

Abstract
Background
Frogeye leaf spot (FLS) is a destructive fungal disease that affects soybean production. The most economical and effective strategy to control FLS is the use of resistant cultivars. However, the use of a limited number of resistant loci in FLS management will be countered by the emergence of new high-virulence Cercospora sojina races. Therefore, we identified quantitative trait loci (QTL) that control resistance to FLS and identified novel resistant genes using a genome-wide association study (GWAS) on 234 Chinese soybean cultivars.
Results
A total of 30,890 single nucleotide polymorphism (SNP) markers were used to estimate linkage disequilibrium (LD) and population structure. The GWAS results showed four loci (p < 0.0001) distributed over chromosomes (Chr.) 5 and 20, that are significantly associated with FLS resistance. No previous studies have reported resistance loci in these regions. Subsequently, 45 genes in the two resistance-related haplotype blocks were annotated. Among them, Glyma20g31630 encoding pyruvate dehydrogenase (PDH), Glyma05g28980, which encodes mitogen-activated protein kinase 7 (MPK7), and Glyma20g31510, Glyma20g31520 encoding calcium-dependent protein kinase 4 (CDPK4) in the haplotype blocks deserves special attention.
Conclusions
This study showed that GWAS can be employed as an effective strategy for identifying disease resistance traits in soybean and narrowing SNPs and candidate genes. The prediction of candidate genes in the haplotype blocks identified by disease resistance loci can provide a useful reference to study systemic disease resistance.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12870-021-03366-y.
Keywords: Soybean, Cercospora sojina, Genome-wide association study, Resistant haplotype, Frogeye leaf spot resistant genes
Background
In most soybean-growing countries, soybeans are prone to many plant diseases. Among these, frogeye leaf spot (FLS) caused by the fungus Cercospora sojina Hara (C. sojina) is one of the most economically harmful [1]. In the main soybean-producing areas of Northeast China [2], the local temperature and leaf wetness periods are very suitable for the occurrence of FLS and epidemics [3]. Because the disease is polycyclic, a suitable environment and improper control measures can result in outbreaks [4]. Although FLS can be controlled by fungicides, there are challenges, such as fungicide resistance and environmental pollution [5]. Thus, the development of resistant cultivars is a preferred disease management strategy. However, the disadvantage of this strategy is that the resistance of cultivars may be rapidly lost. The main reason for this is that resistance mechanisms can be overcome by the emergence of new C. sojina pathotypes [6]. Therefore, high-density markers and methods to counter new pathotype races are key to disease resistance breeding.
Rcs1 [7] (resistant to C. sojina race 1), Rcs2 [8], and Rcs3 [9] have been recognised by the Soybean Committee [1]. Although the selection pressures on C. sojina populations caused by planting resistant cultivars have produced FLS strains that have overcome the Rcs1 and Rcs2 genes [9], the Rcs3 gene continues to confer resistance against most races of C. sojina in the U.S.A. [10]. Quantitative trait loci (QTL) were mapped close to a known resistance gene cluster on the soybean linkage group (LG) J by restriction fragment length polymorphism (RFLP) and simple sequence repeats (SSR) [11]. The gene was mapped in the 2-centimorgan (cM) interval between SSR markers Satt244 and Satt547. Several Phialophora gregata resistance genes, such as Rbs1, Rbs2, and Rbs3, conferring resistance against brown stem rot, have also been mapped within 10 cM of the Rcs3 gene [12, 13]. Two SSR markers associated with Rcs3 were confirmed in 64 soybean cultivars and breeding lines of “Davis” ancestors and progenies [14]. However, SSR markers are less suitable for high-throughput and association studies because they are based on repeat length variants, electrophoresis detection methods, and the occurrence of SSR alleles of identical size but different evolutionary origins [15]. Several single nucleotide polymorphisms (SNPs) and insertions or deletion (indels) associated with Rcs3 have been evaluated. A total of 19 SNPs/Indels were identified and verified, although only 11 SNPs/Indels were located close to Rcs3 in an F2 population of “Davis” × “Blackhawk.” The Rcs3 gene was located close to Satt244 and 0.50 cM from SNPs AZ573TA150 and AZ573CA393. Neither SNPs nor indels had a direct effect on the phenotype of Rcs3; however, 11 SNPs were located in the 3-cM interval around Rcs3 [16]. Subsequently, five plant introductions (PIs) from China (PI594619, PI594661, PI594662A, PI594774, and PI594891) were found to be resistant to FLS in a broad soybean spectrum [17]. Bulked segregant analysis (BSA) results showed that the resistance genes in PI594619 and PI594662A were located near Satt501 on chromosome (Chr.) 18 (LG-G), and near Satt547 and Satt244 on Chr. 16 (LG-J), respectively. A resistance gene in PI594661 was found near Satt244 on Chr.16 (LG-J). In addition, the resistance of PI594891 and PI594774 to FLS was controlled by two dominant genes on Chr. 13 near Satt114, which is different from the Rcs3 allele on Chr. 18. They had a high resistance level similar to the Rcs3 gene in “Davis” among the reported FLS resistance genes [17]. The analysis of lines with key recombination events was used to narrow down the FLS-resistance genomic region of PI594891 from 3.3 Mb to 72.6 kb with five annotated genes. The resistance gene of PI594774 was fine-mapped into a 540-kb region, including 72.6 kb of PI594891. Five candidate genes of Pi594891 were sequenced, and multiple mutations in the promoter, intron, and 5′ and 3′ UTR regions were found [18].
