

Abstract
Growth promoting variants in PIK3CA cause a spectrum of developmental disorders, depending on the developmental timing of the mutation and tissues involved. These phenotypically heterogeneous entities have been grouped as PIK3CA-Related Overgrowth Spectrum disorders (PROS). Deep sequencing technologies have facilitated detection of low-level mosaic, often necessitating testing of tissues other than blood. Since clinical management practices vary considerably among healthcare professionals and services across different countries, a consensus on management guidelines is needed. Clinical heterogeneity within this spectrum leads to challenges in establishing management recommendations, which must be based on patient-specific considerations. Moreover, as most of these conditions are rare, affected families may lack access to the medical expertise that is needed to help address the multi-system and often complex medical issues seen with PROS. In March 2019, macrocephaly-capillary malformation (M-CM) patient organizations hosted an expert meeting in Manchester, United Kingdom, to help address these challenges with regards to M-CM syndrome. We have expanded the scope of this project to cover PROS and developed this consensus statement on the preferred approach for managing affected individuals based on our current knowledge.
Keywords: PIK3CA-related overgrowth spectrum, clinical management, mosaic, expert consensus
INTRODUCTION
Activating variants in PIK3CA cause a phenotypic spectrum of developmental disorders known as the PIK3CA-Related Overgrowth Spectrum (PROS) (Table 1A) (1-10). Deep sequencing technologies have facilitated the identification and clinical testing for PROS-related gain-of-function (GoF) variants that are often either absent or present at very low levels, in peripheral blood. Typically, testing other types of tissues or biopsies is necessary for detection of ultra-low frequency mosaic variants (11-14). As variants are typically growth-promoting, a random and post-zygotic mutational event in PIK3CA may provide the cell of origin with a selective growth advantage. The phenotypic consequences are dependent on when, during development, and where (in which cell) the mutational event occurred, on the distribution of the mutated cell’s progeny as well as on the activating strength of the variant.
Table 1.
PROS classification
| A. Phenotypic spectrum | |
|---|---|
| Localised or tissue-specific, MIM | Multisystemic and with possible brain involvement, MIM |
| Macrodactyly, #155500 | Fibroadipose Overgrowth/Hemihyperplasia-Multiple Lipomatosis (FAO/HHML) |
| Epidermal Nevi, #162900 | Congenital Lipomatous Overgrowth, Vascular Malformations, Epidermal Nevi, Scoliosis/Skeletal and Spinal Abnormalities syndrome (CLOVES), #612918 |
| Seborrheic Keratosis, #182000 | Capillary malformation of the lower lip, Lymphatic malformation of face and neck, Asymmetry of face and limbs, and Partial/generalized Overgrowth syndrome (CLAPO), #613089 |
| PIK3CA-related muscular overgrowth with ectopic accessory muscles (Al-Qattan et al., 2018). | Megalencephaly-Capillary Malformation-Polymicrogyria syndrome (MCAP), #602501 |
| Hemiorofacial asymmetry with peripheral nerve enlargement andperineuriomatous pseudo-onion bulb proliferations (Koutlas et al., 2021) | |
| Facial infiltrating lipomatosis (FIL) (Couto et al., 2017) | |
| Lymphatic malformations (LM) (Zenner et al., 2019) | |
| Klippel-Trenaunay syndrome (KTS), %149000 | |
| B. Management* | |
| MCAP | Non-MCAP PROS |
| Activating, mosaic PIK3CA variant | Activating, mosaic PIK3CAvariant |
| Meg and/or PMG | No significant Meg or PMG or Sx |
| Extra-CNS overgrowth and/or vasc findings | Extra-CNS overgrowth and vasc findings |
rare PROS individuals have been reported with consitutional variants (De Graer et al., 2020)
Meg: Megalencephaly (see Appendix), PMG: Polymicrogyria, Sx: syrinx, CNS: Central Nervous System, vasc: vascular including cutaneous
These mosaic disorders are quite variable in nature, extent, progression, and severity of the clinical presentation. Conversely, deep phenotyping has shown that despite marked diversity, these conditions are also clinically overlapping (15, 16). While many affected individuals have all manifestations of, and are typical for, previously named entities (e.g., MCAP or CLOVES), there are innumerable individuals who manifest only some aspects of these entities and those who manifest attributes of more than one of these entities but within the broad spectrum of PROS. Recently, we have proposed a dyadic approach to the diagnosis and terminology of entities and we believe this may be equally applicable for PROS (17).
This extensive phenotypic overlap warrants a holistic, multidisciplinary approach with expert-driven medical management, and taking into considerations available clinical screening tools in various healthcare settings. Since clinical management practices vary considerably among different professionals, healthcare services, and countries, general management guidelines are needed (18-20). Moreover, as most of these conditions are rare, affected families may lack access to the medical expertise that is needed to help address the multi-system and often complex medical issues seen with PROS. An international collaboration to derive evidence-based management and surveillance guidelines was stimulated and funded by M-CM/MCAP patient-advocacy and support groups from the United Kingdom, the United States and Spain (21).
MATERIALS AND METHODS
These are detailed in Figure 1. The information contained in the publications chosen following literature curation was sent to a panel of experts and assessed using the AGREE II tool (22). We asked each expert to derive management recommendations based on the evidence provided but also on further evidence from their own field if additional recommendations were necessary. The experts were allowed to abstain from scoring any recommendation if it was outside their area of expertise. Recommendations were collated and discussed at the expert consensus meeting held in Manchester, United Kingdom, in March 2019. Following the expert consensus meeting, a first draft of the consensus statement was circulated to the group for review. The final, consensus document is presented here. The majority of the recommendations presented here scored ‘+++’ for Agreement (>75% agree with recommendation) and ‘A’ for Evidence (based on evidence +/− expert consensus) (see the AGREE II tool, Figure 1). We list instead, in Table 6, the recommendations with lower scores.
Figure 1.
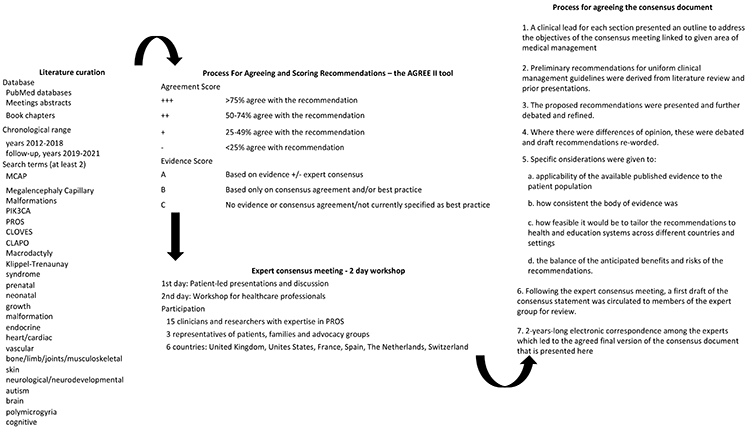
Methods.
Table 6.
