

Abstract
With the rapid increase and accessibility of high-resolution imaging technologies of cells, the interpretation of results relies more and more on the assumption that the three-dimensional integrity of the surrounding cellular landscape is not compromised by the experimental setup. However, the only available technology for directly probing the structural integrity of whole-cell preparations at the nanoscale is electron cryo-tomography, which is time-consuming, costly, and complex. We devised an accessible, inexpensive and reliable screening assay to quickly report on the compatibility of experimental protocols with preserving the structural integrity of whole-cell preparations at the nanoscale. Our Rapid Cell Integrity Assessment (RCIA) assay is executed at room temperature and relies solely on light microscopy imaging. Using cellular electron cryo-tomography as a benchmark, we verify that RCIA1 accurately unveils the adverse impact of reagents and/or protocols such as those used for virus inactivation or to arrest dynamic processes on the cellular nanoarchitecture.
Keywords: cryo-electron tomography, virus inactivation, CLEM, in-situ cellular methods
Graphical Abstract
INTRODUCTION
High resolution imaging methodologies for cells are quickly gaining momentum on both sides of the microscopy spectrum: super-resolution light microscopy (LM) has entered the near-molecular scale enabling the visualization of events previously unresolvable by fluorescence imaging, while cryogenic electron tomography (cryo-ET) is now offering nanometer resolution depiction of three-dimensional cellular landscapes (Turk and Baumeister, 2020) and is capable of reaching near-atomic resolution when averaging is employed (Tegunov et al., 2021). The correct interpretation of these high-resolution data depends on the structural integrity of the cellular context. For whole-cell preparations, it is fundamental to keep the sample’s three-dimensional structural integrity intact, as changes in the intracellular nano-environment may drastically affect the validity of the data interpretation. Currently, cellular cryo-ET is the only available imaging technology that provides three-dimensional (3D) details of the whole-cell preparations with nanometer resolution. While the instrumentation for high-resolution cryo-ET is becoming more readily available, it is still not cost-effective and generally involves elaborate specimen preparations and time-consuming investigations to determine the high-resolution structural integrity of cellular samples. Before embarking on these labor-intensive and costly explorations, it would be greatly beneficial to know beforehand whether the cell preparation has retained its native nanoarchitecture as faithfully as possible.
MATERIAL AND METHODS
Cell culture immunostaining and correlative LM
NIH 3T3, HeLa, 3T3-paxillin (a gift from M.H. Ginsberg and F. Lagarrigue, UCSD and in collaboration with M. Schwartz and A. Kumar, Yale), THP1 and MEF cells were cultured according to standard protocols and used for the assays while in log phase. NIH 3T3 and HeLa cells were plated on carbon-coated glass coverslips coated with poly-d-lysine for the LM assays; 3T3-paxillin and MEF cells were plated on holey carbon-coated gold finder grids (Quantifoil Micro Tools GmbH) for cryo-EM/ET. THP-1 cells were plated also on holey carbon-coated gold finder grids in presence of 160nm PMA in regular growth media and differentiated over the course of 4 days.
For titrating the correct cell concentration to seed on EM grids, we used first carbon-coated glass coverslips (German Glass, EMS) and reserved the more expensive EM grids for the final experiments. For this purpose, we prepared glass coverslips by applying a thin, 2–3 nm carbon layer using a Denton DV-502A (Denton Vacuum LLC) and carbon rods (Ted Pella Inc). The coverslips can be stored at room temperature for prolonged times and were used similarly to EM grids for LM applications.
For validation of the fidelity of the RCIA protocol to report on the nanoarchitecture integrity of the samples, coverslips that are prepared concurrently with the cryo-EM grids. Once cells on EM grids and coverslips had reached the desired density, the samples were treated according to Karen Anderson’s in-house protocols: cells were fixed in 2 – 4% electron microscopy grade paraformaldehyde (Tousimis) from a freshly opened vial, diluted in 1xPBS, 1xDPBS or in a fixative dilution buffer (0.1M Pipes, 1mM EGTA and 1mM MgSO4, pH 6.9). The fixative solution of choice is warmed up to 37°C, the growth media is gently removed from the dish and replaced by the fixative solution, and samples are fixed at 37°C for 30 minutes. Upon completion of the fixation, cells were quickly rinsed with the buffer of choice and then washed three times with room temperature 1xPBS, 1xDPBS or PHEM (60mM Pipes, 25mM HEPES, 10mM EGTA, 2mM MgSO4, pH 6.9). After this point, cells were kept in the selected buffer (1xPBS, 1xDPBS or PHEM) at 4°C for all the steps preceding vitrification or taken through the immunostaining assay described below. Note that the RCIA protocol can be performed solely using the coverslips that are prepared concurrently with the cryo-EM grids. The grids can either be vitrified directly or fixed and then vitrified if fixation is desired or necessary for the experiment. The cells on the coverslip will be subjected to the RCIA protocol to report on the structural integrity of the entire cell preparation, including the ones that were vitrified on the grids without fixation.
For the immunostaining assays, all solutions were made in the selected buffer: if detergent induced permeabilization was used, it was carried out for 5 minutes in 0.1% Triton X100, followed by three rinses. Blocking was done in 2% normal goat serum. Cells that were not exposed to detergent were taken directly to the blocking step. Regardless of whether cells were exposed to detergent or not, after blocking for 30 minutes, cells were rinsed in working buffer, incubated for 1hr with the primary antibody, rinsed three times, incubated for 1hr with the secondary antibody and rinsed three times again. All antibody incubations and rinses were done in working buffer (a 1:5 dilution of the blocking buffer). The stained cells were incubated with DAPI for nuclear counterstaining prior to imaging. Staining with the FluoroNanoGold conjugate was performed similarly, however prolonging the incubations with the antibodies to O/N at 4°C, without counterstaining with DAPI and without mounting media. The antibodies used in these immunostainings were: (LM assays) mouse monoclonal anti clathrin (ab2731; Abcam); rabbit anti giantin (ab80864; Abcam); goat anti mouse, Alexa 488 (R37120; Alexa Fluor 488, goat anti-mouse); goat anti rabbit, Alexa 488 (R37116; Alexa Fluor 488, goat anti rabbit); (EM assays) mouse monoclonal anti paxillin (P135020-050; BD Biosciences); goat anti mouse, FluoroNanogold anti-rabbit, Fab’ AlexaFluor 488 (7204; Nanoprobes).
All correlative LM imaging was done on an inverted light microscope (Eclipse TE 2000-U, Nikon) equipped with manual controlled shutter, filter wheels, and a 14-bit cooled CCD camera (Orca II) controlled by MetaMorph software (Universal Imaging), using a Plan Fluor ELWD 40/0.60 Ph2 or a Plan Fluor 10/0.30 Ph1 objective lens (Nikon). The LM information was used to improve the precision of selecting regions of interest for cryo-tomography data collection.
