

Abstract
Clinical trials of Delta-like 4 (Dll4) neutralizing antibodies (Dll4nAbs) in cancer patients are ongoing. Surprisingly, pulmonary hypertension (PH) occurs in 14-18% of patients treated with Dll4nAbs, but the mechanisms have not been studied. Here, PH progression was measured in mice treated with Dll4nAbs. We detected Notch signaling in lung tissues and analyzed pulmonary vascular permeability and inflammation. Notch target gene array was performed on adult human pulmonary microvascular endothelial cells (HPMECs) after inhibiting Notch cleavage. Similar mechanisms were studied in PH mouse models and PAH patients. The rescue effects of constitutively activated Notch1 in vivo were also measured. We observed that Dll4nAbs induced PH in mice as indicated by significantly increased right ventricular systolic pressure as well as pulmonary vascular and right ventricular remodeling. Mechanistically, Dll4nAbs inhibited Notch1 cleavage and subsequently impaired lung endothelial barrier function and increased immune cell infiltration in vessel walls. In vitro, Notch targeted genes’ expression related to cell growth and inflammation was decreased in HPMECs after the Notch1 inactivation. In lungs of PH mouse models and PAH patients, Notch1 cleavage was inhibited. Consistently, EC cell-cell junction was leaky, and immune cell infiltration increased in PH mouse models. Overexpression activated Notch1 attenuated the progression of PH in mice. In conclusion, Dll4nAbs led to PH development in mice by impaired EC barrier function and increased immune cell infiltration through inhibition of Notch1 cleavage in lung ECs. Reduced Notch1 cleavage in lung ECs could be an underlying mechanism of PH pathogenesis.
Keywords: Dll4 neutralizing antibody, pulmonary arterial hypertension, Dll4-Notch1 signaling, endothelial permeability, inflammation
Summary
Consistently and robustly reduced Notch1 cleavage in pulmonary endothelial cells is a novel molecular mechanism of PH pathogenesis.
Introduction
Cancer remains the second-leading cause of death in the United States. The 5-year relative survival rate for cancer patients has increased from 49% in 1975 through 1977, to 69% in 2008 through 20141. Improvement in survival reflects advances in chemotherapeutic cancer treatments; subsequent adverse effects on the cardiovascular system drives mortality in most cancer survivors 2. Investigating the long-term effects of cancer drugs, such as cardiovascular diseases, is essential for improving cancer patients’ life span and quality of life.
Targeting tumor angiogenesis has become a prominent strategy in cancer therapy. The Delta-like 4 (Dll4)-Notch pathway is a well-recognized mechanism of sprouting angiogenesis. The Notch family consists of highly conserved trans-membrane proteins, including ligands (Dll1, Dll4, Jag1) and receptors (Notch1-4). In the canonical signaling pathway, a ligand binds to the Notch receptor to initiate a series of cleavages. The first cleavage by ADAM10/TACE generates the Notch transmembrane domain (NTM), which consists of a short extracellular juxtamembrane peptide, a transmembrane domain (TM), and an intracellular domain (N-ICD). The second cleavage byγ-secretase releases the N-ICD, which translocates to the nucleus and binds to the recombination signal binding protein for immunoglobulin kappa J region (RBP-J, the transcriptional regulator of Notch signaling) and its coactivator MAM. This triggers the expression of target genes such as Hey and Hes3. Dll4-Notch signaling is upregulated in various human tumors4, 5. Inhibiting this pathway leads to an increase of non-functional sprouting angiogenesis and reduced tumor growth in human xenograft mouse models 6, 7.
Based on these findings, several pharmaceutical companies have developed Dll4 neutralizing antibodies (Dll4nAbs) as a cancer therapy to inhibit Notch activation and thus tumor growth. In the Phase I or II clinical trials, Dll4nAbs have been used as a single agent or in combination with chemotherapy for patients with solid tumors, including breast, colon, ovarian, non-small cell lung and pancreatic cancer7–13. These results indicate Dll4nAbs as a promising therapeutic strategy for cancer in the future8, 14.
Increasing reports have raised concerns about the side effects of Dll4nAb, such as the occurrence of pulmonary hypertension (PH). PH is a progressive disease characterized by vasoconstriction, cell proliferation, and inflammation which leads to elevated pulmonary arterial pressure, often causing right heart failure and death15. Multiple phase I clinical trials demonstrated increased incidence of PH in Dll4nAb treated cancer patients9–13. These data suggest that prolonged high dose treatment of Dll4nAb may cause PH. Dll4 is a membrane bound Notch ligand restricted to endothelial cells (ECs) of small vessels in adult human and mouse lungs (LGEA web portal). The lung is the organ with the highest Dll4 expression (PubMed database, link), implying an essential role in lung EC function and pulmonary vascular diseases. We hypothesize that inactivation of Dll4-Notch1 signaling by Dll4nAbs in lung ECs will lead to the development of PH.
We demonstrate that mice treated with Dll4nAb developed PH by specifically inhibiting Notch1 cleavage in lung ECs. Given that reducing Notch1 cleavage could be a common mechanism of PH pathogenesis, we generated PH mouse models and observed decreased Notch1 cleavage in lungs of PH mice and confirmed evidence for decreased Notch1 cleavage in PAH patients. In addition, overexpressing N1-ICD in lung ECs in vivo using Adeno-Associated Virus (AAV) attenuated the progression of PH in mice.
Methods
The data that support the findings of this study are available from the corresponding author upon reasonable request. Besides sections below, other methods paragraphs are available in the Data Supplement.
Human subjects
Paraffin-embedded lung sections from patients with PAH at lung transplant and control samples from unused donor lungs were provided by Dr. R. James White in the Department of Pulmonary and Critical Care Medicine. Normal lung tissue slides were purchased from Novus Biologicals (NBP2-30182). Informed consent to use the tissue for research purposes was previously obtained. Samples were deidentified. Lungs from both patients with idiopathic PAH (5 subjects) and healthy donors (unused donor lungs without evidence for pulmonary disease from 6 individuals) were studied.
Animal models
All experimental procedures involving animals were in accordance with the guidelines of the National Institutes of Health and American Heart Association for the care and use of laboratory animals and approved by the University of Rochester Committee on Animal Resources and Tongji University School of Medicine. For Dll4nAb injections, we divided 8-week-old male C57 mice into two groups, each containing 20 mice, maintained under normoxic conditions. C57 mice were injected with Dll4nAb (anti-mouse Dll4 antibody generated in Armenian Hamster, HMD4-2) or Armenian Hamster IgG once/week (BioXCell, intraperitoneal injection, i.p.,10μg/g body weight) for 1-4 weeks. At the endpoint, Dll4nAb and placebo carrier-treated mice were euthanized for hemodynamic measurements and gene expression analysis.
For PH animal models, we generated two PH mouse models (C57, 8-12w): sugen+hypoxia, SuHx (SU5416 was subcutaneously injected once/week at 20mg/kg for 3w in conjunction with Hx exposure) and chronic hypoxia, Hx (chronic Hx for 5w). Mice were exposed to either room air (Normoxia, Nx) or chronic normobaric hypoxia in a ventilated plexiglass chamber in which nitrogen was injected to maintain O2 concentration of 10%. CO2 was monitored and ventilation adjusted so as to not exceed 0.5%. Ammonia was removed by ventilation and activated charcoal filtration through an air purifier. PH mouse models were humanely euthanized, and tissues were harvested for western blot, qRT-PCR16, 17, and immunofluorescence staining (IF). DAPT (N-[N-(3,5-difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl ester, a γ-secretase inhibitor (GSI) Millipore, dissolved in ethanol), or vehicle alone (ethanol) were administered subcutaneously at 25 mg/kg per pup at P3 and P4. Pups were humanely euthanized twenty-four hours after DAPT injection (at P5), and lung tissues were harvested for western blot. We only used male mice to avoid variable PH responses often observed in experimental females.
Isolation and en face staining of mouse small pulmonary arteries
We isolated approximately 100-μm-diameter small pulmonary arteries from Hx, Dll4nAb, SuHx and control mice. We harvested whole lungs (without lung perfusion) and submerged them in 4% PFA on ice for 2-3 mins. The fat and connective tissue around and below the small pulmonary artery was gently removed using scissors and forceps under a dissecting microscope. The arteries were separated and split longitudinally to expose the endothelium. Each vessel was fixed with 4% PFA for 10 mins on ice, then washed with PBS for 5 mins. Vessels were immediately double IF stained with cleaved Notch1 and VE-Cadherin, with the entire vascular wall imaged by confocal microscopy.