PROS Recommendations with lower AGREE II Tool scores
| A. Agreement score +++/++, Evidence score B: based only on consensus agreement and/or best practice (Methods and Figure 1) | |
|---|---|
| Section and PROS phenotype (see Table 1) | Recommendation |
| Genetic testing | |
| Klippel Trenaunay syndrome | The confirmation of a PIK3CA pathogenic variant in these patients can be quite challenging and obtaining affected tissue at the time of surgery may offer the best chance. If use of inhibitors is going to be considered, demonstrating the mutation will be necessary, so planning and coordination will be critical |
| Pre- and perinatal diagnosis | |
| PROS | In any pregnancy where the foetus is known to have PROS, pregnancies may be complicated by polyhydramnios, prematurity, and difficult delivery in case of megalencephaly (foetal MRI) and/or breech presentation. The foetal management team should be made aware of these complications and arrangements for delivery made accordingly |
| Critical care | |
| PROS | Request cardiology review in all individuals after diagnosis and follow-up as appropriate in case of a diagnosis of cardiac arrhythmia In the presence of vascular anomalies assess the risk of thrombosis, haemorrhage and infections |
| CNS overgrowth or dysplasia | |
| PROS | The decision to use endoscopic third ventriculostomy, a ventricular diversion procedure (ventriculoperitoneal, ventriculoatrial etc.) vs. suboccipital decompression is beyond the scope of these recommendations. However, a relevant surgical consideration is that these procedures can carry an elevated risk of blood loss and postoperative vascular complications (e.g., stroke). While the incidence of intracranial vascular abnormalities in PROS is unknown, these patients can be at risk for vascular dysregulation. Therefore, surgical management decisions require a well-versed multispecialty team lead by a neurosurgeon to make appropriate recommendations that are individualized on a case-by-case basis |
| MCAP with ventriculomegaly/hydrocephalus | Historically, many children with MCAP and ventriculomegaly were empirically shunted in the absence of evidence for clear hydrocephalus. Given the rapidly progressive nature of megalencephaly in MCAP (with the head OFC crossing centiles especially early in life) combined with the co-occurrence of ventriculomegaly, close monitoring of the child is recommended including (a) close monitoring of OFC progression and trajectory, (b) watching for any signs/symptoms of increasing intracranial pressure/hydrocephalus, (c) close inspection of the brain MRI scan by an experienced neuroradiologist for any signs of increased ICP and/or obstructive ventriculomegaly. The decision of whether to intervene neurosurgically (whether by shunting or a third ventriculostomy) should be ideally made by a multidisciplinary team |
| non-MCAP PROS with truncal involvement | This subset of patients are at risk for spinal lesions including spinal vascular malformations or lipomatous lesions. We recommend a referral to a neurologist for examination and the evaluation of spinal imaging |
| Epilepsy | |
| PROS | We recommend following standard guidelines for treatment such as those established by the International League Against Epilepsy (ILAE) and referral to an epileptologist or epilepsy centre, for additional considerations such as ketogenic diet or epilepsy surgery (Glauser et al., 2013; Wilmhurst et al., 2015 and Kwan et al., 2010) |
| PROS with drug resistant epilepsy | |
| PROS with drug resistant epilepsy and focal cortical dysplasia or unilateral cortical involvement | |
| Neurodevelopment | |
| non-MCAP PROS with disfiguring lesions | These individuals may particularly benefit from psychological support to develop coping strategies in case of lesions that impact their quality of everyday and relational life |
| Endocrinopathies | |
| PROS with hypoglycaemia | In several reported cases hypoglycaemia has gradually improved with age, with progressive increases in fasting tolerance allowing gradual diminution of nutritional support (Leiter et al, 2017), however variable patterns have been reported including later presentations (McDermott et al., 2018). This is unpredictable and means that intermittent re-evaluation of fasting tolerance may be warranted |
| PROS with hypoglycaemia and/or linear growth retardation | A trial of GH may be indicated. In this case careful follow up of linear growth and trajectory of overgrowth should be undertaken, in case pathological growth is exacerbated by GH. Pragmatic delay of GH therapy until after 2 years of age has been suggested, avoiding the major period of brain growth. There exists a theoretical possibility that GH may promote tumourigenesis in the context of PROS in the longer term, as an “oncogenic” PIK3CA variant is already present. Reassuringly, to date no excess risk of neoplasia has been discerned in untreated individuals with MCAP. There are limited reports only of GH therapy in individuals with molecularly proven MCAP to date. It has generally been well tolerated with a single exception, and in at least four cases it proved effective with no discernible adverse effects (Davis et al, 2020). Further evidence is required before a confident statement can be made about relative risks and benefits of GH therapy in GH-deficient PROS |
| Tumourigenesis | |
| PROS | Given the absence of systematic data, we recommend counseling the families on a case-by-case basis |
| Oral/Dental management | |
| PROS with asymmetric facial overgrowth | The most frequent problems are abnormal tooth eruption, dental crowding, gingival and periodontal pathologies and these should be treated as in the general population (see also Appendix) |
| Facial infiltrating lipomatosis | |
| Orofacial presentation by Koutlas et al 2021 | |
| MCAP | |
| B. Evidence score C: no evidence or consensus agreement/not currently specified as best practice (Methods and Figure 1) | |
| Approach to negative testing results | |
| PROS | Currently, there is no evidence to direct how many times re-testing or re-biopsy should be considered where the a priori clinical probability is high. Each case must be assessed individually, taking into account previous experience with each methodology, obtaining new samples, or the need for molecular confirmation in the case of targeted therapies (see also Appendix) |
| Somatic overgrowth (craniofacial, trunk-spine, limb) | |
| MCAP | There are no specific growth charts for MCAP yet, which makes monitoring weight gain challenging |
| Endocrinopathies | The options here are rational but currently untested, and should be undertaken only under endocrine guidance on an experimental basis with due informed consent |
| PROS with neonatal, insulin-dependent hypoglycaemia | Somatostatin/Diazoxide may be of benefit. In intractable cases dependent on intravenous glucose infusion use of an mTOR inhibitor such as sirolimus and/or a PIK3CA inhibitor may be considered |
| PROS with severe, intractable, hypoketotic, hyper insulinemic hypoglycaemia dependent upon IV glucose infusion | Consideration should be given to the exploratory use of an mTOR inhibitor such as sirolimus and/or a PIK3CA inhibitor |
| C. Important issues where there is not yet any consensus | |
| PROS | Surgical management of overgrowth tissue including orthopaedic |
| Surgical management of vascular anomalies and lymphatic malformations | |
| Management of haemorrhage; of inflammation and infections; of pain | |
| Management by interventional radiology | |
| Physiotherapy | |
| Testing of potential pharmacologic therapies | |
RESULTS
Diagnostic criteria and nomenclature
The PROS umbrella term and diagnostic criteria were defined by Keppler-Noreuil et al [2015]. Table 1B exemplifies the distinction we developed to guide the clinical management of these entities, once molecularly confirmed. We have defined two broad phenotypic categories to which these management guidelines should be applied: MCAP and non-MCAP PROS. When one of these terms is used hereafter, we intend that the recommendation applies to that category. If we instead specify “PROS” the recommendation applies to both categories.
It is important to note that we are not suggesting that these descriptors supplant specific clinical terms such as CLOVES, rather, it is a heuristic categorization that we believe is useful to classify individuals into groups for application of our management recommendations. We appreciate that families and healthcare providers are concerned about changes in nomenclature, but the insights in disease pathogenesis obtained from progress in genomics, pathology, and neuroradiology require regular adaptations. Irrespective of any of our preferences, the nearly continuous inter-patient variation of PROS, especially that associated with mosaic alterations, forces us to adopt pragmatic approaches and we acknowledge that these approaches may and will evolve with time.
Genetic testing
1. General discussion
Establishing a molecular diagnosis in PROS is important and applying management strategies to individuals without a molecular diagnosis can be very challenging. A molecular confirmation may guide patient management and monitoring, offer new therapeutic approaches, where available, and allow provision of appropriate recurrence risks. Testing DNA from affected tissue with highly sensitive diagnostic methods offers the best chance of detecting postzygotic/somatic variants in PIK3CA (5, 12, 13).
Cell-free DNA (cfDNA) is now routinely used in cancer applications including testing urine samples in patients with PROS and kidney involvement (23). However, the diagnostic yield may not be as high as in cancer applications as: (a) the probability of cfDNA carrying the mutation may vary with clinical aspects (i.e., presence of a vascular malformation). In PROS, there is probably not as much or any neovascularization as in cancer; (b) the VAF with cfDNA can be very low, and ddPCR is used instead of NGS. ddPCR can only screen about five variants and is thus limited to hotspots screening (or confirmation of NGS screening). Taking into consideration the allelic heterogeneity of PROS (13), false negative tests are expected; (c) it is difficult to interpret the biological significance of very low VAFs (0.001%). Overall, cfDNA testing can be compared, at the moment, with blood or saliva testing against affected tissue testing: it is less invasive, but the diagnostic yield is lower. Further systematic data are needed to advance any recommendations about its use in a diagnostic setting.
2. Recommendations for PROS
Taking into consideration the extensive phenotypic overlap between PROS (Table 1), and the different ranges of mosaicism within tissues and samples, we outline these recommendations in Table 2.
Table 2.
Genetic testing recommendations for PROS
| Method | The molecular diagnostic approach should utilize techniques optimized for the detection of low-level mosaic variants (low variant allele fraction or VAF, see also Appendix). Because many of the variants that cause PROS are typically absent in peripheral blood, Next Generation Sequencing (NGS) or digital droplet polymerase chain reaction (ddPCR) testing |
| Sampling | Affected tissues (i.e., during reduction surgery of macrodactyly or for debulking of ectopic accessory muscles) offer the optimal approach. When tissue samples are not available, a biopsy of affected tissue (e.g., skin with a vascular malformation in a region with soft tissue overgrowth) may be considered. DNA should be extracted without previous culture. Genetic testing on formalin-fixed, paraffin-embedded (FFPE) samples from prior surgeries is also possible. Although, in this type of sample, the DNA may be degraded, reducing the diagnostic yield |
| Optimal depth of NGS | The absolute minimum number of times that a variant must be determined depends on the platform used and its ability to discriminate against background noise. For example, on hybridization-based NGS platforms, a minimum 350x and mean 500x coverage may be sufficient to detect VAFs of 5%. These numbers can be considerably higher on amplicon-based NGS platforms |
| Detection of low-level mosaicism | Previous studies have shown a lower diagnostic rate in individuals presenting with very localized or tissue-specific features (e.g., isolated macrodactyly) (Mirzaa et al., 2016). In a clinical context, diagnostic techniques able to detect mosaic variants with allele frequencies or VAF as low as 5% (0.05), or even lower at 1% (0.01) should be selected |
| Variant validation | Validation by orthogonal methods (e.g., Sanger sequencing for VAFs ≥20%, pyrosequencing for VAFs ≥5%, or ddPCR for VAFs ≤5%) or by a second NGS study of mosaic candidate variants may be needed. Variant validation might not be necessary when the candidate variant is detected in more than one different tissue of the same patient (taking into consideration the limit of detection of the technique). When validating variants in PIK3CA, it is necessary to remember that exons 10 to 14 of this gene share 95% identity with another region or pseudogene of the genome located on chromosome 22. Therefore, validation tests must be designed to discriminate the location of these variants (e.g., using long-range PCR upstream of the final PCR amplification) (Rodriguez-Laguna et al., 2018) |
| Approach to negative testing results | Given that low-level mosaicism is observed in many individuals with PROS, it is recommended to re-evaluate the quality of the sample (e.g., FFPE samples), the sampled tissue, the mosaic detection limit of the technique, and the clinical diagnosis |
3. Recommendations for MCAP
It is possible to detect pathogenic variants in peripheral blood or saliva samples in some individuals. The level of mosaicism in blood or saliva samples is usually low; thus, the diagnostic yield is lower (5, 13) (see also Appendix).
