

Abstract
Lipid microdomains or lipid rafts are dynamic and tightly ordered regions of the plasma membrane. In mammalian cells they are enriched in cholesterol, glycosphingolipids (GSL), GPI-anchored and signaling-related proteins. Several studies have suggested that mammalian pattern recognition receptors (PRRs) are concentrated or recruited to lipid domains during host-pathogen association to enhance the effectiveness of host effector processes. However, pathogens have also evolved strategies to exploit these domains to invade cells and survive. In fungal organisms, a complex cell wall network usually mediates the first contact with the host cells. This cell wall may contain virulence factors that interfere with the host membrane microdomains dynamics, potentially impacting the infection outcome. Indeed, the microdomain disruption can dampen fungus-host cell adhesion, phagocytosis, and cellular immune responses. Here, we provide an overview of regulatory strategies employed by pathogenic fungi to engage with and potentially subvert the lipid microdomains of host cells.
Keywords: lipid microdomains, pathogenic fungi, innate immunity
1. Introduction
The presence of lipid microdomains in the membrane was first suggested by Simon and Van Meer in MDCK cell (1988) [1]. Since then, their existence and participation in vital cellular processes including protein sorting, membrane trafficking, and signal-transduction events have been confirmed [2]. Lipid microdomains are enriched in sphingolipids, cholesterol and specific proteins. The lipid moiety of sphingolipids and sterols are connected through hydrogen bonds and hydrophobic interactions resulting in a highly ordered structure [2,3]. The presence of glycans covalently attached to sphingolipids can also mediate cis interactions, promoting lateral association with other glycosphingolipids and proteins [4]. The major proteins enriched in lipid microdomains include glycosylphosphatidylinositol (GPI) anchored proteins and palmitoylated proteins, making these domains organized platforms for signal transduction [5,6]. They are continuously dynamic, and upon stimulus, can coalesce in larger complexes, aggregating different raft-associated proteins and lipids in intimate association with the cytoskeleton [7]. Consequently, a number of receptors and signal transduction proteins accumulate in these regions increasing signaling efficiency. The internalization of pathogens seems to be one of the events potentially mediated by lipid microdomains [8].
The host-microbe association is a highly dynamic mechanism that involves multiple interactions. In this context, the combination of molecules gathered in the host cell platforms during the cellular response directly impacts the fate of the invaders. It is important to mention that fungal plasma membrane also contains lipid microdomains [9]. In these organisms, the microdomains have been correlated with protein sorting, cell budding and growth, biofilm formation, drug resistance, and infectivity, as extensively reviewed by Farnoud et al [9]. The presence of virulence factors, such as phospholipase B and superoxide dismutase in lipid rafts from C. neoformans [10] and the requirement for their integrity during the infection of macrophages by H. capsulatum [11] suggest that fungal lipid microdomains are also determinant components during the interaction with host cells. Nonetheless, our goal in this review is to discuss the participation of lipid microdomains from host cells during the interaction and invasion of pathogenic fungi.
2. Lipid rafts and the innate immune response to fungal infections
The major pattern recognition receptors (PRRs) involved with recognition of fungi are Toll-like receptors (TLRs) and C-type lectin receptors (CLRs) [12]. PPRs are expressed in many cell types involved in the innate immune response, and, upon activation, they trigger downstream signaling transduction pathways that lead to phagocytosis, microbial killing, and cytokine production. Among TLRs, TLR-2/1, TLR2/6, and TLR4 (surface localization) and TLR3/7 and 9 (endosomal localization) are involved in fungal responses [13–17]. Transmembrane CLRs linked to antifungal immunity include dectin-1, dectin-2, dectin-3, mannose receptor (MR), DC-SIGN, Mincle, and galectin-3 [18].
With the important exception of encapsulated pathogenic fungal species (e.g. Cryptococcus neoformans and C. gattii), the cell wall establishes the first contact of these organisms with PRRs. The precise identification of receptors to fungal pathogen-associated molecular patterns (PAMPs) is challenging since the fungal cell wall is a complex and dynamic structure [18,19], where a diversity of PAMPs, such as glucans, chitin, mannans and mannoproteins, can be exposed to phagocytes PRRs displayed by phagocytes. The cell wall inner layer is relatively conserved among different species, but the outer layers can differ substantially and variably affect the host cell response [20].
The involvement of co-receptors during fungal recognition is associated with the amplification of the immune response. The initial mechanism of cooperation occurs during the engagement of fugal cells to their receptors, where one receptor can capture and present the fungal particle to a co-receptor [21]. In addition, the signaling response mediated after the engagement could be integrated intracellularly [22]. PRRs can cluster on the host cell surface in response to the abundance and presence of distinct ligands, strengthening the immune response. Indeed, the lack of PRRs co-stimulation seems to be the mechanism used by the dematiaceous fungus Fonsecaea pedrosoi to reduce the host immune response, resulting in a chronic infection called chromoblastomycosis [23]. Although leukocytes recognize F. pedrosoi conidia through Mincle, this association is not sufficient to promote a protective inflammatory response [23]. However, the co-stimulation with TLR agonists potentialized the inflammatory response, indicating that a collaborative process involving distinct PRRs is required to induce a protective response. Remarkably, the administration of LPS to mice infected with F. pedrosoi substantially reduced the fungal burden. In addition, fungal burden decrease was also observed in mice infected subcutaneously with F. conidia and topically treated with Imiquimod, a synthetic agonist for TLR7. This imidazoquinolide derivative was successfully used in human chromoblastomycosis [24]. The raft environment seems to be a distinct place where paired receptors could cooperate and amplify the cellular response. Indeed, the recruitment of TLRs to these domains after ligand engagement has been reported [25].
All TLRs, except TLR3, lead to the recruitment of the adaptor molecule MyD88 [26]. MyD88 contains a death domain (DD) at the N terminus that interacts with the kinase IRAK, forming the Myddosome complex, a platform that leads to transcriptional activation through NF-κB and AP-1. TLR4 clustering in lipid rafts produces a large oligomeric assembly of MyD88 with IRAK-1, which is associated with enhanced activation [27]. The importance of rafts for MyD88-dependent TLR activation has been demonstrated in macrophages genetically modified expressing high lipid raft content [28]. These cells displayed a remarkable content of TLR4 in lipid raft regions and were more sensitive to TLR2, 7, and 9, but not to TLR3 stimulation. Other PRRs can collaborate with TLRs, including dectin-1, DC-SIGN, and the scavenger receptor (CD36) [29–32]. Their association could be linked with TLR recruitment to lipid rafts, amplifying the response. For instance, CD36 is a palmitoylated protein usually enriched in lipid rafts, functioning as TLR2 collaborator within the domains [29]. Similarly, CD14 is a GPI-anchored protein present in raft, and its association with LPS stimulates the translocation of TLR4 into lipid microdomains [33,34]. Numerous studies have demonstrated that the association between TLR2, 4, 5, 7 or 9 and dectin-1 strengthens the immune response to achieve appropriate biological responses to pathogenic fungi [22,31,35,36]. Confirming that PRRs collaboration is important to an effective immune response Gantner and colleagues have shown that activation of TLR2 by zymosan particles is not sufficient to induce high levels of reactive oxygen species (ROS) production in macrophages, which requires a collaboration with dectin-1 [35]. In contrast with TLRs, which is able to recognize soluble microbial components, dectin-1 becomes activated only with immobilized forms of β-(1,3)-glucan, leading to a “phagocytic synapse” and a robust antifungal response [37]. The fact that dectin-1 translocates into lipid microdomains of mouse bone marrow-derived DC in response to zymosan stimuli suggests that clustering of these heterologous PRRs takes place in these platforms [38].
The crosstalk between PRRs expressed by DCs has an additional importance for orchestrating the differentiation of T helper (Th) cells. Dectin-1 activation drives DCs maturation and priming of naive CD4 T cells towards Th1 and Th17 phenotypes, commonly involved with protection against pathogenic fungi [39]. Translocation of dectin-1 into lipid microdomains is essential for Syk phosphorylation and consequently the signal transduction until the nuclear factors [38]. Additional evidence shows that the enrichment of dectin-1 and Syk phosphorylation in zymosan contact sites require excluding the inhibitory signal of the phosphatases CD45 and CD148, which strongly reinforces the importance of the membrane domains architecture in immune signaling pathway against fungi [37].
3. Glycosphingolipids-enriched lipid rafts as a platform for fungal recognition.
Glycosphingolipids (GSLs) are a heterogeneous subclass of glycolipids [4]. Lactosylceramide (LacCer) is a remarkable GSL that together with PRRs mediate the immune response to pathogenic fungi. Yeasts species such as C. neoformans, Candida albicans, S. cerevisiae, and the yeast-like Pneumocystis jirovecii as well as the dimorphic fungi Histoplasma capsulatum, P. brasiliensis, and Sporothrix schenckii directly bind to this domain (Fig. 1) [40]. The presence of galactose as a terminal residue of LacCer is linked to β-glucan recognition [41]. LacCer is predominantly increased in human glioma brain cells, and functions as binding sites for C. neoformans [40]. Furthermore, LacCer is enriched in lipid rafts of human neutrophils and their association with β-glucan induces migration, phagocytosis and superoxide generation (Fig. 1) [42–44]. The presence of a β–1,6-long glucosyl side-chain in the β-glucan seems to be crucial for Lyn and PI3K activation and neutrophil migration [45].
