

Abstract
Kir4.1, a glial-specific inwardly rectifying potassium channel, is implicated in astrocytic maintenance of K+ homeostasis. Underscoring the role of Kir4.1 in CNS functioning, genetic mutations in KCNJ10, the gene which encodes Kir4.1, causes seizures, ataxia and developmental disability in humans. Kir4.1 protein and mRNA loss are consistently observed CNS injury, and neurological diseases linked to hyperexcitability and neuronal dysfunction, leading to the notion that Kir4.1 represents an attractive therapeutic target. Despite this, little is understood regarding the mechanisms that underpin this downregulation.
Previous work by our lab revealed that DNA hypomethylation of the Kcnj10 gene functions to regulate mRNA levels during astrocyte maturation whereas hypermethylation in vitro led to decreased promoter activity. In the current study we utilized two vastly different injury models with known acute and chronic loss of Kir4.1 protein and mRNA to evaluate the methylation status of Kcnj10 as a candidate molecular mechanism for reduced transcription and subsequent protein loss. Examining whole hippocampal tissue and isolated astrocytes, in a lithium-pilocarpine model of epilepsy, we consistently identified hypermethylation of CpG island two, which resides in the large intronic region spanning the Kcnj10 gene. Strikingly similar results were observed using a second injury paradigm, a fifth cervical (C5) vertebral hemi-contusion model of spinal cord injury. Our previous work indicates the same gene region is significantly hypomethylated when transcription increases during astrocyte maturation. Our results suggest that DNA methylation can bidirectionally modulate Kcnj10 transcription and may represent a targetable molecular mechanism for the restoring astroglial Kir4.1 expression following CNS insult.
Keywords: Astrocyte, Kir4.1, Kcnj10, DNA Methylation, Pilocarpine
INTRODUCTION
Astrocytes represent the most abundant glial cell type in the central nervous system (CNS). These cells are thought to contribute to the maintenance of extracellular potassium ([K+]e), in part, via the inwardly rectifying potassium channel Kir4.1 (reviewed in Nwaobi et al., 2016). Kir4.1 is a glia-specific potassium channel with the highest expression observed in astrocytes (Zhang et al., 2014). Due to the high open probability of this channel at rest (Ransom and Sontheimer, 1995), Kir4.1 contributes significantly to membrane properties in astrocytes including the hyperpolarized resting membrane, selective potassium conductance, and low input resistance of these cells (Djukic et al., 2007; Kucheryavykh et al., 2007; Olsen et al., 2006; Seifert et al., 2010). Underscoring the importance of this channel for normal CNS functioning, individuals harboring homozygous mutations in Kcnj10, the gene which encodes the Kir4.1 protein, present with several neurological symptoms including early onset tonic clonic seizures, ataxia, sensorineural deafness and developmental delay (Bockenhauer et al., 2009; Reichold et al., 2010; Scholl et al., 2009). Intriguingly, several terrier dog breeds with homozygous mutations in Kir4.1 present with seizures and cerebellar ataxia (Gilliam et al., 2014 2015; Rohdin et al., 2015 2015) and cerebellar ataxia and spongy degernation were also reported in Belgian shepards with homozygous missense mutations in Kir4.1 (Mauri et al., 2017). Kir4.1 was identified as a seizure susceptibility gene in both mice and humans (Buono et al., 2004; Lenzen et al., 2005). Astrocyte specific (Djukic et al., 2007) and more recently oligodendrocyte specific (Larson et al., 2018) murine models have been generated to study the role of Kir4.1 in CNS function. Global and astrocyte specific embryonic knockout of Kir4.1 in mice generates a severe phenotype, including reduced body weight, deficits in balance and coordination of voluntary movements. This phenotype progresses to hind limb paralysis, increased seizure susceptibility and premature death at about 3 weeks of age (Djukic et al., 2007; Kofuji et al., 2000; Neusch et al., 2001). The oligodendrocyte specific Kir4.1 knockout animals also show signs of motor impairment and a lower seizure threshold (Larson et al., 2018). Together, the data from spontaneous loss of function mutations in humans and dogs and embryonic genetic manipulation of Kir4.1 in animals leads to a hyperexcitability phenotype.
Multiple lines of evidence demonstrate consistent downregulation of Kir4.1 as a hallmark of CNS pathology. Kir4.1 downregulation has been observed in many neuropathological conditions, including: neurodegenerative diseases (Scarmeas et al., 2009; Tong et al., 2014; Wilcock et al., 2009), CNS injury (D’Ambrosio et al., 1999; Olsen et al., 2010; Stewart et al., 2010), CNS infection (Lein et al., 2007), animal models of epilepsy (Ivens et al., 2007; Liu and Harada, 2013), human epilepsy tissue (Das et al., 2012; Heuser et al., 2012), and the sclerotic CA1 region of MTLE patients (Bordey and Sontheimer, 1998; Hinterkeuser et al., 2000; Schroder et al., 2000). The alteration in astrocytic biophysical properties and disruption of a key K+ homeostatic mechanism (Kucheryavykh et al., 2007; Neusch et al., 2006; Olsen et al., 2006; Tong et al., 2014) have led to speculation that Kir4.1 represents a potential astrocyte therapeutic target. However, little is known regarding molecular mechanisms which contribute to Kir4.1 channel downregulation.
Kir4.1 expression is strongly developmentally upregulated, a process associated with decreased methylation of the Kir4.1 gene (Nwaobi et al., 2014). In contrast, hypermethylation of the one CpG island in the large intronic region spanning the Kcnj10 gene promoter and transcriptional start site, in vitro, led to transcriptional repression (Nwaobi et al., 2014). These data suggest DNA methylation functions as a powerful transcriptional regulator to bidirectionally modulate Kcnj10 transcription levels. In the current manuscript we evaluated hyper-methylation as a possible molecular mechanism driving reduced levels of Kir4.1 transcription in gliotic astrocytes. Using a lithium-pilocarpine-induced status epilepticus (SE) model we observe reduction in Kcnj10 transcription and subsequent protein expression and function in CA1 and CA3 regions of the hippocampus. This reduction in transcription is associated with hypermethylation of CpG island two which resides in the large intronic region spanning the Kcnj10 gene promoter and transcriptional start site. Importantly, similar results were observed in a second model of CNS spinal cord injury, suggesting this may be a common mechanism of Kcnj10 regulation. These studies suggest that sustained reductions of Kir4.1 post-injury are mediated in part by enhanced DNA methylation. Given the broad clinical implications for both acute and chronic dysregulation of [K] +e in a variety of CNS pathologies, a more comprehensive understanding of the regulation of Kir4.1 expression may prove to be useful in developing therapies for a diverse clinical subset.
MATERIALS AND METHODS
Animals.
All animals were handled in accordance with the National Institutes of Health guidelines. The Animal Care and Use Committee at the University of Alabama at Birmingham and Virginia Polytechnical approved animal use. Animals were housed under reversed 12-h light-dark cycles and were provided with water and food available ad libitum. Male Sprague-Dawley (SD) rats, aged post-natal day 50–60 (200–300g; Charles River, Wilmington, MA) were randomly assigned to two groups, sham or lithium-pilocarpine (LiPilo), and used for tissue collection, molecular studies, and immunohistochemistry. Mixed gender eGFP-S100β adult rats generated by Itakura et al. (Itakura et al., 2007) were utilized for electrophysiology experiments. Here eGFP is driven by the S100β promoter enabling visualization of astrocytes for whole-cell patch clamp recording (Strain W-Tg(S100β-EGFP) Scell, catalogue number 0371, National Bioresource for the Rat, Japan). We have previously demonstrated 95% of GFAP+ cells are also eGFP+ (Nwaobi et al., 2014). No gender differences were detected through western blot analysis (Figure S1).
LiPilo model of Status Epilepticus.
Animals were given an intraperitoneal (IP) injection of either saline or lithium-chloride at 128mg/kg (Sigma), 16–24 hours prior to induction of SE (Pitkänen et al., 2006 2006). Intraperitoneal injection of methyl-scopolamine (5mg/kg; Sigma) was given to all animals and allowed to circulate for 30 minutes prior to IP injection of either saline or pilocarpine (30mg/kg; Sigma). Sham animals did not receive lithium-chloride or pilocarpine. After administration of methyl-scopolamine and pilocarpine, each animal was monitored in individual cages for the 5 stages of the Racine scale. Once animals entered SE (5th stage of the Racine scale) they were allowed to remain in status for 1 hour and then given an intraperitoneal injection of Diazepam (5mg/kg, Hospira) and 10mL of saline (subcutaneous). Animals receive 10mL of saline, 2mL of Nutrical (Vétoquinal), daily for 5 days post SE, at which point all animals demonstrated recovery from initial weight loss due to SE protocol.
C5 Hemi-contusion injury.
Hemi-contusion was induced on the right side of the spinal cord using Infinite Horizon spinal cord injury device (Precision Systems and Instrumentation) as previously described in (Dunham et al., 2010). Briefly, rats were anesthetized using 4% isoflurane. Intraperitoneal injection of ketamine/xylazine at 100/10mg/kg was given. Neck area was shaved and cleaned using beta-iodine and chlorohexidine. Animals were kept on a heating pad and anesthetized using 0.5% isoflurane. A midline incision starting at C2 process down to the T2 was made to expose musculature. Following incision of the trapezius muscle, C4 to C6 paravertebral muscles were removed. A bilateral laminectomy was made at the fifth cervical vertebrate (C5,) exposing the dorsal spinal cord. Spinal cord was stabilized using Adson forceps. A 0.8mm impactor tip was positioned over the right side of the spinal cord. Hemi-contusion injury was induced using a 200kdyn force. Sham-operated animals received no injury. Following injury, musculature layers were sutured using absorbable sutures and skin was sutured. Rats received 3mL of Ringer’s solution containing enrofloxacin (2.5mg/kg) and carprofen (5mg/kg) via subcutaneous injection post-surgery. Animals continued to receive this combination of drugs twice daily for 5 days after injury.
Western blotting.
For LiPilo animals:
Sprague-Dawley rats were euthanized with exposure to carbon dioxide. Following decapitation, brains were removed and the hippocampus was isolated. Protein lysates were prepared by homogenization in lysis buffer (10% SDS, 10% Tris Buffer, pH 7.5 in double distilled water) and sonicated twice at 70% for 10 seconds. Lysates were spun at 12,000 rcf for 5 minutes. Protein concentration was determined by BCA assay (Thermo Scientific). 10μg of protein were loaded and resolved on Biorad mini-protean TGX 4–20% precast gels. Proteins were transferred onto Nitrocellulose membrane (BioRad TransBlot Turbo Transfer Pack) using the BioRad TransBlot Turbo transfer system. Membranes were blocked using 1:1 Odyssey Blocking Buffer (TBS) and TBS for 1 hour. Blots were then probed with primary antibodies, washed (3 times for 10 minutes), and then probed with IR-Dye secondary antibodies (Li-Cor) at 1:20,000 for 1 hour. For primary staining: rabbit anti-Kir4.1 (Alamone) was used at 1:1,500 for 45 minutes, mouse anti-GFAP (Milipore) primary was used at 1:10,000 for 15 minutes, and mouse anti-β-actin (Abcam) was used at 1:5,000 for 45 minutes. Odyssey CLx Imaging System was used for visualization and quantification of the membranes.
For SCI animals:
Sprague-Dawley rats were euthanized with exposure to carbon dioxide. Following decapitation, spinal cords were dissected. Protein lysates were prepared by homogenization in RIPA buffer (10% SDS, 10% Tris Buffer, pH 7.5 in double distilled water) using glass dounce homogenizers, followed by 2 rounds of sonication at 70% for 10 seconds. Lysates were spun at 12,000 rcf for 5 minutes. Protein concentration was determined by BCA assay (Thermo Scientific). 10μg of protein were loaded and resolved on Biorad mini-protean TGX 4–20% precast gels. Proteins were transferred onto PVDF membrane at 100V for 60 minutes. Membranes were blocked using 10% milk in TBS-T. Blots were then probed with primary antibodies, washed (3 times for 10 minutes), and then probed with secondary antibody conjugated to horseradish peroxidase for 1 hour. For primary staining: rabbit anti-Kir4.1 (Alamone) was used at 1:1,500 for 45 minutes, mouse anti-GFAP (Milipore) primary was used at 1:10,000 for 15 minutes, and chicken anti-GAPDH (Milipore) was used at 1:5,000 for 45 minutes. Millipore Luminata Classic Western HRP substrate was used for visualization on autoradiography film.
Quantitative real time PCR (qRT-PCR).
Total mRNA and genomic DNA were isolated using the PureLink Genomic DNA Mini Kit (Invitrogen) and PureLink RNA Mini Kit (Invitrogen) sequentially. 1000 ng of mRNA was converted to cDNA using Invitrogen Superscript VILO cDNA synthesis kit. cDNA was diluted 1:3 using DEPC treated water. Applied Biosystems Taqman probes were used with Taqman Universal Mastermix II, no UNG. qPCR was performed on Applied Biosystems StepOne. Cycling parameters were: 50°C for 2 min, 95°C for 10 min, 40 repeats of 95°C for 15 seconds and 60°C for 1 minute. Actb (β –actin) was used as housekeeping gene for the LiPilo samples while Gapdh was used for the SCI samples. We used multiple housekeeping proteins and genes and while slight differences in total protein, the results of the data analysis were not different for mRNA or protein using β –actin and Gapdh. The ΔΔCt method was utilized to determine Relative Fold Expression of mRNA.
Immunohistochemistry.
Animals were anaesthetized with a peritoneal injection of ketamine (100mg/kg) and perfused with 4% paraformaldehyde solution for 25 minutes. The brain was removed and stored in 4% paraformaldehyde until ready to cut. After washing in phosphate buffered saline, 150μM sections were cut using a Vibratome (PELCO easiSlicer). Sections were placed in blocking buffer (10% goat serum and 0.3% Triton-X100 in phosphate buffered saline (PBS)) for 1h at room temperature. Primary antibodies were made in diluted blocking buffer (1:3 blocking buffer in PBS). Slices were incubated with primary antibody overnight at 4°C with gentle agitation. The sections were then washed three times in diluted phosphate buffered saline incubating with either tetramethylrhodamine isothiocyanate (TRITC) or fluorescein isothiocyanate (FITC) secondary antibodies, obtained from Molecular Probes, for 60 minutes at room temperature. The slices were washed two times with diluted blocking buffer, then incubated with 4’6-diamidino-2-phenylindole (DAPI; 10–4mg/mL; Sigma), and finally washed twice with phosphate buffered saline before being mounted onto glass coverslips. Fluorescent images were acquired with a Nikon A1 confocal system. Fluorescence levels were measured with Image J.
Electrophysiology.
Whole-cell voltage-clamp recordings from 300μm slices were obtained as previously described from coronal brain sections (Campbell et al., 2014; Kahanovitch et al., 2018; Olsen et al., 2006). Briefly, via patch pipettes were made from thin-walled (outer diameter 1.5 mm, inner diameter 1.12 mm) borosilicate glass (TW150F-4, WPI, FL) and had resistances of 5–8 MΩ when filled with K-gluconate pipette solution contained (in mM) 145 K-gluconate, 1 MgCl2, 10 EGTA, 10 Hepes sodium salt, pH adjusted to 7.3 with Tris-base. Recordings were made on the stage of an upright Zeiss Axioobserver.D1 microscope (Zeiss). Current recordings were obtained with an Axopatch 200A amplifier (Axon Instruments) signals were low-pass filtered at 1 kHz and were digitized on-line at 10–20 kHz using a Digidata 1320 digitizing board (Axon Instruments). Data acquisition and storage were conducted with the use of pClamp 10.2 (Axon Instruments). Cell capacitances and series resistances were measured directly from the amplifier, series resistance compensation adjusted to 80% to reduce voltage errors. Cells were continuously superfused with artificial cerebral spinal fluid (ACSF, in mM, NaCl 116, KCl 4.5, MgCl2 0.8, NaHCO3 26.2, glucose 11.1, HEPES 5.0) and all recordings were performed at room temperature. We utilized a reporter rat model expressing eGFP under the promoter of the gene encoding S100β, a predominantly glial Ca2+-binding protein, to assist in identifying hippocampal astrocytes. We have previously identified eGFP expression is confined to GFAP+ (Nwaobi et al., 2014).