Vitrification and cryo-EM screening
RCIA grid samples were maintained in the specific buffer (PBS, DPBS or PHEM) at 4°C prior to vitrification. Although most samples were imaged and vitrified within a day from fixation, we found that those in PHEM could be kept for up to a week at 4°C without noticeable damage to the ultrastructure, as revealed when comparing samples in the following cryo-ET investigations. Although grid samples can be vitrified without prior fixation, the ability of maintaining a good preservation in fixed grid samples over time before vitrification is a particularly desirable feature both when performing large scale correlative studies that involve long LM sessions and when receiving samples from remote collaborators that may require days to reach the laboratory where the cryo experiments are conducted. The vitrification process was performed using a home-designed cryo-plunger to plunge-freeze the samples in liquid nitrogen-cooled liquified ethane. Plunge-frozen samples were stored in liquid nitrogen until they were used. Freezing quality, along with assessment of sample preservation and amenability for cryo-ET investigation was conducted by an initial screening on a T12 Spirit (FEI) cryo-EM. Using SerialEM (Mastronarde, 2005), we generated a montage for each cryo-grid and identified target cells by following the finder markings on the grids, which are visible in both light and electron imaging modalities (Anderson et al., 2018).
Correlative LM/cryo-EM and Cryo-ET
To access the fidelity of RCIA reporting with ultrastructural integrity, cellular cryo-ET was pursued. After screening on a Tecnai Spirit T12 cryo-electron microscope (ThermoFisher Scientific), the grids were transferred into a Titan Krios cryo-electron microscope (ThermoFisher Scientific). An overview image (atlas) of the entire grid was acquired to identify cells using the finder markings on the grids, which are visible in all three imaging modalities (LM, T12, and Titan Krios). Low-resolution images of the grid squares (pixel size 14–28 nm) containing the identified cells were acquired to allow alignment of LM and cryo-tomography images of the same cell. The cryo-ET data was taken with a FEI Titan Krios equipped with field emission gun (XFEG) and operated at 300kV for selected samples from the T12 screening session. Following correlative identification of regions of interest, tilt series (+/− 60°, every 3°) were set up in batch mode using the SerialEM (Mastronarde, 2005) or Tomo package (ThermoFisher Scientific; FEI Company). Data was acquired on a Falcon II or III direct detection imaging device under minimal dose conditions of about 100–120 e−/Å2 and defocus of 8–14 μm with magnification resulting in a pixel size of 0.48 nm in the tilt images. Reconstructions were obtained using real-time automatic reconstruction protocols using patch-based alignment and the simultaneous iterative reconstruction technique (Agulleiro and Fernandez, 2015) run within the pyCoAn package (github.com/pyCoAn/distro), an extended python version of the CoAn package (Volkmann and Hanein, 1999). All volumes were enhanced using a Wiener-like filter accounting for the contrast transfer function at the respective defocus and an estimate of the spectral signal-to-noise ratio (Tegunov and Cramer, 2019). Images of tomogram slices were generated with IMOD (Kremer et al., 1996).
Quantification and statistical analysis
The signal of the fluorescence images was determined by the intensity in the green channel of the image. We used 6 images each from samples with PBS or DPBS buffer and 12 images from samples prepared with PHEM buffer. To quantify the visible damage in tomographic reconstructions, we conducted blinded classification of 40 tomograms (20 PBS and 20 PHEM) into either “visibly damaged” or “not visibly damaged” categories. The classification was blinded in the sense that researchers did not know what tomogram corresponded to what buffer treatment or how many tomograms per buffer were present. Four different researchers conducted the classification independently. Two-sided Student’s T-tests were employed to determine statistical significance.
RESULTS AND DISCUSSION
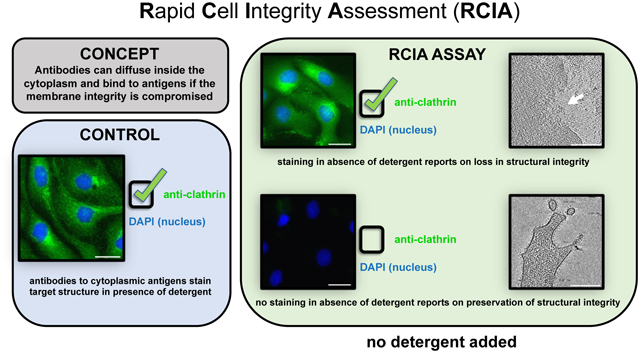
To help evaluate the internal integrity of the cell nanoarchitecture without employing costly workflows, we have designed a simple immunostaining-based screening assay, which we coined Rapid Cell Integrity Assessment (RCIA). The assay quickly and accurately reports on the impact that experimental setups may have had on the nanoscale structural integrity of cell preparations. As a proof of concept, we employed RCIA as a screening method to report on the structural integrity of the cellular nanoarchitecture as a function of buffer composition used during chemical fixation, while employing a cryo-ET workflow for independent validation. One rationale for investigating buffer conditions in conjunction with fixation was the growing need to find optimal conditions to preserve intracellular structural integrity of samples that need to be inactivated by chemical fixation prior to imaging such as samples with intact viruses or virus infected cells, a particularly timely objective during the ongoing COVID19 pandemic (Klein et al., 2020; Wolff et al., 2020). Note, however, that fixation of the cryo-EM grid is in principle optional because the RCIA experiment (which does require fixation) is carried out with the accompanying glass coverslips.
The RCIA concept.
The RCIA screening assay is based on the inability of antibodies, (size approximately 10nm/150kDa) to freely diffuse through the plasma membrane and bind to their specific cytoplasmic antigen target unless the membrane integrity has been compromised. Plasma membrane permeability to antibodies can be introduced for example through treatment with drugs, detergents, or physical/mechanical intervention (such as electrical pulses during electroporation). In the absence of such damage, antibodies are unable to diffuse through the plasma membrane, and thus can only stain cell surface antigens. However, if antibody staining of antigens associated with the cytosolic compartment is detectable, it suggests that not only the plasma membrane integrity of these cells is compromised but that the integrity of the cytosol is also comprised because the antibodies diffused freely within the cytosol, which is densely packed by cytoskeletal and other cytosolic elements in structurally uncompromised cells. In previous work using in-situ cryo-ET (Anderson et al., 2017; Kumar et al., 2018; Marston et al., 2019) we noticed a strong correlation between visible membrane damage, disassembled cytoskeletal elements, and regions within the cytosol that are devoid of discernible macromolecular complexes. Thus, we hypothesized that antibodies diffusing into the cytoplasm represent a hallmark for damage at the nanoscale not only to the plasma membrane but also to intracellular components, in particular cytoskeletal assemblies.
The RCIA protocol and its validation to report on the nanoarchitecture integrity of the samples.
To test our hypothesis and verify the potential and fidelity of our assay, we combined RCIA screening protocol with cryo-ET to directly correlate antibody staining with high-resolution structural integrity of the cell preparations. Towards this goal, we investigated the effects of three different buffer formulations which are used to deliver a chemical fixative. Cryogenic technologies such as cryo-EM, cryo-ET and cryo-correlated light and electron microscopy (cryo-CLEM) have been growing exponentially (Dubochet, 2012; Ognjenović et al., 2019; Turk and Baumeister, 2020; Van Drie and Tong, 2020) and are now the preferred approach to image biological specimens close to their native, fully hydrated state at nanometer scale resolution. In-situ cryo-ET was an excellent test bed and validation approach for the RCIA assay, as this is the only available technology that can directly image in three dimensions with nanometer scale resolution the nanoarchitecture of the cells and thus directly identify the existence and extent of damage. Although cells do not need to be chemically fixed prior to vitrification, there are applications where pathogens like viruses need to be inactivated or where all biological activities must be halted at a precise time point. The latter is generally the case with correlative investigations (Hanein and Volkmann, 2011; Sartori et al., 2007), where subcellular elements imaged with light microscopy may change too quickly to be locked in space and time before vitrification can take place, making an accurate correlation between light and electron microscopy modalities problematic. Rapid fixation with fast penetrating crosslinking aldehydes can prevent changes by quickly blocking vital processes such as cell movement and autolysis (Baker, 1958; Dempster, 1960; Kiernan, 2000). Fixation has been shown to preserve structure to a large extent, both by single particle analysis and cryo-ET (Kastner et al., 2008; Ke et al., 2020; Mageswaran et al., 2021; Oda et al., 2020; Turoňová et al., 2020). While chemical fixation does not disrupt high resolution information, fixation does disrupt the physiological homeostasis of the cell, making the integrity of the cell morphology fully dependent on the buffer that is used to deliver the fixative.
