

Abstract
Taxanes are widely used cancer chemotherapeutics. However, intrinsic resistance limits their efficacy without any actionable resistance mechanism. We have discovered a microtubule (MT) plus-end binding CLIP-170 protein variant, hereafter CLIP-170S, which we found enriched in taxane resistant cell lines and patient samples. CLIP-170S lacks the first Cap-Gly motif, forms longer comets and impairs taxane access to its MT luminal binding site. CLIP-170S knock-down reversed taxane resistance in cells and xenografts, while its re-expression led to resistance, suggesting causation. Using a computational approach in conjunction with the connectivity map we unexpectedly discovered that imatinib was predicted to reverse CLIP-170S-mediated taxane resistance. Indeed, imatinib treatment selectively depleted CLIP-170S, thus, completely reversing taxane resistance. Other RTK inhibitors also depleted CLIP-170S, suggesting a class effect. Herein, we identify CLIP-170S as a clinically prevalent variant that confers taxane resistance while the discovery of imatinib as a CLIP-170S inhibitor, provides novel therapeutic opportunities for future trials.
Graphical Abstract

eTOC blurb:
Taxanes are widely-used cancer chemotherapeutics. Thakkar, Kita et al. discovered a truncated variant of the microtubule plus-end binding protein CLIP-170 that confers resistance to taxanes by binding along the microtubule lattice and disrupting taxane access to the microtubule lumen. RTK inhibitors reverse this mechanism of taxane resistance by depleting the variant.
Background
Gastric cancer (GC) is a highly morbid and prevalent global disease and the second leading cause of cancer related deaths worldwide(Ferlay et al., 2019). Such a dismal prognosis is a result of advanced stage at diagnosis coupled with demonstrated resistance to chemotherapy(Cervera and Flejou, 2011) and few available targeted options(Shah, 2015). Docetaxel is FDA approved as a first line therapy for GC when combined with cisplatin and 5–fluorouracil(Van Cutsem et al., 2006), and paclitaxel is a standard second line treatment option(Shah, 2015). Notably, most patients demonstrate resistance to taxane treatment, with response rates to single agent therapy around 14–25%, and duration of response measured in months(Bang et al., 2010; Power et al., 2010). This is reaffirmed in our post-hoc analysis of the TAX-325 trial(Galletti et al., 2020), in which we identified a significant subset of GC patients who did not derive any clinical benefit from the addition of docetaxel. Thus, there is a dire need to decipher clinically actionable mechanisms of taxane resistance.
Despite the multitude of taxane resistance mechanisms described in preclinical models by us and others, including tubulin mutations, altered MT dynamics or the presence of drug efflux mechanisms, none has impacted clinical care(Dumontet et al., 1996; Giannakakou et al., 2000a; Giannakakou et al., 1997; Giannakakou et al., 2000b; Goncalves et al., 2001; Gonzalez-Garay et al., 1999; Horwitz et al., 1993; Kavallaris et al., 1997; Luduena, 1998; Wang et al., 2005; Wiesen et al., 2007).
Here we describe a truncated variant of the MT plus-end binding protein CLIP-170, hereafter CLIP-170S, which we found to be significantly enriched in taxane-resistant GC cell lines and patient samples. CLIP-170 is the first +TIP protein identified to bind to the plus-end of growing MTs and is involved in the regulation of MT dynamics(Bieling et al., 2008; Bieling et al., 2007; Perez et al., 1999; Pierre et al., 1992; Watson and Stephens, 2006) positioning of MT arrays and signaling (reviewed in(Akhmanova and Steinmetz, 2008; 2015)). We demonstrate a direct role for CLIP-170S in conferring taxane resistance in vitro and in vivo by impairing taxane access into the MT lumen, thus, preventing drug-target engagement. Using a computational approach in conjunction with the connectivity map(Subramanian et al., 2017), we discovered the tyrosine kinase inhibitor, imatinib, as a compound predicted to reverse taxane resistance. Mechanistically we show that imatinib sensitizes resistant GC cell lines to taxane therapy by selective depletion of CLIP-170S. Taken together, this study describes a +TIP protein variant with distinct biological properties, a previously unrecognized mechanism of taxane resistance, and identifies an actionable therapeutic strategy to treat patients with taxane refractory GC.
Results
Taxane-resistant GC cells exhibit reduced taxane residence time on microtubules
We have previously reported that taxane resistance in gastric cancer (GC) is associated with reduced drug target engagement in both cell lines and patient biopsies(Galletti et al., 2020). Using a panel of 12 GC cell lines, we identified a subset (6/12) with intrinsic resistance to DTX due to lack of drug-target engagement, despite unimpaired intracellular drug accumulation, absence of tubulin mutations or other target alterations. To understand the molecular basis of the impaired interaction between taxanes and MTs we incubated native cytoskeletons from sensitive and resistant GC cells with FITC-conjugated paclitaxel (Flutax-2) and determined its binding equilibrium with MTs by live cell imaging, as previously described(Diaz et al., 2003) (Figure 1A). As can be seen, Flutax-2 at saturating concentrations (200 nM) was bound to both sensitive and resistant cell cytoskeletons (Figure 1B, Supplementary Figure 1A, time 0) and was allowed to equilibrate over time. Flutax-2 remained bound to native cytoskeletons from the TMK1 DTX-sensitive cell line for the duration of the experiment (180 min) (Figure 1B). In contrast, in the DTX-resistant Hs746T and SCH cells, Flutax-2 dissociated significantly faster, suggesting transient drug-target interactions (Figure 1B and Supplementary Figure 1A). Next, we determined drug-residence time on MTs by quantifying Flutax-2 fluorescence intensities over time in sensitive and resistant cells (Figure 1C and Supplementary Figure 1B). We identified that Flutax-2 intensities in resistant cell lines decreased at a significantly faster rate (p < 0.0001 for both) as compared to the sensitive cell lines (Figure 1C and Supplementary Figure 1B). Immunofluorescence staining of MTs, under the same experimental conditions, revealed no change in the integrity of the MT network over time and across cell lines (Supplementary Figure 1C, D), suggesting that the Flutax-2 decay in the resistant cells is likely due to lower MT binding affinity.
Figure 1: CLIP-170S, a truncated CLIP-170 variant confers taxane resistance in vitro and in vivo.
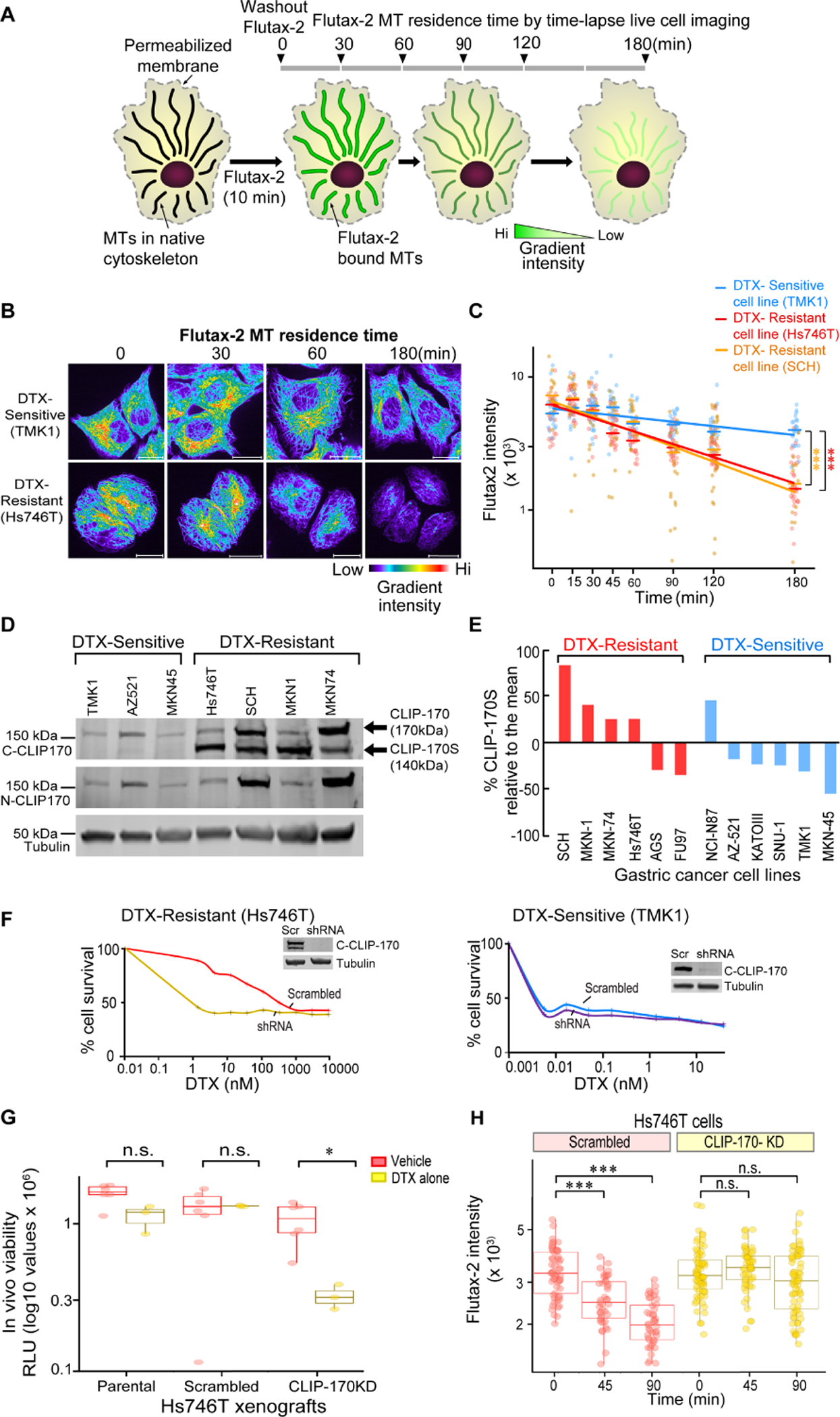
A) Experimental design for quantitation of taxane residence time on MTs. Native cytoskeletons were prepared from docetaxel (DTX)-sensitive and resistant cell lines, treated with FITC-conjugated paclitaxel (Flutax-2) for 10 min, and Flutax-2 residence time on MTs was quantified, following wash-out, by time-lapse live cell imaging for a total of 180 min. Black arrowheads point to 30 min intervals B) Representative images of Flutax-2-labeled MTs in native cytoskeletons from DTX-sensitive (TMK1) and DTX-resistant (Hs746T) cell lines bar = 20 μm. All images illustrate pseudocolor representations of Flutax-2 intensity levels on cellular MTs. Red to blue color gradient represents high to low intensities, respectively C) Linear regression analysis of Flutax-2 fluorescence intensities over time in DTX-sensitive (TMK1, blue) and DTX-resistant (Hs746T, red; SCH, orange) cell lines (n=20–35 individual cells/time point/cell line); mixed effect model was used to calculate the rate of Flutax-2 dissociation following drug washout, *** p<0.001. D) Identification of a CLIP-170S isoform in taxane resistant GC cell lines. CLIP-170 expression was assessed by immunoblot in a panel of GC cell lines grouped according to their intrinsic sensitivity/resistance to DTX. Immunoblot was performed using antibodies against the C-terminus or N-terminus of CLIP-170 or tubulin, as indicated. Immunoblot with anti C-CLIP-170 antibody identified a faster migrating band present only in DTX-resistant cell lines. E) Waterfall plot displaying % CLIP-170S protein expression in an expanded panel of 12 GC cell lines, 6 DTX-resistant (red bars) and 6 DTX-sensitive (blue bars). Percent CLIP-170S was calculated using the densitometry values, CLIP-170S/(CLIP-170 + CLIP-170S)*100, and presented relative to the mean. Negative values indicate absence of CLIP-170S expression. Data are representative of three individual biological repeats; one-tailed Student’s t-test, * p<0.05. F) CLIP-170S knockdown abrogates DTX-resistance in vitro. CLIP-170 and CLIP-170S were stably knocked-down by a pool of shRNAs targeting the common portion of the transcript, in Hs746T cells expressing both isoforms and TMK1 cells expressing only the canonical CLIP-170. Cells transfected with either scrambled (Scr) or CLIP-170 hairpin (shRNA), were plated for a 72 hr cytotoxicity experiment with DTX. Knockdown of CLIP-170 and CLIP-170S completely abrogated the 300-fold DTX resistance in Hs746T cells (top; IC50: Scr = 328 nM, red; shRNA = 1.3 nM, yellow), while knockdown of CLIP-170 alone had no effect on DTX-sensitivity in TMK1 cells (bottom; IC50: Scr = 0.004 nM, blue; shRNA = 0.004 nM, purple). Representative data from one of several independent biological repeats is shown. Inset shows confirmation of knockdown by immunoblot. G) CLIP-170S knockdown reverses taxane resistance in vivo. Hollow fibers containing Hs746T series (parental, scrambled, CLIP-170-KD) were implanted into mice i.p. Mice were treated with vehicle (n=6/cell line) or DTX 10 mg/kg (n=3/cell line) for 14 days and in vivo cell viability was measured by luminescence; red: vehicle, yellow: DTX; Hs746T parental (adjusted p=0.058), Hs746T Scrambled (adjusted p=0.73), Hs746T CLIP-170KD (adjusted p=0.026); Multiple t-test with Holm-Sidak correction). H) Flutax-2 residence time on MTs is restored upon depletion of CLIP-170/CLIP-170S in Hs746T-CLIP170-KD cells. Box-plot representation of Flutax-2 fluorescence intensity in Hs746T-scrambled (red) and Hs746T-CLIP-170-KD cells (yellow) (n=43–71 individual cells/time point/cell line). 5–95% confidence intervals graphs are shown; Mann-Whitney test; n.s.; not significant, *** p<0.001.
