

Abstract
Background
Campylobacter is a genus of bacteria that has been isolated from the gastrointestinal tract of humans and animals, and the environments they inhabit around the world. Campylobacter adapt to new environments by changes in their gene content and expression, but little is known about how they adapt to long-term human colonization. In this study, the genomes of 31 isolates from a New Zealand patient and 22 isolates from a United Kingdom patient belonging to Campylobacter jejuni sequence type 45 (ST45) were compared with 209 ST45 genomes from other sources to identify the mechanisms by which Campylobacter adapts to long-term human colonization. In addition, the New Zealand patient had their microbiota investigated using 16S rRNA metabarcoding, and their level of inflammation and immunosuppression analyzed using biochemical tests, to determine how Campylobacter adapts to a changing gastrointestinal tract.
Results
There was some evidence that long-term colonization led to genome degradation, but more evidence that Campylobacter adapted through the accumulation of non-synonymous single nucleotide polymorphisms (SNPs) and frameshifts in genes involved in cell motility, signal transduction and the major outer membrane protein (MOMP). The New Zealand patient also displayed considerable variation in their microbiome, inflammation and immunosuppression over five months, and the Campylobacter collected from this patient could be divided into two subpopulations, the proportion of which correlated with the amount of gastrointestinal inflammation.
Conclusions
This study demonstrates how genomics, phylogenetics, 16S rRNA metabarcoding and biochemical markers can provide insight into how Campylobacter adapts to changing environments within human hosts. This study also demonstrates that long-term human colonization selects for changes in Campylobacter genes involved in cell motility, signal transduction and the MOMP; and that genetically distinct subpopulations of Campylobacter evolve to adapt to the changing gastrointestinal environment.
Supplementary Information
The online version contains supplementary material available at 10.1186/s13099-021-00469-7.
Keywords: Campylobacter, Genomics, Host adaptation, Phylogenetics
Background
Campylobacter is a genus of bacteria that has been isolated from the gastrointestinal tract of humans and other animals around the world [1], and from the environments these animals inhabit [2]. Some Campylobacter species are host generalists, colonizing a wide range of host species [3], others are host-specific [4], and some are pathogenic to the animals they colonize [5]. Population genomics and animal studies have demonstrated that Campylobacter adapts to hosts by changing its gene content and expression [6, 7]. As long-term Campylobacter colonization in humans is rare, few studies have investigated Campylobacter adaption to human hosts [8, 9].
Previously, a patient in New Zealand was identified that had been excreting the same strain of Campylobacter, C. jejuni sequence type (ST) 45, for over a decade [8]. The patient had been diagnosed with common variable immune deficiency (CVID) and had suffered from daily diarrhea that varied in severity. Previous genomic analysis and antimicrobial susceptibility testing of Campylobacter isolates collected from the patient determined that the patient was consistently colonized with Campylobacter over this time period, and that the Campylobacter had developed resistance to the antimicrobial agents the patient was prescribed. However, it was unclear to what extent the Campylobacter was contributing to the patient’s diarrhea. In this study we compared the genomes of ST45 isolates collected from the patient to those collected from other sources, including another patient from the United Kingdom with long-term colonization, to investigate how Campylobacter had adapted to human hosts, and used 16S rRNA metabarcoding and biochemical markers to investigate changes in microbial communities and biochemical markers over the period of long-term Campylobacter colonization.
Results
Sample and isolate collection
The New Zealand patient submitted three fecal and three serum samples over five months. Three Campylobacter isolates were collected from the first fecal sample, and six were collected from the second and third samples. These 15 isolates were combined with the 16 previously sequenced from this patient. An additional 231 C. jejuni ST45 isolates were downloaded and passed the quality control thresholds, including 22 from a long-term patient from the United Kingdom (Additional file 1).
