

Abstract
Objectives
HPV‐associated (p16+) squamous cell carcinoma of the oropharynx (OPSCC) has improved survival as compared to HPV‐negative, smoking‐associated disease. Intermediate outcomes have been noted in patients with p16+ tumors and smoking exposure. However, the extent of smoking exposure required for outcomes to decrease has not been delineated due to low failure rates and poor availability of quantitative tobacco smoke exposure data. Our primary objective is to characterize the dose‐dependent relationship between recurrence‐free survival (RFS) and tobacco smoke exposure in p16+ OPSCC and secondarily correlate tobacco smoke exposure with genomic alterations.
Methods
Single institution chart review was performed of patients diagnosed with p16+ OPSCC from 2003 to 2015. Patients were excluded if staging, treatment details, recurrence status, or smoking exposure in pack‐years were not available. Two hundred and forty‐four patients were included.
Results
Patients with 25 pack‐years or greater smoking history exhibited a dose‐dependent decrease in RFS compared to never smokers. This was robust to multivariate analysis for including staging and demographic factors. Forty‐three patients with available targeted tumor sequencing data were identified. A strong trend was observed for increased C to A transversion mutations above 25 pack‐years, which are known to be associated with exposure to tobacco smoke. Similarly, the proportion of COSMIC Signature 4 mutations were also found to be more common in patients with more than 25 pack‐years of smoking exposure.
Conclusion
Evidence‐based smoking exposure thresholds are needed to define inclusion criteria for trials of de‐escalation therapy for p16+ OPSCC. Patients with smoking exposure greater than 20 pack‐years have increased risk of recurrence and a distinct pattern of genomic alterations. Further studies are needed to delineate the potential consequences of mild smoking exposure. Smoking‐related mutational signatures may hold potential for biomarker development in p16+ OPSCC.
Level of Evidence
2B
Keywords: HPV, oropharyngeal squamous cell carcinoma, outcomes, smoking exposure
1. INTRODUCTION
Human papilloma virus (HPV)‐associated squamous cell carcinoma of the oropharynx (OPSCC) is well known to be a distinct biological and clinical entity from smoking‐associated HPV‐negative disease. P16 immunohistochemistry is the standard of care biomarker used to assess OPSCC HPV status. The prognostic differences are stark between p16− (smoking‐associated) and p16+ (HPV‐associated) disease. Accordingly, p16+ OPSCC has recently been given a distinct staging system. 1 Based on the favorable outcomes of these patients, our center as well as several others have undertaken clinical trials of de‐intensified chemoradiotherapy for p16+ OPSCC patients in effort to limit morbidity while preserving favorable outcomes. 2 , 3 , 4 , 5 Early results have been promising, demonstrating excellent outcomes with decreased morbidity. 2
However, there is increasing recognition that risk factors for OPSCC are not binary. 6 Some studies have reported that a majority of p16+ patients have some degree of smoking exposure. 5 Several studies have linked tobacco smoke exposure to poor oncologic outcomes in p16+ OPSCC. 7 , 8 , 9 Current trials of de‐intensified therapy for p16+ OPSCC have mostly used smoking exposure cutoffs of <10 pack‐years or greater than 5 years of abstinence. 2 , 3 Although these cut‐offs are reasonable, they are not evidence based. Therefore, further research is needed to validate appropriate thresholds. The effect of cumulative smoking exposure as a prognostic and predictive biomarker has been defined for other cancers 10 , 11 and although there is evidence that overall‐survival is decreased in p16+ OPSCC patients with tobacco use, 12 , 13 , 14 , 15 there is scant only data examining smoking as a continuous variable or recurrence‐free survival (RFS). To allow safe expansion of treatment de‐intensification, more data are needed in order to robustly stratify p16+ OPSCC into low and high‐risk groups.