The administration of AAV-N1-ICD and AAV control in PH mouse models
AAV-N1-ICD and AAV control were generated by VectorBuilder, which include the endothelial-specific AAV2-QuadYF vectors18 that encoded an intracellular fragment of mouse Notch1 (1749–2293aa, lacking the C-terminal PEST domain)19, a T2A self-cleaving peptide sequence, and enhanced GFP (EGFP) under the control of endothelial-specific Tie1 gene promoter (AAV2-QuadYF-mN1ICD-EGFP) or control vectors carrying the CMV promoter and EGFP (AAV2-QuadYF-EGFP). Mice (C57, 6-8w) were anesthetized and injected retro-orbitally at a dose of 2 × 1012 vg/mice in a total volume of 100μl. Two weeks following AAV vectors injection, mice were exposed to either room air or chronic normobaric hypoxia chambers for 5 weeks. At the endpoint, mice were euthanized for hemodynamic measurements and gene expression analysis.
Statistical analysis
All values are expressed as mean ± SEM from three to eight samples. Data was assessed using the Student’s t test, and p < 0.05 was considered statistically significant. The data distribution was analyzed using SPSS software. Because the sample size is between three and eight, we used the Shapiro-Wilk test.
Results
Dll4nAb induces PH in mice
To investigate the role of Dll4nAb in the development of PH, mice were injected with Dll4nAb or IgG control intraperitoneally (i.p., 10μg/g) once/week for up to 4 weeks. This dose was the lowest dose still effective to inhibit Notch signaling as used in other mouse studies 20–22. The occurrence of PH in 4-week Dll4nAb treated mice compared with IgG control mice was demonstrated by a significant increase of RV systolic pressure from 23.0 ± 1.1 to 31.1 ± 1.7 mmHg (Figure 1A), an increase of RV remodeling (Fulton index, Figure 1B) from 0.21 ± 0.01 to 0.27 ± 0.01, and selective, substantial increased muscularization of smaller, distal (<40μm in diameter, ~14-fold) pulmonary arteries (Figure 1C–D). These results indicate that Dll4nAb caused the development of PH in mice.
Figure 1. Dll4nAb treatment induces PH in mice.
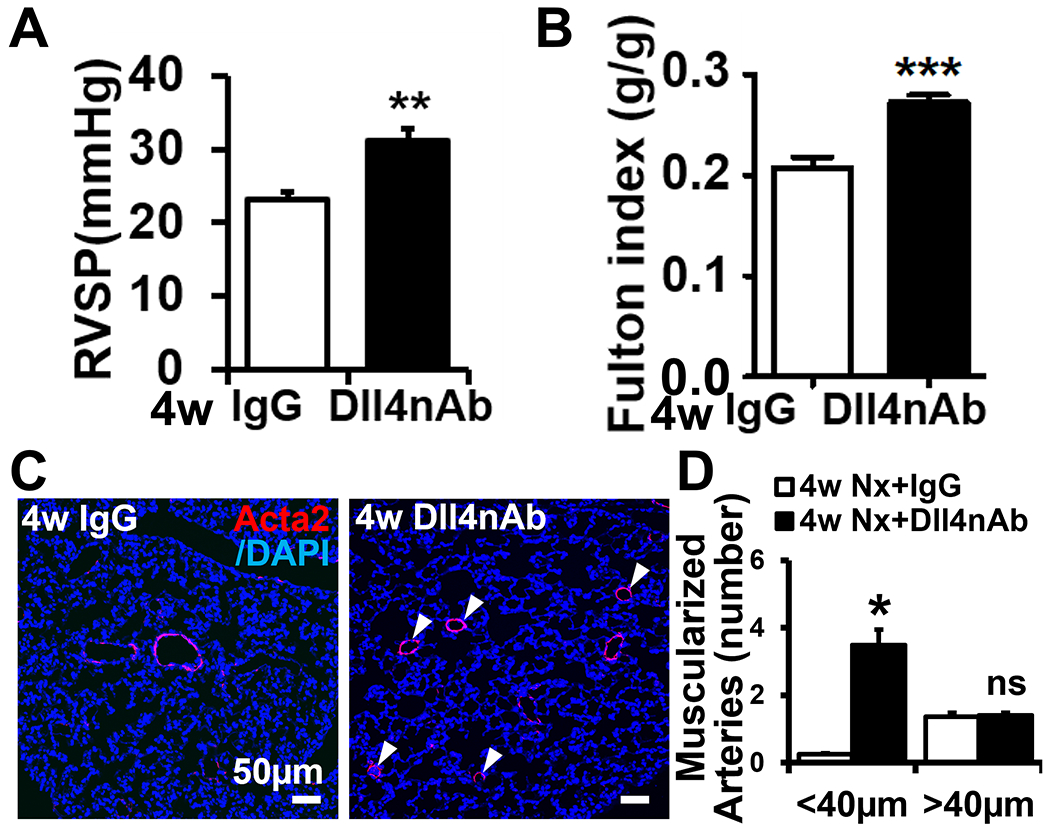
(A-B) RVSP (A) and Fulton index (B) in Dll4nAb and control IgG injected mice (n=15). (C-D) Increased muscularization in Dll4nAb injected mice compared with control IgG mice. (C) Representative micrographs of immunofluorescence staining with anti-α-smooth muscle-actin (Acta2) (red) and (D) quantification of muscularized pulmonary arterial vessels. Arrows, muscularized distal pulmonary arterial vessels. Scale bars: 50μm. Nuclei were counterstained by DAPI (blue). Data shown as Mean±SEM; P values were calculated using the Student’s t-test, and *P<0.05; **P<0.01; ***, P<0.001 compared with control group. ns, not significant.
Dll4nAb specifically inhibits Notch1 signaling in mice
Notch signaling changes in lung after Dll4nAb treatment had not been investigated. In mice treated with Dll4nAb, we found significant decrease of N1-ICD protein (cleaved Notch1 intracellular domain) after 1w (Figure S1A–B in the online-only Data Supplement) and 4w. Compared to IgG control, Notch2-4 ICD proteins in lungs were not changed (Figure 2A–B) and the expression of full length Notch1-4 protein was comparable (quantified data not shown). Protein expression of Hes1, one of the highly conserved transcription factors that are direct Notch target genes, was decreased in the lungs of Dll4nAb treated mice (Figure 2A–B and Figure S1A–B). To confirm the decrease of N1-ICD expression in lung ECs after Dll4nAb injection, we performed IF staining of N1-ICD on mouse lung tissues and co-stained with ETS-related gene (ERG, an EC-specific nuclear marker). In IgG control mouse lungs, N1-ICD colocalized with ERG-positive ECs (Figure 2C). In contrast, N1-ICD expression in ECs of Dll4nAb injected mice was barely detectable. Therefore, Dll4nAb mainly decreases EC N1-ICD expression in lung tissues.
Figure 2. Decrease of N1-ICD expression by 4w Dll4nAb treatment in mice lung tissues and inhibition of Notch1 cleavage induces Notch1 target genes dysregulation in ECs.
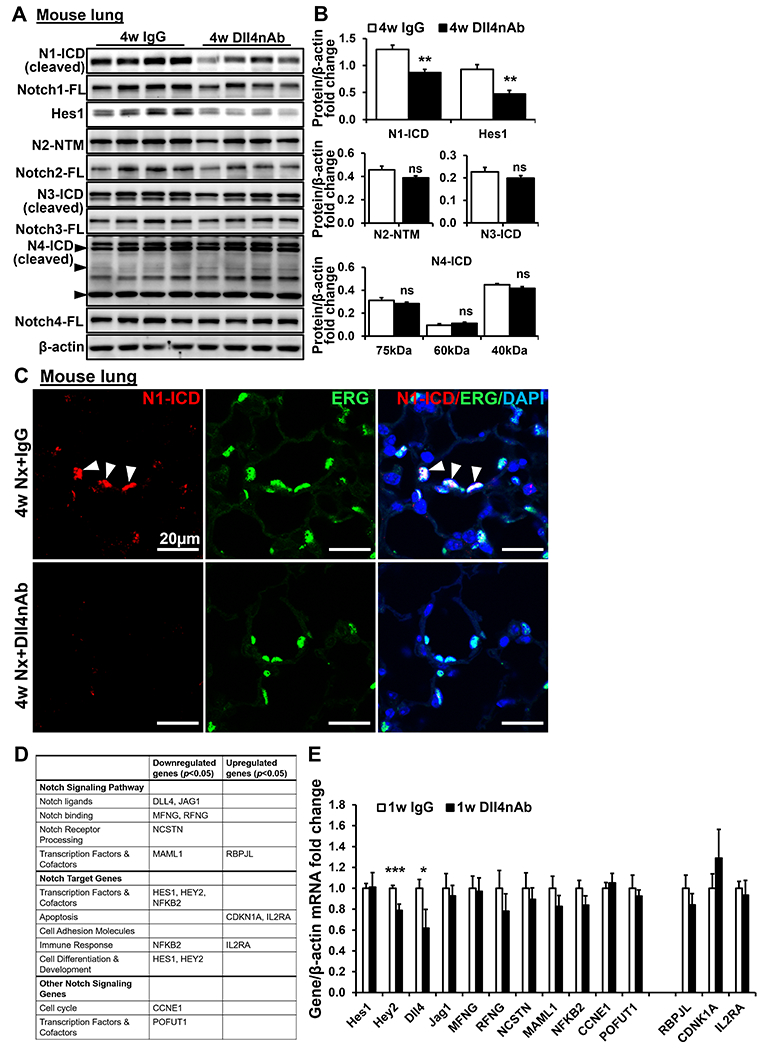
(A-B) N1-ICD, Notch1, Hes1, N2-NTM, Notch2, N3-ICD, Notch3, N4-ICD, and Notch4 were detected in lungs of Dll4nAb and control IgG injected mice by western blot (A) and quantified (B) (n=15). Proteins normalized to β-actin. (C) Double immunofluorescence staining of N1-ICD (red) and ERG (green) on lungs of Dll4nAb and control IgG injected mice(n=5). Arrows indicate N1-ICD staining. Scale bars: 20μm. Nuclei were counterstained by DAPI (blue). (D) EC specific Notch target genes were measured by PCR Arrays(n=3). (E) EC specific Notch target genes were measured in lungs of 1w Dll4nAb injected mice and control mice (n=6) by qPCR. Data shown as Mean±SEM; P values were calculated using the Student’s t-test, and *P<0.05; **, P<0.01; ***, P<0.001 vs control IgG group. ns, not significant.
