

Abstract
Caspase-8 is an apical caspase involved in the programmed form of cell death called apoptosis that is critically important for mammalian development and immunity. Apoptosis was historically described as immunologically silent in contrast to other types of programmed cell death such as necroptosis or pyroptosis. Recent reports suggest considerable crosstalk between these different forms of cell death. It is becoming increasingly clear that caspase-8 has many non-apoptotic roles, participating in multiple processes including regulation of necroptosis (mediated by receptor-interacting serine/threonine kinases, RIPK1-RIPK3), inflammatory cytokine expression, inflammasome activation and cleavage of interleukin-1β and gasdermin D, and protection against shock and microbial infection. In this review, we discuss the involvement of caspase-8 in cell death and inflammation and highlight its role in innate immune responses and the relationship between different forms of cell death. Caspase-8 is one of the central components in this type of crosstalk.
Introduction
Cell death is an essential part of any multicellular organism. It is highly conserved from C. elegans to M. musculus and H. sapiens, and not only plays important roles during development and homeostasis but also has key functions in the immune system (1). In the context of infection in higher order mammals, cell death can be a double-edged sword with potentially both protective and harmful outcomes. The induction of cell death can be a way to limit replication of intracellular bacteria and viruses by eliminating their replication niche. It can also expose intracellular pathogens to other immune cells, as well as facilitate the release of cytokines and danger-associated molecular patterns (DAMPs) to the surroundings. However, elimination of key immune cells can weaken the immune response by reducing the number of cells combating the infection and allowing the pathogen to spread (2, 3). Deregulation or dysfunction of these cell death pathways can lead to inflammation and disease, underscoring the importance of understanding cell death mechanisms. Comprehending how inflammation and cell death are linked will allow us to better grasp disease pathogenesis and help us develop novel therapeutic strategies.
A central component of both cell death and inflammation is the cysteine protease caspase-8. Its original function was described as an initiator of extrinsic apoptosis; however, it has also been found to be involved in other cell death pathways including intrinsic apoptosis and pyroptosis while also playing an inhibitory role in necroptosis. In addition to this, caspase-8 also appears to be mediating optimal transcription of certain inflammatory cytokines independently of cell death. In this review, we give an overview of the many roles of caspase-8 and exemplify how caspase-8 is involved in the increasing amount of crosstalk between cell death and inflammation.
Caspases
A shared modality in many forms of cell death is the role of a group of cysteine proteases called caspases. These are endopeptidases that cleave their substrates at specific amino acid sequences. Unlike many other digestive proteases, they do not degrade their substrates. The different caspases have unique specificities but common to all is that they cleave substrates after an aspartic acid residue; thus giving them the name caspase (the “c” refers to cysteine and the “asp” refers to aspartic acid) (4). They are classified as zymogens, proteins that are inactive at steady state and require cleavage to become active. Cleavage of the caspase exposes a pocket containing a cysteine and an arginine. The arginine keeps the substrate aspartic acid in place allowing the caspase cysteine to cleave the substrate just after its aspartic acid residue.
Caspases are broadly divided into inflammatory caspases and apoptotic caspases. Inflammatory caspases include caspase-1, -11 (-11 in mice, -4 and -5 in humans), and -12, and apoptotic caspases include caspase-2, -3, -6, -7, -8, -9, and -10 (5). An argument can be made that caspase-8 may be classified as both an apoptotic and an inflammatory caspase. The classical view is that both dimerization and cleavage are needed for activation of caspases. Full-length caspases (also called pro-caspases) contain three distinct regions called the pro-domain, the large subunit and the small subunit, with cleavage sites separating the different subunits. For activation of the caspase these sites need to be cleaved, either by another protease or by the caspase itself. On the other hand, the pro-domain can contain binding sites that connect the caspase to signaling complexes or adaptor molecules initiating the pathway (6). Caspase-1, -2, -9, -11 and -12 have a caspase activation and recruitment domain (CARD), whereas caspase-8 and -10 have a death effector domain (DED) (7).
Caspases have differences in the specificity to their substrates. This specificity is determined by the amino acid sequences preceding the aspartic acid cleavage site of the substrate. For cleavage to occur, the substrate must be able to bind in the caspase active pocket in such a way that the substrate aspartic acid residue is exposed. As an example of this specificity, both caspase-1 and -11 can cleave gasdermin D (GSDMD), but only caspase-1 can additionally cleave pro-IL-1β and pro-IL-18 (8). However, recent studies have shown that there is a big overlap between substrates of different caspases and, depending on the concentration of the specific caspase, many caspases can (to different degrees) cleave many of the same substrates (9).
Boucher et al. recently published a study that challenges the dogma that caspases require cleavage to become active. The authors describe how the main catalytically active caspase-1 species, activated after dimerization, is not the cleaved p20/10 subunit containing the active pocket, but rather a transient p33/p10 partly cleaved species and a full-length p46 pro-caspase-1 dimer. The full-length p46 dimer was shown to be able to cleave its substrate GSDMD, the p33/10 heterodimer could cleave both pro-IL-1β/IL-18 and GSDMD, whereas p20/10 rapidly diffused away and became inactive (10).
Like caspase-1, caspase-8 is activated through dimerization. This dimerization triggers pro-caspase-8 autoprocessing activity leading to cleavage and release of active caspase-8 into the cytosol where it can cleave other substrates such as caspase-3 and -7 and induce apoptosis (11–13). Observations also suggest that similar to caspase-1, caspase-8 has certain catalytic properties in its full-length form (14, 15). The activity is gained by homo-oligomerization of two procaspase-8 molecules or by hetero-oligomerization of procaspase-8 with the proteolytically dead caspase-8 homolog cellular FLICE inhibitory protein (cFLIP L) (16, 17). This activity allows autoprocessing in the case of caspase-8 homodimers, inhibits necroptosis (18) possibly through cleavage of RIPK1 and RIPK3 (19, 20), and induces expression of inflammatory cytokines (15).
Once procaspase-8 homo-dimerizes, it is cleaved at Asp373 and Asp387 (mouse caspase-8) between the large and small catalytic subunits, see Figure 1. This cleavage generates a p10 fragment (the small catalytic subunit) and the p41 and p43 fragments. The p41 and p43 fragments are generated from two isoforms of caspase-8. The next step in caspase-8 activation is the cleavage of the p41/p43 fragments at Asp212 and Asp218 leading to the release of the pro-domain and the large catalytic p18 subunit. Catalytically active caspase-8 consists of two large p18 subunits and two small p10 subunits generated as the procaspase-8 dimer is cleaved, and which then cleave caspase-3 and -7 and induce apoptosis (21). The catalytic activity of caspase-8 is critical for embryonic development in mice as caspase-8 null mice as well as caspase-8 catalytically dead mice (C362A or S) die before birth from MLKL-mediated necroptosis (22–24). Interestingly, it appears that the first cleavage step during caspase-8 activation, resulting in the release of the small subunit, has two functions. First, it strongly reduces autoprocessing activity of caspase-8, and second, it allows for increased access to caspase-8 substrates such as caspase-3, caspase-7 and Bid (25).
Figure 1.
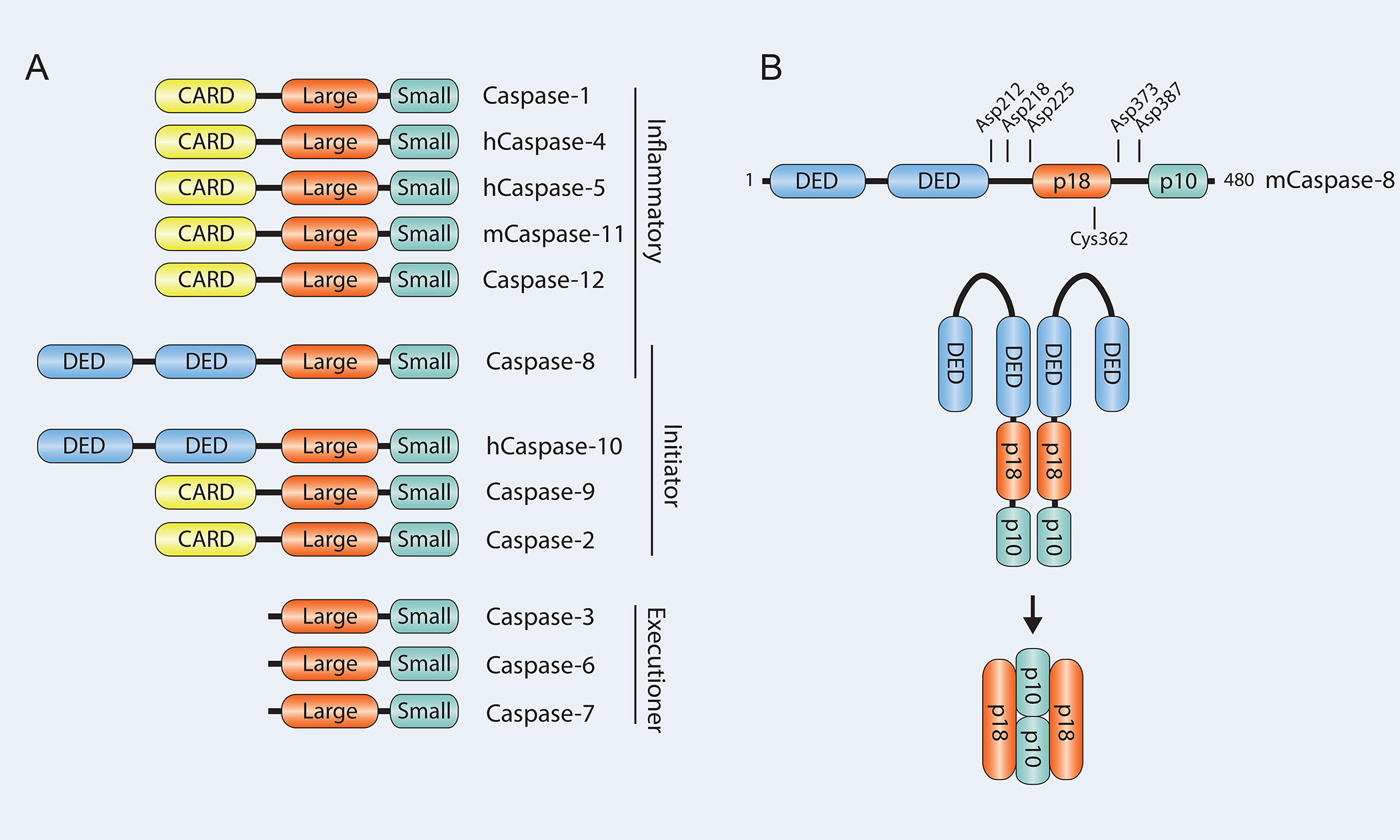
A. Schematic showing the different caspases and their domains. B. Important residues involved in activation of mouse Caspase-8.
Several substrate cleavage sequences are targeted by caspase-8. A typical caspase-8 target sequence found in caspase-3 and utilized in chemical inhibitors is tetrapeptide IETD (cleavage site positions P4-P3-P2-P1, where cleavage occurs after P1 amino acid D/Asp), although it is not very impressive as a specific caspase-8 inhibitor (9). This is a good example of an often-cited problem with specificity in caspase inhibition. Peptides DETD, DEVD and others have also been mentioned as favorable for caspase-8 cleavage (26–28), and it is believed that caspase-8 prefers branched leucine and valine in position P4, in contrast to caspase-1 which prefers bulky, hydrophobic amino acids such as tyrosine and tryptophan (27). Indeed, activities of caspases are often indicated as units measuring activity towards a synthetic tetrapeptide substrate. Optimal substrate sequence predictions are also typically based upon synthetic peptide studies. However, this may not take more complex protein structures into account (8) and considerable variations in the caspase-8 target sequence are observed. For example, the presumed mouse caspase-8 targeted tetrapeptide cleavage site of caspase-3 is IETD, for cFLIP-L: LEVD, for RIPK1: LQHD, for GSDMD: LLSD, pro-IL-1β: LVCD, and pro-IL-18: LESD.
Cell death
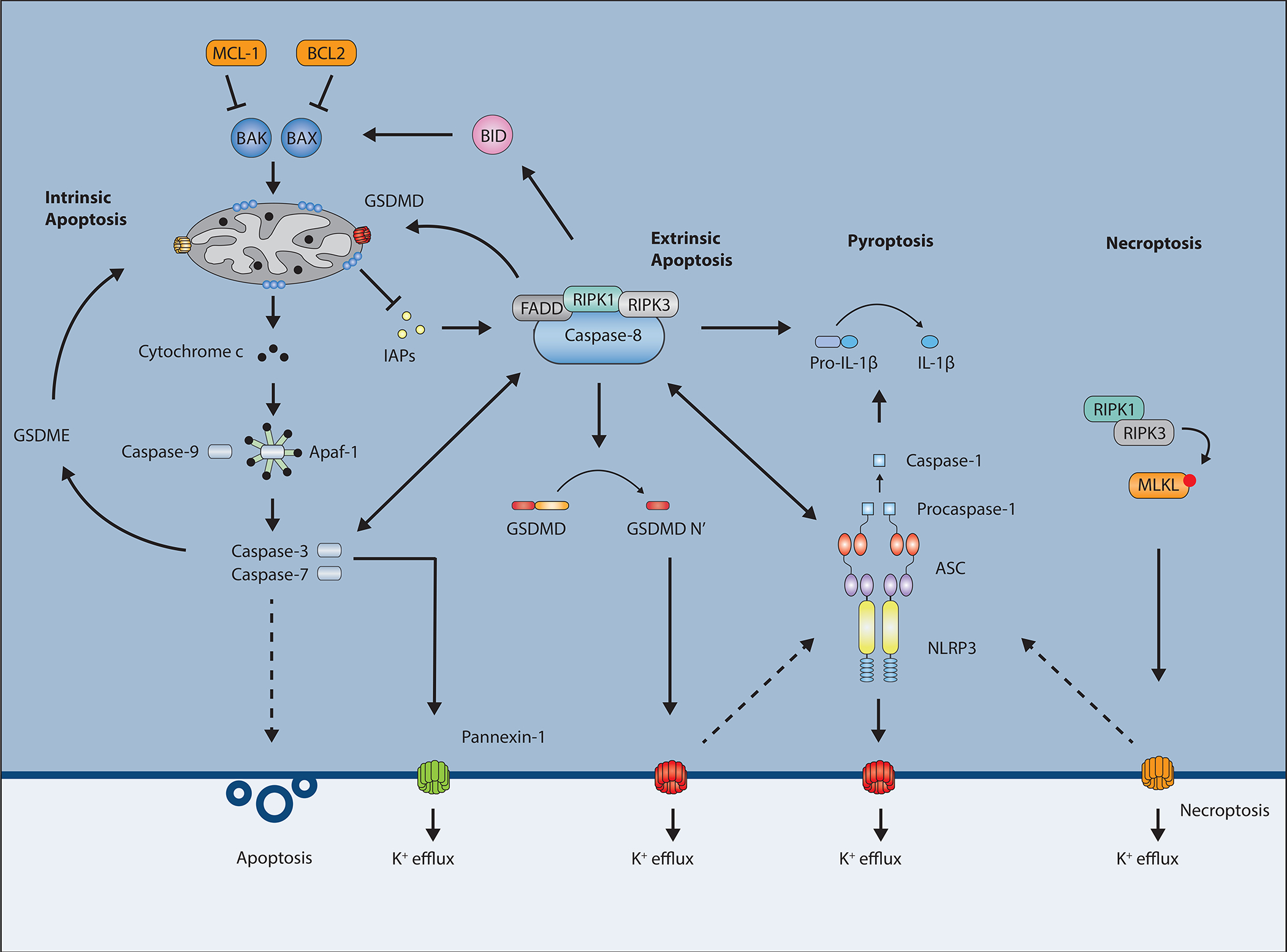
Multiple forms of regulated cell death occur in response to different signals (29, 30). Examples include apoptosis, necroptosis, pyroptosis, ferroptosis, autophagy-dependent cell death, lysosome-dependent cell death and many others. Apoptosis, necroptosis and pyroptosis are three forms of cell death with major involvement in immunity and disease and will be the focus of this review. Apoptosis is the first programmed cell death pathway that was discovered and is an integral part of the development and survival of any eukaryotic organism. Necroptosis and pyroptosis are inflammatory types of death associated with release of the cellular contents that play essential roles in our immune system, see Figure 2.
Figure 2.
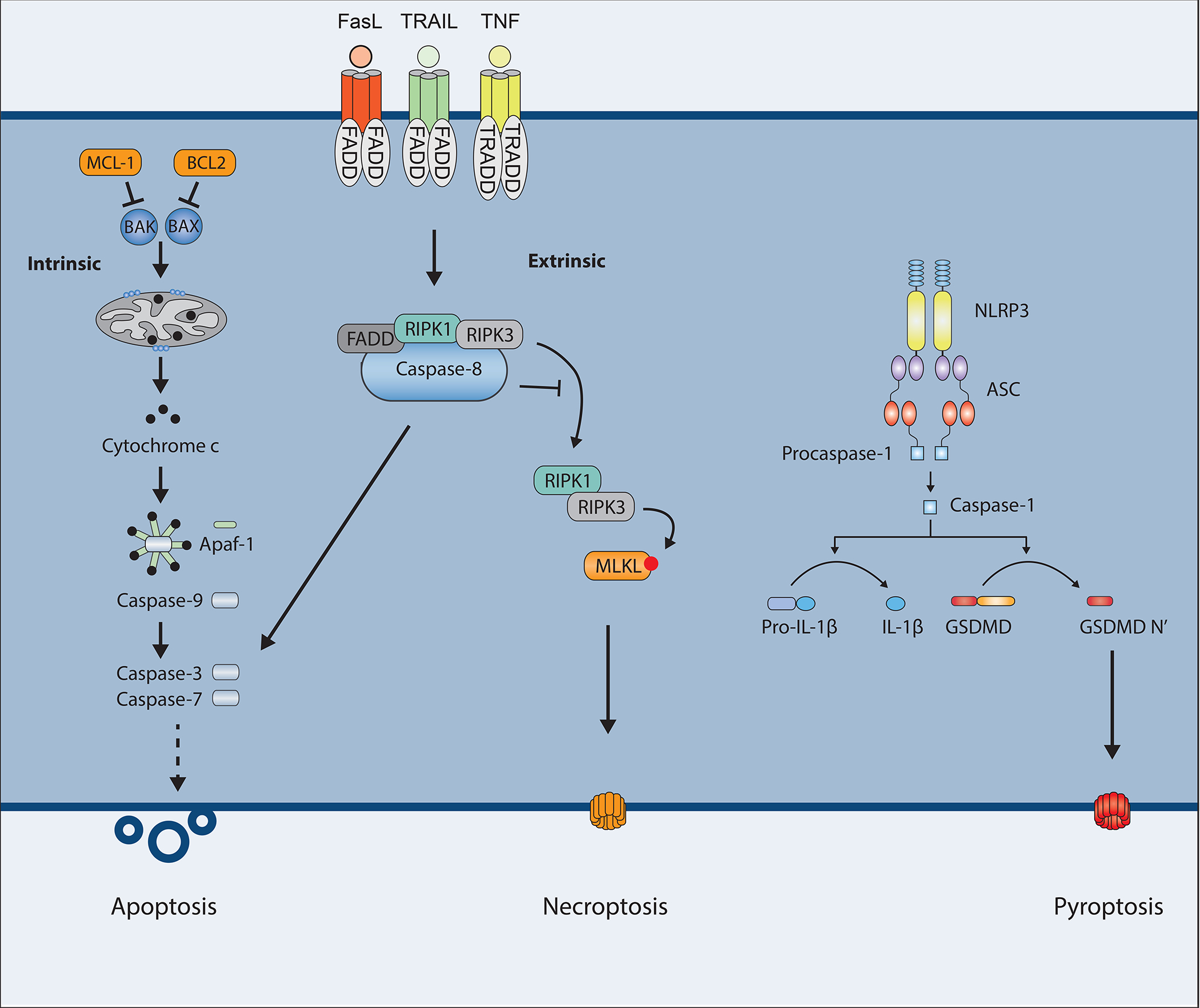
Schematic overview of the different types of programmed cell death. The intrinsic and extrinsic apoptotic pathways culminate in activation of caspase-3 and -7 leading to cell death. Caspase-8 also inhibits RIPK3 and MLKL-driven necroptotic cell death. Activation of the inflammasomes leads to GSDMD-driven pyroptosis.