Genome-wide association studies (GWASs) can mine alleles using phenotypic variation and recombination during population evolution. This is significantly related to the variation of target traits at the whole-genome level without constructing a mapping population [19]. GWASs have been widely used to identify resistance genes in soybeans. Che et al. [20] performed a GWAS on a collection of 219 soybean breeding lines for resistance to the SC3 strain of soybean mosaic virus (SMV). A total of 24 SNPs were identified, which accounted for 25.54–33.60% of the phenotypic variation. Many SNPs were found close to Rsv1, Rsv4, and Rsv5. To study soybean resistance to Sclerotinia stem rot (SSR) by GWASs, Wei et al. [21] genotyped and sequenced 420 soybean lines. Two computational models (compressed mixed linear model with genome association and prediction integrated tool, fixed and random model circulating probability unification) were used to identify SNPs that were significantly associated with disease response. These similar but also different results indicated that the use of different computational models can affect the SNP association with SSR resistance. A total of 125 genes located in the linkage disequilibrium (LD) of Chr. 1, Chr. 11, and Chr. Eighteen were identified using the two models. The candidate genes in these LD blocks can encode isochorismate synthase and regulate host cell death pathways. GWASs can be used to investigate a set of genetically unstructured genotypes. More precise QTLs can be detected in cases of sufficiently used genetic markers. Vuong [22] studied a set of 553 soybean accessions and genotyped and identified 14 loci distributed on different chromosomes, including 60 SNPs significantly associated with soybean cyst nematode (SCN) resistance. GWAS results also confirmed six QTLs and eight novel QTLs that were previously mapped using bi-parental populations. Many studies have shown that GWASs based on LD can be used to examine the genetic variation in soybean disease resistance traits. It is the main tool to identify candidate resistance genes by associating the phenotypic trait with its underlying genetics [23]. However, there are no reports of GWASs being used to study resistance to C. sojina in soybean.
This study analysed 234 soybean accessions using the genotype data of the SoySNP180K BeadChips. We used general linear model and mixed linear model to evaluate the genetic effects of resistance sites and identify resistance-related SNP loci and haplotypes. Possible candidate resistance genes were also annotated. This is an effective approach for molecular breeding and the identification of disease resistance mechanisms.
Methods
Plant material and phenotypic evaluation
A total of 234 soybean accessions collected from three different breeding departments in northeast China was used for GWAS. The accessions were public and available for non-commercial purposes. The FLS strain used was C. sojina Race15, which was identified as the dominant race among Chinese differential cultivars. It was collected from the Jianan Farm in Jiamusi City (N46°4725.64″ N, E130°305.63″ E). Thereafter, the strain was stored in liquid nitrogen after single spore isolation. All soybean accessions were provided by the Beidahuang Kenfeng Seed Co., Ltd., and C. sojina strains were provided by the Jiamusi branch of the Heilongjiang Academy of Agricultural Sciences.
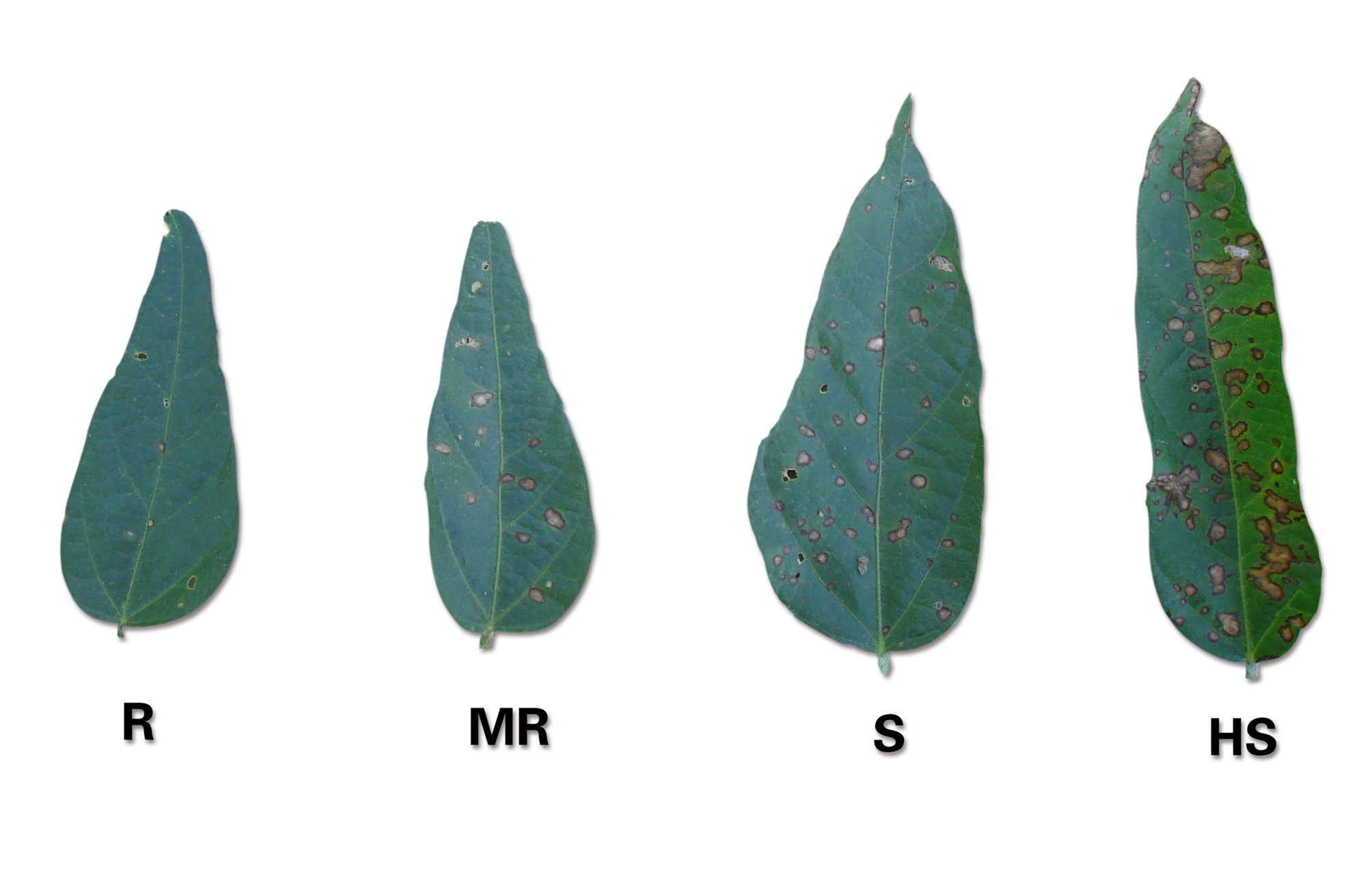
The valuation experiments of FLS incidence were evaluated during the growing season from 2017 to 2019. Before spray inoculation, the strain was purified by single-spore isolation and then cultured on V8 juice agar (V8 juice 200 mL/L, CaCO3 3.0 g/L, agar 15.0 g/L, sterile water to 1 L) [24], and gently washed down from the medium using sterile water and filtering through sterilised multi-layer gauze, the conidial suspensions were adjusted to a concentration of 6 × 104 conidia mL− 1. At the V2–V3 growth stage, one trifoliate leaf per soybean seedling was inoculated with 0.3 mL of the conidial suspension on the upper leaf surface. Thereafter, the inoculated seedlings were transferred to a humidity chamber at 26–28 °C for 72 h. At 14 and 21 d after inoculation, resistance assessment was evaluated primarily on leaf spot size and the number of spots formed on the most severely affected trifoliate leaves. The disease evaluation scale was as follows: spots ≤2 mm diameter, or ≤ 10 spots of 2–3 mm diameter per trifoliate leaf were categorised as a resistant response (R); 10–20 spots of ≤3 mm diameter per trifoliate leaf were categorised as moderately resistant (MR); > 20 spots of 3 mm were categorised as a susceptible response (S); spots that were connected into large groups and most of the leaves dying early due to the disease were categorised as a highly susceptible response (HS) [1](Additional file 1: Fig. S1). To facilitate the later statistical analysis, the resistance grade of soybean accessions expressed in digits, R is represented by 3, MR, S and HS were 5, 7, and 9, respectively. All inoculation tests were repeated at least three times, and 30 plants of each soybean accession were inoculated. Descriptive statistics of phenotypic data were performed using R software [25].