Figure 1-. Interplay of fungal pathogens and host cell lipid microdomains.

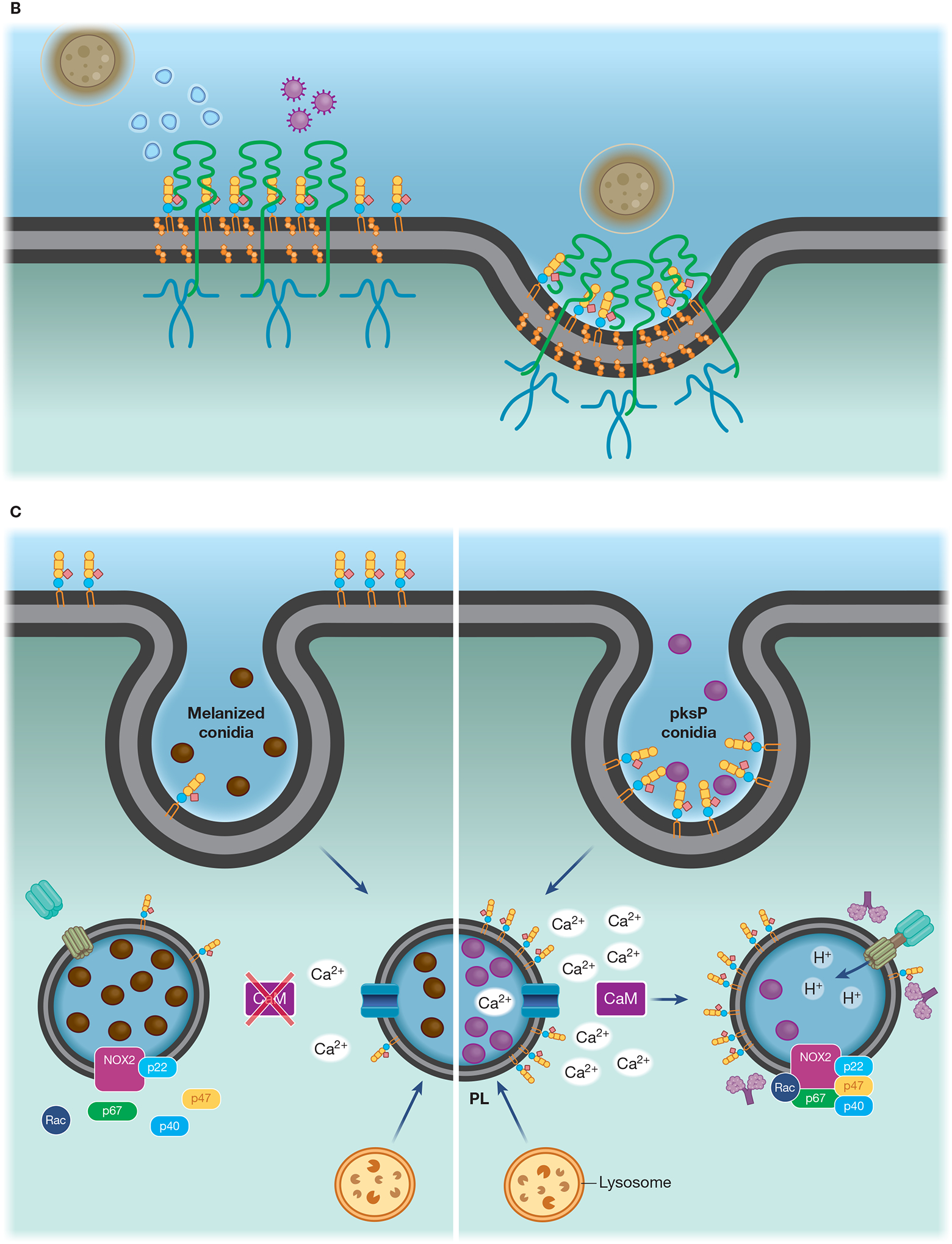
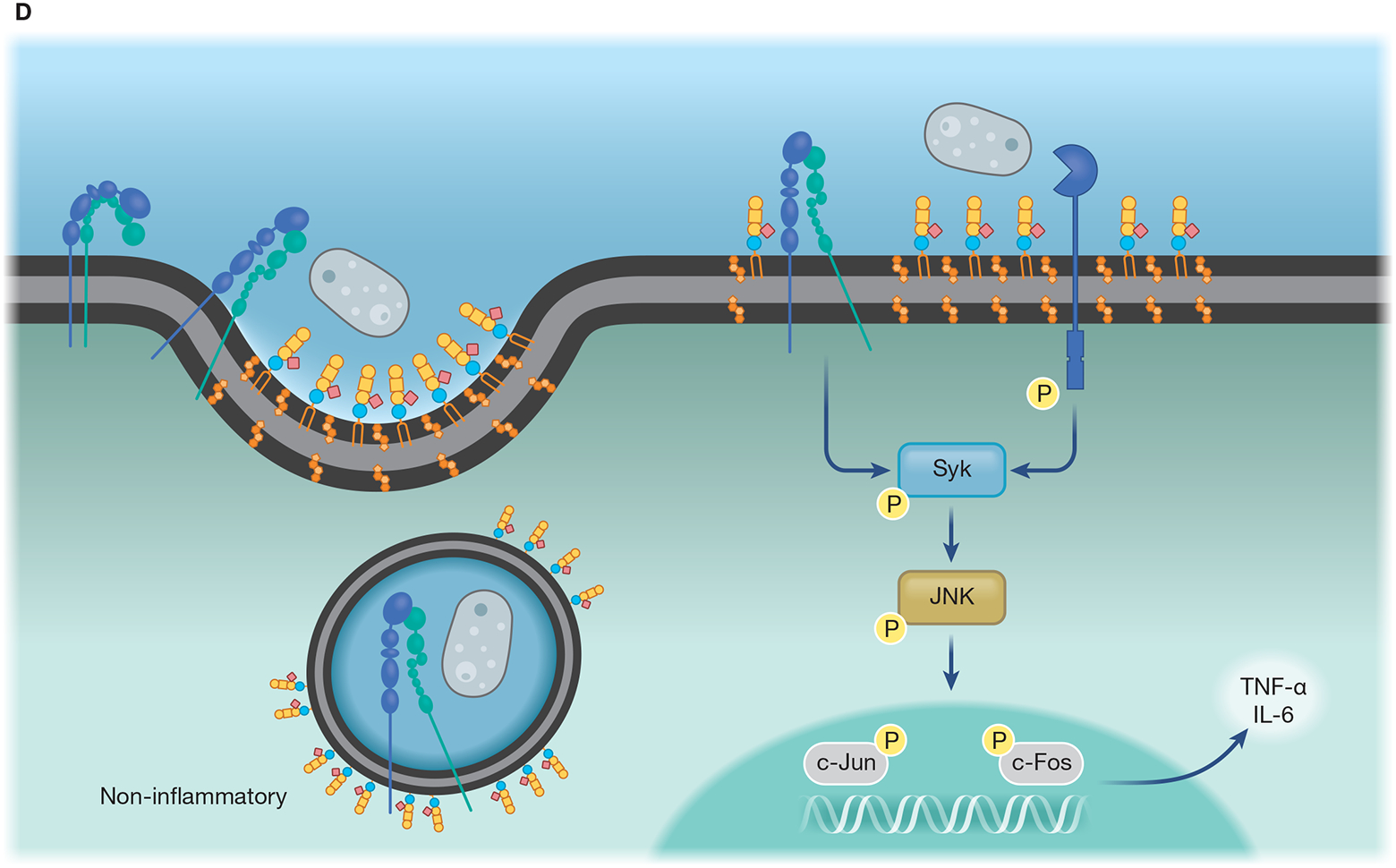
(A) Lipid microdomains enriched in LacCer are binding sites for fungal β-glucan and a signaling platform for CR3 recruitment, allowing this receptor to respond to non-opsonized pathogenic fungi. LacCer domains trigger a signaling pathway through Lyn, PI3K, p38, and PKC, leading to actin polymerization, required for migration, phagocytosis, and ROS production. (B) Caveolae is involved in the invasion of C. neoformans yeast in HBMEC. CD44 is located in caveolin-1-enriched membranes (Cav-1), where it recognizes the fungal hyaluronic acid. The intimate contact between Cav-1 and actin promotes the fungal internalization by HBMEC. The activation of Rho GTPase is correlated with actin remodeling and increased fungal internalization, which could be mediated by Cav-1. In addition, yeast extracellular vesicles and the HIV gp41 stimulate the clustering of CD44 on Cav-1 domain, increasing binding and invasion of C. neoformans yeast. (C) Flotillin domains on phagolysosomal membrane of macrophages are affected by melanin from A. fumigatus conidia. In comparison to non-melanized conidia (pksP), WT conidia block the release of calcium ions from the phagolysosome lumen to the cytoplasm, avoiding the activation of calmodulin (CaM). Conidia melanin impairs the formation of flotillin domains on phagolysosomal membranes and reduces the recruitment and assembly of V-ATPase and NADPH. (D) Recruitment of CR3, the main macrophage receptor for H. capsulatum, to lipid microdomains requires GM1. When β-(1,3)-glucans are exposed on the H. capsulatum cell wall the lipid microdomains allow the cooperation between CR3 and dectin-1, enhancing the inflammatory signaling through Syk, JNK, AP-1 (c-Jun/c-Fos).