Sanger Sequencing for SE.
Three CpG islands were identified in the Kcnj10 gene (Figure 1) using Applied Biosystems Methyl Primer Express software. Genomic DNA was bisulfite converted with the EZ DNA Methylation-Lightning Kit (Zymo Research). Amplification primers (Table 1) were designed and used to amplify bisulfite converted DNA on the Bio-Rad T100 Thermocycler using the TaKaRa EpiTaq™ HS (for bisulfite-treated DNA) kit. Cycling conditions were: 40 repeats of 95°C for 15 seconds, 52–60°C (depending on optimized annealing temperature) for 30 seconds, and 72°C for 45 seconds. Amplicons were cleaned for sequencing using ExoSAP-IT (Affymetrix) reaction. The Sanger sequencing reactions and sequence analysis were performed at the Genomics Sequencing Center (Biocomplexity Institute) at Virginia Polytechnic Institute and State University. The % methylation values were calculated by using the following equation: (observed C’s)/(observed C’s + observed T’s). Change in methylation was calculated by subtracting the average % methylation values of the sham samples from the LiPilo treated samples. The change in % methylation of LiPilo is graphed in grey bars, while standard error of sham is graphed in black.
Figure 1.
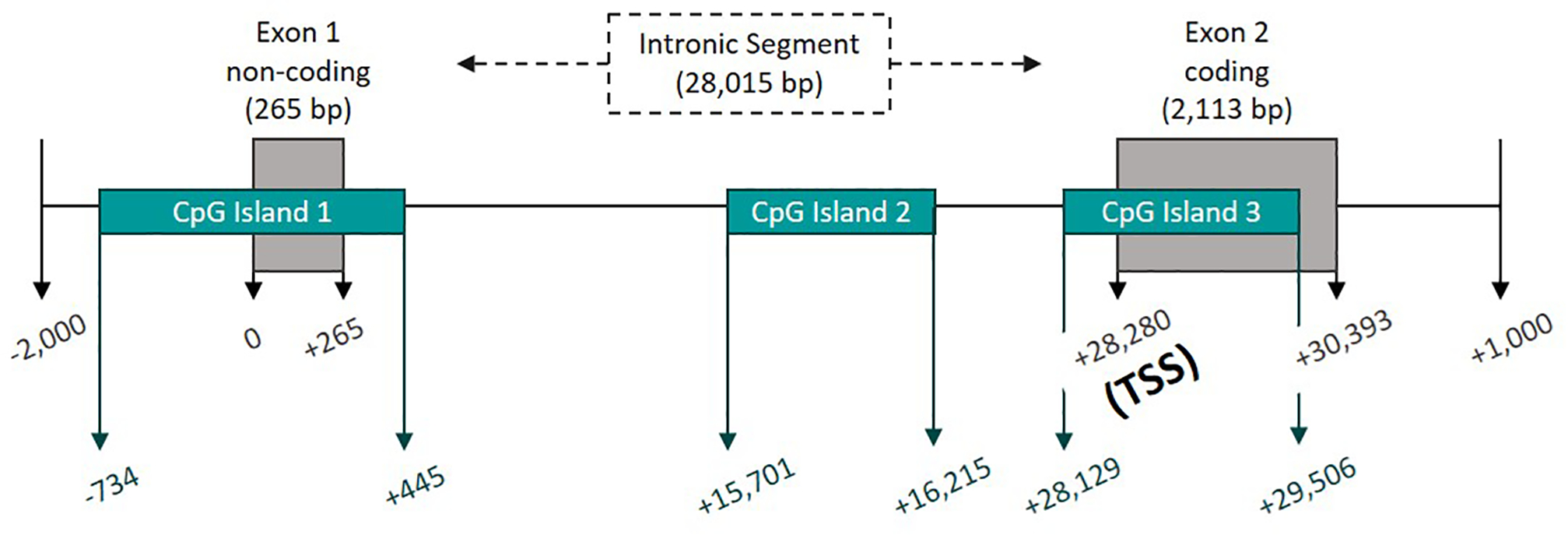
Graphical representation of the Kcnj10 gene and CpG Islands with 0 representing the start of transcription and TSS representing the translational start site.
Table 1:
Primers were designed for bisulfite treated DNA. The sequence of each primer, forward (F) and reverse (R), is listed 5’ to 3’ along with the amplicon length and the CpG sites it amplifies. These primers were used for both the amplification reaction and for Sanger sequencing.
| CpG Site | Amplification/Sequencing Primers | Amplicon Length |
|---|---|---|
|
| ||
| 1 to 2 | F: 5’ GGGGAGGGTATATAGTGGATGGGAAGATTTTG 3’ | 300 |
| R: 5’ AACAACTTCACCTAAAAAAAAAAATAAACCAAAAAAC 3’ | ||
|
| ||
| 2 to 14 | F: 5’ TTGTATYGTTTTTTGGTTTATTTTTTTTTTTAGGTG 3’ | 300 |
| R: 5’ CRACCCTATCAAAATAAAAAAACCAAAAC 3’ | ||
|
| ||
| 8 to 22 | F: 5’ TTAGGAATTTAGGTTAGAATTTAGGTTAAGTTTG 3’ | 274 |
| R: 5’ ACAAAAACATCCRAAAACTAAAATCCAAACAAAATC 3’ | ||
|
| ||
| 21 to 37 | F: 5’ TTTTTTYGGGATTTTGTTTGGATTTTAG 3’ | 258 |
| R: 5’ AATCRCCCCCTCCCTCTACAATACAAAAACC 3’ | ||
|
| ||
| 36 to 45 | F: 5’ TYGGGTTTTTTTATTTATATAAAGTTTGGTTTTTG 3’ | 153 |
| R: 5’ CTCTTAAACCCCCAACTAAAATCAAATAAAAATTTATC 3’ | ||
|
| ||
| 46 to 53 | F: 5’ AGAGTTTTGGTTTTGGTAGTGGTATAG 3’ | 300 |
| R: 5’ AACRTAAATAAAATACTCTTAATAAAAAACCCTTCC 3’ | ||
|
| ||
| 49 to 53 | F: 5’ TATTTTATTTTTGTGTTAAGGTAAGATTTTGGTTTTGG 3’ | 291 |
| R: 5’ ACCCCATACAACTACAAATATACCAAATATC 3’ | ||
|
| ||
| 54 to 66 | F: 5’ AAATATAGGAGAAAATTTTGAGGAGAGGGTATGGG 3’ | 300 |
| R: 5’ TCTATCTCTTTAAATAAAATCTCTTCCCTCC 3’ | ||
|
| ||
| 54 to 68 | F: 5’ ATATAAGGTAAGGTGGAATGTATAGGAAGGGTTG 3’ | 300 |
| R: 5’ ACCTTCCCRATATCAAAATCATATAATCAAACTTAAC 3’ | ||
|
| ||
| 56 to 68 | F: 5’ AGTATTTTTAGGAATGAGAATTAAAGGGGTAG 3’ | 293 |
| R: 5’ ACATCAAAAAACAACTAATAAAAATCAACTCTC 3’ | ||
|
| ||
| 67 to 72 | F: 5’ GAGGGTAGGYGAGTTGGTTTAGTGGGGAATAG 3’ | 275 |
| R: 5’ ATCACAAATATACCAACTCCTCTTAAATTCC 3’ | ||
|
| ||
| 71 to 72 | F: 5’ ATGTAGTAAGYGTTTAAAAGGAGGTAGAGGGAATTTG 3’ | 200 |
| R: 5’ ATCATTTAAATTTCTAAACTTCTTTACCTTTCATC 3’ | ||
|
| ||
| 73 to 77 | F: 5’ AAAGGTATTTGTTATTAAGGTTGGTTATTTGGG 3’ | 227 |
| R: 5’ AACAAAACAACRTAAAAAAAATAATTACCAAAATCC 3’ | ||
|
| ||
| 75 to 86 | F: 5’ AAGAAGGATGATGGTTTTTTTTTTTTTTGGGTGG 3’ | 239 |
| R: 5’ AACRCTTATCAACAATATACTCCATTCTCAC 3’ | ||
|
| ||
| 80 to 93 | F: 5’ GAGAGTYGGTTTTTAGTGGTTTTAGG 3’ | 300 |
| R: 5’ AAATATATACACCTACACCACACAAAAC 3’ | ||
|
| ||
| 90 to 100 | F: 5’ TTTTTGGYGTGGTGTGGTATTTGG 3’ | 296 |
| R: 5’ AATCRTCTCAACCCTCTTCTTTAACC 3’ | ||
|
| ||
| 96 to 106 | F: 5’ ATATTAGYGAGGAATGTTTTTTGGTTATTGTG 3’ | 293 |
| R: 5’ ATTAAACCRAATATTCTCACCCTCC 3’ | ||
|
| ||
| 106 to 115 | F: 5’ AAATTGTTTTAAATTTATTAGATAAAGGAGGGTGAG 3’ | 291 |
| R: 5’ TATAAACTCRTAACCCCAAAAAATCTCCTCC 3’ | ||
|
| ||
| 110 to 122 | F: 5’ GAGTTYGTGTTGATTTTAAGTGGG 3’ | 300 |
| R: 5’ ACTAATACRCACACTAAAAACACTAC 3’ | ||
|
| ||
| 113 to 122 | F: 5’ GAAAAGTTTAAGTTGGAGGAGTTATTAAGAG 3’ | 270 |
| R: 5’ AACACAAAAAAACAATAAAAAAAAAAAATAAAACTCC 3’ | ||
Pyrosequencing for SCI.
Genomic DNA was bisulfite converted with the EZ DNA Methylation-Lightning Kit (Zymo Research). Amplification primers (Table 2) were designed with a biotin-label on either the forward or reverse primer and used to amplify bisulfite converted DNA on the Applied Biosystems 7900HT. Cycling conditions were: 95°C for 10 minutes, 40 repeats of 95°C for 15 seconds and 60°C for 1 minute. 5 μL of each amplified PCR product was immobilized in 70μL of 1X Binding Buffer pH 7.6 (10mM Tris, 2M sodium chloride, 1mM EDTA, and 0.1% Tween 20), and Streptavidin Sepharose™ High Performance beads (GE healthcare). The resulting mixture was then processed with the PyroMark™ Vacuum Prep Workstation. The processed beads and single-stranded DNA were placed in a solution of 1X annealing buffer (20mM Tris, and 2mN Magnesium acetate-tetrahydrate) and specific sequencing primer (20 pmole/μL) respective to the amplification PCR primers used. The pyrosequencing reactions and sequence analyses were performed using the PyroMark™HS96 sequencer (Qiagen) and PyroMark MD software. Methylated standards were run in tandem with all samples as controls for pyrosequencing. All standards were within ±5% of expected percent methylation for all analyzed regions, except for CpG sites 54–63 and CpG sites 85–87 which demonstrated skewing towards more highly methylated states than expected (± 5%).
Table 2:
Amplification primers for pyrosequencing were designed with a biotin-label on either the forward (F) or reverse (R) primer and used to amplify bisulfite converted DNA on the Applied Biosystems 7900HT. The sequence of each primer is listed 5’ to 3’ along with the amplicon length and the CpG sites it amplifies.
| CpG Site | Amplification Primer | Amplicon Length | Pyrosequencing Primer |
|---|---|---|---|
|
| |||
| 1 to 5 | F: 5’ TTTTAGGTGAAGTTGTTGTAGGT 3’ | 108 | F: 5’ AGGTAGAGATGGGTTTTGTA 3’: 1–3 |
| R: 5’ /5biosg/ TCCCRTACAATTTCCAAAATTT 3’ | R: 5’ TATATAGGGAATAA 3’: 4–5 | ||
|
| |||
| 6 to 11 | F: 5’ TTAGGAATTTAGGTTAGAATTTAGGTTAAG 3’ | 143 | F: 5’ AGGTTAAGTTTGTTATAG 3’: 6–7 |
| R: 5 ‘/5biosg/CCTATCAAAATAAAAAAACCAAAAC 3’ | R: 5’ ATTAATTGGGGTTTAGT 3’: 8–11 | ||
|
| |||
| 12 to 25 | Not Targeted | ? | Not Targeted |
|
| |||
| 26 to 34 (26–27; 30–34 not targeted) |
F: 5’ /5biosg/ TGTATTTATTTTTTTAATTTTTAGG 3’ | 156 | F: 5’ TTTTTAGGTTYGGGTTTGGTG 3’: 28–29 |
| R:5’/5biosg/ CCCTCCCTCTACAATACAAAA 3’ | |||
|
| |||
| 35 to 43 | F: 5’ /5-biosg/ TTGGTTTTTGTATTGTAGAGGG 3’ | 135 | F: 5’ GGTTTAATTTTGTTTT 3’: 35–40 |
| R: 5’ /5’biosg/ AAACTCTTAAACCCCCAACTAA 3’ | R: 5’ GTTTTYGGTYGGTTYGATTT 3’: 41–43 | ||
|
| |||
| 44 to 49 | F: 5’ AGTTGGGGGTTTAAGAGTTTTGG 3’ | 288 | F: 5’ GGTTTTGGTAGTGGTATAGT 3’: 44–46 |
| R: 5’ /5’biosg/ AAAACCCTTCCCAAAAAAATCC 3’ | R: 5’ GGTGTTGGGTTTTATTTATTTAG 3’: 47–48 | ||
|
| |||
| 51 | F: 5’/ 5biosg/ GGGAAGGGTTTTTTATTAAGAG 3’ | ? | F: 5’ AATAAAAAACCTAAAATAAC 3’: 51 |
| R: AACCCCATACAACTACAAATATAC 3’ | |||
|
| |||
| 52 to 53 | F: 5’ ATTTGGATATATAAGGTAAGGTGGAA 3’ | 92 | F: 5’ TGATATTGTGGAGATTGG 3’: 52–53 |
| R: 5’ /5biosg/ ACTACCCCTTTAATTCTCATTCC 3’ | |||
|
| |||
| 54 to 63 | F: 5’GATTGGGCGCGTAGTATTTTTAG 3’ | 215 | 5’ TGAGAATTAAAGGGGTAGT 3’: 54–57 |
| R: 5’ /5’biosg/AACTCTATTCCCCACTAAACCAAC 3’ | 5’ TTYGTGYGTTTTYGAGTGYGTG 3’: 58–61 | ||
|
| |||
| 64 to 65 | F: 5’ GGGAAGAGATTTTATTTAAAGAGA 3’ | 145 | F: 5’ TAAAAGTAAATAGAGGGTAG 3’: 64 |
| R: 5’ /5biosg/ AATCAACTCTCTCTTTTCACCAT 3’ | R: 5’ TAAGTTTGATTATATGATTTTGAT 3’: 65 | ||
|
| |||
| 66 to 67 | F: 5’ /5biosg/ ATGGTGAAAAGAGAGAGTTGATT 3’ | 112 | F: 5’ CATTTACTCTATAAATAC 3’: 66 |
| R: 5’ AAATTCCCTCTACCTCCTTTTAA 3’ | R: 5’ CTCTACCTCCTTTTAAAC 3’: 67 | ||
|
| |||
| 68 to 70 | F: 5’ /5biosg/ GGAATGTGATGTTAGGATTGTA 3’ | ? | F: 5’ CCAACTCCTCTTAAATTC 3’: 68–70 |
| R: 5’ CTTTACCTTTCATCTACAAAAA 3’ | |||
|
| |||
| 71 to 73 (71 not targeted) |
F: 5’ /5biosg/ TTTAAGAAGGATGATGGTTTTT 3’ | 101 | F: 5’ AAAATAATTACCAAAATC 3’: 72 |
| R: 5’ TCATCTACAAAAAACAAAACAA 3’ | R: 5’ TCATCTACAAAAAACAAAACAAC 3’: 73 | ||
|
| |||
| 74 to 78 | F: 5’TTGTTTTGTTTTTTGTAGATGA 3’ | 118 | F: 5’ GTTAAGGTTTATTATAGTTAG 3’ : 74–76 |
| R: 5’/5biosg/ CTTTTATCAAAACCCTCCTC 3’ | R: 5’ TTAGTGGTTTTAGGAATA 3’: 77–78 | ||
|
| |||
| 79 to 84 | F: 5’ GAGGAGGGTTTTGATAAAAG 3’ | 162 | F: 5’ GGTTTTGATAAAAGATGG 3’ : 79–80 |
| R: 5’ /5biosg/ AAAAACCAAATACCTACAAAAAT 3’ | R: 5’ GAGTATATTGTTGATAAG 3’: 81 | ||
|
| |||
| 85 to 87 | F: 5’/5biosg/ CGATTTTTATTGATATGTAGTGGC 3’ | ? | F: 5’ ACRACCAAATACCACACCAC 3’: 85 |
| R: TTAACAAAAAATCCCAACTCCAAC 3’ | R: 5’ AACTCCAACAAATCCCC 3’: 86–87 | ||
Acute isolation of astrocytes
Male Sprague-Dawley rats (Charles River, Wilmington, MA) were anesthetized by exposure to CO2 in an enclosed chamber, and decapitated prior to removal of the brain. The brains were isolated and dissected in ice-cold aCSF (in mM): 120 NaCl, 3 KCl, 26.2 NaHCO3, 11.1 glucose, 5 Hepes, 0.2 CaCl2, 1 MgCl2, supplemented with 0.02 mMCNQX (0190, Tocris, UK), 0.02 mM AP5 (0106, Tocris, UK) and equilibrated with gaseous O2:CO2. Isolated hippocampi were minced in 1 mm pieces, dissociated enzymatically with 20 units/ml papain (Worthington Biochemical, Lakewood, NJ) according to the manufacturer’s instructions and then filtered through a 70mm BD Falcon cell strainer. The astrocyte separation procedure was performed using reagents and modified protocols from MiltenyiBiotec (Bergisch Gladbach, Germany) as previously described in Stoica et. al. (2017) and Holt and Olsen (2016). To increase the astrocytic fraction purity, microglia, myelin and oligodendrocyte precursor cells were removed using the CD11b, Myelin Removal Kit, and A2B5 beads, respectively (Miltenyi Biotec). The glutamate transporter GLT-1 was used as a marker for positive selection of astrocytes (Stoica et al., 2017). GLT-1-expressing astrocytes were pulled down from the remaining cell suspension using a polyclonal antibody against an extracellular epitope (AGC-022, Alomone Labs, Israel) and captured by anti-rabbit microbeads on magnetic columns. All samples were used immediately or stored at 80°C for later processing for RNA and genomic DNA extraction.