Apoptosis – intrinsic vs. extrinsic
Apoptosis is essential for the removal of excessive or damaged cells. For instance, the development of fingers in human embryos occur by the cells between the fingers undergoing apoptosis. During microbial infection, apoptosis provides a mechanism for clearing infected cells (31, 32). The contents of an apoptotic cell are contained within the membranes of the cell as the plasma membrane starts to form “blebs” or small membrane-bound apoptotic bodies. Historically, apoptosis was considered a “silent” cell death, not to be associated with inflammation as any inflammatory DAMP is contained within the membranes of the cell; however, this view is quickly becoming outdated. Characteristic features of apoptosis include membrane blebbing, cleavage of DNA that can be visualized as a regular DNA ladder pattern on an agarose gel, nuclear condensation and fragmentation, cell shrinkage, and the appearance of “eat me” signals on the plasma membrane such as the flipping of phosphatidylserine from the inner leaflet of the membrane to the outer (29).
Through the initial studies in C. elegans, it was discovered that a group of caspases mediate apoptosis. As described above, these caspases are synthesized as zymogens and will, upon activation, cleave other proteins after specific residues. They can be grouped into initiator caspases and executioner caspases depending on their position in the cell death cascade. Executioner caspases have many hundreds of substrates within a cell and will cause the cell to die if introduced in an active form into any cell type. The executioner caspases trigger all of the above-mentioned characteristics of apoptosis.
Apoptosis can be categorized as intrinsic or extrinsic apoptosis depending on where the signal to initiate cell death originates. Intrinsic apoptosis is triggered by intracellular stresses such as DNA damage, nutrient deprivation, endoplasmic reticulum (ER) stress, hypoxia or free radicals (33–38). It is intricately regulated by pro- and anti-apoptotic Bcl2 family members, leading to mitochondrial outer membrane permeabilization (MOMP) and subsequent release of cytochrome c resulting in activation of the initiator caspase-9 (39, 40). During the presence of pro-apoptotic Bcl2 members, the proteins Bcl2-associated X (BAX) and Bcl2 homologous antagonist/killer (BAK) will translocate to the mitochondria where they will induce MOMP and trigger the apoptosome, a multimolecular protein complex consisting of apoptotic protease activating factor 1 (APAF-1), cytochrome c and caspase-9, see Figure 2. Once caspase-9 is cleaved and activated in this complex, it cleaves and activates the executioner caspases-3 and -7, resulting in cell death (33, 39, 41–43). Additionally, it appears that mitochondrial DNA can also be released during this process and lead to activation of other inflammatory pathways (44, 45).
In contrast, extrinsic apoptosis is initiated by extracellular signaling through cell surface receptors. Examples of ligands are TNF binding to the TNF receptor (TNFR), TNF-related apoptosis-inducing ligand (TRAIL) binding to the TRAIL receptors (DR4 and DR5), or FAS-ligand (FASL) binding to the FAS receptor (CD95) (46). Ligand binding triggers recruitment of TNFR-associated death domain (TRADD), FAS-associated death domain (FADD), and receptor interacting protein kinase 1 (RIPK1) leading to caspase-8 activation. Like caspase-9, caspase-8 can trigger activation of caspase-3 and -7. Additionally, there is crosstalk between these pathways as in certain cells caspase-8 can trigger the intrinsic pathway through cleavage of BCL2-homology 3-interacting domain death agonist (BID) and, thus, subsequent activation of BAX and MOMP (47). Further crosstalk will be discussed in later chapters.
Although apoptosis is generally considered to be inflammatorily silent, cell death due to infection or tissue injury can lead to accompanying inflammation. This response might be beneficial in helping to repair damaged tissue or battle an invading pathogen. In line with this, excessive apoptosis in certain mouse models can induce inflammation (48–51).
Role of caspase-8 in extrinsic apoptosis
Major contributions to elucidating the mechanisms of extrinsic apoptosis were made in the mid 90’s with the discoveries of caspase-8, RIPK1, FADD, and TRADD (52, 53, 62, 54–61). Studying FAS-induced cell death using yeast two-hybrid screens it was shown that caspase-8, RIPK1, and FADD were critical for this type of apoptosis. The proteins congregated into what was termed the death-inducing signaling complex (DISC) necessary for inducing cell death in response to FAS-activation (63). The assembly of the DISC complex led to rapid cleavage of intracellular proteins mediated by caspase-8 (59, 61), including α-fodrin, gelsolin, PARP, and ICAD among others (64–67). Collectively, these cleaved proteins mediate apoptotic phenotypes such as loss of catalytic activity, DNA fragmentation, membrane blebbing and cell shrinkage, resulting in an orderly breakdown of cellular functions. It became apparent that activation of caspase-8 in the DISC was the major driver of these events, but it was at first not completely clear what proteases were directly activating α-fodrin, gelsolin, PARP, and ICAD. Caspase-3 and -7 were, however, good candidates as they were frequently cleaved and activated during apoptosis. In 1998 it was shown that caspase-8 could cleave pro-caspase-3 providing the link between the DISC complex and apoptosis (68).
Another component of the DISC is called cellular FLICE inhibitory protein (cFLIP) and is closely related to caspase-8 but lacks its catalytic site (69–75). cFLIP was first identified as an inhibitor of FAS-induced cell death as overexpression of cFLIP blocked apoptosis, but was also shown to partly activate caspase-8 at low levels of expression (16, 76). cFLIP comes in three isoforms, two short (cFLIPR and cFLIPS) and one long (cFLIPL) form (77, 78). All three are found in humans while only cFLIPR and cFLIPL are found in mice. cFLIPL will dimerize with caspase-8 and prevent the auto-cleavage of caspase-8, thereby preventing downstream apoptosis, see Figure 3. cFLIPL is an NF-κB target gene and inhibitors of protein translation, such as cycloheximide (Cx), sensitize cells to TNF-induced apoptosis partly by reducing cFLIPL levels.
Figure 3.
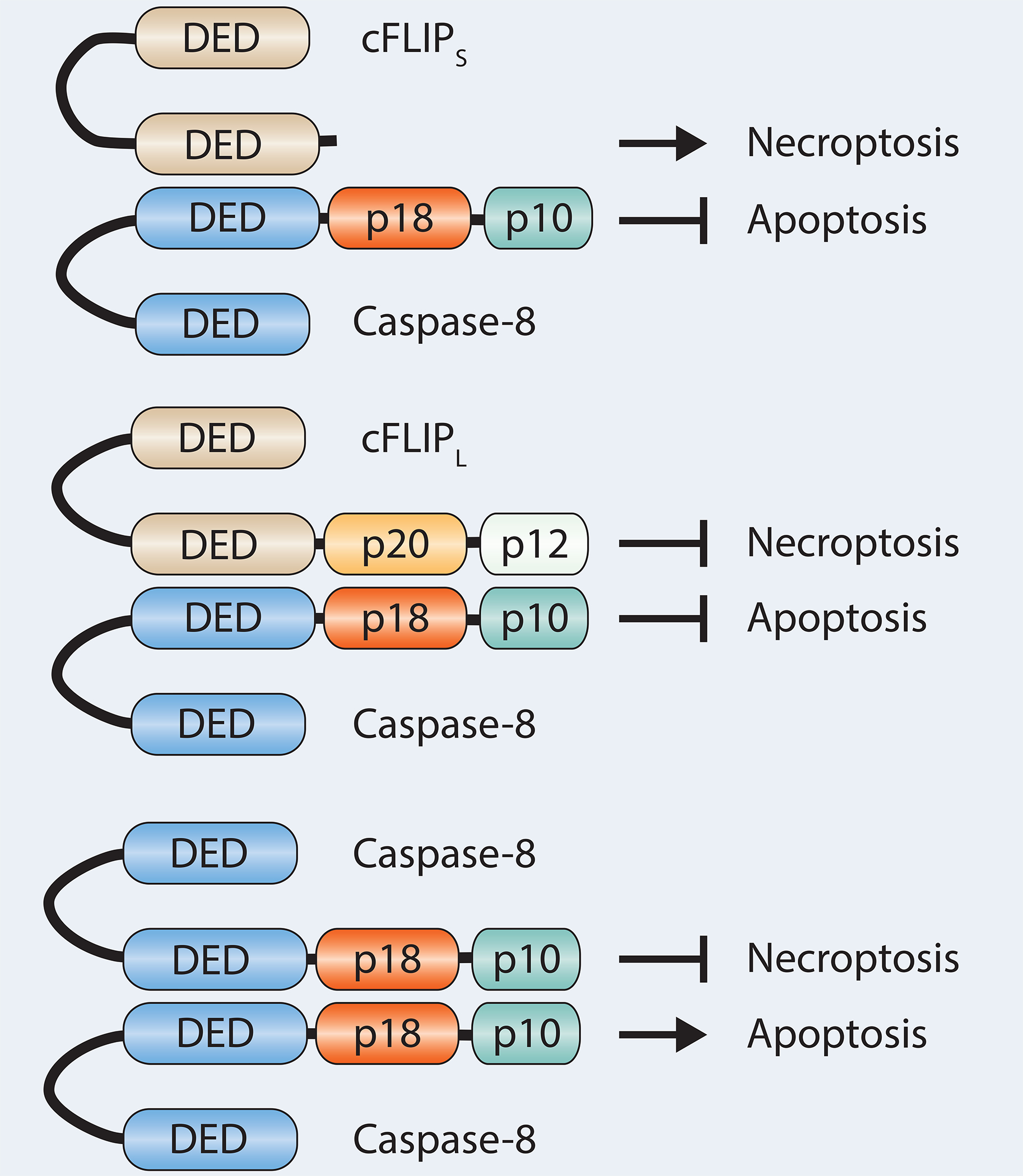
Caspase-8 and cFLIP interactions and possible outcomes. Dimerization of caspase-8 induces apoptosis while inhibiting necroptosis. Caspase-8 dimerization with the proteolytically dead caspase-8 homolog cFLIPL blocks apoptosis but can still inhibit necroptosis. Dimerization with cFLIPS drives necroptosis.
Necroptosis
Necroptosis is programmed inflammatory necrotic cell death typically involving RIPK1 and RIPK3 kinases, and finally the pore-forming pseudokinase MLKL that mediates perforation of the cell membrane. In addition to apoptosis, necroptosis can also be triggered downstream of the TNFR complex. Under certain conditions RIPK1 can initiate RIPK3-induced necroptosis instead of caspase-8-induced apoptosis. RIPK3 phosphorylation results in oligomerization of mixed lineage kinase domain-like protein (MLKL), which inserts itself into the plasma membrane leading to pore formation, ionic instability, water influx, cell swelling and rupture (79–81). The TNFR complex, through RIPK1, can result in both nuclear factor-kappa B (NF-κB)-directed gene transcription and death induction. The gatekeeper RIPK1 is heavily regulated. Many viruses and bacteria have components that target this pathway to block inflammatory gene transcription in an attempt to evade a strong immunological response. As a countermeasure, RIPK1 can direct the cells to die as a way to combat these microbes. In the tug-of-war between host and pathogen, certain pathogens have evolved to counter the induction of death as well. Therefore, necroptosis may function as a back-up death pathway during apoptotic inhibition.
RIPK1 can disassociate from the TNFR complex under certain conditions and instead form a death-inducing signaling complex called complex II (also called the ripoptosome) (82). A number of factors determine if this death complex leads to caspase-8 induced apoptosis or RIPK3-induced necroptosis, as shown in Figure 2. These factors include cell type and microbial effectors and is a topic of intense research in the field. Caspase-8 plays a large role in this signaling fork as once it is recruited to the complex it is capable of cleaving RIPK1 and RIPK3 and, thus, inhibiting necroptosis while at the same time triggering apoptosis (18, 83). Therefore, caspase-8 inhibitors trigger necroptosis during TNF signaling.
cFLIPL is not only important for regulating apoptosis, as discussed above, but is also important for preventing necroptosis. It is thought that cFLIPL-caspase-8 dimers, unable to induce apoptosis, still retain their enzymatic activity and capability to cleave RIPK1 and RIPK3 and blocking necroptosis (18, 84–87). Interestingly, caspase-8 also dimerizes with cFLIPR/S; however, this heterodimer does not show enzymatic activity and cannot cleave RIPK1 and RIPK3. Expression of the short form of cFLIP leads to increased levels of RIPK1 in the ripoptosome and promotion of necroptosis (84). There are also viral isoforms (vFLIP) that resemble cFLIP and inhibit apoptosis (88–90). The ratio between cFLIPL and cFLIPR/S appears to determine whether the cells undergo necroptosis or apoptosis. Higher levels of cFLIPR/S have been shown to protect cells against apoptosis while sensitizing them to necroptosis (84) and in vivo studies have shown that higher levels of cFLIPL protect mice from liver-induced apoptosis and necroptosis (91).
Pyroptosis
The final form of cell death covered in detail here is pyroptosis and can be categorized as a subgroup of necrosis. Mechanistically, pyroptosis is triggered by inflammasome activation, a process bearing some resemblance to the cytochrome c – apaf1 – caspase-9 complex of intrinsic apoptosis, while the end product of pyroptosis more closely resembles necroptosis with pore formation, membrane permeabilization, and cell swelling. Pyroptosis was long regarded as a special type of caspase-1 mediated apoptosis in monocytes in response to certain bacterial challenges. The discovery that caspase-11 (caspase-11 in mice, caspase-4/5 in humans) can also trigger pyroptosis through sensing of intracellular lipopolysaccharide (LPS) (92–95) broadly expanded the role of pyroptosis through its implication in combating intracellular bacteria and mediating septic shock. Pyroptosis is now widely regarded as a crucial pathway in innate immune cells by releasing inflammatory cytokines, removing the pathogen replication niche, and trapping intracellular bacteria within the cellular debris of pyroptotic cells (96, 97). In 2015, the groups of Feng Shao and Vishva Dixit independently identified the gasdermin D (GSDMD) protein as an effector molecule controlling pyroptosis (98, 99). Since other gasdermin family members gasdermin E also can create pores and induce a similar type of cytotoxicity, arguments have been made that pyroptosis can be defined as gasdermin-mediated cell death (29).
As will be discussed in later chapters, even though caspase-8 is not considered the major player during pyroptosis, increasing amounts of evidence suggests that it can play an important role. It appears that caspase-8 can both regulate canonical inflammasomes and directly activate GSDMD.
Gasdermin D
GSDMD was recently discovered to play a role during pyroptosis. GSDMD consists of an N-terminal and C-terminal domain with a caspase-1 and caspase-11 cleavage site in the 44 amino acid linker between the two domains, at D276 (D275 in humans). The C-terminal domain acts as an inhibitor and, once cleaved, the N-terminal is free to oligomerize and insert itself into the plasma membrane leading to pore formation, swelling of the cell, disruption of membrane integrity, violent burst and death (98–101). In line with this, expression of the N-terminal domain by itself is enough to trigger pyroptosis. The N-terminal domain displays strong binding to phosphoinositides and cardiolipin, present in the cell plasma membrane and in the bacterial inner membrane respectively, by recognizing the negatively-charged head group of the lipid (100, 101). This binding causes oligomerization of GSDMD protomers, resulting in pores consisting of an inner diameter of 12–14 nm (100–103); however, recent reports by the groups of Müller, Broz, and Hiller have suggested that these pores can vary in size with diameters closer to around 20 nm (103, 104). In addition to allowing for gradient flow of water and ions such as K+ and Ca2+, the pores are sufficiently large to permit passage of small cytokines such as IL-1β and IL-18. Of note, because of the presence of phosphoinositides largely on the inner leaflet of healthy cells, N-terminal GSDMD only causes pore formation and lysis from within (100, 101).
GSDMD can also be cleaved by caspase-3 in the N-terminal domain at amino acid D88 upstream of the caspase-1 and -11 cleavage site (105, 106). This caspase-3-mediated cleavage is hypothesized to block pyroptosis by inhibiting oligomerization and pore formation of N-terminal GSDMD (105), and macrophages from mice with a D88A mutation in GSDMD have a small but significant increase in cell death induced by certain stimuli (106). Recent studies have also indicated that in neutrophils GSDMD can be cleaved independently of caspases but dependent on the neutrophil-specific serine protease ELANE (107). ELANE is proposed to be released from cytoplasmic granules into the cytosol of aging neutrophils and cleave GSDMD upstream of the caspase-1 cleavage site. This neutrophil-specific GSDMD fragment is also fully capable of inducing lytic cell death. Two reports also demonstrated the involvement of GSDMD in generating neutrophil extracellular traps (NETs) (108, 109). GSDMD can also be cleaved by cathepsin G resulting in inflammation (110).
In addition to this, it appears that under certain conditions GSDMD will oligomerize and form membrane pores, but instead of undergoing pyroptosis, the cell becomes hyperactivated (111). In this scenario, it is proposed that the GSDMD pores are important for release of IL-1β and IL-18, but the concentration of pores is not high enough to cause cell death. This effect may be influenced by active membrane repair mechanisms whereby ESCRT proteins are believed to “snip out” damaged membrane areas in an attempt to counteract pyroptosis and maintain membrane integrity (112). Hence, a dynamic relationship between damage and repair could influence the effects of the pore formation. Interestingly, GSDMD is expressed in a wide range of tissues and cell types, indicating that the role of GSDMD and thus also perhaps the role of pyroptosis might be involved outside of innate immune cells. For instance, GSDMD is highly expressed in gut epithelial cells, and both caspase-1 and -11 have previously been shown to play a role in intestinal epithelial defense against bacteria (113–116). Other gasdermin family members, such as GSDME (also called DFNA5) and GSDMA may also contribute to cell death. As an example, it has been shown that caspase-3 can cleave GSDME and induce pyroptosis in some cells(117, 118).
Inflammasomes
Inflammasomes were discovered by Jürg Tschopp’s group in 2002 (119) and are cytosolic multiprotein complexes that sense pathogen presence and initiate inflammatory responses. They are increasingly recognized to be critical for a well-functioning innate immune response (120, 121). The inflammasomes are central to a wide range of functions in innate immune cells including cytokine production (122), cell death (123), and even regulating gene transcription and metabolism (124).
The canonical inflammasome consists of a sensor, an adaptor, and an executioner. Previously identified sensors include the NOD-like receptor protein 3 (NLRP3) and absent in melanoma 2 (AIM2). The sensor gets activated through recognition of certain PAMPs and DAMPs, and functions as a seed molecule. This recruits the adaptor protein, apoptosis-associated speck-like protein containing CARD (ASC), which binds through pyrin-pyrin domain interactions between the sensor and the adaptor. ASC then oligomerizes and forms long fibrils that through its CARD domain can recruit caspase-1 (125, 126). Dimers of recruited caspase-1 undergo autoproteolysis and activation, and subsequently cleave pro-IL-1β and pro-IL-18 into their mature forms, as well as cleaving GSDMD. Cleaved GSDMD then initiates pyroptosis (98, 99).
Inflammasome-dependent secretion of IL-1β and IL-18 is critical for mounting an adequate immune response to many pathogens (96, 120, 127–130). Inflammasomes may also play an important role in vaccine responses (131–133). However, because of its importance in immunity, deregulation or inappropriate activation of inflammasomes can lead to autoinflammation (134–137) and contribute to autoimmune disorders (138–140). Alzheimer’s disease (141), Parkinson’s disease (142), and many other pathologic processes are associated with deregulation of inflammasomes.
To a certain degree, the functions of IL-1β and IL-18 overlap, as they are both important for the innate immune system. IL-1β functions by recruiting neutrophils to the site of infection, promoting cell adhesion, and activating T helper (TH)17 cells (143). Binding of IL-1β to its receptor leads to NF-κB activation and expression of TNF and RANTES, as well as the chemokines IL-8 and KC that are strong attractants of neutrophils (144–149). In sum, IL-1β (through its effect on neutrophils) has the potential to cause tissue damage. On the other hand, IL-18 functions by activating natural killer (NK) cells and T cells to secrete IFNγ (150). Both IL-1β and IL-18 are critical to control and limit invading pathogens and for efficient activation of adaptive immune responses; however, too large of a response can cause tissue damage and promote autoimmunity.
Because IL-1β and IL-18 are such potent cytokines, it is crucial that activation of the inflammasome and cleavage of caspase-1 is under tight control. This is achieved through a two-step activation mechanism. Several of the inflammasome components such as NLRP3, caspase-11 and pro-IL-1β have a very low basal expression. Thus, they need to be upregulated through NF-κB activation, achieved by, for instance, Toll-like receptor (TLR) signaling after recognition of danger signals such as PAMPs and DAMPs. Once this occurs, assembly of the inflammasomes can be initiated by a second, distinct, trigger signal leading to activation of the inflammasome sensor and subsequent activation of caspase-1. The first signal warns the cells of the presence of pathogens, sensitizing them to further activation, and the second signal is what ultimately leads to production and release of IL-1β and IL-18.