Inhibition of Notch1 cleavage induces Notch1 target genes dysregulation in HPMECs
Our in vivo studies indicated that EC N1-ICD expression in lung tissue was significantly decreased by Dll4nAb. According to single cell sequence of adult human lung tissue (LungMAP database, link), Notch1 and Notch4 are highly expressed in adult human lung ECs whereas Notch2 and Notch3 expression is extremely low (Figure S2A). We treated HPMECs with DAPT for 12h to mimic the Notch1 cleavage inhibition without affecting total Notch1 expression. N1-ICD and downstream target gene Hes1 protein expression were decreased by 84.4% and 49.5%, respectively, while Notch1-FL (full length), N4-ICD and Notch4-FL were not affected (Figure S2B–C). Notch target gene expression was analyzed using the Human Notch Signaling Pathway PCR Array and summarized in Figure 2D. The majority of gene expression was decreased by DAPT, including DLL4, JAG1, HES1, HEY2, MFNG, RFNG, NCSTN, MAML1, NFKB2, CCNE1, and POFUT1. RBPJL, CDKN1A, and IL2RA were significantly increased in HPMECs by DAPT. These findings were consistent with other groups that showed depletion of Notch1 enhanced inflammation and decreased human ECs proliferation 23, 24. To confirm the finding in vivo, we measured these target genes’ expression in lungs of Dll4nAb treated mice by qPCR. Hey2 and Dll4 expression were significantly decreased after 1w Dll4nAb treatment (Figure 2E) while other genes’ expression showed no significance. These findings suggest that Dll4nAb predominantly decreased Notch1 cleavage and Notch1 target gene expression.
Increased EC permeability and inflammation in Dll4nAb injected mice
Dll4-dependent proteolytic activation of Notch1 exposes the transmembrane domain (TM). The TM without N1-ICD attached forms a complex with vascular endothelial cadherin (VE-cadherin) to stabilize EC junctions 25. We proposed that EC barrier function will be impaired by Dll4nAb treatment. As expected, en face staining of VE-cadherin on pulmonary arteries (approximately 100 μm) showed discontinuous cell-cell junctions and formed holes in Dll4nAb treated mice but not in IgG control mice (Figure 3A). Substantial N1-ICD was localized in nuclei of ECs in IgG treated mice but significantly decreased in Dll4nAb injected mice (Figure 3A). The predominant Notch receptors in EC are Notch1 and Notch4. To investigate the effect of decreased N1-ICD expression on EC barrier function, we treated HPMECs (adult) with DAPT for 12h to mimic the decrease of N1-ICD without affecting total Notch1 expression and Notch4 cleavage. HPMECs monolayers were grown to confluence in the upper chamber of a trans-well and assayed for trans-endothelial cell resistance (TEER)26 and peri-cellular permeability after DMSO or DAPT treatment. DAPT significantly reduced TEER in HPMECs compared with DMSO group (−50.25 ± 8.78 vs 5.50 ± 9.29 mΩ, Figure 3B) while increasing peri-cellular permeability by 66.6% (Figure 3C).
Figure 3. Enhanced EC permeability and inflammation by Dll4nAb treatment in normal mice lung tissues.
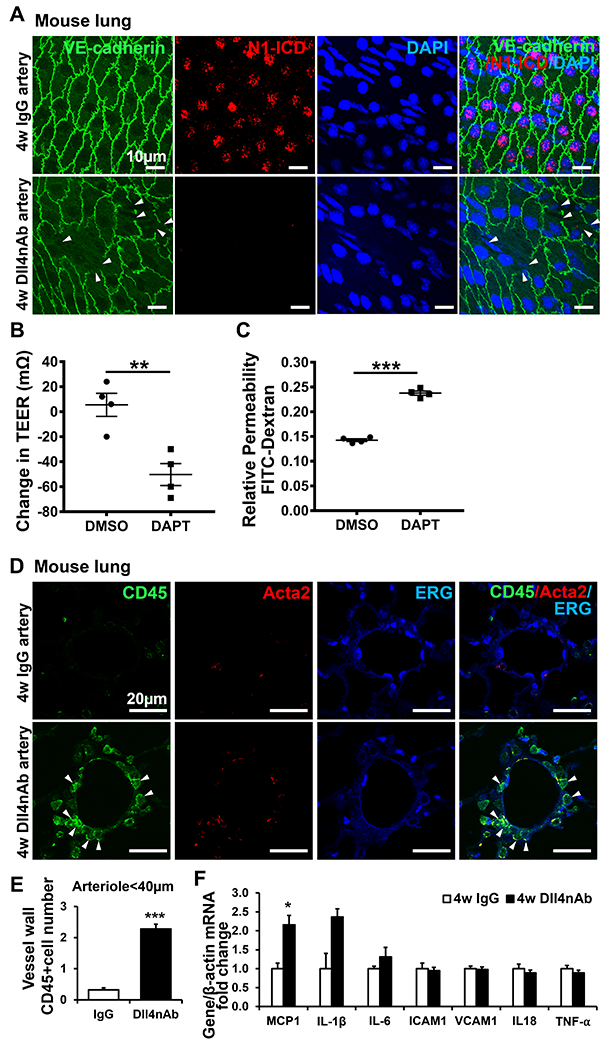
(A) En face staining of N1-ICD (red) and VE-cadherin (green) on ECs of pulmonary arteries of Dll4nAb injected mice. Arrows, impaired adherent junction integrity (n=3 per group). Nuclei were counterstained by DAPI (blue). Scale bars: 10 μm. (B) Quantification of trans-endothelial electrical resistance (TEER) and (C) paracellular permeability in confluent cultures of adult HPMECs monolayers in the absence or presence of DAPT (10μM) treatment for 12h(n=4). (D) Triple immunofluorescence staining of CD45 (green), ERG (blue) and Acata2 (red) on lungs of Dll4nAb and control IgG injected mice(n=5). The green staining indicates CD45-positive leukocyte cells. Arrows indicate infiltrated inflammatory cells adjacent to the pulmonary arteriolar compartment. Scale bars: 20 μm. (E) Quantification of infiltrated CD45-positive cells in both groups (mean±SEM). (F) Inflammatory chemokine and cytokine related genes were measured in lungs of Dll4nAb injected mice and control mice (n=7) by qPCR. Data shown as Mean±SEM; P values were calculated using the Student’s t-test, and *P<0.05; **, P<0.01; ***, P<0.001 compared with control group.
Depletion of Notch1 in induced pluripotent stem cell (iPSC) derived human EC and mouse aortic ECs increases inflammation 24. We hypothesized that immune cell infiltration will increase in lungs of Dll4nAb treated mice. Immune cells in vessel walls were detected by pan-leukocyte antigen CD45, and their location was visualized by co-staining of ERG and α-smooth muscle-actin (Acta2). Similar to other groups’ findings in PAH patients or MCT-induced rat PH model 27, 28, CD45+ cells in the vessel walls of small arteries(≤ 40μm) significantly increased in Dll4nAb injected mice compared with the IgG group (IgG group vs. Ab group: 0.32 ± 0.07 vs. 2.28 ± 0.15, Figure 3D–E). Few CD45+ cells appeared in the vessel walls of medium and large vessels (≥ 200 μm) and were comparable between two groups (data not shown). We also measured chemokine and cytokine levels important for PH pathogenesis by qPCR (Figure 3F and Figure S1C). Monocyte chemoattractant protein 1 (MCP1) showed a 1.2-fold increase in lungs of 4w Dll4nAb injected mice compared with IgG controls. Interleukin 1β (IL-1β) and IL-6 showed a trending increase but no significance. These findings indicate that decreasing EC N1-ICD expression by Dll4nAb increases EC permeability and immune cell infiltration.