Examining a cohort of 102 p16+ OPSCC patients, Maxwell et al demonstrated a clear increase in the risk of recurrence for current as compared to never smokers, as well as strong trends for worse outcomes for former smokers. 12 Smoking exposure of greater than 10 pack‐years was associated with worse prognosis in retrospective analysis of OPSCC patients p16+ and p16− included in RTOG 9003 and 0129. 16 In the same publication, the authors also demonstrated an association with smoking during therapy and tobacco smoke exposure level which persisted in multivariate analysis included p16 status. 13 O'Sullivan et al demonstrated decreased overall survival for p16+ OPSCC patients (n = 189) with greater than 10 pack‐years, but only a trend toward decreased RFS. 14 Lassen et al demonstrated in the MARCH‐HPV project that former or current smokers with p16+ OPSCC treated with radiation had worse progression‐free survival and decreased overall‐survival at 10 years, although no smoking quantification was available. 16
Additionally, several studies suggest an interaction between tobacco smoke exposure and other HPV‐driven disease states. Feng et al have demonstrated increased rates of not only cervical HPV infection, but also cervical intraepithelial neoplasia grade 2. 17 In another report, among females positive for cervical HPV infection, current smokers were found to have increased cervical HPV viral load as compared to nonsmoking or past smokers. 18
Therefore, considering the mounting evidence that smoking both fundamentally modifies HPV biology and alters clinical outcomes in p16+ OPSCC, a key goal in the field is to quantify relevant tobacco smoke exposure levels in p16+ OPSCC. Unfortunately, quantitative smoking exposure data is lacking in many large outcomes databases such as SEER and NCDB. As such, the level of smoke exposure level that is required to alter the risk of adverse outcomes in p16+ OPSCC remains a poorly studied yet key issue in head and neck cancer biology and clinical practice. Although it is clear that smoke exposure is a risk factor for p16+ OPSCC patients, the biological origin of this effect remains unknown.
To address these gaps in knowledge, we have retrospectively analyzed a cohort of patients with p16+ OPSCC at a single institution with the primary endpoint of RFS as a function of smoking exposure in pack‐years. As an adjunct to this analysis, we have analyzed next generation sequencing data which was available for a small subset of these patients. We believe that an analysis of how the degree of smoking exposure affects RFS as well as related mutational signatures will provide a crucial piece of evidence for designing guidelines for treatment de‐intensification as well as help elucidate the biology of HPV‐driven OPSCC.
2. MATERIALS AND METHODS
2.1. Patients and inclusion criteria
Study design was a retrospective chart review at a single high‐volume institution. Upon attainment of appropriate IRB approval for chart review, all available documented cases of oropharyngeal squamous cell carcinoma were extracted from our institutional medical record system in the period between January 1, 2003 to December 31, 2015. Inclusion criteria were patients with squamous cell carcinoma of the anatomic oropharynx, with tumors demonstrating overexpression of p16 by immunohistochemistry. Patients with p16+ OPSCC were identified out of the cases discussed at our institutions multidisciplinary head and neck tumor conference during the study period. Patients were excluded if smoking exposure in pack‐years was not documented. Patients were also excluded if diagnosis date, staging information, follow‐up time, and disease status during follow‐up period were not documented. Although treatment paradigms were evolving over the course of the study, all patients received treatment with intent to cure locoregional disease. A total of 244 patients were identified who met criteria for inclusion.
2.2. Statistical methods
All statistical analyses were performed using the R project v3.5.3. Standard descriptive statistics were reported for demographic variables, chi‐squared test was used for categorical variables and t‐test for continuous variables, unless other specified. Survival data were displayed with the Kaplan‐Meier method and analyzed with the log‐rank test. Patients were stratified by multiple thresholds of smoking exposure in total pack‐years. Patients with smoking exposure level above a given threshold were compared to those with no history of smoking.
2.3. Next generation sequencing and analyses
The UNC‐Seq targeted sequencing platform provides genomic DNA sequencing of approximately 550 human genes in fixed or frozen cancer tissue and matched germline DNA from consenting local patients. Available UNC‐Seq data from the trial LCCC1108: Development of a Tumor Molecular Analyses Program and Its Use to Support Treatment Decisions were queried to identify available molecular data from patient in the above‐described clinical cohort. Forty‐three patients were identified with available sequencing data from both the tumor and matched normal (blood).
2.4. Bioinformatics
Sequencing data were routed through an automated pipeline managed by the Lineberger Bioinformatics Core. The mutation calling workflow used paired tumor and normal libraries to detect somatic single nucleotide variations, large and small indels. Raw sequence was aligned using the BWA‐mem algorithm and refined using an in‐house Assembly Based ReAlignment process to allow for accurate alignment of complex sequence variation.
