholar]
- O’Gorman WE, Dooms H, Thorne SH, Kuswanto WF, Simonds EF, Krutzik PO, Nolan GP, and Abbas AK (2009). The initial phase of an immune response functions to activate regulatory T cells. J. Immunol 183, 332–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ovcinnikovs V, Ross EM, Petersone L, Edner NM, Heuts F, Ntavli E, Kogimtzis A, Kennedy A, Wang CJ, Bennett CL, et al. (2019). CTLA-4-mediated transendocytosis of costimulatory molecules primarily targets migratory dendritic cells. Sci. Immunol 4, eaaw0902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paterson AM, Lovitch SB, Sage PT, Juneja VR, Lee Y, Trombley JD, Arancibia-Cárcamo CV, Sobel RA, Rudensky AY, Kuchroo VK, et al. (2015). Deletion of CTLA-4 on regulatory T cells during adulthood leads to resistance to autoimmunity. J. Exp. Med 212, 1603–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pentcheva-Hoang T, Egen JG, Wojnoonski K, and Allison JP (2004). B7–1 and B7–2 selectively recruit CTLA-4 and CD28 to the immunological synapse. Immunity 21, 401–413. [DOI] [PubMed] [Google Scholar]
- Plitas G, Konopacki C, Wu K, Bos PD, Morrow M, Putintseva EV, Chudakov DM, and Rudensky AY (2016). Regulatory T Cells Exhibit Distinct Features in Human Breast Cancer. Immunity 45, 1122–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price KD, Simutis F, Fletcher A, Ramaiah L, Srour R, Kozlosky J, Sathish J, Engelhardt J, Capozzi A, Crona J, et al. (2018). Abstract LB-B33: Nonclinical safety evaluation of two distinct second generation variants of anti-CTLA4 monoclonal antibody, ipilimumab, in monkeys. Mol. Cancer Ther 17, LB–B33. [Google Scholar]
- Qureshi OS, Zheng Y, Nakamura K, Attridge K, Manzotti C, Schmidt EM, Baker J, Jeffery LE, Kaur S, Briggs Z, et al. (2011). Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science 332, 600–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts EW, Broz ML, Binnewies M, Headley MB, Nelson AE, Wolf DM, Kaisho T, Bogunovic D, Bhardwaj N, and Krummel MF (2016). Critical Role for CD103(+)/CD141(+) Dendritic Cells Bearing CCR7 for Tumor Antigen Trafficking and Priming of T Cell Immunity in Melanoma. Cancer Cell 30, 324–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romano E, Kusio-Kobialka M, Foukas PG, Baumgaertner P, Meyer C, Ballabeni P, Michielin O, Weide B, Romero P, and Speiser DE (2015). Ipilimumab-dependent cell-mediated cytotoxicity of regulatory T cells ex vivo by nonclassical monocytes in melanoma patients. Proc. Natl. Acad. Sci. USA 112, 6140–6145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubtsov YP, Rasmussen JP, Chi EY, Fontenot J, Castelli L, Ye X, Treuting P, Siewe L, Roers A, Henderson WR Jr., et al. (2008). Regulatory T cell-derived interleukin-10 limits inflammation at environmental interfaces. Immunity 28, 546–558. [DOI] [PubMed] [Google Scholar]
- Rubtsov YP, Niec RE, Josefowicz S, Li L, Darce J, Mathis D, Benoist C, and Rudensky AY (2010). Stability of the regulatory T cell lineage in vivo. Science 329, 1667–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi S, Mikami N, Wing JB, Tanaka A, Ichiyama K, and Ohkura N (2020). Regulatory T Cells and Human Disease. Annu. Rev. Immunol 38, 541–566. [DOI] [PubMed] [Google Scholar]
- Salmon H, Idoyaga J, Rahman A, Leboeuf M, Remark R, Jordan S, Casanova-Acebes M, Khudoynazarova M, Agudo J, Tung N, et al. (2016). Expansion and Activation of CD103(+) Dendritic Cell Progenitors at the Tumor Site Enhances Tumor Responses to Therapeutic PD-L1 and BRAF Inhibition. Immunity 44, 924–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salomon B, Lenschow DJ, Rhee L, Ashourian N, Singh B, Sharpe A, and Bluestone JA (2000). B7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity 12, 431–440. [DOI] [PubMed] [Google Scholar]