While the general model of sensor-adapter-executioner is useful for a basic conceptualization of the system, the extent of its diversity is increasingly being recognized and appreciated. For instance, the formation of ASC fibril polymers has been observed through electron microscopy techniques. The structures can be viewed as large molecular organizing centers that can recruit not only caspase-1 but also other molecules such as caspase-8 (151–153). And these structures have also been shown to signal outside of the cell by being released and taken up by neighboring cells (154).
NLRP3
NLRP3 is often regarded as the archetypal inflammasome component. It is a member of the nucleotide-binding domain and leucine rich repeat containing (NLR) family of intracellular pathogen recognition receptors (155). Exactly how NLRP3 is activated is currently under debate but can include events such as efflux of potassium, production of reactive oxygen species (ROS), elevated extracellular ATP levels, bacterial pore-forming toxins, crystallized molecules such as silica or uric acid, oxidized mitochondrial DNA, and many others (156, 157). It has been proposed that perturbation of the mitochondria and the release of mitochondrial DNA causes NLRP3 activation; however, how this occurs is still unknown, and recent genetic studies have argued against the role of mitochondria all together (158, 159). Other groups have suggested that ion flux is the main driver of NLRP3 activation as many events leading to activation also forms pores in the membrane (156). The canonical pore-forming toxin Nigericin, for instance, triggers robust NLRP3 activation (160). It has also been reported that necroptosis can trigger NLRP3 activation through MLKL pores (161). Adding high extracellular concentrations of KCl preventing K+ efflux and depletion of intracellular K+ levels can often block this NLRP3 activation, indicating that NLRP3 is a sensor for intracellular ion concentrations (156). High extracellular concentrations of ATP, another NLRP3 activating event, has been shown to activate the ion transporter P2X7R leading to reduction of intracellular K+ levels by 50%. In this case, however, K+ efflux is not the main driver of NLRP3 activation as inhibitors of the hemichannel protein pannexin-1 blocks NLRP3 activation but not K+ efflux (162). Thus, NLRP3 may have many distinct mechanisms of activation.
A recent study by James Chen and his lab has shed new light on the NLRP3 conundrum (163). In this study, they propose that NLRP3 activation is preceded by disassembly of the trans golgi network (TGN). They show that NLRP3 binds the dispersed TGN (dTGN), gets activated and recruits ASC to form the inflammasome. Specifically, they propose that polybasic regions of the NLRP3 molecule bind to PtdIns4P on the dTGN. This binding is facilitated by changes in ion concentrations through K+ efflux, thus explaining the need for pore formation.
In addition to the sensor, adaptor and executioner, several studies have proposed the kinase NEK7 as a critical participant in the NLRP3-inflammasome (164–166). NEK7 appears to bind to NLRP3 and is required for NLRP3 activation independently of its kinase activity. In addition to this, NLRP3 oligomerization also requires dephosphorylation of certain amino acids by protein phosphatase (167).
Another important NLRP3-activating mechanism occurs through upstream recognition of intracellular LPS by caspase-11. In this pathway, termed the non-canonical inflammasome pathway, LPS-activated caspase-11 cleaves GSDMD directly, leading to pore formation as well as NLRP3 activation through K+ efflux (92, 94, 95, 98, 99, 102, 168). Caspase-11 has a much more narrow substrate specificity than caspase-1 with a strong preference for GSDMD over pro-IL-1β and pro-IL-18 (8). The non-canonical caspase-11 inflammasome plays a critical role in host defense against intracellular Gram-negative bacteria (169–172); however, mice lacking GSDMD or caspase-11 exhibit protection from high-dose LPS-mediated septic shock emphasizing the detrimental role of unchecked inflammation (94, 95, 99).
There are still ways that IL-1β and IL-18 can be processed independently of inflammasomes, as suggested by the fact that mice lacking caspase-1 or ASC have a normal IL-1β response to certain stimuli (173–175). Neutrophil proteases such as serine proteinase-3, cathepsin G and neutrophil elastase can, for instance, process these cytokines directly (176).
Caspase-8 and crosstalk between cell death and inflammation
Caspase-8 has usually been thought of as an initiator caspase for activation of inflammatory silent apoptosis. This idea that apoptotic caspases mediate inflammatory silent cell death has been underscored by the confined nature of apoptosis and by observations that many apoptotic caspases inactivate other more inflammatory pathways. Examples include cleavage and inhibition of GSDMD (105), limiting mitochondrial DNA-dependent STING activation (177), regulation of RIG-I mediated IRF3 activation (178), and cytokine suppression (179). However, recent studies have shown that caspase-8 also has other more inflammatory roles resulting in secretion of inflammatory molecules (180, 181). These findings suggest that in specific cases cells can use caspase-8-dependent extrinsic apoptosis to activate the immune system. An important difference between sterile apoptosis in the context of homeostasis and pathogen-induced apoptosis is that during bacterial infection the cell also encounters a plethora of different bacterial pathogen-associated molecular patterns (PAMPs) not present during sterile extrinsic apoptosis. This also appears to be the case during intrinsic apoptosis. Recent reports have shown that BAX/BAK induced caspase-3/7 cleavage during TLR signaling can trigger NLRP3-dependent inflammation (106, 182, 183). Simultaneous sensing of PAMPs might therefore be the cue determining whether or not the immune cells interpret the conditions as harmless or as a dangerous microbial infection.
Caspase-8 limits RIPK1-mediated cell death
Caspase-8 plays an important role in inhibition of necroptosis. Researchers have used conditional knock-out mice to demonstrate that caspase-8 expression in gut epithelial cells is required to maintain gut integrity by preventing necroptotic cell death in response to microbial challenge (184). Likewise, in many other tissues caspase-8 helps prevent necroptosis and protects against inflammation (185–194). Several microbial inhibitors of caspase-8 as well as mammalian and synthetic inhibitors (CrmA, vICA and cFLIPs, z-VAD-fmk and z-IETD-fmk) trigger necroptosis (195–197). Necroptosis is also triggered by genetic loss of caspase-8 (197), as caspase-8, in cooperation with cFLIPL cleaves and inactivates RIPK1 and RIPK3 (85–87, 198). Caspase-8 deletion by itself leads to embryonic lethality of mice at day 10.5 after conception and this can be rescued by co-deletion of RIPK3 or MLKL (22, 83, 198). Fadd−/− mice are also embryonically lethal and can be rescued by RIPK3 or MLKL ablation (62, 199–201), suggesting that proper activation of caspase-8 is important for preventing necroptosis.
Interestingly, there are a few differences between RIPK3 and MLKL when deleted on a caspase-8 or FADD KO background in mice. Even though both RIPK3 and MLKL ablation rescues caspase-8−/− and Fadd−/− mice from embryonic lethality, caspase-8−/−Mlkl−/− double knockout mice develop morbidities such as lymphadenopathy and systemic autoimmune disease (related to autoimmune lymphoproliferative syndrome, ALPS) more rapidly than caspase-8−/−Ripk3−/− mice (22, 201). This phenotype was associated with an increased proliferation of CD3+B220+ T cells. Additional targeted deletion of caspase-8 in T cells or B cells emphasize the role of caspase-8 in adaptive immunity (202, 203).
Loss of RIPK1 leads to perinatal lethality with mice dying 1–3 days after birth. This lethality is only prevented when both the necroptotic and the apoptotic pathways are blocked (204–206). Loss of RIPK3 or MLKL delays lethality, arguing that RIPK1 can function as both an activator of RIPK3-dependent necroptosis, as well as keeping the RIPK3 machinery in check. This potentially highlights differences between the roles of RIPK1 as a kinase and as a scaffolding protein since RIPK1 kinase activity is required for TNFR1-induced necroptosis, but RIPK1 kinase-dead mice do not induce embryonic mortality (48, 198, 207). The kinase activity of RIPK1 is not only required for necroptosis but also appears to be required for optimal caspase-8 function after TAK1 and IKK inhibition (84, 180, 181, 208). However, to rescue Ripk1−/− -induced lethality completely, both RIPK3 and caspase-8 or FADD need to be deleted (83, 205, 209).
Furthermore, recent reports by Zhang et al., Newton et al., and Lalaoui et al. showed that mice harboring RIPK1 with a mutation in the caspase-8 cleavage site D325 (D324 in humans) die midgestation with signs of aberrant caspase-8 dependent cell death (85–87). This lethality was rescued by combined deletion of RIPK3 and FADD, again indicating that blockage of the necroptotic pathway alone is not sufficient to prevent death. Along these lines, cells from Ripk1D325A/D325ARipk3−/− mice showed activated caspase-3 and -7 after TNF treatment suggesting aberrant caspase-8-activation. Thus, caspase-8, in cooperation with cFLIPL, cleaves and inactivates RIPK1 at D325, limiting spontaneous RIPK1-induced cell death and explaining the crucial role caspase-8 has in maintaining homeostasis. Additionally, uncleaved RIPK1 can direct both RIPK3-mediated necroptosis and caspase-8-mediated apoptosis.
Caspase-8 cleaves GSDMD
The receptors TLR3, TLR4 and TNFR1 recruit the kinase RIPK1 after ligand binding. During certain conditions of pathogenic disturbance, RIPK1 can disassociate from the receptor and form a death complex with caspase-8. One example of such disturbance occurs during Yersinia delivery of the bacterial type III secretion system effector protein YopJ, which inhibits TAK1 and IKKβ kinases through acetylation of key phosphorylation sites (210). TAK1 and IKKβ mediate the phosphorylation of RIPK1 that is necessary to stabilize RIPK1 in an NF-κB-signaling state. Hence, when Yersinia YopJ inhibits TAK1 or IKKβ, RIPK1 activates Caspase-8.
Interestingly, TAK1-inhibition-triggered activation of caspase-8, either by YopJ or small molecule inhibitors, does not lead to inflammatory silent apoptosis but rather an inflammatory type of cell death involving GSDMD and NLRP3 (106, 180, 181, 211, 212). In this scenario, caspase-8 can directly cleave GSDMD at residue D276 into an active form, which, perhaps in cooperation with pannexin-1, leads to pore formation, efflux of K+-ions, NLRP3 activation, and processing and release of IL-1β and IL-18. This process could be viewed analogously to how RIPK3 and MLKL trigger NLRP3 activation. In a contrasting setting, in cells from Lyz2Cre+Tak1−/− mice, RIPK1 and caspase-8 can directly bind to and drive NLRP3 activation (211, 212), suggesting that TAK1 may restrict spontaneous NLRP3-activation. What distinguishes this from acute GSDMD-driven and pannexin1-driven K+-dependent activation of NLRP3 is still unclear. In this type of hybrid response, IL-1β and IL-18 can be cleaved both by caspase-8 and by caspase-8-controlled NLRP3-triggered caspase-1 (180). Low levels of cFLIPL can also be associated with these caspase-8-mediated processes (213). The participation of caspase-8 in GSDMD cleavage after TAK1/IKKß inhibition is a good example of how apoptotic and pyroptotic processed can become intertwined in a form of “apoptotic pyroptosis”.
Furthermore, caspase-8 is still capable of activating caspase-3 leading to membrane blebbing and DNA laddering (214). Hence, it appears that the cell death observed has aspects of both apoptosis and pyroptosis. This could suggest that the conventional categorization of cell death into either apoptosis or pyroptosis is not as strict as previously thought. Under some conditions cells seem to initiate more than one pathway at a time, suggesting that there is a fine-tuning of different cell death pathways depending on stimuli. This also highlights, together with the necroptosis/apoptosis axis downstream of RIPK1, the irreversibility of the cell death pathway once initiated. Activation of the RIPK1-caspase-8 cell death complex has the potential of triggering apoptosis, necroptosis and pyroptosis. And if either one or two of these pathways are blocked, the third pathway will still be able to kill the cell, as shown in Figure 4. This phenomenon emphasizes the complexity of immune cells dying in response to certain infections, possibly to limit the replicative niche as well as warn neighboring cells of impending danger, but also removing these cells from further participation in antimicrobial defenses.
Figure 4.
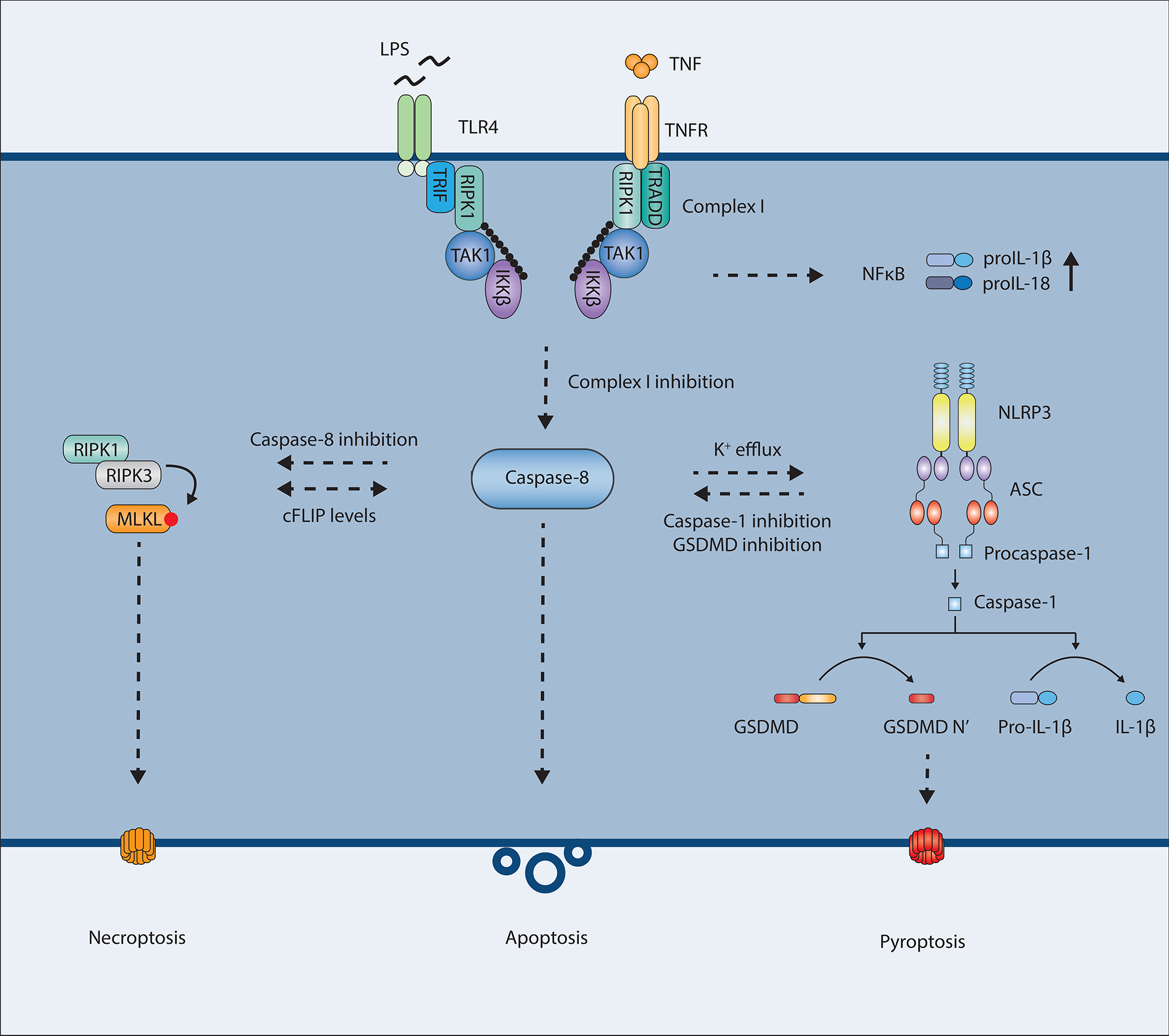
Activated caspase-8 at the focal point between cell death and inflammation.
Several important questions arise following the observation that caspase-8 leads to processing of GSDMD. Is it only RIPK1-dependent extrinsic apoptosis that leads to this pathway or would the same happen after TRAIL- or FasL-induced apoptosis that does not appear to have the same dependency on RIPK1? Is a specific complex required and, if so, is this due to increased structural stability of the active subunit and proximity of the substrate? One possible explanation to these questions could be whether or not the cells detect PAMPs or DAMPs during the induction of cell death. This could signal a need for triggering inflammation as a form of host-directed immunity in response to pathogens trying to shut down inflammatory signaling.
Intrinsic apoptosis leads to caspase-8 activation and IL-1β release
Recent reports by Vince et al., Chauhan et al., and Chen et al. suggested that BAX/BAK-mediated intrinsic apoptosis can also display crosstalk with NLRP3-mediated inflammation (106, 182, 183, 215). As is the case with caspase-8-mediated NLRP3 activation in the context of TAK1 inhibition, intrinsic activation also seems to trigger NLRP3 through pore formation and K+-efflux. In contrast to TAK1 inhibition, however, BAX/BAK-mediated activation of caspase-3 and -7 leads to cell death and NLRP3 activation independently of GSDMD. The levels of IL-1β release are lower than during TAK1 inhibition, but this could suggest that either caspase-3 and -7 are perhaps capable of cleaving caspase-8 leading into the same pathway, or that caspase-3 and -7, under certain conditions of intrinsic apoptosis with concomitant PAMP sensing, can trigger pore formation and K+-efflux through a different mechanism. Chen et al. propose that this happens through caspase-3-dependent cleavage of the channel-forming protein pannexin-1. Pannexin-1 has previously been described as an ATP channel required for release of “find-me” signals during apoptosis (216–218). The question then arises whether caspase-3 and -7 mediated K+-efflux and IL-1β release only occur during concomitant detection of PAMPs as in all of the above-mentioned studies the cells where frequently primed with LPS or TNF prior to stimulation.
One interesting observation is the fact that TAK1-inhibition mediated cell death is rapid, occurring within 2–3 hours, while BAX/BAK-mediated mitochondrial rupture leads to a slower cell death taking 6–8 hours or longer. This could explain the discrepancies between GSDMD-dependencies observed. All three reports observed abrogation of NLRP3-activation and caspase-1 cleavage when blocking K+ efflux, yet Vince et al. and Chauhan et al. still observed mature IL-1β and lactate dehydrogenase (LDH)-release (182, 183). It could be that GSDMD is critical for loss of membrane integrity during rapid onset of caspase-8-mediated cell death, while during intrinsic apoptotic processes, caspase-8 is cleaved at later time points when cell death is already progressing. Other pore-forming molecules or membrane-disrupting mechanisms downstream of caspase-3 and -7 could potentially compensate for the loss of GSDMD, such as pannexin-1 pores. Interestingly, the BAX/BAK studies differ in their attributed role of caspase-8 activating IL-1β. While Chauhan et al. find IL-1β processing and release to be completely dependent on caspase-8, Vince et al. do not.
Further crosstalk is exemplified in a recent paper by Rogers et al. where the authors show that GSDME and GSDMD both target the mitochondria resulting in cytochrome c release and caspase-3 activation (219). GSDME and GSDMD, cleaved downstream of caspase-3 and caspase-1/8/11 respectively, can therefor augment caspase-3 mediated cell death and act as a feedback loop and a back-up pathway, see Figure 5. The role of GSDME in immune cells is however still a bit unclear as reports linking GSDME to cell death has mainly been performed in non-immune cell types.
Figure 5.
Caspase-8 links cell death to inflammation.
Caspase-8 as a key decision maker for apoptosis, necroptosis and pyroptosis
As mentioned above, caspase-8 mediated cleavage of RIPK1 is a main regulatory step in RIPK1-mediated cell death. Two recent reports in Nature by Fritsch et al. and Newton et al. presented evidence that catalytically inactive caspase-8 can also directly nucleate ASC-specks (23, 24) and underscore the fundamental role for caspase-8 in determining type of cell death in various conditions. The authors showed that mouse mutations in the caspase-8 enzymatic site, Casp8C362S/C362S or Casp8C362A/C362A (Casp8C362mut), similar to Casp8−/− mice, is associated with embryonic lethality. Deficiency of caspase-8 could be rescued by Mlkl deletion leading to viable mice, however, Casp8C362mutMlkl−/− mice died perinatally. It was shown by both groups that overexpression of catalytically inactive caspase-8 caused ASC oligomerization and speck formation leading to caspase-1 dependent cleavage of GSDMD and IL-1β. In line with this the premature death seen in Casp8C362mutMlkl−/− mice was rescued in Casp8C362mutMlkl−/− Asc−/− and Casp8C362mutMlkl−/− Casp1−/− mice. Thus, loss of caspase-8 or its catalytic activity leads to MLKL-dependent necroptosis. When this is blocked catalytically inactive caspase-8 can still lead to ASC-dependent pyroptosis. Exactly how CASP8(C362mut) can nucleate ASC leading to cell death is still not fully known. This could be due to conformational changes in caspase-8 driving ASC speck formation, or that the catalytic activity of caspase-8 is constitutively inhibiting this oligomerization. Of note, the CASP8(C362mut)-driven pyroptosis appears to be restricted to innate immune cells, potentially as a safeguard to pathogenic inhibition of caspase-8. Overall, increasing amounts of evidence point to caspase-8 as a molecule able to direct cells towards different types of death.