Genotyping
DNA from 234 accessions was extracted from the samples using the magnetic bead method. The DNA concentration was determined using a Nanodrop 8000 spectrophotometer (Thermo Fisher Scientific). SNP genotyping of the germplasm population was performed at Beidahuang Kenfeng Seed Co., Ltd., using the SoySNP180K Bead Chip (Thermo Fisher Scientific).
GWAS analysis and linkage disequilibrium analysis (LD)
The 234 soybean accessions were genotyped using the SoySNP180K BeadChips. Two models, the general linear model (GLM) and the mixed linear model (MLM) correcting for K-matrix (MLM [K]), were used to reduce errors from relative kinship. The threshold of the SNP locus that was significantly associated with the trait was set at p < 0.0001. The remaining GWAS analyses were also performed using TASSEL 5.0, including the descriptive analysis, genetic distance matrix of soybean varieties of genotype data, principal component analysis, and kinship matrix [26]. Cluster analysis was performed using the UPGMA method, and a phylogenetic tree was drawn using Mega4 software. The Quantile-Quantile (Q-Q) plots and Manhattan were drawn using TASSEL5.0. The degree of linkage disequilibrium (LD) between each pair of SNPs was estimated with the correlation coefficient r2 using Haploview5.0 (minor allele freq: 0.05. The fraction of strong LD informative comparisons must be at least 0.97, and all other parameters used are default parameters).
Candidate genes annotation
According to the LD calculated in the previous step, the LD block was determined, and candidate genes were searched in the LD block. Functional annotation of the genes was searched against the NR [27] and GO [28] databases to identify candidate genes related to FLS resistance. Finally, candidate genes were identified by pathway analysis using the Kyoto Encyclopaedia of Genes and Genomes (KEGG) (https://www.kegg.jp/) [29].
Results
Phenotyping
We initially analysed the genotypes of 382 soybean accessions using the SoySNP180K BeadChips. A total of 180,961 SNP markers were selected across the 20 soybean chromosomes of cultivated (Glycine max) and wild (G. soja) soybean accessions. The number of SNPs is estimated to provide approximately 1 SNP every 6.1 kb. Among these, 58,388 of the SNP markers were of the Poly High Resolution type and accounted for 32.24% of the total markers. SNP markers were selected in the next step. After comparing the data over 3 years, 277 accessions with stable results were selected as the candidate population for the GWAS of the resistance genes. Finally, 234 accessions with both genotype and phenotype data were selected as the GWAS population. All the tested accessions had different degrees of lesions, including the most resistant genotypes, indicating that the infection process was successful. There were seven resistant accessions (R), 119 moderately resistant accessions (MR), 87 susceptible accessions (S), and 21 highly susceptible accessions (HS). (Additional file 2: Table S1).
Genotyping
A total of 58,388 SNP genotypes from 234 accessions were analysed using descriptive analysis. The results showed that 27,302 SNPs had no polymorphism in the association analysis population. After filtering, 30,890 SNPs on 20 soybean chromosomes were selected as genotype data sources for GWAS. The number of SNPs on Chr.18 was the largest (2051), and the number of SNPs on Chr. 1 and Chr.12 had the least (1198) (Fig. 1). On average, there were 1544.5 SNPs on each chromosome, and each SNP covered 31.301 kb of the chromosome (Table 1).
Fig. 1.

Distribution of the 30,890 SNPs on the 20 soybean chromosomes. The graph shows the number of SNPs with a MAF ≥ 0.05 on each chromosome. The red bar from light to dark on the right represents the number of SNPs within 1 cM
Table 1.
Distribution of 30,890 SNPs on 20 chromosomes of soybean
| Chromosome | No. of SNP markers | Starting position (kb) | End position (kb) | SNP coverage (kb) |
Single SNP coverage (kb/SNP) |
|---|---|---|---|---|---|
| 1 | 1198 | 46.209 | 55,901.759 | 55,855.55 | 46.624 |
| 2 | 1930 | 59.603 | 51,642.224 | 51,582.621 | 26.727 |
| 3 | 1436 | 18.316 | 47,723.675 | 47,705.359 | 33.221 |
| 4 | 1606 | 30.661 | 49,194.977 | 49,164.316 | 30.613 |
| 5 | 1274 | 69.827 | 41,932.26 | 41,862.433 | 32.859 |
| 6 | 1665 | 10.299 | 50,630.237 | 50,619.938 | 30.402 |
| 7 | 1389 | 7.129 | 44,605.694 | 44,598.565 | 32.108 |
| 8 | 1777 | 57.34 | 46,925.494 | 46,868.154 | 26.375 |
| 9 | 1719 | 18.921 | 46,832.712 | 46,813.791 | 27.233 |
| 10 | 1259 | 98.244 | 50,913.805 | 50,815.561 | 40.362 |
| 11 | 1434 | 16.255 | 39,163.086 | 39,146.831 | 27.299 |
| 12 | 1198 | 46.664 | 40,103.384 | 40,056.72 | 33.436 |
| 13 | 1714 | 9.149 | 44,339.594 | 44,330.445 | 25.864 |
| 14 | 1243 | 14.558 | 49,706.317 | 49,691.759 | 39.977 |
| 15 | 1903 | 9.353 | 50,860.462 | 50,851.109 | 26.722 |
| 16 | 1540 | 17.629 | 37,327.774 | 37,310.145 | 24.227 |
| 17 | 1551 | 252.274 | 41,878.591 | 41,626.317 | 26.838 |
| 18 | 2051 | 7.193 | 62,264.999 | 62,257.806 | 30.355 |
| 19 | 1504 | 18.1 | 50,558.55 | 50,540.45 | 33.604 |
| 20 | 1499 | 31.395 | 46,769.259 | 46,737.864 | 31.179 |
Population structure analysis of 234 accessions
A total of 30,890 SNPs from 234 accessions were analysed using principal component analysis (PCA). The first and second principal components explained 6.44 and 4.60% of the variance, respectively, and explained 11.04% of the phenotypic variation. A scatter plot of the first and second principal components showed that the soybean genotypes collected from different sources were closer to each other. A subpopulation structure was not observed in this population (Fig. 2). Cluster analysis of 234 soybean accessions based on UPGMA was conducted using 30,890 SNP marker genotypes. There was no obvious classification of the accessions, consistent with the PCA results (Additional file 3: Fig. S2).