The binding affinity of Flutax-2 is a function of its association rate constant (kon), a bimolecular reaction that depends on the drug’s diffusion constant, and dissociation rate constant (koff), a monomolecular reaction. An altered kon in the absence of an altered koff would imply altered diffusion of the drug to the luminal site of MTs, while an altered koff alone would imply that the binding site itself is structurally altered. Quantitation of Flutax- 2 association and dissociation rate constants revealed a 3-fold lower kon value in resistant Hs746T (2 ± 1 × 104 M−1 s−1) versus sensitive TMK1 (6 ± 2 × 104 M−1 s−1) cells (Supplementary Figure 1E). In contrast, no difference was observed in their respective koff values (Hs746T: 4.3 ± 0.5×10−2 s−1 vs TMK1: 3.5 ± 0.4 × 10−2 s−1) (Supplementary Figure 1F). Thus, the lower binding affinity of taxanes for resistant cell cytoskeletons is the result of the slower kon which indicates impaired diffusion of the drug to the luminal binding site in MTs(Nogales et al., 1998).
Discover of CLIP-170S, an N-terminal truncated variant of the +TIP protein CLIP-170
In our initial characterization of DTX sensitive and resistant GC cell lines, we observed that resistant cells, when compared to the sensitive, displayed significantly lower MT dynamics measured by rates of MT growth speed and rescue/repolymerization(Galletti et al., 2020). These results suggested that MT dynamics and the proteins regulating them may underlie taxane resistance in these cells. We focused particularly on one such MT +TIP protein, the CAP-Gly domain containing linker protein, CLIP-170, expression of which shown to inversely correlate with taxane resistance in breast cancer cell lines(Sun et al., 2012). Interrogation of DTX sensitive and resistant cell lines for CLIP-170 protein expression revealed the presence of a faster migrating variant of CLIP-170, hereafter CLIP-170S (for short), in addition to the canonical full-length protein (Figure 1D). This result was specific to the anti-C-terminal CLIP-170 antibody, while anti-N-terminal antibody detected only the canonical protein, suggesting an N-terminal truncation in CLIP-170S. Interestingly, CLIP-170S was preferentially expressed in DTX-resistant cell lines (Figure 1D). Expanding this analysis to the entire panel of 12 GC cell lines identified that CLIP-170S was significantly enriched in DTX-resistant cell lines with 4/6 resistant versus only 1/6 sensitive cell lines expressing CLIP-170S (p< 0.05) (Figure 1E).
CLIP-170S depletion reverses taxane resistance
To determine causation, we stably knocked-down CLIP-170 and CLIP-170S from sensitive and resistant GC cell lines and assessed their DTX sensitivity. Stable knock-down (KD) of both CLIP-170 and CLIP-170S completely reversed DTX resistance in Hs746T cells (~300-fold) (Figure 1F) whereas knockdown of canonical CLIP-170 only in sensitive TMK1 cells had no effect on DTX sensitivity (Figure 1F). We found no difference in the growth rates of cells expressing scramble or the CLIP-specific shRNA (data not shown), ruling out faster growth rates as a contributing factor to enhanced taxane sensitivity. Furthermore, CLIP-170/CLIP-170S KD in Hs746T xenografts also restored taxane sensitivity in vivo (Figure 1G).
To determine the effect of CLIP-170S depletion on MT residence time of Flutax-2, we treated native cytoskeletons from Hs746T cells stably expressing scrambled or CLIP-170 shRNA with Flutax-2 and quantified drug intensities over time (Figure 1H). As previously observed, Flutax-2 intensities reduced significantly in Hs746T cells expressing scrambled control shRNA (p<0.001 for both 0 vs 45 min and 0 vs 90 min). In contrast, in Hs746T-CLIP-170-KD cells, Flutax-2 binding equilibrium remained unchanged for the duration of the experiment suggesting that CLIP-170S depletion restored Flutax-2 MT residence time (Figure 1H).
Taken together, these results demonstrate that depletion of CLIP-170S, restores taxane sensitivity as well as MT taxane residence time.
CLIP-170S lacks the N-terminal 155 amino acids, preferentially associates with MT polymers and confers DTX resistance
To characterize the protein structure of CLIP-170S we performed immunoprecipitation using the anti C-CLIP-170 antibody, followed by LC-MS/MS of both the canonical CLIP-170 and CLIP-170S. Mirror plot representation of normalized peptide signals indicated that CLIP-170S (red) lacks the first N-terminal ~155 amino acids, as compared with the canonical CLIP-170 (blue) (Figure 2A), confirming the size and identity of the faster migrating protein, CLIP-170S. Consistent with these results, 5’-rapid amplification of cDNA ends (5’-RACE) identified two CLIP-170 transcripts in the Hs746T and SCH resistant cells expressing both protein variants. The first transcript corresponded to the canonical CLIP-170 while the second transcript had a start site in exon 3, in the resistant but not in the sensitive GC lines (Supplementary Figure 2). The transcripts that start in exon 3 would utilize the putative start codon at position 620 (amino acid position 156) and therefore, generate a protein of about 140kDa in agreement with the results in Figure 1D. The putative translation start site at methionine 156 for CLIP-170S was further supported by the mass spectrometry data showing that the semitryptic peptide starting at amino acid 156, was more abundant in CLIP-170S. (Supplementary Figure 3).
Figure 2: CLIP-170S lacks the first 155 amino acids, preferentially associates with MT polymers and confers DTX resistance.

A) Mass spectrometry using LC-MS/MS analysis, following immunoprecipitation with the anti C-CLIP-170, antibody was performed for both the canonical CLIP-170 and CLIP-170S isoforms. Mirror plot representation of peptide signals normalized to the main region of CLIP-170 (500–900) indicated that CLIP-170S (red) lacks the first N-terminal ~155 amino acids, as compared with the canonical CLIP-170 (blue). Peptide alignment with the known CLIP-170 structural domains indicates that CLIP-170S is missing the first CAP-Gly motif that mediates proper MT plus-end localization B) The cellular distribution of endogenous CLIP-170 and CLIP-170S was quantitated by confocal microscopy following immunofluorescence staining with the anti C-CLIP-170 (red) and anti-tubulin (green) antibodies. MT plus-end CLIP-170 comet localization (arrows) was observed for the canonical CLIP-170 in TMK1 cells; while CLIP-170 lattice localization (dashed arrows) was observed in the Hs746T cells expressing both canonical and CLIP-170S. The distinct CLIP-170 localization is schematically illustrated next to the corresponding images. C) Measurement of individual comet length using the Imaris software, (TMK1, n=2064 comets; Hs746T, n=3186 comets; Mann-Whitney, ***p=0.0009 D) Ectopic expression of GFP-tagged CLIP-170 and CLIP-170S in TMK1 cells. Single frames were extracted following live cell imaging from cells expressing similarly low levels of each protein. Notice the comet-like pattern of canonical CLIP-170 in contrast to the MT lattice distribution pattern exhibited by CLIP-170S. E) Microtubule co-sedimentation assay of TMK1 cells stably expressing doxycycline (DOX) inducible GFP-CLIP170S or canonical. One mg of precleared cell lysate from each condition was supplemented with exogenous purified tubulin and subjected to a cycle of polymerization in presence of GTP and docetaxel at 37 °C for 30 min or kept at 4°C. The samples were centrifuged at 100,000 × g to separate the microtubule polymers in the pellet (WP or CP) from the soluble tubulin dimers in the supernatant (WS or CS). Equal volumes from each fraction were loaded on adjacent wells, resolved by SDS–PAGE and immunoblotted for GFP and tubulin. Tubulin: 100 ng of purified tubulin was loaded as a control. F) Ectopic expression of GFP-CLIP-170S in TMK1 cells confers DTX resistance. Inset shows immunoblot using the GFP antibody to indicate exogenous expression of each tagged CLIP-170 protein.
The N-terminus of CLIP-170 contains two cytoskeleton-associated protein glycine-rich (CAP-Gly) domains that mediate MT binding and plus-end tracking via binding to End-binding protein-1 (EB1)(Perez et al., 1999; Pierre et al., 1992). Of the two domains the first Cap-Gly binds stronger to EB1, an interaction that is required for plus-end tracking, while the second Cap-Gly motif demonstrates higher affinity for MTs(Gupta et al., 2010; Gupta et al., 2009; Wang et al., 2014; Weisbrich et al., 2007). Peptide alignment with the known CLIP-170 structural domains indicated that CLIP-170S is missing the first CAP-Gly motif (Figure 2A), suggesting potential defects with plus-end tracking. Immunofluorescence staining of endogenous CLIP-170 in DTX sensitive and resistant cell lines identified extended CLIP-170 localization to the MT lattice in cells expressing CLIP-170S, in contrast to the discrete comet like distribution of canonical CLIP-170(Perez et al., 1999) (Figure 2B, bottom vs top panels). Furthermore, quantitation of CLIP-170 comet length identified significantly longer comets in cells expressing CLIP-170S (Figure 2C), which cannot be explained by faster MT growth rates in these cells as we have recently published(Galletti et al., 2020). This phenotype was highly specific to CLIP-170, since immunostaining of EB1, another MT plus-end binding protein, showed no difference in comet length between sensitive and resistant cell lines (Supplementary Figure 4A, B).
To better characterize this CLIP-170 variant, we stably expressed GFP-CLIP-170S or GFP-CLIP-170 in the DTX-sensitive TMK1 cell line, using an inducible promoter. Live cell imaging of GFP-CLIP-170S showed clear MT lattice association in contrast to the discrete MT plus-end comet distribution observed with canonical GFP-CLIP-170 (Figure 2D, Supplementary movie 1). Interestingly, co-expression of GFP-CLIP-170 along with mCherry-CLIP-170S, resulted in lattice localization for the canonical CLIP-170, suggesting a dominant-negative effect of CLIP-170S likely due to heterodimerization (Supplementary Figure 4 C,D).
The extensive MT lattice localization of CLIP-170S suggested higher affinity for MT polymers as compared to canonical CLIP-170. To test this hypothesis, we performed a MT co-sedimentation experiment where we added exogenous purified tubulin to cell lysates and subjected to a polymerization cycle at 37°C, followed by ultracentrifugation to separate MT polymers and MT-binding proteins in the pellet (WP) while the soluble pool of tubulin dimers remains in the supernatant (WS). Immunoblot analysis using the GFP antibody revealed that 49% of CLIP-170S co-sedimented with MT polymers in the WP, in contrast to 24% of canonical CLIP-170. A similar experiment performed under conditions where most of the tubulin remains soluble (4°C incubation) showed that the majority of CLIP-170S remained in the supernatant fraction (CP; 15% vs WP: 49%) (Figure 2E). Together, the results obtained from ectopic expression of CLIP-170 proteins demonstrate preferential association of CLIP-170S with MT polymers in vitro and in vivo. To directly test the hypothesis that CLIP-170S impairs taxane binding, we incubated TMK1 cells expressing GFP-tagged CLIP-170 proteins with a cell permeable docetaxel conjugated with a deep-red fluorophore (tubulin tracker) and quantified its signal intensity. The tubulin tracker fluorescence intensity was significantly lower in cells expressing GFP-CLIP-170S as compared to canonical CLIP-170, indicating that CLIP-170S impairs taxane binding to MTs (Supplementary Figure 5 A,B).