ST45 pan-genome analysis
The 262 ST45 genomes contained a pan-genome of 3041 genes and a core-genome of 1459 genes. The pan-genome also contained 603 pseudogenes, 206 of which the pseudogene or original gene were found in more than 95% of isolates. PlasmidFinder identified a plasmid replicon (IncFII) in one of the ST45 isolates, and RFPlasmid found evidence of plasmid-associated contigs in 109 contigs from 50 ST45 isolates. Amongst the plasmid-associated contigs, three contained antimicrobial resistance (AMR) genes, four contained virulence genes, fourteen were conjugative, six were mobile and 89 were non-mobile. None of the isolates collected from the New Zealand patient contained plasmid-associated contigs, but two of the isolates from the United Kingdom patients did. However, they were non-motile and contained no AMR or virulence genes.
Gene frequency analysis did not find any evidence of specific genes or pseudogenes associated with the 53 long-term Campylobacter patient isolates when compared to the 209 other ST45 isolates or the subset of 145 other human ST45 isolates investigated, and linear regression analysis found that the number of genes was not significantly associated with source (p = 0.2216) or country (p = 0.9765). The number of pseudogenes was also not significantly associated with source (p = 0.101), but was significantly associated with country (p = 0.0114), with isolates from the United States containing more pseudogenes. This difference was small and likely due to sampling (Additional file 2).
All isolates collected from the long-term Campylobacter patients contained the blaOXA-61 beta-lactamase gene [10] and the T86I mutation in the gyrA gene associated with fluoroquinolone resistance [11], as did 152 and two of the 209 other ST45 isolates, respectively. Frameshift and SNP analysis demonstrated that the T86I mutation was the only mutation associated with long-term Campylobacter patient isolates, when compared to all the other ST45 isolates or just the other human ST45 isolated investigated. All isolates collected from the New Zealand patient and two non-patient ST45 isolates contained the A2074T mutation in the 23S rRNA gene associated with macrolide resistance [12]. One of the isolates collected from the United Kingdom patient contained the A2075G mutation in the 23S rRNA gene. No long-term patient-specific virulence genes were identified.
Phylogenetic analysis of the ST45 isolates from different sources and countries demonstrated that the sequence type is very diverse (Fig. 1). Clade analysis identified 29 clades amongst the 262 ST45 isolates. Thirteen of these clades consisted of isolates from multiple sources, whilst the remaining clades consisted of isolates from the same source. However, only three of these clades contained more than one non-human source. The isolates from the New Zealand patient were in the same clade as isolates from water, ovine, poultry and other human isolates (Clade 18), whilst no other isolates were found in the same clade as the United Kingdom long-term patient (Clade 2) (Additional file 3). A closer examination of clade 18 revealed that the isolates collected from the New Zealand patient shared 2–123 SNPs with each other, and 22–239 SNPs with other isolates in this clade. They were most closely related to two genomes from New Zealand human sources. These two genomes were the only other ST45 isolates that contained the A2074T mutation in the 23S rRNA gene.
Fig. 1.

Maximum likelihood tree of 262 ST45 isolates. Colored bars represent isolate metadata and the presence-absence matrix represents antimicrobial resistance genes
New Zealand patient phylogenetics
SNP analysis identified 248 non-recombinant SNPs amongst the 31 isolates collected from the New Zealand patient. TempEst found tip dates and distance were correlated (R2 = 0.92) (Additional file 4). Phylogenetic modeling estimated that these isolates contained a median substitution rate of 3.69 × 10–6 substitutions site−1 year−1 (95% HPD interval: 2.87–4.49 × 10–6 substitutions site−1 year−1) and shared a common ancestor on 09/05/2004 (median, 95% HPD interval: 11/12/2002–16/07/2005). The sampled ancestor model did not identify isolates that represented ancestors of other isolates collected from the patient.
Phylogenetic analysis revealed that two clades had evolved within the New Zealand patient representing separate subpopulations (Fig. 2). These clades shared a common ancestor on 08/07/2010 (median, 95% HPD interval: 19/09/2009–20/03/2011). Comparative genetic analysis revealed that clade A was associated with the rsmD gene, involved in DNA replication and repair; and clade B was associated with the dcuB gene, involved in the transport of C4-dicarboxylates into the cell. These genes were classified as pseudogenes in the other clade due to frameshifts. Two additional frameshifts and eleven non-synonymous SNPs in ten genes differenced between the clades. These genes varied in function (Additional file 5).