Decrease of N1-ICD expression in PH mouse models and PAH patients
We successfully generated two PH mouse models (SuHx or Hx) (Figure S3). To detect Notch activation during PH progression, total Notch and N-ICD protein expression in lungs of SuHx or Hx mice and their littermate controls was measured by western blot (Figure 4A, B, D, Figure S4A–B). We found sustained decrease of N1-ICD in lungs of PH mice over time compared to control lungs. HEY1 mRNA, a specific Notch target gene in ECs, decreased during PH progression (Figure 4C). Total Notch1 expression showed a trending increase but no significant difference in lungs of SuHx (1w) and Hx (1w and 5w) mice, which may be a compensational effect of decreased Notch1 cleavage (Figure 4D). N3-ICD was unchanged in the early stages but significantly increased in 3w SuHx mice, while increased across all time points of Hx mice (Figure S4A–C). Consistent with previous findings29, total Notch3 expression significantly increased after 1w Hx treatment that lasted until 5w. Due to the lack of an anti-mouse antibody for detecting N2-ICD, we used N2-NTM to determine Notch2 cleavage in lungs of mouse models. Although we observed a slight decrease of N2-NTM in the late stage in Hx mice (indicative of increased N2-ICD expression), this change was not significant (Figure S4A–C). Dll4 was highly expressed in lungs of all groups but remained unchanged throughout PH progression (Figure 4A). Dll1 and Jag1 expression was much lower than Dll4 although they showed significant decrease in lungs of 5w Hx mice. Dll1 significantly increased in 3w SuHx mice while Jag1 showed no change in SuHx mice (Figure S4A–C), implying that Dll1 and Jag1 were unlikely to have contributed to the decreasing N1-ICD expression. Hence, we focused on Notch1 due to its sustained and substantial inactivation in PH and significant clinical relevance.
Figure 4. Decrease of N1-ICD expression in PAH patients and PH mouse models.
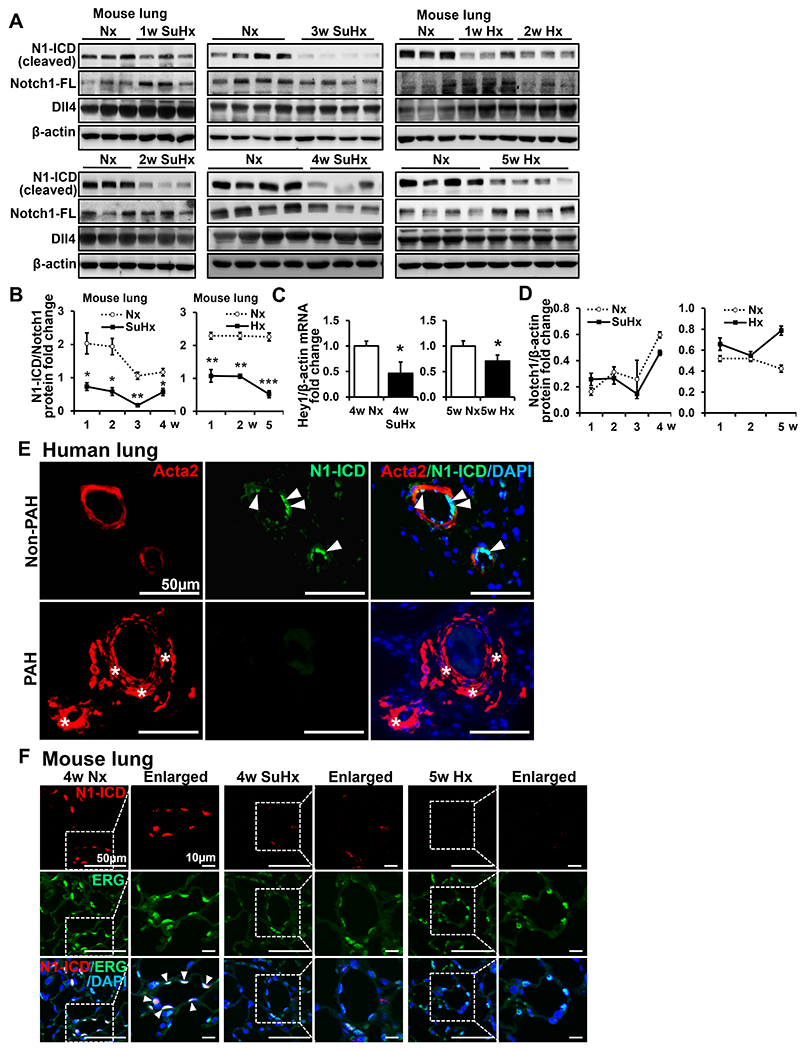
(A-D) N1-ICD, Notch1 and Dll4 were detected in lungs of SU5416 + hypoxia (SuHx)-PH mice, hypoxia (Hx)-PH mice(n=15) and control groups by western blot (A) and N1-ICD quantification (B). Proteins normalized to Notch1. (C) Hey1 was detected in lungs of SuHx-PH, Hx-PH (4w, n=5 and 5w, n=4) and control mice by qPCR. (D) Quantification of Notch1 in lungs of SuHx-PH, Hx-PH and control mice (A). Proteins normalized to β-actin. Data shown as Mean±SEM; P values were calculated using the Student’s t-test, and *, P<0.05. **, P<0.01. ***, P<0.001 compared with control group. (E) Double immunofluorescence staining of N1-ICD (green) and Acta2 (red) on adult PAH or non-PAH patients’ lungs(n=3). Arrows indicate N1-ICD staining and asterisks indicate vascular remodeling. Scale bars: 50 μm. (F) Double IF staining of N1-ICD (red) and ERG (green) on adult mouse lungs (n=4). Arrows indicate merged staining. Dotted line indicates enlarged areas. Scale bars: 50 μm and 10μm, respectively. Nuclei were counterstained by DAPI (blue). N1-ICD, cleaved Notch1 intracellular domain. FL, full length. Nx, normoxia. SuHx, SU5416 + hypoxia. Hx, hypoxia.
To confirm the decrease of N1-ICD expression in PAH patients, we performed IF staining of N1-ICD on PAH and non-PAH patients’ lung tissues and co-stained with Acta2. In non-PAH patients, N1-ICD predominantly localized in EC nuclei. In contrast, in lungs of PAH patients, vessel wall thickness substantially increased and N1-ICD was rarely detectable (Figure 4E). We also confirmed this finding in PH mouse models by IF staining of N1-ICD and co-stained with the EC nuclear marker, ERG. We observed that N1-ICD predominantly colocalized with ERG in the nucleus (Figure 4F) in control groups. Comparably, in PH mouse models, N1-ICD was hardly observed in ECs labeled by ERG nuclear staining. The number of ERG positive ECs were comparable between normoxia and PH models in mice (data not shown), suggesting that impaired Notch1 cleavage in pulmonary ECs was not due to a decrease of total EC numbers. Thus, these findings indicate that decreased N1-ICD expression in ECs is associated with progression of PH.
Increased EC permeability and inflammation in PH mouse models
Decreased N1-ICD expression causes impaired integrity of EC in Dll4nAb treated mice. To confirm this finding in PH mouse models, we performed en face staining of N1-ICD and VE-cadherin on pulmonary arteries from PH and control mice as described in 3.3. We observed N1-ICD predominantly localized in nuclei of ECs in Nx mice but significantly decreased in 1w Hx mice (Figure 5A) as well as in SuHx mice (Figure S5). Additionally, discontinuous cell-cell junctions revealed gaps between ECs in Hx mice (Figure 5A). Infiltration of CD45-positive immune cells increased in the vessel walls of small arteries from 1w PH mouse lung tissues compared with control group (Figure 5B), indicating enhanced inflammation in PH. The in vivo data above suggests that decreased N1-ICD expression in ECs causes increased EC permeability and inflammation which may contribute to the progression of PH.
Figure 5. Decrease of N1-ICD expression in PH increased EC permeability and induced inflammation in vivo.