These results together with the DTX sensitization upon CLIP-170 KD, led us examine the impact of CLIP-170S expression on de novo taxane resistance. Ectopic CLIP-170S expression rendered TMK1 cells resistant to DTX, by 22-fold (DTX IC50 values, TMK1, 1 nM; vs TMK1-GFP-CLIP-170S, 24 nM). In contrast, ectopic expression of canonical CLIP-170 had no effect on taxane sensitivity (Figure 2F and Supplementary Figure 5 C–E). To further demonstrate causation, we took advantage of the high homology between human and rat CLIP-170 together with the fact that the rat sequence is insensitive to the human CLIP-170 targeted shRNAs that we used to knockdown CLIP170 which reversed taxane resistance in Hs746T cells (Figure 1F, left panel). First, we confirmed that the rat CLIP-170S decorated the MT lattice phenocopying the behavior of the human protein (Supplementary Figure 6A, B and Supplementary movie 2), suggesting that CLIP-170S preferential binding to MT polymers is not an exclusive property of the human protein. Importantly, ectopic expression of the rat CLIP-170S restored the drug-resistance phenotype in the human Hs746T-CLIP-170KD cells, across several DTX concentrations (DTX IC50 values: 0.08 nM for CLIP-170KD vs 24 nM for CLIP-170KD+Rat CLIP-170S) (Supplementary Figure 6C, D).
Taken together, these data identify CLIP-170S as a previously unrecognized short isoform of CLIP-170 with high affinity for MT polymers, which in turn results in taxane resistance by impairing effective drug-target engagement.
CLIP-170S impairs taxane access to its MT luminal binding site
The extensive lattice localization, longer comet length and transient taxane MT residence time in cells expressing CLIP-170S, led us to hypothesize that the altered CLIP-170S localization might reduce taxane MT binding thereby impairing effective target engagement. Taxane binding is a two-step process(Buey et al., 2007). The first step involves initial low affinity binding to the outer wall of MTs, which then allows drug diffusion to its kinetically unfavorable, high affinity luminal binding site.
To investigate this hypothesis, we used different chemical probes that interact with the outer versus inner MT surface, and treated native cytoskeletons from sensitive and resistant cells. We first used Hexaflutax, a fluorescently labelled taxoid known to bind to the taxane binding site on the MT surface(Diaz et al., 2005) (Figure 3A). We observed significantly reduced Hexaflutax binding in resistant CLIP-170S expressing cells vs sensitive cell lines expressing only canonical CLIP-170 (p < 0.0001) (Figure 3A). Cyclostreptin is known to covalently bind MTs both at the outer (surface) and inner (luminal) sites, competing with taxane binding(Balaguer et al., 2019; Buey et al., 2007). Pretreatment with cyclostreptin is expected to inhibit Flutax-2 binding (Figure 3B). Indeed, Flutax-2 binding was significantly reduced in TMK1 cells, following cyclostreptin pretreatment (Figure 3C, TMK1 0 vs 1 and 0 vs 2 min; p < 0.0001). CLIP-170 KD in TMK1 cells had no effect on Flutax-2 binding (Figure 3C, TMK-1 vs TMK-1-CLIP-170-KD, n.s across all time points). In contrast, in Hs746T cells, expressing endogenous CLIP-170S, no decrease in Flutax-2 binding was observed following cyclostreptin pre-treatment (Figure 3C, Hs746T, n.s 0 vs 2 min), while CLIP-170 KD significantly reduced Flutax-2 binding (Figure 3C, Hs746T vs Hs746T-CLIP-170-KD, p<0.001 at both 1 and 2 min). Taken together these results, further support a role for CLIP-170S impairing taxane access to its binding site. To investigate the specificity of CLIP-170S effect on taxane-binding, we used peloruside, a MT stabilizing agent that binds to a distinct non-taxoid pocket on the MT surface(Field et al., 2013; Kellogg et al., 2017),(Gaitanos et al., 2004). We found that peloruside was equally effective in both taxane sensitive and resistant GC cells, including the isogenic Hs746T cells stably expressing scrambled or CLIP-170 shRNA (Supplementary Figure 6 E–G).
Figure 3: CLIP-170S impairs taxane binding to MTs in resistant GC cells.
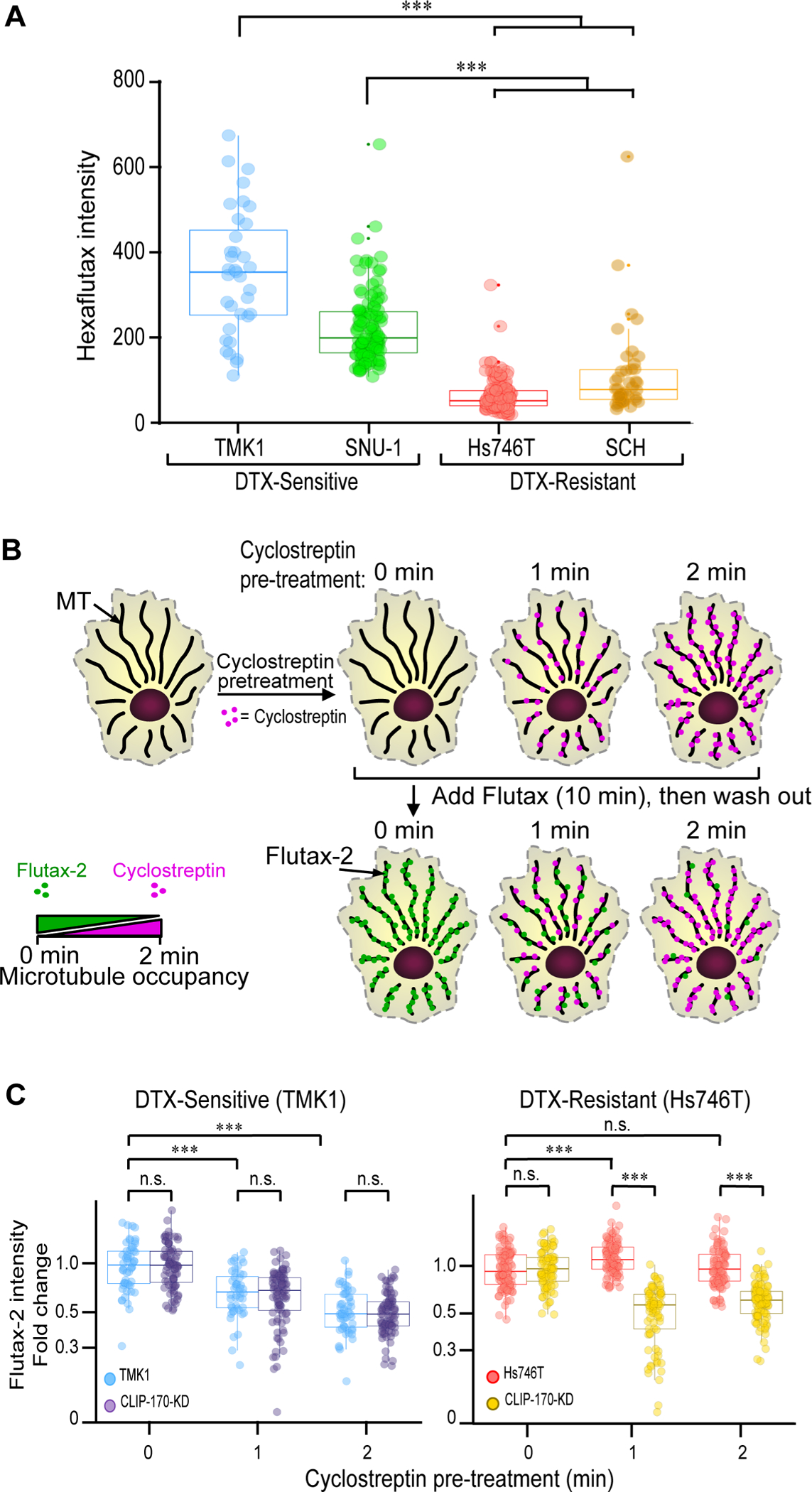
A) DTX-sensitive and DTX-resistant cell lines, were treated with the fluorescently conjugated taxoid, Hexaflutax, and the fluorescence intensity of Hexaflutax binding to MTs was quantified. Sensitive cell lines TMK1 and SNU-1 display higher hexaflutax binding as compared to the resistant GC cells. Results are representative of four individual biological repeats (n=35–145 cells/cell line). Mann-Whitney test, *** P<0.001. B) Schematic illustration of cyclostreptin pre-treatment for 0, 1, or 2 min followed by a 10 min Flutax-2 treatment. Native cytoskeletons were then imaged using a spinning disk confocal microscope. C) Box-plot representation of Flutax-2 fluorescence intensity in cells treated with scrambled or CLIP-170 shRNA (CLIP-170-KD). Significant loss in Flutax-2 MT binding was observed upon CLIP-170 KD in Hs746T (right), but not in TMK1 (left) cells. Mann-Whitney test, *** p<0.001, (n=50–100 cells/time point/cell line).
CLIP-170S expression in GC tumor biopsies correlates with clinical taxane resistance
To determine whether CLIP-170S is expressed in patient tumors, we assessed CLIP-170S expression by immunoblot in tumor biopsies from a total of 37 patients with GC. We identified CLIP-170S expression in 25/37 tumor samples, giving a prevalence estimate of 67.5% (95% CI 52.2 – 82.8 %) (Supplementary Figure 7A,B). Next, we examined CLIP-170S expression in a cohort of 24 patients that received cabazitaxel treatment on a phase II clinical trial of single agent cabazitaxel in the second line therapy for GC (NCT01757171 on www.clinicaltrials.gov). In the 18 patients with disease progression, CLIP-170S was expressed in 13/18 (72%) tumor biopsies (Figure 4a, Supplementary Figure 7B). In contrast, of the 6 patients with stable or responsive disease, only 1/6 (17%) expressed CLIP-170S (p = 0.007) (Figure 4A, Supplementary Figure 7B).
Figure 4: CLIP-170S expression in tumor biopsies is enriched in patients resistant to cabazitaxel therapy and associated with lower progression-free survival; Computational prediction of Imatinib, as a drug that reverses taxane resistance in GC.

A) Swimmers plot shows the duration of cabazitaxel treatment (months) for each patient. Best response status is annotated based on RECIST 1.1 criteria (stable disease, blue circles; progressive disease, red triangles) with the 3-month cutoff marked with the dotted red line. Bars are color coded based on CLIP-170 isoform expression (blue bars, canonical CLIP-170; salmon bars, CLIP-170S isoform). B) Kaplan-Meier curve of PFS for canonical CLIP-170 (blue) versus CLIP-170S (red) with median PFS: 3.01 months [95% CI:1.15–5.72] and PFS: 1.17 months [95% CI:0.95–1.61], respectively; HR=4.35 (95% CI:1.59–11.91), log-rank p=0.002. Number at risk indicates the number of patients without progression at each time point, according to CLIP-170S status (CLIP-170, blue; CLIP-170S, red). C) Schematic representation of computational approach used to identify drugs predicted to reverse taxane resistance. A DTX-resistance gene signature was identified using RNA-Seq data from untreated and docetaxel-treated sensitive and resistant GC cell lines (Accession number: PRJNA594148). This gene signature was used to query the Connectivity Map (CMAP) database, to identify compounds whose transcriptional output was negatively correlated to the DTX-resistance gene signature. D) Scatter density dot plot of 1309 screened small molecules from the CMAP database whose transcriptional outputs were quantified based on their reversal relationship with the DTX-resistance signature. A lower negative Effect Score indicates higher likelihood to reverse the DTX-resistance. E) Imatinib sensitizes HS746T cells to docetaxel. Cytotoxicity assay using DTX-resistant Hs746T cells treated with docetaxel (DTX) or imatinib (IMA) either alone or in combination (DTX + IMA). F) Imatinib selectively inhibits CLIP-170S expression without affecting CLIP-170. Hs746T and TMK1 cells were treated with 1, 25, 50 and 100 μM Imatinib for 3 hr and processed for immunoblotting using antibodies against the C-terminus of CLIP-170 or tubulin. G) Imatinib has no effect on Hs746T-CLIP-170-KD sensitivity to docetaxel. Cytotoxicity assay as in B.
In addition, progression-free survival (PFS) was significantly lower in patients with tumors expressing CLIP-170S versus no CLIP-170S expression (1.17 mo vs 3.01 mo, p=0.002) (Figure 4B). Taken together these results demonstrate that CLIP-170S was enriched in patients resistant to taxane therapy and was significantly associated with shorter PFS, suggesting that CLIP-170S is a target to reverse clinical taxane resistance.
Predictive computational algorithm identifies Imatinib as a candidate compound to reverse taxane resistance.