- Samstein RM, Arvey A, Josefowicz SZ, Peng X, Reynolds A, Sandstrom R, Neph S, Sabo P, Kim JM, Liao W, et al. (2012). Foxp3 exploits a pre-existent enhancer landscape for regulatory T cell lineage specification. Cell 151, 153–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanmamed MF, and Chen L (2018). A Paradigm Shift in Cancer Immunotherapy: From Enhancement to Normalization. Cell 175, 313–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert D, Bode C, Kenefeck R, Hou TZ, Wing JB, Kennedy A, Bulashevska A, Petersen BS, Schäffer AA, Grüning BA, et al. (2014). Autosomal dominant immune dysregulation syndrome in humans with CTLA4 mutations. Nat. Med 20, 1410–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selby MJ, Engelhardt JJ, Quigley M, Henning KA, Chen T, Srinivasan M, and Korman AJ (2013). Anti-CTLA-4 antibodies of IgG2a isotype enhance antitumor activity through reduction of intratumoral regulatory T cells. Cancer Immunol. Res 1, 32–42. [DOI] [PubMed] [Google Scholar]
- Sharma A, Subudhi SK, Blando J, Scutti J, Vence L, Wargo J, Allison JP, Ribas A, and Sharma P (2019). Anti-CTLA-4 Immunotherapy Does Not Deplete FOXP3+ Regulatory T Cells (Tregs) in Human Cancers. Clin. Cancer Res 25, 1233–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson TR, Li F, Montalvo-Ortiz W, Sepulveda MA, Bergerhoff K, Arce F, Roddie C, Henry JY, Yagita H, Wolchok JD, et al. (2013). Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J. Exp. Med 210, 1695–1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smigiel KS, Richards E, Srivastava S, Thomas KR, Dudda JC, Klonowski KD, and Campbell DJ (2014). CCR7 provides localized access to IL-2 and defines homeostatically distinct regulatory T cell subsets. J. Exp. Med 211, 121–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spranger S, Dai D, Horton B, and Gajewski TF (2017). Tumor-Residing Batf3 Dendritic Cells Are Required for Effector T Cell Trafficking and Adoptive T Cell Therapy. Cancer Cell 31, 711–723.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tadokoro CE, Shakhar G, Shen S, Ding Y, Lino AC, Maraver A, Lafaille JJ, and Dustin ML (2006). Regulatory T cells inhibit stable contacts between CD4+ T cells and dendritic cells in vivo. J. Exp. Med 203, 505–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Q, Henriksen KJ, Bi M, Finger EB, Szot G, Ye J, Masteller EL, McDevitt H, Bonyhadi M, and Bluestone JA (2004). In vitro-expanded antigen-specific regulatory T cells suppress autoimmune diabetes. J. Exp. Med 199, 1455–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Q, Adams JY, Tooley AJ, Bi M, Fife BT, Serra P, Santamaria P, Locksley RM, Krummel MF, and Bluestone JA (2006). Visualizing regulatory T cell control of autoimmune responses in nonobese diabetic mice. Nat. Immunol 7, 83–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thauland TJ, Koguchi Y, Dustin ML, and Parker DC (2014). CD28-CD80 interactions control regulatory T cell motility and immunological synapse formation. J. Immunol 193, 5894–5903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vahl JC, Drees C, Heger K, Heink S, Fischer JC, Nedjic J, Ohkura N, Morikawa H, Poeck H, Schallenberg S, et al. (2014). Continuous T cell receptor signals maintain a functional regulatory T cell pool. Immunity 41, 722–736. [DOI] [PubMed] [Google Scholar]
