

Abstract
Hyperuricemia predicts the development of chronic kidney disease (CKD) and metabolic complications, but whether it has a causal role has been controversial. This is especially true given two recently conducted randomized controlled trials that failed to show a benefit of lowering uric acid in type 1 diabetes-associated CKD and subjects with stage 3 to 4 CKD. While these studies suggest that use of urate lowering drugs in unselected patients are unlikely to slow the progression of CKD, there are subsets of subjects with CKD where reducing uric acid synthesis may be beneficial. This may be the case in patients with gout, hyperuricemia (especially associated with increased production), and patients with urate crystalluria. Here we discuss the evidence, and propose that future clinical trials targeting these specific subgroups should be performed.
Keywords: Hyperuricemia, Mendelian randomization, Metabolic Syndrome, Gout
The role of uric acid in chronic kidney disease (CKD) has been a controversial topic for decades. Studies supporting a role for uric acid as an important mediator of CKD is extensive. Epidemiological studies have consistently found that elevated serum uric acid is a predictor for the development of CKD, and early studies documented reduced kidney function in approximately 50 percent of subjects with gout and histologic injury (consisting of glomerulosclerosis, tubulointerstitial fibrosis and arteriolosclerosis) was found in in over 90 percent of autopsies of patients with gout [1,2]. Experimentally raising uric acid could both cause low grade kidney damage and accelerate established CKD [3,4]. Potential mechanisms were also identified driven by both crystalline and non-crystalline uric acid, that included both hemodynamic effects (increasing glomerular hydrostatic pressure and reducing blood flow) as well as non-hemodynamic pathways (stimulating inflammation, activating vasoconstrictive pathways, blocking endogenous vasodilatory pathways, and stimulating vascular cell proliferation and inhibiting endothelial function) [5–9]. Uric acid was also found to affect cellular metabolism, by causing mitochondrial oxidative stress, inhibiting mitochondrial function, and altering cellular energetics [10,11]. Indeed, studies suggested uric acid was not only important in kidney disease, but also in driving acute kidney injury, hypertension, metabolic syndrome and diabetes [12,13]. Uric acid has also been proposed to have potential roles in other conditions, including cancer, behavioral disorders, and Alzheimer’s disease [14–16].
There are also studies that have challenged the pathogenic role of uric acid in CKD. A central argument is that the association of uric acid with CKD is expected because the kidney has a major role in excreting uric acid, and so as kidney function fails, serum uric acid will rise. The finding that an elevated serum uric acid may predict the development of CKD may simply reflect that subtle kidney damage was already present. Indeed, once individuals have CKD, a high uric acid often does not independently predict progression of CKD [1]. Furthermore, a sophisticated method known as Mendelian randomization was used to identify genetic polymorphisms in genes that affect serum uric acid levels, and then a genetic score was developed that could affect approximately 5 to 7 percent of the variation in serum uric acid levels. When such scores were developed, population studies could determine if those who have scores favoring higher serum uric acid levels are at risk for CKD or other metabolic disorders. When these types of analyses were performed, the vast majority concluded that, while genetic polymorphisms could predict gout, they did not predict the development of CKD or other conditions including hypertension, metabolic syndrome, or diabetes [17–20]. More recently, an experimental model of hyperuricemia was reported in which kidney disease also did not develop, challenging the experimental studies [21]. Indeed, while it is accepted that crystalline uric acid is pro-inflammatory, there has even been the suggestion that soluble uric acid may be beneficial, as it can function as an anti-oxidant [22]. Supporting this idea have been several clinical studies suggesting that the acute infusion of uric acid (or its precursor inosine) may confer some benefits in neurological disorders such as Parkinson’s disease and acute ischemic stroke [23,24].
It has been largely thought that the best way to resolve the question of whether uric acid has a causal role in CKD would be to perform clinical trials to determine if lowering uric acid can slow the progression of CKD. Initial studies were positive [25,26], but many of these studies were small, lacked the use of a placebo, and were more consistent with pilot studies. One randomized, placebo-controlled study from India showed a remarkable slowing of CKD with febuxostat (a xanthine oxidase inhibitor) [27], but another larger but similarly designed study from Japan showed no benefit (the FEATHER study) [28]. Our group suggested that the reason might be because the control group in the FEATHER study showed minimal worsening of kidney disease over the two years of the study (−0.47 ml/min/1.73m2 eGFR), and that this prevented the ability for a study drug to show significant benefit even if it were effective [29]. Indeed, when we reviewed over 20 clinical trials that had been published, a benefit was observed in all of the studies in which significant progression occurred in the control group and the negative studies were limited to studies viewed as non-interpretable due to the fact that the kidney function in the control group remained stable [29].
Then, in June 2020, two large randomized, double-blind, placebo-controlled trials were published (Table 1) [30,31]. The first study, known as the Preventing Early Renal Loss in Diabetes (PERL) trial, evaluated type 1 diabetics with mild to moderate CKD (stage 2 and 3) with evidence for worsening kidney function. These subjects were normotensive, with around 90 percent on renin angiotensin system (RAS) blockers, and were randomized to allopurinol or placebo over 3 years. The study even included measurement of true GFR using the iohexol method. The second study, known as the Controlled Trial of Slowing of Kidney Disease Progression from the Inhibition of Xanthine Oxidase (CKD-FIX), studied the effect of allopurinol to slow renal progression in subjects with modest to severe CKD (stages 3 and 4) of all types (including 45 percent who were diabetic). Both studies reported no benefit of allopurinol to slow CKD despite significant worsening of kidney function in the control groups. Indeed, there was a tendency for higher mortality in the allopurinol treated groups when the studies were combined.
Table 1.
Strengths and Weaknesses in the CKD-FIX and PERL Studies
| Study | Characteristics | Participants (n) | Outcome (ΔGFR) | Strengths | Weaknesses |
|---|---|---|---|---|---|
| CKD-FIX | CKD (stage 3 or 4, 45% diabetic, proteinuric or with falling GFR in prior year, any uric acid) | Screened (11,998) Entered (369) Dropped out (93) Final (276) |
(−3.3 vs −3.2 mL/min/1.73 m2 per yr, allopurinol vs placebo) | Randomized, Double-blind Placebo-controlled Two years followup Intention-to-treat analysis |
Excluded Gout Included Normouricemia High dropout rate (25%) |
| PERL | Type 1 diabetes with CKD stage 2 or 3, uric acid ≥4.5 mg/dl) | Screened (1625) Entered (530) Dropped out (108) Final (422) |
(−3.0 vs −2.5 mL/min/1.73 m2 per yr, allopurinol vs placebo) | Randomized, Double-blind Placebo-controlled Three years followup True GFR measured (iohexol) Intention-to-treat analysis |
Excluded Gout Included normouricemia High dropout rate (20%) |
The strength of these two studies include the large number of patients studied, the rigorous design, the relatively long follow-up, the low risk of bias, and the finding of significant worsening of kidney function in the control group (Table 1). There were also weaknesses noted, including the high dropout rate (which affected the final analysis as these were intention-to-treat studies), the inclusion of individuals with normal uric acid levels, and the exclusion of subjects with gout (Table 1). Nevertheless, the findings of these two studies suggested the conclusion that “urate is not causally implicated in the risk for CKD progression in most patients regardless of baseline risk” and that further clinical trials are not warranted [32].