We have previously described how different data types can be combined in a single computational framework to identify protein targets for small molecules in development and to predict new molecules to overcome drug resistance(Madhukar et al., 2019). Here we used RNAseq data from untreated vs DTX-treated sensitive and resistant cell lines in our panel to generate a DTX-resistance gene signature. Using this gene signature as input we queried the Connectivity Map (CMAP) for compounds with transcriptional outputs negatively correlated with DTX-resistance (Figure 4C). We evaluated over 1309 FDA-approved drugs and active small molecules and ranked them based on their potential to reverse the DTX-resistance signature. Unpredictably, this analysis identified imatinib as the top hit predicted to reverse taxane resistance in GC cells (Figure 4D). Imatinib is a BCR-Abl inhibitor FDA approved for the treatment of chronic myelogenous leukemia(Druker et al., 2001; Druker et al., 1996); however, imatinib’s mechanism of action as a receptor-tyrosine kinase inhibitor bears no relevance to taxane activity on the MT cytoskeleton and has never been implicated in taxane resistance. To experimentally validate this prediction, we treated sensitive and resistant cell lines with imatinib alone and in combination with DTX. While treatment with imatinib alone did not show differential activity in sensitive versus resistant cells, when combined with DTX, imatinib completely reversed taxane resistance in Hs746T cells and other CLIP-170S expressing GC cell lines (Figure 4E), indicating a mechanistic interaction between the two drugs. Further, a mere 3 hr treatment with clinically relevant concentrations of imatinib(De Francia et al., 2014), led to selective depletion of CLIP-170S protein without affecting canonical CLIP-170 (Figure 4F). Importantly, we observed that additional RTK inhibitors (RTKi), such as sunitinib, ponatinib, axitinib and cediranib, also selectively deplete CLIP-170S (Supplementary Figure 7C), strongly suggesting a class effect. In the absence of CLIP-170S, imatinib lost its synergistic interaction with DTX (Figure 4G and Supplementary Figure 7D, E), confirming the central role of CLIP-170S in this drug combination and suggesting that CLIP-170S is a previously unrecognized target for imatinib.
Discussion
The microtubule cytoskeleton is an important, validated therapeutic target in oncology, as evidenced by the broad use of taxanes across multiple cancers. Taxanes induce cell death by binding to and stabilizing MTs, thus, blocking essential MT-dependent processes including signaling, trafficking and mitosis(Schiff et al., 1979) and reviewed in(Dumontet and Jordan, 2010; Komlodi-Pasztor et al., 2011). Although widely used, the clinical success of taxane therapy has been marred by intrinsic and acquired resistance, the molecular underpinnings of which have remained elusive.
Here we identify a previously unrecognized variant of the +TIP binding protein CLIP-170, which we named CLIP-170S, as it lacks the first 155 amino acids and results in a shorter (140 kD) protein. The missing N-terminal serine-rich domain and the first Cap-Gly domain are required for effective MT +TIP tracking(Gupta et al., 2009; Weisbrich et al., 2007), thus, CLIP-170S decorates the MT lattice extending beyond the plus-ends and associates preferentially with MT polymers. Interestingly, canonical CLIP-170 was displaced from the MT plus-ends to the lattice in the presence of CLIP-170S, suggesting that CLIP-170S has dominant-negative effect likely due to heterodimerization. The aberrant CLIP-170S cellular distribution is reminiscent of CLIP-170 phosphodeficient mutants that favor its open MT-bound conformation(Lee et al., 2010; Nirschl et al., 2016). Based on these data, we propose a model whereby the N-terminus truncated CLIP-170S adopts an open MT-bound conformation, leading to extensive lattice localization which in turn impairs taxane access to its luminal binding site (Figure 5). In vitro studies have identified that taxanes stabilize MTs at sub-stoichiometric concentrations (Derry et al., 1995; Diaz and Andreu, 1993; Jordan et al., 1993) where taxanes bind β-tubulin along the MT length(Kellogg et al., 2017). Herein, we show that endogenous CLIP-170S is concentrated near the MT plus-end region rather than localized along the entire MT length. Yet, genetic or pharmacologic depletion of CLIP-170S completely reverses taxane resistance suggesting that in vivo taxane anti-tumor activity is mediated by drug binding near the MT plus ends. In support of our data, a recent report identified the presence of taxane-accumulation zones near the MT plus-end which induces cooperative drug binding, leading to downstream stabilization of the entire length of the microtubule lattice(Rai et al., 2020). This finding suggests that taxane-induced MT stabilization may not require contiguous drug binding along the entire MT length. This newly emerged model is consistent with our data where, CLIP-170S localization in a small stretch of the lattice beneath the plus-end is enough to impair drug-target engagement and cause taxane resistance, a paradigm shift in our understanding of taxane mechanism of action in vivo.
Figure 5: Proposed model of the mechanism by which CLIP-170S confers taxane resistance in GC cell lines.

Canonical CLIP-170 (yellow), which contains two Cap-Gly motifs, binds to EB-1 (blue) and is localized to the plus-ends of MTs (Top panel). This expected localization pattern of CLIP-170 does not interfere with the luminal binding of taxanes (pink dots). Thus, cells expressing only the canonical CLIP-170 remain sensitive to taxane treatment. In contrast, CLIP-170S (green), that is missing the first Cap-Gly motif shows extensive MT lattice localization and impairs taxane binding at its luminal site (pink dots) (Bottom panel), which in turn renders cells resistant to taxanes. Inset shows chemical structure of paclitaxel.
We further discovered that Imatinib reverses taxane resistance by selective depletion of CLIP-170S over the canonical CLIP-170. This was an unexpected finding as Imatinib primarily targets BCR-ABL fusion protein in chronic mylogenous leukemia (CML)(Druker et al., 2006; Druker and Lydon, 2000; Druker et al., 2001; Druker et al., 1996) as well as other receptor tyrosine kinases (RTKs) including PDGFR, VEGFR, c-Abl and c-kit(Joensuu et al., 2001; Sadovnik et al., 2014), none of which was expressed in our GC cell line panel (data not shown). Additionally, drugs from the same family of RTK inhibitors (RTKi) also depleted CLIP-170S, not only indicating a class effect but expanding the therapeutic opportunities of taxane combinations with RTKi. We are currently investigating the mechanism of RTKi-induced CLIP-170S downregulation. We envision that this work will pave the way for unprecedented clinical benefits for taxane-refractory patients expressing CLIP-170S in GC and beyond.
Limitations of the study:
Our data suggests that CLIP-170S confers taxane resistance by impairing taxane access to the MT luminal binding site due to its extensive localization to the MT lattice. In addition to our proposed model, it is also possible that taxane resistance in these cells may be the result of an indirect effect of CLIP-170S on taxane binding, where in CLIP-170S binding to MT polymers may cause a conformational change in MT polymers which in turn may effect taxane binding. The exact mechanism by which taxane binding is impaired in cells expressing CLIP-170S can only be determined using techniques such as crystallography and electron microscopy that provide a high structural resolution of the interaction between MTs, CLIP-170S and the taxane molecule. Furthermore, based on 5’RACE data, there is indication that CLIP-170S may be generated using the alternative transcript of CLIP-170. The exact mechanism by which CLIP-170S is expressed endogenously needs to be further corroborated using additional molecular techniques such as H3K4me3 CHIP as well as reporter assays that can help determine the presence of an alternative promoter in CLIP-170 gene.
Star Methods:
RESOURCE AVAILABILITY:
Lead Contact:
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact: Paraskevi Giannakakou: pag2015@med.cornell.edu.
Materials availability:
Any materials generated in the study will be made available by the Lead Contact: Paraskevi Giannakakou: pag2015@med.cornell.edu.
Data and Code availability:
All Data reported in this manuscript will be shared by the Lead Contact upon request. If any further information is required regarding the RNA-seq analysis in conjunction with Connectivity Map, it will be provided by the lead contact upon request. This manuscript does not report original code.
EXPERIMENTAL MODEL AND SUBJECT DETAILS:
Cell Culture:
Gastric cancer cell lines AZ-521, FU97, Hs746T, MKN-7, SCH and TMK-1(Tan et al., 2011) were a gift of Dr. P. Tan (Duke-National University of Singapore, Singapore); KATO-III, MKN-1 and SNU-1 were a gift of Dr. Z. Wainberg (University of California, Los Angeles). AGS, NCI-N87 and MKN-74 cells were kindly provided by Dr. R. Klein (Memorial Sloan Kettering Cancer Center, New York); MKN-45 was a gift of Dr. M. Moehler (Johannes Gutenberg University of Mainz, Germany). Cells were either cultured in Dulbecco’s modified Eagle medium (DMEM) or RPMI 1640 supplemented with 10% FBS and 1% Penicillin-Streptomycin antibiotic solution.
Mouse Models:
For in vivo hollow fiber assay, Female NMRI nude mice (Crl: NMRI-Foxn1nu) were used (Charles River GmbH). Female mice were 4–5 weeks old at the time of delivery. Maximum of 4 animals were housed per individual ventilated cage and were maintained under optimum hygienic conditions, air-conditioned with 10 air changes per hour. The environment was continuously monitored with temperature target ranges of 22 ± 2 °C, relative humidity of 45–65% and 12 hours artificial fluorescent lighting/12 hours darkness. These studies were performed in collaboration with Reaction Biology Europe GmbH, Freiburg, Germany. Experiments with mice were carried out in accordance with the guidelines for animal use issued under IACUC Protocol number 2020-0019 at Weill Cornell Medicine.
Human Tumor Biopsies:
GC samples were collected from two studies. Nine samples were collected under IRB1203012274, the Weill Cornell Medical College Gastric Cancer research database, prior to initiation of therapy, from endoscopic Bard Precisor EXL coated disposable biopsy forceps (Bard International, Murray Hill, NJ). In the second study 28 samples were collected under the Phase II study of Cabazitaxel in refractory metastatic gastric or gastroesophageal adenocarcinoma (NCT01757171) (Shah et al., 2020). Of the 28 samples, four patients failed eligibility screening and did not receive cabazitaxel treatment. Thus, we had 24 evaluable samples with clinical response data from the cabazitaxel trial. In this study (NCT01757171), samples were collected prior to initiation of cabazitaxel, from either the primary tumor or a metastatic site. All collected samples were flash frozen and stored at −80 °C until examination for CLIP-170 and CLIP-170S expression. All subjects provided written informed consent according to the declaration of Helsinki prior to study enrollment. Samples were collected in accordance with the institutional ethical and clinical guidelines under Institutional Review Board approval.
METHOD DETAILS:
Reagents:
For most biochemical experiments, mouse anti-C-CLIP-170 mAb (clone: F-3, Santa Cruz Biotechnology, Dallas, TX) was used. To detect N-terminal of CLIP-170 on western blot, custom developed rabbit anti-N-CLIP-170 polyclonal antibody (Covance, Princeton, NJ) or mouse anti-CLIP-170 (clone: E-8, Santa Cruz Biotechnology, Dallas, TX) were used. For immunofluorescence staining of endogenous CLIP-170, a mixture of mouse monoclonal antibodies 4D6 and 2D6 was used reported(Scheel et al., 1999). Other antibodies used included rabbit anti-GFP polyclonal Ab (Novus Biologicals, Littleton, CO), mouse anti-Myc mAb (clone: 9E10, Santa Cruz Biotechnology), mouse anti-31α-tubulin mAb (clone: DM1α, Sigma-Aldrich, St. Louis, MO), EB-1 Ab (clone: KT51, Abcam, Cambridge, UK) and rat anti-α-tubulin mAb (clone: YL1/2, Bio-Rad, Hercules, CA).
Docetaxel, imatinib, ponatinib, axitinib, Sunitinib and cediranib were purchased from Sigma-Aldrich (St. Louis, MO). Flutax-2 and Hexaflutax were generous gifts from Prof. Wei-Shuo Fang (Institute of Materia Medica, Beijing, China), peloruside was a gift from Dr. John Miller (Victoria University of Wellington, Wellington, New Zealand) and cyclostreptin was a gift from Dr. Chris Vanderwal (University of California, Irvine, CA).
Microscopy:
Zeiss AxiObserver.Z1 coupled with Yokogawa spinning disk confocal head (CSU-X1, Yokogawa, Tokyo, Japan) was used for image acquisition. ORCA-R2 CCD camera (C10600, Hamamatsu Photonics, Hamamatsu, Japan) was used for spinning disk and Flash4.0 (Hamamatsu Photonics) was attached to the other port for epi-fluorescence imaging. 63× oil DIC (N.A.=1.4) Plan-Apochromat (420782–9900) or 40× oil DIC (N.A.=1.3) (UV) VIS-IR Plan-Apochromat (420762–4800) objective lenses were used for imaging. For immunofluorescence staining, LSM700 laser scanning confocal (Zeiss) equipped with 100× oil DIC (N.A.=1.46) Plan-Apochromat objective (420792–9800) was used.
Native cytoskeleton imaging:
Native cytoskeletons were prepared for Flutax-2 imaging as preciously described(Diaz et al., 2003). Briefly, unfixed coverslip-attached GC cells were washed eight times with PEMP buffer (PEM buffer containing 4% polyethylene glycol 8000, pH 6.8) followed by permeabilization with 0.5% Triton X-100 in PEM buffer (100 mM PIPES (pH 6.8), 1 mM EGTA, 1 mM MgCl2) for 60 s at room temperature and finally washed eight times with PEMP buffer.