Fig. 2.

Principal Component Analysis (PCA) of a diverse set of 243 soybean cultivars. PCA scatter plot showing the two main principal components. Dots with different colors and shapes represent different sources. Ken Feng seeds means that soybean accessions come from Beidahuang Keng feng seed Co., Ltd., Certified seeds means that accessions was approved by the Soybean Certification Committee of Heilongjiang Province, Special soybean seeds means that accessions are used for special purposes, such as black coat soybean, brown coat soybean, adzuki soybean and fresh special soybean
GWAS of genes resistant to C. sojina
GLM and MLM models for GWAS were evaluated, and the degree of consistency between the observed and expected p-values was assessed using QQ plots. Both the models controlled the generation of false positives well (Fig. 3), and the significant SNPs associated with disease resistance traits were displayed on Manhattan plots (Fig. 4). In total, four SNPs that were significantly associated with disease resistance traits were detected on Chr. 5 and Chr. 20, respectively, according to both GLM and MLM analyses (Table 2).
Fig. 3.

Quantile-quantile plots for frogeye leaf spot resistance using two models. a General linear model. b Mixed linear model
Fig. 4.

Manhattan plots of the association of SNPs with FLS resistance in soybean identified by GWAS. Chromosomal SNPs can be differentiated by various colours. The red dashed line represents the significance threshold, −log10 (p) = 3.00. a Manhattan plot for resistance by General linear model. b Manhattan plot for resistance by Mixed linear model
Table 2.
SNP loci significantly associated with C. sojina resistance traits by GWAS
| Methods | Marker | Chromosome | Position | Marker F | p value | Add F | Add p | Dom F | Dom p |
|---|---|---|---|---|---|---|---|---|---|
| GLM | Affx-89,062,122 | 5 | 34,658,974 | 11.79327 | 1.33E-05 | 0.21198 | 0.64566 | 23.35405 | 2.46E-06 |
| Affx-89,220,750 | 5 | 34,687,389 | 9.78979 | 8.31E-05 | 0.04618 | 0.83004 | 19.5297 | 1.53E-05 | |
| Affx-89,210,591 | 20 | 40,270,311 | 9.77378 | 8.43E-05 | 2.56054 | 0.11093 | 16.81175 | 5.73E-05 | |
| Affx-89,163,218 | 20 | 40,262,553 | 9.62294 | 9.71E-05 | 2.82916 | 0.09392 | 16.22938 | 7.64E-05 | |
| MLM(K) | Affx-89,062,122 | 5 | 34,658,974 | 10.83494 | 3.19E-05 | 0.11133 | 0.73894 | 21.4606 | 6.05E-06 |
Haplotype analysis for FLS resistance gene
Linkage disequilibrium (LD) between pairs of SNPs on Chr. 5, and Chr. 20 was analysed using the Haploview Software 5.0. C. sojina resistance-related SNPs and adjacent SNPs formed different haplotype blocks. A total of 27 SNPs on Chr. 5 were located in one adjacent haplotype block, forming six haplotypes (Additional file 4: Table S2) (Fig. 5a). A total of 35 SNPs on Chr. 20 were located in one adjacent haplotype block, forming three haplotypes (Additional file 4: Table S2) (Fig. 6a). On Chr. 5, Hap A was significantly more resistant to FLS than Hap D (p = 0.025, p < 0.05). Hap A is the resistant genotype, and Hap D is the susceptible genotype (Fig. 5b). On Chr. 20, Hap A was significantly more resistant to FLS than Hap C (p = 0.016, p < 0.05). Hap B was significantly more resistant to FLS than Hap C (p = 0.019, p < 0.05). Hap A and Hap B are the resistance genotypes, and Hap C is the susceptible genotype (Fig. 6b). The gene information distributed in the three haplotype blocks was extracted, and the positions on the chromosomes were indicated (Fig. 7).
Fig. 5.

Haplotype block for SNPs significantly associated with FLS resistance on Chr. 5. a Numbers in squares indicate 100-fold r2 values of each pair of SNPs. The bars above LD plots represent the physical positions of SNPs. LD blocks are marked with black triangles. b Box plots showing the resistance grade and the distribution of six haplotypes in soybean accessions. The score of the Y axis on the left is the resistance grade of Soybean accessions expressed in digits, resistant response is represented by 3, moderately resistant response is represented by 5, susceptible response is represented by 7, and highly susceptible response is represented by 9. ‘*‘refers to a significant difference in the resistance grade among the haplotypes (p < 0.05)
Fig. 6.

Haplotype block for SNPs significantly associated with FLS resistance on Chr. 20. a Numbers in squares indicate 100-fold r2 values of each pair of SNPs. The bars above LD plots represent the physical positions of SNPs. LD blocks are marked with black triangles. b Box plots showing the resistance grade and the distribution of three haplotypes in soybean accessions. The score of the Y axis on the left is the resistance grade of Soybean accessions expressed in digits, resistant response is represented by 3, moderately resistant response is represented by 5, susceptible response is represented by 7, and highly susceptible response is represented by 9. ‘*‘refers to a significant difference in the resistance grade among the haplotypes (p < 0.05)
Fig. 7.