The roles of caspase-8 during inflammasome activation
Even though caspase-8 is not playing a major role during inflammasome activation in WT cells, several recent reports have shown that if the inflammasome is activated but pyroptosis is blocked caspase-8 can act as a backup and drive cell death. This has been shown for several intracellular bacteria including L. pneumophilia and S. Typhimurium in the absence of either caspase-1 or GSDMD, or in the presence of a proteolytically inactive caspase-1 in response to nigericin (152, 153, 220, 221). Biochemically, this has been proposed to occur through interaction between the ASC PYD domain and the caspase-8 DED domain (222, 223), or through the adaptor molecule FADD (220). There is some discordance about what type of cell death is initiated downstream of caspase-8 activation in this case. In the case of L. pneumophilia and S. Typhimurium (which triggers the NLRC4 inflammasome) it appears to be an apoptotic form of cell death, while in the case of nigericin (which triggers the NLRP3 inflammasome) it appears to be a lytic form of cell death, potentially driven by GSDME. Another question that arises is why caspase-8 is not activated after inflammasome activation in WT cells that are undergoing pyroptosis (221). Even cells treated with glycine to prevent rupture and cell death did not show any caspase-8 activation.
It seems that caspase-8 can affect the NLRP3 inflammasome at the level of priming, activation, and post-assembly. Caspase-8 can control optimal NF-κB signaling and, thus, influence upregulation of the required components of the inflammasome such as NLRP3 and pro-IL-1β (158, 214, 224, 225). Caspase-8 can also activate the NLRP3 inflammasome through K+ efflux, as well as trigger inflammasome activation in response to β-glucans and C. albicans downstream of Dectin-1 (226), potentially through binding to ASC through its DED domain (227). And it can get recruited to the already formed ASC speckle, ready to kick in if other parts are blocked.
In addition to this, caspase-8 can both activate caspase-1 as well as process IL-1β directly independently of caspase-1 (214, 226, 228–231). Treating cells overexpressing caspase-8 and pro-IL-1β with TLR3 and TLR4 ligands showed that caspase-8 could cleave pro-IL-1β at the same site as caspase-1 (231). Finally, caspase-8 also appear to have a role in T-cells by restricting CD8 T-cell hyperaccumulation during murine cytomegalovirus infection (232). Hence, the previous categorization of caspase-8 as mainly an apoptotic caspase is outdated as caspase-8 has several key roles in inflammation and immunity.
Caspase-8 and host defenses against pathogens
Several labs have now shown that caspase-8 can become activated by distinct pathways previously considered independent of caspase-8. Activated caspase-8 can then drive cell death through caspase-3 or GSDMD, and IL-1β-induced inflammation directly or through NLRP3. This puts caspase-8 at the focal point in a complicated crosstalk between different cell death pathways. This could be a way to combat potentially dangerous situations that require activation of inflammation, recruitment of immune cells and clearance of pathogens or damaged cells.
Caspase-8 is also involved in effector-triggered immunity (Many pathogens attempt to circumvent a robust inflammatory response by secreting effector molecules that modify and block host proteins and thus interfere with different cellular processes. Bacteria such as Yersinia and Salmonella use secretion systems to deliver these types of effector proteins to evade an immune response. However, the effectors themselves, or the modification of host proteins by the effectors, can be recognized as pathological by the innate immune system. It is clear that this is an important defense mechanism and a major driving factor in the constant arms race between host and pathogen.
Caspase-8 and cell death are activated in response to modification of MAPKK, IKKβ and TAK1 by Yersinia bacteria, as an apparent back-up mechanism to alert the host of pathogenic activity towards key signaling proteins. Other pathogens can also activate caspase-8 (233). There are reports that pathogens such as Pseudomonas, Vibrio, Porphyromonas, and enteroviruses also target TAK1(234–237). Additionally, as mentioned before, inflammasomes are critical in host defenses against pathogens and are an important target for many microbes to suppress. Reports have suggested that caspase-1 is inhibited by infection with Poxvirus, influenza A virus, and Vibrio parahaemolyticus (238, 239). And induction of intrinsic apoptosis is important during several viral infections as shown by the fact that BAX-deficient mice are susceptible to vesicular stomatitis virus and encephalomyocarditis virus infection (240).
Caspase-8 and influence on the NF-κB system and cytokine expression
Recent work has increased our knowledge about caspase-8 and highlighted its functions as a key player in regulating inflammatory responses. One of the less recognized roles of caspase-8 is in NF-κB signaling where caspase-8 appears to be required for optimal inflammatory induction. It has been shown in vitro that cells from Ripk3−/−Caspase-8−/− as well as Mlkl−/−Fadd−/− and Ripk3−/−Fadd−/− mice display a reduced cytokine response to TLR ligands (158, 201, 214, 241, 242). This also appears to be the case in vivo as LPS treatment in Caspase-8LysMcre mice leads to a dampened response (243), and Ripk3−/−Caspase-8−/− mice were protected in a mouse model of fatal liver damage and endotoxic shock after LPS and D-(+) galactosamine treatment (83). Furthermore, in a study from 2016, Philip et al. showed that pan-caspase inhibition with zVAD-fmk, or specific inhibition of caspase-8 with IETD-fmk, in Ripk3−/− cells also resulted in reduced NF-κB signaling (15). This would suggest that it is not the scaffolding properties of caspase-8 that are required for optimal proinflammatory signaling. In the same study, they made a non-cleavable caspase-8 knock-in mouse (Casp8D387A/D387A) that was unable to induce apoptosis, yet was still capable of restricting necroptosis in a heterodimer with cFLIP (15). These mice showed no defect in TLR signaling, indicating that cleavage of caspase-8 at D387 is not required for optimal NF-κB activation. Another recent paper from the same lab showed that caspase-8 controls c-Rel activity and that Ripk3−/−Caspase-8−/− mice were unable to control Toxoplasma gondii infection due to diminished IL-12 release (244). In contrast, caspase-8 appears to have a scaffolding role during TRAIL-induced pro-inflammatory cytokine expression (245, 246).
Impact on disease
Acute inflammatory responses are essential weapons of our immune system. Inflammasomes are increasingly recognized as critical orchestrators of this defense, and the secretion of the cytokines IL-1β and IL-18 is necessary for efficiently controlling and clearing many microbial infections, as is the induction of different cell death pathways. However, because these pathways are so potent, inappropriate activation of the inflammasomes can lead to autoinflammation and autoimmune diseases (134–137) such as rheumatoid arthritis, type 1 diabetes, gout (247), Alzheimer’s disease (141) and many others. IL-1β functions by recruiting neutrophils to the site of infection, promoting cell adhesion, and activating TH17 cells, while IL-18 activates natural killer (NK) cells and T cells and makes them secrete IFN-γ. Too large of a response can cause tissue damage and promote autoimmunity. Of note, anti-IL-1 therapy such as IL-1 receptor antagonist anakinra is proving to be successful in a variety of diseases (248), hence the value of studying intervention target candidates in related inflammatory pathways is indisputable. As proposed in this review, caspase-8 is a central player in many of these pathways. Caspase-8 inhibitors have been proposed as potential treatments for diseases such as neurodegenerative conditions and cancer (249–253). However, such studies need to be carefully planned and evaluated, as caspase inhibitors are not always specific, and caspase-8 inhibition could trigger necroptosis. The latter mechanism may also be beneficial in some cases, as exemplified by applying caspase inhibitors together with SMAC mimetics to treat acute myeloid leukemia (254).
Sepsis is an example of excessive inflammation causing tissue damage and multiple organ failure. Sepsis and septic shock are lethal complications of bacterial infections responsible for around 250 000 deaths per year in the USA (255). As mentioned above, inflammasome-driven pyroptosis and cytokine processing can be a crucial defense against pathogenic infections leading to release of proinflammatory cytokines and elimination of the replicative niche for many bacteria and viruses. However, it also plays a role in sepsis, and loss of GSDMD confers protection in several mouse models of sepsis (99, 256). Chemical inhibitors of GSDMD are now being investigated as possible treatments towards sepsis and other inflammatory conditions (257).
There is a plethora of evidence suggesting NLRP3 is involved in many autoinflammatory diseases. NLRP3 can, for instance, contribute to atherosclerosis in response to cholesterol crystals (258) and through the type of diet we eat can affect the epigenomic landscape leading to transcriptional reprogramming of our immune cells (124). Its importance in disease is perhaps best reflected by the autoinflammatory condition cryopyrin-associated periodic syndrome (CAPS). This potentially lethal disease is caused by activating mutations in the NLRP3 gene leading to overproduction of IL-1β (259). Better understanding of different pathways leading to activation of NLRP3 and its mechanism of activation are critical to better treat many inflammatory diseases. Treatments options directly targeting NLRP3 are also in development (260).
Furthermore, a number of autoimmune diseases caused by aberrant signaling through death receptors show similarity to the kind of autoinflammation caused by dysfunctional NLRP3 signaling and IL-1β production. For instance, the autoinflammatory disease TNF receptor-associated periodic syndrome (TRAPS) leads to heightened TNF and IL-1β cytokine production. Exactly how IL-1β is generated in this case is unclear, but patients with TRAPS are often treated with IL-1β blockade leading to amelioration of symptoms. The disease is caused by a mutation in the TNFR1 gene leading to abnormal folding and overexpression in the ER and mitochondrial ROS production (259, 261, 262).
Interestingly, there have recently been multiple reports of patients with mutations in the caspase-8 cleavage site of RIPK1 (87, 263). The heterozygous mutations D324N, D324H, D324Y, D324V and D324H have all been shown to prevent caspase-cleavage of RIPK1 resulting in early-onset periodic fever syndrome and severe lymphadenopathy. This condition caused by mutations in RIPK1 D324 has been termed cleavage-resistant RIPK1-induced autoinflammatory syndrome. Patients experienced fevers occurring every 2–4 weeks accompanied with symptoms of extreme chills, severe headaches, and hallucinations. Cells harboring the mutant RIPK1 displayed increased sensitivity to TNF leading to increased S166 phosphorylation of RIPK1, activation of RIPK3 and cleavage of caspase-8 and caspase-3 (85–87, 263). Heterozygous D325A mice also showed elevated levels of TNF and IL-1β in response to non-lethal LPS injection. Unexpectedly, patients did not respond to the TNF antagonist etanercept or the IL-1 receptor antagonist anakinra, but treatment with the IL-6 receptor antagonist tocilizumab strongly reduced symptoms (87, 263). RIPK1 itself is a molecule that has attracted intense interest for anti-inflammatory therapy (264).
Another example is the autoinflammatory disease X-linked lymphoproliferative disease 2 (XLP2) (265). XLP2 results in hemophagocytic lymphohistiocytosis (HLH) in 60–90% of patients, characterized by macrophage hyperactivation, increased levels of IL-18, and similar symptoms to those observed in CAPS patients. The disease is caused by mutations in the X-linked inhibitor of apoptosis (XIAP) gene leading to loss of function. XIAP is involved in controlling ripoptosome formation, and its absence leads to increased cell death and IL-1β secretion in macrophages and dendritic cells (266–268).
Thus, there are many examples of dysregulation of the cell death machinery leading to inflammation and disease. We hypothesize that in some of these cases the underlying mechanism may follow the same crosstalk that we have seen previously involving caspase-8, either directing NLRP3 activation through GSDMD induced pore-formation or by processing IL-1β directly.
Deregulation of apoptosis has also been linked to the development of cancer. Mutations in both the intrinsic and the extrinsic apoptotic pathway contribute to cancer through inhibition of cell death. Additionally, studies by Hakem et al. and Liccardi et al. linked mutations in caspase-8 to cancer through aberrations in chromatin stability (269, 270), an observation that might help explain the contribution of caspase-8 in many other human cancers (271–275). TAK1 inhibitors, that presumably would trigger caspase-8 and subsequent cell death, are also cancer drug candidates (276).
Patients with caspase-8 deficiency, although rare, suffer from a variety of symptoms. Siblings diagnosed with autoimmune lymphoproliferative syndrome (ALPS) were shown to have defects in caspase-8 (277). Another set of siblings presented with life-threatening end organ lymphocytic infiltration in their late thirties had homozygous mutations in CASP8 (278). More recent classifications suggest human ALPS is most often associated with mutations in Fas, and caspase-8 mutants (often called CEDS, caspase-8 deficiency state) have additional defects such as primary immunodeficiency (277, 279, 280). Furthermore, three patients with early onset inflammatory bowel disease had mutations in CASP8 (281). Cells from these patients showed increased IL-1β secretion in response to LPS. Mice completely lacking caspase-8 die prenatally whereas caspase-8 mutant humans do not - although mutations are different, discrepancies could also be explained by humans also expressing the caspase-8 homolog caspase-10. Caspase-10 has been reported to be involved in extrinsic apoptosis and may share some of the same substrates as caspase-8. Other nonoverlapping functions also exist, however, and caspase-10 has been proposed to negatively regulate caspase-8 in certain scenarios (282, 283). Mice do not express caspase-10 and could in theory be more dependent on the function of caspase-8 in cell death and development. In any case, mechanisms related to caspase-8 function are likely playing key roles in a large variety of human diseases.
Conclusions
As we have summarized in this review, caspase-8 is an important player in cell death and inflammation. Caspase-8 can directly cleave caspase-3 to induce apoptosis or GSDMD to induce pyroptosis, while also cleaving RIPK1 and RIPK3 to inhibit necroptosis. Moreover, due to its potential position both upstream and downstream of inflammasome activation caspase-8 has a critical role in inflammation. It can both directly and indirectly trigger inflammasome activation, while also itself become activated downstream of inflammasomes when caspase-1 or GSDMD are blocked. Alternatively, in conditions where caspase-1 activation is limited, caspase-8 can itself cleave IL-1β. This could serve to fine-tune inflammation when only low levels of inflammation are required. And when pathogenic disturbances attempt to block inflammation, caspase-8 can trigger effector-mediated immunity. Finally, it can itself mediate inflammatory cytokine transcription without inducing cell death. We can conclude that caspase-8 is a multifunctional caspase with involvement in many different pathways resulting in essential roles in health and disease.
Acknowledgements
We thank Melanie Trombly for suggestions and critical reading of the manuscript. Our work has been supported by funding from National Institutes of Health (AI146855 and AI128547), The Norwegian Cancer Society (B05035/001) and the Research Council of Norway (Center of Excellence Funding Scheme 223255/F50).
Footnotes
The authors declare no competing financial interests.