For measurement of Flutax-2 residence time on microtubules, native cytoskeletons were incubated with 200 nM Flutax-2 for 10 minutes at 37°C. Following drug washout, cells were rapidly washed 12 times with PEMP buffer and imaged using a spinning disk confocal microscope. As a control for MT network integrity, tubulin was stained using mouse anti-33α-tubulin mAb (clone: DM1α, Sigma-Aldrich, St. Louis, MO) and immunofluorescence was performed using LSM700 laser scanning confocal (Zeiss).
For kon measurements, native cytoskeletons from sensitive and resistant GC cells were incubated with 1 μM Flutax-2 for 10s, 15s, 30s, 60s and 80s as indicated. After incubation with the drug, cells were rapidly washed 12 times with PEMP buffer and mounted for imaging. To measure koff, 13.3 μM of non-labeled docetaxel was added to Flutax-2-stained native cytoskeletons, and replacement of Flutax-2 with label-free DTX was recorded as the function of decreasing fluorescence intensity.
For staining with Hexaflutax, unfixed coverslip-attached GC cells were dipped into 3 μM Hexaflutax for 2 mins and then were washed eight times by dipping the coverslip in a dish containing PEMP buffer (PEM buffer containing 4% polyethylene glycol 8000, pH 6.8) and then mounted for imaging(Diaz et al., 2005).
For staining with cell permeable docetaxel conjugated with a deep-red fluorophore (Tubulin tracker™, Thermo Fisher Scientific, Waltham, MA), DTX-sensitive TMK1 cells expressing inducible GFP-CLIP-170 or GFP-CLIP-170S were induced for expression of GFP-CLIP-170 proteins by treatment with 0.1 μg/ml doxycycline overnight. Next day, after the removal of growth media, cells were washed once with PBS and treated with 200 nM tubulin tracker dye for 30 min at room temperature. Following washout of tubulin tracker, cells were imaged using a spinning disk confocal microscope. Mean fluorescence intensity signal of tubulin tracker was quantified using FIJI software(Schindelin et al., 2012).
Quantitation of Hexaflutax and Flutax-2 staining:
All acquired images were converted to 16-bit TIFF files and analyzed with Metamorph software (Molecular Devices, San Jose, CA). To quantify florescence intensity of cells, region of individual cells was drawn by outlining each cell. Each region was then overlaid to the corresponding raw images to first subtract background, and then average fluorescence intensity of each cell was measured. The results were exported as excel files for further statistical analysis. kon and koff values were calculated using Sigmaplot software (Systat software, San Jose, CA) using the equations f(x) = y0 + a(1−e−bx) and f(x) = y0 + ae−bx, respectively.
Western blotting:
To check the expression of CLIP proteins, cells were directly lysed into appropriate volumes of 1×SDS-PAGE sample buffer. Unless noted, 7.5% Tris-glycine gels were used. For Western blotting, all proteins were transferred to Immobilon® - FL (Millipore, Billerica, MA) membranes to scan and quantify with Odyssey Classic infrared imaging system (LICOR, Lincoln, NE). Non-specific binding was blocked with Tris-HCl buffer (pH 8.0) containing 3% skim milk.
For western blots on patient samples, 5–10 mg of frozen tumor tissue was suspended in 100 μl of RIPA buffer containing cOmplete™ EDTA-free protease inhibitors (Roche, Basel, Switzerland) and Halt™ phosphatase inhibitor (Thermo Fisher Scientific, Waltham, MA), homogenized using a hand-held homogenizer; 80 μg of total cell protein from each sample was resolved by SDS-PAGE and immunoblotted as indicated.
Cytotoxicity assay:
Sulforhodamine B (SRB) cytotoxicity assay was performed as previously described in(Skehan et al., 1990) by seeding the GC cell lines in a 96 well plate (4000 cells/well) and treating with increasing concentrations of the drug for 72 hr. The IC50 was defined as the dose of drug required to inhibit cell growth by 50%. After drug treatment, cells were fixed with trichloroacetic acid (TCA) and stained with Sulforhodamine B (SRB). Percentage of cell viability was calculated as the treatment divided by control for each concentration of drug.
A waterfall plot of relative CLIP-170S expression was created by performing densitometry on immunoblots from DTX-sensitive (n=6) and -resistant (n=6) cell lines. CLIP-170 and CLIP-170S expression was normalized to tubulin and CLIP-170S expression levels was expressed as a percentage of total CLIP-170 in that cell line as CLIP-170S/(CLIP-170+CLIP-170S) *100. The mean-value of %CLIP-170S expression from all the cell lines was calculated and set to 0. The mean value was then subtracted from %CLIP-170S values for each of the individual cell lines, and positive and negative values are plotted along a horizontal line that represents mean %CLIP-170S expression. Positive values project above the horizontal line and represent cell lines with %CLIP170S expression values that exceed the mean (relatively high %CLIP170S expression). Negative values project below the horizontal line and represent cell lines with %CLIP170S expression values that less than the mean (relatively high %CLIP170S expression).
In vivo Hollow Fiber Assay:
Hollow fibers were loaded with ~60–70,000 cells of either the parental Hs746T cells or Hs746T cells containing either Scrambled or CLIP-170 shRNA and incubated overnight in RPMI-1640 with 20% FCS and 1% Penicillin/Streptomycin in a cell culture dish at 37 °C. Next day, one hollow fiber for each cell line was implanted intraperitoneally per mice (n=9), i.e. 3 hollow fibers/mice, and randomized into either the vehicle only group (n=6) or DTX alone group (n=3). Mice were treated with either vehicle only (DMSO) or DTX at 10 mg/kg intravenously once a week of 15 days. At the end of 15 days, hollow fibers were harvested, smashed in RIPA buffer and cell viability was measured by Cell-titer Glo assay (Promega). Luminescence was detected using an Enspire plate reader (Perkin Elmer).
Mass spectrometric analyses:
Cell pellets were suspended in Tris-HCl (pH 7.4) buffer containing 150 mM sodium chloride and 0.5% (w/v) Triton X-100. Cell pellets were homogenized using a Dounce homogenizer for 15 strokes. After lysing cells, the suspension was spun briefly (12,000×g, 5 minutes) to remove cell debris. The supernatant was immunoprecipitated with anti-CLIP-170 mAb (Clone F-3) as described above to concentrate CLIP-170/CLIP-170S proteins. The immunoprecipitates were dissolved in a minimum volume (40 μl) of 1×SDS-PAGE sample buffer, and proteins were separated with a Bolt™ 4–12% Bis-Tris Plus mini-gel (Invitrogen, Carlsbad, CA) using MOPS buffer (200V). Separated proteins were visualized with NOVEX® colloidal blue staining kit (Invitrogen, Carlsbad, CA). Bands corresponding to the molecular weight of CLIP-170 and CLIP-170S were excised and digested using either trypsin (Promega), Endopeptidase Lys C (Wako), or modified by propionic anhydride (Garcia et al., 2007) followed by trypsination. Digests of proteins were analyzed by C18 based nano LC-MS/MS using either an OrbitrapXL mass spectrometer operated in High/Low mode or a Q-Exactive Plus (Thermo Fisher Scientific). LC-MS/MS data were queried against Uniprot’s Human database (2013) using Proteome Discoverer 1.4 (Thermo Fisher Scientific) /Mascot 2.4 (Matrix Science). Semi-tryptic constraints, peptide N-termini acetylation and oxidation of methionine’s were allowed as variable modifications. For samples treated with propionic anhydride, propionylation of N-termini and lysine’s were allowed as variable modifications. Matched peptides were filtered using <5% FDR (Percolator (Kall et al., 2007) and mass accuracy of < 5 ppm. Tandem mass spectra of peptides near residue 155 were also manually validated. Extracted peptide area obtained by the three strategies were normalized to the total peptide signal from residue 500 to 900 of CLIP-170 (P30622–2), summed and plotted as a function of the CLIP-170 (P30622–2) sequence using PepEx (Guerrero et al., 2014). Using Skyline (MacLean et al., 2010) MS1 signals for potential semi-tryptic peptides for covering residue 152 through 177 (P30622–2) were extracted for both CLIP-170 and CLIP-170S.
Immunofluorescence staining:
To stain endogenous CLIP-170 proteins and MTs, GC cells grown on coverslips were rapidly fixed using ice cold methanol (stored at −20°C) in a glass jar(Pierre et al., 1992). Cells were first fixed for 5 minutes in PHEM buffer (60mM PIPES, 25mM HEPES, 10mM EGTA, 2mM MgSO4, pH7.0) containing 0.5% glutaraldehyde/0.5% formaldehyde/0.5% Triton X-100, followed by incubation for 10 minutes in PHEM containing 1% formaldehyde(Waterman-Storer and Salmon, 1997). PHEM buffer containing boiled 5% donkey serum was used for blocking and antibody incubation steps. To stain for endogenous CLIP-170 we used a mixture of anti-CLIP-170 antibodies (clones 2D6 and 4D3) as previously described (Scheel et al., 1999). Endogenous tubulin and GFP were detected by rat-anti-tubulin Ab (Clone: YL1/2 from Abcam, Cambridge, MA), and rabbit anti-GFP polyclonal Ab (Novus Biologicals, Centennial, CO). As a control, immunofluorescence staining of endogenous EB-1 protein was performed using anti-EB1 antibody (clone: KT51, Abcam, Cambridge, UK). EB-1 comet lengths quantified by segmentation of EB-1 comets using ‘Curve trace 0.3.5: Sterger’s algorithm’ plugin in FIJI software.
Live cell imaging of EGFP-CLIP-170 and EGFP-CLIP-170-S:
To conduct live cell imaging of TMK1 cells stably expressing ectopic doxycycline inducible GFP-CLIP-170 or GFP-CLIP-170S, cells were grown on a coverslip and treated with 0.1μg/ml doxycycline for 24 hrs. Next day, coverslips were placed on a slide glass-based custom chamber and the GFP signal was recorded using the spinning disk confocal imaging system as described above. Similarly, for Hs746T CLIP-170KD cells expression Rat GFP-CLIP-170 or Rat GFP-CLIP-170S, cells were treated with 0.5 μg/ml doxycycline overnight before imaging. TMK1 cells were co-transfected with plasmids encoding GFP-CLIP-170 or mCherry-CLIP-170S Imaging was performed using spinning disk confocal microscopy and GFP and mCherry signal intensities were recorded in individual cells.
5’-RACE:
5’-RACE (Rapid Amplification of cDNA Ends) was performed using SMARTer RACE 5’/3’ kit as per manufacturer’s instructions (Cat no: 634858 Takara/Clontech, Mountain View, CA). Briefly, poly-A RNA was extracted using 100 μg total RNA from Hs746T, SCH and TMK1 cells using the Oligotex mRNA isolation kit (Qiagen, Hilden, Germany; Cat no. 70022). Integrity of the purified total RNA and mRNA was assessed by gel electrophoresis. Approximately, 400–500 ng of poly-A RNA was used for first strand cDNA synthesis primed using oligodT primers. The GC tailing mechanism by SMARTScribe RT enzyme allows addition of the Smarter Oligonucloetide II A, which then serves as an adapter. This first strand cDNA can then be directly used for RACE. 5’-RACE PCR was performed, using adapter specific UPM forward primer (5’ CTAATACGACTCACTATAGGGCAAGCAGTGGTATCAACGCAGAGT) and a CLIP1 (the gene encoding CLIP-170) specific exon 3 reverse primer (5’ GATTACGCCAAGCTTTTCTGTGATGGTTTCTGAGGGATGTTTGAAGG 3’), to generate 5’RACE fragments. To facilitate In-fusion HD cloning of RACE fragments, the sequence 5’ GATTACGCCAAGCTT 3’ sequence was added to the 5’ end of CLIP1 specific reverse primer. These RACE fragments were then purified using a PCR purification column and cloned into the linearized pRACE vector provided with the kit using the In-fusion HD cloning system. Approximately, 20 – 40 individual clones for each cell line were first screened for presence of the insert using PCR, and were then sequenced to identify transcripts corresponding to either CLIP-170 or CLIP-170S.
Microtubule co-sedimentation assay:
Microtubule co-sedimentation was performed as previously described(Galletti et al., 2014). In brief, TMK1 cells stably expressing GFP-CLIP-170 (TMK1-GFP-CLIP-170) or GFP-CLIP-170S (TMK1-GFP-CLIP-170S), under the influence of doxycycline inducible promoter, were treated with 0.1ug/ml doxycycline for 24 hrs. Cells were harvested the next day in PEM buffer (100mM PIPES (pH 6.8), 1mM EGTA, 1mM MgCl2) and 1 mg of whole cell lysate from each cell line was supplemented with exogenous purified bovine brain tubulin (Cytoskeleton) at a final concentration of 10μM, reconstituted in PEM buffer in the presence of 1 mM GTP, and 10 μM docetaxel (DTX) and subjected to a cycle of polymerization for 30 min at 37 °C. Samples were then centrifuged at 100,000 × g for 30 min at room temperature in Airfuge™ (Beckman Coulter, CA), and the warm supernatant (WS) was separated from warm pellet (WP). WP was then resuspended in an equal volume of PEM buffer. In parallel, a similar reaction was set up where the reactions were incubated on ice for 30 mins and cold pellet (CP) was separated from cold supernatant (CP) post-centrifugation. Equal volumes from each respective fraction were loaded onto an SDS-PAGE and transferred and immunoblotted with antibodies against GFP or tubulin. 1 μg of purified Tubulin was also loaded on SDS-PAGE gel alongside all other samples to test the purity of tubulin from other microtubule associated proteins.