Variant effects were derived using the Variant Effect Predictor software. 19 Copy number calls were generated with the SynthEx algorithm using the tumor sequencing data and a library of 200 unmatched normal samples sequenced with the same technique. 20 A conservative approach was taken. Thirty replicates varying parameter k (number of nearest neighbor) were done per‐tumor and the model with the fewest deviations from the expected copy number of 2 was selected. Sex chromosomes were excluded.
Tumor mutational burden was approximated by reporting the total number of high‐quality variants (Phred quality score > = 30) per tumor. Tumor copy number alteration burden was estimated by the number of distinct copy‐altered genomic segments as determined by the SynthEx pipeline. 20 Tumor genomic heterogeneity was estimated by taking the median absolute deviation (MAD) of the variant allele frequency of all high‐quality mutations (presented as 100 times the MAD for clarity). 21 We used the R packages MAFtools and deconstructSigs to perform mutational analysis based on C to A single nucleotide polymorphisms and COSMIC signatures. 22 , 23
3. RESULTS
Sixty‐nine percent of patients in our p16+ OPSCC cohort reported some level of smoking exposure. Patient demographics are summarized in Table 1. Stage at presentation and treatment strategies selected were similar between patients with and without smoking exposure. Considering our institution has been involved in de‐intensification trials for OPSCC, it is expected that a majority of patients involved in these trials were in the no smoking exposure group. All de‐intensification studies at our institution have excluded patients with high levels of smoking exposure. There were no recurrences in patients involved in clinical trials who did not have any tobacco smoke exposure.
TABLE 1.
Clinical and demographic patient factors
| Non‐smoker | Ever smoker | P value | |
|---|---|---|---|
| n = 76 | n = 168 | ||
| Age (mean [SD]) | 55.51 (10.16) | 57.11 (9.29) | .229 |
| Male (%) | 65 (85.5) | 143 (85.1) | 1 |
| Race (%) | .47 | ||
| Black | 8 (10.5) | 14 (8.3) | |
| White | 67 (88.2) | 147 (87.5) | |
| Other | 1 (1.3) | 2 (1.2) | |
| Unknown | 0 (0.0) | 5 (3.0) | |
| T‐stage (%) | .78 | ||
| Tis | 0 (0.0) | 1 (0.6) | |
| T1‐2 | 51 (67.1) | 114 (67.9) | |
| T3‐4b | 25 (32.9) | 53 (31.5) | |
| N‐stage (%) | .16 | ||
| 0 | 9 (11.8) | 20 (11.9) | |
| 1 | 13 (17.1) | 13 (7.7) | |
| 2a‐c | 48 (63.2) | 116 (69.0) | |
| 3 | 6 (7.9) | 19 (11.3) | |
| M0 (%) | 76 (100.0) | 164 (97.6) | .42 |
| Treatment strategy (%) | .63 | ||
| Chemo‐radiation | 52 (68.4) | 102 (60.7) | |
| Radiation therapy | 3 (3.9) | 12 (7.1) | |
| Surgery alone | 2 (2.6) | 12 (7.1) | |
| Surgery with adjuvant treatment | 10 (13.2) | 25 (14.9) | |
| Induction chemo., chemo‐radiation | 5 (6.6) | 9 (5.4) | |
| Induction chemo., surgery | 4 (5.3) | 8 (4.8) | |
| Documented recurrence (%) | 0.12 (0.33) | 0.19 (0.39) | .17 |
| Pack‐years smoking (mean [SD]) | 0 (0.0) | 27.27 (22.24) | |
| Clinical trial participation (%) | .03 | ||
| Yes | 33 (43.4) | 45 (26.8) | |
| No | 13 (17.1) | 45 (26.8) | |
| Unknown | 30 (39.5) | 78 (46.4) |
Note: Patients were stratified by history of smoking exposure. Staging variables are reported according to AJCC7.