4. Recommendations for non-MCAP PROS
In highly focal disorders within the PROS spectrum (e.g., CLOVES, fibroadipose hyperplasia), pathogenic variants are detectable in affected tissues with wide ranges of mosaicism. There is conflicting evidence about correlation between diagnostic rate and localised or tissue-specific presentations (5, 9, 12, 13). This problem, however, could be restricted to certain cases of PROS, e.g., PIK3CA-related muscular overgrowth, and due to the very restricted distribution of affected tissue. In such cases, NGS following sampling of affected tissue is the best option (e.g., during reduction surgery of macrodactyly or for debulking of ectopic accessory muscles).
Pre-and perinatal diagnosis
1. General discussion
The increase in the numbers of diagnosed individuals has broadened our knowledge of the variability and natural history of PROS. While most individuals with PROS present at or shortly after birth, prenatal findings of Central Nervous System (CNS) and/or somatic overgrowth have been reported in affected individuals (24-26). However, prenatal NGS genetic testing for PIK3CA variants should take into consideration the fact that these are frequently mosaic disorders which may limit the ability to detect genomic variants and to provide prognostic information.
2. Recommendations for PROS
PROS should be included as a differential diagnosis in cases of foetal overgrowth with (a) antenatal megalencephaly; (b) presence of fibroadipose overgrowth; (c) foetal pleural effusion/ascites (3, 24-26).
Somatic overgrowth (craniofacial, trunk-spine, limb)
1. General discussion
Overgrowth in PROS individuals is typically congenital in onset, static or progressive with manifestations that include either two of a broad spectrum including: 1) adipose, muscle, nerve, brain, or skeletal overgrowth, 2) vascular malformations, and 3) epidermal nevi; or isolated features that may include any of the two of: large lymphatic malformation, macrodactyly, truncal adipose overgrowth, hemimegalencephaly, epidermal nevi, seborrheic keratoses, or benign lichenoid keratoses.
Facial infiltrating lipomatosis (FIL) is a congenital disorder that causes overgrowth of one side of the face. Couto et al [2017] evaluated tissues from three individuals and detected PIK3CA variants in every tissue and cell type tested with VAFs ranging from 1.5% to 53%. Klippel–Trenaunay syndrome (KTS), is a slow-flow combined vascular malformation characterized by the triad of capillary malformation, venous malformation with or without lymphatic malformation, and limb overgrowth, which is also caused by activating PIK3CA variants (6). Lymphatic malformations (LM) are congenital, non-neoplastic vascular malformations associated with a narrow spectrum of postzygotic activating PIK3CA variants. Zenner et al [2019] found pathogenic variants in affected LM tissue in 64 individuals (79%) with isolated LMs, with VAFs ranging from 0.1% to 13%. Luks et al [2015] detected somatic PIK3CA variants in individuals with lymphatic and other vascular malformation and overgrowth conditions: isolated LM (16 of 17), KTS with LM (19 of 21), fibroadipose vascular anomaly (5 of 8), or congenital lipomatous overgrowth with vascular, epidermal, and skeletal anomalies syndrome (31 of 33), five specific variants accounting for ~80% of affected individuals. CLAPO syndrome (Capillary malformation of the lower lip, Lymphatic malformation predominant on the face and neck, Asymmetry, and Partial/generalized Overgrowth) is associated with PIK3CA mosaic activating variants (7). Upper limb muscle overgrowth with hypoplasia of the index finger is an overgrowth syndrome caused by a specific somatic PIK3CA variant (c.3140A>G) (8). These examples illustrate the variability of PROS and the need for individual management based on symptoms.
2. Recommendations for PROS
Regular growth measurements as part of a routine neonatal/paediatric assessment are recommended in all individuals with PROS. Growth assessment should specifically seek asymmetry of the legs, and all individuals with leg length discrepancy (LLD) should be referred to an orthopaedic surgeon for review and management. Of note, LLD may be either true or anatomical because of asymmetric overgrowth of the lower limbs, or functional because of involvement of the hips (by hip dysplasia or contractures). Where rapid growth of a specific body area is observed referral should be made for consideration of other monitoring techniques such as volumetric studies. Imaging studies, including angiography should also be considered. Referral for advice to a feeding team is appropriate if there are concerns, especially where the CNS is involved in any of the three categories of PROS.
3. Recommendations for MCAP
Individuals with MCAP may have complex feeding difficulties that should be assessed and taken into consideration in monitoring weight gain.
4. Recommendations for non-MCAP PROS
In individuals with non-MCAP PROS, segmental overgrowth may be extreme, with concomitant paucity of adipose tissue in non-overgrown areas. Liposuction has been used to debulk fat overgrowth rather than open resection, however only subjective assessment of outcomes is available. Some cases have been complicated by post-operative weeping of lymphatic fluid (Biesecker, unpublished observations). Surgical debulking is an option for localised overgrowth but the efficacy of surgical debulking is not proven. There are outstanding questions about the possibility and speed of regrowth following surgery (30).
CNS overgrowth or dysplasia
1. General discussion
In a cohort of 72 individuals with PROS, Mirzaa et al [2016] evidenced a phenotypic range including two individuals with non syndromic megalencephaly (Meg, see also Appendix), 58 with MCAP, eight with body overgrowth and vascular malformations without Meg, two individuals with Hemi-Meg or dysplastic Meg and two with CLOVES syndrome. Including previously published data, 41 PIK3CA variants were identified including three hotspots (p.Glu542Lys, p.Glu545Lys, p.His1047Arg), two strong (p.Cys420Arg, p.His1047Leu), and 36 known or presumed moderate GoF variants, several of them recurrent. This data evidences the phenotypic variability and allelic heterogeneity regarding the CNS overgrowth and dysplasia in individuals with PROS who will mostly benefit from a coordinated and multidisciplinary clinical approach tailored to the individual's specific needs and manifestations.
2. Recommendations for PROS
A cranial MRI scan should be performed in all individuals with PROS in the presence of (a) rapidly enlarging occipital-frontal circumference (OFC) and/or (b) moderate-to-severe developmental delay or intellectual disability (ID) (c) epilepsy or (d) facial or craniofacial neurological involvement. In all affected individuals with brain involvement there is potential for hydrocephalus which can be due to venous hypertension/congestion or cerebellar tonsillar herniation (CBTE). Table 3A summarises our recommendations for regular monitoring of PROS individuals with CNS overgrowth or dysplasia including critical care recommendations (Table 3B and 6).
Table 3.
PROS critical care recommendations
| A. Recommendations for regular monitoring of diagnosed CNS overgrowth or dysplasia | |
|---|---|
| From birth until age 8 years | Cranial MRI every 6 months until age 2 and then annually until age 8 years to monitor specifically for progressive ventriculomegaly/hydrocephalus and CBTE or Chiari malformations. Brain MRIs may necessitate sedation in children and the risk to benefit ratio need to be weighed on an individual basis. An exception can be made for children over 4 years of age who are asymptomatic or have no signs suggesting raised ICP or Chiari and whose OFCs are more or less parallel to the standard growth curve (albeit large). In such instances, we recommend yearly follow-up with a paediatric neurologist for routine monitoring. |
| If an individual is diagnosed between 2 and 8 years of age | A baseline brain MRI is recommended at the time of diagnosis with yearly MRI scans to monitor for progressive ventriculomegaly/hydrocephaly and CBTE. Additionally, a brain MRI should be considered if the head growth trajectory rapidly accelerates (for e.g., with crossing of OFC centiles on the growth chart). |
| Children with ventriculomegaly or CBTE | Should be referred for neurosurgical evaluation, management, and monitoring for progression. The decision for neurosurgical intervention, including placement of a ventriculoperitoneal shunt, endoscopic third ventriculostomy or posterior fossa decompression, requires careful consideration by a genetics/paediatric neurosurgical team and is beyond the scope of the current recommendations. |
| Management of CBTE | Nonsurgical management for stable, asymptomatic CBTE is appropriate, and decompression should be considered on a case-by-case basis especially when symptoms of brainstem compression appear or syringomyelia develops. This requires the input of a multi-disciplinary team including a genetics/paediatric neurosurgical team familiar with the care of this complex syndrome. |
| B. CNS critical care and mortality risk recommendations | |
| Look for signs or symptoms of raised ICP | 1. Consider surgical intervention for hydrocephalus or cerebellar tonsillar herniation if symptomatic |
| 2. Carry out nocturnal polysomnography if raised ICP is suspected | |
| Request a brain MRI in patients with MCAP PROS | Patients with ventriculomegaly and cerebellar tonsillar herniation should be considered for ventriculoperitoneal shunting initially |
| Inquire about symptoms of sleep apnoea at each medical check. | All patients with PROS with head and neck involvement should have a sleep study |
| C. Other critical care and mortality risk recommendations | |
| Be aware of the risk of pleural effusions | 1. Increased risk for effusions occurs with/after pneumonia. Check for this specifically, clinically and by imaging, if signs are suggestive 2. Effusions should be ruled out by chest imaging if there are significant respiratory difficulties in patients with PROS at any age |
MRI, Magnetic Resonance Imaging; ICP, IntraCranial Pressure; OFC, OccipitoFrontal Circumference; CBTE, cerebellar tonsillar herniation.