Generation of cell lines with stable CLIP-170 and CLIP-170S knockdown:
Approximately 1 × 106 cells of the HS746T and TMK1 cell lines were infected with CLIP-170 shRNA lentiviral particles (Cat no: sc-43281-V, Santacruz biotechnology, Dallas, TX) at a multiplicity of infection of 1. A scrambled shRNA control was also set up in parallel. Infected cells were then selected with puromycin (1 μg/ml for HS746T and 0.5 μg/ml for TMK1). Individual clones were selected by plating approximately 100 cells in a 100 mm dish and allowed to grow in presence of puromycin for two weeks. The clones were then picked using cloning cylinders, grown individually, and screened using western blot for a complete knockdown of CLIP-170 and CLIP-170S in HS746T cells and that of CLIP-170 in TMK1 cells. Specific clones that showed a complete knockdown of the relevant form of CLIP-170 at protein level were then used for further experiments including SRB cytotoxicity assays as well as immunofluorescence staining of CLIP-170 and EB1.
Generation of TMK1 cell lines with stably expressing Rat GFP-CLIP-170 and Rat GFP-CLIP-170S:
Approximately 1 × 106 cells of the TMK1 cell lines were infected with lentiviral containing either the GFP-CLIP-170 or GFP-CLIP-170S construct at a multiplicity of infection of 1. Infected cells were then selected with Puromycin (1 μg/ml) for two weeks. Once the stable cell lines were established, induced expression of the GFP-CLIP-170 proteins was assessed using 0.1 μg/ml doxycycline for 24 hr using western blot against GFP and C-terminal CLIP-170 antibody (CLIP-170 F-3 clone, Santacruz). Cells were treated 0.1 μg/ml doxycycline for 24 hr prior to addition of docetaxel in SRB cytotoxicity assays.
Generation of Hs746T cell lines with stably expressing Rat GFP-CLIP-170 or Rat GFP-CLIP-170S:
Approximately 1 × 106 cells of the Hs746T CLIP-170KD cells were infected with lentiviral containing either the Rat GFP-CLIP-170 or Rat GFP-CLIP-170S construct at a multiplicity of infection of 1. Infected cells were then selected with Hygromycin (50 μg/ml) for two weeks. Once the stable cell lines were established, induced expression of the Rat GFP-CLIP-170 proteins was assessed using 0.5 μg/ml doxycycline for 24 hr using western blot against GFP antibody. Cells were treated 0.5 μg/ml doxycycline for 24 hr prior to addition of docetaxel in SRB cytotoxicity assays.
Computational Identification of Gleevec:
Gene expression fold change values were calculated for the resistant and sensitive cell lines after treatment with DTX (Accession number: PRJNA594148). In order to identify a signature corresponding to taxane resistance, we calculated the differences in fold changes between the DTX-resistant and DTX-sensitive cells. To find a drug capable of reversing the resistance specific signature, we used the Connectivity Map (CMAP) database (Lamb, 2007; Subramanian et al., 2017) to test 1309 compounds. We calculated the average fold change induced by each compound across different cancer cell lines, and averaged the fold change values across all tested lines to obtain an overall transcriptional response for each drug. A Pearson correlation was calculated between each drug’s transcriptional response and the earlier calculated DTX resistance signature. Negative Pearson correlations indicate compounds, whose induced transcriptional response was opposite to resistance specific signature and thus could be used to reverse resistance in cells.
Combination Index of Imatinib and Docetaxel:
To assess potential synergy or antagonism between the combination of DTX and Imatinib we performed the combination index analysis, as previously described using the CompuSyn algorithm (Chou, 2006). Experimentally, Hs746T and Hs746T CLIP-170-KD cells were plated in 96 well plates (4000 cells per well) and were treated with either DTX alone, Imatinib alone or DTX+Imatinib respectively for 72 hrs. The IC50 was defined as the dose of drug required to inhibit cell growth by 50%. The CI was then computed using Compusyn algorithm by using values for different dose combinations and their respective effect levels to generate Fa vs CI plots. Combination index is calculated using the Chou-Talay method based on the median-effect principle (MEP), and is represented by the equation fa/fu = (D/Dm)m, where fa and fu is the fraction affected and unaffected at dose, Dm is the dose required to achieve the median effect level of 50% and m is the coefficient indicating the sigmoidicity of the dose effect curve(Chou and Talalay, 1984). A combination index value (CI) below 1 indicates that the combination of the two drugs have a synergistic effect, CI equal to 1 indicate that the effect is additive and CI value above 1 would indicate antagonism(Chou and Talalay, 1984).
Plasmids:
Full-length or CLIP-170S DNA fragments were amplified using PrimeSTAR MAX DNA polymerase (Takara) and cloned into EcoRI-SalI sites of pEGFP-C2 vector (Clontech Laboratories, Mountain View, CA) to generate GFP-CLIP-170 and GFP-CLIP-170S, respectively. GFP-CLIP-170/CLIP-170S were amplified and first cloned into pENTR1A using BamHI and NotI sites and was later sub-cloned into pCW57.1 vector containing doxycycline inducible promoter and a puromycin resistance gene, using the LR clonase reaction. Rat GFP-CLIP-170/CLIP-170S were amplified and cloned into pCW57-MSC1-P2A-MCS2 vector containing doxycycline inducible promoter and a hygromycin resistance gene, using AgeI and BamHI sites. mCherry-CLIP-170S was amplified and cloned using Eco RI and Sal I sites. All plasmids were verified for sequence integrity before they were used for the experiments.
QUANTIFICATION AND STATISTICAL ANALYSIS:
Statistical analyses and graphic presentation were done by Prism software (Graphpad) and R (version 3.5.2). For kon/koff kinetic analyses and curve fitting, Sigmaplot (Systat Software, San Jose, CA) was used. For statistical analysis of KM curve and the swimmer’s plot, the time-to-event variables were summarized descriptively. In particular, the Kaplan-Meier estimator was used to generate estimates and curves for patients with different CLIP-170 status. To compare PFS curves between patients with different CLIP-170 status, log-rank test was applied. Median PFS was provided along with 95% confidence interval. Cox proportional hazard model was utilized to estimate hazard ratio of disease progression between patients with different CLIP-170 status. Besides, swimmers’ plot for patients with known CLIP-170 status was also provided with best response for each patient shown at the beginning of each bar. To quantify Flutax-2 fluorescence in DTX-sensitive and resistant cells, raw fluorescence intensity values were first log-transformed (log10) to improve normality. This transformation, along with the large sample size, ensured that a linear model was appropriate analysis for these data. A linear mixed model analysis on log transformed Intensity values as outcome was performed using R version 3.6.3, with cell-batch as the random effect, which is nested within the cell-line type (Hs746T vs TMK1). Time, cell-line type and their interaction, were used as independent variables.
Supplementary Material
Supplementary movies 1: Ectopic expression of GFP-CLIP-170 in TMK1 cells. Related to Figure 2, Supplementary Figure 4, Supplementary Figure 5 and Supplementary Figure 6: Live cell imaging of TMK1 cells with ectopic expression of similarly low levels of either GFP-tagged CLIP-170 (movie 1, left panel) or CLIP-170S (movie 1, right panel).
Supplementary movies 2: Ectopic expression of GFP- Rat CLIP-170 in Hs746T CLIP-170 KD cells. Related to Figure 1, Figure 2, Supplementary Figure 5, Supplementary Figure 6: Live cell imaging of Hs746T CLIP-170 KD cells with ectopic expression of similarly low levels of either GFP-tagged Rat CLIP-170 (movie 2, left panel) or CLIP-170S (movie 2, right panel).
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CLIP-170 antibody (C-terminal/F-3 clone) | Santacruz biotechnology | Cat No: sc-28325, RRID:AB_671001 |
| CLIP-170 antibody (N-terminal/E-8 clone) | Santacruz biotechnology | Cat No: sc-166801, RRID:AB_2082236 |
| Mouse anti-16α-tubulin mAb antibody (DM1-α clone) | Millipore-Sigma | Cat No: T6199-100UL, RRID:AB_477583 |
| Rabbit anti-GFP antibody | Novus Biological | Cat No: NB600-303, RRID:AB_10001300 |
| Rat anti-α-tubulin monoclonal antibody (YL1/2 clone) | Bio-rad | Cat No: MCA77G, RRID:AB_325003 |
| EB-1 antibody (KT51 clone) | Abcam | Cat No: ab53358, RRID:AB_881313 |
| Mouse anti-Myc antibody (9E10 clone) | Santacruz biotechnology | Cat No: sc-40, RRID:AB_2857941 |
| Bacterial and virus strains | ||
| Biological samples | ||
| Human tumor endoscopic biopsies | Shah et. al, 2020 and this paper | ClinicalTrials.gov Identifier: NCT01757171 and internally approved study IRB1203012274 |
| Chemicals, peptides, and recombinant proteins | ||
| RPMI 1640 | Corning | Cat No: 10-040-CV |
| Hexaflutax-2 | Diaz et. al., 2005 | doi: 10.1074/jbc.M407816200 |
| Flutax-2 | Diaz et. al. 2003 | doi: 10.1074/jbc.M211163200 |
| Docetaxel | Millipore-Sigma | Cat No: 01885-5MG-F |
| Imatinib | Millipore-Sigma | Cat No: SML1027-100MG |
| Cediranib | Millipore-Sigma | Cat No: SML2850-5MG |
| Sunitinib | Millipore-Sigma | Cat No: PZ0012-5MG |
| Ponatinib | Millipore-Sigma | Cat No: ADV465748635-25MG |
| Axitinib | Millipore-Sigma | Cat No: PZ0193-5MG |
| Tubulin tracker™ | Thermo Fisher Scientific | Cat No: T34077 |
| Doxycycline | Millipore-Sigma | Cat No: D9891-10G |
| Puromycin | Thermo Fisher Scientific | Cat No: A1113803 |
| Hygromycin B | Thermo Fisher Scientific | Cat No: 10687010 |
| Tubulin protein (>99% pure): porcine brain | Cytoskeleton | Cat No: T240-B |
| GTP: 100mM stock | Cytoskeleton | Cat No: BST06-001 |
| cOmplete™ EDTA-free protease inhibitors | Roche | Cat No: 11836170001 |
| Halt™ phosphatase inhibitor | Thermo Fisher Scientific | Cat No: 78420 |
| Sulforhodamine B (SRB) | Millipore-Sigma | Cat No: S1402-25G |
| Cell-titer Glo assay | Promega | Cat No: G9241 |
| Bolt™ 4–12% Bis-Tris Plus mini-gel | Thermo Fisher Scientific | Cat No: NW04120BOX |
| NOVEX® colloidal blue staining kit | Thermo Fisher Scientific | Cat No: LC6025 |
| 7.5% Mini-PROTEAN® TGX™ Precast Protein Gels | Bio-rad | Cat No: 4561024 |
| SMARTer RACE 5’/3’ kit | Takara/Clontech | Cat No: 634858 |
| Oligotex mRNA isolation kit | Qiagen | Cat no: 70022 |
| CLIP-170 shRNA lentiviral particles | Santacruz biotechnology | Cat no: sc-43281-V |
| Gateway™ LR Clonase™ Enzyme mix | Thermo Fisher Scientific | Cat no: 11791019 |
| Critical commercial assays | ||
| Deposited data | ||
| RNA sequencing data for untreated and DTX-treated resistant and sensitive GC cell lines | Galletti et. al., 2020 | Accession number: PRJNA594148 |
| Experimental models: Cell lines | ||
| AZ-521 | Tan et al., 2011 | doi: 10.1053/j.gastro.2011.04.042 |
| FU97 | Tan et al., 2011 | doi: 10.1053/j.gastro.2011.04.042 |
| Hs746T | Tan et al., 2011 | doi: 10.1053/j.gastro.2011.04.042 |
| MKN-7 | Tan et al., 2011 | doi: 10.1053/j.gastro.2011.04.042 |
| SCH | Tan et al., 2011 | doi: 10.1053/j.gastro.2011.04.042 |
| TMK-1 | Tan et al., 2011 | doi: 10.1053/j.gastro.2011.04.042 |
| KATO-III | Laboratory of Dr. Z. Wainberg, University of California, Los Angeles | N/A |
| MKN-1 | Laboratory of Dr. Z. Wainberg, University of California, Los Angeles | N/A |
| SNU-1 | Laboratory of Dr. Z. Wainberg, University of California, Los Angeles | N/A |
| AGS | Laboratory of Dr. R. Klein, Memorial Sloan Kettering Cancer Center, New York | N/A |
| NCI-N87 | Laboratory of Dr. R. Klein, Memorial Sloan Kettering Cancer Center, New York | N/A |
| MKN-74 | Laboratory of Dr. R. Klein, Memorial Sloan Kettering Cancer Center, New York | N/A |
| MKN-45 | Laboratory of Dr. M. Moehler, Johannes Gutenberg University of Mainz, Germany | N/A |
| Experimental models: Organisms/strains | ||
| Mus Musculus; Female NMRI nude (Crl: NMRI-Foxn1nu) |
Charles River GmbH | MGI Cat# 5653040, RRID:MGI:5653040 |
| Oligonucleotides | ||
| Recombinant DNA | ||
| pEGFP-C2 vector | Clontech/Takara | Cat No: 6083-1 |
| pENTR1A | Thermo Fisher Scientific | Cat No: 11813-011 |
| pCW57.1 | Addgene | https://www.addgene.org/41393/, RRID:Addgene_41393 |
| pCW57-MCS1-P2A-MCS2 | Addgene | https://www.addgene.org/89180/, RRID:Addgene_89180 |
| Software and algorithms | ||
| Microsoft Excel | Microsoft | https://www.microsoft.com/en-us/microsoft-365/excel |
| Skyline | MacLean et. al., 2010 | https://skyline.ms/project/home/software/skyline/begin.view |
| GraphPad Prism 9 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| FIJI | Schindelin et al., 2012 | Fiji, RRID:SCR_002285 https://fiji.sc/ |
| Sigmaplot v11 | Systat Software | https://sigmaplot.fileplanet.com/ |
| R (version 3.5.2) | R Core Team | https://cran.r-project.org/bin/windows/base/old/3.5.2/ |
| CompuSyn algorithm | Chou T.C. et. al., 2006 | https://www.combosyn.com/ |
| R (version 3.6.3) | R Core Team | https://cran.r-project.org/bin/windows/base/old/3.6.3/ |
| Other | ||
Highlights:
CLIP-170S is a truncated variant of the microtubule plus-end binding protein CLIP-170.