Examination of Kaplan‐Meier curves revealed an incremental decrease in RFS with increasing cigarette smoke exposure (Figure 1B). RFS hazard ratio estimates of smoking exposure greater than the indicated amount are shown in Figure 1A, demonstrating a strong correlation with decreasing RFS and smoking above 20 pack‐years (P = .003, Spearman). The association of >25 pack‐years of smoking to decreased RFS was found to be robust to multivariate analysis including known prognostic factors including male sex, black race, clinical node positivity, and advanced T‐stage (see Table 2).
FIGURE 1.
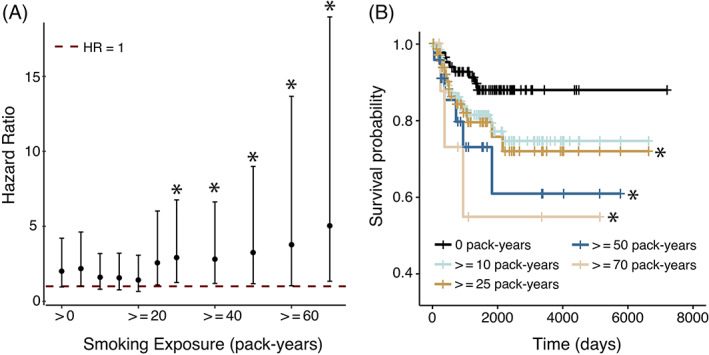
RFS among p16+ OPSCC patients by smoking exposure level. (A) Hazard ratio of patients with smoking exposure above the threshold given by x‐axis (ie, exposure is greater than or equal to (>=) than this threshold, or strictly greater than (>) zero) as compared to non‐smoking patients (ie, exposure equals 0). Error bars represent the 95% confidence interval. All subgroups of patients with 25 pack‐years or greater were found to have significantly decreased RFS. (B) Kaplan‐Meier plots showing survival as stratified by smoking exposure groups. The symbol “*” indicates a significant decrease in RFS (log‐rank test, P < .05)
TABLE 2.
Univariate and multivariate Cox regression analysis
| HR | 95% CI | P value | |
|---|---|---|---|
| Univariate | |||
| >25 pack‐years | 2.56 | 1.08‐5.96 | .03 |
| Multivariate | |||
| >25 pack‐years | 2.53 | 1.03‐6.2 | .042 |
| Male sex | 0.46 | 0.15‐1.4 | .16 |
| Black race | 1.15 | 0.25‐5.3 | .85 |
| T‐stage III‐IV | 1.72 | 0.7‐4.1 | .21 |
| N‐stage I‐III | 0.65 | 0.21‐2.0 | .44 |
Note: Non‐smokers are compared to those with greater than 25 pack‐years exposure.
No level of smoking exposure was found to be statistically significant for decreased RFS when adding clinical trial participation to the model. This is likely due to patient selection bias leading to very‐good outcomes for non‐smoking patients enrolled in clinical trials, as well as the fact that trial participation is highly confounded with selection for low tobacco smoke exposure. In the 45 patient, non‐smoking clinical trial subgroup no recurrences were observed. Furthermore, the correlation between increasing hazard‐ratio of recurrence remained highly correlated above 20 pack‐years after removing all clinical trial participants from the analysis (P = .003, Spearman).
In a subset of patients (n = 43), available next generation sequencing data from the UNCseq program was analyzed for genomic factors associated with smoking exposure. These sequenced tumors were all documented to be HPV+ by the presence of sequencing reads mapping to the HPV+ viral genome. There were no differences in specific SNP or structural variants between patients with and without smoking exposure above 25 pack‐years (data not shown). We also examined tumor mutational burden, burden of copy‐number alterations, and a gross estimate of intra‐tumor genomic heterogeneity (median‐absolute deviation of the tumor variant allele frequencies) none of which were found to be different when comparing tumors from patients with or without smoking exposure above or below 25 pack years (see Figure 2A).
FIGURE 2.