References
- 1.Jorgensen I, Rayamajhi M, Miao EA, Programmed cell death as a defence against infection. Nat. Publ. Gr 17, 151–164 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ashida H, Mimuro H, Ogawa M, Kobayashi T, Sanada T, Kim M, Sasakawa C, Cell death and infection : A double-edged sword for host and pathogen survival. 195, 931–942 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lamkanfi M, Dixit VM, Manipulation of host cell death pathways during microbial infections. Cell Host Microbe. 8 (2010), pp. 44–54. [DOI] [PubMed] [Google Scholar]
- 4.Alnemri ES, Livingston DJ, Nicholson DW, Salvesen G, Thornberry NA, Wong WW, Yuan J, Human ICE/CED-3 Protease Nomenclature. Cell. 87, 171 (1996). [DOI] [PubMed] [Google Scholar]
- 5.Man SM, Kanneganti T-D, Converging roles of caspases in inflammasome activation, cell death and innate immunity. Nat. Rev. Immunol 16, 7–21 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Green DR, Means to an End : Apoptosis and Other Cell Death Mechanisms by (2010; isbn: 9780879698874). [Google Scholar]
- 7.Creagh EM, Caspase crosstalk: Integration of apoptotic and innate immune signalling pathways. Trends Immunol. 35 (2014), pp. 631–640. [DOI] [PubMed] [Google Scholar]
- 8.Gonzalez Ramirez ML, Poreba M, Snipas SJ, Groborz K, Drag M, Salvesen GS, Extensive peptide and natural protein substrate screens reveal that mouse caspase-11 has much narrower substrate specificity than caspase-1. J. Biol. Chem 293, 7058–7067 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Poreba M, Groborz K, Navarro M, Snipas SJ, Drag M, Salvesen GS, Caspase selective reagents for diagnosing apoptotic mechanisms. Cell Death Differ., 229–244 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boucher D, Monteleone M, Coll RC, Chen KW, Ross CM, Teo JL, Gomez GA, Holley CL, Bierschenk D, Stacey KJ, Yap AS, Bezbradica JS, Schroder K, Caspase-1 self-cleavage is an intrinsic mechanism to terminate inflammasome activity. J. Exp. Med 215, 827–840 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Muzio M, Stockwell BR, Stennicke HR, Salvesen GS, Dixit VM, An induced proximity model for caspase-8 activation. J. Biol. Chem (1998), doi: 10.1074/jbc.273.5.2926. [DOI] [PubMed] [Google Scholar]
- 12.Martin DA, Siegel RM, Zheng L, Lenardo MJ, Membrane oligomerization and cleavage activates the caspase-8 (FLICE/MACHα1) death signal. J. Biol. Chem (1998), doi: 10.1074/jbc.273.8.4345. [DOI] [PubMed] [Google Scholar]
- 13.Yang X, Chang HY, Baltimore D, Autoproteolytic activation of pro-caspases by oligomerization. Mol. Cell (1998), doi: 10.1016/S1097-2765(00)80032-5. [DOI] [PubMed] [Google Scholar]
- 14.Kang T-B, Oh G-S, Scandella E, Bolinger B, Ludewig B, Kovalenko A, Wallach D, Mutation of a Self-Processing Site in Caspase-8 Compromises Its Apoptotic but Not Its Nonapoptotic Functions in Bacterial Artificial Chromosome-Transgenic Mice. J. Immunol 181, 2522–2532 (2008). [DOI] [PubMed] [Google Scholar]
- 15.Philip NH, DeLaney A, Peterson LW, Santos-Marrero M, Grier JT, Sun Y, Wynosky-Dolfi MA, Zwack EE, Hu B, Olsen TM, Rongvaux A, Pope SD, López CB, Oberst A, Beiting DP, Henao-Mejia J, Brodsky IE, Activity of Uncleaved Caspase-8 Controls Anti-bacterial Immune Defense and TLR-Induced Cytokine Production Independent of Cell Death. PLoS Pathog. 12, 1–30 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chang DW, Xing Z, Pan Y, Algeciras-Schimnich A, Barnhart BC, Yaish-Ohad S, Peter ME, Yang X, C-FLIPLis a dual function regulator for caspase-8 activation and CD95-mediated apoptosis. EMBO J. (2002), doi: 10.1093/emboj/cdf356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Micheau O, Thome M, Schneider P, Holler N, Tschopp J, Nicholson DW, Briand C, Grütter MG, The long form of FLIP is an activator of caspase-8 at the Fas death-inducing signaling complex. J. Biol. Chem (2002), doi: 10.1074/jbc.M206882200. [DOI] [PubMed] [Google Scholar]
- 18.Oberst A, Dillon CP, Weinlich R, Mccormick LL, Pop C, Hakem R, Salvesen GS, Green DR, Catalytic activity of the caspase-8-FLIPL complex inhibits RIPK3- dependent necrosis. Nature. 471, 363–367 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lin Y, Devin A, Rodriguez Y, Liu ZG, Cleavage of the death domain kinase RIP by Caspase-8 prompts TNF-induced apoptosis. Genes Dev. 13, 2514–2526 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Feng S, Yang Y, Mei Y, Ma L, Zhu D. e., Hoti N, Castanares M, Wu M, Cleavage of RIP3 inactivates its caspase-independent apoptosis pathway by removal of kinase domain. Cell. Signal 19, 2056–2067 (2007). [DOI] [PubMed] [Google Scholar]
- 21.Chang DW, Xing Z, Capacio VL, Peter ME, Yang X, Interdimer processing mechanism of procaspase-8 activation. EMBO J. (2003), doi: 10.1093/emboj/cdg414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Alvarez-Diaz S, Dillon CP, Lalaoui N, Tanzer MC, Rodriguez DA, Lin A, Lebois M, Hakem R, Josefsson EC, O’Reilly LA, Silke J, Alexander WS, Green DR, Strasser A, The Pseudokinase MLKL and the Kinase RIPK3 Have Distinct Roles in Autoimmune Disease Caused by Loss of Death-Receptor-Induced Apoptosis. Immunity. 45, 513–526 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Newton K, Wickliffe KE, Maltzman A, Dugger DL, Reja R, Roose-girma M, Modrusan Z, Sagolla MS, Webster JD, Dixit VM, Activity of caspase-8 determines plasticity between cell death pathways. Nature (2019), doi: 10.1038/s41586-019-1752-8. [DOI] [PubMed] [Google Scholar]
- 24.Fritsch M, Günther SD, Schwarzer R, Albert M, Schorn F, Werthenbach JP, Schiffmann LM, Stair N, Stocks H, Seeger JM, Lamkanfi M, Krönke M, Pasparakis M, Kashkar H, Caspase-8 is the molecular switch for apoptosis, necroptosis and pyroptosis. Nature. 575, 683–687 (2019). [DOI] [PubMed] [Google Scholar]
- 25.Hughes MA, Harper N, Butterworth M, Cain K, Cohen GM, MacFarlane M, Reconstitution of the Death-Inducing Signaling Complex Reveals a Substrate Switch that Determines CD95-Mediated Death or Survival. Mol. Cell (2009), doi: 10.1016/j.molcel.2009.06.012. [DOI] [PubMed] [Google Scholar]
- 26.Song J, Tan H, Perry AJ, Akutsu T, Webb GI, Whisstock JC, Pike RN, PROSPER: An Integrated Feature-Based Tool for Predicting Protease Substrate Cleavage Sites. PLoS One. 7, 1–23 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Porȩba M, Strózyk A, Salvesen GS, Dra̧ g M, Caspase substrates and inhibitors. Cold Spring Harb. Perspect. Biol (2013), doi: 10.1101/cshperspect.a008680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Van De Craen M, Van Loo G, Declercq W, Schotte P, Van Den Brande I, Mandruzzato S, Van Der Bruggen P, Fiers W, Vandenabeele P, Molecular cloning and identification of murine caspase-8. J. Mol. Biol 284, 1017–1026 (1998). [DOI] [PubMed] [Google Scholar]
- 29.Galluzzi L, Vitale I, Molecular mechanisms of cell death : recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 25, 486–541 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tang D, Kang R, Vanden Berghe T, Vandenabeele P, Kroemer G, The molecular machinery of regulated cell death. Cell Res. (2019), doi: 10.1038/s41422-019-0164-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Speir M, Lawlor KE, Glaser SP, Abraham G, Chow S, Vogrin A, Schulze KE, Schuelein R, OReilly LA, Mason K, Hartland EL, Lithgow T, Strasser A, Lessene G, Huang DCS, Vince JE, Naderer T, Eliminating Legionella by inhibiting BCL-XL to induce macrophage apoptosis. Nat. Microbiol 1, 1–9 (2016). [DOI] [PubMed] [Google Scholar]
- 32.Ebert G, Allison C, Preston S, Cooney J, Toe JG, Stutz MD, Ojaimi S, Baschuk N, Nachbur U, Torresi J, Silke J, Begley CG, Pellegrini M, Eliminating hepatitis B by antagonizing cellular inhibitors of apoptosis. Proc. Natl. Acad. Sci 112, 5803–5808 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Czabotar PE, Lessene G, Strasser A, Adams JM, Control of apoptosis by the BCL-2 protein family: Implications for physiology and therapy. Nat. Rev. Mol. Cell Biol 15 (2014), pp. 49–63. [DOI] [PubMed] [Google Scholar]
- 34.Pihán P, Carreras-Sureda A, Hetz C, BCL-2 family: integrating stress responses at the ER to control cell demise. Cell Death Differ. 24, 1478–1487 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Roos WP, Thomas AD, Kaina B, DNA damage and the balance between survival and death in cancer biology. Nat. Rev. Cancer. 16, 20–33 (2015). [DOI] [PubMed] [Google Scholar]
- 36.Vitale I, Manic G, De Maria R, Kroemer G, Galluzzi L, DNA Damage in Stem Cells. Mol. Cell 66 (2017), pp. 306–319. [DOI] [PubMed] [Google Scholar]
- 37.Nunez G, London L, Hockenbery D, Alexander M, McKearn JP, Korsmeyer SJ, Deregulated Bcl-2 gene expression selectively prolongs survival of growth factor-deprived hemopoietic cell lines. J Immunol 144, 3602–3610 (1990). [PubMed] [Google Scholar]
- 38.Brumatti G, Salmanidis M, Ekert PG, Crossing paths: interactions between the cell death machinery and growth factor survival signals. Cell Mol Life Sci. 67, 1619–1630 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tait SW, Green DR, Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol. 11, 621–632 (2010). [DOI] [PubMed] [Google Scholar]
- 40.Galluzzi L, Kepp O, Kroemer G, Mitochondrial regulation of cell death: a phylogenetically conserved control. Microb. Cell 3, 101–108 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Moldoveanu T, Follis AV, Kriwacki RW, Green DR, Many players in BCL-2 family affairs. Trends Biochem. Sci 39 (2014), pp. 101–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shamas-Din A, Kale J, Leber B, Andrews DW, Mechanisms of Action of Bcl-2 Family Proteins. Cold Spring Harb Perspect Biol. 5, 1–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X, Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 91, 479–489 (1997). [DOI] [PubMed] [Google Scholar]
- 44.McArthur K, Whitehead LW, Heddleston JM, Li L, Padman BS, Oorschot V, Geoghegan ND, Chappaz S, Davidson S, Chin HS, Lane RM, Dramicanin M, Saunders TL, Sugiana C, Lessene R, Osellame LD, Chew TL, Dewson G, Lazarou M, Ramm G, Lessene G, Ryan MT, Rogers KL, Van Delft MF, Kile BT, BAK/BAX macropores facilitate mitochondrial herniation and mtDNA efflux during apoptosis. Science (80-. ). (2018), doi: 10.1126/science.aao6047. [DOI] [PubMed] [Google Scholar]
- 45.Riley JS, Quarato G, Cloix C, Lopez J, O’Prey J, Pearson M, Chapman J, Sesaki H, Carlin LM, Passos JF, Wheeler AP, Oberst A, Ryan KM, Tait SW, Mitochondrial inner membrane permeabilisation enables mt DNA release during apoptosis. EMBO J. (2018), doi: 10.15252/embj.201899238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wilson NS, Dixit V, Ashkenazi A, Death receptor signal transducers: Nodes of coordination in immune signaling networks. Nat. Immunol 10 (2009), pp. 348–355. [DOI] [PubMed] [Google Scholar]
- 47.Li H, Zhu H, Xu CJ, Yuan J, Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 94, 491–501 (1998). [DOI] [PubMed] [Google Scholar]
- 48.Berger SB, Kasparcova V, Hoffman S, Dare L, Schaeffer M, Capriotti C, Cook M, Finger J, Hughes-earle A, Harris A, Kaiser WJ, Mocarski ES, Gough PJ, Berger SB, Kasparcova V, Hoffman S, Swift B, Dare L, Schaeffer M, Capriotti C, Cook M, Finger J, Hughes-earle A, Harris PA, Kaiser WJ, Mocarski ES, Bertin J, Gough PJ, Cutting Edge: RIP1 Kinase Activity Is Dispensable for Normal Development but Is a Key Regulator of Inflammation in SHARPIN-Deficient Mice. J Immunol 192, 5476–5480 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Panayotova-Dimitrova D, Feoktistova M, Ploesser M, Kellert B, Hupe M, Horn S, Makarov R, Jensen F, Porubsky S, Schmieder A, Zenclussen AC, Marx A, Kerstan A, Geserick P, He YW, Leverkus M, CFLIP Regulates Skin Homeostasis and Protects against TNF-Induced Keratinocyte Apoptosis. Cell Rep. 5, 397–408 (2013). [DOI] [PubMed] [Google Scholar]
- 50.Kumari S, Redouane Y, Lopez-Mosqueda J, Shiraishi R, Romanowska M, Lutzmayer S, Kuiper J, Martinez C, Dikic I, Pasparakis M, Ikeda F, Sharpin prevents skin inflammation by inhibiting TNFR1-induced keratinocyte apoptosis. Elife. 3, 1–20 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.a Rickard J, Anderton H, Etemadi N, Nachbur U, Darding M, Peltzer N, Lalaoui N, Lawlor KE, Vanyai H, Hall C, Bankovacki A, Gangoda L, Wong WW-L, Corbin J, Huang C, Mocarski ES, Murphy JM, Alexander WS, Voss AK, Vaux DL, Kaiser WJ, Walczak H, Silke J, TNFR1-dependent cell death drives inflammation in Sharpin-deficient mice. Elife. 3, 1–23 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stanger BZ, Leder P, Lee T-H, Kim E, Seed B, RIP: A novel protein containing a death domain that interacts with Fas/APO-1 (CD95) in yeast and causes cell death. Cell. 81, 513–523 (1995). [DOI] [PubMed] [Google Scholar]
- 53.Chinnaiyan AM, O’Rourke K, Tewari M, Dixit VM, FADD, a novel death domain-containing protein, interacts with the death domain of fas and initiates apoptosis. Cell. 81, 505–512 (1995). [DOI] [PubMed] [Google Scholar]
- 54.Hsu H, Xiong J, Goeddel DV, The TNF receptor 1-associated protein TRADD signals cell death and NF-κB activation. Cell. 81, 495–504 (1995). [DOI] [PubMed] [Google Scholar]
- 55.Grimm S, Stanger BZ, Leder P, RIP and FADD: two “death domain”-containing proteins can induce apoptosis by convergent, but dissociable, pathways. Proc. Natl. Acad. Sci 93, 10923–10927 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kelliher MA, Grimm S, Ishida Y, Kuo F, Stanger BZ, Leder P, The Death Domain Kinase RIP Mediates the TNF-Induced NF-κB Signal. Immunity. 8, 297–303 (1998). [DOI] [PubMed] [Google Scholar]
- 57.Varfolomeev EE, A potential mechanism of “cross-talk” between the p55 tumor necrosis factor receptor and Fas/APO1: proteins binding to the death domains of the two receptors also bind to each other. J. Exp. Med 183, 1271–1275 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Boldin MP, Varfolomeev EE, Pancer Z, Mett IL, Camonis JH, Wallach D, A Novel Protein That Interacts with the Death Domain of Fas/APO1 Contains a Sequence Motif Related to the Death Domain. J. Biol. Chem 270, 7795–7798 (1995). [DOI] [PubMed] [Google Scholar]
- 59.Boldin MP, Goncharov TM, V Goltseve Y, Wallach D, Involvement of MACH, a Novel MORT1/FADD-Interacting Protease, in Fas/APO-1- and TNF Receptor–Induced Cell Death. Cell. 85, 803–815 (1996). [DOI] [PubMed] [Google Scholar]
- 60.Srinivasula SM, Ahmad M, Fernandes-Alnemri T, Litwack G, Alnemri ES, Molecular ordering of the Fas-apoptotic pathway: The Fas/APO-1 protease Mch5 is a CrmA-inhibitable protease that activates multiple Ced-3/ICE-like cysteine proteases. Proc. Natl. Acad. Sci 93, 14486–14491 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Muzio M, Chinnaiyan AM, Kischkel FC, O’Rourke K, Shevchenko A, Ni J, Scaffidi C, Bretz JD, Zhang M, Gentz R, Mann M, Krammer PH, Peter ME, Dixit VM, FLICE A Novel FADD-Homologous ICE/CED-3–like Protease, Is Recruited to the CD95 (Fas/APO-1) Death-Inducing Signaling Complex. Cell. 85, 817–827 (1996). [DOI] [PubMed] [Google Scholar]
- 62.Yeh WC, De La Pompa JL, McCurrach ME, Shu HB, Elia AJ, Shahinian A, Ng M, Wakeham A, Khoo W, Mitchell K, El-Deiry WS, Lowe SW, Goeddel DV, Mak TW, FADD: Essential for embryo development and signaling from some, but not all, inducers of apoptosis. Science. 279, 1954–1958 (1998). [DOI] [PubMed] [Google Scholar]
- 63.Kischkel FC, Hellbardt S, Behrmann I, Germer M, Pawlita M, Krammer PH, Peter ME, Cytotoxicity-dependent APO-1 (Fas/CD95)-associated proteins form a death-inducing signaling complex (DISC) with the receptor. EMBO J. 14, 5579–5588 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tewari M, Beidler DR, Dixit VM, CrmA-inhibitable Cleavage of the 70-kDa Protein Component of the U1 Small Nuclear Ribonucleoprotein during Fas- and Tumor Necrosis Factor-induced Apoptosis. J. Biol. Chem 270, 18738–18741 (1995). [DOI] [PubMed] [Google Scholar]
- 65.Tewari M, Quan LT, O’Rourke K, Desnoyers S, Zeng Z, Beidler DR, Poirier GG, Salvesen GS, Dixit VM, Yama/CPP32β, a mammalian homolog of CED-3, is a CrmA-inhibitable protease that cleaves the death substrate poly(ADP-ribose) polymerase. Cell. 81, 801–809 (1995). [DOI] [PubMed] [Google Scholar]
- 66.Martin SJ, O’Brien GA, Nishioka WK, McGahon AJ, Mahboubi A, Saido TC, Green DR, Proteolysis of Fodrin (Non-erythroid Spectrin) during Apoptosis. J. Biol. Chem 270, 6425–6428 (1995). [DOI] [PubMed] [Google Scholar]
- 67.Porter AG, Jänicke RU, Emerging roles of caspase-3 in apoptosis. Cell Death Differ. 6, 99–104 (1999). [DOI] [PubMed] [Google Scholar]
- 68.Stennicke HR, Jürgensmeier JM, Shin H, Deveraux Q, Wolf BB, Yang X, Zhou Q, Ellerby HM, Ellerby LM, Bredesen D, Green DR, Reed JC, Froelich CJ, Salvesen GS, Pro-caspase-3 is a major physiologic target of caspase-8. J. Biol. Chem 273, 27084–27090 (1998). [DOI] [PubMed] [Google Scholar]
- 69.Goltsev YV, Kovalenko AV, Arnold E, Varfolomeev EE, Brodianskii VM, Wallach D, CASH, a novel caspase homologue with death effector domains. J. Biol. Chem (1997), doi: 10.1074/jbc.272.32.19641. [DOI] [PubMed] [Google Scholar]
- 70.Han DKM, Chaudhary PM, Wright ME, Friedman C, Trask BJ, Riedel RT, Baskin DG, Schwartz SM, Hood L, MRIT, a novel death-effector domain-containing protein, interacts with caspases and BclXL and initiates cell death. Proc. Natl. Acad. Sci. U. S. A (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hu S, Vincenz C, Ni J, Gentz R, Dixit VM, I-FLICE, a novel inhibitor of tumor necrosis factor receptor-1- and CD- 95-induced apoptosis. J. Biol. Chem (1997), doi: 10.1074/jbc.272.28.17255. [DOI] [PubMed] [Google Scholar]
- 72.Inohara N, Koseki T, Hu Y, Chen S, Núñez G, CLARP, a death effector domain-containing protein interacts with caspase-8 and regulates apoptosis. Proc. Natl. Acad. Sci. U. S. A (1997), doi: 10.1073/pnas.94.20.10717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Irmler M, Thome M, Hahne M, Schneider P, Hofmann K, Steiner V, Bodmer JL, Schröter M, Burns K, Mattmann C, Rimoldi D, French LE, Tschopp J, Inhibition of death receptor signals by cellular FLIP. Nature (1997), doi: 10.1038/40657. [DOI] [PubMed] [Google Scholar]
- 74.Shu HB, Halpin DR, Goeddel DV, Casper is a FADD- and caspase-related inducer of apoptosis. Immunity (1997), doi: 10.1016/S1074-7613(00)80450-1. [DOI] [PubMed] [Google Scholar]
- 75.Srinivasula SM, Ahmad M, Ottilie S, Bullrich F, Banks S, Wang Y, Fernandes-Alnemri T, Croce CM, Litwack G, Tomaselli KJ, Armstrong RC, Alnemri ES, FLAME-1, a novel FADD-like anti-apoptotic molecule that regulates Fas/TNFR1-induced apoptosis. J. Biol. Chem (1997), doi: 10.1074/jbc.272.30.18542. [DOI] [PubMed] [Google Scholar]
- 76.Kavuri SM, Geserick P, Berg D, Dimitrova DP, Feoktistova M, Siegmund D, Gollnick H, Neumann M, Wajant H, Leverkus M, Cellular FLICE-inhibitory Protein (cFLIP) isoforms block CD95- and TRAIL death receptor-induced gene induction irrespective of processing of caspase-8 or cFLIP in the death-inducing signaling complex. J. Biol. Chem 286, 16631–16646 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ueffing N, Keil E, Freund C, Kühne R, Schulze-Osthoff K, Schmitz I, Mutational analyses of c-FLIP R, the only murine short FLIP isoform, reveal requirements for DISC recruitment. 15, 773–782 (2008). [DOI] [PubMed] [Google Scholar]