LacCer enriched domains could still interact with immune receptors, such as CR3, a complement receptor composed of integrins CD11b (Mac-1) and CD18 [41]. CR3 co-localizes with LacCer-enriched membrane microdomains when human neutrophils are activated with the CD11b monoclonal antibody VIM12 [46]. The signaling transmitted by the stimulation is mediated through the interaction of LacCer and the C-terminal portion of CD18, which leads to Lyn phosphorylation (Fig. 1).
4. Role of lipid rafts in fungal infection.
In this section, we will review studies that have focused on the contribution of lipid microdomains to host interaction with the following fungal pathogens: C. albicans, C. neoformans, Pneumocystis jirovecii, Aspergillus fumigatus, P. brasiliensis, and H. capsulatum (for a brief summary of molecular interactions involving host cell lipid rafts, see Table 1).
Table 1:
Examples of interaction between host lipid microdomains and pathogenic fungi
| Fungal pathogen | Host cell | Molecular Interaction (pathogen/host) | Functional Response | References |
|---|---|---|---|---|
| Candida albicans | Monocytes | β- (1,3)- glucan | Phagocytosis | [47] |
| Neutrophils | β-(1,6)-side chain-branched β-glucan | Chemotaxis | [45] | |
| HA1 | [48][49][50] | |||
| Cryptococcus neoformans | HBMEC1 | EVs1 | Adhesion and brain invasion | [51] |
| AEC1 | β- (1,3)- glucan | MIP-2 synthesis1 | [52][53] | |
| Pneumocystis jirovecii | Human dendritic cells | β- (1,3)- glucan | Activation of the IL-23/IL-17 axis | [54] |
| Aspergillus fumigatus | Macrophages | DHN1 melanin | Inhibition of acidification of PL1 | [55] |
| Paracoccidioides brasiliensis | AEC | Not determined | IL-6 and IL-8 secretion | [56] |
| Histoplasma | AEC | Not determined | IL-6 and IL-8 secretion | [57] |
| capsulatum | Macrophages | HSP60* and β- (1,3)- glucan* | Phagocytosis and cytokine synthesis | [58][59] |
HA – Hyaluronic acid, HBMEC - Brain microvascular endothelial cells; EVs - Extracellular Vesicles; DHN - Dihydroxynaphthalene; PL – Phagolysosome; AEC – Alveolar Epithelial Cells, MIP-2 - Macrophage inflammatory protein-2, HSP60 - Heat Shock Protein 60.
possible ligands according to literature findings
4.1. Candida albicans
One of the common strategies to investigate the role of lipid rafts in PRRs crosstalk is the disruption of host cell lipid domains using methyl-β-cyclodextrin (m-β-CD) and Deoxycholate Amphotericin B (DAmb), drugs that extract and immobilize, respectively, sterols from the plasma membrane [60]. Although DAmb and other compounds that immobilize membrane sterol in mammalian cells have been extensively used in lipid domains studies, we cannot rule out their steric interference that could impact fungal binding. Pre-treatment of monocytes with both drugs directly affected C. albicans engulfment, reducing lipid raft coalescence and dectin-1 recruitment to C. albicans binding sites (Fig. 2) [47]. However, treatment of host cells with m-β–CD did not prevent the production of cytokines, indicating that the co-participation of non-raft PRRs could bypass the correct lipid raft assembly and trigger the stimulus required for cytokine synthesis. In addition, raft disruption on host cells drastically dampened Th17 and Th1 responses, suggesting that the preservation of dectin-1 dynamics in lipid-raft of antigen presenting cells is crucial for specific T-cell responses in this model [47]. Thus, although the recognition of C. albicans by human monocytes takes place independently of the integrity of lipid microdomains, a correct and balanced response by these leukocytes rely on the membrane organization provided by the rafts. Further studies are necessary to investigate the impact of host lipid domains during interaction with C. albicans in other cells from the immune system.
Figure 2-. Lipid domains from host cells are involved with association of fungal organisms.
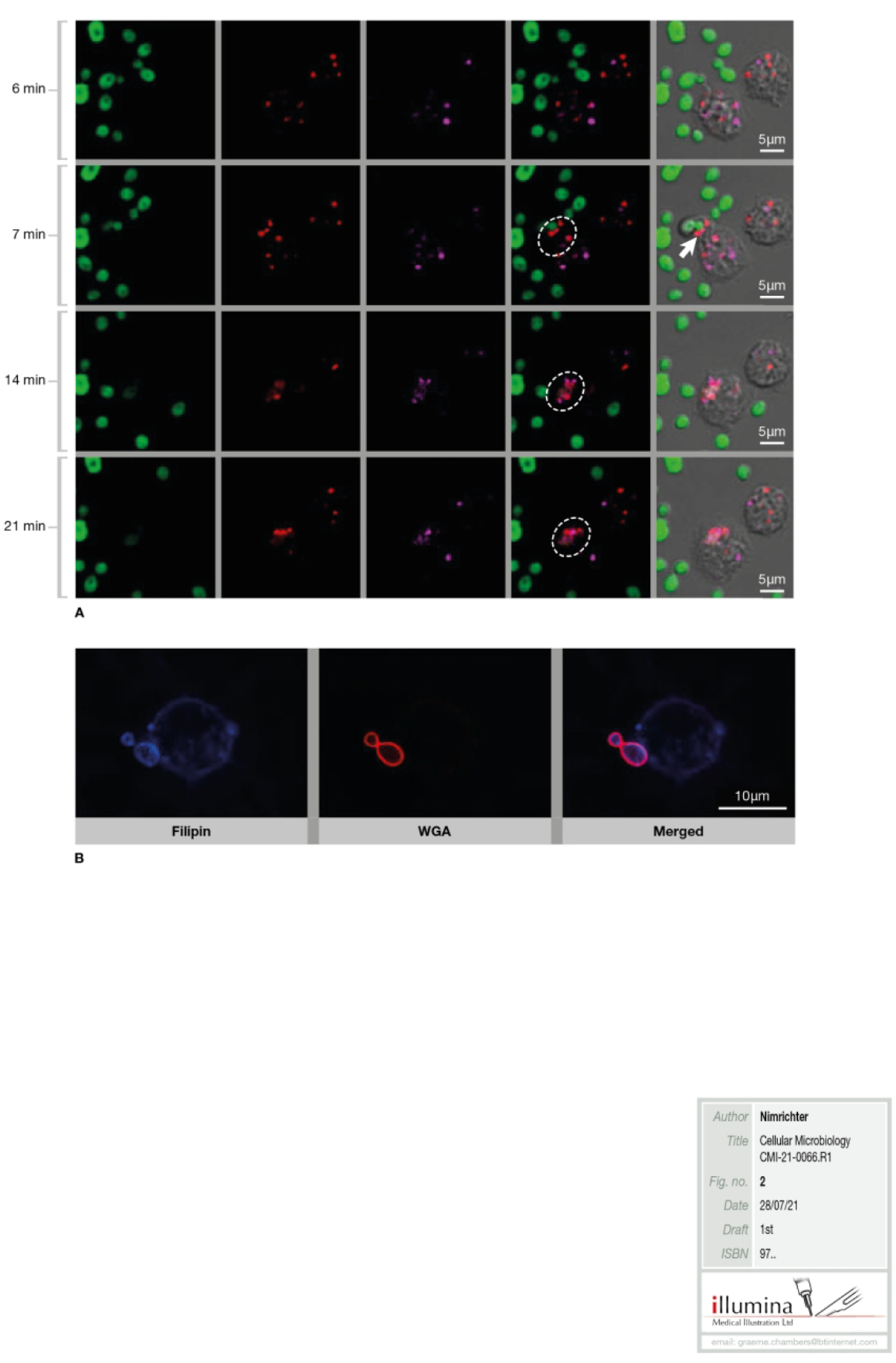
(A) Involvement of lipid rafts in Dectin-1-mediated phagocytosis of C. albicans (Ca). Time lapse analysis of Dectin-1 (red) and lipid raft components (cholerae toxin B/GM1, magenta) during the initial steps of association between C. albicans (green) and human monocytes. The timescale shows the recruitment of the glycosphingolipid GM1 and the PRR dectin-1 to the point of monocyte-fungal cell contact (white arrow and dashed circles). Scale bars, 5 μm. Adapted from Turris et al [47]. (B) Importance of lipid rafts in the association of Histoplasma capsulatum (Hc) with macrophages. Filipin staining (cholesterol/blue) shows the enrichment of sterol on the adhesion area indicating that cholesterol within lipid microdomains could be involved in Hc- macrophages recognition. Merged images denoting the co-localization of filipin staining and WGA-Alexa546-Hc (red). Bar, 10 μm. Obtained from Guimarães et al [58).