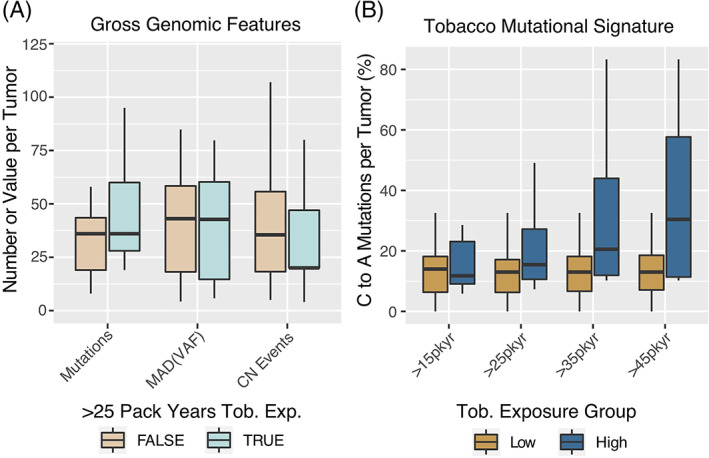
Genomic features of p16+ OPSCC tumors by smoking exposure level. (A) Genomic Features identified by the UNCseq targeted exome sequencing platform. Mutations—total number of high confidence variants per tumor. CN Events—total number of genomic copy‐number events detected by the SynthEx pipeline. MAD (VAF)—the MAD of the variant allele frequencies of all high confidence mutations per tumor. MAD (VAF)*100 is displayed for clarity. (B) C to A transversion mutations—as a function of smoking exposure. Boxplots—box represents IQR and whiskers include data within 1.5 (IQR)
We also examined the percentage of C to A transversion mutations which have been associated with mucosal exposure to tobacco smoke. 24 Although differences were not statistically significant, a strong trend was seen toward more C to A mutation in tumors with smoking exposure above 25 pack‐years. Interestingly, this followed a dose‐dependent pattern above 25 pack‐years, similar to the survival result (see Figure 2B). To more broadly include any mutation types associated with tobacco smoke exposure, we examined the COSMIC signature 4 (associated with tobacco‐related mutagenesis) and found a corresponding dose‐dependent increase in the signature strength with greater smoking exposure (Figure 3A), which was statistically significant difference (t‐test) starting at an exposure level of 25 pack‐years. 23 We found the reverse trend for COSMIC signature 5 (Figure 3B) for which the origin is unclear, is thought to be influenced by an interaction between smoking and age. 23
FIGURE 3.
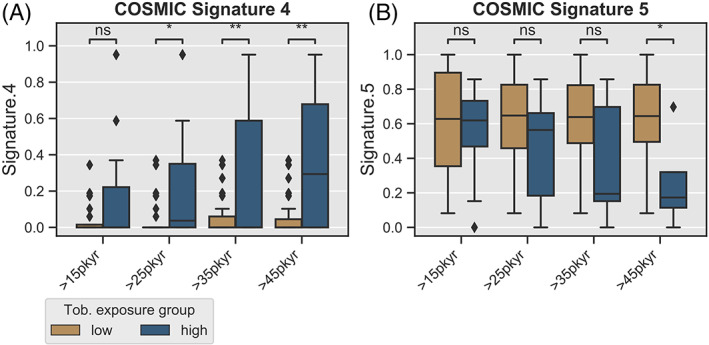
COSMIC Signatures of p16+ OPSCC tumors by smoking exposure level. (A) Proportion of COSMIC signature 4 (associated with tobacco exposure) related mutations (y‐axis) as identified by R package deconstructSigs package, as a function of smoking exposure. X‐axis represents sliding cut‐off separating samples into “high” and “low” categories. (B) COSMIC signature 5 (mutagenic mechanism unknown) vs smoking exposure. Boxplots—box represents IQR and whiskers include data within 1.5 (IQR)
4. DISCUSSION
Considering the clinical importance of risk stratifying HPV+ OPSCC patients for treatment planning and the lack of quantitative smoking annotation in most large cancer outcomes data repositories (including NCDB and SEER), there is therefore a gap to be filled by single center studies. Our results demonstrate a dose‐dependent worsening of RFS above 25 pack‐years of smoking exposure in p16+ OPSCC. In addition, there was a trend toward worse RFS in patients with any smoke exposure as compared to never‐smokers, although this did not achieve significance. Larger prospective studies are needed to better control for treatment strategy and determine more precisely the minimum level of smoking exposure which is clinically relevant. However, based on these results, patients with greater than 20 pack‐years are certainly at some significant increased risk of poor outcome and should probably not be considered as candidates for de‐escalation therapy. Our findings are highly consistent with Mirghani et al who also studied a cohort of p16+ OPSCC patients. 5 Their cohort was 56% smokers with 24% having greater than 20 pack‐years of smoking exposure. 5 More than 20 pack‐years was high associated with poor survival. 5