Statistics
All statistical tests were performed using Graphpad Prism software (San Diego, CA) and exact value of N, degrees of freedom, test value and exact p-value when >0.001 are reported in the text or appropriate figure legend. A Kruskal–Wallis was used for ANOVA and Mann–Whitney for T-tests for non-parametric data and a Welch’s corrected T-test was utilized when two groups being compared displayed unequal variances. Additionally, a Kruskal-Wallis test was performed on pyrosequencing data. A Kruskal-Wallis was chosen to consider that data represented percentages that fell outside of 30–70%. The test used for individual data sets are stated in the text. All values are reported as means ± SE with n indicating the number of animals sampled in all cases except electrophysiology where n represents the number of individual cells recording data was obtained and reported.
RESULTS
Kir4.1 undergoes acute and chronic reductions in expression following Status Epilepticus (SE)
Kir4.1 expression is significantly reduced in models of epilepsy (Ivens et al., 2007; Kinboshi et al., 2017; Nagao et al., 2013; Ohno, 2018; Zurolo et al., 2012) and after traumatic CNS injury (Olsen et al., 2010; Pivonkova et al., 2010; Stewart et al., 2010) but little is known regarding the underlying molecular mechanism contributing to Kir4.1 protein and mRNA reductions. We used a LiPilo model of SE to examine changes in Kir4.1 expression in the hippocampus during the development of epilepsy after a primary insult. We examined protein expression at acute (1 days post status (DPSE)), during the latent period (7 DPSE), and chronic (30 DPSE) time points (Kim et al., 2017). Western blot densitometric analysis using β –actin as a loading control shows significant reductions in Kir4.1 protein compared to sham treated animals at both acute time points which persisted through chronic time point (representative Western blots shown in Figure 2A, densiometric analysis shown in Figure 2B). We evaluated GFAP expression as a marker for reactive gliosis in the same tissue homogenates. While a simultaneous upregulation of GFAP protein was observed at 7 DPSE through 30 DPSE (representative Western blots shown in Figure 2A, densiometric analysis shown in Figure 2C) our data indicates that reduced expression of Kir4.1 protein is an early event and proceeds upregulation at the 24 hour time point. We next performed quantitative PCR (qPCR) to examine mRNA expression levels of the Kir4.1 gene, Kcnj10. Kir4.1 mRNA was significantly decreased by approximately 77%, 38%, and 25% at 1 DPSE, 7 DPSE, and 30 DPSE, respectively coinciding with increased GFAP mRNA at all time points (Figure 2 D and E). Given the role of Kir4.1 in astrocyte cell function and the role in maintaining neuronal excitability our data suggests early and sustained loss of Kir4.1 protein expression may contribute to the process of epileptogenesis during the latent period. Moreover, the parallel reduction in Kir4.1 protein and Kcnj10 mRNA expression levels suggests a transcriptional mechanism of regulation.
Figure 2. Reductions in Kir4.1 protein and mRNA occurs following SE both acutely and chronically.
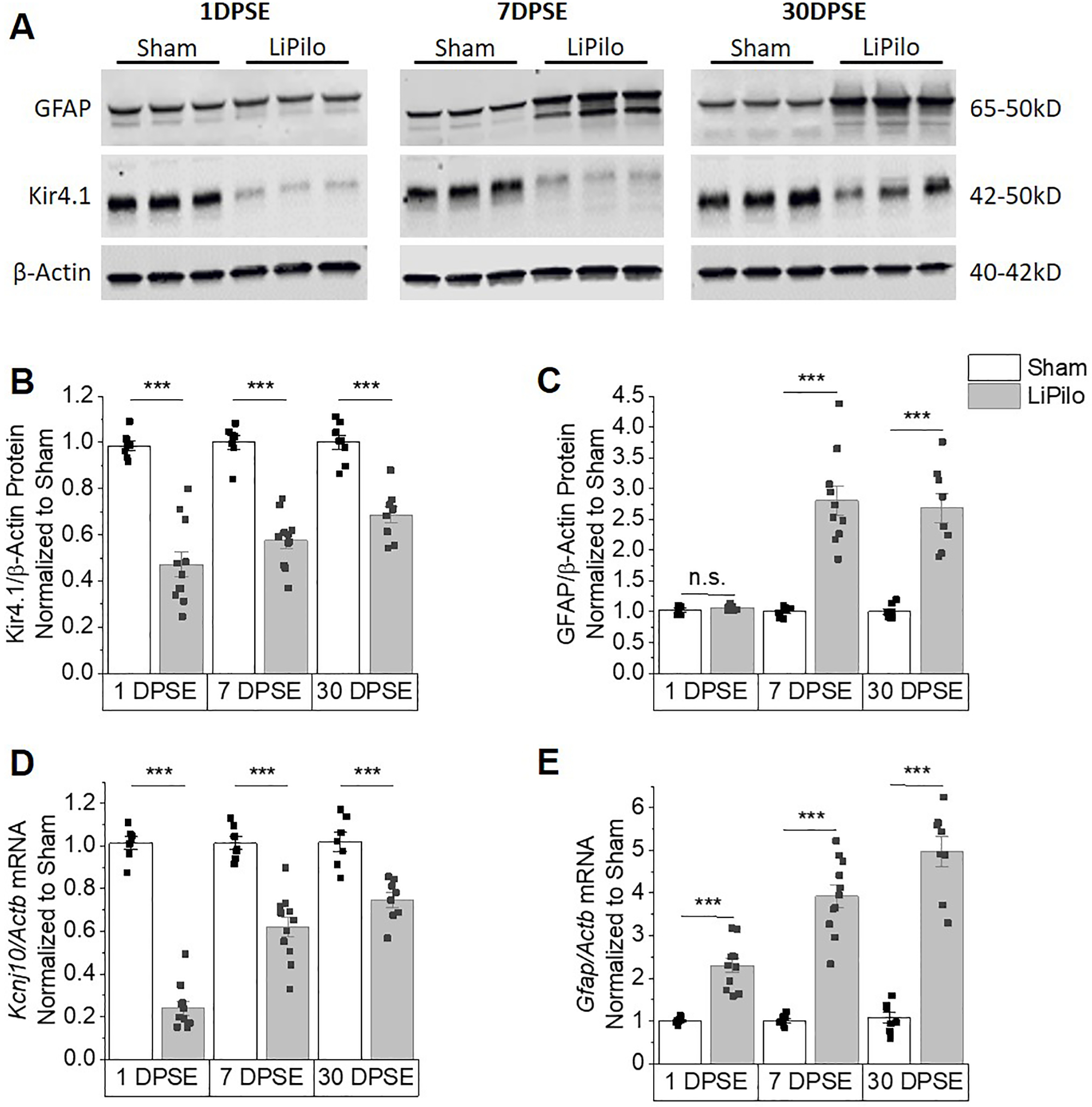
(A) Representative western blots of Kir4.1 and GFAP expression. Kir4.1 appears as a monomer at 50kDa. (B and C) Densitomertric analysis of western blots reveals significant reductions in Kir4.1 protein at 1, 7 and 30 DPSE (1DPSE WelchT12.93=8.828, p<0.001, n=7,11; 7DPSE T16=8.642, p<0.001, n=7,11 and 30DPSE T15=6.664, p<0.001, n=8,9). Upregulation of GFAP confirms post-injury gliotic response (1DPSE T13=1.212, p=0.247, n=6,9; 7DPSE WelchT9.309=7.549, p<0.001, n=7,10 and 30DPSE WelchT7.379=7.005, p<0.001, n=8,8). β –actin was used as a loading control. (D and E) Kcnj10 mRNA decreases in parallel with loss of protein expression. Loss of Kcnj10 transcripts occurs both acutely and chronically (1DPSE T16=16.92, p<0.001, n=7,11; 7DPSE T16=6.119, p<0.001, n=7,11 and 30DPSE T13=4.930, p<0.001, n=7,8). Gfap mRNA increases in parallel with the gain of protein expression both acutely and chronically (1DPSE WelchT10.57=7.680, p<0.001, n=7,11; 7DPSE WelchT10.68=10.92, p<0.001, n=7,11 and 30DPSE WelchT8.959=10.25, p<0.001, n=7,8). Error bars represent S.E.M.
Previous studies have shown various cellular abnormalities in the CA1 and CA3 regions of the hippocampus after SE, including loss of pyramidal neurons (Parent and Kron, 2012) and increases in IBA1 positive cells (Wyatt-Johnson et al., 2017). Here we demonstrate reductions in Kir4.1 protein expression in the CA1 and CA3 regions of the hippocampus. Representative images of immunostaining for Kir4.1 and GFAP in hippocampal slices at 1 DPSE, 7 DPSE, and 30 DPSE are given in Figure 3A–C (Kir4.1) and S3A–C (GFAP) respectively. For analysis the CA1, CA2 and CA3 were broken down into the pyramidal layer (PL) and the stratum (Str) while the dentate gyrus (DG) was broken down in to the granule cell layer (GCL) and the molecular layer (ML) as shown in Figure S2A. ImageJ was used to determine median pixel intensity of each region. Kir4.1 staining is decreased in the CA1 and CA3 regions but not in the CA2 or DG as early as 1DPSE continuing through 30DPSE (Figure 3 A–C; quantified in S2B). In contrast, the hippocampus shows global gliosis based on GFAP staining at 7 and 30 DPSE with the most robust increases seen in the CA1 (Figure S3A–C; quantified in S3D).
Figure 3. Kir4.1 expression is reduced in the CA1 and CA3 regions of the hippocampus post SE mirrored by reduced Kir4.1 functionality in the CA1.
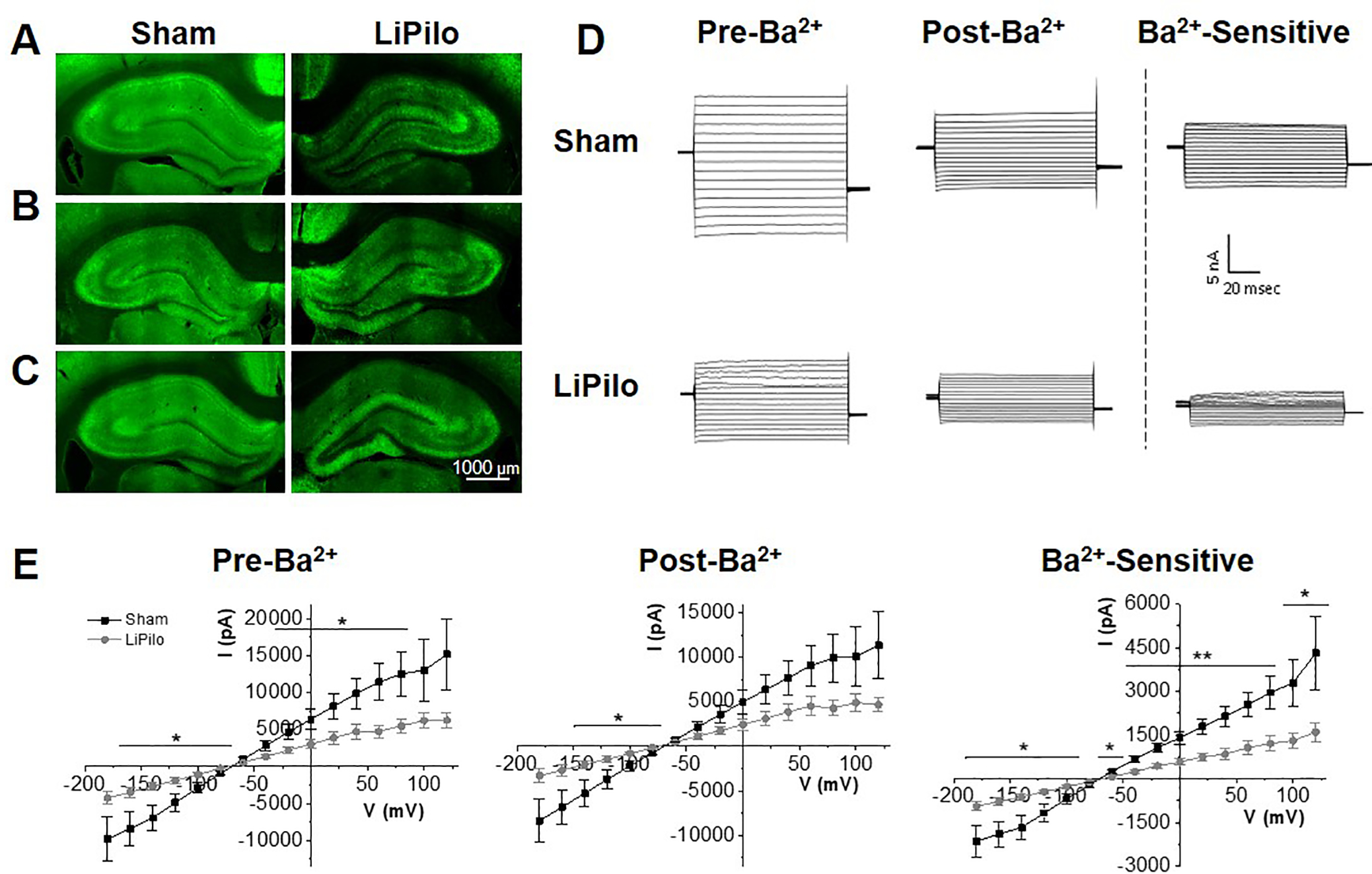
Representative images of sham and pilocarpine treated animals, stained against Kir4.1 (green). Expression of Kir4.1 at 1 DPSE (A), 7 DPSE (B), 30 DPSE (C). (D) Representative current recordings in control ACSF (pre- Ba2+), ACSF to 100 mM Ba2+ (post- Ba2+ ) and Ba2+ -sensitive traces (Ba2+-sensitive) in response to a voltage step protocol (−180 mV to +80 mV, Δ20 mV) for astrocytes in the CA1 of Sham and LiPilo animals 7–14 DPSE. (E) I-V curve of control ACSF (pre- Ba2+), ACSF to 100 mM Ba2+ (post- Ba2+ ) and Ba2+-sensitive traces (Ba2+-sensitive) in response to a voltage step protocol (−180 mV to +80 mV, Δ20 mV) for each Sham and LiPilo animals. Insets – conductance as measured by the slope between −140 mV to −60 mV (Sham n=19 cells; LiPilo n=18 cells, p<0.05) Error bars represent S.E.M.