4.2. Cryptococcus neoformans
A set of studies demonstrates that C. neoformans penetration into human BMEC (HBMEC) is a lipid raft-dependent process (Fig. 1) [48–50]. Adhesion to HBMEC seems to be mediated by hyaluronic acid (HA), a fibrous structure outside the C. neoformans cell wall [61,62]. Treatment with hyaluronidase and deletion of CPS1, the coding gene of hyaluronic synthase, significantly reduced the binding of C. neoformans to HBMEC [61]. In addition, C. neoformans HA is recognized by CD44 at the HBMEC surface, a step blocked by anti-CD44 antibodies and CD44 shRNA treatments [62]. Association of C. neoformans with HBMEC induces the redistribution of CD44 to lipid rafts at the binding sites. Remarkably, treatment of host cells with filipin, a polyene antibiotic that immobilizes and reduces sterol availability, impacting the lateral mobility of host cell membranes, significantly decreases yeast association with HBMEC, suggesting that lipid raft integrity is required.
The colocalization of CD44 with caveolin-1 (Cav-1) also supports the role of these platforms during the internalization of C. neoformans by HBMEC [48,50]. Cav-1 is the main component of caveolae, a subset of microdomains involved with signal transduction and endocytosis (Fig. 1) [63]. The anchorage of caveolae on the plasma membrane is sustained by the connection of Cav-1 with actin cytoskeleton [64]. Thus, caveolae-dependent endocytosis is regulated through reorganization of the actin cytoskeleton, and triggered by phosphorylation of Cav-1 [65]. When Cav-1 expression is down-regulated in HBMEC, the number of internalized yeasts is reduced, with a small impact on fungal adhesion, suggesting that Cav-1 is not involved with binding but has a strong impact in the fungus’ internalization [50]. The binding of C. neoformans and HA to HBMEC also induces Cav-1 phosphorylation, a change that has been linked to caveolae-mediated internalization [50]. Cytoskeleton rearrangement was further investigated during C. neoformans internalization. Inhibition of actin polymerization and tyrosine kinase activity reduced the association and blood-brain barrier (BBB) transmigration of C. neoformans, confirming that both events are required for invasion [66]. Actin rearrangement appears to be initiated by C. neoformans’ binding to receptors at the endothelial plasma membrane, triggering the activation of Rho GTPases (RhoA, Rac1, and Cdc42) and phosphorylation of focal adhesion kinase (FAK), protein kinase C α (PKCα), and ezrin, promoting yeast transmigration across the BBB (Fig. 1) [66].
A pre-adhesion step could be mediated by extracellular vesicles (EVs) released by C. neoformans (Fig. 1) [51]. EVs are lipid bilayer membrane compartments used by cells to export a number of molecules, including proteins, polysaccharides, pigments, lipids and nucleic acids [67]. Their participation during disease development has been suggested for different fungi. Treatment of HBMEC with C. neoformans-derived EVs induces membrane ruffling, a response usually caused by cytoskeletal rearrangement [51]. These results were also observed when C. neoformans or HA were incubated with HBMEC [48]. However, the response mediated by EVs is CD44-independent [51]. Incubation with EVs promoted the redistribution of lipid raft components recruiting CD44, Cav-1, and up-shifted the intracellular distribution of β-actin. Changes induced by EVs could enhance C. neoformans recognition and transcytosis. Corroborating with the cellular changes, intravenous administration of EVs enhanced the ability of C. neoformans to reach the brain in a murine mice model of cryptococcosis.
Finally, the interplay between HIV infection and invasion of C. neoformans into HBMECs seems to be also mediated by lipid rafts [68,69]. It has been reported that a specific domain of the glycoprotein 41 (gp41), induces the recruitment of CD44 to HBMECs lipid rafts, and potentiate the C. neoformans invasion. The protein gp41 is a transmembrane subunit of the HIV envelope protein complex that mediates virus fusion [70]. The recombinant peptide, encompassing the specific sequence of amino acids of gp41 (between 579 and 611), was able to stimulate C. neoformans invasion in vitro and its intravenous injection enhanced brain infection in mice by C. neoformans [68]. In addition, treatment with gp41-I90 and C. neoformans synergistically enhanced the transmigration of monocytes in HBMEC and in vivo [71]. These findings contribute novel perspectives about the role that lipid rafts play in comorbidity between HIV and fungal neuro-infection.
4.3. Pneumocystis jirovecii
A balanced inflammatory response in pneymocystosis (PcP) is crucial for a patient to resolve Pneumocystis infection [72]. Lipid rafts are involved in the response of alveolar epithelial cell (AEC) to the β-glucans of Pneumocystis (PCBG). LacCer was responsible for stimulating the release of MIP-2 from AEC in a process dependent of PKC [52,53]. Treatment of AECs with monoclonal antibodies against LacCer, but not GM1, impairs internalization of PCBG [53]. In DCs, PCBG stimulates IL-23 and IL-6 secretion, culminating in the activation of the IL-23/IL-17 axis [54]. This activation is lipid raft-mediated, and dectin-1 is the main receptor, leading to the downstream activation of Syk and NF-κB. However, there is evidence that suggests that LacCer works as a co-receptor in this process, as β-glucan stimulation acts to mobilize and concentrate LacCer within the lipid microdomains [54]. These findings are clinically relevant because these cytokines are crucial for a Th17 phenotype, which plays a major role in fungal clearance and defense [73,74].
Overall, the major problem in patients with PcP is the overstimulation of the inflammatory response, which is induced by the interaction of host cells and fungal β-glucans [75]. Interestingly, an array of studies show that this response is significantly dampened by the inhibition of host cells lipid rafts [54,76], not only evidencing their role in the pathology but also suggesting that they could be a novel therapeutic target to pharmacological agents such as nystatin, which is capable of inhibiting the assembly of glycosphingolipid-cholesterol rich microdomains in host cells, interrupting PCBG recognition and potentially impairing fungal internalization [52].
4.4. Aspergillus fumigatus
For airborne pathogens, AECs and resident alveolar macrophages are the first cells involved with fungal recognition in the lung [77]. Recently, the ability of bronchial epithelial cells to attenuate virulence of A. fumigatus conidia was also linked to a mechanism of internalization involving lipid rafts [78]. The presence of flotillin-2 and caveolin in a ring-like structure surrounding internalized conidia, along with other host cell proteins, were demonstrated. Similar to Cav-1, flotillin-2 is also a protein enriched in lipid rafts and associated with endocytosis [78]. Remarkably, treatment of host cells with filipin reversed the ability of bronchial epithelial cells to decrease the conidia’s virulence [78].
The pigment melanin is an important virulence factor that protects fungal cells against host oxidative responses [79]. A recent study using a melanized-wild type conidia and a melanin-free conidia (pksP mutant) suggested that the presence of this pigment also helps A. fumigatus to control lipid rafts’ composition at the initial steps of fungal internalization and during the phagolysosomal maturation [55]. Remarkably, GM1 and flotillin-1 were significatively reduced in the phagolysosomal membranes containing melanized conidia. Since the recruitment of the proton pump V-ATPase is dependent on the presence of flotillin-1, a substantial reduction on phagolysosome acidification was observed, allowing conidia germination [55,80]. Phagosome maturation involves gradual changes to membrane composition, including the assembly of subunits of V-ATPase, and NADPH oxidase [81]. In line with that, NADPH oxidase complex assembly at flotillin-containing membrane microdomains was also compromised by melanin [55]. The signaling pathway that triggers this cascade of events is not fully elucidated, but the same study shows that formation of lipid rafts at the phagolysosomal membrane is dependent on calmodulin (CaM) activity. In this context, A. fumigatus’ melanin mediates Ca2+ sequestration inside the phagosome lumen, which reduces the availability of these ions in the peri-phagosomal area where calmodulin activation takes place [55,82]. Accordingly, treatment with calmodulin inhibitors significantly reduced the co-localization of raft marker the ganglioside GM1 in phagolysosomes containing conidia [55].
The role of melanin is complex, and can up or downregulate fungal phagocytosis depending on its structure [79]. An anti-phagocytic role was observed for melanin in A. fumigatus conidia, and this was correlated to modulation of flotillin-associated microdomains [55]. The pksP mutant conidia were more phagocytosed by macrophages, but in the absence of flotillin, the phagocytosis of this mutant was reduced. The presence of melanin and a hydrophobin rodlet at conidia cell wall prevents immune recognition [20]. However, the pksP conidia expose β-glucan, which activates LC3-associated phagocytosis (LAP), promoting fungal killing through NADPH oxidase activity in the phagosome [82]. LAP activation was recently shown to enhance the antimicrobial activity in macrophages and DCs infected by C. albicans, A. fumigatus or H. capsulatum, but the association between LAP and membrane microdomains has not been evaluated [82,83]. Purified melanin inhibits NADPH oxidase activity through exclusion of its subunit p22phox from the phagosome membrane [82]. Indeed, it is important to note that inhibition of Ca2+–CaM signaling by fungal melanin was also correlated with the blocking of LC3 recruitment to the phagosomal membrane of macrophages, which suggests a possible role of membrane microdomains in phagocytic routes involved with phagolysosomal activity [84].