- 78.Budd RC, Yeh WC, Tschopp J, cFLIP regulation of lymphocyte activation and development. Nat. Rev. Immunol 6, 196–204 (2006). [DOI] [PubMed] [Google Scholar]
- 79.He S, Wang L, Miao L, Wang T, Du F, Zhao L, Wang X, Receptor Interacting Protein Kinase-3 Determines Cellular Necrotic Response to TNF-α. Cell (2009), doi: 10.1016/j.cell.2009.05.021. [DOI] [PubMed] [Google Scholar]
- 80.Dondelinger Y, Declercq W, Montessuit S, Roelandt R, Goncalves A, Bruggeman I, Hulpiau P, Weber K, Sehon CA, Marquis RW, Bertin J, Gough PJ, Savvides S, Martinou JC, Bertrand MJM, Vandenabeele P, MLKL Compromises Plasma Membrane Integrity by Binding to Phosphatidylinositol Phosphates. Cell Rep. 7, 971–981 (2014). [DOI] [PubMed] [Google Scholar]
- 81.Xia B, Fang S, Chen X, Hu H, Chen P, Wang H, Gao Z, MLKL forms cation channels. Cell Res. 26, 517–528 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wajant H, Siegmund D, TNFR1 and TNFR2 in the Control of the Life and Death Balance of Macrophages. Front. Cell Dev. Biol 7, 1–14 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kaiser WJ, Upton JW, Long AB, Livingston-Rosanoff D, Daley-Bauer LP, Hakem R, Caspary T, Mocarski ES, RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature. 471, 368–373 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Feoktistova M, Geserick P, Kellert B, Dimitrova DP, Langlais C, Hupe M, Cain K, MacFarlane M, G. H??cker, M. Leverkus, CIAPs Block Ripoptosome Formation, a RIP1/Caspase-8 Containing Intracellular Cell Death Complex Differentially Regulated by cFLIP Isoforms. Mol. Cell 43, 449–463 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhang X, Dowling JP, Zhang J, RIPK1 can mediate apoptosis in addition to necroptosis during embryonic development. Cell Death Dis. 10, 245 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Newton K, Wickliffe KE, Dugger DL, Maltzman A, Roose-Girma M, Dohse M, Kőműves L, Webster JD, Dixit VM, Cleavage of RIPK1 by caspase-8 is crucial for limiting apoptosis and necroptosis. Nature. 8 (2019), doi: 10.1038/s41586-019-1548-x. [DOI] [PubMed] [Google Scholar]
- 87.Lalaoui N, Boyden SE, Oda H, Wood GM, Stone DL, Chau D, Liu L, Stoffels M, Kratina T, Lawlor KE, Zaal KJM, Hoffmann PM, Etemadi N, Shield-Artin K, Biben C, Tsai WL, Blake MD, Kuehn HS, Yang D, Anderton H, Silke N, Wachsmuth L, Zheng L, Moura NS, Beck DB, Gutierrez-Cruz G, Ombrello AK, Pinto-Patarroyo GP, Kueh AJ, Herold MJ, Hall C, Wang H, Chae JJ, Dmitrieva NI, McKenzie M, Light A, Barham BK, Jones A, Romeo TM, Zhou Q, Aksentijevich I, Mullikin JC, Gross AJ, Shum AK, Hawkins ED, Masters SL, Lenardo MJ, Boehm M, Rosenzweig SD, Pasparakis M, Voss AK, Gadina M, Kastner DL, Silke J, Mutations that prevent caspase cleavage of RIPK1 cause autoinflammatory disease. Nature. 577, 103–108 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hu S, Vincenz C, Buller M, Dixit VM, A Novel Family of Viral Death Effector Domain-containing Molecules That Inhibit Both CD-95- and Tumor Necrosis Factor Receptor-1-induced Apoptosis. J. Biol. Chem. 272, 9621–9625 (1997). [DOI] [PubMed] [Google Scholar]
- 89.Thome M, Schneider P, Hofmann K, Fickenscher H, Meinl E, Neipel F, Mattmann C, Burns K, Bodmer JL, Schröter M, Scaffidi C, Krammer PH, Peter ME, Tschopp J, Viral FLICE-inhibitory proteins (FLIPs) prevent apoptosis induced by death receptors. Nature. 386, 517–521 (1997). [DOI] [PubMed] [Google Scholar]
- 90.Chaudhary PM, Jasmin A, Eby MT, Hood L, Modulation of the NF-κB pathway by virally encoded death effector domains-containing proteins. Oncogene. 18, 5738–5746 (1999). [DOI] [PubMed] [Google Scholar]
- 91.Ram DR, Ilyukha V, Volkova T, Buzdin A, Tai A, Smirnova I, Poltorak A, Balance between short and long isoforms of cFLIP regulates Fas-mediated apoptosis in vivo. Proc. Natl. Acad. Sci 113, 1606–1611 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, Hu L, Shao F, Inflammatory caspases are innate immune receptors for intracellular LPS. Nature. 514, 187–192 (2014). [DOI] [PubMed] [Google Scholar]
- 93.Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, Newton K, Qu Y, Liu J, Heldens S, Zhang J, Lee WP, Roose-Girma M, Dixit VM, Non-canonical inflammasome activation targets caspase-11. Nature. 479, 117–121 (2011). [DOI] [PubMed] [Google Scholar]
- 94.Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, Akashi-Takamura S, Miyake K, Zhang J, Lee WP, Muszyński A, Forsberg LS, Carlson RW, Dixit VM, Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science. 341, 1246–9 (2013). [DOI] [PubMed] [Google Scholar]
- 95.Hagar JA, Powell DA, Aachoui Y, Ernst RK, Miao EA, Cytoplasmic LPS Activates Caspase-11: Implications in TLR4-Independent Endotoxic Shock. Science. 341, 1250–1253 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Miao EA, Leaf IA, Treuting PM, Mao DP, Dors M, Sarkar A, Warren SE, Wewers MD, Aderem A, Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat. Immunol 11, 1136–1142 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Jorgensen I, Zhang Y, Krantz BA, Miao EA, Pyroptosis triggers pore-induced intracellular traps (PITs) that capture bacteria and lead to their clearance by efferocytosis. J. Exp. Med 213, 2113–2128 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, Zhuang Y, Cai T, Wang F, Shao F, Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 526, 660–665 (2015). [DOI] [PubMed] [Google Scholar]
- 99.Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, Cuellar T, Haley B, Roose-Girma M, Phung QT, Liu PS, Lill JR, Li H, Wu J, Kummerfeld S, Zhang J, Lee WP, Snipas SJ, Salvesen GS, Morris LX, Fitzgerald L, Zhang Y, Bertram EM, Goodnow CC, Dixit VM, Caspase-11 cleaves gasdermin D for non-canonical inflammasome signaling. Nature. 526, 666–671 (2015). [DOI] [PubMed] [Google Scholar]
- 100.Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, Sun H, Wang D-C, Shao F, Pore-forming activity and structural autoinhibition of the gasdermin family. Nature. 535, 111–116 (2016). [DOI] [PubMed] [Google Scholar]
- 101.Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, Lieberman J, Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature. 535, 153–158 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Aglietti RA, Estevez A, Gupta A, Ramirez MG, Liu PS, Kayagaki N, Ciferri C, Dixit VM, Dueber EC, GsdmD p30 elicited by caspase-11 during pyroptosis forms pores in membranes. Proc. Natl. Acad. Sci 113, 7858–7863 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sborgi L, Rühl S, Mulvihill E, Pipercevic J, Heilig R, Farady CJ, Müller DJ, Broz P, Hiller S, GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. 35, 1766–1778 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Mulvihill E, Sborgi L, Mari SA, Pfreundschuh M, Hiller S, Müller DJ, Mechanism of membrane pore formation by human gasdermin‐D. EMBO J. 37, e98321 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Taabazuing CY, Okondo MC, Bachovchin DA, Pyroptosis and Apoptosis Pathways Engage in Bidirectional Crosstalk in Monocytes and Macrophages. Cell Chem. Biol 24, 507–514 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Chen KW, Demarco B, Heilig R, Shkarina K, Boettcher A, Farady CJ, Pelczar P, Broz P, Extrinsic and intrinsic apoptosis activate pannexin‐1 to drive NLRP3 inflammasome assembly. EMBO J, e101638 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kambara H, Liu F, Zhang X, Liu P, Bajrami B, Teng Y, Zhao L, Zhou S, Yu H, Zhou W, Silberstein LE, Cheng T, Han M, Xu Y, Luo HR, Gasdermin D Exerts Anti-inflammatory Effects by Promoting Neutrophil Death. Cell Rep. 22, 2809–2817 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Chen KW, Monteleone M, Boucher D, Sollberger G, Ramnath D, Condon ND, von Pein JB, Broz P, Sweet MJ, Schroder K, Noncanonical inflammasome signaling elicits gasdermin D–dependent neutrophil extracellular traps. Sci. Immunol 3, eaar6676 (2018). [DOI] [PubMed] [Google Scholar]
- 109.Sollberger G, Choidas A, Burn GL, Habenberger P, Di Lucrezia R, Kordes S, Menninger S, Eickhoff J, Nussbaumer P, Klebl B, Krüger R, Herzig A, Zychlinsky A, Gasdermin D plays a vital role in the generation of neutrophil extracellular traps. Sci. Immunol 3, eaar6689 (2018). [DOI] [PubMed] [Google Scholar]
- 110.Burgener SS, Leborgne NGF, Snipas SJ, Salvesen GS, Bird PI, Benarafa C, Cathepsin G Inhibition by Serpinb1 and Serpinb6 Prevents Programmed Necrosis in Neutrophils and Monocytes and Reduces GSDMD-Driven Inflammation. Cell Rep. 27, 3646–3656.e5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Evavold CL, Ruan J, Tan Y, Xia S, Wu H, Kagan JC, The Pore-Forming Protein Gasdermin D Regulates Interleukin-1 Secretion from Living Macrophages. Immunity. 48, 1–10 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Rühl S, Shkarina K, Demarco B, Heilig R, Santos JC, Broz P, ESCRT-dependent membrane repair negatively regulates pyroptosis downstream of GSDMD activation. Science (80-. ). 362, 956–960 (2018). [DOI] [PubMed] [Google Scholar]
- 113.Saeki N, Kuwahara Y, Sasaki H, Satoh H, Shiroishi T, Gasdermin (Gsdm) localizing to mouse chromosome 11 is predominantly expressed in upper gastroiatestinal tract but significantly suppressed in human gastric cancer cells. Mamm. Genome 11, 718–724 (2000). [DOI] [PubMed] [Google Scholar]
- 114.Saeki N, Sasaki H, Gasdermin Superfamily : A Novel Gene Family Functioning in Epithelial Cells (2012). [Google Scholar]
- 115.Knodler LA, Crowley SM, Sham HP, Yang H, Wrande M, Ma C, Ernst RK, Steele-Mortimer O, Celli J, Vallance BA, Noncanonical inflammasome activation of caspase-4/caspase-11 mediates epithelial defenses against enteric bacterial pathogens. Cell Host Microbe. 16, 249–256 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Sellin ME, Müller AA, Felmy B, Dolowschiak T, Diard M, Tardivel A, Maslowski KM, Hardt WD, Epithelium-intrinsic NAIP/NLRC4 inflammasome drives infected enterocyte expulsion to restrict salmonella replication in the intestinal mucosa. Cell Host Microbe. 16, 237–248 (2014). [DOI] [PubMed] [Google Scholar]
- 117.Rogers C, Fernandes-Alnemri T, Mayes L, Alnemri D, Cingolani G, Alnemri ES, Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat. Commun 8, 1–14 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Wang Y, Gao W, Shi X, Ding J, Liu W, He H, Wang K, Shao F, Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature. 547, 99–103 (2017). [DOI] [PubMed] [Google Scholar]
- 119.Martinon F, Burns K, Tschopp J, The Inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-β. Mol. Cell 10, 417–426 (2002). [DOI] [PubMed] [Google Scholar]
- 120.Vladimer GI, Marty-Roix R, Ghosh S, Weng D, Lien E, Inflammasomes and host defenses against bacterial infections. Curr. Opin. Microbiol 16, 23–31 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Davis BK, Wen H, Ting JP, The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu Rev Immunol. 29, 707–735 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Maslanik T, Mahaffey L, Tannura K, Beninson L, Greenwood BN, Fleshner M, The inflammasome and danger associated molecular patterns (DAMPs) are implicated in cytokine and chemokine responses following stressor exposure. Brain. Behav. Immun 28, 54–62 (2013). [DOI] [PubMed] [Google Scholar]
- 123.Bergsbaken T, Fink SL, Cookson BT, Pyroptosis: Host cell death and inflammation. Nat. Rev. Microbiol 7 (2009), pp. 99–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Christ A, Günther P, Lauterbach MAR, Duewell P, Biswas D, Pelka K, Scholz CJ, Oosting M, Haendler K, Baßler K, Klee K, Schulte-Schrepping J, Ulas T, Moorlag SJCFM, Kumar V, Park MH, Joosten LAB, Groh LA, Riksen NP, Espevik T, Schlitzer A, Li Y, Fitzgerald ML, Netea MG, Schultze JL, Latz E, Western Diet Triggers NLRP3-Dependent Innate Immune Reprogramming. Cell. 172, 162–175 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Proell M, Gerlic M, Mace PD, Reed JC, Riedl SJ, The CARD plays a critical role in ASC foci formation and inflammasome signalling. Biochem. J 449, 613–621 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Fernandes-Alnemri T, Wu J, Yu J-W, Datta P, Miller B, Jankowski W, Rosenberg S, Zhang J, Alnemri ES, The pyroptosome: a supramolecular assembly of ASC dimers mediating inflammatory cell death via caspase-1 activation. Cell Death Differ. 14, 1590–1604 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Maltez VI, Miao EA, Reassessing the Evolutionary Importance of Inflammasomes. J. Immunol 196, 956–962 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Tsuji NM, Tsutsui H, Seki E, Kuida K, Okamura H, Nakanishi K, Flavell RA, Roles of caspase-1 in Listeria infection in mice. Int. Immunol 16, 335–343 (2004). [DOI] [PubMed] [Google Scholar]
- 129.Zheng H, Fletcher D, Kozak W, Jiang M, Hofmann KJ, Corn CA, Soszynski D, Grabiec C, Trumbauer ME, Shaw A, Kostura MJ, Stevens K, Rosen H, North RJ, Chen HY, Tocci MJ, Kluger MJ, Van der Ploeg LHT, Resistance to fever induction and impaired acute-phase response in interleukin-1β-deficient mice. Immunity. 3, 9–19 (1995). [DOI] [PubMed] [Google Scholar]
- 130.Case CL, Shin S, Roy CR, Asc and Ipaf inflammasomes direct distinct pathways for caspase-1 activation in response to Legionella pneumophila. Infect. Immun 77, 1981–1991 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Sharp FA, Ruane D, Claass B, Creagh E, Harris J, Malyala P, Singh M, O’Hagan DT, Petrilli V, Tschopp J, O’Neill LAJ, Lavelle EC, Uptake of particulate vaccine adjuvants by dendritic cells activates the NALP3 inflammasome. Proc. Natl. Acad. Sci 106, 870–875 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Li H, Nookala S, Re F, Aluminum Hydroxide Adjuvants Activate Caspase-1 and Induce IL-1 and IL-18 Release. J. Immunol 178, 5271–5276 (2007). [DOI] [PubMed] [Google Scholar]
- 133.Pisarev VM, Parajuli P, Mosley RL, Chavez J, Zimmerman D, Winship D, Talmadge JE, Flt3 ligand and conjugation to IL-1β peptide as adjuvants for a type 1, T-cell response to an HIV p17 gag vaccine. Vaccine. 20, 2358–2368 (2002). [DOI] [PubMed] [Google Scholar]
- 134.Yang JL, Xu H, Shao F, The immunological function of familial Mediterranean fever disease protein Pyrin. Sci. China Life Sci 57 (2014), pp. 1156–1161. [DOI] [PubMed] [Google Scholar]
- 135.Medlej-Hashim M, Loiselet J, Lefranc G, Mégarbané A, Familial Mediterranean Fever (FMF): From diagnosis to treatment. Cah. Sante 14 (2004). [PubMed] [Google Scholar]
- 136.Stack JH, Beaumont K, Larsen PD, Straley KS, Henkel GW, Randle JCR, Hoffman HM, IL-Converting Enzyme/Caspase-1 Inhibitor VX-765 Blocks the Hypersensitive Response to an Inflammatory Stimulus in Monocytes from Familial Cold Autoinflammatory Syndrome Patients. J. Immunol 175, 2630–2634 (2005). [DOI] [PubMed] [Google Scholar]
- 137.Hoffman HM, Scott P, Mueller JL, Misaghi A, Stevens S, Yancopoulos GD, Murphy A, Valenzuela DM, Liu-Bryan R, Role of the leucine-rich repeat domain of cryopyrin/NALP3 in monosodium urate crystal-induced inflammation in mice. Arthritis Rheum. 62, 2170–2179 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Jha S, Srivastava SY, Brickey WJ, Iocca H, Toews A, Morrison JP, Chen VS, Gris D, Matsushima GK, Ting JP-Y, The Inflammasome Sensor, NLRP3, Regulates CNS Inflammation and Demyelination via Caspase-1 and Interleukin-18. J. Neurosci 30, 15811–15820 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Gris D, Ye Z, Iocca HA, Wen H, Craven RR, Gris P, Huang M, Schneider M, Miller SD, Ting JP-Y, NLRP3 Plays a Critical Role in the Development of Experimental Autoimmune Encephalomyelitis by Mediating Th1 and Th17 Responses. J. Immunol 185, 974–981 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Inoue M, Williams KL, Gunn MD, Shinohara ML, NLRP3 inflammasome induces chemotactic immune cell migration to the CNS in experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci 109, 10480–10485 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, Fitzgerald KA, Latz E, Moore KJ, Golenbock DT, The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat. Immunol 9, 857–865 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Yan Y, Jiang W, Liu L, Wang X, Ding C, Tian Z, Zhou R, Dopamine controls systemic inflammation through inhibition of NLRP3 inflammasome. Cell. 160, 62–73 (2015). [DOI] [PubMed] [Google Scholar]
- 143.Sims JE, Smith DE, The IL-1 family: Regulators of immunity. Nat. Rev. Immunol 10 (2010), pp. 89–102. [DOI] [PubMed] [Google Scholar]
- 144.Lebovic DI, Chao VA, Martini JF, Taylor RN, IL-1β induction of RANTES (regulated upon activation, normal T cell expressed and secreted) chemokine gene expression in endometriotic stromal cells depends on a nuclear factor-κB site in the proximal promoter. J. Clin. Endocrinol. Metab 86, 4759–4764 (2001). [DOI] [PubMed] [Google Scholar]
- 145.Saperstein S, Chen L, Oakes D, Pryhuber G, Finkelstein J, IL-1beta augments TNF-alpha-mediated inflammatory responses from lung epithelial cells. J. Interferon Cytokine Res. 29, 273–84 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Saperstein S, Huyck H, Kimball E, Johnston C, Finkelstein J, Pryhuber G, The effects of interleukin-1beta in tumor necrosis factor-alpha-induced acute pulmonary inflammation in mice. Mediators Inflamm. 2009, 1–10 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Matsukawa A, Yoshimura T, Maeda T, Ohkawara S, Takagi K, Yoshinaga M, Neutrophil accumulation and activation by homologous IL-8 in rabbits. IL-8 induces destruction of cartilage and production of IL-1 and IL-1 receptor antagonist in vivo. J Immunol. 154, 5418–5425 (1995). [PubMed] [Google Scholar]
- 148.Matsukawa A, Yoshimura T, Fujiwara K, Maeda T, Ohkawara S, Yoshinaga M, Involvement of growth-related protein in lipopolysaccharide-induced rabbit arthritis: cooperation between growth-related protein and IL-8, and interrelated regulation among TNFalpha, IL-1, IL-1 receptor antagonist, IL-8, and growth-related protein. Lab. Invest 79, 591–600 (1999). [PubMed] [Google Scholar]
- 149.De Filippo K, Henderson RB, Laschinger M, Hogg N, Neutrophil Chemokines KC and Macrophage-Inflammatory Protein-2 Are Newly Synthesized by Tissue Macrophages Using Distinct TLR Signaling Pathways. J. Immunol 180, 4308–4315 (2008). [DOI] [PubMed] [Google Scholar]
- 150.Akira S, The role of IL-18 in innate immunity. Curr. Opin. Immunol 12 (2000), pp. 59–63. [DOI] [PubMed] [Google Scholar]