We investigated whether the loss of Kir4.1 expression translated into functional loss of Kir4.1 channel activity. For these experiments we performed whole cell voltage-clamp recording from coronal tissue slices from either Sham or LiPilo treated animals in the CA1 region. Previous studies demonstrate that Kir4.1 mediates contributes significantly to the potassium permeability, hyperpolarized resting membrane potential and low input resistance of astrocytes (for review see, Nwaobi et al., 2016). Astrocytes were identified by their small soma size, EGFP+ expression, hyperpolarized resting membrane potential, and low input resistance. To isolate Kir4.1 currents, we stepped hippocampal CA1 astrocytes from a holding potential of –80 to 0 mV, and then from –180 to 100 mV in 20-mV increments as previously described (Campbell et al., 2014; Kahanovitch et al., 2018; Olsen et al., 2010). We washed on 100 μM BaCl2, a concentration that specifically blocks Kir channels (Ransom and Sontheimer, 1995), and performed a point by point subtraction of Ba2+ sensitive from total currents. Representative current responses (Sham, LiPilo) in control, 100 μM Ba2+ and Ba2+ sensitive currents are shown in Figure 3D. Mean data was utilized to generate a current-voltage plot (Sham: n = 19 cells, LiPilo: n = 18 cells) where a significant reduction in baseline, Ba2+-insensitive and Ba2+ -sensitive current amplitude was observed (Figure 3E). The Ba2+-sensitive conductance of the astrocytes (as represented by the slope between −140mV and −60mV) was significantly lower in the LiPilo astrocytes (Sham: 24.01 ± 3.84 nS, LiPilo: 8.60 ± 1.09 nS, F(1,181)=14, p<0.001, n=19,18, insets Figure 3E).
Although whole cell and Ba2+-sensitive current amplitude was significantly reduced in LiPilo vs. Sham animals, we observed no difference in resting membrane potential (−73.3 ± 1.5 mV and −73.9 ± 2.1 mV, T35=0.2601, p = 0.7963, n=19,18) and no significant difference in input resistance (28.5 ± 7.1 MΩ and 64.15 ± 22.7 MΩ, T34=1.499, p=0.1431, n = 19,18) between the two groups. This may be due to the remaining ~50% of Kir4.1 channels still present in astrocytes from LiPilo treated or other K+ leak channels expressed in the astrocyte membrane (Du et al., 2015; Savtchenko et al., 2018; Schools et al., 2006; Zhou et al., 2009)}. Taken together, we observed changes in Kir4.1 protein expression and function and mRNA in the hippocampus following SE in rats. Changes in gene expression occurred early and were sustained through 30 days. Our next set of experiments focus on DNA methylation as a potential regulator for loss of Kir4.1 mRNA in the LiPilo model.
Changes in DNA methylation of Kir4.1 CpG islands occurs 30 days following SE.
Our group previously demonstrated DNA methylation functions as a powerful negative regulator of Kir4.1 transcription, whereby enhanced DNA methylation at Kir4.1 CpG islands functions to decrease Kir4.1 transcription (Nwaobi et al., 2014). Given the loss of Kir4.1 protein and mRNA we posited that DNA hypermethylation may serve to negatively regulate gene transcription post SE. Thus, we examined DNA methylation levels of sham animals and animals that underwent LiPilo-induced SE at gene regions previously described to be affected by DNA methylation, CpG islands 1, 2 and 3 in whole hippocampal homogenates at 30 DPSE (diagramed in Figure 1). A schematic of each CpG island is shown with arrows denoting CpG sites that demonstrated significant changes in methylation post-injury (Figure 4 A, C and E). CpG island 1 spans the Kcnj10 gene promoter. It is generally accepted the promoters of actively transcribed genes are lowly methylated (Deaton and Bird, 2011; Maunakea et al., 2010) and this is the case for Kcnj10 (Nwaobi et al., 2014). When comparing LiPilo to Sham animals, all significant changes in the methylation status of CpG island 1 demonstrated a decrement in methylation. These data argue against methylation induced transcriptional silencing of the Kcnj10 gene promoter as a mechanism for reducing Kir4.1 transcription within our injury model. On the other hand, previous work revealed robust DNA de-methylation changes occurring in the second half of CpG island 2 (CpG sites 66 – 72) during development when transcription levels increase dramatically with advancing age (Nwaobi et al., 2014). Associated with this de-methylation is a significant reduction in a physical interaction with DNMT1 as demonstrated by ChIP analysis. Furthermore, hypermethylation of CpG island 2 of the Kcnj10 gene is sufficient to inhibit transcription (Nwaobi et al., 2014). Here we observed maximal % increase in methylation of the second half of CpG island 2 (Figure 4D, CpG sites 59 and 66–72), supporting previous work indicating this region is critical for Kcnj10 transcription. Methylation changes associated with CpG Island 3 which span the transcriptional start site and almost the entire coding region of the gene shows minimal changes in methylation (maximal change of approximately 10%) (Figure 4F).
Figure 4. Changes in methylation of Kir4.1 CpG islands occur following status epilepticus in whole hippocampal homogenate.
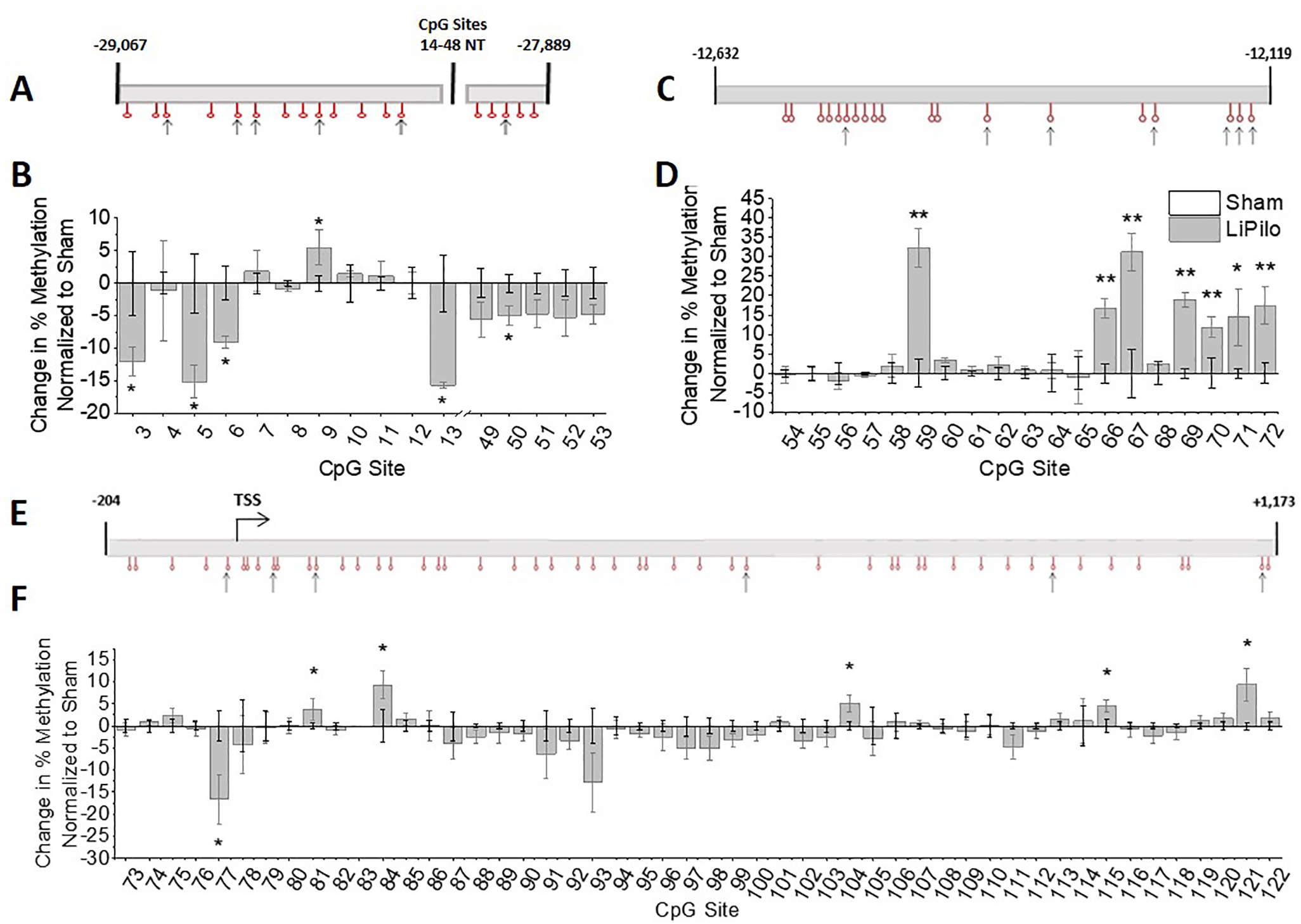
(A) Schematic of Kcnj10 gene is diagrammed for CpG island 1 with sites that undergo significant changes in methylation denoted with an arrow. (B) Change in percent methylation normalized to sham animals is graphed (CpG Site 5 T4=2.908, p=0.0438, n=3,3; CpG Site 6 T4=3.226, p=0.0321, n=3,3; Site 13 T4=6.107, p=0.004, n=3,3). (C) Schematic of Kcnj10 gene is diagrammed for CpG island 2 with sites that undergo significant changes in methylation denoted with an arrow. (D) Change in percent methylation normalized to sham animals is graphed (CpG Site 59 T10=5.346, p<0.001, n=6,6; CpG Site 66 T10=4.752, p<0.001, n=6,6; CpG Site 67 T10=3.997, p=0.0025, n=6,6; CpG Site 69 T10=8.179, p<0.001, n=6,6; CpG Site 70 T10=2.485, p=0.0323, n=6,6; CpG Site 71 WelchT4.092=2.976, p=0.0398, n=6,5; CpG Site 72 T4=3.209, p=0.0326, n=3,3). (E) Schematic of Kcnj10 gene is diagrammed for CpG island 3 with sites that undergo significant changes in methylation denoted with an arrow. (F) Change in percent methylation normalized to sham animals is graphed (CpG Site 77 T8.242=2.509, p=0.0356, n=6,6; CpG Site 84 T8=2.527, p=0.0354, n=5,5; CpG Site 104 T9=2.869, p=0.0185, n=5,6; CpG Site 115 T10=2.749, p=0.0205, n=6,6; CpG Site 121 WelchT5.542=2.641, p=0.0415, n=6,6). Error bars represent S.E.M. Note: Changes in % are calculated as: (average % methylation of experiment group) – (average % methylation of control group). Bars for the sham animals are not visible because the average changes in % methylation are 0 with the error bars representing S.E.M.
Kir4.1 downregulation in isolated astrocytes following LiPilo is accompanied by hypermethylation of CpG Island 2
Kir4.1 is also expressed in oligodendrocytes and oligodendrocyte precursor cells. Therefore, we used a modified magnetic cell separation technique to isolate astrocytes from the hippocampus of Sham and LiPilo animals 7 and 30 DPSE to assess a more astrocyte specific response as we have previously described (Holt and Olsen, 2016). Expression of cell type specific markers for astrocytes (Slc1a3, Slc1a2, and Gfap), neurons (Rbfox3), myelin (Mbp), and microglia (Itgam) were analyzed through qPCR to assess the purity of the astrocyte isolation. Astrocytic markers were enriched in isolated astrocytes compared to whole hippocampal samples while neuronal, myelin, and microglial markers were depleted in both Sham (Figure 5A) and LiPilo treated animals (Figure 5B) suggesting a relatively pure astrocyte collection. The isolated astrocytes demonstrated a significant decrease in the expression of Kcnj10 mRNA at 7 DPSE (~43%) (Figure S4A) and 30 DPSE (~75%) (Figur 5C) as well as a significant increase in Gfap mRNA expression in LiPilo treated animals at 7 and 30 DPSE compared to sham animals (Figure S4B and 5C respectively). Next, we analyzed astrocyte specific changes in methylation of CpG island 2. Based on the data obtained from whole hippocampal samples we focused on CpG island 2. Again, methylation analysis revealed significant hypermethylation (~5–15%) at 6 CpG sites (66, 67, and 69–72) compared to sham animal at 7 (Figure S4) and significant hypermethylation DPSE (~19–25% ) of 7 CpG sites (66–72) at 30 DPSE (Figure 5E), mimicking the changes seen at 30 days in whole hippocampal tissue. Interestingly, we observed hypermethylation of CpG site 68 at 30 DPSE that was not observed in whole hippocampal tissue. This may be due to methylation changes that occur exclusively in astrocytes that are masked by other CNS cell types in whole hippocampal homogenates. Together, our data indicate early and sustained loss of Kir4.1 function, protein and mRNA expression in astrocytes is associated hypermethylation of CpG island 2 of Kcnj10 in following SE injury in rats.
Figure 5. Isolated astrocytes show changes in Kir4.1 mRNA expression and changes in methylation patterns as early as 7 DPSE.