RCIA shows clear differences for different buffer formulations.
We tested two formulations of the common PBS buffer along with a more complex PIPES-based PHEM buffer. PBS buffers owe their popularity in microscopy and cell biology applications to being well tolerated by tissues and, unlike Tris buffers, having good buffering abilities at low temperatures (Gillespie and McKnight, 1976; Mazur, 1970). The PIPES-based PHEM buffer is a more finely tuned formulation, characterized by a relatively high concentration of EGTA that was originally formulated to support the preservation of cytoskeletal components in fixed and detergent-extracted cells (Schliwa and van Blerkom, 1981; Schliwa et al., 1981). We determined that diluting the fixative in either 1x PBS (PBS without calcium and magnesium) or DPBS (PBS with calcium and magnesium) permitted the penetration and specific binding of antibodies despite the absence of detergents. Detergents in immunostaining protocols serve as membrane permeabilizing agents allowing diffusion of molecules such as antibodies into the cytoplasm (Melan, 1994). As shown in Figure 1, a mouse monoclonal antibody against a cytosolic epitope of the vesicular protein clathrin efficiently and specifically labeled the antigen when the sample was fixed in presence of PBS (Figure 1B) or DPBS (Figure 1D) in the absence of cell permeabilizing detergents. In contrast, by using our fixative dilution buffer in the preparation of the fixative (see Methods) and then PHEM in the absence of detergent during the staining, almost no labeling was seen in the sample (Figure 1F). When the DPBS or PBS buffers were modified by adding EGTA and used to deliver the fixative, labeling in the absence of detergents was still observable (Figure S1). A quantitative analysis of the clathrin signals reveals that the difference is highly significant (PBS vs PHEM p-value = 3.40 × 10−5; DBPS vs PHEM p-value = 7.16 × 10−5), while the difference between PBS and DPBS is not statistically significant (p-value = 0.382). Addition of detergent showed efficient labeling of clathrin vesicles with all three buffers (Figure 1A, C, E). The permissiveness of PBS was not limited to one antibody: a more discrete cytosolic antigen (the N-terminus of the Golgi protein Giantin, exposed to the cytosol) was specifically labeled by a rabbit polyclonal antibody in absence of detergent-induced permeabilization in most of the cell population (Figures 2A–B, S2) while no staining was observed when the PHEM condition was used during fixation (Figure 2C).
Figure 1. The buffer formulation affects the plasma membrane permeability.
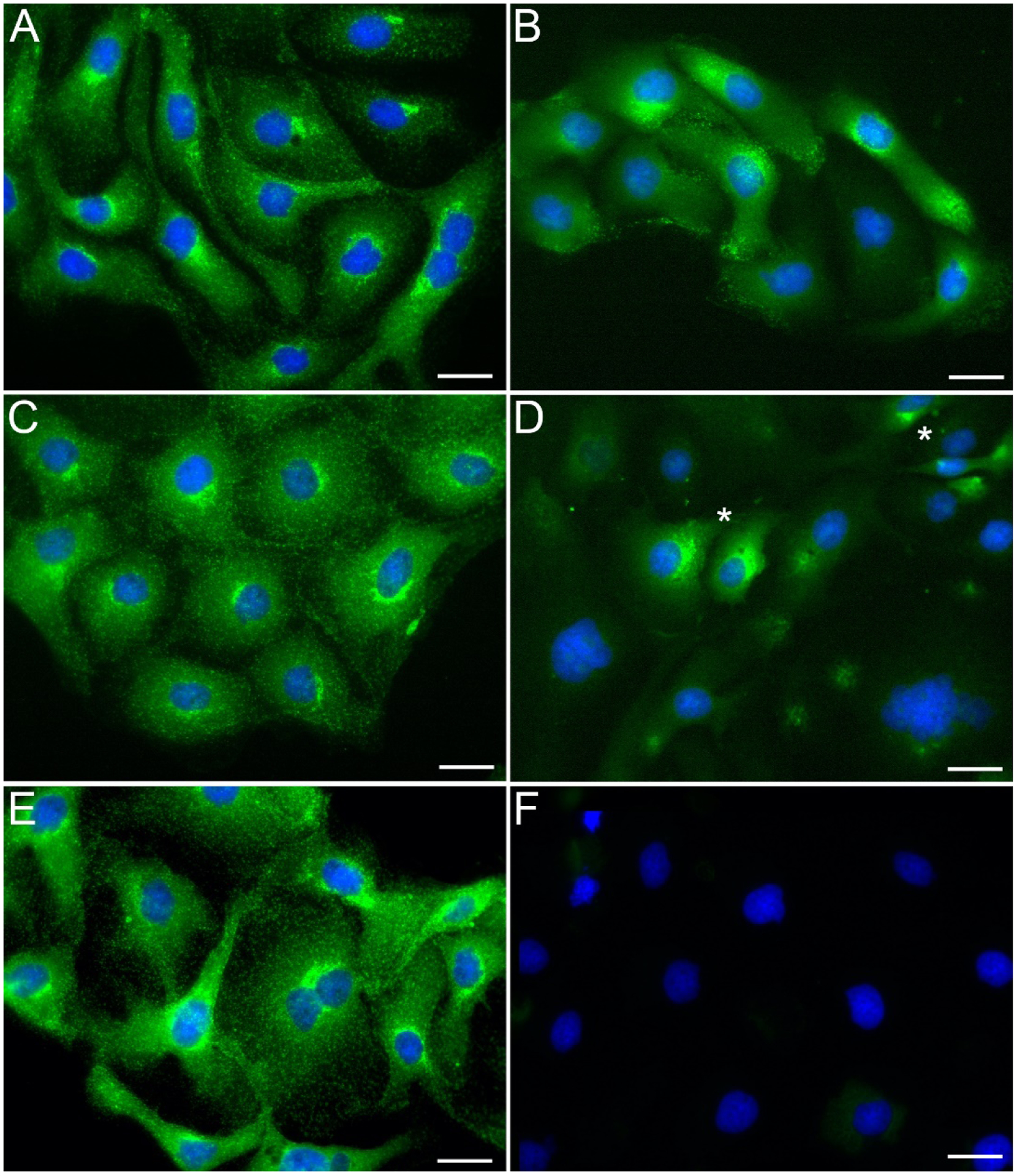
Following our RCIA protocol, we fixed PTK-1 cells in a 4% solution of paraformaldehyde prepared in PBS (A, B), DPBS (C, D) or PHEM buffer (E, F; see Methods for formulation) and then immunostained them in the respective buffer after treatment with detergent (0.1% Triton X-100, 5min before adding primary antibody; A, C, E) or without detergent added (B, D, F) using a mouse monoclonal antibody against the cytosolic epitope of clathrin, followed by a goat anti mouse, Alexa 488 (displayed in green). Cells were counterstained with DAPI after completion of the immunostaining (displayed in blue). All three buffers supported specific binding of the primary antibody when used in conjunction to Triton X-100 (A, C, E). When the detergent was omitted, specific binding to the antigen was observed in most cells in presence of PBS (B), and to a lesser extent in presence of DPBS, along with occasional polarized localization in few cells (D; asterisks); in presence of PHEM however, the staining was non-detectable (F). Scale bars, 10μm.