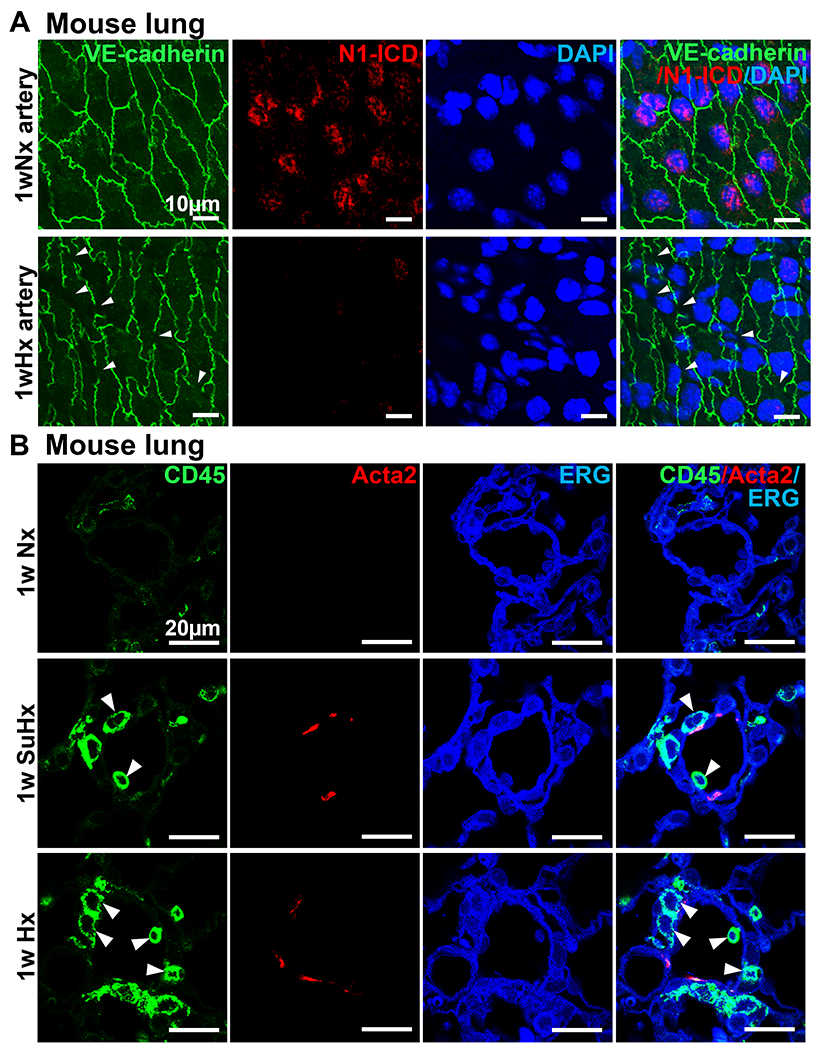
(A) En face staining of N1-ICD (red) and VE-cadherin (green) on ECs of pulmonary arteries after 1w hypoxia treatment. Arrows, impaired adherent junction integrity. (n=3 per group). Nuclei were counterstained by DAPI (blue). Scale bars: 10 μm. (B) Triple immunofluorescence staining of CD45 (green), ERG (blue) and Acta2 (red) on lungs of 1w PH and control mice(n=3). The green staining indicates CD45-positive leukocyte cells. Arrows indicate infiltrated inflammatory cells adjacent to the pulmonary arteriolar compartment. Scale bars: 20 μm. Nx, normoxia. SuHx, SU5416 + hypoxia. Hx, hypoxia.
EC specific AAV N1-ICD mitigates PH development in PH mice
We found a significant decrease of N1-ICD in lung ECs during the progression of PH and we speculate that inhibition of Notch1 activation may lead to PH. To test this hypothesis, we performed gain-of-function experiments. The continuous activated Notch1(AAV-N1-ICD) and AAV control were generated using AAV2-QuadYF vector, which exhibits strong tropism to ECs and facilitates transduction to the vasculature after systemic administration18, and administrated to control and PH mice. We observed that AAV-N1-ICD treatments attenuated the progression of PH in mice compared to the AAV control group. The effects of AAV-N1-ICD were demonstrated by a significant decrease of RVSP from 37.5 ± 1.2 to 33.3 ± 1.0 mmHg (Figure 6C), a decrease of RV remodeling (Fulton index, Figure 6D) from 0.40 ± 0.01 to 0.32 ± 0.01, and a selective reduction in the number of muscularized smaller, distal arteries (<40μm in diameter, ~53.6%) (Figure 6E–F). There were no significant effects of the exogenous N1-ICD on normoxia treated mice. The successful delivery of AAV N1-ICDs to lung ECs was detected by IF staining of N1-ICD co-stained with the EC specific marker Von Willebrand Factor (VWF) and was detected by western blot. We observed that EGFP predominantly colocalized with VWF in the cytoplasm of mouse lung ECs (Figure 6A) and observed an abundant expression of the exogenous N1-ICD by western blot in the lungs of AAV-N1-ICD injected mice (using the antibody that specifically recognizes Leu1745 to Lys1770 of murine N1-ICD) compared to AAV control lungs. Thus, these findings demonstrated that restoration of N1-ICD functionality partially attenuated the progression of hypoxic PH in male mice.
Figure 6. AAV- N1-ICD attenuated the progression of PH in mice.
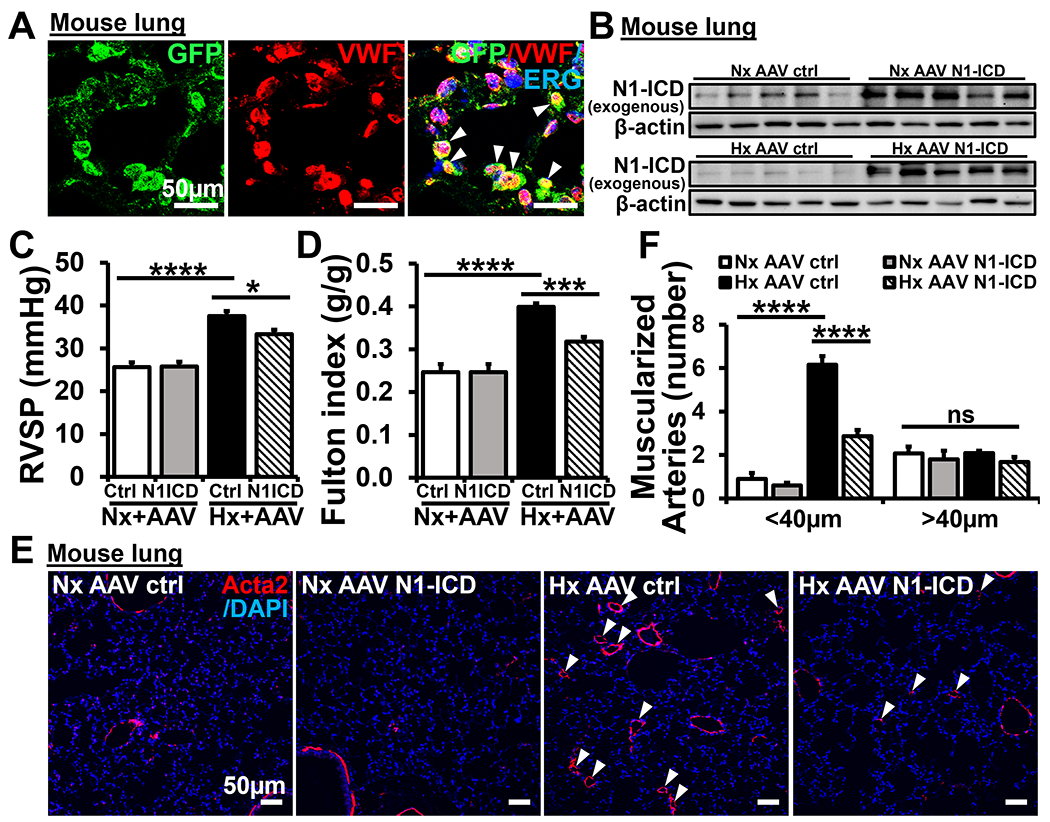
(A) Double IF staining of GFP (green) and VWF (red) on ECs of pulmonary arteries after 5w normoxia or hypoxia treatment (7weeks post injection of AAV, n=5-10 per group). Arrows indicate merged staining. (B) The exogenous N1-ICD was detected in lungs of 5wNx AAV ctrl (n=6), 5wNx AAV N1-ICD (n=9), 5wHx AAV ctrl (n=5) and 5wHx AAV N1-ICD (n=10) by western blot. Proteins normalized to β-actin. (C) RVSP and (D) Fulton index in AAV-N1-ICD and AAV control injected mice. (E-F) Decreased muscularization in AAV-N1-ICD injected mice compared with AAV control mice. (E) Representative micrographs of IF staining with anti-α-smooth muscle-actin (Acta2) (red) and (F) quantification of muscularized pulmonary arterial vessels. Arrows, muscularized distal pulmonary arterial vessels. Scale bars: 50μm. Nuclei were counterstained by DAPI (blue). Data shown as Mean±SEM; P values were calculated using the Student’s t-test, and *P<0.05; ***P<0.001; ****, P<0.0001 compared with control group. ns, not significant. Nx, normoxia. Hx, hypoxia.
Discussion
The major finding of this study is that robustly reduced Notch1 cleavage in pulmonary ECs is a novel molecular mechanism of PH pathogenesis. We found that a specific decrease of EC N1-ICD expression (reduced Notch1cleavage) by Dll4nAb treatment in mice led to the progression of PH through impaired EC junction integrity and increased inflammation. N1-ICD expression is consistently and robustly decreased in lung tissues of PAH patients and mouse models during the progression of PH. This N1-ICD reduction impairs EC barrier function and enhances infiltration of inflammatory cells. EC-specific N1-ICD overexpression attenuated PH in mice.