There is a large and robust literature describing the biochemical effects of smoking at the molecular level, particularly as it relates to carcinogenesis. Tumors arising in smoking individuals tend to have a higher total levels of somatic mutations 22 as well as indels and copy number rearrangements. 25 It has also been shown across a range of cohorts that smoking results in DNA methylation changes that persist for decades 24 and may contribute to certain tumorigenesis processes. 26 , 27 More recently, tumors from individuals with a smoking history have been shown to have an increased level of C to A transversions, consistent with nucleotide excision repair of bulky DNA adducts. 24 We found a similar trend of increased C to A transversions in the subset of our patients with tumor sequencing data available. Interestingly, this trend closely mirrored the survival data, increasing in a dose‐dependent fashion above 20 pack‐years. A positive, graduated relationship between smoking and mutational signature was also recently demonstrated in lung adenocarcinoma. 28 These results provides initial evidence that these tumors may be genetically distinct as compared to those from non‐smoking patients. However, the otherwise similarity of genomic features of HPV+ tumors from smokers and non‐smokers raises the question of whether biological factors intrinsic to the tumor or extrinsic (tumor immune interactions) may be more important for determining treatment outcome. Indeed, tumor immune infiltrate characteristics are known to be prognostic in p16+ OPSCC.
Early success of de‐intensification trials for p16+ OPSCC have been encouraging. For example, Chera et al report 100% 3‐year cause‐specific survival and locoregional control in a study of de‐intensified chemo‐radiation consisting of 60 Gray to high‐risk regions and 54 Gray to sub‐clinical regions with 30 mg/m2 Cisplatin weekly. 2 Additionally, they report 0% rate of feeding tube dependence at 1 year. Garden et al has also reported retrospectively on patients treated at a single center with less than 10 pack‐years of smoking exposure, de‐intensified to radiation alone, and report 2 and 5 year progression‐free survival rates of 90% and 80%, respectively. However, evidence also supports the need for to be careful patient selection prior to de‐intensification. For example, Cheraghlou et al reviewed the NCDB and found decreased survival of patients with AJCC8 stage II HPV+ OPSCC treated with a single modality. 20
The present study is limited by its retrospective nature, inclusion of heterogeneous treatment protocols, clinical trial enrollment, and somewhat limited size. However, the demonstration of a dose‐dependent relationship between smoking exposure above 20 pack‐years and RFS is a key finding which may contribute to robust and safe patient selection criteria for de‐intensification in the future. Larger studies are needed to examine the effects of low‐level smoking exposure where changes in clinical outcome measures are also small. Hopefully, the field will continue to work toward molecular biomarkers which may more clearly risk stratify patients and allow optimal treatment selection based on mechanistic insight.
5. CONCLUSION
Evidence‐based smoking exposure thresholds are needed to define inclusion criteria for further trials of de‐escalation therapy for p16+ OPSCC patients. Patients with p16+ tumors and smoking exposure greater than 20 pack‐years have increased risk of recurrence. Mutational signature analysis may hold potential for biomarker of smoking exposure in p16+ OPSCC. Further studies are needed to delineate the potential consequences of smoking exposure of <20 pack‐years.
CONFLICT OF INTEREST
The authors declare that they have no potential conflict of interest.