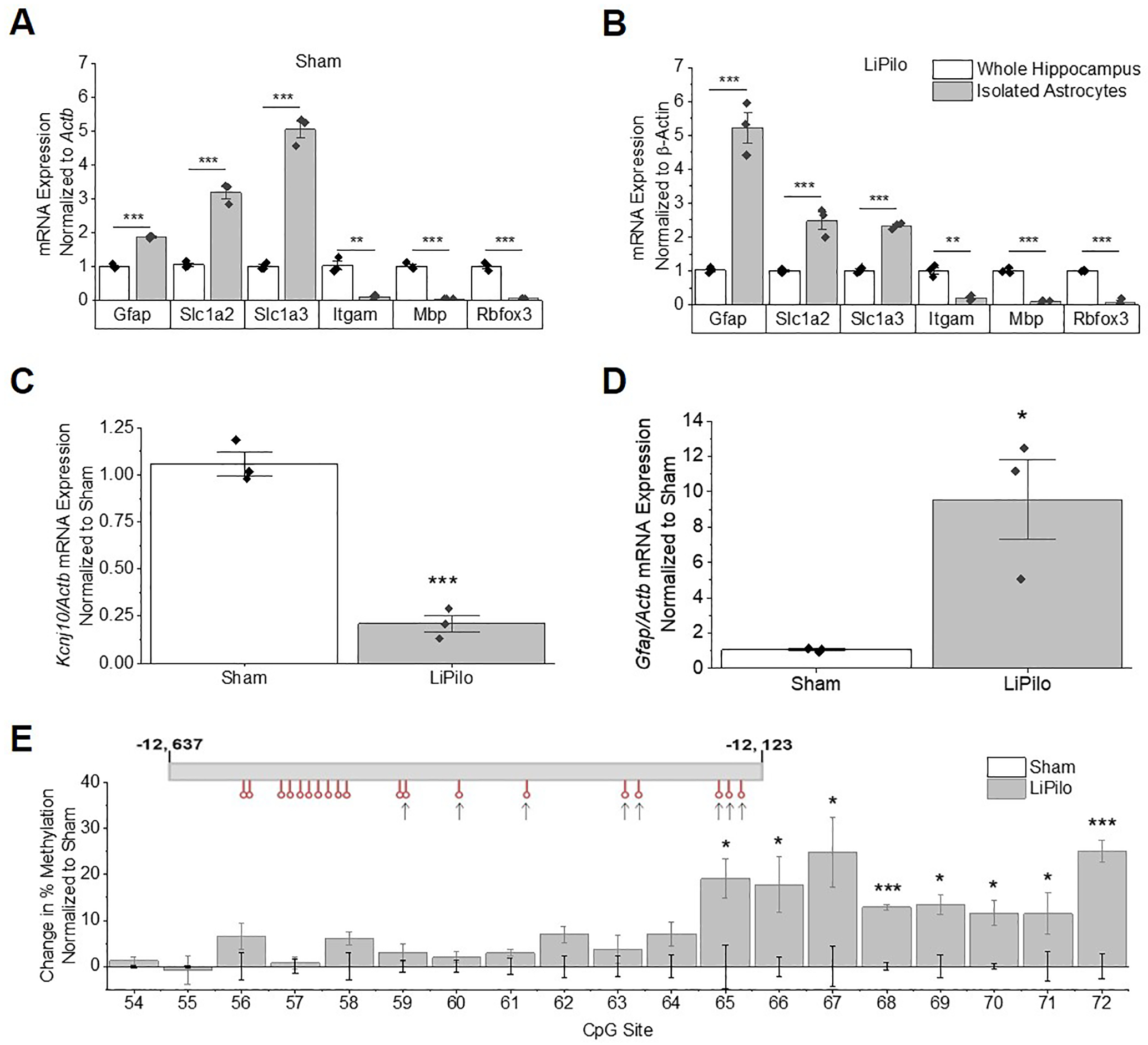
(A and B) mRNA expression of astrocytic markers (Slc1a3, Slc1a2, and Gfap) are enriched, while mRNA expression of neuronal (Rbfox3), myelin (Mbp), and microglial (Itgam) markers are depleted relative to whole hippocampal isolates of Sham (A: Gfap T4=18.77, p<0.0001, n=3,3; Slc1a2 T4=11.67, p<0.001, n=3,3; Slc1a3 T4=16.30, p<0.0001, n=3,3; Itgam T4=7.014, p=0.0022, n=3,3; Mbp WelchT2.013=17.32, p=0.0032, n=3,3; Rbfox WelchT2.011=13.50, p=0.0053, n=3,3) and LiPilo (B: Gfap T4=9.391, p<0.001, n=3,3; Slc1a2 T4=5.949, p=0.0040, n=3,3; Slc1a3 T4=20.21, p<0.0001, n=3,3; Itgam T4=7.492, p=0.0017, n=3,3; Mbp T4=15.28, p<0.001, n=3,3; Rbfox T4=15.47, p<0.001, n=3,3). (C) qPCR analysis shows Kcnj10 transcripts are reduced in isolated astrocytes at 7 DPSE (T4=10.86, p<0.001, n=3,3). (D) qPCR analysis shows Gfap transcripts are increased in isolated astrocytes at 7 DPSE (WelchT2.004=4.530, p=0.0453, n=3,3). (E) Percent change in methylation normalized to sham animals is graphed (CpG Site 65 T4=3.010, p=0.0395, n=3,3; CpG Site 66 T4=2.924, p=0.0431, n=3,3;CpG Site 67 T4=2.809, p=0.0483, n=3,3; CpG Site 68 T4=12.10, p<0.001, n=3,3; CpG Site 69 T4=4.049, p=0.0155, n=3,3; CpG Site 70 T4=4.209, p=0.0136, n=3,3; CpG Site 71 T4=2.936, p=0.0425, n=3,3; CpG Site 72 T4=7.084, p=0.0021, n=3,3). Error bars represent S.E.M.
Downregulation of Kir4.1 in a second injury model is associated with hypermethylation of CpG island 2.
As indicated above, reduced levels of Kcnj10 are commonly observed in models of epilepsy and CNS injury and are often associated with reactive gliosis. To ascertain if DNA hypermethylation may serve as a common mechanism of regulation of the Kcnj10 gene in injury we next evaluated the methylation status of Kcnj10 following trauma in a different CNS region. We and others have shown Kir4.1 expression is highest in caudal brain structures, particularly spinal cord gray matter (Kelley et al., 2018; Olsen et al., 2010; Olsen et al., 2006). During rodent postnatal development, Kcnj10 mRNA expression is higher in spinal cord relative to cortical regions and shows earlier developmental upregulation. These changes associate with earlier and more significant hypomethylation of the Kcnj10 gene (Nwaobi et al., 2014). Notably, robust demethylation associated with increased expression during development was observed in the last 8 CpG sites of CpG island 2 (Nwaobi et al., 2014). Also relevant to the current study, Kir4.1 mRNA and protein are significantly and persistently downregulated in spinal cord injury (Min et al., 2012; Najafi et al., 2016; Olsen et al., 2010). Here we utilized a clinically relevant model of SCI, a fifth cervical vertebral (C5) hemi-contusion injury (Dunham et al., 2010), to determine if DNA hypermethylation represents common mechanism of Kir4.1 down regulation between injury paradigms. Adult male rats received a moderate C5 hemi-contusion (~200 kdyn, see methods) which results in both gray and white matter damage as well as functional deficits (Dunham et al., 2010). Significant tissue loss as well as infiltration of inflammatory cells has been demonstrated to occur at the lesion (Gwak et al., 2012), which could presumably skew reductions in Kir4.1 protein expression or mRNA levels due to altered ratios of astrocytic to non-astrocytic cell populations. Thus, we examined Kir4.1 expression 3mm caudal to the lesion epicenter at 28 DPI, a region where both temporally and spatially, there are reduced numbers of proliferating cells and injury stabilization had occurred (Dunham et al., 2010). We have previously demonstrated that the loss of Kir4.1 expression following SCI lateralizes to both rostral and caudal several spinal segments (Olsen et al., 2010). A cartoon depicting the injured spinal cord and the regions examined is shown in Figure 6A. As seen in the cartoon, a 0.8mm section of the cord was injured (denoted by an “*”). 3mm of tissue was collected at the lesion epicenter as well as tissue 3mm caudal to the lesion epicenter. Western blotting demonstrates reduced Kir4.1 protein 3mm caudal to the lesion at 28 DPI. Densitometric analysis confirms significant reductions in Kir4.1 protein (~30% reduction) with as well as an upregulation of GFAP (~70% increase) (Figure 6 B and C). We observed a significant loss of Kcnj10 mRNA (~30%) (Figure 6D) and significant increases in Gfap mRNA (Figure 6E) which correlated with protein changes in this same region. The parallel reductions in both Kir4.1 protein and mRNA suggest again, a transcriptional rather than post-translational mechanism mediating altered protein expression.
Figure 6. Reductions in Kir4.1 expression extending caudally from lesion epicenter coincides with hyper-methylation of CpG island 2 of the Kcnj10 gene.
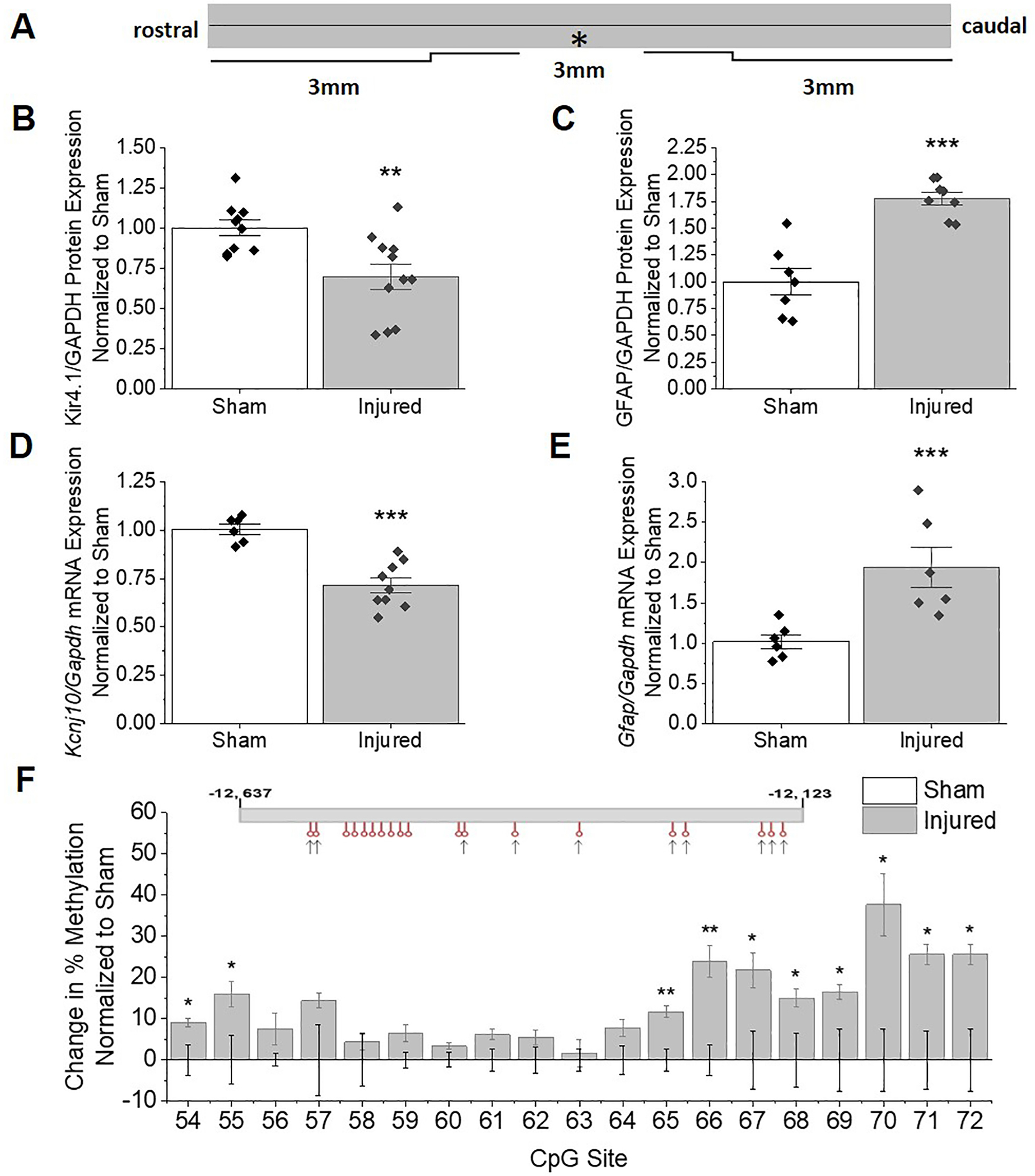
(A) Schematic of spinal cord demonstrates location of injury in relation to areas collected for analysis. (B) Western blot analysis demonstrates loss of Kir4.1 protein at 3mm section caudal to the lesion in injured animals 28 DPI compared to sham controls (T19=3.179, p=0.0049, n=10,11). (C) GFAP remains elevated at 28 DPI at 3 mm caudal from lesion (T13=5.903, p<0.0001, n=7,8). Gapdh was used as a loading control. (D and E) qPCR analysis shows Kcnj10 transcripts remain reduced at 3mm caudal to lesion at 28 DPI (T6.140=3.437, p=0.0134, n=6,6) while Gfap transcripts remain elevated (T13=5.449, p<0.001, n=6,9). Error bars represent S.E.M. (F) Schematic of Kcnj10 gene is diagrammed for CpG island 2 with sites that undergo significant increases in methylation denoted with an arrow. (G) Percent increase in methylation normalized to sham animals is graphed (CpG Site 54 T6=3.011, p=0.0237, n=3,5; CpG Site 55 T6=2.674, p=0.0368, n=3,5; CpG Site 65 T6=4.386, p=0.0046, n=3,5; CpG Site 66 T6=4.073, p=0.0066, n=3,5; CpG Site 67 T6=2.815, p=0.0307, n=3,5; CpG Site 68 T6=2.667, p=0.0372, n=3,5; CpG Site 69 WelchT2.615=3.564, p=0.0469, n=3,5; CpG Site 70 T4=3.534, p=0.0241, n=3,3; CpG Site 71 T4=3.447, p=0.0261, n=3,3; CpG Site 72 T4=3.201, p=0.0329, n=3,3). Maximal increase in % methylation of CpG island 2 occurs at CpG sites 66–70 (~25–37% increase).
We next examined DNA methylation levels of Kcnj10 via pyrosequencing of bisulfite converted DNA in sham operated and SCI injured animals at 28 days post injury rather than Sanger sequencing as performed above. Our goal in using a second method of DNA methylation analysis was to validate the robustness of the methylation response. Further, the SCI injury model represents a markedly different injury with a presumably different gliotic response. Utilizing this different injury paradigm and method of methylation analysis, we observed similar changes in post-injury methylation patterns with significant hypermethylation of the last 8 CpG sites of CpG island 2 (Figure 6F) in injured animals compared to sham-operated. CpG sites 66–72 (CpG island 2) demonstrated the largest % increase in methylation (~25–37% increase); CpG sites 75–79 (CpG island 3) demonstrated the largest % increase in methylation (~13–23% increase) (Figure S5D). Similar to the LiPilo injury, CpG island 1 underwent demethylation post SCI injury, while CpG island 3 exhibited hypermethylation (Figure S5B). A schematic of each CpG island is shown with arrows denoting CpG sites that demonstrated significant changes in methylation post-injury. Together, results obtained from these vastly different injuries, both resulting in chronic loss of Kir4.1 mRNA and protein expression with concomitant marked gliosis, indicate that sustained reductions of Kir4.1 post-injury are mediated in part by enhanced DNA methylation.