Impaired Notch1 cleavage in pulmonary ECs is an important molecular mechanism for PH pathogenesis. The results from preclinical studies and clinical trials support our findings. Dll4nAb treatment increased pulmonary vascular remodeling in monkeys and rats30, 31. Phase I clinical trials demonstrated increased incidence of PH in Dll4nAb treated cancer patients9–11. As a single agent, Demcizumab (OMP-21M18) 2.5mg/kg once weekly or 10 mg/kg once every other week resulted in PH in two of five or one of ten patients, respectively9. Enoticumab (REGN421) caused the development of grade II or III PH in 14% of patients10. Navicixizumab (OMP-305B83)-related pulmonary hypertension occurred in 18.2% of patients and the patients experienced more severe symptoms at a higher dose of 7.5 to 12.5 mg/kg12. Consistently, we demonstrated that Dll4nAb treatment resulted in PH development by specifically decreasing N1-ICD expression but not Notch2-4. Moreover, the dose we used in mice is the lowest dose used in clinical trials of cancer patients, suggesting PH occurrence increases by systemic and prolonged high dose treatment of Dll4nAb in cancer patients 32. A localized treatment of Dll4nAb in cancer patients to disrupt tumor angiogenesis should be considered in future clinical application.
The role of Notch signaling in PH progression is very controversial. Notch1 is enriched in EC of adult human and mouse lungs23. Depletion of Notch1 in human lung EC increases cell apoptosis 23. Two groups demonstrated enhanced inflammatory gene expression after depletion of Notch1 in human induced pluripotent stem cell (iPSC)-derived endothelial cells (ECs) or specific knock down of EC Notch1 in mice 24, 33. Polacheck et al revealed the non-canonical Notch1 signaling pathway affects EC barrier function through releasing cleaved transmembrane domain (TMD) to stabilize the EC barrier by binding to VE-cadherin 25. Human genetic mutations in Dll4 or Notch1 (causing Notch1 haploinsufficiency) lead to Adams-Oliver Syndrome (AOS), and PAH is one of the manifestations, suggesting Dll4-Notch1 signaling may play a critical role in the pathogenesis of PAH 34–38. Miyagawa et al demonstrated that deletion of endothelial Notch1 exacerbates hypoxia-induced PH in mice39, which supports the key role of Notch1 in PH. However, opposite effects of Notch1 in PAH have also been reported. Dabral et al demonstrated that Notch1 expression is restricted to adult lung EC and Notch1 activation (Notch1 cleavage, N1-ICD) increases in lungs of PAH patients and a rat PH model23. Notch2, 3 activation was increased during the PAH progression, and mice with PH that were treated with the γ-secretase inhibitor showed reversal of the disease29, 40–42. We believe the discrepancies between some literature and our data can be explained by the following. 1) Differences in antibody specificity. We and other groups verify the specificity of N1-ICD antibody (CST, #4147) (Figure S6) 25, 33, 43–45. Dabral et al measured N1-ICD with a different antibody from Santa Cruz (S-6014), but the data sheet states that it detects Notch1 transmembrane domain (NTM)23. When Notch1 is activated, the NTM is cleaved and NTM expression decreases. Their data showing increased amounts of NTM actually indicates decreased N1-ICD expression, making it consistent with our findings. In addition, their in vitro data determined that increased N1-NTM expression induced EC proliferation via upregulation of p21 and inhibited EC apoptosis via Bcl-2 and Survivin, supporting our finding of decreased N1-ICD expression inducing EC arrest in PH and Dll4nAb treated mice. 2) Treatment with γ-secretase inhibition (GSI) and Dll4nAb may have differential impact on Notch signaling. Dabral et al analyzed the impact of Notch signaling on EC proliferation, finding that GSI significantly reduced the RVSP and right heart hypertrophy in SuHx rats23. Our findings demonstrated that Dll4nAb specifically blocks Notch1 cleavage while Li et al showed GSIs block cleavage of Notch3 in lungs29. 3) Differences in time of treatment. Li et al showed that injecting mice with DAPT for 2w after 2w hypoxia exposure (and thus observing mice at 4w Hx) ameliorates PH 29. According to our data, Notch1 activation is high in normal conditions and decreases during PH progression. Notch1 activation is extremely low after 2w hypoxia whereas Notch3 activation is increased after 2w hypoxia 29. Therefore, DAPT injection had minimal effects on Notch1, mainly reducing Notch3 activation with therapeutic benefits. However, the role of DAPT on PH progression in normal mice was not investigated nor Notch1 activation measured.
Decreased Notch1 cleavage in ECs affects pulmonary vascular functions through Notch canonical and non-canonical pathways (Figure S7). 1) Canonical pathway. We detected Notch target genes regulated by inhibiting Notch1 activation (DAPT treatment) in ECs. Consistent with the role of N1-ICD as a transcriptional coactivator, the majority of gene expression was decreased after inhibiting Notch1 activation. Significantly changed genes were related to cell cycle regulation, vessel network formation, and inflammation. Recent evidence indicates that senescence plays a role in the pathobiology of PAH 46. Knockdown of Hes1 blocks the inhibitory effect of Jagged1-Notch activation on cellular senescence in mesenchymal stem cells 47. Thereby, dysregulation of Notch target genes in EC may contribute to PAH progression by increasing cell senescence characterized by inhibition of cell proliferation and cell cycle arrest. Although depletion of Notch1 in human lung ECs increases cell apoptosis in vitro23, we did not observe a significant increase in EC apoptosis in lung tissues of Dll4nAb or the PH mouse model in vivo (data not shown). This is possibly attributable to the missing time point at which this occurs. In addition, Dll4-Notch1 canonical pathway can also inhibit EC transcytosis, the transport of macromolecules larger than 3nm via transcellular pathways48. EC permeability includes transcellular and paracellular pathways48. In a mouse retinal hypertension model, Yang et al reported that increase of Notch1 target gene, VEGFR2 and BREBP1, contribute to EC transcytosis-mediated vascular leakage 49. 2) Non-canonical pathway. Polacheck et al revealed that activation of Notch1 releases the transmembrane domain in ECs, which subsequently forms a complex with VE-cadherin to stabilize EC junctions 25. EC Notch1 deficient mice have augmentation of EC permeability compared with littermate controls 25. In our study, in vitro inhibition of Notch1 cleavage robustly decreased TEER and increased permeability in HPMECs. In vivo, en face staining simultaneously showed the impaired EC barrier function and decreased N1-ICD expression in small pulmonary arteries of both the PH mouse model and Dll4nAb treated mice, which indicated deactivation of this non-canonical Notch1 pathway destroys EC junctions during PAH progression. Moreover, we measured the lung masses of IgG or Dll4nAb treated mice for 4 weeks (Figure S1D). No obvious differences were observed, suggesting that increased RVSP in Dll4nAb-treated mice is not due to pulmonary edema but instead is related to the identified muscularization. We also observed increased infiltration of inflammatory cells in vessel walls of the PH mouse model and Dll4nAb treated mice. Although hyperpermeability was first observed in MCT-induced rat PH model50, the contribution of endothelial hyperpermeability to PH progression has perhaps been underappreciated.
In summary, we discovered Dll4nAb treatment in mice led to the development of PH by specifically inhibiting EC Notch1 cleavage, indicating that Dll4nAb in cancer patients may be mechanistically attributable to reduced N1-ICD mediated signaling in the pulmonary endothelium. We believe that this mechanistic data could provide an opportunity to titrate the dose to reduce toxicity or perhaps to avoid using the drug in more vulnerable patients. Localized application of Dll4nAb may be optimal to potentially reduce the risk of PH. In addition, PH animal models phenotypically resemble Dll4nAb treated mice, which suggests that the decrease of EC Notch1 cleavage could be a common mechanism of PH pathogenesis. This mechanism is further confirmed in tissue from PAH patients with advanced disease. Thus, our findings detailing the importance of N1-ICD mediated signaling in the pulmonary endothelium may apply broadly to lung health and disease.
Perspectives
Dll4nAb treatment in mice led to the development of PH by specifically inhibiting Notch1 cleavage, indicating that systemic usage of Dll4nAb in cancer patients will increase the risk of PH. The necessity for a localized treatment of Dll4nAb in cancer patients has tremendous clinical significance. Resembling Dll4nAb treated mice, Notch1 cleavage was sustained and robustly inhibited in lung ECs of PH animal models and PAH patients. Specific enhancing Notch1 cleavage could be a potential therapeutic strategy for PH.
Supplementary Material
Novelty and Significance.
What Is New?
Dll4nAb led to the progression of PH in mice by specifically inhibiting Notch1 activation through impairing EC barrier function and increasing immune cell infiltration in the vessel wall.
A sustained and significant inhibition of Notch1 activation occurred in lung ECs of PH mouse models and PAH patients. EC-specific N1-ICD overexpression can reverse PH in PH mouse models.