But, are there no caveats in this conclusion? One potential explanation is that uric acid is causal, but that the use of RAS inhibitors may be effectively mimicking the effect of lowering uric acid, as there are experimental and clinical studies that show that many of the effects of hyperuricemia appear to act by stimulation of the RAS [33–35]. If true, however, this would not provide any rationale for lowering of uric acid since RAS inhibition is standard fare in the subject with CKD.
There is another, important question. Lowering uric acid in the average patient with advanced CKD may not be of benefit, and lowering uric acid in an average type 1 diabetic subject with CKD may not show overall benefit, but can we generalize these studies to all patients with CKD? Here we discuss specific groups where we suggest clinical trials for lowering uric acid to preserve kidney function are needed (Figure 1).
Figure 1. Chronic Kidney Disease and Uric acid: Subpopulations to Consider for Clinical Trials.
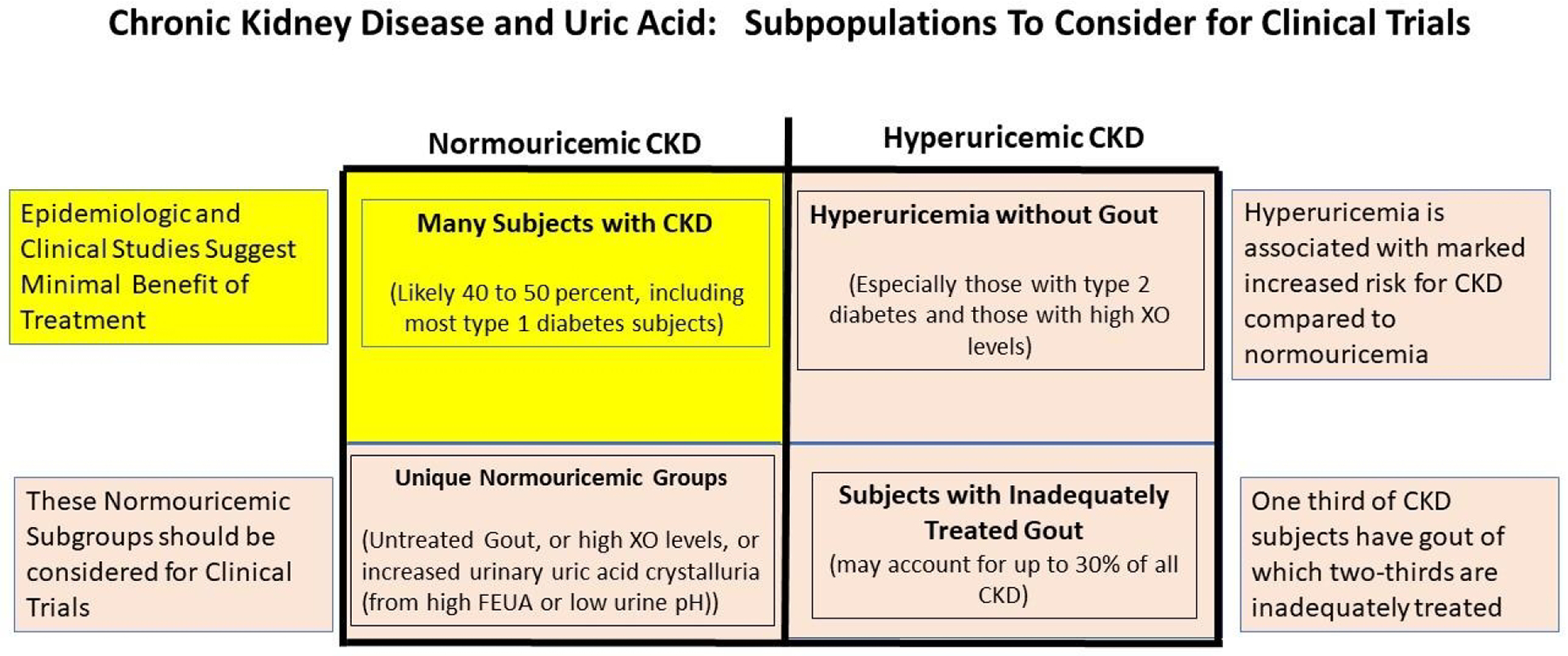
We do not recommend uric acid lowering treatment for normouricemic subjects with CKD, that includes the majority of type 1 diabetic subjects (yellow blocked section) based on the results of the CKD-FIX and PERL studies. This also includes many subjects with CKD who may have hypertension or proteinuria. However, there are certain subsets of CKD for which we would recommend clinical trials (brown boxes). This would include subjects with gout, subjects with hyperuricemia (especially those with type 2 diabetes or those with high plasma xanthine oxidase levels), and those relatively unique normouricemic subjects with untreated gout, high plasma XO levels, or with evidence of urate crystalluria (with low urine pH or high fractional excretion of uric acid).
Subgroups of CKD where Lowering Uric acid may be of Benefit
Gout and CKD
Gout is common in subjects with CKD, being present in about one-third of subjects with CKD stages 3 or 4 [36]. Unfortunately, the vast majority are either not treated or inadequately treated, and hence only 25 percent of subjects with gout and CKD have serum uric acid levels at the target goal of 6 mg/dl or less [37]. The PERL and CKD-FIX studies excluded all subjects with a history of gout, even if it was remote, assuming that they would already be on treatment for their arthritis. Therefore, the conclusions of these studies do not apply to patients with gout in whom lowering uric acid might theoretically be the most beneficial at reducing the progression of kidney disease.
Subjects with CKD and gout have more tophi than gouty subjects with normal kidney function (sixfold greater in stage 3 CKD)[38], suggesting they carry higher risk for extra-articular crystal deposits. Recently it has been recognized that urate crystals can deposit in many locations, including the kidney, blood vessels, and spine [39]. One study reported that urate crystals are present in the aorta or coronary vessels in 80 percent of subjects with gout [40], where it can localize to plaques[41]. Urate crystals may also act as a nidus for calcification [42], and many urate deposits correspond to sites of vascular calcification [40]. Urate crystals also deposit in the kidney, and one study suggests this may occur in up to one-third of subjects with gout [43]. Finally, urate crystals can also occur in the blood vessels and kidneys of subjects with hyperuricemia in the absence of gout, although it is much less frequent than that which occurs in subjects with gout [40].
Thus, it is quite possible that subjects with a history of gout and CKD might represent a unique group in which lowering uric acid might benefit not simply reducing gout attacks, but with potential benefits on CKD and vascular and heart disease.