3. Recommendations for MCAP
In individuals with MCAP, regular neurodevelopmental assessments during infancy are recommended, including routine and frequent monitoring of brain overgrowth by serial Occipitofrontal Circumference (OFC) measurements. This is ideally performed by a consistent operator or in the same centre and documented on general population OFC charts. Most individuals with MCAP have OFCs that are either > 2SD above the mean or within the upper normal range (96th - 99.6th centile) at birth that may rapidly progress to OFCs above two standard deviations or more, usually within the first 1-2 years of life.
Progressive ventriculomegaly and hydrocephalus in these children is multifactorial and may be a result of both obstructive and communicating hydrocephalus, especially when the child develops cerebellar tonsillar herniation with posterior fossa cerebellar overgrowth. We caution the use of ventriculoperitoneal shunting in an asymptomatic child without progressive ventriculomegaly. We recommend that a paediatric neurosurgeon familiar with this condition is involved in the decision making and management of ventriculomegaly in these children (see also Table 6A).
A subset of children with MCAP have cortical malformations, the most common being perisylvian polymicrogyria (PMG). The presence of PMG, in general, increases the risk for developmental delay/intellectual disability, epilepsy, and muscle tone abnormalities. Bilateral, perisylvian PMG in particular can be associated with oromotor weakness leading to feeding, swallowing, and expressive language difficulties. Anecdotal data suggest that younger children with PMG (specifically of the frontal region) have hypotonia, are not spastic, and may have pseudobulbar problems, whereas older children may have spasticity in addition to pseudobulbar problems, although the frequency of these complications in MCAP specifically, and PROS in general, remain to be determined (31-37).
Chiari malformation and syringohydromyelia are known features of MCAP, but clinicians should be aware of the possibility that syringomyelia, if undetected or untreated in MCAP, may develop relatively rapidly over the course of 2-3 years, potentially involving the entire cord, possibly in conjunction with progressive CBTE and cerebrospinal fluid outflow obstruction (36). In addition to following standard guidelines for cranial MRI surveillance in MCAP, clinicians should monitor closely for neurological signs/symptoms that are suggestive of Chiari malformation or syringohydromyelia, with low threshold for considering MRI of the cranium and spine if Chiari malformation is a differential diagnostic consideration.
3. Recommendations for non-MCAP PROS
The assessment, management, and follow-up of individuals with spinal lesions requires a multidisciplinary team evaluation (neurologist, neurosurgeon, vascular surgeon, and/or interventional radiologist), individualised on a case-by-case basis. Individuals with spine curvature abnormalities (e.g., kyphoscoliosis) require monitoring and imaging (by spinal radiographs or MRI) as clinically indicated. Spinal canal ultrasonography is of limited use after the age of 2 months and should only be used as a screening study in infants with truncal involvement because of risk of tethered cord and lipo-meningocele (30).
Epilepsy
1. General discussion
Epilepsy may be a significant concern for individuals with PROS with brain involvement. Cortical malformations seen in individuals with PROS may affect the severity and frequency of epilepsy. For example, epilepsy associated with PMG (not specifically PROS-related) is often focal in origin, though it can also have generalized onset (38). The precise incidence of epilepsy is not known but has been estimated to be as high as 50% in children with bilateral cortical malformations (38, 39). The effect of seizures on cognitive outcomes in PROS is not known. In general, uncontrolled seizures with earlier age of onset (i.e., infancy) is associated with poorer neurocognitive outcomes (40, 41), but this observation has not been tested specifically with respect to PROS. The average age of onset of epilepsy in association with PROS is also not known. Seizures can occur anytime from the neonatal period to adulthood.
2. Recommendations for PROS
A referral to a paediatric neurologist/epileptologist is recommended if seizures occur in any individual with features of PROS. There is insufficient evidence for individuals with PROS who develop epilepsy, to recommend a specific antiepileptic drug (AED) or a management mainstay. Accordingly, we recommend following standard guidelines as outlined in Table 6A (42-44).
Neurodevelopment
1. General discussion
There is a spectrum of neurodevelopmental impairment associated with PROS that includes ID, autism spectrum disorder (ASD), communication disorder, attention deficit hyperactivity disorder (ADHD), anxiety, other behavioural issues, and motor dysfunction. The range of cognitive developmental ability varies from normal to mildly affected to severe cognitive problems. Yeung et al [2017] performed exome sequencing in 21 individuals with megalencephaly and developmental delay/autism spectrum disorder and identified pathogenic variants in PTEN (n=4), PIK3CA (n = 3), MTOR (n = 1) and PPP2R5D (n = 2). Individuals who tested positive for pathogenic variants in the PI3K-AKT-mTOR pathway had a lower developmental quotient than the rest of the cohort (DQ = 62.8 vs. 76.1, p = 0.021). Their presentation was non-specific, except for megalencephaly (45).
2. Recommendations for PROS
Neurodevelopmental concerns are of particular importance for individuals with PROS with brain involvement. We recommend a formal neuropsychological assessment in all individuals with PROS and with possible brain involvement (Table 1). In contrast, individuals without brain involvement do not always need neuropsychological assessment. Detailed recommendations are outlined in Table 4.
Table 4.
Neurodevelopment: Recommendations for individuals with PROS with brain involvement
| Neuropsychological assessment | All individuals should have a formal neuropsychological assessment. Upon initial diagnosis, individuals should undergo a comprehensive assessment of cognitive, communication, socialization, behavioural, and motor needs. This assessment would identify baseline neurodevelopmental deficits and strategize a plan for ongoing monitoring and treatment |
| Treatment plans | These should target each individual neurodevelopmental disorder (e.g., ID, ASD, ADHD, anxiety, etc.) and should be based on standard of care (e.g., applied behavioural analysis therapy for autism). For affected infants and toddlers, some locales provide early intervention programs which evaluate for developmental delay and provide developmental services pertaining to different developmental domains |
| For school age children | Parents/caretakers should consider requesting an individualized education plan (IEP) or an Education and Healthcare Plan (EHCP) in order to optimize learning and development. Depending on the individual’s developmental profile, possible developmental services that may be warranted include physical therapy, occupational therapy, and speech and language therapy. In individuals who are nonverbal, augmentative and alternative communication may be helpful |
| Transition | Assessments should be offered around normal transition periods (i.e., puberty/move to high school and transition to adult care). These often entail a change in behaviour or routines that need be taken in consideration along with the individual’s (a) range of cognitive abilities, (b) coping/social strategies in view of possible disfiguring lesions, (c) possible co-existing autistic traits and (d) size/overgrowth |
| Neurological decline/developmental regression | The range of developmental levels is wide, and individuals can be expected to progress along their own learning curve. Loss of developmental milestones should prompt evaluation for treatable causes of developmental regression (e.g., shunt malfunction or tethered cord). A neurological decline may be caused by worsening of a Chiari malformation with the associated risk of developing a syrinx or hydrocephaly, and primary or repeat MRI imaging should be considered. Specific complaints to consider are (paroxysmal) neck pain, limb weakness, swallowing problems, and sleep apnoea |
Tumourigenesis
1. General discussion
The underlying pathogenetic mechanism for this spectrum of disorders suggests a possible increased tumourigenesis risk. MCAP is primarily associated with somatic mosaicism for PIK3CA pathogenic variants not commonly identified in tumours, whereas non-MCAP PROS disorders are associated with somatic mosaicism for PIK3CA pathogenic variants at the cancer-related mutational hotspots. Anecdotal observations so far suggest a 1-3.3% Wilms Tumour (WT) risk in cases with a confirmed genetic diagnosis, which is lower than the Beckwith-Wiedemann syndrome (BWS) tumour risk (46, 47). There have been four case reports of individuals with PROS who developed cancer including leukemia, vestibular schwannoma, retinoblastoma, and meningioma (48). Gripp et al. [2016] reported the fourth individual with asymmetric overgrowth due to a somatic PIK3CA variant who had a Wilms tumour or nephroblastomatosis. Similarly to two previously reported individuals with renal tumours, the somatic PIK3CA variant affects the commonly mutated codon 1047. Codon 1047 is most commonly affected by somatic variants in PROS, and fibroadipose overgrowth is common in individuals with codon 1047. Because some individuals with CLOVES develop Wilms tumour, Michel et al [2018] tested urine as a source of DNA for variant detection. They extracted DNA from the urine of 17 and 24 individuals with CLOVES and KTS, respectively, and screened five common PIK3CA mutational hotspots using ddPCR. Six of 17 CLOVES participants (35%) had mutant PIK3CA alleles in urine, while no urine sample from a participant with KTS had detectable variants (49). Biderman Waberski et al. [2018] also evaluated the utility of samples other than affected tissue biopsies for molecular diagnosis of PROS using ddPCR with variant detection at levels as low as 0.001% and found high levels of PIK3CA variant in urine cell-free DNA (cfDNA) that correlated with a history of nephroblastomatosis or Wilms tumour compared with individuals without known renal involvement (p <0.05). Otherwise, the most common benign tumours in PROS are lipomas which may be multiple and usually remain stable in size (30, 31).