Figure 2. PBS renders the cells’ membrane permissive to antibody penetration in the absence of detergent-induced permeabilization.
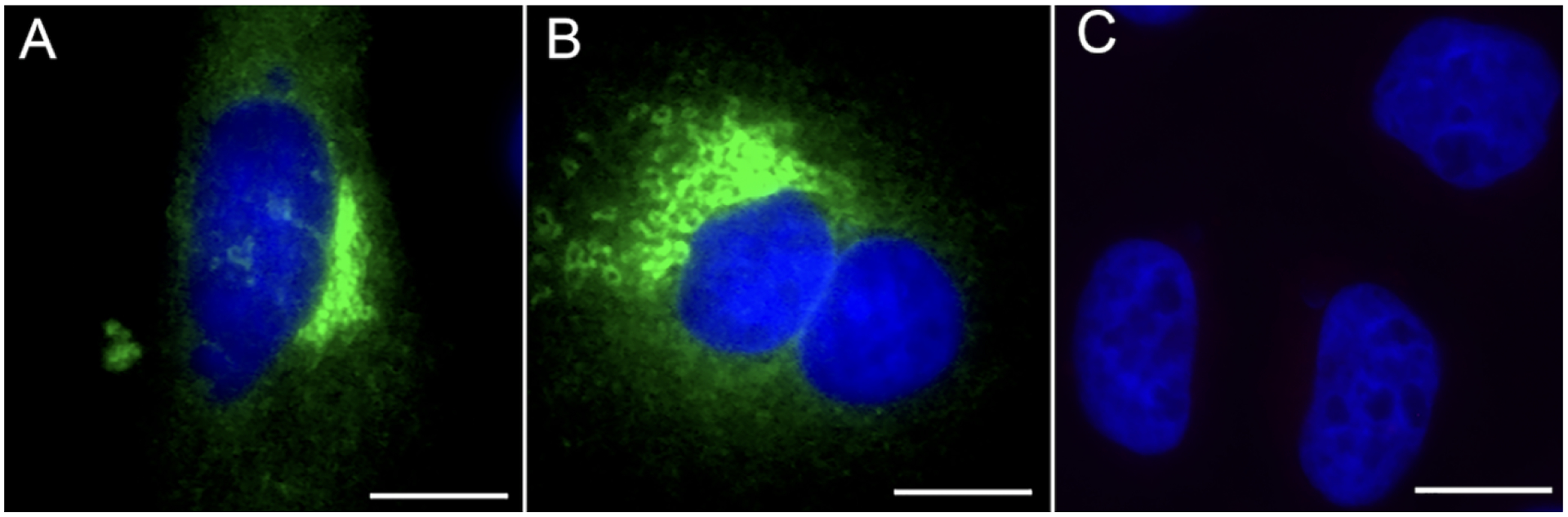
HeLa cells were either fixed in 4% paraformaldehyde diluted in PBS (A, B) or prepared with fixative dilution buffer and PHEM (C). Then, following our RCIA protocol, the samples were immunostained without detergent-induced permeabilization with a rabbit polyclonal antibody against the N terminus of the Golgi protein Giantin followed by a goat anti rabbit, Alexa488 conjugate secondary antibody (A, B; green stain in figure; A and B are two different examples of cells from the same kind of immunostaining performed in absence of detergent). No staining was observed in the PHEM condition (C). The nuclear stain DAPI (displayed in blue) was applied to demark cell locations. Scale bars, 5μm.
RCIA allows optimization of experimental protocols independent of cell types.
The cell type used in these experiments was of no relevance to the differential response to PBS, DPBS and PHEM: NIH 3T3 (Figure 1) and HeLa cells (Figure 2) immunostaining was similar in their response to the buffer formulation in the absence of detergent. Based on these immunostainings, we concluded that PHEM was superior in maintaining the plasma membrane integrity during and post fixation and was thereby preventing efficiently the diffusion of molecules such as antibodies into the cytoplasm. We then established that the fidelity of RCIA’s reporting correlates with nanoscale integrity by employing in situ cryo-ET of cells subjected to the three buffers in the step preceding vitrification. NIH 3T3 (Figure 3A–L), THP-1 (Figure 3M, N) and MEF cells (Figure 3O, P) were processed as described in the Materials and Methods section and used in these investigations. We chose and compared for each sample classic subcellular features such as plasma membrane, cytoskeletal filaments and internal membranous compartments. The three-dimensional cryo-ET reconstructions clearly showed that the lipid bilayer was disrupted in multiple areas in samples treated with PBS (Figure 3A) and DPBS (Figure 3B), leaving large gaps opened to the extracellular environment, where, at times, cellular material appeared to have spilled out (Figure 3A and B, white asterisk). Cells fixed in the presence of the dilution buffer (see Methods) and then maintained in PHEM for up to a week prior to vitrification, instead showcased an intact plasma membrane, often decorated with elements relatable to membrane receptors (Figure 3C and M, white arrows). Portions of the cytoplasm devoid of discernible macromolecules in the PBS condition (Figure 3D, white asterisks) were often visible in the samples: these voids are unusual in cell preparations and may point to a substantial deterioration of larger macromolecular assembly due to poor sample preservation. The DPBS condition did not display the same extent of regions devoid of discernible macromolecules but was still somewhat diminished in complexity (Figure 3E). In contrast, the cytoplasm in the PHEM condition was richly populated with subcellular components (Figure 3F and N) and clearly discernible macromolecular assemblies. While cytoskeletal elements such as actin filaments were visible in both phosphate buffers (Figure 3G and H, white arrows), their appearance lacked definition, with frequent shorter entities rather than the characteristic, long actin filaments found in the PHEM condition (Figure 3I and O, white arrows). Internal membranous compartments appeared similarly preserved in both phosphate buffers (Figure 3J and K, white arrows). The PHEM condition allowed us to identify comparable structures (Figure 3L, white arrows) along with well-preserved mitochondria (Figure 3P, white asterisk), a subcellular component notoriously difficult to maintain intact during microscopy preparations.
Figure 3. The composition of the buffer used affects the quality of preservation of the sample’s nanoarchitecture.