Inhibition of Notch1 cleavage could be a common mechanism of PH pathogenesis.
What Is Relevant?
Specific decrease of EC N1-ICD expression (reduced Notch1cleavage) by Dll4nAb treatment in mice led to the progression of PH.
N1-ICD expression consistently and robustly decreased in lung tissues of PAH patients and mouse models during the progression of PH.
Inhibition of Notch1 cleavage in lung ECs impaired EC barrier function and increased infiltration of immune cells which promotes pulmonary vascular remodeling during the progression of PH.
Acknowledgments
We appreciate donor families and Human Tissue Core (HTC) staff and investigators for making this work possible. We are grateful to Dr. Elaine M. Smolock of Center for Professional Development for generously providing assistance with manuscript writing.
Sources of Funding
This work was supported by Jinjiang Pang’s grants from the National Institutes of Health (R01 HL122777-05, R01 HL122777-06A1), American Heart Association Innovative Project Award (19IPLOI34760446) and Lung Biology Strategic Plan Pilot Project (University of Rochester). This original work was also funded by NIH/NHLBI 1U01HL122700 in support of the developing lung molecular atlas program (LungMAP) HTC at University of Rochester Medical Center.
Footnotes
Conflict of Interest Statement
None.
References
- 1.ACS Cancer facts & figures 2019. <http://www.cancer.org/research/cancer-facts-statistics/all-cancer-facts-figures/cancer-facts-figures-2019>. Accessed June 22 2020
- 2.Abe J, Martin JF, Yeh ET. The future of onco-cardiology: We are not just “side effect hunters”. Circ Res. 2016;119:896–899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Krebs LT, Xue Y, Norton CR, Shutter JR, Maguire M, Sundberg JP, Gallahan D, Closson V, Kitajewski J, Callahan R, Smith GH, Stark KL, Gridley T. Notch signaling is essential for vascular morphogenesis in mice. Genes Dev. 2000;14:1343–1352 [PMC free article] [PubMed] [Google Scholar]
- 4.Aster JC, Blacklow SC. Targeting the notch pathway: Twists and turns on the road to rational therapeutics. J Clin Oncol. 2012;30:2418–2420 [DOI] [PubMed] [Google Scholar]
- 5.Dufraine J, Funahashi Y, Kitajewski J. Notch signaling regulates tumor angiogenesis by diverse mechanisms. Oncogene. 2008;27:5132–5137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Noguera-Troise I, Daly C, Papadopoulos NJ, Coetzee S, Boland P, Gale NW, Lin HC, Yancopoulos GD, Thurston G. Blockade of dll4 inhibits tumour growth by promoting non-productive angiogenesis. Nature. 2006;444:1032–1037 [DOI] [PubMed] [Google Scholar]
- 7.Ridgway J, Zhang G, Wu Y, Stawicki S, Liang WC, Chanthery Y, Kowalski J, Watts RJ, Callahan C, Kasman I, Singh M, Chien M, Tan C, Hongo JA, de Sauvage F, Plowman G, Yan M. Inhibition of dll4 signalling inhibits tumour growth by deregulating angiogenesis. Nature. 2006;444:1083–1087 [DOI] [PubMed] [Google Scholar]
- 8.Takebe N, Nguyen D, Yang SX. Targeting notch signaling pathway in cancer: Clinical development advances and challenges. Pharmacol Ther. 2014;141:140–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Smith DC, Eisenberg PD, Manikhas G, Chugh R, Gubens MA, Stagg RJ, Kapoun AM, Xu L, Dupont J, Sikic B. A phase i dose escalation and expansion study of the anticancer stem cell agent demcizumab (anti-dll4) in patients with previously treated solid tumors. Clin Cancer Res. 2014;20:6295–6303 [DOI] [PubMed] [Google Scholar]
- 10.Chiorean EG, LoRusso P, Strother RM, Diamond JR, Younger A, Messersmith WA, Adriaens L, Liu L, Kao RJ, DiCioccio AT, Kostic A, Leek R, Harris A, Jimeno A. A phase i first-in-human study of enoticumab (regn421), a fully human delta-like ligand 4 (dll4) monoclonal antibody in patients with advanced solid tumors. Clin Cancer Res. 2015;21:2695–2703 [DOI] [PubMed] [Google Scholar]
- 11.McKeage MJ, Kotasek D, Markman B, Hidalgo M, Millward MJ, Jameson MB, Harris DL, Stagg RJ, Kapoun AM, Xu L, Hughes BGM. Phase ib trial of the anti-cancer stem cell dll4-binding agent demcizumab with pemetrexed and carboplatin as first-line treatment of metastatic non-squamous nsclc. Target Oncol. 2018;13:89–98 [DOI] [PubMed] [Google Scholar]
- 12.Jimeno A, Moore KN, Gordon M, Chugh R, Diamond JR, Aljumaily R, Mendelson D, Kapoun AM, Xu L, Stagg R, Smith DC. A first-in-human phase 1a study of the bispecific anti-dll4/anti-vegf antibody navicixizumab (omp-305b83) in patients with previously treated solid tumors. Invest New Drugs. 2019;37:461–472 [DOI] [PubMed] [Google Scholar]
- 13.Coleman RL, Handley KF, Burger R, Molin GZD, Stagg R, Sood AK, Moore KN. Demcizumab combined with paclitaxel for platinum-resistant ovarian, primary peritoneal, and fallopian tube cancer: The sierra open-label phase ib trial. Gynecol Oncol. 2020;157:386–391 [DOI] [PubMed] [Google Scholar]
- 14.Hoey T, Yen WC, Axelrod F, Basi J, Donigian L, Dylla S, Fitch-Bruhns M, Lazetic S, Park IK, Sato A, Satyal S, Wang X, Clarke MF, Lewicki J, Gurney A. Dll4 blockade inhibits tumor growth and reduces tumor-initiating cell frequency. Cell Stem Cell. 2009;5:168–177 [DOI] [PubMed] [Google Scholar]
- 15.Humbert M, Guignabert C, Bonnet S, Dorfmuller P, Klinger JR, Nicolls MR, Olschewski AJ, Pullamsetti SS, Schermuly RT, Stenmark KR, Rabinovitch M. Pathology and pathobiology of pulmonary hypertension: State of the art and research perspectives. Eur Respir J. 2019;53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pang J, Yan C, Natarajan K, Cavet ME, Massett MP, Yin G, Berk BC. Git1 mediates hdac5 activation by angiotensin ii in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2008;28:892–898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pang J, Xu X, Getman MR, Shi X, Belmonte SL, Michaloski H, Mohan A, Blaxall BC, Berk BC. G protein coupled receptor kinase 2 interacting protein 1 (git1) is a novel regulator of mitochondrial biogenesis in heart. J Mol Cell Cardiol. 2011;51:769–776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lipinski DM, Reid CA, Boye SL, Peterson JJ, Qi X, Boye SE, Boulton ME, Hauswirth WW. Systemic vascular transduction by capsid mutant adeno-associated virus after intravenous injection. Hum Gene Ther. 2015;26:767–776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Murtaugh LC, Stanger BZ, Kwan KM, Melton DA. Notch signaling controls multiple steps of pancreatic differentiation. Proc Natl Acad Sci U S A. 2003;100:14920–14925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yamanda S, Ebihara S, Asada M, Okazaki T, Niu K, Ebihara T, Koyanagi A, Yamaguchi N, Yagita H, Arai H. Role of ephrinb2 in nonproductive angiogenesis induced by delta-like 4 blockade. Blood. 2009;113:3631–3639 [DOI] [PubMed] [Google Scholar]
- 21.Oishi H, Sunamura M, Egawa S, Motoi F, Unno M, Furukawa T, Habib NA, Yagita H. Blockade of delta-like ligand 4 signaling inhibits both growth and angiogenesis of pancreatic cancer. Pancreas. 2010;39:897–903 [DOI] [PubMed] [Google Scholar]
- 22.Fukuda D, Aikawa E, Swirski FK, Novobrantseva TI, Kotelianski V, Gorgun CZ, Chudnovskiy A, Yamazaki H, Croce K, Weissleder R, Aster JC, Hotamisligil GS, Yagita H, Aikawa M. Notch ligand delta-like 4 blockade attenuates atherosclerosis and metabolic disorders. Proc Natl Acad Sci U S A. 2012;109:E1868–1877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dabral S, Tian X, Kojonazarov B, Savai R, Ghofrani HA, Weissmann N, Florio M, Sun J, Jonigk D, Maegel L, Grimminger F, Seeger W, Savai Pullamsetti S, Schermuly RT. Notch1 signalling regulates endothelial proliferation and apoptosis in pulmonary arterial hypertension. Eur Respir J. 2016;48:1137–1149 [DOI] [PubMed] [Google Scholar]