- 151.Man SM, Tourlomousis P, Hopkins L, Monie TP, Fitzgerald KA, Bryant CE, Salmonella infection induces recruitment of Caspase-8 to the inflammasome to modulate IL-1beta production. J Immunol 191, 5239–5246 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Lee BL, Mirrashidi KM, Stowe IB, Kummerfeld SK, Watanabe C, Haley B, Cuellar TL, Reichelt M, Kayagaki N, ASC- And caspase-8-dependent apoptotic pathway diverges from the NLRC4 inflammasome in macrophages. Sci. Rep 8, 1–12 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Mascarenhas DPA, Cerqueira DM, Pereira MSF, Fernanda V, Castanheira S, Fernandes TD, Manin GZ, Cunha LD, Zamboni S, Inhibition of caspase-1 or gasdermin-D enable caspase-8 activation in the Naip5 / NLRC4 / ASC inflammasome. PLoS Pathog. 13, 1–28 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Franklin BS, Bossaller L, De Nardo D, Ratter JM, Stutz A, Engels G, Brenker C, Nordhoff M, Mirandola SR, Al-Amoudi A, Mangan MS, Zimmer S, Monks BG, Fricke M, Schmidt RE, Espevik T, Jones B, Jarnicki AG, Hansbro PM, Busto P, Marshak-Rothstein A, Hornemann S, Aguzzi A, Kastenmuller W, Latz E, The adaptor ASC has extracellular and “prionoid” activities that propagate inflammation. Nat. Immunol 15, 727–737 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Martinon F, Mayor A, Tschopp J, The inflammasomes: guardians of the body. Annu. Rev. Immunol 27, 229–265 (2009). [DOI] [PubMed] [Google Scholar]
- 156.Muñoz-Planillo R, Kuffa P, Martínez-Colón G, Smith B, Rajendiran T, Núñez G, K+ Efflux Is the Common Trigger of NLRP3 Inflammasome Activation by Bacterial Toxins and Particulate Matter. Immunity. 38, 1142–1153 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Latz E, Xiao TS, Stutz A, Activation and regulation of the inflammasomes. Nat. Rev. Immunol 13, 397–411 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Allam R, Lawlor KE, Yu EC-W, Mildenhall AL, Moujalled DM, Lewis RS, Ke F, Mason KD, White MJ, Stacey KJ, Strasser A, a O’Reilly L, Alexander W, Kile BT, Vaux DL, Vince JE, Mitochondrial apoptosis is dispensable for NLRP3 inflammasome activation but non-apoptotic caspase-8 is required for inflammasome priming. EMBO Rep. 15, 1–9 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Lawlor KE, Vince JE, Ambiguities in NLRP3 inflammasome regulation: is there a role for mitochondria? Biochim Biophys Acta. 1840, 1433–1440 (2014). [DOI] [PubMed] [Google Scholar]
- 160.Mariathasan S, Weiss DS, Newton K, McBride J, O’Rourke K, Roose-Girma M, Lee WP, Weinrauch Y, Monack DM, Dixit VM, Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. 440, 228–232 (2006). [DOI] [PubMed] [Google Scholar]
- 161.Gutierrez KD, Davis MA, Daniels BP, Olsen TM, Ralli-Jain P, Tait SWG, Gale M, Oberst A, MLKL Activation Triggers NLRP3-Mediated Processing and Release of IL-1β Independently of Gasdermin-D. J. Immunol 198, 2156–2164 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Pelegrin P, Surprenant A, Pannexin-1 mediates large pore formation and interleukin-1β release by the ATP-gated P2X7 receptor. EMBO J. 25, 5071–5082 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Chen J, Chen ZJ, PtdIns4P on dispersed trans-Golgi network mediates NLRP3 inflammasome activation. Nature. 564 (2018), doi: 10.1038/s41586-018-0761-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Schmid-Burgk JL, Chauhan D, Schmidt T, Ebert TS, Reinhardt J, Endl E, Hornung V, A genome-wide CRISPR screen identifies NEK7 as an essential component of NLRP3 inflammasome activation. J. Biol. Chem 291, 103–109 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.He Y, Zeng MY, Yang D, Motro B, Núñez G, Nek7 is an essential mediator of NLRP3 oligomerization and inflammasome activation downstream of potassium efflux. Nature. 530, 354–357 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Shi H, Wang Y, Li X, Zhan X, Tang M, Fina M, Su L, Pratt D, Bu CH, Hildebrand S, Lyon S, Scott L, Quan J, Sun Q, Russell J, Arnett S, Jurek P, Chen D, V Kravchenko V, Mathison JC, Moresco EMY, Monson NL, Ulevitch RJ, Beutler B, NLRP3 activation and mitosis are mutually exclusive events coordinated by NEK7, a new inflammasome component. Nat. Immunol 17, 250–258 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Stutz A, Kolbe C-C, Stahl R, Horvath GL, Franklin BS, van Ray O, Brinkschulte R, Geyer M, Meissner F, Latz E, NLRP3 inflammasome assembly is regulated by phosphorylation of the pyrin domain. J. Exp. Med 214, 1725–1736 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Rühl S, Broz P, Caspase-11 activates a canonical NLRP3 inflammasome by promoting K+efflux. Eur. J. Immunol 45, 2927–2936 (2015). [DOI] [PubMed] [Google Scholar]
- 169.Rathinam VAK, Vanaja SK, Waggoner L, Sokolovska A, Becker C, Stuart LM, Leong JM, Fitzgerald KA, TRIF licenses caspase-11-dependent NLRP3 inflammasome activation by gram-negative bacteria. Cell. 150, 606–619 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Aachoui Y, Kajiwara Y, Leaf IA, Mao D, Ting JPY, Coers J, Aderem A, Buxbaum JD, Miao EA, Canonical Inflammasomes Drive IFN-γ to Prime Caspase-11 in Defense against a Cytosol-Invasive Bacterium. Cell Host Microbe. 18, 320–332 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Aachoui Y, Leaf I. a., Hagar J. a., Fontana MF, Campos CG, Zak DE, Tan MH, Cotter P. a., Vance RE, Aderem A, Miao E. a., Caspase-11 Protects Against Bacteria That Escape the Vacuole. Science. 339, 975–978 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Akhter A, Caution K, Abu Khweek A, Tazi M, Abdulrahman BA, Abdelaziz DHA, Voss OH, Doseff AI, Hassan H, Azad AK, Schlesinger LS, Wewers MD, Gavrilin MA, Amer AO, Caspase-11 Promotes the Fusion of Phagosomes Harboring Pathogenic Bacteria with Lysosomes by Modulating Actin Polymerization. Immunity. 37, 35–47 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Kono H, Orlowski GM, Patel Z, Rock KL, The IL-1-Dependent Sterile Inflammatory Response Has a Substantial Caspase-1-Independent Component That Requires Cathepsin C. J. Immunol (2012), doi: 10.4049/jimmunol.1200136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Provoost S, Joos GF, Pauwels NS, Vandenabeele P, Tournoy KG, Vanden Berghe T, Maes T, Lambrecht BN, NLRP3/Caspase-1-Independent IL-1 Production Mediates Diesel Exhaust Particle-Induced Pulmonary Inflammation. J. Immunol (2011), doi: 10.4049/jimmunol.1004062. [DOI] [PubMed] [Google Scholar]
- 175.Mayer-Barber KD, Barber DL, Shenderov K, White SD, Wilson MS, Cheever A, Kugler D, Hieny S, Caspar P, Nunez G, Schlueter D, Flavell RA, Sutterwala FS, Sher A, Núñez G, Schlueter D, Flavell RA, Sutterwala FS, Sher A, Cutting Edge: Caspase-1 Independent IL-1 Production Is Critical for Host Resistance to Mycobacterium tuberculosis and Does Not Require TLR Signaling In Vivo. J. Immunol (2010), doi: 10.4049/jimmunol.0904189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Guma M, Ronacher L, Liu-Bryan R, Takai S, Karin M, Corr M, Caspase 1-independent activation of interleukin-1β in neutrophil-predominant inflammation. Arthritis Rheum. (2009), doi: 10.1002/art.24959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.White MJ, McArthur K, Metcalf D, Lane RM, Cambier JC, Herold MJ, Van Delft MF, Bedoui S, Lessene G, Ritchie ME, Huang DCS, Kile BT, Apoptotic caspases suppress mtDNA-induced STING-mediated type i IFN production. Cell. 159, 1549–1562 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Rajput A, Kovalenko A, Bogdanov K, Yang SH, Kang TB, Kim JC, Du J, Wallach D, RIG-I RNA helicase activation of irf3 transcription factor is negatively regulated by caspase-8-mediated cleavage of the RIP1 protein. Immunity (2011), doi: 10.1016/j.immuni.2010.12.018. [DOI] [PubMed] [Google Scholar]
- 179.Lüthi AU, Cullen SP, McNeela EA, Duriez PJ, Afonina IS, Sheridan C, Brumatti G, Taylor RC, Kersse K, Vandenabeele P, Lavelle EC, Martin SJ, Suppression of Interleukin-33 Bioactivity through Proteolysis by Apoptotic Caspases. Immunity (2009), doi: 10.1016/j.immuni.2009.05.007. [DOI] [PubMed] [Google Scholar]
- 180.Orning P, Weng D, Starheim K, Ratner D, Best Z, Lee B, Brooks A, Xia S, Wu H, Kelliher MA, Berger SB, Gough PJ, Bertin J, Proulx MM, Goguen JD, Kayagaki N, Fitzgerald KA, Lien E, Pathogen blockade of tak1 triggers caspase-8–dependent cleavage of gasdermin d and cell death. Science (80-. ). (2018), , doi: 10.1126/science.aau2818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Sarhan J, Liu BC, Muendlein HI, Li P, Nilson R, Tang AY, Rongvaux A, Bunnell SC, Shao F, Green DR, Poltorak A, Caspase-8 induces cleavage of gasdermin D to elicit pyroptosis during Yersinia infection [Immunology and Inflammation]. Proc. Natl. Acad. Sci. U. S. A (2018), doi: 10.1073/pnas.1809548115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Vince JE, De Nardo D, Gao W, Vince AJ, Hall C, McArthur K, Simpson D, Vijayaraj S, Lindqvist LM, Bouillet P, Rizzacasa MA, Man SM, Silke J, Masters SL, Lessene G, Huang DCS, Gray DHD, Kile BT, Shao F, Lawlor KE, The Mitochondrial Apoptotic Effectors BAX/BAK Activate Caspase-3 and −7 to Trigger NLRP3 Inflammasome and Caspase-8 Driven IL-1β Activation. Cell Rep. 25, 2339–2353.e4 (2018). [DOI] [PubMed] [Google Scholar]
- 183.Chauhan D, Bartok E, Gaidt MM, Bock FJ, Herrmann J, Seeger JM, Broz P, Beckmann R, Kashkar H, Tait SWG, Müller R, Hornung V, BAX/BAK-Induced Apoptosis Results in Caspase-8-Dependent IL-1β Maturation in Macrophages. Cell Rep. 25, 2354–2368.e5 (2018). [DOI] [PubMed] [Google Scholar]
- 184.Günther C, Buchen B, He GW, Hornef M, Torow N, Neumann H, Wittkopf N, Martini E, Basic M, Bleich A, Watson AJM, Neurath MF, Becker C, Caspase-8 controls the gut response to microbial challenges by Tnf-α-dependent and independent pathways. Gut. 64, 601–610 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Kovalenko A, Kim J-C, Kang T-B, Rajput A, Bogdanov K, Dittrich-Breiholz O, Kracht M, Brenner O, Wallach D, Caspase-8 deficiency in epidermal keratinocytes triggers an inflammatory skin disease. J. Exp. Med 206, 2161–2177 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Kang T-B, Ben-Moshe T, Varfolomeev EE, Pewzner-Jung Y, Yogev N, Jurewicz A, Waisman A, Brenner O, Haffner R, Gustafsson E, Ramakrishnan P, Lapidot T, Wallach D, Caspase-8 Serves Both Apoptotic and Nonapoptotic Roles. J. Immunol (2004), doi: 10.4049/jimmunol.173.5.2976. [DOI] [PubMed] [Google Scholar]
- 187.Liedtke C, Bangen JM, Freimuth J, Beraza N, Lambertz D, Cubero FJ, Hatting M, Karlmark KR, Streetz KL, Krombach GA, Tacke F, Gassler N, Riethmacher D, Trautwein C, Loss of caspase-8 protects mice against inflammation-related hepatocarcinogenesis but induces non-apoptotic liver injury. Gastroenterology (2011), doi: 10.1053/j.gastro.2011.08.037. [DOI] [PubMed] [Google Scholar]
- 188.Ben Moshe T, Barash H, Kang TB, Kim JC, Kovalenko A, Gross E, Schuchmann M, Abramovitch R, Galun E, Wallach D, Role of caspase-8 in hepatocyte response to infection and injury in mice. Hepatology (2007), doi: 10.1002/hep.21495. [DOI] [PubMed] [Google Scholar]
- 189.Kang TB, Jeong JS, Yang SH, Kovalenko A, Wallach D, Caspase-8 deficiency in mouse embryos triggers chronic RIPK1-dependent activation of inflammatory genes, independently of RIPK3. Cell Death Differ. (2018), doi: 10.1038/s41418-018-0104-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Rauch I, Deets KA, Ji DX, Brodsky IE, Gronert K, Vance RE, Rauch I, Deets KA, Ji DX, Von Moltke J, Tenthorey JL, Lee AY, NAIP-NLRC4 Inflammasomes Coordinate Intestinal Epithelial Cell Expulsion with Eicosanoid and IL-18 Release via Activation of Caspase-1 and -8 Article NAIP-NLRC4 Inflammasomes Coordinate Intestinal Epithelial Cell Expulsion with Eicosanoid and IL-18 Releas. Immunity. 46, 649–659 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Kang TB, Yang SH, Toth B, Kovalenko A, Wallach D, Caspase-8 Blocks Kinase RIPK3-Mediated Activation of the NLRP3 Inflammasome. Immunity. 38, 27–40 (2013). [DOI] [PubMed] [Google Scholar]
- 192.Li C, Lasse S, Lee P, Nakasaki M, Chen SW, Yamasaki K, Gallo RL, Jamora C, Development of atopic dermatitis-like skin disease from the chronic loss of epidermal caspase-8. Proc. Natl. Acad. Sci. U. S. A (2010), doi: 10.1073/pnas.1009751108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Günther C, Martini E, Wittkopf N, Amann K, Weigmann B, Neumann H, Waldner MJ, Hedrick SM, Tenzer S, Neurath MF, Becker C, Caspase-8 regulates TNF-α-induced epithelial necroptosis and terminal ileitis. Nature. 477, 335–9 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Lee P, Gund R, Dutta A, Pincha N, Rana I, Ghosh S, Witherden D, Kandyba E, MacLeod A, Kobielak K, Havran WL, Jamora C, Stimulation of hair follicle stem cell proliferation through an IL-1 dependent activation of γδT-cells. Elife (2017), doi: 10.7554/eLife.28875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Mocarski ES, Guo H, Kaiser WJ, Necroptosis: The Trojan horse in cell autonomous antiviral host defense. Virology. 479–480 (2015), pp. 160–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.He S, Liang Y, Shao F, Wang X, Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3-mediated pathway. Proc. Natl. Acad. Sci 108, 20054–20059 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Varfolomeev EE, Schuchmann M, Luria V, Chiannilkulchai N, Beckmann JS, Mett IL, Rebrikov D, Brodianski VM, Kemper OC, Kollet O, Lapidot T, Soffer D, Sobe T, Avraham KB, Goncharov T, Holtmann H, Lonai P, Wallach D, Targeted disruption of the mouse Caspase 8 gene ablates cell death induction by the TNF receptors, Fas/Apo1, and DR3 and is lethal prenatally. Immunity. 9, 267–276 (1998). [DOI] [PubMed] [Google Scholar]
- 198.Newton K, Dugger DL, Wickliffe KE, Kapoor N, Cristina de‐Almagro M, Vucic D, Komuves L, Ferrando RE, French DM, Webster J, Roose‐Girma M, Warming S, Dixit VM, Activity of Protein Kinase RIPK3 Determines whether Cells Die by Necroptosis or Apoptosis. Sci. express 343, 1357–1360 (2014). [DOI] [PubMed] [Google Scholar]
- 199.Zhang J, Cado D, Chen A, Kabra NH, Winoto A, Fas-mediated apoptosis and activation-induced T-cell proliferation are defective in mice lacking FADD/Mort1. Nature. 392, 296–299 (1998). [DOI] [PubMed] [Google Scholar]
- 200.Dillon CP, Oberst A, Weinlich R, Janke LJ, Kang T-B, Ben-Moshe T, Mak TW, Wallach D, Green DR, Survival Function of the FADD-CASPASE-8-cFLIPL Complex. Cell Rep. 1, 401–407 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Zhang X, Fan C, Zhang H, Zhao Q, Liu Y, Xu C, Xie Q, Wu X, Yu X, Zhang J, Zhang H, MLKL and FADD Are Critical for Suppressing Progressive Lymphoproliferative Disease and Activating the NLRP3 Inflammasome. Cell Rep. 16, 3247–3259 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Beisner DR, Ch’en IL, Kolla RV, Hoffmann A, Hedrick SM, Cutting Edge: Innate Immunity Conferred by B Cells Is Regulated by Caspase-8. J. Immunol (2005), doi: 10.4049/jimmunol.175.6.3469. [DOI] [PubMed] [Google Scholar]
- 203.Salmena L, Lemmers B, Hakem A, Matysiak-Zablocki E, Murakami K, Billie Au PY, Berry DM, Tamblyn L, Shehabeldin A, Migon E, Wakeham A, Bouchard D, Yeh WC, McGlade JC, Ohashi PS, Hakem R, Essential role for caspase 8 in T-cell homeostasis and T-cell-mediated immunity. Genes Dev. 17, 883–895 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Dillon CP, Weinlich R, Rodriguez DA, Cripps JG, Quarato G, Gurung P, Verbist KC, Brewer TL, Llambi F, Gong YN, Janke LJ, Kelliher MA, Kanneganti TD, Green DR, RIPK1 blocks early postnatal lethality mediated by caspase-8 and RIPK3. Cell. 157, 1189–1202 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Kaiser WJ, Daley-Bauer LP, Thapa RJ, Mandal P, Berger SB, Huang C, Sundararajan A, Guo H, Roback L, Speck SH, Bertin J, Gough PJ, Balachandran S, Mocarski ES, RIP1 suppresses innate immune necrotic as well as apoptotic cell death during mammalian parturition. Proc. Natl. Acad. Sci 111, 7753–7758 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Rickard JA, O’Donnell JA, Evans JM, Lalaoui N, Poh AR, Rogers T, Vince JE, Lawlor KE, Ninnis RL, Anderton H, Hall C, Spall SK, Phesse TJ, Abud HE, Cengia LH, Corbin J, Mifsud S, Di Rago L, Metcalf D, Ernst M, Dewson G, Roberts AW, Alexander WS, Murphy JM, Ekert PG, Masters SL, Vaux DL, Croker BA, Gerlic M, Silke J, RIPK1 regulates RIPK3-MLKL-driven systemic inflammation and emergency hematopoiesis. Cell. 157, 1175–1188 (2014). [DOI] [PubMed] [Google Scholar]
- 207.Polykratis A, Hermance N, Zelic M, Roderick J, Kim C, Van T-M, Lee TH, Chan FKM, Pasparakis M, Kelliher MA, Cutting Edge: RIPK1 Kinase Inactive Mice Are Viable and Protected from TNF-Induced Necroptosis In Vivo. J. Immunol 193, 1539–1543 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Tenev T, Bianchi K, Darding M, Broemer M, Langlais C, Wallberg F, Zachariou A, Lopez J, MacFarlane M, Cain K, Meier P, The Ripoptosome, a Signaling Platform that Assembles in Response to Genotoxic Stress and Loss of IAPs. Mol. Cell 43, 432–448 (2011). [DOI] [PubMed] [Google Scholar]
- 209.Zhang H, Zhou X, McQuade T, Li J, Chan FKM, Zhang J, Functional complementation between FADD and RIP1 in embryos and lymphocytes. Nature. 471, 373–377 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Paquette N, Conlon J, Sweet C, Rus F, Wilson L, Pereira A, Rosadini CV, Goutagny N, Weber ANR, Lane WS, Shaffer SA, Maniatis S, Fitzgerald KA, Stuart L, Silverman N, Serine/threonine acetylation of TGF -activated kinase (TAK1) by Yersinia pestis YopJ inhibits innate immune signaling. Proc. Natl. Acad. Sci 109, 12710–12715 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Malireddi RKS, Gurung P, Mavuluri J, Dasari TK, Klco JM, Chi H, TAK1 restricts spontaneous NLRP3 activation and cell death to control myeloid proliferation. J. Exp. Med 215, 1023–1034 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Malireddi RKS, Gurung P, Kesavardhana S, Samir P, Burton A, Mummareddy H, Vogel P, Pelletier S, Burgula S, Kanneganti TD, Innate immune priming in the absence of TAK1 drives RIPK1 kinase activity-independent pyroptosis, apoptosis, necroptosis, and inflammatory disease. J. Exp. Med 217, 1–13 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Muendlein HI, Jetton D, Connolly WM, Eidell KP, Magri Z, Smirnova I, Poltorak A, CFLIPL protects macrophages from LPS-induced pyroptosis via inhibition of complex II formation. Science (80-. ). 367, 1379–1384 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Weng D, Marty-Roix R, Ganesan S, Proulx MK, Vladimer GI, Kaiser WJ, Mocarski ES, Pouliot K, Chan FK-M, a Kelliher M, a Harris P, Bertin J, Gough PJ, Shayakhmetov DM, Goguen JD, a Fitzgerald K, Silverman N, Lien E, Caspase-8 and RIP kinases regulate bacteria-induced innate immune responses and cell death. Proc. Natl. Acad. Sci. U. S. A 111, 7391–7396 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Chen KW, Demarco B, Broz P, Eur. J. Immunol, in press, doi: 10.1002/eji.201948254. [DOI] [PubMed] [Google Scholar]
- 216.Qu Y, Misaghi S, Newton K, Gilmour LL, Louie S, Cupp JE, Dubyak GR, Hackos D, Dixit VM, Pannexin-1 Is Required for ATP Release during Apoptosis but Not for Inflammasome Activation. J. Immunol 186, 6553–6561 (2011). [DOI] [PubMed] [Google Scholar]