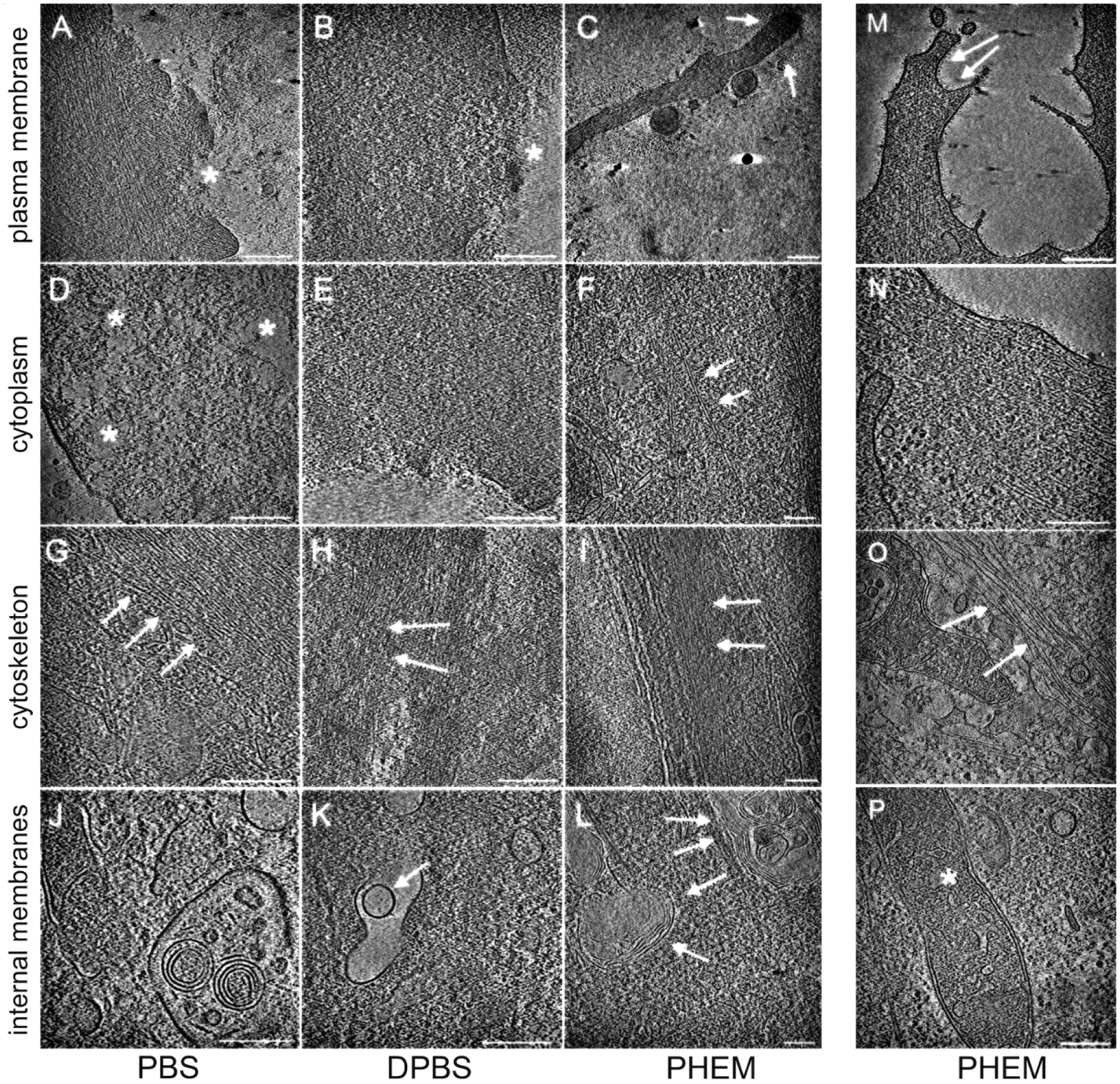
NIH 3T3 cells were grown on EMBRA finder grids, fixed and treated as described in PBS (A, D, G, J), DPBS (B, E, H, K), or PHEM (C, F, I, L) until vitrification. Following a similar preparation protocol, additional samples were generated for the PHEM condition, this time using THP-1 (M, N) and MEF (O, P) cells. Cryo-ET investigations focused on plasma membrane and neighboring areas (A, B, C, M; white asterisks in A and B point to damage to the plasma membrane and spillage of cellular matter into the extracellular environment; white arrows in C and M point to intact plasma membrane); cytoplasm (D, E, F, N; white asterisks in D mark areas void of discernible macromolecular assemblies; arrows in F point to actin filaments), areas rich in cytoskeletal elements (G, H, I, O; actin filaments, white arrows); areas with membranous compartment (white arrows in J, K and L) and mitochondria (white asterisk in P). Scale bars, 200 nm.
PHEM buffer optimizes RCIA fidelity.
Because damage can occur for different reasons during the sample preparation and the imaging process, some of which are not related to homeostasis, we quantified the extent of damage for PHEM versus PBS buffer. When PBS buffer is used, 67.5 (± 11.5)% of the tomograms show visible damage while only 23.8 (± 5.5)% of tomograms show visible damage when PHEM buffer was used. Thus, PBS leads to nearly three times as much visible damage than PHEM buffer. This difference is statistically highly significant (p-value = 9.95 × 10−4). The results correlate very well with the quantified differences in fluorescence intensity when these different buffers are used (Figure 4). As an additional and direct corroboration of RCIA reporting on membrane permeability in the presence of PBS and absence of detergent at high resolution, we used anti-paxillin antibodies followed by a secondary antibody conjugated to FluoroNanoGold, a fluorescent compound covalently bound to a 1.4-nm gold particle that, owing to its high density, is identifiable in cryo-ET volumes when clustered. Although no detergent was used throughout, we were able to observe FluoroNanoGold at locations with fluorescence 10 to 20 nm above the basal membrane within the cryo-ET volumes of PBS-treated cells (Figure 5), consistent with the specific height of paxillin above the basal membrane (about 11 ± 9 nm) at the end of protrusions as determined by super-resolution microscopy (Kanchanawong et al., 2010). Thus, if combined with PHEM buffer, the RCIA assay provides the means to detect degradation of structural integrity in a fast and reliable manner. A step-by-step protocol for the use of the RCIA/PHEM protocol is provided in the supplementary material.
Figure 4. Quantification of fluorescence intensities and visible damage as a function of buffer conditions.