4.5. Paracoccidioides brasiliensis
P. brasiliensis is a thermally dimorphic fungus, and infection is initiated by the inhalation of conidia or arthroconidia that differentiate to yeasts in the host’s lung [85]. Incubation of human AEC line A549 with yeasts of P. brasiliensis showed an enrichment of the raft marker GM1 at the fungal-host cell adhesion points. Binding was followed by a rapid activation of SRC family kinase (SFK) and ERK1/2. Treatment of A549 cells with nystatin and m-β-CD resulted in reduced fungal association and impaired activation of AECs. The lipid raft composition analysis confirmed that the association of P. brasiliensis yeasts with A549 cells induced the phosphorylation of SFK in lipid rafts. Pre-treatment with m-β-CD also abolished activation of SFK. Further studies demonstrated that P. brasiliensis recognition by A549 also promotes an upregulation of α3 and α5 integrins. Both integrins were recruited to caveolin-associated lipid rafts at the P. brasiliensis-A549 point of contact, suggesting the involvement of caveolae in fungal endocytosis [56]. In fact, treatment of A549 cells with genistein, a tyrosine kinase inhibitor used to block caveolae-dependent endocytosis, inhibited the adhesion and invasion of the fungus, demonstrating the importance of caveolae domains on the internalization of P. brasiliensis [86]. The role of AEC in driving an inflammatory response to an invasive fungal infection has been addressed and linked to a correct raft assembly [87,88]. The recruitment of α3 and α5 integrins to these domains induces IL-6 and IL-8 secretion by A549 cells [56]. IL-8 release by epithelial cells is essential for neutrophil recruitment and inflammatory response orchestration [89].
The enrichment of GM1 at P. brasiliensis binding sites in A549 cells and the previous studies showing that other fungi recognize LacCer at the plasma membrane of neutrophils suggest that this pathogen could bind to other GSL at the host cell surface. In fact, GalCer, LacCer, CTH (Galα1-4Galα1-4Glcα1-1Cer), GD3, GM1, and GD1a supported P. brasiliensis adhesion [90]. In addition, pre-incubation of P. brasiliensis yeast with GM1 made these cells reactive to the GM1-glycan ligand cholerae toxin subunit B (CTxB). Steric blocking of GM1 with CTxB, and GM3 with the antibody DH2, significantly reduced the adhesion of P. brasiliensis to human lung fibroblasts, confirming that these GSL could support fungal binding [90].
4.6. Histoplasma capsulatum
H. capsulatum is another thermally dimorphic fungus that affects primarily the lungs. The mechanism of interaction of yeasts with AEC is similar to described for P. brasiliensis. Translocation of α3 and α5 integrins and signaling machinery to lipid domains are associated with secretion of IL-6 and IL-8 by these cells. However, the role of AEC in fungal control remains elusive [57]. On the other hand, infection of macrophages is known as a critical step during histoplasmosis development. In some virulent strains of H. capsulatum, yeasts have the ability to block macrophage activation by inhibiting the recognition of the β-(1,3)-glucan by dectin-1 [91].
The role of PRRs on the innate immune response to H. capsulatum yeast has been a focus of several studies [92–94]. H. capsulatum yeasts use the β2 integrin (CD18) of CR3 to mediate recognition by macrophages through the surface exposure of fungal 60 kDa heat shock protein (HSP60) [95]. CR3 might be the exclusive phagocytic receptor involved in the internalization of this fungus (non-opsonized) by macrophages suggesting that a phagocytic synapse involving other PRRs at the lipid membrane domain is not required during host cell invasion [92]. However, when sialic acids are removed from host cells by enzymatic treatment, an impairment of macrophage association with H. capsulatum was observed. These results indicate that sialylated GSL could be involved in fungal interaction with immune cells [92]. Results from our group suggest that GSLs are involved in lipid rafts organization during macrophage infection by H. capsulatum (Fig. 1) [58]. We demonstrated the enrichment of cholesterol at the binding sites of H. capsulatum to macrophages (Fig 2). Depletion of cholesterol after treatment of macrophages with m-β-CD reduces H. capsulatum adhesion and internalization. Since m-β-CD can also extract other host membrane components it is important to confirm that the PRRs or their co-receptors are still at regular levels [60]. In addition, peritoneal macrophages from mice deficient for complex gangliosides production, such as GM1 and GD1a (B4galnt1−/) have a reduced ability to recognize H. capsulatum [58]. GM1-associated microdomains appear to be extremely important for H. capsulatum’ adhesion to host cells, although not to opsonized yeast cells. Our data also suggest an interesting interplay between GM1 and the CD11b/CD18 integrin. We demonstrated that both GM1 and the CD18/CD11b integrin are recruited to the macrophage/H. capsulatum interaction site. Furthermore, CD18 recruitment to lipid microdomains fractions was reduced in macrophages from B4galnt1−/−, indicating that GM1 could be an accessory molecule that facilitates the recruitment of CR3 into macrophage lipid rafts [58]. Remarkably, macrophage-H. capsulatum association induced recruitment of laterally organized lipid domains first at the site of fungus–phagocyte contact and then along the entire surface of the host cell.
As previously mentioned, the CR3 pathway involves lipid microdomains, and the signaling is mediated by LacCer-associated Lyn that activates cytoskeleton rearrangement [96]. HSP60 is expressed on the fungal surface in clusters, which may facilitate the CR3 binding process [95]. However, CR3 contains multiple binding sites, including a lectin site at the C-terminal domain that recognizes β-glucans [97]. H. capsulatum lacking α-(1,3)-glucan induces activation of the dectin-1 signaling pathway in macrophages, and the colocalization of dectin-1 and CR3 into GM1 microdomains was observed at the cell interface of interaction between host cells and heat-killed yeasts (Fig. 1) [59]. The collaborative work between dectin-1 and CR3 into lipid raft was crucial to trigger the cytokine response through downstream signaling of Syk, JNK, and transcriptional activity of AP-1 [59]. These results collectively suggest that H. capsulatum yeasts can avoid the recruitment of specific receptors to the space of lipids rafts in the plasma membrane of macrophages to more effectively facilitate the yeasts’ infection of macrophages.
5. Conclusion
Understanding lipid raft assembly during fungal-host cell interaction is an interesting paradigm that reveals other mechanisms by which each fungus can modify the host cell response and promote a variety of clinical conditions. It is a multistep process that impacts changes in the whole plasma membrane and, although the players are well-known, the dynamics behind the molecular events remain largely opaque. Because these pathways are tightly involved in fungal recognition and its intracellular fate and the elaboration of immune responses, they pay pose as attractive targets for therapeutics and pharmacological management of mycoses.
Take Away.
Lipid microdomains are ordered regions of the plasma membrane enriched in cholesterol, glycosphingolipids (GSL), GPI-anchored and signaling-related proteins.
Pathogen recognition by host immune cells can involve lipid microdomain participation. During this process these domains can coalesce in larger complexes recruiting receptors and signaling proteins, significantly increasing their signaling abilities.
The antifungal innate immune response is mediated by the engagement of pathogen-associated molecular patterns to pattern recognition receptors (PRRs) at the plasma membrane of innate immune cells. Lipid microdomains can concentrate or recruit PRRs during host cell-fungi association through a multi-interactive mechanism. This association can enhance the effectiveness of host effector processes. However, virulence factors at the fungal cell surface and extracellular vesicles can re-assembly these domains, compromising the downstream signaling and favoring the disease development.
Lipid microdomains are therefore very attractive targets for novel drugs to combat fungal infections.
Funding:
This work was supported by grants from the Brazilian agency Conselho Nacional de Desenvolvimento Científico e Tecnológico (311179/2017-7 and 408711/2017-7 to L.N.; 311470/2018-1 to AJG), FAPERJ (E-26/202.809/2018, E-26/202.696/2018 and E-26/202.760/2015). TNS was supported by Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES, Finance Code 001). JR was supported by Pasteur-Roux-Cantarini fellowship from the Institut Pasteur. J.D.N. was supported in part by NIH R21 AI124797.
Footnotes
Conflicts of Interest: The authors declare no conflict of interest.
Data availability statement:
The data that support the findings of this study (displayed at Figure 2) are openly available in https://doi.org/10.1371/journal.pone.0142531.g003 and at http://doi.org/[10.1371/journal.pone.0142531] and http://doi.org/10.1111/cmi.12976, reference numbers [47, 58].