Location map of the gene information in the two haplotype blocks on chromosomes 5 and 20
Candidate genes for FLS resistance at GWAS loci
A total of 45 genes within the two haplotype blocks of Chr. 5 and Chr. 20 were annotated with Glyma1.0 in the NR, GO, and KEGG databases (Additional file 5: Table S3). These genes were separated into 29 GO terms, including mitochondrial outer membrane (GO:0005741), calcium-dependent protein serine/threonine kinase activity (GO:0009931), calcium-dependent protein kinase activity (GO:0010857), MAP kinase activity (GO:0004707), protoxylem development (GO:0090059), and xylan metabolic process (GO:0045491) (Additional file 6: Table S4). The enriched KEGG pathway is involved in plant–pathogen interaction (gmx04626), MAPK signalling pathway–plant interaction (gmx04016), and biosynthesis of secondary metabolites (gmx01110) (Additional file 7: Table S5). Among these genes, Glyma05g28980 encodes mitogen-activated protein kinase 7 (MPK7). Glyma20g31510 and Glyma20g31520 encode calcium-dependent protein kinase (CDPK4) family proteins, and Glyma20g31630 encodes pyruvate dehydrogenase (PDH), which may be involved in plant disease resistance. These genes were predicted to be candidate resistance genes.
Discussion
GWASs have made significant progress in soybean genetics research; however, this study is limited to a few characters [30]. Compared with traditional QTL mapping, whole-genome association analysis has the advantages of a wide detection range [31], high resolution [32, 33], and more material sources [34]. However, the GWAS analysis has limitations. Association mapping is complementary to traditional bi-parental linkage mapping; however, bi-parental linkage mapping cannot be replaced. The interaction effects of genes and the environment will affect GWAS analysis results; therefore, rigorous phenotyping is required. Due to the scale of the study, more complex traits controlled by multiple loci with relatively small phenotypic effects will require large populations [35]. Population structure is a significant factor in correlation studies; therefore, it is necessary to select germplasm resources and evaluate the population structure carefully. This study used more than three replications to increase the accuracy of phenotype identification. In addition, GWAS is only a prediction of candidate genetic sites, and further research is needed in combination with other experimental methods to explore their biological functions.
Lee [36] reduced the number of SNPs related to target traits by adjusting p-value selectively. Only the most significant SNP in each LD block was selected as the representative location and listed by trait, environment, and analytical method. Selecting a low threshold p-value increases the possibility of false positives. In the present study, we selected two models for association analysis and considered the number of SNP loci associated with disease resistance traits, when p < 0.0001 was selected as the threshold. In contrast, the GLM was found slightly better than the MLM. However, this study shows relatively less statistical power, therefore we retained the two models assuming to obtain more reliable correlation sites. Although the statistical power was improved, we found that most of the observed significance was still lower than expected and therefore, the results of this study are considered to have relatively weak statistical power. This may be due to the linkage disequilibrium between a large number of SNP loci in the population, and the number of significant loci (loci without linkage disequilibrium) is significantly lower than the actual number of loci, and the expected p-value is underestimated. However, a small number of loci reached the threshold. Therefore, these loci remain very important and need to be verified.
We obtained SNP sites at the physical positions Gm5:34561008–34,707,609, Gm20:40140684–40,353,461. Lin et al. [37] used QTL mapping and GWAS to identify loci conferring partial resistance to Pythium sylvaticum in soybean. They found that a significant SNP marker overlapped with the QTL identified for partial resistance to Pythium sylvaticum at Gm20:2245263. Hu et al. [38] studied the resistance loci of pod dehiscence and Jing et al. [39] to the soybean sclerotinia stem rot by GWAS, and found that the resistance loci were located at Gm20:8202869 and Gm20:33803317, respectively. Both of these loci found were significantly away from the loci found in the present study. Che et al. [20] used genome-wide association to study soybean mosaic virus SC3 resistance and found four SNP sites located in Gm20:41544070–41,680,482, which were also confirmed to be related to disease resistance in anti-SCN (soybean cyst nematode) studies. Sara et al. [40] used a diverse panel to reveal the genetic architecture of charcoal rot (Macrophomina phaseolina) resistance in soybean and found the resistance loci at Gm20:42356434, which are very close to the distance found in this study.
C. sojina races used in previous sequencing studies are unique to the US, and we used the latest variant race of China. Different countries adopt their own local differential soybean cultivars to identify C. sojina races by phenotype, making the races unable to be unified [6]. Genetic studies of many plant-pathogen interactions indicate that plants often contain single locus that confers resistance to specific races of a pathogen containing a complementary avirulence gene [41]. Race-specific interactions in the FLS of soybeans have been shown to follow a gene-for-gene model. Sharma et al. detected some additional minor FLS resistance loci in LG A1 (Chr.5) and I (Chr.20) [5]. Among these loci, one was associated with Satt440 on LG I (Chr. 20) at 112.7 cM. The locus explained 15% of the variation in FLS at 42 dpi (days after manual infestation) (p = 0.001) with the “Essex” allele reducing disease severity by up to 0.95 units [42]. However, its genetic distance cannot be converted, therefore it cannot be compared with this study. When the selected loci were compared with the known loci, no coverage was found. It is inferred that the four SNPs associated with FLS resistance may represent new loci that require further verification.