Hyperuricemia (especially when unrelated to renal retention)
The CKD-FIX and PERL studies were not designed to study the benefit of treating hyperuricemia, therefore, both trials included subjects with normal uric acid levels. The risk for developing CKD, however, increases dramatically after serum uric acid levels reach the hyperuricemic range (typically at around 8 mg/dl), especially in women [44–46]. For example, in one population-based study, the risk for ESKD was 1.22 for men without hyperuricemia and 4.64 for men with hyperuricemia and 0.87 for women without hyperuricemia and 9.03 for women with hyperuricemia [44]. In the PERL study, the mean serum uric acid was in the normal range (6.1 mg/dl) documenting that the trial largely included normouricemic individuals. Any entry serum uric acid level was acceptable for the CKD-FIX trial, and since approximately half of CKD subjects are hyperuricemic at the time of dialysis initiation [47], it is likely that there were significant numbers of patients with normal uric acid levels despite the mean uric acid at entry being in the hyperuricemic range.
It is also possible that the type of hyperuricemia may be important. Many of the metabolic effects of uric acid appear to be mediated by intracellular uric acid levels. Factors that stimulate the production of uric acid, such as fructose, increase intracellular uric acid that then secondarily increases circulating (extracellular) uric acid [48,49]. This is then associated with a variety of metabolic effects, including the stimulation of oxidative stress, inflammation, and mitochondrial dysfunction [11,50,51]. Similarly, increasing extracellular uric acid can also lead to an increase in intracellular uric acid associated with metabolic activation [52]. However, recent studies suggest that high levels of extracellular uric acid may turn down the urate transport system reducing their ability to increase intracellular uric acid, at least in the monocyte [53].
This would suggest that hyperuricemia due to increased production might be more likely to mediate inflammatory effects compared to passive retention of uric acid such as from worsening kidney function. This might explain why hyperuricemia in the setting of normal renal function (which may be more likely from increased production) predicts the development of CKD while elevated uric acid in subjects with preexisting CKD does not predict worsening of kidney function [1].
One indirect way to assess intracellular uric acid is by measuring plasma xanthine oxidase activity by enzyme activity assays or western blot analysis. Indeed, plasma xanthine oxidase activity correlates with metabolic disorders better than serum uric acid [54]. Plasma xanthine oxidase activity have been linked with insulin resistance even when serum uric acid levels are low (<4 mg/dl) [55]. Plasma xanthine oxidase activity is high in many subjects with CKD, but it does not correlate well with serum uric acid [56]. Interestingly, subjects with CKD and elevated plasma xanthine oxidase activity have the greatest risk for developing cardiovascular events [56]. Thus, future studies may need to specifically test whether lowering uric acid benefits CKD subjects with hyperuricemia associated with or without elevated plasma xanthine oxidase activity.
Elevated Fractional Excretion of Uric acid.
While the the role of uric acid has generally focused on serum and intracellular uric acid, elevations in urinary uric acid may also predispose to kidney injury via crystalline and non-crystalline effects. The best example is tumor lysis syndrome in which massive production of uric acid leads to marked hyperuricemia with urate crystallization in the urinary tubules, leading to local obstruction and inflammation and an acute kidney injury (AKI) [57,58]. Similar types of AKI have been reported following rhabdomyolysis, grand mal seizures, and following cardiovascular surgery (especially in children). Urate crystalluria may also occur with heat stress and could potentially have a role in the CKD of unknown etiology observed in Central America [59]. AKI can also occur with the acute administration of uricosuric agents in hyperuricemic individuals [60]. Finally, AKI can occur in hypouricemic individuals with genetic absence of key urate transporters, such as SLC2A9 or URAT1 [61]. In the latter cases, this usually occurs after exercise in which uricosuria is worse and dehydration may be more common. One study reported that administering xanthine oxidase inhibitors to a hypouricemic individual with recurrent AKI led to a resolution of the AKI [62]. This is an example of how uric acid could be causing AKI even when the serum uric acid is <2 mg/dl.
Therefore, uric acid could potentially be causing CKD in some patients through a crystalluria mechanism. Indeed, we found that administering bicarbonate to type 1 diabetic subjects could alkalinize the urine, reduce urate crystals, and decrease tubular biomarkers of injury [63].
Today numerous studies are evaluating the effect of bicarbonate supplementation as a means to slow CKD progression[64]. While the focus has been on correcting metabolic acidosis, a simpler mechanism might be that it is alkalinizing the urine and decreasing urate crystal formation. Indeed, acid urine pH is now recognized as a risk factor for CKD [65]. It would be of interest if the benefit of bicarbonate administration was primarily in subjects who have acid urine and high urinary uric acid levels. Again, a clinical study investigating this might be warranted.
Hyperuricemia associated with Non-alcoholic liver disease (NAFLD) or Type 2 Diabetes
Experimental studies suggest that intake of added sugars containing fructose have a major role in driving both nonalcoholic fatty liver disease (NAFLD) and type 2 diabetes, and in both conditions there is evidence that elevations in intracellular uric acid has a contributory role [66,67]. Hyperuricemia is also a predictor for CKD in the subject with type 2 diabetes [68]. Likewise, the prevalence of CKD is higher in NAFLD patients, and it has been reported a relationship between the severity of NAFLD and the presence of decreased eGFR or proteinuria [69]. Moreover, incident CKD is also common in patients having NAFLD diagnosed by ultrasound at baseline [70]. Xanthine oxidase is shed into the circulation mainly from liver cells and its serum activity is increased in NAFLD patients [71]. These observations raise the possibility that hyperuricemia subjects with type 2 diabetes or NAFLD may benefit from XO inhibitors to prevent or slow CKD progression.
Biomarkers and Mendelian Randomization
Today we tend to assess the role of uric acid in disease by evaluating serum uric acid, and while it generally correlates with gout, metabolic disease, and kidney disease, we should recognize that serum uric acid level, may not reflect the systemic urate burden/pool, nor the intracellular uric acid level. As mentioned, experimental studies suggest that it is the intracellular uric acid level - better reflected by plasma xanthine oxidase activity - that drives most metabolic and renal diseases, and it is the fractional excretion of uric acid and the urinary pH that dictate urinary crystallization and acute kidney injury in crystal-dependent models of injury.
Even gout is imperfectly predicted by serum uric acid level. The classic teaching is that the risk for gout occurs when uric acid levels are greater than 6.8 mg/dl, since this represents the saturation concentration when uric acid crystallizes in water containing 140 mM sodium at pH 7.4. [72] Nevertheless, of the 30 million people in the United States with serum uric acid levels above this concentration, only 9 million have gout [73]. Thus, one can have super-saturable uric acid concentrations without having gout. Likewise, one can have gout attacks despite uric acid levels being < 6 mg/dl.[74,75] One possibility is that serum uric acid was high but that it was lower when measured during an acute gout attack due to transient increased urinary excretion[76]. There is also often an inappropriate reliance on fasting serum uric acid, as this may not accurately reflect postprandial levels following intake of purines or fructose, or with the acute effects of exercise or heat [59,77,78]. Despite these concerns, some subjects always have normal uric acid levels when tested on multiple occasions despite suffering from gout [74].