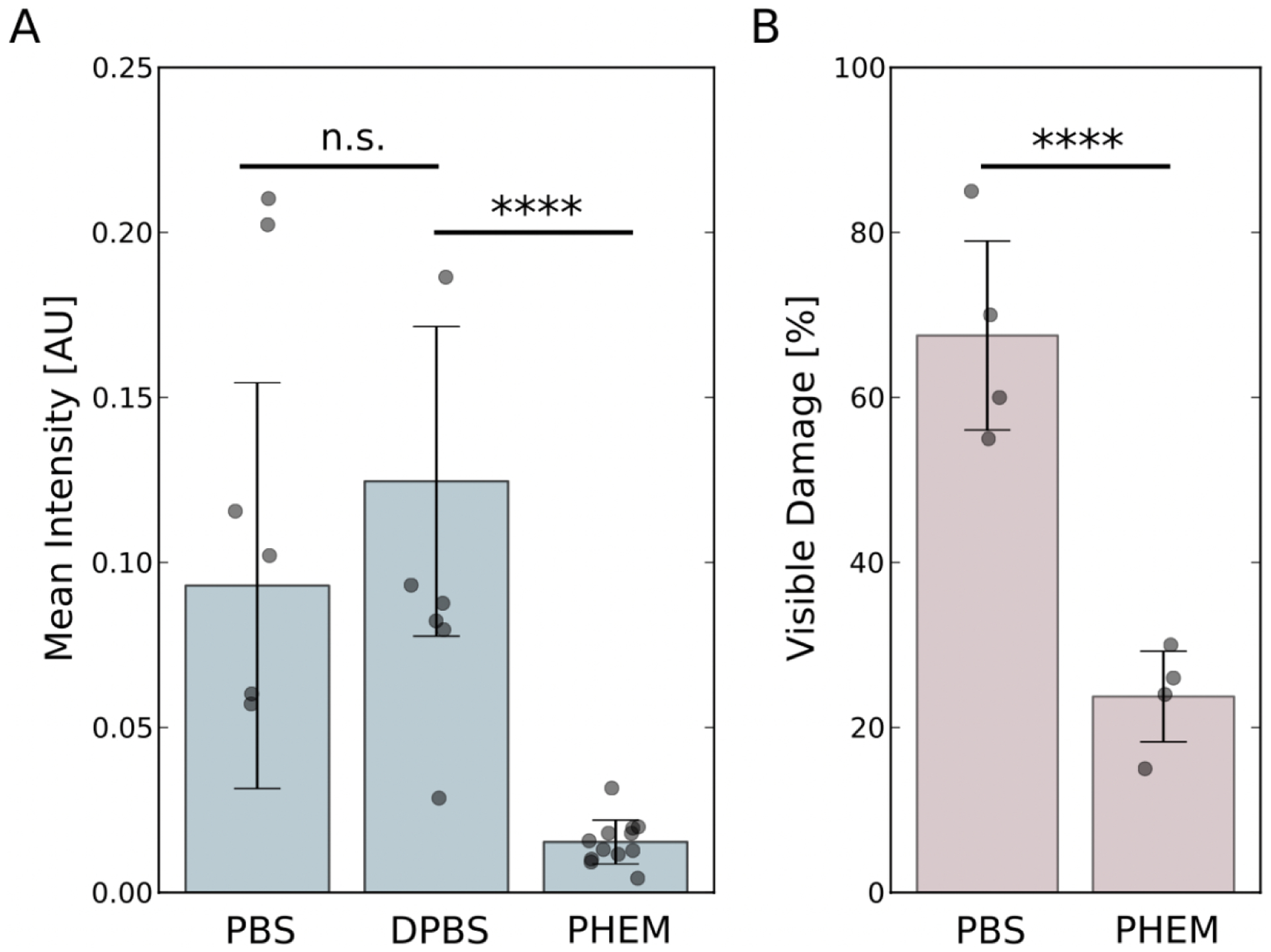
Comparisons were carried out using two-tailed t-tests. The dots show individual data points, and the bars indicate the mean of the distributions. The error bars represent one standard deviation. (A) Intensity distributions as a function of buffer conditions. There is no statistical difference between the PBS and DPBS conditions (p-value = 0.382) but the differences between PBS and PHEM (p-value = 3.40 × 10−5) as well as DPBS and PHEM buffers (p-value = 7.16 × 10−5) are highly significant. (B). Distribution of visible damage as a function of buffer conditions. The difference between PBS and PHEM buffer conditions is highly significant (p-value = 9.95 × 10−4).
Figure 5. FluoroNanogold antibody conjugates diffuse through the plasma membrane in absence of detergent-induced permeabilization.
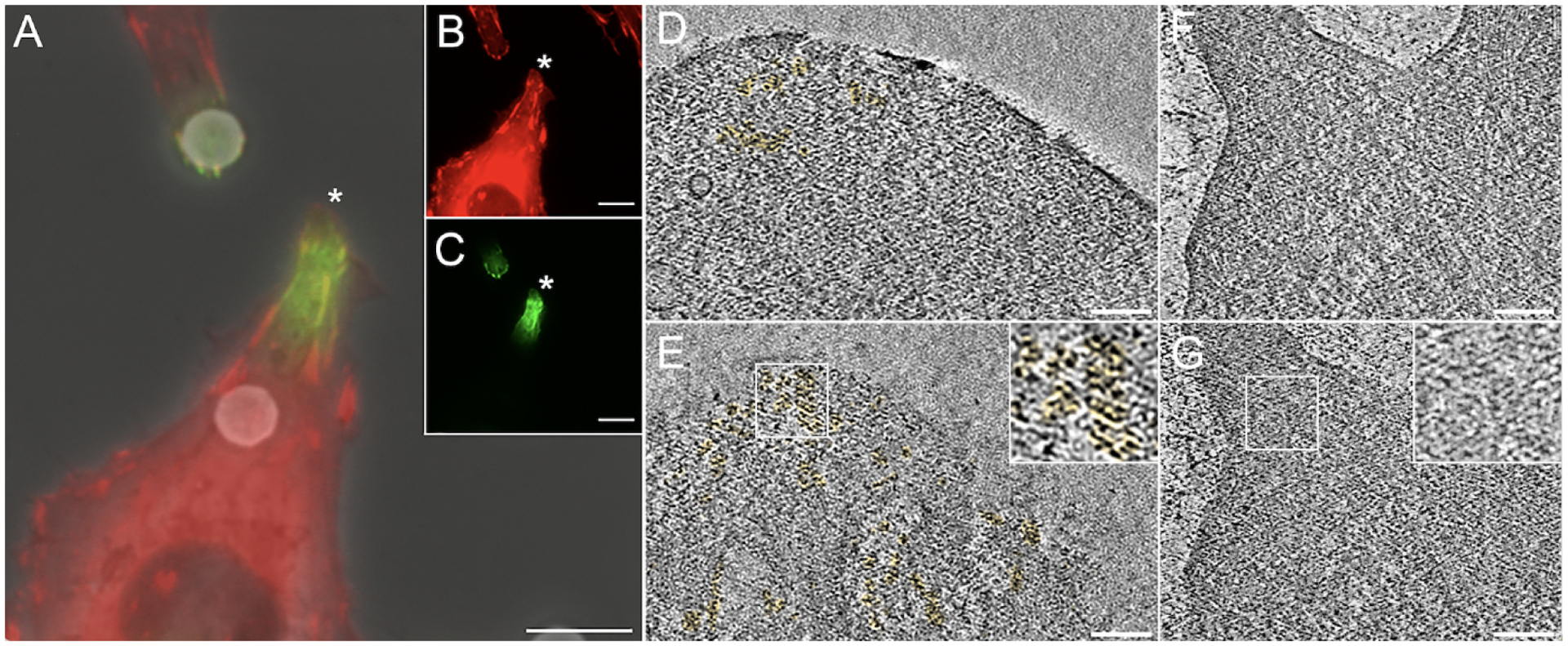
3T3 cells stably expressing mRuby-paxillin fixed with 4% paraformaldehyde diluted in PBS and immunostained with mouse anti-paxillin followed by goat anti-mouse, FluoroNanoGold. Although no detergent was used throughout, we detected a robust signal at the tip of cellular projections, where the mRuby-Paxillin transgene was congregating. The fluorescent images of the two channels were overlaid to the phase contrast image (A). The individual channels are displayed in the inset (B, mRuby paxillin; C, antibody staining). The area marked by the white asterisk was chosen for cryo-ET investigation. Although FluoroNanoGold is 1.4 nm wide and thereby difficult to detect directly, we were able to take advantage of paxillin concentrating at the tips and the known height distribution from the basal membrane of about 11 ± 9 nm (Kanchanawong et al., 2010) to identify areas with gold clusters. (D) single 2-nm thick slice from a tomogram of the area indicated by asterisk in C at a distance of 20 nm from the basal membrane, gold clusters are pseudo-colored in yellow. (E) as in D but at a distance of 10 nm from the basal membrane. (F) 2-nm thick slice from a tomogram of a control cell fixed with PHEM buffer and without nanogold/antibody staining. Distance from basal membrane is 20 nm. (G) same as F at a distance of 10 nm from the basal membrane. The insets in E and G show enlarged views of the areas indicated by the white boxes. Note the absence of high-density clusters in the control cell. Scale bars, 5μm (A-C); 100 nm (D-G).