References
- [1].Simons K, Van Meer G. Lipid sorting in epithelial cells. Biochemistry 1988;27:6197–202. 10.1021/bi00417a001. [DOI] [PubMed] [Google Scholar]
- [2].Simons K, Ikonen E. Functional rafts in cell membranes. Nature 1997;387:569–72. 10.1038/42408. [DOI] [PubMed] [Google Scholar]
- [3].Mukherjee S, Maxfield FR. Membrane domains. Annu Rev Cell Dev Biol 2004;20:839–66. 10.1146/annurev.cellbio.20.010403.095451. [DOI] [PubMed] [Google Scholar]
- [4].D’Angelo G, Capasso S, Sticco L, Russo D. Glycosphingolipids: synthesis and functions. FEBS J 2013;280:6338–53. 10.1111/febs.12559. [DOI] [PubMed] [Google Scholar]
- [5].Westerlund B, Grandell P-M, Isaksson YJE, Slotte JP. Ceramide acyl chain length markedly influences miscibility with palmitoyl sphingomyelin in bilayer membranes. Eur Biophys J 2010;39:1117–28. 10.1007/s00249-009-0562-6. [DOI] [PubMed] [Google Scholar]
- [6].Komura N, Suzuki KGN, Ando H, Konishi M, Koikeda M, Imamura A, et al. Raft-based interactions of gangliosides with a GPI-anchored receptor. Nat Chem Biol 2016;12:402–10. 10.1038/nchembio.2059. [DOI] [PubMed] [Google Scholar]
- [7].Gulbins E, Dreschers S, Wilker B, Grassmé H. Ceramide, membrane rafts and infections. J Mol Med 2004;82:357–63. 10.1007/s00109-004-0539-y. [DOI] [PubMed] [Google Scholar]
- [8].Bukrinsky MI, Mukhamedova N, Sviridov D. Lipid rafts and pathogens: the art of deception and exploitation. J Lipid Res 2020;61:601–10. 10.1194/jlr.TR119000391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Farnoud AM, Toledo AM, Konopka JB, Del Poeta M, London E. Raft-Like Membrane Domains in Pathogenic Microorganisms, 2015, p. 233–68. 10.1016/bs.ctm.2015.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Siafakas AR, Wright LC, Sorrell TC, Djordjevic JT. Lipid rafts in Cryptococcus neoformans concentrate the virulence determinants phospholipase B1 and Cu/Zn superoxide dismutase. Eukaryot Cell 2006;5:488–98. 10.1128/EC.5.3.488-498.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Tagliari L, Toledo MS, Lacerda TG, Suzuki E, Straus AH, Takahashi HK. Membrane microdomain components of Histoplasma capsulatum yeast forms, and their role in alveolar macrophage infectivity. Biochim Biophys Acta 2012;1818:458–66. 10.1016/j.bbamem.2011.12.008. [DOI] [PubMed] [Google Scholar]
- [12].Kirkland TN, Fierer J. Innate Immune Receptors and Defense Against Primary Pathogenic Fungi. Vaccines 2020;8:303. 10.3390/vaccines8020303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Miyazato A, Nakamura K, Yamamoto N, Mora-Montes HM, Tanaka M, Abe Y, et al. Toll-Like Receptor 9-Dependent Activation of Myeloid Dendritic Cells by Deoxynucleic Acids from Candida albicans. Infect Immun 2009;77:3056–64. 10.1128/IAI.00840-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Netea MG, Van De Veerdonk F, Verschueren I, Van Der Meer JWM, Kullberg BJ. Role of TLR1 and TLR6 in the host defense against disseminated candidiasis. FEMS Immunol Med Microbiol 2008;52:118–23. 10.1111/j.1574-695X.2007.00353.x. [DOI] [PubMed] [Google Scholar]
- [15].Kasperkovitz PV, Cardenas ML, Vyas JM. TLR9 Is Actively Recruited to Aspergillus fumigatus Phagosomes and Requires the N-Terminal Proteolytic Cleavage Domain for Proper Intracellular Trafficking. J Immunol 2010;185:7614–22. 10.4049/jimmunol.1002760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Biondo C, Malara A, Costa A, Signorino G, Cardile F, Midiri A, et al. Recognition of fungal RNA by TLR7 has a nonredundant role in host defense against experimental candidiasis. Eur J Immunol 2012;42:2632–43. 10.1002/eji.201242532. [DOI] [PubMed] [Google Scholar]
- [17].Rubino I, Coste A, Le Roy D, Roger T, Jaton K, Boeckh M, et al. Species-Specific Recognition of Aspergillus fumigatus by Toll-like Receptor 1 and Toll-like Receptor 6. J Infect Dis 2012;205:944–54. 10.1093/infdis/jir882. [DOI] [PubMed] [Google Scholar]
- [18].Patin EC, Thompson A, Orr SJ. Pattern recognition receptors in fungal immunity. Semin Cell Dev Biol 2019;89:24–33. 10.1016/j.semcdb.2018.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Kang X, Kirui A, Muszyński A, Widanage MCD, Chen A, Azadi P, et al. Molecular architecture of fungal cell walls revealed by solid-state NMR. Nat Commun 2018;9:2747. 10.1038/s41467-018-05199-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].The Fungal Cell Wall: Structure, Biosynthesis, and Function. The Fungal Kingdom, American Society of Microbiology; 2017, p. 267–92. 10.1128/microbiolspec.FUNK-0035-2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Hoebe K, Georgel P, Rutschmann S, Du X, Mudd S, Crozat K, et al. CD36 is a sensor of diacylglycerides. Nature 2005;433:523–7. 10.1038/nature03253. [DOI] [PubMed] [Google Scholar]
- [22].Dennehy KM, Ferwerda G, Faro-Trindade I, Pyż E, Willment JA, Taylor PR, et al. Syk kinase is required for collaborative cytokine production induced through Dectin-1 and Toll-like receptors. Eur J Immunol 2008;38:500–6. 10.1002/eji.200737741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].da Glória Sousa M, Reid DM, Schweighoffer E, Tybulewicz V, Ruland J, Langhorne J, et al. Restoration of Pattern Recognition Receptor Costimulation to Treat Chromoblastomycosis, a Chronic Fungal Infection of the Skin. Cell Host Microbe 2011;9:436–43. 10.1016/j.chom.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].de Sousa M da GT, Belda W, Spina R, Lota PR, Valente NS, Brown GD, et al. Topical Application of Imiquimod as a Treatment for Chromoblastomycosis. Clin Infect Dis 2014;58:1734–7. 10.1093/cid/ciu168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Płóciennikowska A, Hromada-Judycka A, Borzęcka K, Kwiatkowska K. Co-operation of TLR4 and raft proteins in LPS-induced pro-inflammatory signaling. Cell Mol Life Sci 2015;72:557–81. 10.1007/s00018-014-1762-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Kawasaki T, Kawai T. Toll-like receptor signaling pathways. Front Immunol 2014;5:461. 10.3389/fimmu.2014.00461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Motshwene PG, Moncrieffe MC, Grossmann JG, Kao C, Ayaluru M, Sandercock AM, et al. An Oligomeric Signaling Platform Formed by the Toll-like Receptor Signal Transducers MyD88 and IRAK-4. J Biol Chem 2009;284:25404–11. 10.1074/jbc.M109.022392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Zhu X, Owen JS, Wilson MD, Li H, Griffiths GL, Thomas MJ, et al. Macrophage ABCA1 reduces MyD88-dependent Toll-like receptor trafficking to lipid rafts by reduction of lipid raft cholesterol. J Lipid Res 2010;51:3196–206. 10.1194/jlr.M006486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Triantafilou M, Gamper FGJ, Haston RM, Mouratis MA, Morath S, Hartung T, et al. Membrane Sorting of Toll-like Receptor (TLR)-2/6 and TLR2/1 Heterodimers at the Cell Surface Determines Heterotypic Associations with CD36 and Intracellular Targeting. J Biol Chem 2006;281:31002–11. 10.1074/jbc.M602794200. [DOI] [PubMed] [Google Scholar]
- [30].Gringhuis SI, den Dunnen J, Litjens M, van het Hof B, van Kooyk Y, Geijtenbeek TBH. C-Type Lectin DC-SIGN Modulates Toll-like Receptor Signaling via Raf-1 Kinase-Dependent Acetylation of Transcription Factor NF-κB. Immunity 2007;26:605–16. 10.1016/j.immuni.2007.03.012. [DOI] [PubMed] [Google Scholar]
- [31].Inoue M, Moriwaki Y, Arikawa T, Chen Y-H, Oh YJ, Oliver T, et al. Cutting Edge: Critical Role of Intracellular Osteopontin in Antifungal Innate Immune Responses. J Immunol 2011;186:19–23. 10.4049/jimmunol.1002735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Inoue M, Shinohara ML. Clustering of Pattern Recognition Receptors for Fungal Detection. PLoS Pathog 2014;10:e1003873. 10.1371/journal.ppat.1003873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Triantafilou M, Miyake K, Golenbock DT, Triantafilou K. Mediators of innate immune recognition of bacteria concentrate in lipid rafts and facilitate lipopolysaccharide-induced cell activation. J Cell Sci 2002;115:2603–11. [DOI] [PubMed] [Google Scholar]