Thus, while generally one can ascribe gout as a condition associated with an elevated serum uric acid, a high serum uric acid is not requisite for gout nor does a low serum uric acid exclude it. It is similar to the story of the sugar cubes. If one sees a series of white sugar cubes, one cannot say that there are only white sugar cubes, but if one sees a series of white sugar cubes and one brown cube, one can say with certainty that sugar cubes do not always have to be white. The implication is that serum uric acid is not directly causal of gout, for one can have high uric acid levels without gout and low uric acid levels with gout. Rather, serum uric acid is a risk factor and the true causal risk factor is the net effect of the various factors driving urate crystallization that are important in determining whether a gout attack occurs.
A problem with the Mendelian randomization studies is that they assume the serum uric acid is not only the most important uric acid marker, but that it represents causality when it relates to uric acid disorders. While serum uric acid may commonly reflect intracellular uric acid, we know from the earlier discussions that this does not need to be true.
Mendelian randomization studies also depend on genetic score based on gene polymorphisms that influence serum uric acid. Most of these genes influence transport in and out of cells rather than affecting uric acid production. One of the strongest genetic polymorphisms involves SLC2A9, which has opposing functions depending on where it is expressed in the body, such that it eliminate serum uric acid via the gut but increases serum uric acid via the kidney [79]. This is why knocking out SLC2A9 in the kidney, or systemically, causes massive uricosuria and hypouricemia [80], but knocking out SCL2A9 in the liver causes hyperuricemia [79]. Interestingly, there is evidence that the metabolic effects of fructose and uric acid primarily result from metabolism in the liver [81]. Evidence supporting this comes from the gut-specific SLC2A9 knockout that develops metabolic abnormalities (increased body fat, hyperinsulinemia, hypertriglyceridemia, elevated blood pressure)[79]. Thus a polymorphism in SLC2A9 that raises serum uric acid via its effects on the kidney would not be expected to cause metabolic effects since there would be enhanced excretion of uric acid in the gut, reducing portal vein and hepatic levels. This could account for the negative Mendelian studies [17,20,82]. Nevertheless, more recently a number of Mendelian randomization studies have been able to associated genetic uric acid scores with a variety of outcomes, including hypertension, cardiovascular events, and kidney disease [83–86].
In summary, while the PERL and CKD-FIX studies have given important information, it is not a time to abandon all studies on the role of uric acid in CKD. Rather, it is an exciting time to test whether targeting specific subpopulations to test whether lowering uric acid in these groups can provide benefit (Figure 1). Among these specific subgroups, are the patients with gout, with hyperuricemia, with high plasma xanthine oxidase, and patients with high urinary excretion of uric acid.
Funding.
Supported in part by VA Merit (BXI01BX004501) (RJJ), NIH DK125351 (RJJ), and DK121496 (RJJ and MAL).
Footnotes
Conflict of Interest. RJJ has received honoraria from Horizon Pharma and Danone, has stocks with XORTX Therapeutics, and has equity with Colorado Research Partners LLC (CRP). LGSL and MAL also have equity with CRP. No other conflicts exist.
References
- 1.Johnson RJ, Nakagawa T, Jalal D, Sanchez-Lozada LG, Kang DH, Ritz E: Uric acid and chronic kidney disease: which is chasing which? Nephrol Dial Transplant 2013;28:2221–2228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Talbott JH, Terplan KL: The kidney in gout. Medicine (Baltimore) 1960;39:405–467. [PubMed] [Google Scholar]
- 3.Mazzali M, Hughes J, Kim YG, Jefferson JA, Kang DH, Gordon KL, Lan HY, Kivlighn S, Johnson RJ: Elevated uric acid increases blood pressure in the rat by a novel crystal-independent mechanism. Hypertension 2001;38:1101–1106. [DOI] [PubMed] [Google Scholar]
- 4.Kang DH, Nakagawa T, Feng L, Watanabe S, Han L, Mazzali M, Truong L, Harris R, Johnson RJ: A role for uric acid in the progression of renal disease. J Am Soc Nephrol 2002;13:2888–2897. [DOI] [PubMed] [Google Scholar]
- 5.Yu MA, Sanchez-Lozada LG, Johnson RJ, Kang DH: Oxidative stress with an activation of the renin-angiotensin system in human vascular endothelial cells as a novel mechanism of uric acid-induced endothelial dysfunction. J Hypertens 2010;28:1234–1242. [PubMed] [Google Scholar]
- 6.Sanchez-Lozada LG, Tapia E, Santamaria J, Avila-Casado C, Soto V, Nepomuceno T, Rodriguez-Iturbe B, Johnson RJ, Herrera-Acosta J: Mild hyperuricemia induces vasoconstriction and maintains glomerular hypertension in normal and remnant kidney rats. Kidney Int 2005;67:237–247. [DOI] [PubMed] [Google Scholar]
- 7.Kim SM, Lee SH, Kim YG, Kim SY, Seo JW, Choi YW, Kim DJ, Jeong KH, Lee TW, Ihm CG, Won KY, Moon JY: Hyperuricemia-induced NLRP3 activation of macrophages contributes to the progression of diabetic nephropathy. Am J Physiol Renal Physiol 2015;308:F993–F1003. [DOI] [PubMed] [Google Scholar]
- 8.Xiao J, Zhang XL, Fu C, Han R, Chen W, Lu Y, Ye Z: Soluble uric acid increases NALP3 inflammasome and interleukin-1beta expression in human primary renal proximal tubule epithelial cells through the Toll-like receptor 4-mediated pathway. Int J Mol Med 2015;35:1347–1354. [DOI] [PubMed] [Google Scholar]
- 9.Khosla UM, Zharikov S, Finch JL, Nakagawa T, Roncal C, Mu W, Krotova K, Block ER, Prabhakar S, Johnson RJ: Hyperuricemia induces endothelial dysfunction. Kidney Int 2005;67:1739–1742. [DOI] [PubMed] [Google Scholar]