RCIA is applicable to a wide variety of methods relying on 3D cellular integrity.
While our optimized RCIA screening assay itself inherently includes the use of PHEM buffer and fixative, it serves only as a reporter for the intracellular structural integrity of the samples studied. The RCIA assay is performed using glass coverslips and not the cryo-EM grids themselves, so instead of fixing the cells on the cryo-EM grid, the grid can be plunge-frozen while only the cells on the coverslips from the same sample are processed for RCIA. For example, if one wants to evaluate the structural consequences of drug treatment at high resolution in unfixed cells, the RCIA/PHEM protocol can be used to determine whether the drug regiment adversely affects the structural integrity of the treated cells using glass coverslips only and refine the treatment until structural damage is minimized. Once the drug treatment has been optimized this way, one would commence the analysis focused on the functional aspects of the drug treatment using cryo-ET (or other high-resolution techniques) of unfixed cells and in parallel run the RCIA/PHEM protocol with fixed cells from the same sample on glass coverslips. The RCIA results report if the high-resolution structural integrity of the cell preparation imaged by cryo-ET was compromised or not. It should be noted that the RCIA protocol can in principle also be applied directly to live cells without fixation because antibody staining in the presence of membrane damage in live cells (for example by electroporation) has been demonstrated (Conic et al., 2018).
Note that the information provided by RCIA is complementary to more traditional cell viability tests that probe membrane integrity such as dye exclusion assays (Bowman et al., 2010; Strober, 2001). While these types of tests have proven useful for assessments of necrotic and apoptotic cell death with membrane damage, interpretation of results in terms of different types of membrane damage is not straightforward (Ainla et al., 2012; Kim et al., 2009; Šatkauskas S. Jakštys B., 2016) and none of these exclusion methods has been shown to report on the intracellular high-resolution structural integrity of the tested cells in terms of cytoskeletal components and other cytosolic elements. Conventional antibody labeling protocols require the presence of detergent, which disrupts membranes and facilitates lysis of cells, allowing the relatively large antibodies (10-nm range) to cross the membrane and to freely diffuse to their destination. RCIA is conducted in the absence of detergent. Thus, the ability of the antibodies to enter and to diffuse within the cytosol is related to membrane and intracellular damage. Because of the inverse relationship between particle size and diffusion coefficient (Nenninger et al. 2010), antibodies are more restricted in diffusion than the much smaller dyes if the dense cytosolic components are fully intact. Thus, their signal within the RCIA protocol is more closely reporting intracellular damage than dyes would. In addition, the fact that antibodies can easily be labeled with electron dense probes directly identifiable in cryo-ET allows direct verification and validation by cryo-ET. RCIA can be performed with any cell culture promoting substrates. In fact, the assay can be used to test the effect of more complex substrates, such as the silicone pillar arrays used for certain types of traction force microscopy, on the 3D integrity by comparing RCIA results using these substrates with RCIA results from more standard cell culture substrates like glass coverslips. In the case of cryo-ET, carbon-coated glass coverslips can be used for the RCIA screening step as these unique substates in our hand emulate the cell culture conditions on cryo-EM grid substrates well and the RCIA screening step does not require the costly cryo-EM substrates to be present.
CONCLUSIONS
The RCIA assay facilitates and expedites screening conditions to ensure high-resolution intracellular integrity in the context of techniques where the interpretation of results depends on an intact intracellular environment including cellular cryo-ET, water-window X-ray microscopy, cellular thermal shift assays, in-cell nuclear magnetic resonance spectroscopy, and cellular super-resolution light microscopy. The RCIA assay provides the means to quickly identify perturbations of high-resolution structural integrity of cells caused by reagents, experimental setup, biophysical manipulations, virus inactivation protocols, or drug treatments, increasing the reliability and reproducibility of imaging and biophysical assays that involve cell preparations.
Supplementary Material
HIGHLIGHTS.
We present a rapid, accessible, inexpensive, and reliable light microscopy-based screening assay that quickly reports on the intracellular high-resolution structural integrity of whole-cell preparations.
This protocol will benefit all whole-cell preparation studies where the validity of conclusions relies on retaining the three-dimensional integrity of the intracellular nano-environment.
The Rapid Cell Integrity Assessment (RCIA) screening assay can be used to test the effects on structural integrity for any type of cell perturbations including drug treatments, virus inactivation, fixation protocols, and biophysical manipulations.
RCIA reporting of high-resolution integrity was verified by cellular cryogenic tomography.
ACKNOWLEDGEMENTS
We are grateful to M.H. Ginsberg and F. Lagarrigue, UCSD, and M. Schwartz and A. Kumar, Yale, for contributing some of the cell lines. This work was supported by National Institutes of Health grants R01 AI132378 (NV, DH) and P01 GM121203 (NV). NIH grant S10-OD012372 (DH) and PEW innovation funds 864K625 (DH) funded the purchase of the Titan Krios electron cryo-microscope (ThermoFisher Scientific), Falcon direct detector, and Eagle CMOS imaging device (ThermoFisher Scientific). The authors declare no competing financial interests.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
ABBREVIATIONS
RCIA: Rapid Cell Integrity Assessment; cryo-ET: cryogenic electron tomography; cryo-EM: cryogenic electron microscopy; LM: light microscopy; 3D: three-dimensional; cryo-CLEM: cryogenic correlative light and electron microscopy; NIH 3T3: mouse fibroblast cell line “3-day transfer, inoculum 3×105 cells”; HeLa: human cervix carcinoma cell line (origin: Henrietta Lacks); THP1: human monocytic cell line TIB202™; MEF: Mouse Embryonic Fibroblasts; EM: Electron Microscopy; PMA: Phorbol 12-Myristate 13-Acetate; PHEM: Pipes Hepes EGTA Magnesium; DAPI: 4’,6-diamidino-2-phenylindole; CCD: Charge-Coupled Device; PBS: Phosphate Buffered Saline; DPBS: Dulbecco’s formula Phosphate-Buffered Saline, with Calcium and Magnesium; Tris: tris(hydroxymethyl)aminomethane.