- Vignali DAA, Collison LW, and Workman CJ (2008). How regulatory T cells work. Nat. Rev. Immunol 8, 523–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villarreal DO, L’Huillier A, Armington S, Mottershead C, Filippova EV, Coder BD, Petit RG, and Princiotta MF (2018). Targeting CCR8 induces protective antitumor immunity and enhances vaccine-induced responses in colon cancer. Cancer Res. 78, 5340–5348. [DOI] [PubMed] [Google Scholar]
- Walker LSK, and Sansom DM (2015). Confusing signals: recent progress in CTLA-4 biology. Trends Immunol. 36, 63–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walunas TL, Lenschow DJ, Bakker CY, Linsley PS, Freeman GJ, Green JM, Thompson CB, and Bluestone JA (1994). CTLA-4 can function as a negative regulator of T cell activation. Immunity 1, 405–413. [DOI] [PubMed] [Google Scholar]
- Wang C, Lee JH, and Kim CH (2012). Optimal population of FoxP3+ T cells in tumors requires an antigen priming-dependent trafficking receptor switch. PLoS ONE 7, e30793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welm BE, Dijkgraaf GJP, Bledau AS, Welm AL, and Werb Z (2008). Lentiviral transduction of mammary stem cells for analysis of gene function during development and cancer. Cell Stem Cell 2, 90–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, Nomura T, and Sakaguchi S (2008). CTLA-4 control over Foxp3+ regulatory T cell function. Science 322, 271–275. [DOI] [PubMed] [Google Scholar]
- Wing JB, Tanaka A, and Sakaguchi S (2019). Human FOXP3+ Regulatory T Cell Heterogeneity and Function in Autoimmunity and Cancer. Immunity 50, 302–316. [DOI] [PubMed] [Google Scholar]
- Wolchok JD, Chiarion-Sileni V, Gonzalez R, Rutkowski P, Grob JJ, Cowey CL, Lao CD, Wagstaff J, Schadendorf D, Ferrucci PF, et al. (2017). Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med 377, 1345–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright GP, Notley CA, Xue S-A, Bendle GM, Holler A, Schumacher TN, Ehrenstein MR, and Stauss HJ (2009). Adoptive therapy with redirected primary regulatory T cells results in antigen-specific suppression of arthritis. Proc. Natl. Acad. Sci. USA 106, 19078–19083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Borde M, Heissmeyer V, Feuerer M, Lapan AD, Stroud JC, Bates DL, Guo L, Han A, Ziegler SF, et al. (2006). FOXP3 controls regulatory T cell function through cooperation with NFAT. Cell 126, 375–387. [DOI] [PubMed] [Google Scholar]
- Yamazaki C, Sugiyama M, Ohta T, Hemmi H, Hamada E, Sasaki I, Fukuda Y, Yano T, Nobuoka M, Hirashima T, et al. (2013). Critical roles of a dendritic cell subset expressing a chemokine receptor, XCR1. J. Immunol 190, 6071–6082. [DOI] [PubMed] [Google Scholar]
- Yan D, Farache J, Mingueneau M, Mathis D, and Benoist C (2015). Imbalanced signal transduction in regulatory T cells expressing the transcription factor FoxP3. Proc. Natl. Acad. Sci. USA 112, 14942–14947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Huynh A, Whitcher G, Chang J, Maltzman JS, and Turka LA (2013). An obligate cell-intrinsic function for CD28 in Tregs. J. Clin. Invest 123, 580–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng C, Zheng L, Yoo JK, Guo H, Zhang Y, Guo X, Kang B, Hu R, Huang JY, Zhang Q, et al. (2017). Landscape of Infiltrating T Cells in Liver Cancer Revealed by Single-Cell Sequencing. Cell 169, 1342–1356.e16. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. An NFAT-GFP reporter system to study Treg cell activation, related to Figure 1 (A), Scheme of the retroviral vector MW NFAT-GFP-IRES-H2B-RFP for coordinate expression of NFAT-GFP and H2B-RFP. (B), Protocol to transduce polyclonal Treg and Th cells with MW NFAT-GFP-IRES-H2B-RFP. C-D, Expression of Foxp3 (C) and of NFAT-GFP and H2B-RFP (D) in Treg and Th cells at the end of the 6-day culture and transduction protocol. (E), Comparable NFAT deactivation kinetics in Treg and Th cells. Purified splenic CD4+ T cells from Foxp3GFP mice were stimulated with ionomycin, which caused accumulation of NFAT in the nucleus. Cells were then exposed to cyclosporine A (CsA), which blocks NFAT activation through inhibition of calcineurin, fixed at various time points thereafter, and stained with αNFAT1 mAb and DAPI. Kinetics of NFAT relocation to the cytoplasm were similar in GFP+ Treg (arrows) and GFP− Th cells. Scale bar = 10 μm. (F), Illustration of a previously published algorithm for quantification of NFAT activation from 2-dimensional (2D) images of T cells in vivo (left), its limitations (center), and a 3D-algorithm newly developed for this study (right, see also Method details). G-I, Performance of NFAT SI generated by each algorithm as a binary classifier of cells as “signaling” (NFAT SI ≥ 0.5) or “non-signaling” (NFAT SI < 0.5). Correlation of NFAT SI computed by each algorithm in one representative movie out of five (G), confusion matrices of all five movies analyzed (H), and quantification of accuracy, specificity, sensitivity, alpha-, and beta-errors (I). n = 1210 cells from 5 recording of HA-specific NFAT-GFP and H2B-RFP co-expressing CTL in tumor tissue or tdLN of CT26HA tumor-implanted BALB/c mice (dataset from Marangoni et al., 2013). Bars in (I) represent the mean value and symbols individual movies. p value calculated by paired Student t test. (J), Approach to validate each algorithm against a “ground truth” reference method. K-L, Correlation of instantaneous NFAT SI generated by each algorithm with the reference method (K) and distribution of median deviation of individual data points (L). In (K), data points highlighted by squares represent an instance where the 3D Central accumulation algorithm provided an accurate NFAT SI, while the 2D radial profile method provided a much higher, incorrect NFAT SI for the same cell, relative to the reference method. The deviation of the calculated NFAT SI from the reference value for that data point is indicated at the top of each plot. p value in (L) calculated by Kolmogorov-Smirnov test. See STAR Methods for additional details.
Figure S2. Effects of cDC ablation on Treg cell activation, related to Figure 2 A-B, Identification of mCherry-expressing cells in CD11cmCherry mice. The gating scheme and population frequencies are reported in A, and the percentage of each population among mCherry-expressing tumor-associated cells is shown in B. C-D, Selective ablation of cDC in zDCDTR and control bone marrow chimeras treated with DT. Gating scheme and gate frequencies are shown in (C), quantification of cDC1, cDC2, and TAM in (D). n = 4–5 per group from two experiments. E-F, Treg cell frequencies and expression of various activation markers in tumors (E) and tdLN (F) following administration of FTY720. n = 7–13 per group from two independent experiments. G-I, Effects of cDC ablation in zDCDTR BMCs on the frequency of Th and CD8+ T cells in the TME and tdLN (G), expression of activation markers in Treg cells in tdLN (H), frequency of CD44hi CD62L− eTreg cells and their expression of Ki67 in tdLN (I). Data in (G-I) (n = 8 mice per group) are pooled from two independent experiments. MFI were normalized to the mean of the control groups. Bars represent medians, each symbol represents an individual animal. Numbers in parentheses represent fold change. p values were calculated by Mann-Whitney U test.