DISCUSSION
Previous work indicates DNA methylation functions to bidirectionally modulate Kir4.1 gene transcription (Nwaobi et al., 2014). Considering the ability of DNA methylation to dynamically change in the CNS following physiological and pathological stimuli (Conerly and Grady, 2010; Thompson et al., 2018) as well as the role of DNA methylation in mediating developmental increases in Kcnj10 transcription, we questioned whether DNA hypermethylation may serve as a candidate molecular mechanism for the commonly observed downregulation of Kir4.1 post CNS insult. In the current study we use two very different injury paradigms affecting two different CNS regions associated with chronic reactive gliosis to ask the question if downregulation of Kir4.1 protein and mRNA is associate with Kcnj10 hypermethylation. Using two different techniques to evaluate DNA methylation we provide evidence that chronic loss of Kir4.1 post-injury is mediated by enhanced DNA methylation of CpG island 2 and, in particular, the last 6 CpG sites (Figure 4D). Loss or downregulation of Kir4.1 in CNS pathologies is common in both astrocytes, OPCs, and oligodendrocytes (Schirmer et al., 2014; Zurolo et al., 2012). Kir4.1 in oligodendrocytes is primarily located in the perinodal areas and the inner myelin tongue allowing for juxta-axonal K+ removal (Schirmer et al., 2018). Loss of Kir4.1 in oligodendrocytes and OPCs has been shown to contribute to late onset mitochondrial damage, demyelination, axonal degeneration as well as lowering the seizure threshold (Larson et al., 2018; Neusch et al., 2001; Schirmer et al., 2018). While we do not discount the role that oligodendrocyte or OPC derived Kir4.1 may contribute to epileptogenesis our particular focus was astrocytic Kir4.1 for this study. To specifically evaluate astrocyte alterations in Kir4.1, we used a modified magnetic cell separation technique (Holt and Olsen, 2016) to isolate astrocytes from sham and SE hippocampi. These isolated astrocytes demonstrated significant hypermethylation at the same 6 CpG sites (sites 66–72) as early as 7 DPSE (Figure S4C) that continues through 30 DPSE (Figure 5E). Interestingly, these are the same CpG sites that show significant hypomethylation across development (Nwaobi et al., 2014) suggesting a mechanism of bi-directional regulation via DNA methylation on Kcnj10 transcription. More studies are needed to verify if these same changes occur in other Kir4.1 expressing cells like OPCs and oligodendrocytes, but given the consistent loss of Kir4.1 in a variety of injury models, this mechanism of regulation may be applicable to other CNS insults and, thus conceivably exploited for therapeutic benefit in a wide array of traumatic CNS injuries.
DNA methylation represents a dynamic therapeutic target in CNS injuries
DNA methylation consists of methylation of cytosines at the carbon 5 position (5-methylcytosine, 5-mC) and is one of the most common covalent modification of vertebrate genomic DNA (Bird, 2002; Shin et al., 2014). Three DNA methyltransferase (DNMT) proteins generate cytosine methylation specifically on CpG dinucleotide sites. In mammalian cells roughly 80–90% of standalone CpG sites are methylated (Hon et al., 2013; Shin et al., 2014) with little methylation of CpG sites within the confines of CpG islands (Bird, 2002; Deaton and Bird, 2011; Shin et al., 2014; Teter et al., 1996). CpG methylation (mCpG) has been shown to play critical roles in genomic imprinting, development, cellular differentiation, and X-chromosome inactivation (Gabel et al., 2015; Kinde et al., 2015). DNA methylation can physically block the binding of transcriptional regulator proteins such as transcription factors or enhancers to the gene, or increase the affinity for methyl-CpG-binding domain (MBD) proteins that can recruit additional proteins such as histone deacetylases and other chromatin remodeling proteins to affect chromatin structure (Kobow and Blumcke, 2011). In addition, the disruption of DNA methylation has been linked to human disease, including multiple cancers (Huang and Rao, 2014; Kinde et al., 2015; Kulis and Esteller, 2010), memory formation, drug addiction, and several neurodegenerative diseases (Massart et al., 2015; Morris and Monteggia, 2014; Sanchez-Mut et al., 2016). DNA methylation is chemically stable with a half-life of over a thousand years (Shen et al., 1994; Shin et al., 2014) making an attractive candidate for mediating long-term changes in the epigenome.
Several studies have observed global and gene specific changes in DNA methylation following both traumatic brain injury and models of neuropathic pain. Sagarkar et. al. showed that increases anxiety-like behaviors and decreases brain-derived neurotrophic factor (BDNF) expression after a minimal traumatic brain injury (MTBI) corresponds with increases expression of DNMT3a and DNMT3b as well as increased mCpG levels in the BDNF promotor. Importantly, blockade of DNA methylation via DNA methyltransferase inhibitor 5-aza-cytidine (5-aza) corrected the deficits in BDNF expression and reduced anxiety-like behaviors (Sagarkar et al., 2017). Focusing on another injury paradigm, several lines of evidence suggest epigenetic mechanisms such as DNA methylation play a role in neuropathic pain (Denk and McMahon, 2012). Using a chronic constriction injury of the sciatic nerve in rats to induce neuropathic pain, Wang et al., observed global increases in DNA methylation of the lumbar spinal cord of injured animals compared to sham operated animals (Wang et al., 2011). Additionally, intrathecal administration of 5-aza blocked increases in DNA methylation and was associated with concomitant reductions in neuropathic pain following injury (Wang et al., 2011). The role of epigenetics in epilepsy development is a new and emerging research area (Garriga-Canut et al., 2006; Henshall and Kobow, 2015; Kobow and Blumcke, 2011; Lubin, 2012; Qureshi and Mehler, 2010). These studies demonstrate a key role for DNA methylation in regulating the pathophysiological outcome of various injuries as well as emphasize the therapeutic benefit in targeting DNA methylation because unlike genetic mutations, epigenetic changes are potentially reversible.
DNA methylation as mechanism of Kcnj10 transcriptional regulation
While numerous studies have observed post-injury loss of Kir4.1 (Gupta and Prasad, 2013; Olsen et al., 2010; Pivonkova et al., 2010; Stewart et al., 2010), few studies have examined the mechanism which regulates Kir4.1. Recent work has suggested that Kir4.1 may be regulated by microRNAs by reducing Kcnj10 transcription in glioblastoma cell lines (Thuringer et al., 2017) and in injured human corneal epithelial cells during wound healing (Lin et al., 2013). Previous studies have also linked inflammatory cytokines to Kir4.1 expression (Arisi et al., 2015; Zurolo et al., 2012). Zurolo et al. (2012), demonstrated an IL-1β induced down regulation of Kir4.1 at the acute (1 DPSE) but not the latent time point (7 DPSE) in the cortical tissue of a model of TLE in rats. While we cannot discount the role of cytokines in the acute (1 DPSE) regulation of Kir4.1 expression, multiple cytokines, including IL-1β, are differentially regulated across multiple brain regions after pilocarpine-induced seizures (Arisi et al., 2015). IL-1β is significantly increased from sham animals at 2, 6 and 24 hours in the piriform and neocortex, but returns to sham levels by 24 hours post-SE in the hippocampus (Arisi et al., 2015). Combined, the data from Zurolo (2012) and Arisi (2015) suggest that IL-1β as well as other cytokines may be playing more of an acute role in Kir4.1 regulation. This may contribute to the striking differences we observed in Kir4.1 protein and mRNA expression at the 24 hour time point (Figure 2). Of note- we did not evaluate DNA methylation until 7 days post SE.
Our current work supports a previous indicated role for epigenetic regulation of Kir4.1 in epileptic tissue whereby Kcnj10 reductions were accompanied by histone dimethylation of the euchromatic histone-lysine N-methyltransferase 2 (G9a) enzyme (Zhang et al., 2018). It has also been reported that DNA demethylation of Kcnj10 occurs in late stage neural precursor cells specifically at the promoter allowing for transcription during astrocyte differentiation development (Hatada et al., 2008). Additionally our previous work indicates demethylation in CpG islands, including robust changes in CpG island 2 occurs during early postnatal development when Kcnj10 mRNA expression increases, while hyper-methylation of the promoter and CpG island 2 was sufficient to significantly decrease transcription (Nwaobi et al., 2014). DNA methylation patterns occurring in CpG islands have been highly studied and are considered an essential mechanism in regulating gene expression (Bird, 2002; Teter et al., 1996).
In silico analysis of KCNJ10 – Localizing regions of interest
In-silico analysis of the CpG Island 2 sequence using Promo (http://alggen.lsi.upc.es/cgi-bin/promo_v3/promo/promoinit.cgi?dirDB=TF_8.3), a virtual laboratory for the identification of presumed transcription factor binding sites (TFBS) sequences, identified over 200 potential TFBS that directly interact within the hypermethylated region of CpG island 2. Among the list of transcription factors were inflammatory mediator, NFκB; histone acetyltransferase, p300; and signal mediators STAT4 and STAT5A. Interestingly we did not identify binding sites for STAT3, a known player in astrogliosis and GFAP expression (Wang et al., 2014). Further investigation into potential transcriptional regulation of the Kcnj10 gene with the Ensembl genome browser revealed little to no annotation of the Kcnj10 gene in rats forcing us to study the mouse gene. We discovered that the region of the mouse intron that aligns with rat CpG Island 2 sits in between a promotor flanking region, and a binding site for transcriptional repressor, CTCF. CTCF or 11 zinc finger transcriptional repressor is a chromatin binding factor which mediates transcriptional regulation via binding to insulators to inhibit interactions between various DNA regulatory elements including promoters, enhancers and silencers. Furthermore, this region of DNA is adjacent to an open chromatin region. Open chromatin regions are regions in the DNA that are devoid of nucleosomes, demonstrate significant protein binding that among other functions, regulates gene transcription. Blast analysis of these two regions showed 85% homology between the rat and mouse sequences suggesting a potential mechanism for DNA methylation at CpG island 2. It is known that DNA methylation can prohibit the binding of an enhancer complexes as well or transcription factor binding resulting in reduced gene expression (Blattler et al., 2014; Hoivik et al., 2011). Future studies are needed to determine exactly how the observed increases in methylation of CpG Island 2 affects Kir4.1 mRNA expression.
Limitations of functional studies
The data presented herein indicates a loss of functional Kir4.1, as indicated by an approximate 50% decrease in Ba2+ sensitive currents in CA1 hippocampal astrocytes post pilocarpine injection, supporting decreased mRNA and protein expression. It should be noted that there are limations to astrocyte electrophysiological studies, most notably, the low input resistance of the astrocyte membrane leads to a poor voltage clamp, a well recognized problem, particularly in the slice prepartation (Larsen and MacAulay, 2014; Ma et al., 2014a; Nwaobi et al., 2016; Olsen, 2012). The instrinsic leakiness of the plama membrane, due to high expression of Kir4.1 and other K+ leak channels leads to significant differences between the command potential (Vc) and the actual holding potential (Vh) realized at distant sites on the membrane. Previous dual-patch studies have found that the actual Vh,, a mere 2.0 μm across the soma of the astrocytes is approcimately 19% of the Vc (Ma et al., 2014b)}. Thus, each 20 mV change in Vc corresponds to an actual ~3.8 mV change in voltage across the soma. In the presented experiments using a Vc from −180 mV to +100 mV corresponds to an estimated Vh of −95 mV to −46 mV (presuming EK of −80 mV). The resulting current-voltage relationship is linear over this narrow voltage range and the charactreristic inward rectification at depolarized potentials (> +40 mV) and inactivation at hyperpolarized potentials (< −140 mV) of astrocyte Kir channels (Ransom and Sontheimer, 1995)} are not observed. This voltage drop across distance of the astrocyte membrane also predicts significant space clamp issues leading us to underestimate changes in membrane current occuring at sites distal to our recording pipette.
Therapeutically targeting Kir4.1 expression in pathology
The loss of Kir4.1 protein expression in the context of CNS pathology has led to the notion that this protein represents a potential glial cell therapeatuic target. Notably, astrocyte targeted viral rescue of Kir4.1 in a mouse model of Huntington disease (R6/2), restored the astrocyte hyperpolarized RMP, normalized dysregulated extracelluar K+, decreased neuronal dysfunction in medium spiny neurons, resulted in a concomitant upregulation of the primary astrocyte glutamate transporter GLT-1 and prolonged survival in mice (Tong et al., 2014). To date- this is only targeted rescue experimental data. However, in the context of epilepsy, current antiepileptic drugs, including valproate, dose dependently increase, Kir4.1 expression in control animals (Mukai et al., 2018). It has yet to be demonstrated that this occurs in animal models of epilepsy and perhaps contributes to the therapeutic efficacy of these drugs. To date, no study has rescued Kir4.1 in an injury model or in epileptic tissue to determine if restoration of this single astrocytic protein is sufficient to confer protection or benefit. The current work indicates that manipulation of the methylation status of Kcnj10, utilizing DNA methyltransferase inhibitors, which increases Kcnj10 transcription in cultured astrocytes (Nwaobi et al., 2014) may serve as a plausible mechanism to ‘rescue’ Kir4.1 expression post CNS insult. However, this manipulation is likely to impact hundreds of genes in all CNS cell types and thus, specific manipulation of Kir4.1, via viral mediated approaches or CRISPR technology is needed.
Supplementary Material
Figure S2. Statistical analysis of Kir4.1 immunohistochemistry. (A) For analysis, the CA1, CA2 and CA3 were regions were broken down into either the pyramidal layer (PL) or stratum (Str), while the dentate gyrus (DG) was broken down into either the granual cell layer (GCL) and molecular layer (ML) for analysis. (B) Table representing median pixel intensity (A.U) ± SE of Kir4.1 staining for each region.
{kind=link}
Figure S3: GFAP expression is increased in all regions of the hippocampus at 7 and 30 DPSE. Representative images of sham and pilocarpine treated animals, stained against GFAP (red) at 1 DPSE (A), 7 DPSE (B), 30 DPSE (C). (D) Table representing median pixel intensity (A.U) ± SE of GFAP staining for each region.
{kind=link}
Figure S1: Both male and female rats show significant changes in Kir4.1 at 1 and 30 DPSE. (A) Representative western blots of Kir4.1 and GFAP expression at 1 DPSE in male and female rats. Kir4.1 appears as a monomer at 50kDa. (B-C) Densitomertric analysis of western blots reveals significant reductions in Kir4.1 protein at 1DPSE in both male and female rats (Male T4=4.351, p=0.0122, n=3,3; Female T4=3.265, p=0.0309, n=3,3) β –actin was used as a loading control. (D) Representative western blots of Kir4.1 and GFAP expression at 30 DPSE in male and female rats. Kir4.1 appears as a monomer at 50kDa. (E-F) Densitomertric analysis of western blots reveals significant changes in Kir4.1 (Male T4=6.687, p=0.0026, n=3,3; Female T4=13.42, p<0.001, n=3,3) and GFAP (Male T4=4.351, p=0.0122, n=3,3; Female T4=3.265, p=0.0309, n=3,3) protein expression at 30DPSE in both male and female rats. β –actin was used as a loading control.
{kind=link}
Figure S4: Isolated astrocytes show changes in Kir4.1 mRNA expression and changes in methylation patterns as early as 7DPSE. (A) qPCR analysis shows Kcnj10 transcripts are reduced in isolated astrocytes at 7 DPSE (WelchT4.102=3.758, p=1.0189, n=3,5). (B) qPCR analysis shows Gfap transcripts are increased in isolated astrocytes at 7 DPSE (WelchT4.024=3.101, p=0.0359, n=3,5). (C) Percent change in methylation normalized to sham animals is graphed (CpG Site 58 T5=2.962, p=0.0314, n=3,4; CpG Site 66 T4=3.104, p=0.0361, n=3,3;CpG Site 67 T5=2.739, p=0.0408, n=3,4; CpG Site 69 T5=5.466, p=0.0028, n=3,4; CpG Site 70 T4=4.633, p=0.0098, n=3,3; CpG Site 71 T4=5.663, p=0.0048, n=3,3; CpG Site 72 T4=7.020, p=0.0022, n=3,3) Error bars represent S.E.M.