Taxane-resistant GC cell lines and tumor biopsies both express CLIP-170S.
CLIP-170S confers taxane resistance by impairing taxane access to the MT lumen.
Depletion of CLIP-170S by RTK inhibitors can reverse taxane resistance.
Acknowledgements:
This work was supported in part by the NIH T32 Training grant 5T32CA062948 (to KK), the NIH T32 Training grant 5T32CA062948 (to GG), the NIH T32 Training grant 5T32CA203702 (to UDC) by Clinical and Translational Science Center at Weill Cornell Medicine NIH/NCATS grant ULTR00457 (to GG), the NIH/NCI R01CA228512 (to P.G., M.A.S. and O.E.), NIH/NCI R21 CA216800 (to P.G), NIH/NCI R01 CA179100 (to P.G), DoD PC180637 (P.G., ARC) and by the Ministerio de Economía y Competitividad grant BFU2016-75319-R and European Union H2020-MSCA-ITN-ETN/0582 ITN TUBINTRAIN (Awarded to J.F.D.). D.S. was supported by the Intramural Research Program of the Eunice Kennedy Shriver National Institute of Child Health and Human Development, NIH. Yao Lu, MS was supported in part by funds from the Clinical and Translational Science Center (CTSC), National Center for Advancing Translational Sciences (NCATS) of the National Institutes of Health, award # UL1-TR002384-01. The content is solely the responsibility of the authors and does not necessarily represent the official views of the funding source NCATS based in Rockville, MD. We would like to thank Dr. Erica Holzbaur for Rat CLIP-170 cDNA. We would also like to thank Prerna Mahtani, MS and Trevor Lee for technical assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests: Drs. Neel Madhukar and Olivier Elemento are the co-founders and equity stakeholders of OneThree Biotech, an artificial intelligence-driven drug discovery and development company. All other authors have no competing interests and have nothing to declare.
Data availability:
The data that support this study are available from the corresponding authors upon request.
References:
- Akhmanova A, and Steinmetz MO (2008). Tracking the ends: a dynamic protein network controls the fate of microtubule tips. Nat Rev Mol Cell Biol 9, 309–322. 10.1038/nrm2369. [DOI] [PubMed] [Google Scholar]
- Akhmanova A, and Steinmetz MO (2015). Control of microtubule organization and dynamics: two ends in the limelight. Nat Rev Mol Cell Biol 16, 711–726. 10.1038/nrm4084. [DOI] [PubMed] [Google Scholar]
- Balaguer FA, Muhlethaler T, Estevez-Gallego J, Calvo E, Gimenez-Abian JF, Risinger AL, Sorensen EJ, Vanderwal CD, Altmann KH, Mooberry SL, et al. (2019). Crystal Structure of the Cyclostreptin-Tubulin Adduct: Implications for Tubulin Activation by Taxane-Site Ligands. Int J Mol Sci 20. 10.3390/ijms20061392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bang YJ, Van Cutsem E, Feyereislova A, Chung HC, Shen L, Sawaki A, Lordick F, Ohtsu A, Omuro Y, Satoh T, et al. (2010). Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet 376, 687–697. 10.1016/S0140-6736(10)61121-X. [DOI] [PubMed] [Google Scholar]
- Bieling P, Kandels-Lewis S, Telley IA, van Dijk J, Janke C, and Surrey T (2008). CLIP-170 tracks growing microtubule ends by dynamically recognizing composite EB1/tubulin-binding sites. J Cell Biol 183, 1223–1233. 10.1083/jcb.200809190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieling P, Laan L, Schek H, Munteanu EL, Sandblad L, Dogterom M, Brunner D, and Surrey T (2007). Reconstitution of a microtubule plus-end tracking system in vitro. Nature 450, 1100–1105. 10.1038/nature06386. [DOI] [PubMed] [Google Scholar]
- Buey RM, Calvo E, Barasoain I, Pineda O, Edler MC, Matesanz R, Cerezo G, Vanderwal CD, Day BW, Sorensen EJ, et al. (2007). Cyclostreptin binds covalently to microtubule pores and lumenal taxoid binding sites. Nat Chem Biol 3, 117–125. 10.1038/nchembio853. [DOI] [PubMed] [Google Scholar]
- Cervera P, and Flejou JF (2011). Changing pathology with changing drugs: tumors of the gastrointestinal tract. Pathobiology 78, 76–89. 10.1159/000315535. [DOI] [PubMed] [Google Scholar]
- Chou TC (2006). Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev 58, 621–681. 10.1124/pr.58.3.10. [DOI] [PubMed] [Google Scholar]
- Chou TC, and Talalay P (1984). Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul 22, 27–55. [DOI] [PubMed] [Google Scholar]
- De Francia S, D’Avolio A, Ariaudo A, Pirro E, Piccione F, Simiele M, Fava C, Calcagno A, Di Perri G, and Saglio G (2014). Plasma and intracellular imatinib concentrations in patients with chronic myeloid leukemia. Ther Drug Monit 36, 410–412. 10.1097/FTD.0000000000000013. [DOI] [PubMed] [Google Scholar]
- Derry WB, Wilson L, and Jordan MA (1995). Substoichiometric binding of taxol suppresses microtubule dynamics. Biochemistry 34, 2203–2211. 10.1021/bi00007a014. [DOI] [PubMed] [Google Scholar]
- Diaz JF, and Andreu JM (1993). Assembly of purified GDP-tubulin into microtubules induced by taxol and taxotere: reversibility, ligand stoichiometry, and competition. Biochemistry 32, 2747–2755. 10.1021/bi00062a003. [DOI] [PubMed] [Google Scholar]
- Diaz JF, Barasoain I, and Andreu JM (2003). Fast kinetics of Taxol binding to microtubules. Effects of solution variables and microtubule-associated proteins. J Biol Chem 278, 8407–8419. 10.1074/jbc.M211163200. [DOI] [PubMed] [Google Scholar]
- Diaz JF, Barasoain I, Souto AA, Amat-Guerri F, and Andreu JM (2005). Macromolecular accessibility of fluorescent taxoids bound at a paclitaxel binding site in the microtubule surface. J Biol Chem 280, 3928–3937. 10.1074/jbc.M407816200. [DOI] [PubMed] [Google Scholar]
- Druker BJ, Guilhot F, O’Brien SG, Gathmann I, Kantarjian H, Gattermann N, Deininger MW, Silver RT, Goldman JM, Stone RM, et al. (2006). Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med 355, 2408–2417. 10.1056/NEJMoa062867. [DOI] [PubMed] [Google Scholar]
- Druker BJ, and Lydon NB (2000). Lessons learned from the development of an abl tyrosine kinase inhibitor for chronic myelogenous leukemia. J Clin Invest 105, 3–7. 10.1172/JCI9083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Druker BJ, Talpaz M, Resta DJ, Peng B, Buchdunger E, Ford JM, Lydon NB, Kantarjian H, Capdeville R, Ohno-Jones S, and Sawyers CL (2001). Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med 344, 1031–1037. 10.1056/NEJM200104053441401. [DOI] [PubMed] [Google Scholar]
- Druker BJ, Tamura S, Buchdunger E, Ohno S, Segal GM, Fanning S, Zimmermann J, and Lydon NB (1996). Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat Med 2, 561–566. [DOI] [PubMed] [Google Scholar]
- Dumontet C, Duran GE, Steger KA, Beketic-Oreskovic L, and Sikic BI (1996). Resistance mechanisms in human sarcoma mutants derived by single-step exposure to paclitaxel (Taxol). Cancer Res 56, 1091–1097. [PubMed] [Google Scholar]
- Dumontet C, and Jordan MA (2010). Microtubule-binding agents: a dynamic field of cancer therapeutics. Nat Rev Drug Discov 9, 790–803. 10.1038/nrd3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferlay J, Colombet M, Soerjomataram I, Mathers C, Parkin DM, Pineros M, Znaor A, and Bray F (2019). Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int J Cancer 144, 1941–1953. 10.1002/ijc.31937. [DOI] [PubMed] [Google Scholar]
- Field JJ, Diaz JF, and Miller JH (2013). The binding sites of microtubule-stabilizing agents. Chem Biol 20, 301–315. 10.1016/j.chembiol.2013.01.014. [DOI] [PubMed] [Google Scholar]
- Gaitanos TN, Buey RM, Diaz JF, Northcote PT, Teesdale-Spittle P, Andreu JM, and Miller JH (2004). Peloruside A does not bind to the taxoid site on beta-tubulin and retains its activity in multidrug-resistant cell lines. Cancer Res 64, 5063–5067. 10.1158/0008-5472.CAN-04-0771. [DOI] [PubMed] [Google Scholar]
- Galletti G, Matov A, Beltran H, Fontugne J, Miguel Mosquera J, Cheung C, MacDonald TY, Sung M, O’Toole S, Kench JG, et al. (2014). ERG induces taxane resistance in castration-resistant prostate cancer. Nat Commun 5, 5548. 10.1038/ncomms6548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galletti G, Zhang C, Gjyrezi A, Cleveland K, Zhang J, Powell S, Thakkar PV, Betel D, Shah MA, and Giannakakou P (2020). Microtubule Engagement with Taxane Is Altered in Taxane-Resistant Gastric Cancer. Clin Cancer Res 26, 3771–3783. 10.1158/1078-0432.CCR-19-3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia BA, Mollah S, Ueberheide BM, Busby SA, Muratore TL, Shabanowitz J, and Hunt DF (2007). Chemical derivatization of histones for facilitated analysis by mass spectrometry. Nat Protoc 2, 933–938. 10.1038/nprot.2007.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannakakou P, Gussio R, Nogales E, Downing KH, Zaharevitz D, Bollbuck B, Poy G, Sackett D, Nicolaou KC, and Fojo T (2000a). A common pharmacophore for epothilone and taxanes: molecular basis for drug resistance conferred by tubulin mutations in human cancer cells. Proc Natl Acad Sci USA 97, 2904–2909. 10.1073/pnas.040546297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannakakou P, Sackett DL, Kang YK, Zhan Z, Buters JT, Fojo T, and Poruchynsky MS (1997). Paclitaxel-resistant human ovarian cancer cells have mutant beta-tubulins that exhibit impaired paclitaxel-driven polymerization. J Biol Chem 272, 17118–17125. 10.1074/jbc.272.27.17118. [DOI] [PubMed] [Google Scholar]
- Giannakakou P, Sackett DL, Ward Y, Webster KR, Blagosklonny MV, and Fojo T (2000b). p53 is associated with cellular microtubules and is transported to the nucleus by dynein. Nat Cell Biol 2, 709–717. 10.1038/35036335. [DOI] [PubMed] [Google Scholar]
- Goncalves A, Braguer D, Kamath K, Martello L, Briand C, Horwitz S, Wilson L, and Jordan MA (2001). Resistance to Taxol in lung cancer cells associated with increased microtubule dynamics. Proc Natl Acad Sci USA 98, 11737–11742. 10.1073/pnas.191388598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Garay ML, Chang L, Blade K, Menick DR, and Cabral F (1999). A beta-tubulin leucine cluster involved in microtubule assembly and paclitaxel resistance. J Biol Chem 274, 23875–23882. 10.1074/jbc.274.34.23875. [DOI] [PubMed] [Google Scholar]
- Guerrero A, Dallas DC, Contreras S, Chee S, Parker EA, Sun X, Dimapasoc L, Barile D, German JB, and Lebrilla CB (2014). Mechanistic peptidomics: factors that dictate specificity in the formation of endogenous peptides in human milk. Mol Cell Proteomics 13, 3343–3351. 10.1074/mcp.M113.036194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta KK, Joyce MV, Slabbekoorn AR, Zhu ZC, Paulson BA, Boggess B, and Goodson HV (2010). Probing interactions between CLIP-170, EB1, and microtubules. J Mol Biol 395, 1049–1062. 10.1016/j.jmb.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta KK, Paulson BA, Folker ES, Charlebois B, Hunt AJ, and Goodson HV (2009). Minimal plus-end tracking unit of the cytoplasmic linker protein CLIP-170. J Biol Chem 284, 6735–6742. 10.1074/jbc.M807675200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horwitz SB, Cohen D, Rao S, Ringel I, Shen HJ, and Yang CP (1993). Taxol: mechanisms of action and resistance. J Natl Cancer Inst Monogr, 55–61. [PubMed] [Google Scholar]