In this study, one encoding the PDH gene, one encoding the MPK7 gene, and two encoding CDPK4 genes in the haplotype block were worthy of attention. They are all related to the biological pathway of salicylic acid (SA). The plant systemic acquired resistance (SAR) is an inducible immune system. The cells in the infected parts of plants can produce signal molecules such as salicylic acid (SA), lipids, peptides, and nitric oxide. These signal molecules diffuse to the normal tissues and cells of plants through the vascular system and then activate the expression of stress resistance genes and the regulation of physiological metabolism in normal cells, thereby enhancing the immune ability of cells and effectively restricting disease spread [43]. SA can induce the expression of various pathogenesis-related (PR) genes and help plants resist the invasion of disease organisms, such as viruses, bacteria, and fungi. SA is involved in the formation of plant innate immunity and effector-triggered immunity in plants. SA can also activate the suicide process of plant cells and form necrotic spots in the infected parts to prevent the invasion and spread of pathogens [44]. The gene encoding MPK7 (GhMPK7) cloned from cotton belongs to the C-MAPK group. It plays an important role in broad-spectrum resistance to fungi and viruses regulated by SA and is also involved in the regulation of plant growth and development [45]. MPK7 is co-expressed with MKK3 and promotes strong expression of Pseudomonas syringae resistance genes in plants [46]. In addition, this study proved that the MKK3 pathway plays a role in pathogen defence and further underscores the importance and complexity of MAPK signalling in plant stress responses. MKK3 plays a role in jasmonate (JA)-mediated developmental signalling and generates H2O2 to activate MPK7, which acts as a secondary signal to activate defence genes [47]. Among phytohormones, JA plays an important role in resisting biological stress [48]. However, reverse genetic studies have indicated that MAPKs, SA-induced protein kinase (SIPK), and wound-induced protein kinase (WIPK), are rapidly activated by fatty acid–amino acid conjugates. MAPKs and calcium-dependent protein kinase (CDPK) are necessary for the induction of JA in response to biological stress [49]. When CDPK4 and CDPK5 in Nicotiana attenuata were simultaneously silenced, transgenic plants (IRcdpk4/5) induced high levels of defense metabolites. A study by Yang [49] found that CDPK4 and CDPK5 affect plant resistance to biological stress in a JA- and JA-signalling-dependent manner. Transgenic plants showed over-activation of SIPK, a MAPK involved in various stress responses, and genetic analysis indicated that the increased SIPK activity in IRcdpk4/5 plants leads to exceptionally high JA levels. Some studies have found that CPK4/11 and CPK5/6 play an important role in the resistance of Arabidopsis to Pseudomonas syringae in a MAPK-independent manner [50]. The effector in Heterodera avenae can interact specifically with an Arabidopsis pyruvate dehydrogenase subunit, which might interfere with the SA signalling pathway and suppress plant defence responses [50]. We identified candidate genes for soybean resistance to FLS using an association analysis [17]. The effective use of these QTLs requires functional verification combined with proteomics when effective markers are identified for use in resistance breeding.
Conclusion
Based on the GWAS results of the GLM and MLM models, a total of four SNPs were associated with FLS resistance, of which were located on Chr. 5 and Chr. 20, respectively. Resistance-related SNPs and adjacent SNPs formed two haplotype blocks. Then, 45 candidate genes in the haplotype blocks were annotated in the NR, GO, and KEGG databases. Four of these are worthy of special attention, these proteins are directly or indirectly involved in the biological pathway of salicylic acid (SA) and jasmonic acid (JA). These two plant hormones may induce the expression of disease resistance-related genes and are essential for plant systemic acquired resistance (SAR). Our study provides useful information on the underlying mechanisms of FLS resistance.
Supplementary Information
{kind=link}
Additional file 1: Figure S1. Scale of disease resistance grade
Additional file 2: Table S1. Phenotypic evaluation of 234 soybean accessions
Additional file 3: Figure S2. Dendrogram analysis of 234 soybean accessions by UPGMA
Additional file 4: Table S2. Resistance-related SNPs and adjacent SNPs formed two haplotype blocks on chromosomes 5 and 20
Additional file 5: Table S3. Functional classification of genes in two haplotype blocks on chromosomes 5 and 20
Additional file 6: Table S4. GO enrichment analysis of genes in the two haplotype blocks
Additional file 7: Table S5. KEGG pathway analysis of genes in the two haplotype blocks
Acknowledgements
We thank LetPub (www.letpub.com) for linguistic assistance during manuscript preparation.
Abbreviations
- GWAS
Genome-wide association analysis
- FLS
Frogeye leaf spot
- QTL
Quantitative trait loci
- SNP
Single nucleotide polymorphism
- LD
Linkage disequilibrium
- PCA
Principal component analysis
- MLM
Mixed linear model
Authors’ contributions
GX and HSS both participated in the conception and design of the study, performed the experimental work and data analysis, and drafted the manuscript. DJJ and HXP supervised the study and wrote the commentary. ZZG, LW, WC, MYS, WQS, ZHH and YXH assisted with data analysis and biological interpretation, and revised the manuscript. LZM, LZJ, YLL, QL, MQY and GXD participated in the experimental design. ZMM and YS participated in the conception and experimental design of the study and assisted with biological interpretation. All authors have reviewed and accepted the final version of the manuscript.
Funding
This work was supported by the Natural Science Foundation of Heilongjiang Province (LH2021C091) and the Agricultural Science and Technology Innovation Leaping Project of Heilongjiang Academy of Agricultural Sciences (HNK2019CX14). The funding bodies played no role in the design of the study, collection, analysis, and interpretation of data and in writing the manuscript.
Availability of data and materials
Data generated or analysed during this study are included in this published article and its supplementary information files.
Declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Xin Gu and Shanshan Huang contributed equally to this work.
Contributor Information
Xiping Hu, Email: huxiping-888@163.com.
Junjie Ding, Email: me999@126.com.