2. Recommendations for PROS
Further longitudinal studies are necessary to confidently assess these risks and no recommendation for periodic tumor surveillance can be made at this time (Table 6).
Endocrinopathies
1. General discussion
A range of endocrinopathies have been reported in PROS, affecting a minority of individuals. They include (a) hypoglycaemia, (b) growth hormone (GH) deficiency, and (c) other forms of hypothalamic-pituitary dysfunction. Several non-mutually exclusive mechanisms may contribute to these. The PIK3CA protein mediates signaling by many cell surface hormone or growth factor receptors, including insulin and insulin-like growth factor 1 (IGF1). Pathologically activated PIK3CA may thus mimic insulin and/or IGF1 action. For this to translate into clinically important endocrinopathy, target tissues need to harbour a high variant burden.
When the effect of insulin is mimicked, fasting hypoglycaemia with suppressed ketones and free fatty acids but preserved liver glycogen are seen (50). This resembles congenital hyperinsulinism but plasma insulin concentrations at the time of hypoglycaemia are undetectable. This profile has been reported in MCAP and overlapping forms of PROS with brain involvement (51, 52) and also in people with genetic activation of AKT2, another insulin signaling pathway component (53).
Cell autonomous activation of IGF1 signaling, analogously, may activate tissue growth without stimulation by IGF1. Because negative feedback within the GH-IGF endocrine axis is mediated by IGF1 action on hypothalamus and pituitary, activation of IGF1 signaling at these sites can be erroneously sensed as hyperactivation of the axis, leading to suppression of GH production (50, 51). This poses the dilemma of whether to replace GH even though some tissues are already experiencing increased GH-like activity. Suppressed production of GH also increases susceptibility to hypoglycaemia, as GH is an important “stress” or counter-regulatory hormone that defends glycaemia during severe illness.
Finally, PIK3CA activation may cause endocrinopathy through non-specific developmental dysregulation. This may affect pancreatic islets, dysregulating insulin secretion, or the neuronal connectivity of the hypothalamus and/or pituitary. In keeping with this, some infants with PROS and hypoglycaemia have been reported to have detectable insulin at the time of severe hypoglycaemia (54), while several types of pituitary dysfunction have been reported in PROS with brain involvement (5, 50, 51, 55).
2. Recommendations for PROS
Episodic severe hypoglycaemia mandates emergency rescue. Preventative measures depend on whether it is insulin-dependent or insulin-independent and whether there is underlying deficiency of GH or cortisol. GH deficiency is found in a significant minority of cases of MCAP and other forms of PROS with brain involvement (50, 51, 55). It confers susceptibility to stress hypoglycaemia and/or postnatal linear growth retardation, and may be ameliorated by GH therapy. Nevertheless, cautions and caveats apply. In a small minority of individuals, additional pituitary deficits have been reported, including central hypothyroidism (5, 50, 51, 55), central hypoadrenalism and cryptorchidism, which sometimes denotes central hypogonadism (51). Hypothyroidism has important neurodevelopmental effects if untreated, while hypoadrenalism can cause stress-related hypoglycaemia and hypotension. Our recommendations for screening, diagnosis, treatment and monitoring of endocrinopathies in PROS are summarised in Table 5.
Table 5.
Endocrinopathies: Recommendations for PROS
| Hypoglycaemia | Growth Hormone and other Pituitary Deficiencies | ||
|---|---|---|---|
| Screening and Diagnosis | |||
| Suggestive signs and symptoms | It is important to raise awareness among parents and caregivers that episodic irritability, loss of tone, loss of consciousness, or seizures may be wholly attributable to, or exacerbated by, hypoglycaemia. These are most likely to occur as feeding intervals increase, or during intercurrent illness or other stress | Baseline screening | Baseline screening of thyroid function (critically including both TSH and free thyroxine determination) in all patients with MCAP or other forms of PROS with brain involvement (see Table 1) |
| Endocrine supervision | Symptoms suggestive of periodic hypoglycaemia should trigger an inpatient fast under endocrine supervision. Attention should be paid to obtaining the correct diagnostic samples at the time of symptoms. Confirmed hypoglycaemia should be followed up by screening of GH and cortisol secretion | Monitoring of linear growth | Where there is evidence of linear growth retardation, IGF1 and IGFBP3 should be measured. If these are low, then a provocative test of GH secretion should be undertaken in accord with local endocrine practice and taking into consideration clinical indicators for use of insulin in epilepsy |
| Inpatient fasting | There is insufficient evidence to justify inpatient fasting without suggestive symptoms. However, hypoglycaemia is a particular risk in some circumstances, when less invasive screening using continuous glucose monitoring (CGM) is warranted. If CGM is not accessible or suitable, capillary plasma glucose monitoring during symptoms can be considered. We suggest that specialist endocrine opinion is sought during (a) evaluation of seizures and (b) prior to sedation, e.g. for neuroimaging, as some CGM devices are incompatible with MRI. | Patients with proven hypoglycaemia | The above evaluation of the GH axis should also be undertaken in patients with proven hypoglycaemia, in which case a dynamic test of the hypothalamic-pituitary adrenal axis should also be undertaken |
| Treatment and monitoring | |||
| Emergency management | This is conventional, depending on severity of hypoglycemia. It may range from infusion of intravenous glucose to provision of sugar-containing drinks or snacks. Glucagon injection may also be of use | Deficiency of free thyroid hormone levels | Given the importance of thyroid function for neurodevelopment, any deficiency of free thyroid hormone should be corrected with L-thyroxine. TSH is not a suitable marker of hormone replacement in central hypothyroidism. Monitoring should rely instead on free thyroxine concentrations. There is a lack of data on the use of T3 in this group |
| Long term management | Preventative measures depend on aetiology of hypoglycaemia, and duration of fasting tolerated. In severe cases regular enteral or parenteral nutrition should be instituted with inclusion of overnight feeding, although modified starch preparations may progressively be used instead or as well in older patients | Deficiency of the adrenal axis | Should be corrected with cortisol. Note, however, that mild blunting of the axis may be a consequence rather than the cause of chronic hypoglycaemia due to resetting of counter-regulatory responses |
3. Recommendations for non-MCAP PROS
To date endocrinopathy in PROS has solely been reported in people with brain involvement. Although in principle physiologically inappropriate activation of PIK3CA in insulin-responsive tissues such as liver, muscle or adipose tissue could lower blood glucose, this has not been reported to date, even with extreme regional overgrowth of muscle and adipose tissue (50).
Vascular anomalies and risk of thrombosis
1. General discussion
A range of developmental vascular anomalies (abnormal vasculogenesis) comprise part of the diagnostic criteria of PROS. Vascular malformations are frequent in PROS, occurring in 59% in one recent cohort (56). These include (a) cutaneous capillary malformations that may be midline facial (i.e., persistent salmon patch) or widespread on the entire body (including “reticulated” capillary malformation reminiscent of cutis marmorata), (b) venous aneurysms / prominent venous pattern, (c) other low flow vascular malformations (venous, lymphatic) typically overlying truncal overgrowth and d) high-flow vascular malformations (arteriovenous; especially spinal-paraspinal). Vascular malformations may be superficial or deep (visceral). All of these vascular anomalies have been described as a component feature in MCAP although they occur less frequently. The majority of these lesions may only be identified by MRA/V scans (57, 58).
Individuals with PROS have an increased risk for deep venous thrombosis (DVT)/pulmonary embolism (PE), especially those with combined capillary-lymphatic-venous malformations. Incidence of DVT and PE has been reported to be 17% and 14-22% for DVT and PE, respectively (64% occurring after surgery or sclerotherapy). Venous thrombosis and dural sinus stasis has occurred in MCAP. Predisposition to thrombosis is likely to be multifactorial with risk factors including chronic stasis within vascular malformations, stasis from impaired mobility (e.g., following surgery or dehydration), decreased anticoagulant proteins, and effects of the PIK3CA variant on vascular endothelium (58).