- 24.Theodoris CV, Li M, White MP, Liu L, He D, Pollard KS, Bruneau BG, Srivastava D. Human disease modeling reveals integrated transcriptional and epigenetic mechanisms of notch1 haploinsufficiency. Cell. 2015;160:1072–1086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Polacheck WJ, Kutys ML, Yang J, Eyckmans J, Wu Y, Vasavada H, Hirschi KK, Chen CS. A non-canonical notch complex regulates adherens junctions and vascular barrier function. Nature. 2017;552:258–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ye L, Martin TA, Parr C, Harrison GM, Mansel RE, Jiang WG. Biphasic effects of 17-beta-estradiol on expression of occludin and transendothelial resistance and paracellular permeability in human vascular endothelial cells. J Cell Physiol. 2003;196:362–369 [DOI] [PubMed] [Google Scholar]
- 27.Majka SM, Skokan M, Wheeler L, Harral J, Gladson S, Burnham E, Loyd JE, Stenmark KR, Varella-Garcia M, West J. Evidence for cell fusion is absent in vascular lesions associated with pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol. 2008;295:L1028–1039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Florentin J, Coppin E, Vasamsetti SB, Zhao J, Tai YY, Tang Y, Zhang Y, Watson A, Sembrat J, Rojas M, Vargas SO, Chan SY, Dutta P. Inflammatory macrophage expansion in pulmonary hypertension depends upon mobilization of blood-borne monocytes. J Immunol. 2018;200:3612–3625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li X, Zhang X, Leathers R, Makino A, Huang C, Parsa P, Macias J, Yuan JX, Jamieson SW, Thistlethwaite PA. Notch3 signaling promotes the development of pulmonary arterial hypertension. Nat Med. 2009;15:1289–1297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Couch JA, Zhang G, Beyer JC, de Zafra CL, Gupta P, Kamath AV, Lewin-Koh N, Tarrant J, Allamneni KP, Cain G, Yee S, Ross S, Cook R, Tsai SP, Ruppel J, Ridgway JB, Paluch M, Hass PE, Franklin J, Yan M. Balancing efficacy and safety of an anti-dll4 antibody through pharmacokinetic modulation. Clin Cancer Res. 2016;22:1469–1479 [DOI] [PubMed] [Google Scholar]
- 31.Yan M, Callahan CA, Beyer JC, Allamneni KP, Zhang G, Ridgway JB, Niessen K, Plowman GD. Chronic dll4 blockade induces vascular neoplasms. Nature. 2010;463:E6–7 [DOI] [PubMed] [Google Scholar]
- 32.Nair AB, Jacob S. A simple practice guide for dose conversion between animals and human. J Basic Clin Pharm. 2016;7:27–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mack JJ, Mosqueiro TS, Archer BJ, Jones WM, Sunshine H, Faas GC, Briot A, Aragon RL, Su T, Romay MC, McDonald AI, Kuo CH, Lizama CO, Lane TF, Zovein AC, Fang Y, Tarling EJ, de Aguiar Vallim TQ, Navab M, Fogelman AM, Bouchard LS, Iruela-Arispe ML. Notch1 is a mechanosensor in adult arteries. Nat Commun. 2017;8:1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lehman A, Wuyts W, Patel MS. Adams-oliver syndrome. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A, eds. Genereviews((r)). Seattle (WA); 1993. [Google Scholar]
- 35.Dadzie OE, Tyszczuk L, Holder SE, Teixeira F, Charakida A, Scarisbrick J, Chu A. Adams-oliver syndrome with widespread cmtc and fatal pulmonary vascular disease. Pediatr Dermatol. 2007;24:651–653 [DOI] [PubMed] [Google Scholar]
- 36.Stittrich AB, Lehman A, Bodian DL, Ashworth J, Zong Z, Li H, Lam P, Khromykh A, Iyer RK, Vockley JG, Baveja R, Silva ES, Dixon J, Leon EL, Solomon BD, Glusman G, Niederhuber JE, Roach JC, Patel MS. Mutations in notch1 cause adams-oliver syndrome. Am J Hum Genet. 2014;95:275–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Meester JA, Southgate L, Stittrich AB, Venselaar H, Beekmans SJ, den Hollander N, Bijlsma EK, Helderman-van den Enden A, Verheij JB, Glusman G, Roach JC, Lehman A, Patel MS, de Vries BB, Ruivenkamp C, Itin P, Prescott K, Clarke S, Trembath R, Zenker M, Sukalo M, Van Laer L, Loeys B, Wuyts W. Heterozygous loss-of-function mutations in dll4 cause adams-oliver syndrome. Am J Hum Genet. 2015;97:475–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Southgate L, Sukalo M, Karountzos ASV, Taylor EJ, Collinson CS, Ruddy D, Snape KM, Dallapiccola B, Tolmie JL, Joss S, Brancati F, Digilio MC, Graul-Neumann LM, Salviati L, Coerdt W, Jacquemin E, Wuyts W, Zenker M, Machado RD, Trembath RC. Haploinsufficiency of the notch1 receptor as a cause of adams-oliver syndrome with variable cardiac anomalies. Circ Cardiovasc Genet. 2015;8:572–581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miyagawa K, Shi M, Chen PI, Hennigs JK, Zhao Z, Wang M, Li CG, Saito T, Taylor S, Sa S, Cao A, Wang L, Snyder MP, Rabinovitch M. Smooth muscle contact drives endothelial regeneration by bmpr2-notch1-mediated metabolic and epigenetic changes. Circ Res. 2019;124:211–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hurst LA, Dunmore BJ, Long L, Crosby A, Al-Lamki R, Deighton J, Southwood M, Yang X, Nikolic MZ, Herrera B, Inman GJ, Bradley JR, Rana AA, Upton PD, Morrell NW. Tnfalpha drives pulmonary arterial hypertension by suppressing the bmp type-ii receptor and altering notch signalling. Nat Commun. 2017;8:14079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xiao Y, Gong D, Wang W. Soluble jagged1 inhibits pulmonary hypertension by attenuating notch signaling. Arterioscler Thromb Vasc Biol. 2013;33:2733–2739 [DOI] [PubMed] [Google Scholar]
- 42.Morris HE, Neves KB, Montezano AC, MacLean MR, Touyz RM. Notch3 signalling and vascular remodelling in pulmonary arterial hypertension. Clin Sci (Lond). 2019;133:2481–2498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hellstrom M, Phng LK, Hofmann JJ, Wallgard E, Coultas L, Lindblom P, Alva J, Nilsson AK, Karlsson L, Gaiano N, Yoon K, Rossant J, Iruela-Arispe ML, Kalen M, Gerhardt H, Betsholtz C. Dll4 signalling through notch1 regulates formation of tip cells during angiogenesis. Nature. 2007;445:776–780 [DOI] [PubMed] [Google Scholar]
- 44.Pitulescu ME, Schmidt I, Giaimo BD, Antoine T, Berkenfeld F, Ferrante F, Park H, Ehling M, Biljes D, Rocha SF, Langen UH, Stehling M, Nagasawa T, Ferrara N, Borggrefe T, Adams RH. Dll4 and notch signalling couples sprouting angiogenesis and artery formation. Nat Cell Biol. 2017;19:915–927 [DOI] [PubMed] [Google Scholar]
- 45.Jin ZG, Wong C, Wu J, Berk BC. Flow shear stress stimulates gab1 tyrosine phosphorylation to mediate protein kinase b and endothelial nitric-oxide synthase activation in endothelial cells. J Biol Chem. 2005;280:12305–12309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.van der Feen DE, Berger RMF, Bartelds B. Converging paths of pulmonary arterial hypertension and cellular senescence. Am J Respir Cell Mol Biol. 2019;61:11–20 [DOI] [PubMed] [Google Scholar]
- 47.Tian Y, Xu Y, Xue T, Chen L, Shi B, Shu B, Xie C, Max Morandi M, Jaeblon T, Marymont JV, Dong Y. Notch activation enhances mesenchymal stem cell sheet osteogenic potential by inhibition of cellular senescence. Cell Death Dis. 2017;8:e2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sukriti S, Tauseef M, Yazbeck P, Mehta D. Mechanisms regulating endothelial permeability. Pulm Circ. 2014;4:535–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang JM, Park CS, Kim SH, Noh TW, Kim JH, Park S, Lee J, Park JR, Yoo D, Jung HH, Takase H, Shima DT, Schwaninger M, Lee S, Kim IK, Lee J, Ji YS, Jon S, Oh WY, Kim P, Uemura A, Ju YS, Kim I. Dll4 suppresses transcytosis for arterial blood-retinal barrier homeostasis. Circ Res. 2020;126:767–783 [DOI] [PubMed] [Google Scholar]
- 50.Sugita T, Hyers TM, Dauber IM, Wagner WW, McMurtry IF, Reeves JT. Lung vessel leak precedes right ventricular hypertrophy in monocrotaline-treated rats. J Appl Physiol Respir Environ Exerc Physiol. 1983;54:371–374 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.