REFERENCES
- Agulleiro JI, Fernandez JJ, 2015. Tomo3D 2.0--exploitation of advanced vector extensions (AVX) for 3D reconstruction. J Struct Biol 189, 147–152. [DOI] [PubMed] [Google Scholar]
- Ainla A, Xu S, Sanchez N, Jeffries GD, Jesorka A, 2012. Single-cell electroporation using a multifunctional pipette. Lab Chip 12, 4605–4609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson KL, Page C, Swift MF, Hanein D, Volkmann N, 2018. Marker-free method for accurate alignment between correlated light, cryo-light, and electron cryo-microscopy data using sample support features. J Struct Biol 201, 46–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson KL, Page C, Swift MF, Suraneni P, Janssen MEW, Pollard TD, Li R, Volkmann N, Hanein D, 2017. Nano-scale actin-network characterization of fibroblast cells lacking functional Arp2/3 complex. J Struct Biol 197, 312–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker JR, 1958. Principles of biological microtechnique; a study of fixation and dyeing. Methuen, Wiley, London, New York. [Google Scholar]
- Bowman AM, Nesin OM, Pakhomova ON, Pakhomov AG, 2010. Analysis of plasma membrane integrity by fluorescent detection of Tl(+) uptake. J Membr Biol 236, 15–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conic S, Desplancq D, Tora L, Weiss E, 2018. Electroporation of Labeled Antibodies to Visualize Endogenous Proteins and Posttranslational Modifications in Living Metazoan Cell Types. Bio Protoc 8, 10.21769/BioProtoc.3069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dempster WT, 1960. Rates of penetration of fixing fluids. Am J Anat 107, 59–72. [DOI] [PubMed] [Google Scholar]
- Dubochet J, 2012. Cryo-EM--the first thirty years. J Microsc 245, 221–224. [DOI] [PubMed] [Google Scholar]
- Gillespie JS, McKnight AT, 1976. Adverse effects of tris hydrochloride, a commonly used buffer in physiological media. J Physiol 259, 561–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanein D, Volkmann N, 2011. Correlative light-electron microscopy. Adv Protein Chem Struct Biol 82, 91–99. [DOI] [PubMed] [Google Scholar]
- Kanchanawong P, Shtengel G, Pasapera AM, Ramko EB, Davidson MW, Hess HF, Waterman CM, 2010. Nanoscale architecture of integrin-based cell adhesions. Nature 468, 580–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastner B, Fischer N, Golas MM, Sander B, Dube P, Boehringer D, Hartmuth K, Deckert J, Hauer F, Wolf E, Uchtenhagen H, Urlaub H, Herzog F, Peters JM, Poerschke D, Lührmann R, Stark H, 2008. GraFix: sample preparation for single-particle electron cryomicroscopy. Nat Methods 5, 53–55. [DOI] [PubMed] [Google Scholar]
- Ke Z, Oton J, Qu K, Cortese M, Zila V, McKeane L, Nakane T, Zivanov J, Neufeldt CJ, Cerikan B, Lu JM, Peukes J, Xiong X, Kräusslich HG, Scheres SHW, Bartenschlager R, Briggs JAG, 2020. Structures and distributions of SARS-CoV-2 spike proteins on intact virions. Nature 588, 498–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiernan JA, 2000. Formaldehyde, Formalin, Paraformaldehyde And Glutaraldehyde: What They Are And What They Do. Microscopy Today 8, 8–13. [Google Scholar]
- Kim H, Yoon SC, Lee TY, Jeong D, 2009. Discriminative cytotoxicity assessment based on various cellular damages. Toxicol Lett 184, 13–17. [DOI] [PubMed] [Google Scholar]
- Klein S, Cortese M, Winter SL, Wachsmuth-Melm M, Neufeldt CJ, Cerikan B, Stanifer ML, Boulant S, Bartenschlager R, Chlanda P, 2020. SARS-CoV-2 structure and replication characterized by in situ cryo-electron tomography. Nat Commun 11, 5885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremer JR, Mastronarde DN, McIntosh JR, 1996. Computer visualization of three-dimensional image data using IMOD. J Struct Biol 116, 71–76. [DOI] [PubMed] [Google Scholar]
- Kumar A, Anderson KL, Swift MF, Hanein D, Volkmann N, Schwartz MA, 2018. Local Tension on Talin in Focal Adhesions Correlates with F-Actin Alignment at the Nanometer Scale. Biophys J 115, 1569–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mageswaran SK, Yang WY, Chakrabarty Y, Oikonomou CM, Jensen GJ, 2021. A cryo-electron tomography workflow reveals protrusion-mediated shedding on injured plasma membrane. Sci Adv 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marston DJ, Anderson KL, Swift MF, Rougie M, Page C, Hahn KM, Volkmann N, Hanein D, 2019. High Rac1 activity is functionally translated into cytosolic structures with unique nanoscale cytoskeletal architecture. Proc Natl Acad Sci U S A 116, 1267–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastronarde DN, 2005. Automated electron microscope tomography using robust prediction of specimen movements. J Struct Biol 152, 36–51. [DOI] [PubMed] [Google Scholar]
- Mazur P, 1970. Cryobiology: the freezing of biological systems. Science 168, 939–949. [DOI] [PubMed] [Google Scholar]
- Melan MA, 1994. Overview of cell fixation and permeabilization. Methods Mol Biol 34, 55–66. [DOI] [PubMed] [Google Scholar]
- Nenninger A, Mastroianni G, Mullineaux CW, 2010. Size dependence of protein diffusion in the cytoplasm of Escherichia coli. J Bacteriol 192, 4535–4540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda T, Yanagisawa H, Wakabayashi T, 2020. Cryo-EM structures of cardiac thin filaments reveal the 3D architecture of troponin. J Struct Biol 209, 107450. [DOI] [PubMed] [Google Scholar]
- Ognjenović J, Grisshammer R, Subramaniam S, 2019. Frontiers in Cryo Electron Microscopy of Complex Macromolecular Assemblies. Annu Rev Biomed Eng 21, 395–415. [DOI] [PubMed] [Google Scholar]
- Sartori A, Gatz R, Beck F, Rigort A, Baumeister W, Plitzko JM, 2007. Correlative microscopy: bridging the gap between fluorescence light microscopy and cryo-electron tomography. J Struct Biol 160, 135–145. [DOI] [PubMed] [Google Scholar]
- Šatkauskas S Jakštys B, R.P., Jakutavičiūtė M, 2016. Different Cell Viability Assays Following Electroporation In Vitro. Springer International [Google Scholar]
- Schliwa M, van Blerkom J, 1981. Structural interaction of cytoskeletal components. J Cell Biol 90, 222–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schliwa M, van Blerkom J, Porter KR, 1981. Stabilization and the cytoplasmic ground substance in detergent-opened cells and a structural and biochemical analysis of its composition. Proc Natl Acad Sci U S A 78, 4329–4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strober W, 2001. Trypan blue exclusion test of cell viability. Curr Protoc Immunol Appendix 3, Appendix 3B. [DOI] [PubMed] [Google Scholar]
- Tegunov D, Cramer P, 2019. Real-time cryo-electron microscopy data preprocessing with Warp. Nat Methods 16, 1146–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tegunov D, Xue L, Dienemann C, Cramer P, Mahamid J, 2021. Multi-particle cryo-EM refinement with M visualizes ribosome-antibiotic complex at 3.5 Å in cells. Nat Methods 18, 186–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turk M, Baumeister W, 2020. The promise and the challenges of cryo-electron tomography. FEBS Lett. [DOI] [PubMed] [Google Scholar]
- Turoňová B, Sikora M, Schürmann C, Hagen WJH, Welsch S, Blanc FEC, von Bülow S, Gecht M, Bagola K, Hörner C, van Zandbergen G, Landry J, de Azevedo NTD, Mosalaganti S, Schwarz A, Covino R, Mühlebach MD, Hummer G, Krijnse Locker J, Beck M, 2020. In situ structural analysis of SARS-CoV-2 spike reveals flexibility mediated by three hinges. Science 370, 203–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Drie JH, Tong L, 2020. Cryo-EM as a powerful tool for drug discovery. Bioorg Med Chem Lett 30, 127524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkmann N, Hanein D, 1999. Quantitative fitting of atomic models into observed densities derived by electron microscopy. J Struct Biol 125, 176–184. [DOI] [PubMed] [Google Scholar]
- Wolff G, Limpens R, Zevenhoven-Dobbe JC, Laugks U, Zheng S, de Jong AWM, Koning RI, Agard DA, Grünewald K, Koster AJ, Snijder EJ, Bárcena M, 2020. A molecular pore spans the double membrane of the coronavirus replication organelle. Science 369, 1395–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.