- [34].Nakahira K, Kim HP, Geng XH, Nakao A, Wang X, Murase N, et al. Carbon monoxide differentially inhibits TLR signaling pathways by regulating ROS-induced trafficking of TLRs to lipid rafts. J Exp Med 2006;203:2377–89. 10.1084/jem.20060845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Gantner BN, Simmons RM, Canavera SJ, Akira S, Underhill DM. Collaborative Induction of Inflammatory Responses by Dectin-1 and Toll-like Receptor 2. J Exp Med 2003;197:1107–17. 10.1084/jem.20021787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Ferwerda G, Meyer-Wentrup F, Kullberg B-J, Netea MG, Adema GJ. Dectin-1 synergizes with TLR2 and TLR4 for cytokine production in human primary monocytes and macrophages. Cell Microbiol 2008;10:2058–66. 10.1111/j.1462-5822.2008.01188.x. [DOI] [PubMed] [Google Scholar]
- [37].Goodridge HS, Reyes CN, Becker CA, Katsumoto TR, Ma J, Wolf AJ, et al. Activation of the innate immune receptor Dectin-1 upon formation of a ‘phagocytic synapse’. Nature 2011;472:471–5. 10.1038/nature10071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Xu S, Huo J, Gunawan M, Su I-H, Lam K-P. Activated Dectin-1 Localizes to Lipid Raft Microdomains for Signaling and Activation of Phagocytosis and Cytokine Production in Dendritic Cells. J Biol Chem 2009;284:22005–11. 10.1074/jbc.M109.009076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Agrawal S, Gupta S, Agrawal A. Human Dendritic Cells Activated via Dectin-1 Are Efficient at Priming Th17, Cytotoxic T and B Cell ResponsesCD8. PLoS One 2010;5:e13418. 10.1371/journal.pone.0013418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Jimenez-Lucho V, Ginsburg V, Krivan HC. Cryptococcus neoformans, Candida albicans, and other fungi bind specifically to the glycosphingolipid lactosylceramide (Gal beta 1–4Glc beta 1–1Cer), a possible adhesion receptor for yeasts. Infect Immun 1990;58:2085–90. 10.1128/IAI.58.7.2085-2090.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Goodridge HS, Wolf AJ, Underhill DM. β-glucan recognition by the innate immune system. Immunol Rev 2009;230:38–50. 10.1111/j.1600-065X.2009.00793.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Iwabuchi K, Masuda H, Kaga N, Nakayama H, Matsumoto R, Iwahara C, et al. Properties and functions of lactosylceramide from mouse neutrophils. Glycobiology 2015;25:655–68. 10.1093/glycob/cwv008. [DOI] [PubMed] [Google Scholar]
- [43].Iwabuchi K, Prinetti A, Sonnino S, Mauri L, Kobayashi T, Ishii K, et al. Involvement of very long fatty acid-containing lactosylceramide in lactosylceramide-mediated superoxide generation and migration in neutrophils. Glycoconj J 2008;25:357–74. 10.1007/s10719-007-9084-6. [DOI] [PubMed] [Google Scholar]
- [44].Nakayama H, Iwahara C, Takamori K, Ogawa H, Iwabuchi K. Lactosylceramide is a Pattern Recognition Receptor that Forms Lyn-Coupled Membrane Microdomains on Neutrophils. Immunol Endocr Metab Agents Med Chem 2008;8:327–35. 10.2174/187152208787169251. [DOI] [Google Scholar]
- [45].Sato T, Iwabuchi K, Nagaoka I, Adachi Y, Ohno N, Tamura H, et al. 25331130. J Leukoc Biol 2006;80:204–11. 10.1189/jlb.0106069. [DOI] [PubMed] [Google Scholar]
- [46].Nakayama H, Yoshizaki F, Prinetti A, Sonnino S, Mauri L, Takamori K, et al. Lyn-coupled LacCer-enriched lipid rafts are required for CD11b/CD18-mediated neutrophil phagocytosis of nonopsonized microorganisms. J Leukoc Biol 2008;83:728–41. 10.1189/jlb.0707478. [DOI] [PubMed] [Google Scholar]
- [47].de Turris V, Teloni R, Chiani P, Bromuro C, Mariotti S, Pardini M, et al. Candida albicans Targets a Lipid Raft/Dectin-1 Platform to Enter Human Monocytes and Induce Antigen Specific T Cell Responses. PLoS One 2015;10:e0142531. 10.1371/journal.pone.0142531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Jong A, Wu C-H, Shackleford GM, Kwon-Chung KJ, Chang YC, Chen H-M, et al. Involvement of human CD44 during Cryptococcus neoformans infection of brain microvascular endothelial cells. Cell Microbiol 2008;10:1313–26. 10.1111/j.1462-5822.2008.01128.x. [DOI] [PubMed] [Google Scholar]
- [49].Huang S-H, Long M, Wu C-H, Kwon-Chung KJ, Chang YC, Chi F, et al. Invasion of Cryptococcus neoformans into Human Brain Microvascular Endothelial Cells Is Mediated through the Lipid Rafts-Endocytic Pathway via the Dual Specificity Tyrosine Phosphorylation-regulated Kinase 3 (DYRK3). J Biol Chem 2011;286:34761–9. 10.1074/jbc.M111.219378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Long M, Huang S-H, Wu C-H, Shackleford GM, Jong A. Lipid raft/caveolae signaling is required for Cryptococcus neoformans invasion into human brain microvascular endothelial cells. J Biomed Sci 2012;19:19. 10.1186/1423-0127-19-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Huang S-H, Wu C-H, Chang YC, Kwon-Chung KJ, Brown RJ, Jong A. Cryptococcus neoformans-Derived Microvesicles Enhance the Pathogenesis of Fungal Brain Infection. PLoS One 2012;7:e48570. 10.1371/journal.pone.0048570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Evans SE, Kottom TJ, Pagano RE, Limper AH. Primary alveolar epithelial cell surface membrane microdomain function is required for Pneumocystis β-glucan-induced inflammatory responses. Innate Immun 2012;18:709–16. 10.1177/1753425912436763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Hahn PY, Evans SE, Kottom TJ, Standing JE, Pagano RE, Limper AH. Pneumocystis carinii Cell Wall β-Glucan Induces Release of Macrophage Inflammatory Protein-2 from Alveolar Epithelial Cells via a Lactosylceramide-mediated Mechanism. J Biol Chem 2003;278:2043–50. 10.1074/jbc.M209715200. [DOI] [PubMed] [Google Scholar]
- [54].Carmona EM, Kottom TJ, Hebrink DM, Moua T, Singh R-D, Pagano RE, et al. Glycosphingolipids Mediate Pneumocystis Cell Wall β-Glucan Activation of the IL-23/IL-17 Axis in Human Dendritic Cells. Am J Respir Cell Mol Biol 2012;47:50–9. 10.1165/rcmb.2011-0159OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Schmidt F, Thywißen A, Goldmann M, Cunha C, Cseresnyés Z, Schmidt H, et al. Flotillin-Dependent Membrane Microdomains Are Required for Functional Phagolysosomes against Fungal Infections. Cell Rep 2020;32:108017. 10.1016/j.celrep.2020.108017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Maza PK, Straus AH, Toledo MS, Takahashi HK, Suzuki E. Interaction of epithelial cell membrane rafts with Paracoccidioides brasiliensis leads to fungal adhesion and Src-family kinase activation. Microbes Infect 2008;10:540–7. 10.1016/j.micinf.2008.02.004. [DOI] [PubMed] [Google Scholar]
- [57].Maza PK, Suzuki E. Histoplasma capsulatum-Induced Cytokine Secretion in Lung Epithelial Cells Is Dependent on Host Integrins, Src-Family Kinase Activation, and Membrane Raft Recruitment. Front Microbiol 2016;7. 10.3389/fmicb.2016.00580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Guimarães AJ, Cerqueira MD, Zamith-Miranda D, Lopez PH, Rodrigues ML, Pontes B, et al. Host membrane glycosphingolipids and lipid microdomains facilitate Histoplasma capsulatum internalisation by macrophages. Cell Microbiol 2018:e12976. 10.1111/cmi.12976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Huang J-H, Lin C-Y, Wu S-Y, Chen W-Y, Chu C-L, Brown GD, et al. CR3 and Dectin-1 Collaborate in Macrophage Cytokine Response through Association on Lipid Rafts and Activation of Syk-JNK-AP-1 Pathway. PLOS Pathog 2015;11:e1004985. 10.1371/journal.ppat.1004985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Mahammad S, Parmryd I. Cholesterol depletion using methyl-β-cyclodextrin. Methods Mol Biol 2015;1232:91–102. 10.1007/978-1-4939-1752-5_8. [DOI] [PubMed] [Google Scholar]
- [61].Jong A, Wu C-H, Chen H-M, Luo F, Kwon-Chung KJ, Chang YC, et al. Identification and Characterization of CPS1 as a Hyaluronic Acid Synthase Contributing to the Pathogenesis of Cryptococcus neoformans Infection. Eukaryot Cell 2007;6:1486–96. 10.1128/EC.00120-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Jong A, Wu C-H, Gonzales-Gomez I, Kwon-Chung KJ, Chang YC, Tseng H-K, et al. Hyaluronic Acid Receptor CD44 Deficiency Is Associated with Decreased C ryptococcus neoformans Brain Infection. J Biol Chem 2012;287:15298–306. 10.1074/jbc.M112.353375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Kiss AL, Botos E. Endocytosis via caveolae: alternative pathway with distinct cellular compartments to avoid lysosomal degradation? J Cell Mol Med 2009;13:1228–37. 10.1111/j.1582-4934.2009.00754.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Mundy DI. Dual control of caveolar membrane traffic by microtubules and the actin cytoskeleton. J Cell Sci 2002;115:4327–39. 10.1242/jcs.00117. [DOI] [PubMed] [Google Scholar]