Figure S3. Effects of CnB deletion on Treg cell activation, related to Figure 2 (A), PCR analysis of CnB deletion in Treg cells in tdLN (lanes 5/6) and tumors (lanes 7/8) of tamoxifen treated Foxp3creERT2 or Foxp3creERT2 × CnBf/f mice. Foxp3cre and Foxp3cre × CnBf/f mice, in which CnB is constitutively deleted in Treg cells were used as controls (lanes 3/4). The ~3000-bp bands correspond to the non-rearranged CnBf/f or the WT CnB loci, whereas the 200-bp band corresponds to the rearranged CnBf/f locus, indicating deletion of CnB. Vertical lines denote non-adjacent lanes, stitched together for display purposes. B-C, Treg cell frequencies and expression of various activation markers in tumors (B) and tdLN (C) following tamoxifen-induced deletion of CnB without concurrent FTY720 treatment. n = 11–14 per group from two independent experiments. MFI were normalized to the mean of the control groups. Bars represent medians, each symbol represents an individual mouse. Numbers in parentheses represent fold change. p values were calculated by Mann-Whitney U test.
Figure S4. Analysis of non-signaling segments of migratory tracks of HA-Treg, HA-Th, and HA-Th cells in the presence of non-visualized HA-Treg cells, related to Figure 3 (A), Gating on non-signaling segments for motility analysis shown in (B-G). B-C, Duration of non-signaling segments (B) and percentage of segments with a duration of 20 minutes or less (C). n = 42 (HA-Treg), 45 (HA-Th) and 67 segments (HA-Th in the presence of HA-Treg cells) recorded in 2 independent experiments. (D), Instantaneous 3D velocities in non-signaling segments. n = 1409 (HA-Treg), 1520 (HA-Th) and 2408 (HA-Th in the presence of HA-Treg cells). (E), Arrest coefficients of non-signaling segments. n = 42 (HA-Treg), n = 45 (HA-Th) and n = 67 (HA-Th in presence of HA-Treg cells). (F), 10-min displacements in non-signaling segments. n = 700 (HA-Treg), n = 711 (HA-Th), and n = 1230 (HA-Th in presence of HA-Treg cells). (G), Migratory tracks of non-overlapping 10-minute non-signaling segments with normalized starting coordinates. n = 89 (HA-Treg), 92 (HA-Th) and 155 (HA-Th in the presence of HA-Treg cells). Data were collected in 6 (HA-Treg), 6 (HA-Th), and 5 (HA-Th in the presence of HA-Treg cells) movies, recorded in two independent experiments. Bars in (B, C, E) depict medians, and symbols represent individual non-signaling segments. p values calculated by Mann-Whitney U test. Fold change relative to the HA-Treg cell group median is shown in parenthesis.
Figure S5. Increase of co-stimulatory ligands on APC and Treg cell hyper-proliferation in tdLN of mice treated with αCTLA-4 mAb, related to Figure 4 (A), Expression of CD80 and 86 as well as various markers of general DC activation on tumor-associated cDC1 and cDC2, 24 hours after administration of αCTLA-4 (clone 4F10), αPD-1, or an isotype control mAb. n = 8–9 per group from two independent experiments. (B), Gating strategy to quantify CD80 and CD86 expression within cDC1 and cDC2 subsets among resident DC (rDC) and migratory DC (mDC) in tdLN. Numbers indicate gate frequencies. (C), Normalized MFI of CD80 and CD86 on indicated subsets 24 h after either ablation of all Treg cells (gray versus white bars) or CTLA-4 blockade (red versus black bars). n = 10 (DT treatment in WT mice), n = 5 (DT treatment in Foxp3DTR mice), n = 10 (isotype control), n = 10 (αCTLA-4 4F10), pooled from two separate experiments. (D), Normalized MFI of CD40, MHC-I and MHC-II on the indicated DC subsets 24 h after CTLA-4 blockade or treatment with isotype control Abs. n = 8–9 per group from two independent experiments. (E), Ki67 expression by Th and CD8+T cells in tumors and tdLN. Data represent 7–10 mice per group from two independent experiments. F-G, Frequency (F) and absolute numbers (G) of indicated T cell types in tdLN 6 days after treatment with αCTLA-4 mAb clone 4F10 (n = 8) or isotype-matched control mAb (n = 10), and FTY720. Data are pooled from two independent experiments. H-L, Frequencies of eTreg cells (H), their expression of Ki67 (I), and of various activation markers (J-L) in tdLN 6 days after treatment with αCTLA-4 mAb or isotype-matched control mAb. Data represent n = 10 per group pooled from two independent experiments. Numbers within gates represent cell percentages. Bars indicate medians, each symbol represents an individual animal. p values were calculated by Mann-Whitney U test. Fold change relative to the median of the control group is shown in parentheses. MFI were normalized to mean values in control groups.