{kind=link}
Figure S5: Changes in methylation occur on Kir4.1 CpG island 1 and 3 sites following SCI. (A) Schematic of CpG island 1 of Kcnj10 gene is diagrammed with sites that undergo significant decreases in methylation denoted with an arrow. (B) Percent decrease in methylation normalized to sham animals is graphed (CpG Site 5 T5=3.884, p=0.0116, n=3,4; CpG Site 46 T5=4.164, p=0.0088, n=3,4; CpG Site 47 T5=4.872, p=0.0046, n=3,4; CpG Site 51 T5=7.263, p<0.001, n=3,4); maximal decrease (~20% decrease) occurs in CpG site 51. Error bars represent S.E.M. (C) Schematic of CpG island 3 of Kcnj10 gene is diagrammed with sites that undergo significant changes in methylation denoted with an arrow. (D) Percent increase in methylation normalized to mock animals is graphed. Maximal increase in % methylation of CpG island 3 occurs at CpG sites 75–78 (~13–23% increase). (CpG Site 75 T5=4.209, p=0.0084, n=3,4; CpG Site 76 T5=6.513, p=0.0013, n=3,4; CpG Site 77 WelchT2.179=4.114, p=0.0013, n=3,4; CpG Site 78 T5=3.372, p=0.0198, n=3,4; CpG Site 79 T5=3.122, p=0.0262, n=3,4; CpG Site 80 T5=4.753, p=0.0051, n=3,4; CpG Site 81 T5=5.126, p=0.0037, n=3,4; CpG Site 82 T5=3.167, p=0.0249, n=3,4; CpG Site 83 T5=2.821, p=0.0371, n=3,4; CpG Site 84 T5=4.710, p=0.0053, n=3,4). Error bars represent S.E.M.
Key Points.
Kir4.1 mRNA and protein are chronically down regulated in the hippocampus after pilocarpine induced SE as well as in the spinal cord after SCI. Hypermethylation of CpG sites 66–72 corresponds with decreased expression of Kir4.1 in both injury models.
ACKNOWLEDGEMENTS
The authors wish to thank Kelley Dunhum, Betty Pat and the T.J. Atchinson Spinal Cord Injury Research Program for their assistance in performing spinal injuries and E. Lin at the Heflin Center for Genomic Sciences at UAB for her work in designing primers utilized in pyrosequencing and running pyrosequencing reactions and sequence analysis. This work was supported in part by NINDS R01NS075062. We would like to thank the National Bioresource Project (NBRP) for the Rat (Kyoto, Japan for generously providing S100β-eGFP+ rats.
REFERENCES
- Arisi GM, Foresti ML, Katki K, and Shapiro LA (2015). Increased CCL2, CCL3, CCL5, and IL-1beta cytokine concentration in piriform cortex, hippocampus, and neocortex after pilocarpine-induced seizures. Journal of neuroinflammation 12, 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird A (2002). DNA methylation patterns and epigenetic memory. Genes Dev 16, 6–21. [DOI] [PubMed] [Google Scholar]
- Blattler A, Yao L, Witt H, Guo Y, Nicolet CM, Berman BP, and Farnham PJ (2014). Global loss of DNA methylation uncovers intronic enhancers in genes showing expression changes. Genome Biol 15, 469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bockenhauer D, Feather S, Stanescu HC, Bandulik S, Zdebik AA, Reichold M, Tobin J, Lieberer E, Sterner C, Landoure G, et al. (2009). Epilepsy, ataxia, sensorineural deafness, tubulopathy, and KCNJ10 mutations. N. Engl. J Med 360, 1960–1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordey A, and Sontheimer H (1998). Properties of human glial cells associated with epileptic seizure foci. Epilepsy Res 32, 286–303. [DOI] [PubMed] [Google Scholar]
- Buono RJ, Lohoff FW, Sander T, Sperling MR, O’Connor MJ, Dlugos DJ, Ryan SG, Golden GT, Zhao H, Scattergood TM, et al. (2004). Association between variation in the human KCNJ10 potassium ion channel gene and seizure susceptibility. Epilepsy Res 58, 175–183. [DOI] [PubMed] [Google Scholar]
- Campbell SL, Hablitz JJ, and Olsen ML (2014). Functional changes in glutamate transporters and astrocyte biophysical properties in a rodent model of focal cortical dysplasia. Front Cell Neurosci 8, 425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conerly M, and Grady WM (2010). Insights into the role of DNA methylation in disease through the use of mouse models. Dis Model Mech 3, 290–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Ambrosio R, Maris DO, Grady MS, Winn HR, and Janigro D (1999). Impaired K(+) homeostasis and altered electrophysiological properties of post-traumatic hippocampal glia. J. Neurosci 19, 8152–8162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das A, Wallace GC, Holmes C, McDowell ML, Smith JA, Marshall JD, Bonilha L, Edwards JC, Glazier SS, Ray SK, et al. (2012). Hippocampal tissue of patients with refractory temporal lobe epilepsy is associated with astrocyte activation, inflammation, and altered expression of channels and receptors. Neuroscience 220, 237–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deaton AM, and Bird A (2011). CpG islands and the regulation of transcription. Genes Dev 25, 1010–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denk F, and McMahon SB (2012). Chronic pain: emerging evidence for the involvement of epigenetics. Neuron 73, 435–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djukic B, Casper KB, Philpot BD, Chin LS, and McCarthy KD (2007). Conditional knock-out of Kir4.1 leads to glial membrane depolarization, inhibition of potassium and glutamate uptake, and enhanced short-term synaptic potentiation. J. Neurosci 27, 11354–11365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Y, Ma B, Kiyoshi CM, Alford CC, Wang W, and Zhou M (2015). Freshly dissociated mature hippocampal astrocytes exhibit passive membrane conductance and low membrane resistance similarly to syncytial coupled astrocytes. J Neurophysiol 113, 3744–3750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunham KA, Siriphorn A, Chompoopong S, and Floyd CL (2010). Characterization of a graded cervical hemicontusion spinal cord injury model in adult male rats. J. Neurotrauma 27, 2091–2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabel HW, Kinde B, Stroud H, Gilbert CS, Harmin DA, Kastan NR, Hemberg M, Ebert DH, and Greenberg ME (2015). Disruption of DNA-methylation-dependent long gene repression in Rett syndrome. Nature 522, 89–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garriga-Canut M, Schoenike B, Qazi R, Bergendahl K, Daley TJ, Pfender RM, Morrison JF, Ockuly J, Stafstrom C, Sutula T, et al. (2006). 2-Deoxy-D-glucose reduces epilepsy progression by NRSF-CtBP-dependent metabolic regulation of chromatin structure. Nat Neurosci 9, 1382–1387. [DOI] [PubMed] [Google Scholar]
- Gilliam D, O’Brien DP, Coates JR, Johnson GS, Johnson GC, Mhlanga-Mutangadura T, Hansen L, Taylor JF, and Schnabel RD (2014). A homozygous KCNJ10 mutation in Jack Russell Terriers and related breeds with spinocerebellar ataxia with myokymia, seizures, or both. J Vet Intern Med 28, 871–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta RK, and Prasad S (2013). Early down regulation of the glial Kir4.1 and GLT-1 expression in pericontusional cortex of the old male mice subjected to traumatic brain injury. Biogerontology 14, 531–541. [DOI] [PubMed] [Google Scholar]
- Hatada I, Namihira M, Morita S, Kimura M, Horii T, and Nakashima K (2008). Astrocyte-specific genes are generally demethylated in neural precursor cells prior to astrocytic differentiation. PLoS One 3, e3189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henshall DC, and Kobow K (2015). Epigenetics and Epilepsy. Cold Spring Harb Perspect Med 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuser K, Eid T, Lauritzen F, Thoren AE, Vindedal GF, Tauboll E, Gjerstad L, Spencer DD, Ottersen OP, Nagelhus EA, et al. (2012). Loss of perivascular Kir4.1 potassium channels in the sclerotic hippocampus of patients with mesial temporal lobe epilepsy. J Neuropathol. Exp Neurol 71, 814–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinterkeuser S, Schroder W, Hager G, Seifert G, Blumcke I, Elger CE, Schramm J, and Steinhauser C (2000). Astrocytes in the hippocampus of patients with temporal lobe epilepsy display changes in potassium conductances. Eur. J. Neurosci 12, 2087–2096. [DOI] [PubMed] [Google Scholar]
- Hoivik EA, Bjanesoy TE, Mai O, Okamoto S, Minokoshi Y, Shima Y, Morohashi K, Boehm U, and Bakke M (2011). DNA methylation of intronic enhancers directs tissue-specific expression of steroidogenic factor 1/adrenal 4 binding protein (SF-1/Ad4BP). Endocrinology 152, 2100–2112. [DOI] [PubMed] [Google Scholar]
- Holt LM, and Olsen ML (2016). Novel Applications of Magnetic Cell Sorting to Analyze Cell-Type Specific Gene and Protein Expression in the Central Nervous System. PLoS One 11, e0150290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hon GC, Rajagopal N, Shen Y, McCleary DF, Yue F, Dang MD, and Ren B (2013). Epigenetic memory at embryonic enhancers identified in DNA methylation maps from adult mouse tissues. Nat Genet 45, 1198–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, and Rao A (2014). Connections between TET proteins and aberrant DNA modification in cancer. Trends Genet 30, 464–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itakura E, Odaira K, Yokoyama K, Osuna M, Hara T, and Inoue K (2007). Generation of transgenic rats expressing green fluorescent protein in S-100beta-producing pituitary folliculo-stellate cells and brain astrocytes. Endocrinology 148, 1518–1523. [DOI] [PubMed] [Google Scholar]
- Ivens S, Kaufer D, Flores LP, Bechmann I, Zumsteg D, Tomkins O, Seiffert E, Heinemann U, and Friedman A (2007). TGF-beta receptor-mediated albumin uptake into astrocytes is involved in neocortical epileptogenesis. Brain 130, 535–547. [DOI] [PubMed] [Google Scholar]
- Kahanovitch U, Cuddapah VA, Pacheco NL, Holt LM, Mulkey DK, Percy AK, and Olsen ML (2018). MeCP2 Deficiency Leads to Loss of Glial Kir4.1. eNeuro 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley KW, Ben Haim L, Schirmer L, Tyzack GE, Tolman M, Miller JG, Tsai HH, Chang SM, Molofsky AV, Yang Y, et al. (2018). Kir4.1-Dependent Astrocyte-Fast Motor Neuron Interactions Are Required for Peak Strength. Neuron 98, 306–319 e307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Choi Y, Joung HY, Choi YS, Kim HJ, Joo Y, Oh JH, Hann HJ, Cho ZH, and Lee HW (2017). Structural and Functional Alterations at Pre-Epileptic Stage Are Closely Associated with Epileptogenesis in Pilocarpine-induced Epilepsy Model. Exp Neurobiol 26, 287–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinboshi M, Mukai T, Nagao Y, Matsuba Y, Tsuji Y, Tanaka S, Tokudome K, Shimizu S, Ito H, Ikeda A, et al. (2017). Inhibition of Inwardly Rectifying Potassium (Kir) 4.1 Channels Facilitates Brain-Derived Neurotrophic Factor (BDNF) Expression in Astrocytes. Frontiers in molecular neuroscience 10, 408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinde B, Gabel HW, Gilbert CS, Griffith EC, and Greenberg ME (2015). Reading the unique DNA methylation landscape of the brain: Non-CpG methylation, hydroxymethylation, and MeCP2. Proceedings of the National Academy of Sciences of the United States of America 112, 6800–6806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobow K, and Blumcke I (2011). The methylation hypothesis: do epigenetic chromatin modifications play a role in epileptogenesis? Epilepsia 52 Suppl 4, 15–19. [DOI] [PubMed] [Google Scholar]
- Kofuji P, Ceelen P, Zahs KR, Surbeck LW, Lester HA, and Newman EA (2000). Genetic Inactivation of an Inwardly Rectifying Potassium Channel (Kir4.1 Subunit) in Mice: Phenotypic Impact in Retina. Journal of Neuroscience 20, 5733–5740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucheryavykh YV, Kucheryavykh LY, Nichols CG, Maldonado HM, Baksi K, Reichenbach A, Skatchkov SN, and Eaton MJ (2007). Downregulation of Kir4.1 inward rectifying potassium channel subunits by RNAi impairs potassium transfer and glutamate uptake by cultured cortical astrocytes. Glia 55, 274–281. [DOI] [PubMed] [Google Scholar]
- Kulis M, and Esteller M (2010). DNA methylation and cancer. Adv Genet 70, 27–56. [DOI] [PubMed] [Google Scholar]
- Larsen BR, and MacAulay N (2014). Kir4.1-mediated spatial buffering of K(+): experimental challenges in determination of its temporal and quantitative contribution to K(+) clearance in the brain. Channels (Austin) 8, 544–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson VA, Mironova Y, Vanderpool KG, Waisman A, Rash JE, Agarwal A, and Bergles DE (2018). Oligodendrocytes control potassium accumulation in white matter and seizure susceptibility. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, Boe AF, Boguski MS, Brockway KS, Byrnes EJ, et al. (2007). Genome-wide atlas of gene expression in the adult mouse brain. Nature 445, 168–176. [DOI] [PubMed] [Google Scholar]
- Lenzen KP, Heils A, Lorenz S, Hempelmann A, Hofels S, Lohoff FW, Schmitz B, and Sander T (2005). Supportive evidence for an allelic association of the human KCNJ10 potassium channel gene with idiopathic generalized epilepsy. Epilepsy Res 63, 113–118. [DOI] [PubMed] [Google Scholar]
- Lin D, Halilovic A, Yue P, Bellner L, Wang K, Wang L, and Zhang C (2013). Inhibition of miR-205 impairs the wound-healing process in human corneal epithelial cells by targeting KIR4.1 (KCNJ10). Invest Ophthalmol Vis Sci 54, 6167–6178. [DOI] [PubMed] [Google Scholar]
- Liu X, and Harada S (2013). RNA isolation from mammalian samples. Curr. Protoc. Mol. Biol Chapter 4, Unit. [DOI] [PubMed] [Google Scholar]
- Lubin FD (2012). Epileptogenesis: can the science of epigenetics give us answers? Epilepsy Curr 12, 105–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma B, Xu G, Wang W, Enyeart JJ, and Zhou M (2014a). Dual patch voltage clamp study of low membrane resistance astrocytes in situ. Mol Brain 7, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma B, Xu G, Wang W, Enyeart JJ, and Zhou M (2014b). Dual patch voltage clamp study of low membrane resistance astrocytes in situ. Mol. Brain 7, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massart R, Barnea R, Dikshtein Y, Suderman M, Meir O, Hallett M, Kennedy P, Nestler EJ, Szyf M, and Yadid G (2015). Role of DNA methylation in the nucleus accumbens in incubation of cocaine craving. J Neurosci 35, 8042–8058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maunakea AK, Nagarajan RP, Bilenky M, Ballinger TJ, D’Souza C, Fouse SD, Johnson BE, Hong C, Nielsen C, Zhao Y, et al. (2010). Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature 466, 253–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauri N, Kleiter M, Leschnik M, Hogler S, Dietschi E, Wiedmer M, Dietrich J, Henke D, Steffen F, Schuller S, et al. (2017). A Missense Variant in KCNJ10 in Belgian Shepherd Dogs Affected by Spongy Degeneration with Cerebellar Ataxia (SDCA1). G3 (Bethesda) 7, 663–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min KJ, Jeong HK, Kim B, Hwang DH, Shin HY, Nguyen AT, Kim JH, Jou I, Kim BG, and Joe EH (2012). Spatial and temporal correlation in progressive degeneration of neurons and astrocytes in contusion-induced spinal cord injury. J. Neuroinflammation 9, 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris MJ, and Monteggia LM (2014). Role of DNA methylation and the DNA methyltransferases in learning and memory. Dialogues Clin Neurosci 16, 359–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukai T, Kinboshi M, Nagao Y, Shimizu S, Ono A, Sakagami Y, Okuda A, Fujimoto M, Ito H, Ikeda A, et al. (2018). Antiepileptic Drugs Elevate Astrocytic Kir4.1 Expression in the Rat Limbic Region. Front Pharmacol 9, 845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagao Y, Harada Y, Mukai T, Shimizu S, Okuda A, Fujimoto M, Ono A, Sakagami Y, and Ohno Y (2013). Expressional analysis of the astrocytic Kir4.1 channel in a pilocarpine-induced temporal lobe epilepsy model. Front Cell Neurosci 7, 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Najafi E, Stoodley MA, Bilston LE, and Hemley SJ (2016). Inwardly rectifying potassium channel 4.1 expression in post-traumatic syringomyelia. Neuroscience 317, 23–35. [DOI] [PubMed] [Google Scholar]