2. Recommendations for PROS
Treatment for individuals with PROS will be dependent on the findings in each individual, which are variable in onset and course, affected tissue, and location of affected lesions. A complete history re DVT and PE and assessment of the extent of the vascular findings with follow-up of abnormal findings is appropriate. An ultrasound can be a very effective tool at initially defining whether the malformations are high flow, low flow, or non-vascular. MRI/A/V scans of the chest, abdomen, pelvis and extremities at the time of initial diagnosis depending on the involvement of clinical findings (including tissue overgrowth and /or superficial vascular anomalies) and Doppler ultrasounds of affected limbs are recommended. This helps define deeper components (e.g., lymphatic and venous malformations, gastrointestinal and genitourinary, spine involvement). Because this disorder may be associated with progression, the frequency of follow up should be based on clinical history and physical examination. For instance, monitoring progression of the lesions would be important in cases of (a) superficial craniofacial vascular anomalies/lesions with concomitant ophthalmic involvement, (b) symptomatic spinal/trunk/limb involvement, (c) CLAPO syndrome, (d) varicose veins.
The lateral marginal vein is an anomalous clinical entity found more frequently in individuals with KTS and other PROS which has been associated with significant morbidity and mortality attributable to venous hypertension and potentially lethal thromboembolic events. Although limited literature exists regarding the diagnosis and management of this rare anomaly, its treatment is warranted once an intact deep venous system has been identified. In the absence of consensus, treatment algorithms are used (59).
Individuals with PROS with venous malformations may benefit from a haematology evaluation, and basic clinical coagulation laboratory studies, including baseline D-dimer test, which is a useful screen for DVT and PE. To date, there is no specific biomarker of the thrombosis risk of PROS individuals (58). The Vascular Anomalies Special Interest Group of the American Society of Paediatric Haematology and Oncology recommends perioperative and pre sclerotherapy correction of D-dimer that are more than 5 times the normal with at least 2 weeks of therapeutic Low-Molecular-Weight Heparin (LMWH) pre-and post-procedure (60). With lower D-dimers levels, DVT anticoagulant prophylaxis and management similar to that recommended for Proteus syndrome with 0.5 mg/kg enoxaparin, sequential compression devices until the patient is ambulatory, and mobilization postoperatively to reduce the risk of thrombosis is suggested (60). There is not sufficient evidence to support routine anticoagulation. Those who are positive for DVT or PE on imaging should undergo acute anticoagulation according to the standard American College of Chest Physicians’ guidelines (61).
DISCUSSION
This expert consensus document constitutes a formal attempt to align professional efforts with patient need, to safeguard transfer of knowledge of existing medical practice and to create models for the sustainable care and management of individuals with PROS. Our effort represents an international viewpoint to address the significant challenge of a clinical management pathway for affected individuals. Our approach also identified the areas of care that require further investigation or development. Further longitudinal studies and clinical trials are necessary to provide ad hoc management recommendations for further aspects (Table 6).
One of the main challenges to our group lay in deciding on ‘splitting’ or ‘lumping’ of the management guidelines according to distinct clinical presentations of overlapping molecular cause (MCAP, CLOVES). We agreed on a unified approach for a number of reasons: (1) these conditions are characterized by a clinical variability or progression that frequently defies the borders of their clinical or molecular definition; (2) many of the affected individuals present or are tested at neonatal or paediatric age when the morbidity may only be hinted at, rather than be manifest; (3) new phenotypes of the same group continue to emerge; (4) what is common for most of the affected individuals is a mosaic, gain-of-function, growth-promoting pathogenetic variant.
We also believe that our approach is complementary to condition-specific guidelines (62). A few of the recommendations made in this manuscript and especially the ones concerning the CNS-related risks may also be applicable for individuals with Megalencephaly-Polymicrogyria-Polydactyly-Hydrocephalus (MPPH) syndrome (see Appendix). Most of the recommendations are also applicable for the rare PROS individuals with a germline variant (25) taking into consideration that (a) testing DNA from peripheral blood may be sufficient for confirmation of the diagnosis and (b) germline mosaicism in a healthy parent cannot be ruled out so these families should be referred for genetic counseling. On the other hand, our recommendations regarding CNS and neurodevelopmental monitoring, epilepsy and endocrinopathy may only apply in cases of PROS with brain involvement and rare cases of localised or tissue-specific PROS (Tables 1A, 4 and 5). Any conclusions stated in this manuscript are indicative of a preferred practice plan agreed by experts but may not be applicable for specific single cases of individuals with PROS.
The PI3K/AKT/mTOR signaling pathway is one of the main regulators of cell proliferation and a target of multiple therapeutic strategies (63). A nonrandomized open-label pilot treatment study (intra-patient cross-over) using low dose Sirolimus of 39 individuals with PROS (three with MCAP) and progressive overgrowth suggests that low-dose Sirolimus can modestly reduce overgrowth, but that the side-effect profile is significant, mandating individualized risk-benefit evaluations for Sirolimus treatment in PROS. This study did not have outcome measures for the CNS-related malformations and neurologic complications associated with MCAP (56). An open-label, phase 1/2 study of miransertib in 15 individuals with PROS showed encouraging results in lesion stability, movement fluidity, fatiguability, pain relief and on the Karnofsky/Lansky performance scale (64). The administration of alpelisib under compassionate care protocols in 19 non-MCAP PROS individuals who had life-threatening complications and/or were scheduled for debulking surgery evidenced encouraging results in reduction of vascular tumour size, hemihypertrophy and scoliosis and improvement of congestive heart failure improvement. However, the type of care protocol does not allow unequivocal conclusions about drug safety or efficacy (65). At the time of submission of this manuscript, a prospective Phase II multi-center study with an upfront 16-week, randomized, double-blind, placebo-controlled period, and extension periods, to assess the efficacy, safety and pharmacokinetics of alpelisib in paediatric and adult participants with PROS, is open (Identifier: NCT04589650, 66).
We believe that outlining clinical management guidelines is essential in view of the on-going clinical trials and up-and-coming treatments for PROS. A standard of care can be useful for monitoring the effects of such trials or caring for non-responders.
ACKNOWLEDGEMENTS
We are grateful for the support of Christy Collins on behalf of the M-CM Network USA and Lorraine Yeomans, M-CM UK. We thank our patients and their families for all they have taught us.
FUNDING
The expert meeting was funded by the M-CM Network and M-CM UK with support from AMCME España. GMM is supported by funding from Jordan’s Guardian Angels, the Brotman Baty Institute, and the Sunderland Foundation. LGB was supported by NHGRI intramural research support via HG200388-07. RKS is supported by the Wellcome Trust [grant 210752/Z/18/Z].
FINANCIAL DISCLOSURES
LGB receives honoraria from Cold Spring Harbor Press and receives in-kind research support from ArQule Inc., (now wholly owned by Merck, Inc.) for treatment protocols of individuals with overgrowth syndromes. SS has received consulting fees from GLG, Guidepoint (which connected to a client, Fortress Biotech), Novartis, ExpertConnect, Orchard Therapeutics. RS has received consulting fees from Novartis. VEP is an employee and stockholder at AstraZeneca.
APPENDIX
Megalencephaly:
The term has been introduced as it implies a pathological feature not covered by the previously defined term ‘macrocephaly’, which may or may not be associated with megalencephaly. Here we define megalencephaly as brain volume >2.5 S.D. above the mean for age.
Variant Allele Fraction (VAF):
The fraction of the times the candidate variant is detected (i.e., reads in NGS or droplets in ddPCR). Since there are two alleles in each cell, the percentage of cells with the variant is twice the VAF. A heterozygous germline variant, present in all cells, has a VAF of 0.5 (50% of all alleles)
Genetic testing sampling:
Our recommendations for MCAP include a two-step process that is to request biopsies of affected tissue only if the result is negative in blood or saliva samples. This however also depends on the family views and on whether they prefer to wait for different steps. A skin biopsy is an easy process, and some families may prefer a first step skin biopsy to have an answer more quickly. Ideally, this should be discussed with the families.
Genetic testing results:
These may have considerable importance for personal and medical decisions. Thus, the molecular report must be understood by non-specialist healthcare professionals. There is a high proportion of mutational hot spots in PIK3CA that facilitate reporting. It also should be accompanied by a clinical report explaining the implications and consequences of the results for that specific patient, where possible. In fact many laboratories do not always receive detailed clinical data linked to referrals so they may be limited in the ability to comment on the implications and consequences for each tested individual. As with other pathologies with a genetic cause, a negative test result does not rule out the clinical diagnosis.
Epilepsy and seizure:
Though sometimes used interchangeably, the terms seizure and epilepsy are not the same. A seizure represents abnormal synchronous electrical activity in the brain, leading to neurological symptoms. In contrast, epilepsy is defined as having two or more unprovoked seizures occurring more than 24 hours apart or having one unprovoked seizure with a significantly elevated risk for having a future unprovoked seizure (1).
Cerebellar tonsillar ectopia/herniation (CBTE):
This includes Chiari I malformations but not types II, III, IV.
Oral/Dental Management:
Individuals can have dental anomalies secondary to asymmetric facial overgrowth, FIL or the orofacial presentation described by Koutlas et al. (2021) (2, 3). PIK3CA variants were identified by testing DNA of the pulp of an extracted tooth of an individual with MCAP as well as through gingival biopsy (4, 5).