Figure S6. CTLA-4 does not regulate Treg cells in cis in tdLN, related to Figure 5 (A), Experimental protocol. (B), Tamoxifen-induced loss of CTLA-4 expression in CD45.2+ Treg cells in tdLN. C-D, Expression of Ki67 in CTLA-4-sufficient or -deficient CD45.2+ Treg cells (C) and their frequency (D) in tdLN. In (B-C) numbers represent cell percentages within gates. Ten mice per group were analyzed in two experiments. E-F, Expression of activation markers in CTLA-4-sufficient or -deficient Treg cells within tumors (E) or tdLN (F). n = 7–8 (E) and 10 per group (F). G-H, Production of IL-2 by Treg, Th, and CD8+ T cells from tumors and tdLN upon ex vivo re-stimulation with PMA and ionomycin, following 6 days of αCTLA-4 or isotype mAb treatment, as described for Figure 4D. Background IL-2 production (4h culture in absence of PMA/ionomycin) was subtracted from IL-2 production after 4h stimulation with PMA/ionomycin for each individual mouse to determine the frequencies of IL-secreting cells (G). Based on these and absolute cell counts, the absolute numbers of IL-2 producing cells were calculated (H). n = 14–16 per group in two separate experiments. (I), Gating strategy to identify Treg cells in tumors and tdLN using surface markers. J-K, In situ transcriptional activity of the Il2 gene in Treg, Th, and CD8+ T cells from tumors and tdLN in Il2GFP mice after 6 days of αCTLA-4 treatment, as described for Figure 4D. The gating strategy in (I) was used to avoid loss of GFP signal during cell fixation/permeabilization for Foxp3 staining. n = 3 mice per group. Numbers indicate gate frequencies. Bars show median values, each symbol represents an individual animal. MFIs were normalized to the mean values in control groups. p values calculated by Mann-Whitney U test. Fold change compared to the median of the control group in parentheses.
Figure S7. Teff cell functions are not increased after CTLA-4 blockade whereas Treg cell expansion depends on CD28, related to Figures 6 and 7 A-B, Expression of granzyme B, and of the effector cytokines IFNγ and TNF in PMA/ionomycin re-stimulated Th and CD8+ T cells in the TME (A) and in tdLN (B) following 6 days of CTLA-4 blockade. n = 7–9 per group from two independent experiments. (C), Frequency of Treg cells among live hematopoietic cells and of Ki67+ eTreg cells in tdLN after blockade of CTLA-4 and/or IL-2. n = 10–11 per group in two separate experiments. (D), Experimental protocol. (E), Frequency and expression of Ki67 in CD28hi and CD28lo eTreg cells in tdLN after CTLA-4 blockade. F-G, Expression of the indicated activation markers on CD28hi and CD28lo Treg cells in the TME (F) and in eTreg cells in tdLN (G) after CTLA-4 blockade. Data represent 9–10 mice per group pooled from two independent experiments. Bars show medians, each symbol represents an individual animal. Numbers represent cell frequencies within gates. MFIs were normalized to the mean values in control groups. p values calculated by Mann-Whitney U test. Fold change as compared to the median of the control group in parentheses. H-I, Number of Treg cells contacting each APC per hour (H) and duration of individual contacts (I) at various time points in the same regions of interests of the TME. In (H), symbols represent individual APC and bars show means. In (I), symbols represent individual Treg:APC contacts and bars show medians. (J), Example of an APC (oval) located at the tumor/stroma border (dashed line), identified both before and following 18h of αCTLA-4 treatment. Scale bar = 50μm.
Data Availability Statement
Imaris scripts to calculate NFAT SI using the 3D central accumulation algorithm, as well as the MATLAB cell motility analysis scripts are available upon request.