- Neusch C, Papadopoulos N, Muller M, Maletzki I, Winter SM, Hirrlinger J, Handschuh M, Bahr M, Richter DW, Kirchhoff F, et al. (2006). Lack of the Kir4.1 channel subunit abolishes K+ buffering properties of astrocytes in the ventral respiratory group: impact on extracellular K+ regulation. J. Neurophysiol 95, 1843–1852. [DOI] [PubMed] [Google Scholar]
- Neusch C, Rozengurt N, Jacobs RE, Lester HA, and Kofuji P (2001). Kir4.1 Potassium Channel Subunit Is Crucial for Oligodendrocyte Development and In Vivo Myelination. Journal of Neuroscience 21, 5429–5438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nwaobi SE, Cuddapah VA, Patterson KC, Randolph AC, and Olsen ML (2016). The role of glial-specific Kir4.1 in normal and pathological states of the CNS. Acta Neuropathol 132, 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nwaobi SE, Lin E, Peramsetty SR, and Olsen ML (2014). DNA methylation functions as a critical regulator of Kir4.1 expression during CNS development. Glia 62, 411–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno Y (2018). Astrocytic Kir4.1 potassium channels as a novel therapeutic target for epilepsy and mood disorders. Neural Regen Res 13, 651–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen M (2012). Examining potassium channel function in astrocytes. Methods Mol Biol 814, 265–281. [DOI] [PubMed] [Google Scholar]
- Olsen ML, Campbell SC, McFerrin MB, Floyd CL, and Sontheimer H (2010). Spinal cord injury causes a wide-spread, persistent loss of Kir4.1 and glutamate transporter 1: benefit of 17+¦-oestradiol treatment. Brain 133, 1013–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen ML, Higashimori H, Campbell SL, Hablitz JJ, and Sontheimer H (2006). Functional expression of Kir4.1 channels in spinal cord astrocytes. Glia 53, 516–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parent JM, and Kron MM (2012). Neurogenesis and Epilepsy. In Jasper’s Basic Mechanisms of the Epilepsies, th, Noebels JL, Avoli M, Rogawski MA, Olsen RW, and Delgado-Escueta AV, eds. (Bethesda (MD)). [PubMed] [Google Scholar]
- Pitkänen A, Schwartzkroin PA, and Moshé SL (2006). Models of seizures and epilepsy (Amsterdam;: Elsevier Academic; ). [Google Scholar]
- Pivonkova H, Benesova J, Butenko O, Chvatal A, and Anderova M (2010). Impact of global cerebral ischemia on K+ channel expression and membrane properties of glial cells in the rat hippocampus. Neurochemistry International 57, 783–794. [DOI] [PubMed] [Google Scholar]
- Qureshi IA, and Mehler MF (2010). Emerging role of epigenetics in stroke: part 1: DNA methylation and chromatin modifications. Arch. Neurol 67, 1316–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransom CB, and Sontheimer H (1995). Biophysical and pharmacological characterization of inwardly rectifying K+ currents in rat spinal cord astrocytes. Journal of Neurophysiology 73, 333–346. [DOI] [PubMed] [Google Scholar]
- Reichold M, Zdebik AA, Lieberer E, Rapedius M, Schmidt K, Bandulik S, Sterner C, Tegtmeier I, Penton D, Baukrowitz T, et al. (2010). KCNJ10 gene mutations causing EAST syndrome (epilepsy, ataxia, sensorineural deafness, and tubulopathy) disrupt channel function. Proc Natl Acad Sci U. S. A 107, 14490–14495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohdin C, Gilliam D, O’Leary CA, O’Brien DP, Coates JR, Johnson GS, and Jaderlund KH (2015). A KCNJ10 mutation previously identified in the Russell group of terriers also occurs in Smooth-Haired Fox Terriers with hereditary ataxia and in related breeds. Acta Vet Scand 57, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagarkar S, Bhamburkar T, Shelkar G, Choudhary A, Kokare DM, and Sakharkar AJ (2017). Minimal traumatic brain injury causes persistent changes in DNA methylation at BDNF gene promoters in rat amygdala: A possible role in anxiety-like behaviors. Neurobiol Dis 106, 101–109. [DOI] [PubMed] [Google Scholar]
- Sanchez-Mut JV, Heyn H, Vidal E, Moran S, Sayols S, Delgado-Morales R, Schultz MD, Ansoleaga B, Garcia-Esparcia P, Pons-Espinal M, et al. (2016). Human DNA methylomes of neurodegenerative diseases show common epigenomic patterns. Transl Psychiatry 6, e718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savtchenko LP, Bard L, Jensen TP, Reynolds JP, Kraev I, Medvedev N, Stewart MG, Henneberger C, and Rusakov DA (2018). Disentangling astroglial physiology with a realistic cell model in silico. Nat Commun 9, 3554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarmeas N, Honig LS, Choi H, Cantero J, Brandt J, Blacker D, Albert M, Amatniek JC, Marder K, Bell K, et al. (2009). Seizures in Alzheimer disease: who, when, and how common? Arch. Neurol 66, 992–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schirmer L, Mobius W, Zhao C, Cruz-Herranz A, Ben Haim L, Cordano C, Shiow LR, Kelley KW, Sadowski B, Timmons G, et al. (2018). Oligodendrocyte-encoded Kir4.1 function is required for axonal integrity. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schirmer L, Srivastava R, Kalluri SR, Bottinger S, Herwerth M, Carassiti D, Srivastava B, Gempt J, Schlegel J, Kuhlmann T, et al. (2014). Differential loss of KIR4.1 immunoreactivity in multiple sclerosis lesions. Ann Neurol 75, 810–828. [DOI] [PubMed] [Google Scholar]
- Scholl UI, Choi M, Liu T, Ramaekers VT, Hausler MG, Grimmer J, Tobe SW, Farhi A, Nelson-Williams C, and Lifton RP (2009). Seizures, sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance (SeSAME syndrome) caused by mutations in KCNJ10. Proc. Natl. Acad. Sci. U. S. A 106, 5842–5847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schools GP, Zhou M, and Kimelberg HK (2006). Development of gap junctions in hippocampal astrocytes: evidence that whole cell electrophysiological phenotype is an intrinsic property of the individual cell. J Neurophysiol 96, 1383–1392. [DOI] [PubMed] [Google Scholar]
- Schroder W, Hinterkeuser S, Seifert G, Schramm J, Jabs R, Wilkin GP, and Steinhauser C (2000). Functional and molecular properties of human astrocytes in acute hippocampal slices obtained from patients with temporal lobe epilepsy. Epilepsia 41 Suppl 6, S181–S184. [DOI] [PubMed] [Google Scholar]
- Seifert G, Carmignoto G, and Steinhauser C (2010). Astrocyte dysfunction in epilepsy. Brain Res Rev 63, 212–221. [DOI] [PubMed] [Google Scholar]
- Shen JC, Rideout WM 3rd, and Jones PA (1994). The rate of hydrolytic deamination of 5-methylcytosine in double-stranded DNA. Nucleic Acids Res 22, 972–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin J, Ming GL, and Song H (2014). DNA modifications in the mammalian brain. Philos Trans R Soc Lond B Biol Sci 369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart TH, Eastman CL, Groblewski PA, Fender JS, Verley DR, Cook DG, and D’Ambrosio R (2010). Chronic dysfunction of astrocytic inwardly rectifying K+ channels specific to the neocortical epileptic focus after fluid percussion injury in the rat. J. Neurophysiol 104, 3345–3360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoica A, Larsen BR, Assentoft M, Holm R, Holt LM, Vilhardt F, Vilsen B, Lykke-Hartmann K, Olsen ML, and MacAulay N (2017). The alpha2beta2 isoform combination dominates the astrocytic Na(+) /K(+) -ATPase activity and is rendered nonfunctional by the alpha2.G301R familial hemiplegic migraine type 2-associated mutation. Glia 65, 1777–1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teter B, Rozovsky I, Krohn K, Anderson C, Osterburg H, and Finch C (1996). Methylation of the glial fibrillary acidic protein gene shows novel biphasic changes during brain development. Glia 17, 195–205. [DOI] [PubMed] [Google Scholar]
- Thompson EE, Nicodemus-Johnson J, Kim KW, Gern JE, Jackson DJ, Lemanske RF, and Ober C (2018). Global DNA methylation changes spanning puberty are near predicted estrogen-responsive genes and enriched for genes involved in endocrine and immune processes. Clin Epigenetics 10, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thuringer D, Chanteloup G, Boucher J, Pernet N, Boudesco C, Jego G, Chatelier A, Bois P, Gobbo J, Cronier L, et al. (2017). Modulation of the inwardly rectifying potassium channel Kir4.1 by the pro-invasive miR-5096 in glioblastoma cells. Oncotarget 8, 37681–37693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong X, Ao Y, Faas GC, Nwaobi SE, Xu J, Haustein MD, Anderson MA, Mody I, Olsen ML, Sofroniew MV, et al. (2014). Astrocyte Kir4.1 ion channel deficits contribute to neuronal dysfunction in Huntington’s disease model mice. Nat. Neurosci 17, 694–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Liu C, Guo QL, Yan JQ, Zhu XY, Huang CS, and Zou WY (2011). Intrathecal 5-azacytidine inhibits global DNA methylation and methyl- CpG-binding protein 2 expression and alleviates neuropathic pain in rats following chronic constriction injury. Brain Res 1418, 64–69. [DOI] [PubMed] [Google Scholar]
- Wang YN, Yang M, Yu LH, Guo J, Chen N, and He L (2014). [Leptin play the key role in astroglial differentiation of mouse neural stem cells and regulated the STAT3 signaling through Jak-STAT3 pathway]. Sichuan Da Xue Xue Bao Yi Xue Ban 45, 552–556, 562. [PubMed] [Google Scholar]
- Wilcock DM, Vitek MP, and Colton CA (2009). Vascular amyloid alters astrocytic water and potassium channels in mouse models and humans with Alzheimer’s disease. Neuroscience 159, 1055–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyatt-Johnson SK, Herr SA, and Brewster AL (2017). Status Epilepticus Triggers Time-Dependent Alterations in Microglia Abundance and Morphological Phenotypes in the Hippocampus. Front Neurol 8, 700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SP, Zhang M, Tao H, Luo Y, He T, Wang CH, Li XC, Chen L, Zhang LN, Sun T, et al. (2018). Dimethylation of Histone 3 Lysine 9 is sensitive to the epileptic activity, and affects the transcriptional regulation of the potassium channel Kcnj10 gene in epileptic rats. Mol Med Rep 17, 1368–1374. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, et al. (2014). An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci 34, 11929–11947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M, Xu G, Xie M, Zhang X, Schools GP, Ma L, Kimelberg HK, and Chen H (2009). TWIK-1 and TREK-1 are potassium channels contributing significantly to astrocyte passive conductance in rat hippocampal slices. J Neurosci 29, 8551–8564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zurolo E, de GM, Iyer A, Anink J, van Vliet EA, Heimans JJ, Reijneveld JC, Gorter JA, and Aronica E (2012). Regulation of Kir4.1 expression in astrocytes and astrocytic tumors: a role for interleukin-1 beta. J. Neuroinflammation 9, 280. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S2. Statistical analysis of Kir4.1 immunohistochemistry. (A) For analysis, the CA1, CA2 and CA3 were regions were broken down into either the pyramidal layer (PL) or stratum (Str), while the dentate gyrus (DG) was broken down into either the granual cell layer (GCL) and molecular layer (ML) for analysis. (B) Table representing median pixel intensity (A.U) ± SE of Kir4.1 staining for each region.
Figure S3: GFAP expression is increased in all regions of the hippocampus at 7 and 30 DPSE. Representative images of sham and pilocarpine treated animals, stained against GFAP (red) at 1 DPSE (A), 7 DPSE (B), 30 DPSE (C). (D) Table representing median pixel intensity (A.U) ± SE of GFAP staining for each region.
Figure S1: Both male and female rats show significant changes in Kir4.1 at 1 and 30 DPSE. (A) Representative western blots of Kir4.1 and GFAP expression at 1 DPSE in male and female rats. Kir4.1 appears as a monomer at 50kDa. (B-C) Densitomertric analysis of western blots reveals significant reductions in Kir4.1 protein at 1DPSE in both male and female rats (Male T4=4.351, p=0.0122, n=3,3; Female T4=3.265, p=0.0309, n=3,3) β –actin was used as a loading control. (D) Representative western blots of Kir4.1 and GFAP expression at 30 DPSE in male and female rats. Kir4.1 appears as a monomer at 50kDa. (E-F) Densitomertric analysis of western blots reveals significant changes in Kir4.1 (Male T4=6.687, p=0.0026, n=3,3; Female T4=13.42, p<0.001, n=3,3) and GFAP (Male T4=4.351, p=0.0122, n=3,3; Female T4=3.265, p=0.0309, n=3,3) protein expression at 30DPSE in both male and female rats. β –actin was used as a loading control.
Figure S4: Isolated astrocytes show changes in Kir4.1 mRNA expression and changes in methylation patterns as early as 7DPSE. (A) qPCR analysis shows Kcnj10 transcripts are reduced in isolated astrocytes at 7 DPSE (WelchT4.102=3.758, p=1.0189, n=3,5). (B) qPCR analysis shows Gfap transcripts are increased in isolated astrocytes at 7 DPSE (WelchT4.024=3.101, p=0.0359, n=3,5). (C) Percent change in methylation normalized to sham animals is graphed (CpG Site 58 T5=2.962, p=0.0314, n=3,4; CpG Site 66 T4=3.104, p=0.0361, n=3,3;CpG Site 67 T5=2.739, p=0.0408, n=3,4; CpG Site 69 T5=5.466, p=0.0028, n=3,4; CpG Site 70 T4=4.633, p=0.0098, n=3,3; CpG Site 71 T4=5.663, p=0.0048, n=3,3; CpG Site 72 T4=7.020, p=0.0022, n=3,3) Error bars represent S.E.M.
Figure S5: Changes in methylation occur on Kir4.1 CpG island 1 and 3 sites following SCI. (A) Schematic of CpG island 1 of Kcnj10 gene is diagrammed with sites that undergo significant decreases in methylation denoted with an arrow. (B) Percent decrease in methylation normalized to sham animals is graphed (CpG Site 5 T5=3.884, p=0.0116, n=3,4; CpG Site 46 T5=4.164, p=0.0088, n=3,4; CpG Site 47 T5=4.872, p=0.0046, n=3,4; CpG Site 51 T5=7.263, p<0.001, n=3,4); maximal decrease (~20% decrease) occurs in CpG site 51. Error bars represent S.E.M. (C) Schematic of CpG island 3 of Kcnj10 gene is diagrammed with sites that undergo significant changes in methylation denoted with an arrow. (D) Percent increase in methylation normalized to mock animals is graphed. Maximal increase in % methylation of CpG island 3 occurs at CpG sites 75–78 (~13–23% increase). (CpG Site 75 T5=4.209, p=0.0084, n=3,4; CpG Site 76 T5=6.513, p=0.0013, n=3,4; CpG Site 77 WelchT2.179=4.114, p=0.0013, n=3,4; CpG Site 78 T5=3.372, p=0.0198, n=3,4; CpG Site 79 T5=3.122, p=0.0262, n=3,4; CpG Site 80 T5=4.753, p=0.0051, n=3,4; CpG Site 81 T5=5.126, p=0.0037, n=3,4; CpG Site 82 T5=3.167, p=0.0249, n=3,4; CpG Site 83 T5=2.821, p=0.0371, n=3,4; CpG Site 84 T5=4.710, p=0.0053, n=3,4). Error bars represent S.E.M.