- 10.Sanchez-Lozada LG, Lanaspa MA, Cristobal-Garcia M, Garcia-Arroyo F, Soto V, Cruz-Robles D, Nakagawa T, Yu MA, Kang DH, Johnson RJ: Uric Acid-Induced Endothelial Dysfunction Is Associated with Mitochondrial Alterations and Decreased Intracellular ATP Concentrations. Nephron Exp Nephrol 2012;121:e71–e78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lanaspa MA, Sanchez-Lozada LG, Choi YJ, Cicerchi C, Kanbay M, Roncal-Jimenez CA, Ishimoto T, Li N, Marek G, Duranay M, Schreiner G, Rodriguez-Iturbe B, Nakagawa T, Kang DH, Sautin YY, Johnson RJ: Uric acid induces hepatic steatosis by generation of mitochondrial oxidative stress: potential role in fructose-dependent and -independent fatty liver. J Biol Chem 2012;287:40732–40744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ejaz AA, Nakagawa T, Kanbay M, Kuwabara M, Kumar A, Garcia Arroyo FE, Roncal-Jimenez C, Sasai F, Kang DH, Jensen T, Hernando AA, Rodriguez-Iturbe B, Garcia G, Tolan DR, Sanchez-Lozada LG, Lanaspa MA, Johnson RJ: Hyperuricemia in Kidney Disease: A Major Risk Factor for Cardiovascular Events, Vascular Calcification, and Renal Damage. Semin Nephrol 2020;40:574–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Feig DI, Madero M, Jalal DI, Sanchez-Lozada LG, Johnson RJ: Uric acid and the origins of hypertension. J Pediatr 2013;162:896–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nakagawa T, Lanaspa MA, Millan IS, Fini M, Rivard CJ, Sanchez-Lozada LG, Andres-Hernando A, Tolan DR, Johnson RJ: Fructose contributes to the Warburg effect for cancer growth. Cancer Metab 2020;8:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fini MA, Elias A, Johnson RJ, Wright RM: Contribution of uric acid to cancer risk, recurrence, and mortality. Clin Transl Med 2012;1:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Johnson RJ, Gomez-Pinilla F, Nagel M, Nakagawa T, Rodriguez-Iturbe B, Sanchez-Lozada LG, Tolan DR, Lanaspa MA: Cerebral Fructose Metabolism as a Potential Mechanism Driving Alzheimer’s Disease. Front Aging Neurosci 2020;12:560865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jordan DM, Choi HK, Verbanck M, Topless R, Won HH, Nadkarni G, Merriman TR, Do R: No causal effects of serum urate levels on the risk of chronic kidney disease: A Mendelian randomization study. PLoS Med 2019;16:e1002725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pfister R, Barnes D, Luben R, Forouhi NG, Bochud M, Khaw KT, Wareham NJ, Langenberg C: No evidence for a causal link between uric acid and type 2 diabetes: a Mendelian randomisation approach. Diabetologia 2011;54:2561–2569. [DOI] [PubMed] [Google Scholar]
- 19.Sedaghat S, Pazoki R, Uitterlinden AG, Hofman A, Stricker BH, Ikram MA, Franco OH, Dehghan A: Association of uric acid genetic risk score with blood pressure: the Rotterdam study. Hypertension 2014;64:1061–1066. [DOI] [PubMed] [Google Scholar]
- 20.Yang Q, Kottgen A, Dehghan A, Smith AV, Glazer NL, Chen MH, Chasman DI, Aspelund T, Eiriksdottir G, Harris TB, Launer L, Nalls M, Hernandez D, Arking DE, Boerwinkle E, Grove ML, Li M, Linda Kao WH, Chonchol M, Haritunians T, Li G, Lumley T, Psaty BM, Shlipak M, Hwang SJ, Larson MG, O’Donnell CJ, Upadhyay A, van Duijn CM, Hofman A, Rivadeneira F, Stricker B, Uitterlinden AG, Pare G, Parker AN, Ridker PM, Siscovick DS, Gudnason V, Witteman JC, Fox CS, Coresh J: Multiple genetic loci influence serum urate levels and their relationship with gout and cardiovascular disease risk factors. Circ Cardiovasc Genet 2010;3:523–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sellmayr M, Hernandez Petzsche MR, Ma Q, Kruger N, Liapis H, Brink A, Lenz B, Angelotti ML, Gnemmi V, Kuppe C, Kim H, Bindels EMJ, Tajti F, Saez-Rodriguez J, Lech M, Kramann R, Romagnani P, Anders HJ, Steiger S: Only Hyperuricemia with Crystalluria, but not Asymptomatic Hyperuricemia, Drives Progression of Chronic Kidney Disease. J Am Soc Nephrol 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ames BN, Cathcart R, Schwiers E, Hochstein P: Uric acid provides an antioxidant defense in humans against oxidant- and radical-caused aging and cancer: a hypothesis. Proc Natl Acad Sci U S A 1981;78:6858–6862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schwarzschild MA, Macklin EA, Bakshi R, Battacharyya S, Logan R, Espay AJ, Hung AY, Bwala G, Goetz CG, Russell DS, Goudreau JL, Parashos SA, Saint-Hilaire MH, Rudolph A, Hare JM, Curhan GC, Ascherio A, Parkinson Study Group S-PDI: Sex differences by design and outcome in the Safety of Urate Elevation in PD (SURE-PD) trial. Neurology 2019;93:e1328–e1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chamorro A, Amaro S, Castellanos M, Gomis M, Urra X, Blasco J, Arenillas JF, Roman LS, Munoz R, Macho J, Canovas D, Marti-Fabregas J, Leira EC, Planas AM, Investigators U-I: Uric acid therapy improves the outcomes of stroke patients treated with intravenous tissue plasminogen activator and mechanical thrombectomy. Int J Stroke 2017;12:377–382. [DOI] [PubMed] [Google Scholar]
- 25.Siu YP, Leung KT, Tong MK, Kwan TH: Use of allopurinol in slowing the progression of renal disease through its ability to lower serum uric acid level. Am J Kidney Dis 2006;47:51–59. [DOI] [PubMed] [Google Scholar]
- 26.Goicoechea M, Garcia de Vinuesa S, Verdalles U, Verde E, Macias N, Santos A, Perez de Jose A, Cedeno S, Linares T, Luno J: Allopurinol and Progression of CKD and Cardiovascular Events: Long-term Follow-up of a Randomized Clinical Trial. Am J Kidney Dis 2015 [DOI] [PubMed] [Google Scholar]
- 27.Sircar D, Chatterjee S, Waikhom R, Golay V, Raychaudhury A, Chatterjee S, Pandey R: Efficacy of Febuxostat for Slowing the GFR Decline in Patients With CKD and Asymptomatic Hyperuricemia: A 6-Month, Double-Blind, Randomized, Placebo-Controlled Trial. Am J Kidney Dis 2015;66:945–950. [DOI] [PubMed] [Google Scholar]