References
- 1.Mian MAR, Missaoui AM, Walker DR, Phillips DV, Boerma HR. Frogeye leaf spot of soybean: a review and proposed race designations for isolates of Cercospora sojina Hara. Crop Sci. 2008;48(1):14–24. [Google Scholar]
- 2.Gu X, Ding JJ, Liu W, Yang XH, Yao LL, Gao XD, et al. Comparative genomics and association analysis identifies virulence genes of Cercospora sojina in soybean. BMC Genomics. 2020;21(1):172. doi: 10.1186/s12864-020-6581-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kim H, Newell AD, Cota-Sieckmeyer RG, Rupe JC, Fakhoury AM, Bluhm BH. Mating-type distribution and genetic diversity of Cercospora sojina populations on soybean from Arkansas: evidence for potential sexual reproduction. Phytopathology. 2013;103(10):1045–1051. doi: 10.1094/PHYTO-09-12-0229-R. [DOI] [PubMed] [Google Scholar]
- 4.Camera JN, Ghissi VC, Reis EM, Deuner CC. The combined effects of temperature and leaf wetness periods on soybean frogeye leaf spot intensity. Semina Cienc Agrarias. 2016;37(1):77–84. [Google Scholar]
- 5.Shrestha SK, Cochran A, Mengistu A, Lamour K, Castro-Rocha A, Young-Kelly H. Genetic diversity, QoI fungicide resistance, and mating type distribution of Cercospora sojina—implications for the disease dynamics of frogeye leaf spot on soybean. PLoS One. 2017;12(5):e0177220. doi: 10.1371/journal.pone.0177220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gu X, Yang S, Yang XH, Yao LL, Gao XD, Zhang M, et al. Comparative transcriptome analysis of two Cercospora sojina strains reveals differences in virulence under nitrogen starvation stress. BMC Microbiol. 2020;20(1):166. doi: 10.1186/s12866-020-01853-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Athow K, Probst AH. The inheritance of resistance to frogeye leaf spot of soybeans. Phytopathology. 1952;42(12):660–662. [Google Scholar]
- 8.Athow KL, Probst AH, Kurtzman CP, Laviolette FA. A newly identified physiological race of Cercospora sojina on soybean. Phytopathology. 1962;52(7):712–714. [Google Scholar]
- 9.Phillips DV, Boerma H. Two genes for resistance to race 5 of Cercospora sojina in soybeans. Phytopathology. 1982;72(7):764–766. [Google Scholar]
- 10.Baker WA, Weaver DB, Qui J, Pace PF. Genetic analysis of frogeye leaf spot resistance in PI54610 and Peking soybean. Crop Sci. 1999;39(4):1021–1025. [Google Scholar]
- 11.Mian MAR, Wang T, Phillips DV, Alvernaz J, Boerma HR. Molecular mapping of the Rcs3 gene for resistance to frogeye leaf spot of soybean. Crop Sci. 1999;39(6):1687–1691. [Google Scholar]
- 12.Bachman MS, Tamulonis JP, Nickell CD, Bent AF. Molecular markers linked to brown stem rot resistance genes Rbs1 and Rbs2 in soybean. Crop Sci. 2001;41(2):527–535. [Google Scholar]
- 13.Webb DM. Brown stem rot resistance in soybeans. Pioneer Hi-Bred Int. Inc., Johnston, IA. U.S. 1997;Patent No. 5689035.
- 14.Missaoui AM, Phillips DV, Boerma HR. DNA marker analysis of ‘Davis’ soybean and its descendants for the Rcs3 gene conferring resistance to Cercospora sojina. Crop Sci. 2007;47(3):1263–1270. [Google Scholar]
- 15.Viard F, Franck P, Dubois MP, Estoup A, Jarne P. Variation of microsatellite size homoplasy across electromorphs, loci, and populations in three invertebrate species. J Mol Evol. 1998;47(1):42–51. doi: 10.1007/pl00006361. [DOI] [PubMed] [Google Scholar]
- 16.Missaoui AM, Ha BK, Phillips DV, Boerma HR. Single nucleotide polymorphism detection of the Rcs3 gene for resistance to frogeye leaf spot in soybean. Crop Sci. 2007;47(4):1681–1690. [Google Scholar]
- 17.Hoskin A Genetic mapping of soybean resistance genes to frogeye leaf spot in five Chinese Plant Introductions and efficiency of early generation selection for low phytate soybean lines. Institute of Plant Breeding, Genetics, and Genomics. University of Georgia. 2011.
- 18.Pham AT, Harris DK, Buck J, Hoskins A, Serrano J, Abdel-Haleem H, Cregan P, Song Q, Boerma HR, Li Z. Fine mapping and characterization of candidate genes that control resistance to Cercospora sojina K. Hara in two soybean germplasm accessions. PLOS ONE. 2015;10(5):e0126753. doi: 10.1371/journal.pone.0126753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nordborg M, Weigel D. Next-generation genetics in plants. Nature. 2008;456(7223):720–723. doi: 10.1038/nature07629. [DOI] [PubMed] [Google Scholar]
- 20.Che Z, Yan H, Liu H, Yang H, Du H, Yang Y, Liu B, Yu D. Genome-wide association study for soybean mosaic virus SC3 resistance in soybean. Mol Breed. 2020;40(7):69. [Google Scholar]
- 21.Wei W, Oliveira MAC, Figueiró ADA, Wet X, Shilpa M, Wickland DP, et al. Genome-wide association mapping of resistance to a Brazilian isolate of Sclerotinia sclerotiorum in soybean genotypes mostly from Brazil. BMC Genomics. 2017;18(1):849. doi: 10.1186/s12864-017-4160-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vuong TD, Sonah H, Meinhardt CG, Deshmukh R, Kadam S, Nelson RL, et al. Genetic architecture of cyst nematode resistance revealed by genome-wide association study in soybean. BMC Genomics. 2015;16:593. doi: 10.1186/s12864-015-1811-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wei L, Jian H, Lu K, Filardo F, Yin N, Liu L, et al. Genome-wide association analysis and differential expression analysis of resistance to Sclerotinia stem rot in Brassica napus. Plant Biotechnol J. 2016;14(6):1368–1380. doi: 10.1111/pbi.12501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim JS, Lee YS, Kim SK, Kim KD, Kim J. Differential responses of soybean cultivars to Cercospora sojina isolates, the causal agent of frogeye leaf spot in Korea. Plant Pathol J. 2011;27(2):183–186. [Google Scholar]
- 25.R Core Team . R: A language and environment for statistical computing. Vienna: R Foundation for Statistical Computing; 2018. [Google Scholar]
- 26.Yu J, Pressoir G, Briggs WH, Vroh Bi I, Yamasaki M, Doebley JF, McMullen MD, Gaut BS, Nielsen DM, Holland JB, Kresovich S, Buckler ES. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat Genet. 2006;38(2):203–208. doi: 10.1038/ng1702. [DOI] [PubMed] [Google Scholar]
- 27.Li W, Jaroszewski L, Godzik A. Tolerating some redundancy significantly speeds up clustering of large protein databases. Bioinformatics. 2002;18(1):77–82. doi: 10.1093/bioinformatics/18.1.77. [DOI] [PubMed] [Google Scholar]