MPPH:
Megalencephaly-polymicrogyria-polydactyly-hydrocephalus (MPPH) syndrome is a rare disorder that primarily affects the development of the brain. It is caused by germline variants in AKT3, CCND2 or PIK3R2. Affected individuals share a number of overlapping features with individuals with PROS. Usually they are born with megalencephaly which may increase rapidly during the first 2 years of life; they may also present with PMG and/or polydactyly. Unlike individuals with PROS, they do not manifest extra-CNS overgrowth and/or vascular findings.
APPENDIX REFERENCE LIST
- 1.Fisher RS, Acevedo C, Arzimanoglou A, et al. ILAE official report: a practical clinical definition of epilepsy. Epilepsia. 2014;55:475–482. [DOI] [PubMed] [Google Scholar]
- 2.Koutlas IG, Anbinder A-L, Alshagroud R, et al. Hemiorofacial asymmetry with peripheral nerve enlargement and associated perineurial hyperplasia and/or perineuriomatous pseudo-onion bulb proliferations is part of the PIK3CA-related overgrowth spectrum (PROS). Hum Genet Genomics Adv 2020. 1:100009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maclellan RA, Luks VL, Vivero MP, et al. PIK3CA activating mutations in facial infiltrating lipomatosis. Plast. Reconstr. Surg 2014;133:12e–19e. [DOI] [PubMed] [Google Scholar]
- 4.McDermott JH, Byers H, Clayton-Smith J Detection of a mosaic PIK3CA mutation in dental DNA from a child with megalencephaly capillary malformation syndrome. Clin Dysmorphol. 2016;25:16–18. [DOI] [PubMed] [Google Scholar]
- 5.Marty M, Bonnaud C, Jones N, et al. Gingival Biopsy to Detect Mosaicism in Overgrowth Syndromes: Report of Two Cases of Megalencephaly-Capillary Malformation Syndrome with Periodontal Anomalies. Case Rep Dent. 2020. September;2020:8826945. [DOI] [PMC free article] [PubMed] [Google Scholar]
Footnotes
CONFLICT OF INTEREST STATEMENT
LGB is a member of the Illumina ethics advisory board. RKS consults for Novartis. The rest of the authors declare no conflict of interest.
DATA AVAILABILITY STATEMENT
All data generated or analysed during this study are included in this published article.
REFERENCES
- 1.Rivière JB, Mirzaa GM, O’Roak BJ,et al. De novo germline and postzygotic mutations in AKT3, PIK3R2 and PIK3CA cause a spectrum of related megalencephaly syndromes. Nat Genet. 2012. June;44:934–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lindhurst MJ, Parker VE, Payne F, et al. Mosaic overgrowth with fibroadipose hyperplasia is caused by somatic activating mutations in PIK3CA. Nat Genet. 2012;44:928–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Keppler-Noreuil KM, Sapp JC, Lindhurst MJ, et al. Clinical delineation and natural history of the PIK3CA-related overgrowth spectrum. Am J Med Genet A. 2014;164A:1713–1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Keppler-Noreuil KM, Rios JJ, Parker VE, et al. PIK3CA-related overgrowth spectrum (PROS): diagnostic and testing eligibility criteria, differential diagnosis, and evaluation. Am J Med Genet A. 2015;167A:287–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mirzaa G, Timms AE, Conti V, et al. PIK3CA-associated developmental disorders exhibit distinct classes of mutations with variable expression and tissue distribution. JCI Insight. 2016;1:e87623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vahidnezhad H, Youssefian L, Uitto J. Klippel-Trenaunay syndrome belongs to the PIK3CA-related overgrowth spectrum (PROS). Exp Dermatol. 2016;25:17–19. [DOI] [PubMed] [Google Scholar]
- 7.Rodriguez-Laguna L, Ibañez K, Gordo G, et al. CLAPO syndrome: identification of somatic activating PIK3CA mutations and delineation of the natural history and phenotype. Genet Med 2018;20:882–889. [DOI] [PubMed] [Google Scholar]
- 8.Al-Qattan MM, Hadadi A, Al-Thunayan AM, et al. Upper limb muscle overgrowth with hypoplasia of the index finger: a new over-growth syndrome caused by the somatic PIK3CA mutation c.3140A>G. BMC Med Genet. 2018; 19: 158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Frisk S, Taylan F, Blaszczyk I, et al. Early activating somatic PIK3CA mutations promote ectopic muscle development and upper limb overgrowth. Clin Genet. 2019;96:118–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Koutlas IG, Anbinder A-L, Alshagroud R, et al. Hemiorofacial asymmetry with peripheral nerve enlargement and associated perineurial hyperplasia and/or perineuriomatous pseudo-onion bulb proliferations is part of the PIK3CA-related overgrowth spectrum (PROS). Hum Genet Genomics Adv 2020. 1:100009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Martínez-Glez V, Romanelli V, Mori MA, et al. Macrocephaly-capillary malformation: Analysis of 13 patients and review of the diagnostic criteria. Am J Med Genet A. 2010;152A:3101–3106. [DOI] [PubMed] [Google Scholar]
- 12.Tapper WJ, Foulds N, Cross NC, et al. Megalencephaly syndromes: exome pipeline strategies for detecting low-level mosaic mutations. PloS One. 2014;9:e86940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kuentz P, St-Onge J, Duffourd Y, et al. Molecular diagnosis of PIK3CA-related overgrowth spectrum (PROS) in 162 patients and recommendations for genetic testing. Genet Med. 2017;19:989–997. [DOI] [PubMed] [Google Scholar]
- 14.Chang F, Liu L, Fang E et al. Molecular Diagnosis of Mosaic Overgrowth Syndromes Using a Custom-Designed Next-Generation Sequencing Panel. J Mol Diagn. 2017;19:613–624. [DOI] [PubMed] [Google Scholar]
- 15.Robinson PN. Deep phenotyping for precision medicine. Hum Mutat. 2012;33:777–80. [DOI] [PubMed] [Google Scholar]
- 16.Sapp JC, Buser A, Burton-Akright J, et al. A Dyadic Genotype-Phenotype Approach to Diagnostic Criteria for Proteus Syndrome. Am J Med Genet C. 2019;181:565–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Biesecker LG, Adam MP, Alkuraya FS, et al. A Dyadic Approach to the Delineation of Diagnostic Entities in Clinical Genomics. AJHG.2021;108:8–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rare Disease Info: Guidelines & Manual. EURORDIS. https://www.eurordis.org/publication/rare-disease-info-guidelines-manual Last accessed 10 June 2021. [Google Scholar]
- 19.Pavan S, Rommel K, Mateo Marquina ME, et al. Clinical Practice Guidelines for Rare Diseases: The Orphanet Database. PloS One. 2017;12:e0170365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pai M, Yeung CHT, Akl EA, et al. Strategies for eliciting and synthesizing evidence for guidelines in rare diseases. BMC Med Res Methodol. 2019;19:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.M-CM Network, https://www.m-cm.net/. Last accessed 10 June 2021.