- 28.Kimura K, Hosoya T, Uchida S, Inaba M, Makino H, Maruyama S, Ito S, Yamamoto T, Tomino Y, Ohno I, Shibagaki Y, Iimuro S, Imai N, Kuwabara M, Hayakawa H, Ohtsu H, Ohashi Y, Investigators FS: Febuxostat Therapy for Patients With Stage 3 CKD and Asymptomatic Hyperuricemia: A Randomized Trial. Am J Kidney Dis 2018;72:798–810. [DOI] [PubMed] [Google Scholar]
- 29.Sato Y, Feig DI, Stack AG, Kang DH, Lanaspa MA, Ejaz AA, Sanchez-Lozada LG, Kuwabara M, Borghi C, Johnson RJ: The case for uric acid-lowering treatment in patients with hyperuricaemia and CKD. Nat Rev Nephrol 2019;15:767–775. [DOI] [PubMed] [Google Scholar]
- 30.Doria A, Galecki AT, Spino C, Pop-Busui R, Cherney DZ, Lingvay I, Parsa A, Rossing P, Sigal RJ, Afkarian M, Aronson R, Caramori ML, Crandall JP, de Boer IH, Elliott TG, Goldfine AB, Haw JS, Hirsch IB, Karger AB, Maahs DM, McGill JB, Molitch ME, Perkins BA, Polsky S, Pragnell M, Robiner WN, Rosas SE, Senior P, Tuttle KR, Umpierrez GE, Wallia A, Weinstock RS, Wu C, Mauer M, Group PS: Serum Urate Lowering with Allopurinol and Kidney Function in Type 1 Diabetes. N Engl J Med 2020;382:2493–2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Badve SV, Pascoe EM, Tiku A, Boudville N, Brown FG, Cass A, Clarke P, Dalbeth N, Day RO, de Zoysa JR, Douglas B, Faull R, Harris DC, Hawley CM, Jones GRD, Kanellis J, Palmer SC, Perkovic V, Rangan GK, Reidlinger D, Robison L, Walker RJ, Walters G, Johnson DW, Investigators C-FS: Effects of Allopurinol on the Progression of Chronic Kidney Disease. N Engl J Med 2020;382:2504–2513. [DOI] [PubMed] [Google Scholar]
- 32.Pyne L, Walsh M: Allopurinol: Good for Gout But Not for Preventing Loss of Kidney Function. Am J Kidney Dis 2021;77:459–461. [DOI] [PubMed] [Google Scholar]
- 33.Mazzali M, Kanellis J, Han L, Feng L, Xia YY, Chen Q, Kang DH, Gordon KL, Watanabe S, Nakagawa T, Lan HY, Johnson RJ: Hyperuricemia induces a primary renal arteriolopathy in rats by a blood pressure-independent mechanism. Am J Physiol Renal Physiol 2002;282:F991–997. [DOI] [PubMed] [Google Scholar]
- 34.Talaat KM, El-Sheikh AR: The Effect of Mild Hyperuricemia on Urinary Transforming Growth Factor Beta and the Progression of Chronic Kidney Disease. Am J Nephrol 2007;27:435–440. [DOI] [PubMed] [Google Scholar]
- 35.Feig DI, Soletsky B, Johnson RJ: Effect of allopurinol on blood pressure of adolescents with newly diagnosed essential hypertension: a randomized trial. JAMA 2008;300:924–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Krishnan E: Reduced glomerular function and prevalence of gout: NHANES 2009–10. PLoS One 2012;7:e50046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fuldeore MJ, Riedel AA, Zarotsky V, Pandya BJ, Dabbous O, Krishnan E: Chronic kidney disease in gout in a managed care setting. BMC Nephrol 2011;12:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dalbeth N, House ME, Horne A, Taylor WJ: Reduced creatinine clearance is associated with early development of subcutaneous tophi in people with gout. BMC Musculoskelet Disord 2013;14:363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Khanna P, Johnson RJ, Marder B, LaMoreaux B, Kumar A: Systemic Urate Deposition: An Unrecognized Complication of Gout? J Clin Med 2020;9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Klauser AS, Halpern EJ, Strobl S, Gruber J, Feuchtner G, Bellmann-Weiler R, Weiss G, Stofferin H, Jaschke W: Dual-Energy Computed Tomography Detection of Cardiovascular Monosodium Urate Deposits in Patients With Gout. JAMA Cardiol 2019;4:1019–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Patetsios P, Song M, Shutze WP, Pappas C, Rodino W, Ramirez JA, Panetta TF: Identification of uric acid and xanthine oxidase in atherosclerotic plaque. Am J Cardiol 2001;88:188–191, A186. [DOI] [PubMed] [Google Scholar]
- 42.Coe FL, Raisen L: Allopurinol treatment of uric-acid disorders in calcium-stone formers. Lancet 1973;1:129–131. [DOI] [PubMed] [Google Scholar]
- 43.Bardin T, Nguyen QD, Tran KM, Le NH, D MD, Richette P, Letavernier E, Correas JM, Resche-Rigon M: Diffuse hyperechoic renal medulla pattern in gout: a crosssectional study of 502 patients. Kidney Int 2020;in press [DOI] [PubMed] [Google Scholar]
- 44.Iseki K, Ikemiya Y, Inoue T, Iseki C, Kinjo K, Takishita S: Significance of hyperuricemia as a risk factor for developing ESRD in a screened cohort. Am J Kidney Dis 2004;44:642–650. [PubMed] [Google Scholar]
- 45.Iseki K, Iseki C, Ikemiya Y, Fukiyama K: Risk of developing end-stage renal disease in a cohort of mass screening. Kidney Int 1996;49:800–805. [DOI] [PubMed] [Google Scholar]
- 46.Obermayr RP, Temml C, Gutjahr G, Knechtelsdorfer M, Oberbauer R, Klauser-Braun R: Elevated uric acid increases the risk for kidney disease. J Am Soc Nephrol 2008;19:2407–2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Suliman ME, Johnson RJ, Garcia-Lopez E, Qureshi AR, Molinaei H, Carrero JJ, Heimburger O, Barany P, Axelsson J, Lindholm B, Stenvinkel P: J-shaped mortality relationship for uric acid in CKD. Am J Kidney Dis 2006;48:761–771. [DOI] [PubMed] [Google Scholar]
- 48.Kim KM, Henderson GN, Ouyang X, Frye RF, Sautin YY, Feig DI, Johnson RJ: A sensitive and specific liquid chromatography-tandem mass spectrometry method for the determination of intracellular and extracellular uric acid. J Chromatogr B Analyt Technol Biomed Life Sci 2009;877:2032–2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.van den Berghe G, Bronfman M, Vanneste R, Hers HG: The mechanism of adenosine triphosphate depletion in the liver after a load of fructose. A kinetic study of liver adenylate deaminase. Biochem J 1977;162:601–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Glushakova O, Kosugi T, Roncal C, Mu W, Heinig M, Cirillo P, Sanchez-Lozada LG, Johnson RJ, Nakagawa T: Fructose induces the inflammatory molecule ICAM-1 in endothelial cells. J Am Soc Nephrol 2008;19:1712–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cirillo P, Gersch MS, Mu W, Scherer PM, Kim KM, Gesualdo L, Henderson GN, Johnson RJ, Sautin YY: Ketohexokinase-dependent metabolism of fructose induces proinflammatory mediators in proximal tubular cells. J Am Soc Nephrol 2009;20:545–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sautin YY, Nakagawa T, Zharikov S, Johnson RJ: Adverse effects of the classic antioxidant uric acid in adipocytes: NADPH oxidase-mediated oxidative/nitrosative stress. Am J Physiol Cell Physiol 2007;293:C584–596. [DOI] [PubMed] [Google Scholar]