- [65].Zimnicka AM, Husain YS, Shajahan AN, Sverdlov M, Chaga O, Chen Z, et al. Src-dependent phosphorylation of caveolin-1 Tyr-14 promotes swelling and release of caveolae. Mol Biol Cell 2016;27:2090–106. 10.1091/mbc.E15-11-0756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Kim J-C, Crary B, Chang YC, Kwon-Chung KJ, Kim KJ. Cryptococcus neoformans Activates RhoGTPase Proteins Followed by Protein Kinase C, Focal Adhesion Kinase, and Ezrin to Promote Traversal across the Blood-Brain Barrier. J Biol Chem 2012;287:36147–57. 10.1074/jbc.M112.389676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Rizzo J, Rodrigues ML, Janbon G. Extracellular Vesicles in Fungi: Past, Present, and Future Perspectives. Front Cell Infect Microbiol 2020;10. 10.3389/fcimb.2020.00346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Jong AY, Wu C-H, Jiang S, Feng L, Chen H-M, Huang S-H. HIV-1 gp41 ectodomain enhances Cryptococcus neoformans binding to HBMEC. Biochem Biophys Res Commun 2007;356:899–905. 10.1016/j.bbrc.2007.03.100. [DOI] [PubMed] [Google Scholar]
- [69].Huang S-H, Wu C-H, Jiang S, Bahner I, Lossinsky AS, Jong AY. HIV-1 gp41 ectodomain enhances Cryptococcus neoformans binding to human brain microvascular endothelial cells via gp41 core-induced membrane activities. Biochem J 2011;438:457–66. 10.1042/BJ20110218. [DOI] [PubMed] [Google Scholar]
- [70].Pan C, Liu S, Jiang S. HIV-1 gp41 Fusion Intermediate: A Target for HIV Therapeutics. J Formos Med Assoc 2010;109:94–105. 10.1016/S0929-6646(10)60029-0. [DOI] [PubMed] [Google Scholar]
- [71].He X, Shi X, Puthiyakunnon S, Zhang L, Zeng Q, Li Y, et al. CD44-mediated monocyte transmigration across Cryptococcus neoformans-infected brain microvascular endothelial cells is enhanced by HIV-1 gp41-I90 ectodomain. J Biomed Sci 2016;23:28. 10.1186/s12929-016-0247-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Limper AH, Offord KP, Smith TF, Martin WJ. Pneumocystis carinii Pneumonia: Differences in Lung Parasite Number and Inflammation in Patients with and without AIDS. Am Rev Respir Dis 1989;140:1204–9. 10.1164/ajrccm/140.5.1204. [DOI] [PubMed] [Google Scholar]
- [73].Ma CS, Chew GYJ, Simpson N, Priyadarshi A, Wong M, Grimbacher B, et al. Deficiency of Th17 cells in hyper IgE syndrome due to mutations in STAT3. J Exp Med 2008;205:1551–7. 10.1084/jem.20080218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Milner JD, Brenchley JM, Laurence A, Freeman AF, Hill BJ, Elias KM, et al. Impaired TH17 cell differentiation in subjects with autosomal dominant hyper-IgE syndrome. Nature 2008;452:773–6. 10.1038/nature06764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Sokulska M, Kicia M, Wesołowska M, Hendrich AB. Pneumocystis jirovecii—from a commensal to pathogen: clinical and diagnostic review. Parasitol Res 2015;114:3577–85. 10.1007/s00436-015-4678-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Kottom TJ, Hebrink DM, Jenson PE, Gudmundsson G, Limper AH. Evidence for Proinflammatory β–1,6 Glucans in the Pneumocystis carinii Cell Wall. Infect Immun 2015;83:2816–26. 10.1128/IAI.00196-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Hartl D, Tirouvanziam R, Laval J, Greene CM, Habiel D, Sharma L, et al. Innate Immunity of the Lung: From Basic Mechanisms to Translational Medicine. J Innate Immun 2018;10:487–501. 10.1159/000487057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Clark HR, Powell AB, Simmons KA, Ayubi T, Kale SD. Endocytic Markers Associated with the Internalization and Processing of Aspergillus fumigatus Conidia by BEAS-2B Cells. MSphere 2019;4. 10.1128/mSphere.00663-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Smith DFQ, Casadevall A. The Role of Melanin in Fungal Pathogenesis for Animal Hosts, 2019, p. 1–30. 10.1007/82_2019_173. [DOI] [PubMed] [Google Scholar]
- [80].Dermine J-F, Duclos S, Garin J, St-Louis F, Rea S, Parton RG, et al. Flotillin-1-enriched Lipid Raft Domains Accumulate on Maturing Phagosomes. J Biol Chem 2001;276:18507–12. 10.1074/jbc.M101113200. [DOI] [PubMed] [Google Scholar]
- [81].Pauwels A-M, Trost M, Beyaert R, Hoffmann E. Patterns, Receptors, and Signals: Regulation of Phagosome Maturation. Trends Immunol 2017;38:407–22. 10.1016/j.it.2017.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Akoumianaki T, Kyrmizi I, Valsecchi I, Gresnigt MS, Samonis G, Drakos E, et al. Aspergillus Cell Wall Melanin Blocks LC3-Associated Phagocytosis to Promote Pathogenicity. Cell Host Microbe 2016;19:79–90. 10.1016/j.chom.2015.12.002. [DOI] [PubMed] [Google Scholar]
- [83].Huang J-H, Liu C-Y, Wu S-Y, Chen W-Y, Chang T-H, Kan H-W, et al. NLRX1 Facilitates Histoplasma capsulatum-Induced LC3-Associated Phagocytosis for Cytokine Production in Macrophages. Front Immunol 2018;9. 10.3389/fimmu.2018.02761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Kyrmizi I, Ferreira H, Carvalho A, Figueroa JAL, Zarmpas P, Cunha C, et al. Calcium sequestration by fungal melanin inhibits calcium–calmodulin signalling to prevent LC3-associated phagocytosis. Nat Microbiol 2018;3:791–803. 10.1038/s41564-018-0167-x. [DOI] [PubMed] [Google Scholar]
- [85].Martinez R New Trends in Paracoccidioidomycosis Epidemiology. J Fungi 2017;3:1. 10.3390/jof3010001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Monteiro da Silva JL, Andreotti PF, Benard G, Soares CP, Miranda ET, Mendes-Giannini MJS. Epithelial cells treated with genistein inhibit adhesion and endocytosis of Paracoccidioides brasiliensis. Antonie Van Leeuwenhoek 2007;92:129–35. 10.1007/s10482-006-9129-z. [DOI] [PubMed] [Google Scholar]
- [87].Wang J, Gigliotti F, Maggirwar S, Johnston C, Finkelstein JN, Wright TW. Pneumocystis carinii Activates the NF-κB Signaling Pathway in Alveolar Epithelial Cells. Infect Immun 2005;73:2766–77. 10.1128/IAI.73.5.2766-2777.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Bigot J, Guillot L, Guitard J, Ruffin M, Corvol H, Balloy V, et al. Bronchial Epithelial Cells on the Front Line to Fight Lung Infection-Causing Aspergillus fumigatus. Front Immunol 2020;11. 10.3389/fimmu.2020.01041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Kunkel SL, Standiford T, Kasahara K, Strieter RM. Interleukin-8 (IL-8): The Major Neutrophil Chemotactic Factor in the Lung. Exp Lung Res 1991;17:17–23. 10.3109/01902149109063278. [DOI] [PubMed] [Google Scholar]
- [90].Ywazaki CY, Maza PK, Suzuki E, Takahashi HK, Straus AH. Role of Host Glycosphingolipids on Paracoccidioides brasiliensis Adhesion. Mycopathologia 2011;171:325–32. 10.1007/s11046-010-9376-4. [DOI] [PubMed] [Google Scholar]
- [91].Brown GD. Trimming Surface Sugars Protects Histoplasma from Immune Attack. MBio 2016;7. 10.1128/mBio.00553-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Lin J-S, Huang J-H, Hung L-Y, Wu S-Y, Wu-Hsieh BA. Distinct roles of complement receptor 3, Dectin-1, and sialic acids in murine macrophage interaction with Histoplasma yeast. J Leukoc Biol 2010;88:95–106. 10.1189/jlb.1109717. [DOI] [PubMed] [Google Scholar]
- [93].Van Prooyen N, Henderson CA, Hocking Murray D, Sil A. CD103+ Conventional Dendritic Cells Are Critical for TLR7/9-Dependent Host Defense against Histoplasma capsulatum, an Endemic Fungal Pathogen of Humans. PLOS Pathog 2016;12:e1005749. 10.1371/journal.ppat.1005749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Chang T-H, Huang J-H, Lin H-C, Chen W-Y, Lee Y-H, Hsu L-C, et al. Dectin-2 is a primary receptor for NLRP3 inflammasome activation in dendritic cell response to Histoplasma capsulatum. PLOS Pathog 2017;13:e1006485. 10.1371/journal.ppat.1006485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Long KH, Gomez FJ, Morris RE, Newman SL. Identification of Heat Shock Protein 60 as the Ligand on Histoplasma capsulatum That Mediates Binding to CD18 Receptors on Human Macrophages. J Immunol 2003;170:487–94. 10.4049/jimmunol.170.1.487. [DOI] [PubMed] [Google Scholar]
- [96].Nakayama H, Ogawa H, Takamori K, Iwabuchi K. GSL-Enriched Membrane Microdomains in Innate Immune Responses. Arch Immunol Ther Exp (Warsz) 2013;61:217–28. 10.1007/s00005-013-0221-6. [DOI] [PubMed] [Google Scholar]
- [97].Ehlers MRW. CR3: a general purpose adhesion-recognition receptor essential for innate immunity. Microbes Infect 2000;2:289–94. 10.1016/S1286-4579(00)00299-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study (displayed at Figure 2) are openly available in https://doi.org/10.1371/journal.pone.0142531.g003 and at http://doi.org/[10.1371/journal.pone.0142531] and http://doi.org/10.1111/cmi.12976, reference numbers [47, 58].