Schrank T, Weir W, Lal A, Landess L, Lenze N, Hackman T. Quantifying smoking exposure, genomic correlates, and related risk of treatment failure in p16+ squamous cell carcinoma of the oropharynx. Laryngoscope Investigative Otolaryngology. 2021;6(6):1376‐1382. doi: 10.1002/lio2.695
Funding information AAO_HNS Translational Innovator Award to T.S.; National Cancer Institute, Grant/Award Number: T32CA009156
BIBLIOGRAPHY
- 1. Zhan KY, Eskander A, Kang SY, et al. Appraisal of the AJCC 8th edition pathologic staging modifications for HPV\textminuspositive oropharyngeal cancer, a study of the National Cancer Data Base. Oral Oncol. 2017;73(Suppl 1):152‐159. doi: 10.1016/j.oraloncology.2017.08.020 [DOI] [PubMed] [Google Scholar]
- 2. Chera BS, Amdur RJ, Tepper JE, et al. Mature results of a prospective study of deintensified chemoradiotherapy for low‐risk human papillomavirus‐associated oropharyngeal squamous cell carcinoma. Cancer. 2018;124(11):2347‐2354. doi: 10.1002/cncr.31338 [DOI] [PubMed] [Google Scholar]
- 3. Samuels SE, Tao Y, Lyden T, et al. Comparisons of dysphagia and quality of life (QOL) in comparable patients with HPV‐positive oropharyngeal cancer receiving chemo‐irradiation or cetuximab‐irradiation. Oral Oncol. 2016;54(C):68‐74. doi: 10.1016/j.oraloncology.2015.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dabas S, Gupta K, Ranjan R, Sharma AK, Shukla H, Dinesh A. Oncological outcome following de‐intensification of treatment for stage I and II HPV negative oropharyngeal cancers with transoral robotic surgery (TORS): a prospective trial. Oral Oncol. 2017;69:80‐83. doi: 10.1016/j.oraloncology.2017.04.010 [DOI] [PubMed] [Google Scholar]
- 5. Mirghani H, Leroy C, Chekourry Y, et al. Smoking impact on HPV driven head and neck cancer's oncological outcomes? Oral Oncol. 2018;82(4):131‐137. doi: 10.1016/j.oraloncology.2018.05.007 [DOI] [PubMed] [Google Scholar]
- 6. Kumar B, Cordell KG, Lee JS, et al. EGFR, p16, HPV Titer, Bcl‐xL and p53, sex, and smoking as indicators of response to therapy and survival in oropharyngeal cancer. J Clin Oncol off J Am Soc Clin Oncol. 2008;26(19):3128‐3137. doi: 10.1200/JCO.2007.12.7662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Peterson LA, Bellile EL, Wolf GT, et al. Cigarette use, comorbidities, and prognosis in a prospective head and neck squamous cell carcinoma population. Head Neck. 2016;38(12):1810‐1820. doi: 10.1002/hed.24515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Anantharaman D, Billot A, Waterboer T, et al. Predictors of oropharyngeal cancer survival in Europe. Oral Oncol. 2018;81:89‐94. doi: 10.1016/j.oraloncology.2018.04.016 [DOI] [PubMed] [Google Scholar]
- 9. Chaturvedi AK, Anderson WF, Lortet‐Tieulent J, et al. Worldwide trends in incidence rates for oral cavity and oropharyngeal cancers. J Clin Oncol off J Am Soc Clin Oncol. 2013;31(36):4550‐4559. doi: 10.1200/JCO.2013.50.3870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kim C, Gao R, Sei E, et al. Chemoresistance evolution in triple‐negative breast cancer delineated by single‐cell sequencing. Cell. 2018;173(4):879‐893.e13. doi: 10.1016/j.cell.2018.03.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rink M, Furberg H, Zabor EC, et al. Impact of smoking and smoking cessation on oncologic outcomes in primary non–muscle‐invasive bladder cancer. Eur Urol. 2013;63(4):724‐732. doi: 10.1016/j.eururo.2012.08.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Maxwell JH, Kumar B, Feng FY, et al. Tobacco use in human papillomavirus‐positive advanced oropharynx cancer patients related to increased risk of distant metastases and tumor recurrence. Clin Cancer Res Off J Am Assoc Cancer Res. 2010;16(4):1226‐1235. doi: 10.1158/1078-0432.CCR-09-2350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gillison ML, Zhang Q, Jordan R, et al. Tobacco smoking and increased risk of death and progression for patients with p16‐positive and p16‐negative oropharyngeal cancer. J Clin Oncol Off J Am Soc Clin Oncol. 2012;30(17):2102‐2111. doi: 10.1200/JCO.2011.38.4099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. O'Sullivan B, Huang SH, Siu LL, et al. Deintensification candidate subgroups in human papillomavirus‐related oropharyngeal cancer according to minimal risk of distant metastasis. J Clin Oncol Off J Am Soc Clin Oncol. 2013;31(5):543‐550. doi: 10.1200/JCO.2012.44.0164 [DOI] [PubMed] [Google Scholar]