- 28.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene ontology: tool for the unification of biology. Nat Genet. 2000;25(1):25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kanehisa M, Goto S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28(1):27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Contreras-Soto RI, Mora F, de Oliveira MA, Higashi W, Scapim CA, Schuster I. A genome-wide associationstudy for agronomic traits in soybean using SNP markers and SNP-based haplotype analysis. PLoS One. 2017;12(2):e0171105. doi: 10.1371/journal.pone.0171105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yu J, Holland JB, McMullen MD, Buckler ES. Genetic design and statistical power of nested association mapping in maize. Genetics. 2008;178(1):539–551. doi: 10.1534/genetics.107.074245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang J, Jiang H, Yeh CT, Yu J, Jeddeloh JA, Nettleton D, et al. Extreme-phenotype genome-wide association study (XP-GWAS): a method for identifyingtrait-associated variants by sequencing pools of individuals selected from a diversity panel. Plant J. 2015;84(3):587–596. doi: 10.1111/tpj.13029. [DOI] [PubMed] [Google Scholar]
- 33.Huang X, Wei X, Sang T, Zhao Q, Feng Q, Zhao Y, et al. Genome-wide association studies of 14 agronomic traits in rice landraces. Nat Genet. 2010;42(11):961–967. doi: 10.1038/ng.695. [DOI] [PubMed] [Google Scholar]
- 34.Winham SJ, Biernacka JM. Gene-environment interactions in genome-wide association studies: current approaches and new directions. J Child Psychol Psychiatry. 2013;54(10):1120–1134. doi: 10.1111/jcpp.12114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang Q, Wu HS, Guo CY, Fox CS. Analyze multivariate phenotypes in genetic association studies by combining univariate association tests. Genet Epidemiol. 2010;34(5):444–454. doi: 10.1002/gepi.20497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee S, Van K, Sung M, et al. Genome-wide association study of seed protein, oil and amino acid contents in soybean from maturity groups I to IV. Theor Appl Genet. 2019;132(6):1639–1659. doi: 10.1007/s00122-019-03304-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lin F, Wani SH, Collins PJ, Paul J, Collins, Wen Z, Li W, et al. QTL mapping and GWAS for identification of loci conferring partial resistance to Pythium sylvaticum in soybean (Glycine max (L.) Merr) Molecular Breeding. 2020;40(6):54. [Google Scholar]
- 38.Hu D, Kan G, Hu W, Li Y, Hao D, Li X, et al. Identification of loci and candidate genes responsible for pod dehiscence in soybean via genome-wide association analysis across multiple environments. Front Plant Sci. 2019;10:811–1. [DOI] [PMC free article] [PubMed]
- 39.Jing Y, Teng W, Qiu L, Zheng H, Li W, Han Y, Zhao X. Genetic dissection of soybean partial resistance to sclerotinia stem rot through genome wide association study and high throughout single nucleotide polymorphisms. Genomics. 2021;113:1262–1271. doi: 10.1016/j.ygeno.2020.10.042. [DOI] [PubMed] [Google Scholar]
- 40.Coser SM, Chowda Reddy RV, Zhang J, Mueller DS, Mengistu A, Wise KA, Allen TW, Singh A, Singh AK. Genetic architecture of charcoal rot (Macrophomina phaseolina) resistance in soybean revealed using a diverse panel. Frontiers in Plant Science. 2017;8:1626. doi: 10.3389/fpls.2017.01626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Flor HH. Current status of the gene-for-gene concept. Annu Rev Phytopathol. 1971;9(1):275–296. [Google Scholar]
- 42.Sharma H, Lightfoot DA. Quantitative trait loci underlying partial resistance to Cercospora sojina race 2 detected in soybean seedlings in greenhouse assays. Atlas J Biol. 2014;3(1):175–182. [Google Scholar]
- 43.Park SW, Kaimoyo E, Kumar D, Mosher S, Klessig DF. Methyl salicylate is a critical mobile signal for plant systemic acquired resistance. Science. 2007;318(5847):113–116. doi: 10.1126/science.1147113. [DOI] [PubMed] [Google Scholar]
- 44.Bari R, Jones JDG. Role of plant hormones in plant defence responses. Plant Mol Biol. 2009;69(4):473–488. doi: 10.1007/s11103-008-9435-0. [DOI] [PubMed] [Google Scholar]
- 45.Shi J, An HL, Zhang L, Gao Z, Guo XQ. GhMPK7, a novel multiple stress-responsive cotton group C MAPK gene, has a role in broad spectrum disease resistance and plant development. Plant Mol Biol. 2010;74(1–2):1–17. doi: 10.1007/s11103-010-9661-0. [DOI] [PubMed] [Google Scholar]
- 46.R’bert D’c, Brader G, Pettkó-Szandtner A, et al. The Arabidopsis mitogen-activated protein kinase kinase MKK3 is upstream of group C mitogen-activated protein kinases and participates in pathogen signaling. Am Soc Plant Biol. 2007;19:3266–3279. doi: 10.1105/tpc.106.050039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Orozco-Cardenas M, Ryan CA. Hydrogen peroxide is generated systemically in plant leaves by wounding and systemin via the octadecanoid pathway. Proc Natl Acad Sci U S A. 1999;96(11):6553–6557. doi: 10.1073/pnas.96.11.6553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhu SY, Yu XC, Wang XJ, Zhao R, Li Y, Fan RC, et al. Two calcium-dependent protein kinases, CPK4 and CPK11, regulate abscisic acid signal transduction in Arabidopsis. Plant Cell. 2007;19(10):3019–3036. doi: 10.1105/tpc.107.050666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang SS, Pan LL, Chen YP, Yang D, Liu Q, Jian H. Heterodera avenae GLAND5 effector interacts with pyruvate dehydrogenase subunit of plant to promote nematode parasitism. Front Microbiol. 2019;10:1241. doi: 10.3389/fmicb.2019.01241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yang DH, Hettenhausen C, Baldwin IT, Wu JQ. Silencing Nicotiana attenuata calcium-dependent protein kinases, CDPK4 and CDPK5, strongly up-regulates wound- and herbivory-induced jasmonic acid accumulations. Plant Physiol. 2012;159(4):1591–1607. doi: 10.1104/pp.112.199018. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Figure S1. Scale of disease resistance grade
Additional file 2: Table S1. Phenotypic evaluation of 234 soybean accessions
Additional file 3: Figure S2. Dendrogram analysis of 234 soybean accessions by UPGMA
Additional file 4: Table S2. Resistance-related SNPs and adjacent SNPs formed two haplotype blocks on chromosomes 5 and 20
Additional file 5: Table S3. Functional classification of genes in two haplotype blocks on chromosomes 5 and 20
Additional file 6: Table S4. GO enrichment analysis of genes in the two haplotype blocks
Additional file 7: Table S5. KEGG pathway analysis of genes in the two haplotype blocks
Data Availability Statement
Data generated or analysed during this study are included in this published article and its supplementary information files.