- 22.Brouwers MC, Kho ME, Browman GP, et al. AGRE E II: advancing guideline development, reporting and evaluation in healthcare. CMAJ. 2010;182:E839–E842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Biderman Waberski M, Lindhurst M, Keppler-Noreuil KM, et al. Urine cell-free DNA is a biomarker for nephroblastomatosis or Wilms tumour in PIK3CA-related overgrowth spectrum (PROS). Genet Med. 2018;20:1077–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Emrick LT, Murphy L, Shamshirsaz AA et al. Prenatal Diagnosis of CLOVES Syndrome Confirmed by Detection of a Mosaic PIK3CA Mutation in Cultured Amniocytes. Am J Med Genet A 2014; 0: 2633–2637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Quinlan-Jones E, Williams D, Bell C, et al. Prenatal Detection of PIK3CA-related Overgrowth Spectrum in Cultured Amniocytes Using Long-range PCR and Next-generation Sequencing. Pediatr Dev Pathol Off J Soc Pediatr Pathol Paediatr Pathol Soc 2017; 20: 54–57. [DOI] [PubMed] [Google Scholar]
- 26.De Graer C, Marangoni M, Romnée S et al. Novel features of PIK3CA-Related Overgrowth Spectrum: Lesson from an aborted fetus presenting a de novo constitutional PIK3CA mutation. Eur J Med Genet 2020; 63: 103775. [DOI] [PubMed] [Google Scholar]
- 27.Couto JA, Konczyk DJ, Vivero MP, et al. Somatic PIK3CA Mutations are Present in Multiple Tissues of Facial Infiltrating Lipomatosis. Pediatr Res. 2017; 82: 850–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zenner K, Cheng CV, Jensen DM , et al. Genotype correlates with clinical severity in PIK3CA-associated lymphatic malformations. JCI Insight. 2019; 4: e129884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Luks VL, Kamitaki N, Vivero MP et al. Lymphatic and other vascular malformative/overgrowth disorders are caused by somatic mutations in PIK3CA. J Pediatr. 2015;166:1048–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Keppler-Noreuil KM, Parker VE, Darling TN, Martinez-Agosto JA. Somatic overgrowth disorders of the PI3K/AKT/mTOR pathway & therapeutic strategies. Am J Med Genet C Semin Med Genet. 2016;172: 402–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mirzaa G, Conway R, Graham JM Jr, Dobyns WB. PIK3CA-Related Segmental Overgrowth. 2013. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2019. Available from http://www.ncbi.nlm.nih.gov/books/NBK153722/ Last accessed 10 June 2021. [Google Scholar]
- 32.Conway RL, Pressman BD, Dobyns WB, et al. Neuroimaging findings in macrocephaly-capillary malformation: a longitudinal study of 17 patients. Am J Med Genet A. 2007b;143A:2981–3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mirzaa GM, Conway RL, Gripp KW, et al. Megalencephaly-capillary malformation (MCAP) and megalencephaly-polydactyly-polymicrogyria-hydrocephalus (MPPH) syndromes: two closely related disorders of brain overgrowth and abnormal brain and body morphogenesis. Am J Med Genet A. 2012;158A:269–291. [DOI] [PubMed] [Google Scholar]
- 34.Mirzaa GM, Rivière JB, Dobyns WB. Megalencephaly syndromes and activating mutations in the PI3K-AKT pathway: MPPH and MCAP. Am J Med Genet C Semin Med Genet. 2013;163C:122–130. [DOI] [PubMed] [Google Scholar]
- 35.Jansen LA, Mirzaa GM, Ishak GE, et al. PI3K/AKT pathway mutations cause a spectrum of brain malformations from megalencephaly to focal cortical dysplasia. Brain. 2015;138:1613–1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Segal D, Heary RF, Sabharwal S, et al. Severe holocord syrinx in a child with megalencephaly-capillary malformation syndrome. J Neurosurg Pediatr. 2016;18:79–82. [DOI] [PubMed] [Google Scholar]
- 37.D’Gama AM, Woodworth MB, Hossain AA, et al. Somatic Mutations Activating the mTOR Pathway in Dorsal Telencephalic Progenitors Cause a Continuum of Cortical Dysplasias. Cell Rep. 2017;21:3754–3766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shain C, Ramgopal S, Fallil Z, et al. Polymicrogyria-associated epilepsy: a multicentre phenotypic study from the Epilepsy Phenome/Genome Project. Epilepsia. 2013;54:1368–1375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Garde A, Guibaud L, Goldenberg A, et al. Clinical and neuroimaging findings in 33 patients with MCAP syndrome: A survey to evaluate relevant endpoints for future clinical trials. Clin Genet. 2021;99:650–661. [DOI] [PubMed] [Google Scholar]
- 40.Kwan P, Arzimanoglou A, Berg AT, et al. Definition of drug resistant epilepsy: consensus proposal by the ad hoc Task Force of the ILAE Commission on Therapeutic Strategies. Epilepsia. 2010;51:1922 and published correction in Epilepsia 2010;51:1069-1077. [DOI] [PubMed] [Google Scholar]
- 41.Berg AT, Zelko FA, Levy SR, Testa FM. Age at onset of epilepsy, pharmacoresistance, and cognitive outcomes: a prospective cohort study. Neurology. 2012;79:1384–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Seizure First Aid, Centres for Disease Control and prevention. https://www.cdc.gov/epilepsy/about/first-aid.htm Last accessed 10 June 2021.
- 43.Glauser T, Ben-Menachem E, Bourgeois B, et al. Updated ILAE evidence review of antiepileptic drug efficacy and effectiveness as initial monotherapy for epileptic seizures and syndromes. Epilepsia. 2013;54:551–563. [DOI] [PubMed] [Google Scholar]
- 44.Wilmshurst JM, Gaillard WD, Vinayan KP, et al. Summary of recommendations for the management of infantile seizures: Task Force Report for the ILAE Commission of Paediatrics. Epilepsia. 2015;56:1185–1197. [DOI] [PubMed] [Google Scholar]
- 45.Yeung KS, Tso WWY, Ip JJK, et al. Identification of mutations in the PI3K-AKT-mTOR signalling pathway in patients with macrocephaly and developmental delay and/or autism. Mol Autism. 2017; 8: 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gripp KW, Baker L, Kandula V, et al. Nephroblastomatosis or Wilms Tumour in a Fourth Patient With a Somatic PIK3CA Mutation. Am J Med Genet A. 2016; 170: 2559–2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Peterman CM, Fevurly RD, Alomari AI, et al. Sonographic screening for Wilms tumour in children with CLOVES syndrome. Pediatr Blood Cancer. 2017:64:12. [DOI] [PubMed] [Google Scholar]
- 48.Griff JR, Duffy KA, Kalish JM. Characterization and Childhood Tumour Risk Assessment of Genetic and Epigenetic Syndromes Associated With Lateralized Overgrowth. Front Pediatr. 2020;8:613260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Michel ME, Konczyk DJ, Yeung KS et al. Causal somatic mutations in urine DNA from persons with the CLOVES subgroup of the PIK3CA-related overgrowth spectrum. Clin Genet. 2018;93:1075–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Leiter SM, Parker VER, Welters A, et al. Hypoinsulinaemic, hypoketotic hypoglycaemia due to mosaic genetic activation of PI3-kinase. Eur J Endocrinol. 2017;177:175–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Davis S, Ware MA, Zeiger J, et al. Growth hormone deficiency in megalencephaly-capillary malformation syndrome: An association with activating mutations in PIK3CA. Am J Med Genet A. 2020;182:162–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stutterd C, McGillivray G, Stark Z, et al. Polymicrogyria in association with hypoglycemia points to mutation in the mTOR pathway. Eur J Med Genet. 2018;61:738–740. [DOI] [PubMed] [Google Scholar]
- 53.Hussain K, Challis B, Rocha N, et al. An activating mutation of AKT2 and human hypoglycemia. Science. 2011;334:474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McDermott JH, Hickson N, Banerjee I, et al. Hypoglycaemia represents a clinically significant manifestation of PIK3CA- and CCND2-associated segmental overgrowth. Clin Genet. 2018;93:687–692. [DOI] [PubMed] [Google Scholar]
- 55.Minagawa M, Nagai F, Kanazawa M et al. A case of Macrocephaly-Cutis Marmorata Teleangiectasica Congenita complicated with growth hormone (gh)- deficiency and hypothyroidism. Clin Pediatr Endocrinol. 2005;14: 53–56. [Google Scholar]
- 56.Parker VER, Keppler-Noreuil KM, Faivre L, et al. Safety and efficacy of low-dose sirolimus in the PIK3CA-related overgrowth spectrum. Genet Med. 2019;21:1189–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Limaye N, Kangas J, Mendola A, et al. Somatic Activating PIK3CA Mutations Cause Venous Malformation. Am J Hum Genet. 2015;97:914–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Keppler-Noreuil KM, Lozier J et al. Thrombosis risk factors in PIK3CA-related overgrowth spectrum and Proteus syndrome. Am J Med Genet C Semin Med Genet. 2019;181:571–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhuo KY, Russell S, Wargon O, Adams S. Localised intravascular coagulation complicating venous malformations in children: Associations and therapeutic options. J Paediatr Child Health. 2017;53:737–741. [DOI] [PubMed] [Google Scholar]
- 60.Fereydooni A, Nassiri N. Evaluation and management of the lateral marginal vein in Klippel-Trénaunay and other PIK3CA-related overgrowth syndromes. J Vasc Surg Venous Lymphat Disord. 2020;8:482–493. [DOI] [PubMed] [Google Scholar]
- 61.Kearon C, Akl EA, Ornelas J, Blaivas A, et al. Antithrombotic Therapy for VTE Disease: CHEST Guideline and Expert Panel Report. Chest. 2016;149:315–352. [DOI] [PubMed] [Google Scholar]
- 62.French guidelines for CLOVES and Klippel-Trenaunay syndromes. Available at https://www.has-sante.fr/upload/docs/application/pdf/2020-12/pnds_pros_hors_mcap_dec2020_1.pdf Last accessed 10 June 2021. accessed 10 June 2021.
- 63.De Santis MC, Sala V, Martini M, et al. PI3K Signaling in Tissue Hyper-Proliferation: From Overgrowth Syndromes to Kidney Cysts. Cancers (Basel). 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zampino G, Leoni C, Buonuomo PS et al. An open-label, phase 1/2 study of miransertib (ARQ 092), an oral pan-AKT inhibitor, in patients with PIK3CA-related Overgrowth Spectrum (PROS) and Proteus Syndrome (PS): study design and preliminary results (NCT03094832). European Society of Human Genetics Conference; June 15-18, 2019; Gothenburg, Sweden. Abstract C 18.6. [Google Scholar]
- 65.Venot Q, Blanc T, Rabia SH, et al. Targeted therapy in patients with PIK3CA-related overgrowth syndrome. Nature. 2018;558:540–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.ClinicalTrials.gov, https://clinicaltrials.gov/ct2/show/NCT04589650 Last accessed 10 June 2021.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated or analysed during this study are included in this published article.