- 15. Vawda N, Banerjee RN, Debenham BJ. Impact of smoking on outcomes of HPV‐related oropharyngeal cancer treated with primary radiation or surgery. Int J Radiat Oncol. 2019;103(5):1125‐1131. doi: 10.1016/j.ijrobp.2018.11.046 [DOI] [PubMed] [Google Scholar]
- 16. Lassen P, Lacas B, Pignon J‐P, et al. Prognostic impact of HPV‐associated p16‐expression and smoking status on outcomes following radiotherapy for oropharyngeal cancer: the MARCH‐HPV project. Radiother Oncol J Eur Soc Ther Radiol Oncol. 2018;126(1):107‐115. doi: 10.1016/j.radonc.2017.10.018 [DOI] [PubMed] [Google Scholar]
- 17. Feng RM, Hu SY, Zhao FH, et al. Role of active and passive smoking in high‐risk human papillomavirus infection and cervical intraepithelial neoplasia grade 2 or worse. J Gynecol Oncol. 2017;28(5):e47. doi: 10.3802/jgo.2017.28.e47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Xi LF, Koutsky LA, Castle PE, et al. Relationship between cigarette smoking and human papilloma virus types 16 and 18 DNA load. Cancer Epidemiol Biomark Prev Publ Am Assoc Cancer Res Cosponsored Am Soc Prev Oncol. 2009;18(12):3490‐3496. doi: 10.1158/1055-9965.EPI-09-0763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. The Ensembl Variant Effect Predictor | Genome Biology | Full Text. Accessed June 11, 2021. https://genomebiology.biomedcentral.com/articles/10.1186/s13059-016-0974-4
- 20. Silva GO, Siegel MB, Mose LE, et al. SynthEx: a synthetic‐normal‐based DNA sequencing tool for copy number alteration detection and tumor heterogeneity profiling. Genome Biol. 2017;18(1):66. doi: 10.1186/s13059-017-1193-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rocco JW. Mutant allele tumor heterogeneity (MATH) and head and neck squamous cell carcinoma. Head Neck Pathol. 2015;9(1):1‐5. doi: 10.1007/s12105-015-0617-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mayakonda A, Lin D, Assenov Y, Plass C, Koeffler PH. Maftools: efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018;28:1747‐1756. doi: 10.1101/gr.239244.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rosenthal R. DeconstructSigs: Identifies Signatures Present in a Tumor Sample; 2016. https://CRAN.R-project.org/package=deconstructSigs
- 24. Alexandrov LB, Ju YS, Haase K, et al. Mutational signatures associated with tobacco smoking in human cancer. Science. 2016;354(6312):618‐622. doi: 10.1126/science.aag0299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gibbons DL, Byers LA, Kurie JM. Smoking, p53 mutation, and lung cancer. Mol Cancer Res. 2014;12(1):3‐13. doi: 10.1158/1541-7786.MCR-13-0539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chen Y, Widschwendter M, Teschendorff AE. Systems‐epigenomics inference of transcription factor activity implicates aryl‐hydrocarbon‐receptor inactivation as a key event in lung cancer development. Genome Biol. 2017;18(1):236‐218. doi: 10.1186/s13059-017-1366-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Herceg Z, Ambatipudi S. Smoking‐associated DNA methylation changes: no smoke without fire. Epigenomics. 2019;11(10):1117‐1119. doi: 10.2217/epi-2019-0136 [DOI] [PubMed] [Google Scholar]
- 28. Chen Z, Wen W, Cai Q, et al. From tobacco smoking to cancer mutational signature: a mediation analysis strategy to explore the role of epigenetic changes. BMC Cancer. 2020;20(1):880‐811. doi: 10.1186/s12885-020-07368-1 [DOI] [PMC free article] [PubMed] [Google Scholar]