Fig. 2.

Maximum clade credibility tree of 31 ST45 isolates collected from the same New Zealand patient
Linear regression modeling found that date of collection was not associated with the number of genes (p = 0.0635) or pseudogenes (p = 0.0501) (Additional file 6). Clades were not associated with different number of genes (p = 0.566) or pseudogenes (p = 0.539).
United Kingdom patient phylogenetics
SNP analysis identified 122 non-recombinant SNPs amongst the 22 isolates collected from the United Kingdom patient. Phylogenetic modeling estimated that these isolates contained a median substitution rate of 1.96 × 10–6 substitutions site−1 year−1 (95% HPD interval: 1.51–2.44 × 10–6 substitutions site−1 year−1) and shared a common ancestor on 17/04/1999 (median, 95% HPD interval: 29/11/1997–23/05/2000) (Additional file 7). The sampled ancestor model identified three isolates (median, 95% HPD interval: 1–4) that represented ancestors of other isolates collected from the patient. Linear regression modeling found that date of collection was negatively associated with the number of genes (p = 0.0129) and positively associated with the number of pseudogenes (p = 0.00175) (Additional file 8).
Campylobacter DNA replication and repair
DNA replication and repair mechanisms in Campylobacter involves 27 genes. 54 mutations were identified that differed between isolates collected from the New Zealand and United Kingdom patients (Additional file 9). The gene with the largest number of mutations was the mutS gene (n = 20).
Long-term within human selection
Isolates from the New Zealand patient had a pangenome of 1722 genes and an accessory genome of 86 genes, whilst the isolates collected from the United Kingdom patient had a pangenome of 1743 genes and an accessory genome of 103 genes. Analysis of gene loss over the course of colonization identified six genes from the New Zealand patient isolates and five from the United Kingdom patient isolates that were stably-lost over time. However, neither these genes nor the rest of the accessory genome were associated with any functional group (Additional file 10).
The isolates collected from the New Zealand patient shared 134 core non-synonymous SNPs and 23 core frameshifts in 92 genes, whilst the isolates collected from the United Kingdom patient shared 88 core non-synonymous SNPs and 31 core frameshifts in 81 genes. Of these, 23 non-synonymous SNPs and two frameshifts from the New Zealand patient isolates, and 15 non-synonymous SNPs and six frameshifts from the United Kingdom patient isolates were stably-inherited. In both patients’ isolates, cell motility (COG group N) and signal transduction mechanisms (COG group T) were the functional groups with the highest proportion of genes containing these mutations (Fig. 3). In addition, some genes contained multiple non-synonymous SNPs and frameshifts (Additional file 11). The porA and ccmL genes were the only gene that had more than five of these mutations amongst isolates collected from both patients.
Fig. 3.

Bar plots of the proportion of genes of each functional group that contained non-synonymous SNPs or frameshifts (A, B) and stably-inherited SNPs or frameshifts (C, D) in the New Zealand (A, C) and United Kingdom (B, D) patients
16S rRNA metabarcoding of bacterial species
The three fecal microbiota samples from the New Zealand patients displayed a large amount of variation in the proportion of each bacterial taxa present (Fig. 4). The Campylobacter concentrations were not directly correlated to the microbiota makeup, likely due to variations in the bacterial concentration of fecal samples.
Fig. 4.

Bar plot of the proportion of the New Zealand patient’s microbiota, identified through 16S rRNA profiling, comprised of detectable bacterial phyla (A) and genera (B). Bacterial genera that represented less than one percent of the microbiota were placed in the ‘other’ category
Biochemical and serological tests
The New Zealand patient tested negative for anti-Campylobacter antibodies (Table 1), and had low immunoglobulin levels that were consistent with their immunosuppressive disease [13] and prior history [8]. The concentration of calprotectin was directly proportional to the number of isolates from clade B (Fig. 5). No other associations between the biochemical results, microbiota results, Campylobacter concentration and the proportion of each subpopulation were identified.