- 53.Ma Q, Honarpisheh M, Li C, Sellmayr M, Lindenmeyer M, Bohland C, Romagnani P, Anders HJ, Steiger S: Soluble Uric Acid Is an Intrinsic Negative Regulator of Monocyte Activation in Monosodium Urate Crystal-Induced Tissue Inflammation. J Immunol 2020;205:789–800. [DOI] [PubMed] [Google Scholar]
- 54.Furuhashi M, Matsumoto M, Tanaka M, Moniwa N, Murase T, Nakamura T, Ohnishi H, Saitoh S, Shimamoto K, Miura T: Plasma Xanthine Oxidoreductase Activity as a Novel Biomarker of Metabolic Disorders in a General Population. Circ J 2018;82:1892–1899. [DOI] [PubMed] [Google Scholar]
- 55.Furuhashi M, Mori K, Tanaka M, Maeda T, Matsumoto M, Murase T, Nakamura T, Koyama M, Moniwa N, Ohnishi H, Saitoh S, Shimamoto K, Miura T: Unexpected high plasma xanthine oxidoreductase activity in female subjects with low levels of uric acid. Endocr J 2018;65:1083–1092. [DOI] [PubMed] [Google Scholar]
- 56.Gondouin B, Jourde-Chiche N, Sallee M, Dou L, Cerini C, Loundou A, Morange S, Berland Y, Burtey S, Brunet P, Guieu R, Dussol B: Plasma Xanthine Oxidase Activity Is Predictive of Cardiovascular Disease in Patients with Chronic Kidney Disease, Independently of Uric Acid Levels. Nephron 2015;131:167–174. [DOI] [PubMed] [Google Scholar]
- 57.Shimada M, Johnson RJ, May WS Jr., Lingegowda V, Sood P, Nakagawa T, Van QC, Dass B, Ejaz AA: A novel role for uric acid in acute kidney injury associated with tumour lysis syndrome. Nephrol Dial Transplant 2009;24:2960–2964. [DOI] [PubMed] [Google Scholar]
- 58.Kim YG, Huang XR, Suga S, Mazzali M, Tang D, Metz C, Bucala R, Kivlighn S, Johnson RJ, Lan HY: Involvement of macrophage migration inhibitory factor (MIF) in experimental uric acid nephropathy. Mol Med 2000;6:837–848. [PMC free article] [PubMed] [Google Scholar]
- 59.Roncal-Jimenez C, García-Trabanino R, Barregard L, Lanaspa MA, Wesseling C, Harra T, Aragón A, Grases F, Jarquin ER, González MA, Weiss I, Glaser J, Sánchez-Lozada LG, Johnson RJ: Heat Stress Nephropathy From Exercise-Induced Uric Acid Crystalluria: A Perspective on Mesoamerican Nephropathy. Am J Kidney Dis 2016;67:20–30. [DOI] [PubMed] [Google Scholar]
- 60.Sanchez-Nino MD, Zheng-Lin B, Valino-Rivas L, Sanz AB, Ramos AM, Luno J, Goicoechea M, Ortiz A: Lesinurad: what the nephrologist should know. Clin Kidney J 2017;10:679–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shen H, Feng C, Jin X, Mao J, Fu H, Gu W, Liu A, Shu Q, Du L: Recurrent exercise-induced acute kidney injury by idiopathic renal hypouricemia with a novel mutation in the SLC2A9 gene and literature review. BMC Pediatr 2014;14:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bhasin B, Stiburkova B, De Castro-Pretelt M, Beck N, Bodurtha JN, Atta MG: Hereditary renal hypouricemia: a new role for allopurinol? Am J Med 2014;127:e3–4. [DOI] [PubMed] [Google Scholar]
- 63.Bjornstad P, Maahs DM, Roncal CA, Snell-Bergeon JK, Shah VN, Milagres T, Ellis SL, Hatch M, Chung LT, Rewers MJ, Garg S, Cherney DZ, Pyle L, Nadeau KJ, Johnson RJ: Role of bicarbonate supplementation on urine uric acid crystals and diabetic tubulopathy in adults with type 1 diabetes. Diabetes Obes Metab 2018;20:1776–1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Goraya N, Wesson DE: Clinical evidence that treatment of metabolic acidosis slows the progression of chronic kidney disease. Curr Opin Nephrol Hypertens 2019;28:267–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nakanishi N, Fukui M, Tanaka M, Toda H, Imai S, Yamazaki M, Hasegawa G, Oda Y, Nakamura N: Low urine pH Is a predictor of chronic kidney disease. Kidney Blood Press Res 2012;35:77–81. [DOI] [PubMed] [Google Scholar]
- 66.Jensen T, Abdelmalek MF, Sullivan S, Nadeau KJ, Green M, Roncal C, Nakagawa T, Kuwabara M, Sato Y, Kang DH, Tolan DR, Sanchez-Lozada LG, Rosen HR, Lanaspa MA, Diehl AM, Johnson RJ: Fructose and sugar: A major mediator of non-alcoholic fatty liver disease. J Hepatol 2018;68:1063–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Johnson RJ, Nakagawa T, Sanchez-Lozada LG, Shafiu M, Sundaram S, Le M, Ishimoto T, Sautin YY, Lanaspa MA: Sugar, uric acid, and the etiology of diabetes and obesity. Diabetes 2013;62:3307–3315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.De Cosmo S, Viazzi F, Pacilli A, Giorda C, Ceriello A, Gentile S, Russo G, Rossi MC, Nicolucci A, Guida P, Feig D, Johnson RJ, Pontremoli R, Group AM-AS: Serum Uric Acid and Risk of CKD in Type 2 Diabetes. Clin J Am Soc Nephrol 2015;10:1921–1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Targher G, Chonchol MB, Byrne CD: CKD and nonalcoholic fatty liver disease. Am J Kidney Dis 2014;64:638–652. [DOI] [PubMed] [Google Scholar]
- 70.Targher G, Francque SM: A fatty liver leads to decreased kidney function? J Hepatol 2017;67:1137–1139. [DOI] [PubMed] [Google Scholar]
- 71.Zhang J, Xu C, Zhao Y, Chen Y: The significance of serum xanthine oxidoreductase in patients with nonalcoholic fatty liver disease. Clin Lab 2014;60:1301–1307. [DOI] [PubMed] [Google Scholar]
- 72.Loeb JN: The influence of temperature on the solubility of monosodium urate. Arthritis Rheum 1972;15:189–192. [DOI] [PubMed] [Google Scholar]
- 73.Singh G, Lingala B, Mithal A: Gout and hyperuricaemia in the USA: prevalence and trends. Rheumatology (Oxford) 2019;58:2177–2180. [DOI] [PubMed] [Google Scholar]