- 217.Sandilos JK, Walk SF, Ravichandran KS, Chiu Y-H, Bayliss DA, Armstrong AJ, Chekeni FB, Pannexin 1, an ATP Release Channel, Is Activated by Caspase Cleavage of Its Pore-associated C-terminal Autoinhibitory Region. J. Biol. Chem 287, 11303–11311 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Chekeni FB, Elliott MR, Sandilos JK, Walk SF, Kinchen JM, Lazarowski ER, Armstrong AJ, Penuela S, Laird DW, Salvesen GS, Isakson BE, Bayliss DA, Ravichandran KS, Pannexin 1 channels mediate ‘find-me’ signal release and membrane permeability during apoptosis. Nature. 467, 863–867 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Rogers C, Erkes DA, Nardone A, Aplin AE, Fernandes-Alnemri T, Alnemri ES, Gasdermin pores permeabilize mitochondria to augment caspase-3 activation during apoptosis and inflammasome activation. Nat. Commun 10, 1689 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Van Opdenbosch N, Van Gorp H, Verdonckt M, Saavedra PHV, de Vasconcelos NM, Gonçalves A, Vande Walle L, Demon D, Matusiak M, Van Hauwermeiren F, D’Hont J, Hochepied T, Krautwald S, Kanneganti TD, Lamkanfi M, Caspase-1 Engagement and TLR-Induced c-FLIP Expression Suppress ASC/Caspase-8-Dependent Apoptosis by Inflammasome Sensors NLRP1b and NLRC4. Cell Rep. 21, 3427–3444 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Schneider KS, Groß CJ, Dreier RF, Saller BS, Mishra R, Gorka O, Heilig R, Meunier E, Dick MS, Ćiković T, Sodenkamp J, Médard G, Naumann R, Ruland J, Kuster B, Broz P, Groß O, The Inflammasome Drives GSDMD-Independent Secondary Pyroptosis and IL-1 Release in the Absence of Caspase-1 Protease Activity. Cell Rep. 21, 3846–3859 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Sagulenko V, Thygesen SJ, Sester DP, Idris A, Cridland JA, Vajjhala PR, Roberts TL, Schroder K, Vince JE, Hill JM, Silke J, Stacey KJ, AIM2 and NLRP3 inflammasomes activate both apoptotic and pyroptotic death pathways via ASC. Cell Death Differ. 20, 1149–1160 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Vajjhala PR, Lu A, Brown DL, Pang SW, Sagulenko V, Sester DP, Cridland SO, Hill JM, Schroder K, Stow JL, Wu H, Stacey KJ, The inflammasome adaptor ASC induces procaspase-8 death effector domain filaments. J. Biol. Chem 290, 29217–29230 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Lemmers B, Salmena L, Bidère N, Su H, Matysiak-Zablocki E, Murakami K, Ohashi PS, Jurisicova A, Lenardo M, Hakem R, Hakem A, Essential role for caspase-8 in toll-like receptors and NFκB signaling. J. Biol. Chem (2007), doi: 10.1074/jbc.M606721200. [DOI] [PubMed] [Google Scholar]
- 225.Su H, Su H, Bidere N, Bidere N, Zheng L, Zheng L, Cubre A, Cubre A, Sakai K, Sakai K, Dale J, Dale J, Salmena L, Salmena L, Hakem R, Hakem R, Straus S, Straus S, Lenardo M, Lenardo M, Requirement for Caspase-8 in NF-{kappa}B Activation by Antigen Receptor. Science (80-. ). 307, 1465–1468 (2005). [DOI] [PubMed] [Google Scholar]
- 226.Ganesan S, Rathinam V. a K., Bossaller L, Army K, Kaiser WJ, Mocarski ES, Dillon CP, Green DR, Mayadas TN, Levitz SM, Hise AG, Silverman N, Fitzgerald KA, Caspase-8 Modulates Dectin-1 and Complement Receptor 3-Driven IL-1β Production in Response to β-Glucans and the Fungal Pathogen, Candida albicans. J. Immunol 193, 2519–2530 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Vajjhala PR, Lu A, Brown DL, Pang SW, Sagulenko V, Sester DP, Cridland SO, Hill JM, Schroder K, Stow JL, Wu H, Stacey KJ, The inflammasome adaptor ASC induces procaspase-8 death effector domain filaments. J. Biol. Chem 290, 29217–29230 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Philip NH, Dillon CP, Snyder AG, Fitzgerald P, a Wynosky-Dolfi M, Zwack EE, Hu B, Fitzgerald L, a Mauldin E, Copenhaver AM, Shin S, Wei L, Parker M, Zhang J, Oberst A, Green DR, Brodsky IE, Caspase-8 mediates caspase-1 processing and innate immune defense in response to bacterial blockade of NF-κB and MAPK signaling. Proc. Natl. Acad. Sci. U. S. A 111, 7385–7390 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Antonopoulos C, El Sanadi C, Kaiser WJ, Mocarski ES, Dubyak GR, Proapoptotic Chemotherapeutic Drugs Induce Noncanonical Processing and Release of IL-1 via Caspase-8 in Dendritic Cells. J. Immunol 191, 4789–4803 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Gringhuis SI, Kaptein TM, Wevers BA, Theelen B, Van Der Vlist M, Boekhout T, Geijtenbeek TBH, Dectin-1 is an extracellular pathogen sensor for the induction and processing of IL-1β via a noncanonical caspase-8 inflammasome. Nat. Immunol (2012), doi: 10.1038/ni.2222. [DOI] [PubMed] [Google Scholar]
- 231.Maelfait J, Vercammen E, Janssens S, Schotte P, Haegman M, Magez S, Beyaert R, Stimulation of Toll-like receptor 3 and 4 induces interleukin-1beta maturation by caspase-8. J. Exp. Med 205, 1967–73 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Feng Y, Daley-Bauer LP, Roback L, Guo H, Koehler HS, Potempa M, Lanier LL, Mocarski ES, Caspase-8 restricts antiviral CD8 T cell hyperaccumulation. Proc. Natl. Acad. Sci (2019), doi: 10.1073/pnas.1904319116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Gurung P, Kanneganti TD, Novel Roles for Caspase-8 in IL-1β and Inflammasome Regulation. Am. J. Pathol 185, 17–25 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Zhou X, Gewurz BE, Ritchie JM, Takasaki K, Greenfeld H, Kieff E, Davis BM, Waldor MK, A Vibrio parahaemolyticus T3SS Effector Mediates Pathogenesis by Independently Enabling Intestinal Colonization and Inhibiting TAK1 Activation. Cell Rep. 3, 1690–1702 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Lei X, Han N, Xiao X, Jin Q, He B, Wang J, Enterovirus 71 3C Inhibits Cytokine Expression through Cleavage of the TAK1/TAB1/TAB2/TAB3 Complex. J. Virol 88, 9830–9841 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.He C, Zhou Y, Liu F, Liu H, Tan H, Jin S, Wu W, Ge B, Bacterial nucleotidyl cyclase inhibits the host innate immune response by suppressing TAK1 activation. Infect. Immun 85 (2017), doi: 10.1128/IAI.00239-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Barth K, Genco CA, Microbial Degradation of Cellular Kinases Impairs Innate Immune Signaling and Paracrine TNFα Responses. Sci. Rep 6, 34656 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Taxman DJ, Huang MTH, Ting JPY, Inflammasome inhibition as a pathogenic stealth mechanism. Cell Host Microbe (2010), , doi: 10.1016/j.chom.2010.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Cunha LD, Zamboni DS, Subversion of inflammasome activation and pyroptosis by pathogenic bacteria. Front. Cell. Infect. Microbiol 3, 1–14 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Chattopadhyay S, Yamashita M, Zhang Y, Sen GC, The IRF-3/Bax-Mediated Apoptotic Pathway, Activated by Viral Cytoplasmic RNA and DNA, Inhibits Virus Replication. J. Virol 85, 3708–3716 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Gurung P, Anand PK, Malireddi RKS, Vande Walle L, Van Opdenbosch N, Dillon CP, Weinlich R, Green DR, Lamkanfi M, Kanneganti T-D, FADD and Caspase-8 Mediate Priming and Activation of the Canonical and Noncanonical Nlrp3 Inflammasomes. J. Immunol 192, 1835–1846 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Su H, Bidère N, Zheng L, Cubre A, Sakai K, Dale J, Salmena L, Hakem R, Straus S, Lenardo M, Requirement for caspase-8 in NF-κB activation by antigen receptor. Science (80-. ). 307, 1465–1468 (2005). [DOI] [PubMed] [Google Scholar]
- 243.Cuda CM, Misharin AV, Khare S, Saber R, Tsai FN, Archer AM, Homan PJ, Haines GK, Hutcheson J, Dorfleutner A, Budinger GRS, Stehlik C, Perlman H, Conditional deletion of caspase-8 in macrophages alters macrophage activation in a RIPK-dependent manner. Arthritis Res. Ther 17, 1–16 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.DeLaney AA, Berry CT, Christian DA, Hart A, Bjanes E, Wynosky-Dolfi MA, Li X, Tummers B, Udalova IA, Chen YH, Hershberg U, Freedman BD, Hunter CA, Brodsky IE, Caspase-8 promotes c-Rel–dependent inflammatory cytokine expression and resistance against Toxoplasma gondii. Proc. Natl. Acad. Sci, 201820529 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Henry CM, Martin SJ, Caspase-8 Acts in a Non-enzymatic Role as a Scaffold for Assembly of a Pro-inflammatory “FADDosome” Complex upon TRAIL Stimulation. Mol. Cell (2017), doi: 10.1016/j.molcel.2017.01.022. [DOI] [PubMed] [Google Scholar]
- 246.Hartwig T, Montinaro A, von Karstedt S, Sevko A, Surinova S, Chakravarthy A, Taraborrelli L, Draber P, Lafont E, Arce Vargas F, El-Bahrawy MA, Quezada SA, Walczak H, The TRAIL-Induced Cancer Secretome Promotes a Tumor-Supportive Immune Microenvironment via CCR2. Mol. Cell (2017), doi: 10.1016/j.molcel.2017.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Menu P, Vince JE, The NLRP3 inflammasome in health and disease: the good, the bad and the ugly. Clin. Exp. Immunol 166, 1–15 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Dinarello CA, Simon A, Van Der Meer JWM, Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat. Rev. Drug Discov (2012), , doi: 10.1038/nrd3800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Stupack DG, Caspase-8 as a therapeutic target in cancer. Cancer Lett. (2013), doi: 10.1016/j.canlet.2010.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Rohn TT, Head E, Nesse WH, Cotman CW, Cribbs DH, Activation of caspase-8 in the Alzheimer’s disease brain. Neurobiol. Dis (2001), doi: 10.1006/nbdi.2001.0449. [DOI] [PubMed] [Google Scholar]
- 251.Fricker M, Vilalta A, Tolkovsky AM, Brown GC, Caspase inhibitors protect neurons by enabling selective necroptosis of inflamed microglia. J. Biol. Chem (2013), doi: 10.1074/jbc.M112.427880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Hartmann A, Troadec JD, Hunot S, Kikly K, Faucheux BA, Mouatt-Prigent A, Ruberg M, Agid Y, Hirsch EC, Caspase-8 is an effector in apoptotic death of dopaminergic neurons in Parkinson’s disease, but pathway inhibition results in neuronal necrosis. J. Neurosci (2001), doi: 10.1523/jneurosci.21-07-02247.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Sánchez I, Chi-Jie X, Peter J, Kakizaka A, Blenis J, Yuan J, Caspase-8 is required for cell death induced by expanded polyglutamine repeats. Neuron (1999), doi: 10.1016/S0896-6273(00)80716-3. [DOI] [PubMed] [Google Scholar]
- 254.Brumatti G, Ma C, Lalaoui N, Nguyen NY, Navarro M, Tanzer MC, Richmond J, Ghisi M, Salmon JM, Silke N, Pomilio G, Glaser SP, De Valle E, Gugasyan R, Gurthridge MA, Condon SM, Johnstone RW, Lock R, Salvesen G, Wei A, Vaux DL, Ekert PG, Silke J, The caspase-8 inhibitor emricasan combines with the SMAC mimetic birinapant to induce necroptosis and treat acute myeloid leukemia. Sci. Transl. Med (2016), doi: 10.1126/scitranslmed.aad3099. [DOI] [PubMed] [Google Scholar]
- 255.Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, Bellomo R, Bernard GR, Chiche J, Coopersmith CM, Hotchkiss RS, Levy MM, Marshall JC, Martin GS, Opal SM, Rubenfeld GD, Van Der T, Vincent J, Angus DC, The third international consensus definitions for sepsis and septic shock. Jama. 315, 801–810 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Kang R, Zeng L, Zhu S, Xie Y, Liu J, Wen Q, Cao L, Xie M, Ran Q, Kroemer G, Wang H, Billiar TR, Jiang J, Tang D, Lipid Peroxidation Drives Gasdermin D-Mediated Pyroptosis in Lethal Polymicrobial Sepsis. Cell Host Microbe. 24, 97–108.e4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Hu JJ, Liu X, Zhao J, Xia S, Ruan J, Luo X, Kim J, Lieberman J, Wu H, Identification of pyroptosis inhibitors that target a reactive cysteine in gasdermin D. bioRxiv (2018), doi: 10.1101/365908. [DOI] [Google Scholar]
- 258.Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, Abela GS, Franchi L, Nuñez G, Schnurr M, Espevik T, Lien E, Fitzgerald KA, Rock KL, Moore KJ, Wright SD, Hornung V, Latz E, NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 464, 1357–1361 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Aksentijevich I, Kastner DL, Genetics of monogenic autoinflammatory diseases: Past successes, future challenges. Nat. Rev. Rheumatol 7 (2011), pp. 469–478. [DOI] [PubMed] [Google Scholar]
- 260.Zahid A, Li B, Kombe AJK, Jin T, Tao J, Pharmacological inhibitors of the nlrp3 inflammasome. Front. Immunol (2019), , doi: 10.3389/fimmu.2019.02538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.and R. M. S. Bulua Ariel C., Simon Anna, Maddipati Ravikanth, Pelletier Martin, Park Heiyoung, Kim Kye-Young, Sack Michael N., Kastner Daniel L., Mitochondrial reactive oxygen species promote production of proin ammatory cytokines and are elevated in TNFR1-associated periodic syndrome. J. Exp. Med 208, 519–533 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Simon A, Park H, Maddipati R, Lobito AA, Bulua AC, Jackson AJ, Chae JJ, Ettinger R, de Koning HD, Cruz AC, Kastner DL, Komarow H, Siegel RM, Concerted action of wild-type and mutant TNF receptors enhances inflammation in TNF receptor 1-associated periodic fever syndrome. Proc. Natl. Acad. Sci 107, 9801–9806 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Tao P, Sun J, Wu Z, Wang S, Wang J, Li W, A dominant autoinflammatory disease caused by non-cleavable variants of RIPK1. Nature (2019), doi: 10.1038/s41586-019-1830-y. [DOI] [PubMed] [Google Scholar]
- 264.Degterev A, Ofengeim D, Yuan J, Targeting RIPK1 for the treatment of human diseases. Proc. Natl. Acad. Sci 116, 201901179 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Yang X, Miyawaki T, Kanegane H, SAP and XIAP deficiency in hemophagocytic lymphohistiocytosis. Pediatr. Int 54 (2012), pp. 447–454. [DOI] [PubMed] [Google Scholar]
- 266.Lawlor KE, Khan N, Mildenhall A, Gerlic M, a Croker B, a D’Cruz A, Hall C, Kaur Spall S, Anderton H, Masters SL, Rashidi M, Wicks IP, Alexander WS, Mitsuuchi Y, a Benetatos C, Condon SM, Wong WW-L, Silke J, Vaux DL, Vince JE, RIPK3 promotes cell death and NLRP3 inflammasome activation in the absence of MLKL. Nat. Commun 6, 1–19 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Vince JE, Wong WWL, Gentle I, Lawlor KE, Allam R, O’Reilly L, Mason K, Gross O, Ma S, Guarda G, Anderton H, Castillo R, H??cker G, Silke J, Tschopp J, Inhibitor of Apoptosis Proteins Limit RIP3 Kinase-Dependent Interleukin-1 Activation. Immunity. 36, 215–227 (2012). [DOI] [PubMed] [Google Scholar]
- 268.Yabal M, Müller N, Adler H, Knies N, Groß CJ, Damgaard RB, Kanegane H, Ringelhan M, Kaufmann T, Heikenwälder M, Strasser A, Groß O, Ruland J, Peschel C, Gyrd-Hansen M, Jost PJ, XIAP Restricts TNF- and RIP3-Dependent Cell Death and Inflammasome Activation. Cell Rep. 7, 1796–1808 (2014). [DOI] [PubMed] [Google Scholar]
- 269.Hakem A, El Ghamrasni S, Maire G, Lemmers B, Karaskova J, Jurisicova A, Sanchez O, Squire J, Hakem R, Caspase-8 is essential for maintaining chromosomal stability and suppressing B-cell lymphomagenesis. Blood. 119, 3495–3502 (2012). [DOI] [PubMed] [Google Scholar]
- 270.Liccardi G, Ramos Garcia L, Tenev T, Annibaldi A, Legrand AJ, Robertson D, Feltham R, Anderton H, Darding M, Peltzer N, Dannappel M, Schünke H, Fava LL, Haschka MD, Glatter T, Nesvizhskii A, Schmidt A, Harris PA, Bertin J, Gough PJ, Villunger A, Silke J, Pasparakis M, Bianchi K, Meier P, RIPK1 and Caspase-8 Ensure Chromosome Stability Independently of Their Role in Cell Death and Inflammation. Mol. Cell 73, 413–428.e7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Teitz T, Wei T, Valentine MB, Vanin EF, Grenet J, Valentine VA, Behm FG, Look AT, Lahti JM, Kidd VJ, Caspase 8 is deleted or silenced preferentially in childhood neuroblastomas with amplification of MYCN. Nat. Med (2000), doi: 10.1038/75007. [DOI] [PubMed] [Google Scholar]
- 272.Hopkins-Donaldson S, Ziegler A, Kurtz S, Bigosch C, Kandioler D, Ludwig C, Zangemeister-Wittke U, Stahel R, Silencing of death receptor and caspase-8 expression in small cell lung carcinoma cell lines tumors by DNA methylation. Cell Death Differ. (2003), doi: 10.1038/sj.cdd.4401157. [DOI] [PubMed] [Google Scholar]
- 273.Martinez R, Setien F, Voelter C, Casado S, Quesada MP, Schackert G, Esteller M, CpG island promoter hypermethylation of the pro-apoptotic gene caspase-8 is a common hallmark of relapsed glioblastoma multiforme. Carcinogenesis (2007), doi: 10.1093/carcin/bgm014. [DOI] [PubMed] [Google Scholar]
- 274.Stupack DG, Teitz T, Potter MD, Mikolon D, Houghton PJ, Kidd VJ, Lahti JM, Cheresh DA, Potentiation of neuroblastoma metastasis by loss of caspase-8. Nature (2006), doi: 10.1038/nature04323. [DOI] [PubMed] [Google Scholar]
- 275.Boege Y, Malehmir M, Healy ME, Bettermann K, Lorentzen A, Vucur M, Ahuja AK, Böhm F, Mertens JC, Shimizu Y, Frick L, Remouchamps C, Mutreja K, Kähne T, Sundaravinayagam D, Wolf MJ, Rehrauer H, Koppe C, Speicher T, Padrissa-Altés S, Maire R, Schattenberg JM, Jeong JS, Liu L, Zwirner S, Boger R, Hüser N, Davis RJ, Müllhaupt B, Moch H, Schulze-Bergkamen H, Clavien PA, Werner S, Borsig L, Luther SA, Jost PJ, Weinlich R, Unger K, Behrens A, Hillert L, Dillon C, Di Virgilio M, Wallach D, Dejardin E, Zender L, Naumann M, Walczak H, Green DR, Lopes M, Lavrik I, Luedde T, Heikenwalder M, Weber A, A Dual Role of Caspase-8 in Triggering and Sensing Proliferation-Associated DNA Damage, a Key Determinant of Liver Cancer Development. Cancer Cell (2017), doi: 10.1016/j.ccell.2017.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Singh A, Sweeney MF, Yu M, Burger A, Greninger P, Benes C, Haber DA, Settleman J, TAK1 inhibition promotes apoptosis in KRAS-dependent colon cancers. Cell. 148, 639–650 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Chun HJ, Zheng L, Ahmad M, Wang J, Speirs CK, Siegel RM, Dale JK, Puck J, Davis J, Hall CG, Skoda-Smith S, Atkinson TP, Straus SE, Lenardo MJ, Pleiotropic defects in lymphocyte activation caused by caspase-8 mutations lead to human immunodeficiency. Nature (2002), doi: 10.1038/nature01063. [DOI] [PubMed] [Google Scholar]
- 278.Niemela J, Kuehn HS, Kelly C, Zhang M, Davies J, Melendez J, Dreiling J, Kleiner D, Calvo K, Oliveira JB, Rosenzweig SD, Caspase-8 Deficiency Presenting as Late-Onset Multi-Organ Lymphocytic Infiltration with Granulomas in two Adult Siblings. J. Clin. Immunol 35, 348–355 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Su HC, Lenardo MJ, Genetic Defects of Apoptosis and Primary Immunodeficiency. Immunol. Allergy Clin. North Am (2008), , doi: 10.1016/j.iac.2008.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Teachey D, Bride K, Autoimmune lymphoproliferative syndrome: More than a FAScinating disease. F1000Research (2017), , doi: 10.12688/f1000research.11545.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Lehle AS, Farin HF, Marquardt B, Michels BE, Magg T, Li Y, Liu Y, Ghalandary M, Lammens K, Hollizeck S, Rohlfs M, Hauck F, Conca R, Walz C, Weiss B, Lev A, Simon AJ, Groß O, Gaidt MM, Hornung V, Clevers H, Yazbeck N, Hanna-Wakim R, Shouval DS, Warner N, Somech R, Muise AM, Snapper SB, Bufler P, Koletzko S, Klein C, Kotlarz D, Intestinal Inflammation and Dysregulated Immunity in Patients With Inherited Caspase-8 Deficiency. Gastroenterology. 156, 275–278 (2019). [DOI] [PubMed] [Google Scholar]
- 282.Fischer U, Stroh C, Schulze-Osthoff K, Unique and overlapping substrate specificities of caspase-8 and caspase-10. Oncogene. 25, 152–159 (2006). [DOI] [PubMed] [Google Scholar]
- 283.Horn S, Hughes MA, Schilling R, Sticht C, Tenev T, Ploesser M, Meier P, Sprick MR, MacFarlane M, Leverkus M, Caspase-10 Negatively Regulates Caspase-8-Mediated Cell Death, Switching the Response to CD95L in Favor of NF-κB Activation and Cell Survival. Cell Rep. (2017), doi: 10.1016/j.celrep.2017.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]