- Joensuu H, Roberts PJ, Sarlomo-Rikala M, Andersson LC, Tervahartiala P, Tuveson D, Silberman S, Capdeville R, Dimitrijevic S, Druker B, and Demetri GD (2001). Effect of the tyrosine kinase inhibitor STI571 in a patient with a metastatic gastrointestinal stromal tumor. N Engl J Med 344, 1052–1056. 10.1056/NEJM200104053441404. [DOI] [PubMed] [Google Scholar]
- Jordan MA, Toso RJ, Thrower D, and Wilson L (1993). Mechanism of mitotic block and inhibition of cell proliferation by taxol at low concentrations. Proc Natl Acad Sci USA 90, 9552–9556. 10.1073/pnas.90.20.9552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kall L, Canterbury JD, Weston J, Noble WS, and MacCoss MJ (2007). Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nat Methods 4, 923–925. 10.1038/nmeth1113. [DOI] [PubMed] [Google Scholar]
- Kavallaris M, Kuo DY, Burkhart CA, Regl DL, Norris MD, Haber M, and Horwitz SB (1997). Taxol-resistant epithelial ovarian tumors are associated with altered expression of specific beta-tubulin isotypes. J Clin Invest 100, 1282–1293. 10.1172/JCI119642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellogg EH, Hejab NMA, Howes S, Northcote P, Miller JH, Diaz JF, Downing KH, and Nogales E (2017). Insights into the Distinct Mechanisms of Action of Taxane and Non-Taxane Microtubule Stabilizers from Cryo-EM Structures. J Mol Biol 429, 633–646. 10.1016/j.jmb.2017.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komlodi-Pasztor E, Sackett D, Wilkerson J, and Fojo T (2011). Mitosis is not a key target of microtubule agents in patient tumors. Nat Rev Clin Oncol 8, 244–250. 10.1038/nrclinonc.2010.228. [DOI] [PubMed] [Google Scholar]
- Lamb J (2007). The Connectivity Map: a new tool for biomedical research. Nat Rev Cancer 7, 54–60. 10.1038/nrc2044. [DOI] [PubMed] [Google Scholar]
- Lee HS, Komarova YA, Nadezhdina ES, Anjum R, Peloquin JG, Schober JM, Danciu O, van Haren J, Galjart N, Gygi SP, et al. (2010). Phosphorylation controls autoinhibition of cytoplasmic linker protein-170. Mol Biol Cell 21, 2661–2673. 10.1091/mbc.E09-12-1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luduena RF (1998). Multiple forms of tubulin: different gene products and covalent modifications. Int Rev Cytol 178, 207–275. [DOI] [PubMed] [Google Scholar]
- MacLean B, Tomazela DM, Shulman N, Chambers M, Finney GL, Frewen B, Kern R, Tabb DL, Liebler DC, and MacCoss MJ (2010). Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26, 966–968. 10.1093/bioinformatics/btq054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madhukar NS, Khade PK, Huang L, Gayvert K, Galletti G, Stogniew M, Allen JE, Giannakakou P, and Elemento O (2019). A Bayesian machine learning approach for drug target identification using diverse data types. Nat Commun 10, 5221. 10.1038/s41467-019-12928-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nirschl JJ, Magiera MM, Lazarus JE, Janke C, and Holzbaur EL (2016). alpha-Tubulin Tyrosination and CLIP-170 Phosphorylation Regulate the Initiation of Dynein-Driven Transport in Neurons. Cell Rep 14, 2637–2652. 10.1016/j.celrep.2016.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogales E, Wolf SG, and Downing KH (1998). Structure of the alpha beta tubulin dimer by electron crystallography. Nature 391, 199–203. 10.1038/34465. [DOI] [PubMed] [Google Scholar]
- Perez F, Diamantopoulos GS, Stalder R, and Kreis TE (1999). CLIP-170 highlights growing microtubule ends in vivo. Cell 96, 517–527. 10.1016/s0092-8674(00)80656-x. [DOI] [PubMed] [Google Scholar]
- Pierre P, Scheel J, Rickard JE, and Kreis TE (1992). CLIP-170 links endocytic vesicles to microtubules. Cell 70, 887–900. 10.1016/0092-8674(92)90240-d. [DOI] [PubMed] [Google Scholar]
- Power DG, Kelsen DP, and Shah MA (2010). Advanced gastric cancer--slow but steady progress. Cancer Treat Rev 36, 384–392. 10.1016/j.ctrv.2010.01.005. [DOI] [PubMed] [Google Scholar]
- Rai A, Liu T, Glauser S, Katrukha EA, Estevez-Gallego J, Rodriguez-Garcia R, Fang WS, Diaz JF, Steinmetz MO, Altmann KH, et al. (2020). Taxanes convert regions of perturbed microtubule growth into rescue sites. Nat Mater 19, 355–365. 10.1038/s41563-019-0546-6. [DOI] [PubMed] [Google Scholar]
- Sadovnik I, Lierman E, Peter B, Herrmann H, Suppan V, Stefanzl G, Haas O, Lion T, Pickl W, Cools J, et al. (2014). Identification of Ponatinib as a potent inhibitor of growth, migration, and activation of neoplastic eosinophils carrying FIP1L1-PDGFRA. Exp Hematol 42, 282–293 e284. 10.1016/j.exphem.2013.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheel J, Pierre P, Rickard JE, Diamantopoulos GS, Valetti C, van der Goot FG, Haner M, Aebi U, and Kreis TE (1999). Purification and analysis of authentic CLIP-170 and recombinant fragments. J Biol Chem 274, 25883–25891. 10.1074/jbc.274.36.25883. [DOI] [PubMed] [Google Scholar]
- Schiff PB, Fant J, and Horwitz SB (1979). Promotion of microtubule assembly in vitro by taxol. Nature 277, 665–667. 10.1038/277665a0. [DOI] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat Methods 9, 676–682. 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah MA (2015). Update on metastatic gastric and esophageal cancers. J Clin Oncol 33, 1760–1769. 10.1200/JCO.2014.60.1799. [DOI] [PubMed] [Google Scholar]
- Shah MA, Enzinger P, Ko AH, Ocean AJ, Philip PA, Thakkar PV, Cleveland K, Lu Y, Kortmansky J, Christos PJ, et al. (2020). Multicenter Phase II Study of Cabazitaxel in Advanced Gastroesophageal Cancer: Association of HER2 Expression and M2-Like Tumor-Associated Macrophages with Patient Outcome. Clin Cancer Res 26, 4756–4766. 10.1158/1078-0432.CCR-19-3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skehan P, Storeng R, Scudiero D, Monks A, McMahon J, Vistica D, Warren JT, Bokesch H, Kenney S, and Boyd MR (1990). New colorimetric cytotoxicity assay for anticancer-drug screening. J Natl Cancer Inst 82, 1107–1112. 10.1093/jnci/82.13.1107. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Narayan R, Corsello SM, Peck DD, Natoli TE, Lu X, Gould J, Davis JF, Tubelli AA, Asiedu JK, et al. (2017). A Next Generation Connectivity Map: L1000 Platform and the First 1,000,000 Profiles. Cell 171, 1437–1452 e1417. 10.1016/j.cell.2017.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Li D, Yang Y, Ren Y, Li J, Wang Z, Dong B, Liu M, and Zhou J (2012). Microtubule-binding protein CLIP-170 is a mediator of paclitaxel sensitivity. J Pathol 226, 666–673. 10.1002/path.3026. [DOI] [PubMed] [Google Scholar]
- Tan IB, Ivanova T, Lim KH, Ong CW, Deng N, Lee J, Tan SH, Wu J, Lee MH, Ooi CH, et al. (2011). Intrinsic subtypes of gastric cancer, based on gene expression pattern, predict survival and respond differently to chemotherapy. Gastroenterology 141, 476–485, 485 e471–411. 10.1053/j.gastro.2011.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Cutsem E, Moiseyenko VM, Tjulandin S, Majlis A, Constenla M, Boni C, Rodrigues A, Fodor M, Chao Y, Voznyi E, et al. (2006). Phase III study of docetaxel and cisplatin plus fluorouracil compared with cisplatin and fluorouracil as first-line therapy for advanced gastric cancer: a report of the V325 Study Group. J Clin Oncol 24, 4991–4997. 10.1200/JCO.2006.06.8429. [DOI] [PubMed] [Google Scholar]
- Wang Q, Crevenna AH, Kunze I, and Mizuno N (2014). Structural basis for the extended CAP-Gly domains of p150(glued) binding to microtubules and the implication for tubulin dynamics. Proc Natl Acad Sci USA 111, 11347–11352. 10.1073/pnas.1403135111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, O’Brate A, Zhou W, and Giannakakou P (2005). Resistance to microtubule-stabilizing drugs involves two events: beta-tubulin mutation in one allele followed by loss of the second allele. Cell Cycle 4, 1847–1853. 10.4161/cc.4.12.2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterman-Storer CM, and Salmon ED (1997). Actomyosin-based retrograde flow of microtubules in the lamella of migrating epithelial cells influences microtubule dynamic instability and turnover and is associated with microtubule breakage and treadmilling. J Cell Biol 139, 417–434. 10.1083/jcb.139.2.417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson P, and Stephens DJ (2006). Microtubule plus-end loading of p150(Glued) is mediated by EB1 and CLIP-170 but is not required for intracellular membrane traffic in mammalian cells. J Cell Sci 119, 2758–2767. 10.1242/jcs.02999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisbrich A, Honnappa S, Jaussi R, Okhrimenko O, Frey D, Jelesarov I, Akhmanova A, and Steinmetz MO (2007). Structure-function relationship of CAP-Gly domains. Nat Struct Mol Biol 14, 959–967. 10.1038/nsmb1291. [DOI] [PubMed] [Google Scholar]
- Wiesen KM, Xia S, Yang CP, and Horwitz SB (2007). Wild-type class I beta-tubulin sensitizes Taxol-resistant breast adenocarcinoma cells harboring a beta-tubulin mutation. Cancer Lett 257, 227–235. 10.1016/j.canlet.2007.07.022. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary movies 1: Ectopic expression of GFP-CLIP-170 in TMK1 cells. Related to Figure 2, Supplementary Figure 4, Supplementary Figure 5 and Supplementary Figure 6: Live cell imaging of TMK1 cells with ectopic expression of similarly low levels of either GFP-tagged CLIP-170 (movie 1, left panel) or CLIP-170S (movie 1, right panel).
Supplementary movies 2: Ectopic expression of GFP- Rat CLIP-170 in Hs746T CLIP-170 KD cells. Related to Figure 1, Figure 2, Supplementary Figure 5, Supplementary Figure 6: Live cell imaging of Hs746T CLIP-170 KD cells with ectopic expression of similarly low levels of either GFP-tagged Rat CLIP-170 (movie 2, left panel) or CLIP-170S (movie 2, right panel).
Data Availability Statement
All Data reported in this manuscript will be shared by the Lead Contact upon request. If any further information is required regarding the RNA-seq analysis in conjunction with Connectivity Map, it will be provided by the lead contact upon request. This manuscript does not report original code.
The data that support this study are available from the corresponding authors upon request.