- 74.McCarty DJ: Gout without hyperuricemia. JAMA 1994;271:302–303. [PubMed] [Google Scholar]
- 75.Hall AP, Barry PE, Dawber TR, McNamara PM: Epidemiology of gout and hyperuricemia. A long-term population study. Am J Med 1967;42:27–37. [DOI] [PubMed] [Google Scholar]
- 76.Urano W, Yamanaka H, Tsutani H, Nakajima H, Matsuda Y, Taniguchi A, Hara M, Kamatani N: The inflammatory process in the mechanism of decreased serum uric acid concentrations during acute gouty arthritis. J Rheumatol 2002;29:1950–1953. [PubMed] [Google Scholar]
- 77.Cox CL, Stanhope KL, Schwarz JM, Graham JL, Hatcher B, Griffen SC, Bremer AA, Berglund L, McGahan JP, Keim NL, Havel PJ: Consumption of fructose- but not glucose-sweetened beverages for 10 weeks increases circulating concentrations of uric acid, retinol binding protein-4, and gamma-glutamyl transferase activity in overweight/obese humans. Nutr Metab (Lond) 2012;9:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Clifford AJ, Riumallo JA, Youn VR, Scrimshaw NS: Effect of Oral Purines on Serum and Urinary Uric Acid of Normal, Hyperuricemic and Gouty Humans. J Nutr 1976;106:428–450. [Google Scholar]
- 79.DeBosch BJ, Kluth O, Fujiwara H, Schurmann A, Moley K: Early-onset metabolic syndrome in mice lacking the intestinal uric acid transporter SLC2A9. Nat Commun 2014;5:4642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Preitner F, Bonny O, Laverriere A, Rotman S, Firsov D, Da Costa A, Metref S, Thorens B: Glut9 is a major regulator of urate homeostasis and its genetic inactivation induces hyperuricosuria and urate nephropathy. Proc Natl Acad Sci U S A 2009;106:15501–15506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Andres-Hernando A, Orlicky DJ, Kuwabara M, Ishimoto T, Nakagawa T, Johnson RJ, Lanaspa MA: Deletion of Fructokinase in the Liver or in the Intestine Reveals Differential Effects on Sugar-Induced Metabolic Dysfunction. Cell Metab 2020;32:117–127 e113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tin A, Marten J, Halperin Kuhns VL, Li Y, Wuttke M, Kirsten H, Sieber KB, Qiu C, Gorski M, Yu Z, Giri A, Sveinbjornsson G, Li M, Chu AY, Hoppmann A, O’Connor LJ, Prins B, Nutile T, Noce D, Akiyama M, Cocca M, Ghasemi S, van der Most PJ, Horn K, Xu Y, Fuchsberger C, Sedaghat S, Afaq S, Amin N, Arnlov J, Bakker SJL, Bansal N, Baptista D, Bergmann S, Biggs ML, Biino G, Boerwinkle E, Bottinger EP, Boutin TS, Brumat M, Burkhardt R, Campana E, Campbell A, Campbell H, Carroll RJ, Catamo E, Chambers JC, Ciullo M, Concas MP, Coresh J, Corre T, Cusi D, Felicita SC, de Borst MH, De Grandi A, de Mutsert R, de Vries APJ, Delgado G, Demirkan A, Devuyst O, Dittrich K, Eckardt KU, Ehret G, Endlich K, Evans MK, Gansevoort RT, Gasparini P, Giedraitis V, Gieger C, Girotto G, Gogele M, Gordon SD, Gudbjartsson DF, Gudnason V, German Chronic Kidney Disease S, Haller T, Hamet P, Harris TB, Hayward C, Hicks AA, Hofer E, Holm H, Huang W, Hutri-Kahonen N, Hwang SJ, Ikram MA, Lewis RM, Ingelsson E, Jakobsdottir J, Jonsdottir I, Jonsson H, Joshi PK, Josyula NS, Jung B, Kahonen M, Kamatani Y, Kanai M, Kerr SM, Kiess W, Kleber ME, Koenig W, Kooner JS, Korner A, Kovacs P, Kramer BK, Kronenberg F, Kubo M, Kuhnel B, La Bianca M, Lange LA, Lehne B, Lehtimaki T, Lifelines Cohort S, Liu J, Loeffler M, Loos RJF, Lyytikainen LP, Magi R, Mahajan A, Martin NG, Marz W, Mascalzoni D, Matsuda K, Meisinger C, Meitinger T, Metspalu A, Milaneschi Y, Program VAMV, O’Donnell CJ, Wilson OD, Gaziano JM, Mishra PP, Mohlke KL, Mononen N, Montgomery GW, Mook-Kanamori DO, Muller-Nurasyid M, Nadkarni GN, Nalls MA, Nauck M, Nikus K, Ning B, Nolte IM, Noordam R, O’Connell JR, Olafsson I, Padmanabhan S, Penninx B, Perls T, Peters A, Pirastu M, Pirastu N, Pistis G, Polasek O, Ponte B, Porteous DJ, Poulain T, Preuss MH, Rabelink TJ, Raffield LM, Raitakari OT, Rettig R, Rheinberger M, Rice KM, Rizzi F, Robino A, Rudan I, Krajcoviechova A, Cifkova R, Rueedi R, Ruggiero D, Ryan KA, Saba Y, Salvi E, Schmidt H, Schmidt R, Shaffer CM, Smith AV, Smith BH, Spracklen CN, Strauch K, Stumvoll M, Sulem P, Tajuddin SM, Teren A, Thiery J, Thio CHL, Thorsteinsdottir U, Toniolo D, Tonjes A, Tremblay J, Uitterlinden AG, Vaccargiu S, van der Harst P, van Duijn CM, Verweij N, Volker U, Vollenweider P, Waeber G, Waldenberger M, Whitfield JB, Wild SH, Wilson JF, Yang Q, Zhang W, Zonderman AB, Bochud M, Wilson JG, Pendergrass SA, Ho K, Parsa A, Pramstaller PP, Psaty BM, Boger CA, Snieder H, Butterworth AS, Okada Y, Edwards TL, Stefansson K, Susztak K, Scholz M, Heid IM, Hung AM, Teumer A, Pattaro C, Woodward OM, Vitart V, Kottgen A: Target genes, variants, tissues and transcriptional pathways influencing human serum urate levels. Nat Genet 2019;51:1459–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gill D, Cameron AC, Burgess S, Li X, Doherty DJ, Karhunen V, Abdul-Rahim AH, Taylor-Rowan M, Zuber V, Tsao PS, Klarin D, Program VAMV, Evangelou E, Elliott P, Damrauer SM, Quinn TJ, Dehghan A, Theodoratou E, Dawson J, Tzoulaki I: Urate, Blood Pressure, and Cardiovascular Disease: Evidence From Mendelian Randomization and Meta-Analysis of Clinical Trials. Hypertension 2021;77:383–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kleber ME, Delgado G, Grammer TB, Silbernagel G, Huang J, Kramer BK, Ritz E, Marz W: Uric Acid and Cardiovascular Events: A Mendelian Randomization Study. J Am Soc Nephrol 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yan D, Wang J, Jiang F, Zhang R, Wang T, Wang S, Peng D, He Z, Chen H, Bao Y, Hu C, Jia W: A causal relationship between uric acid and diabetic macrovascular disease in Chinese type 2 diabetes patients: A Mendelian randomization analysis. Int J Cardiol 2016;214:194–199. [DOI] [PubMed] [Google Scholar]
- 86.Ge JY, Ji Y, Zhu ZY, Li X: Genetically Elevated Serum Uric Acid and Renal Function in an Apparently Healthy Population. Urol Int 2019:1–6. [DOI] [PubMed] [Google Scholar]