Table 1.
Biochemical, serological and Campylobacter parameters for three samples collected over five months from the same patient
| Sample | Measurement | 1 | 2 | 3 | Normal range |
|---|---|---|---|---|---|
| Date | 03/09/2016 | 09/11/2016 | 21/02/2017 | ||
|
Fecal consistency Calprotectin |
Bristol stool scale µg/g |
6–7 231 |
6–7 155 |
7 87 |
3–4 < 112 |
| C-reactive protein | mg/L | 0.84 | 1.45 | 1.13 | < 5.0 |
| IgA | g/L | < 0.2 | < 0.2 | < 0.2 | 0.7–4.0 |
| IgG | g/L | 3.9 | 4.5 | 4.7 | 7.0–16.0 |
| IgM | g/L | < 0.2 | < 0.2 | < 0.2 | 0.4–2.3 |
| anti-Campylobacter IgA | Titer | Negative | Negative | Negative | Negative |
| anti-Campylobacter IgG | Titer | Negative | Negative | Negative | Negative |
| anti-Campylobacter IgM | Titer | Negative | Negative | Negative | Negative |
| Campylobacter concentration | cfu/g | 1.38 × 107 | 1.48 × 107 | 1.08 × 106 | 0 |
| Proportion Campylobacter | 0.111 | 0.053 | 0.001 | 0 | |
| Clade A isolates | 0/3 | 3/6 | 5/6 | 0 | |
Fig. 5.
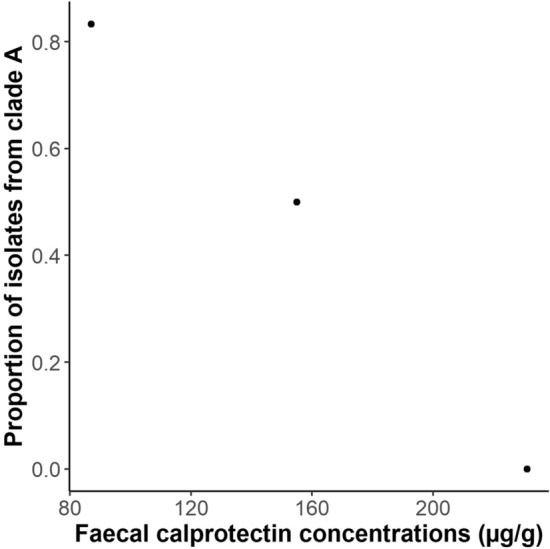
Scatterplots of the number of the proportion of isolates collected from clade A versus the fecal calprotectin concentration for three samples collected from the New Zealand patient
Discussion
Long-term colonization of human patients places selective pressure on Campylobacter. Non-synonymous SNPs and frameshifts were found in multiple genes from Campylobacter isolates collected from the New Zealand and United Kingdom patients, especially in genes involved in motility and signal transduction mechanisms. Two genes were found that had more than five of these mutations in both patients: the porA gene that encodes the major outer membrane protein (MOMP) and the ccmL gene that encodes the multi-ligand binding chemoreceptor (CcmL). Previous work on isolates from the New Zealand patient demonstrated that they varied in motility, possibly to evade phagocytes [8]; whilst studies on Campylobacter collected from the same patients within 24-h demonstrated variation in motility and chemotaxis genes [14, 15]. This suggests that long-term colonization is selecting for changes in Campylobacter motility, signal transduction (particularly chemotaxis) and membrane proteins, possibly to evade host defenses.
Populations of bacteria are continuously accumulating mutations: canonically, if they are beneficial then those containing them will thrive and the mutation will increase in frequency, whilst if they are deleterious those containing them will struggle and the mutation will eventually disappear. However, Ramiro et al. [16] investigated Escherichia coli colonizing the gut of mice over 190 days and found that beneficial mutations increased in frequency but were not fixed in the population, and slightly deleterious mutations remained in the population for extended periods of time, possibly by the variable mouse gut buffering their deleterious effects. In this study, mutations were observed that were stably-inherited within the Campylobacter populations of the long-term patients, especially in those involved in cell motility and signal transduction. These Campylobacter were collected over longer time periods than Ramiro et al. experiments, possibly allowing these beneficial mutations to be fixed within the population or the Campylobacter were exposed to larger selective sweeps than the E. coli. However, a large number of mutations were observed in isolates that were not fixed within the Campylobacter population, suggesting that the human gastrointestinal tract allows for variation in many Campylobacter genes, but over time selective sweeps select for specific genetic variants. Nevertheless, we cannot rule out the possibility that many of the stably-inherited mutations were the result of “genetic hitchhiking” [17].
The isolates collected from the New Zealand and United Kingdom patients both belonged to ST45. However, within this sequence type the isolates from each patient were distantly related, allowing us to identify mutations specific to long-term colonization by comparing isolates from these patients to ST45 from other sources. Genetic comparisons identified one mutation associated with long-term patient colonization, T86I in the gyrA gene. This mutation is the most common cause of quinolone resistance in C. jejuni [11], and the New Zealand patient [8] and likely the United Kingdom patient [9] were prescribed quinolones, selecting for this mutation. A large number of other genes contained mutations in the New Zealand and United Kingdom patient isolates. However, the mutations either differed between isolates from each patient or were found in multiple other ST45 isolates. This suggests that apart from quinolone resistance, long-term human colonization selected for changes in particular genes, but not specific mutations.
Bacteria can adapt to environments by both gene loss and gene acquisition. Gene loss may increase the fitness of the bacteria by decreasing resource expenditure on unnecessary cellular processes [18], whilst gene acquisition may allow the bacteria to gain cellular processes to thrive in the environment [19]. Previous studies on long-term Pseudomonas aeruginosa cystic fibrosis (CF) [20] and Salmonella enterica [21] infections found that gene loss outweighed gene acquisition over the course of these infections. However, Bayjaynov et al. [22] investigated long-term colonizing E. faecium strains and found similar amounts of gene loss and gene acquisition. In our study, several genes were identified that were stably-lost over the course of colonization in the New Zealand and United Kingdom patients, and there was some evidence of the total number of genes decreasing over time. However, no functional group was overrepresented in the accessory genome, suggesting that genome reduction is not the main way Campylobacter adapts to long-term colonization. Gene loss could have occurred between acquisition and isolation of the first isolate, but there was no evidence that the long-term patients had smaller number of genes compared to the other human ST45 isolates. This is a similar observation to long-term E. faecium colonization rather than to P. aeruginosa or S. enterica infections, namely evidence of similar amounts of gene loss and gene gain. Further research is required to determine if this is due to the different biology of these bacteria, different environments, or different types of infection.
In the New Zealand patient, we identified two genetically distinct clades or subpopulations of Campylobacter. In CF infections, divergence of P. aeruginosa subpopulations has been reported prior to infection [23] and within the host [24]. Analysis of long-term E. faecium colonization has demonstrated the presence of multiple lineages within the gastrointestinal tract of humans, but these lineages diverged prior to colonization [22]. Phylogenetic analysis suggests that the two Campylobacter subpopulations diverged between 2009 and 2011, after the first isolate was collected from the patient in 2006, indicating that the subpopulations diverged within the host. There is evidence of multiple subpopulations coexisting prior to this divergence in the New Zealand patient, and in the isolates collected from the United Kingdom patient, but these patients were not sampled frequently during these time periods, preventing further analysis of these subpopulations.
The competitive exclusion principle states that no two species can occupy the same niche [25]. Accordingly, the subpopulations of Campylobacter found within the patient are likely to have distinct niches. In CF patients, the presence of multiple subpopulations has been attributed to the bacterium accumulating mutations and phenotypically adapting to different environments within the human lung [26]. There is evidence of this when looking at the genetic differences between the two Campylobacter lineages found within the human host, specifically the dcuB gene that encodes a fumarate/succinate active antiporter under low oxygen conditions that can also import aspartate [27]. Succinate concentrations in the gastrointestinal tract have been shown to increase during gut dysbiosis [28] and inflammation [29], but it remains to be determined if these conditions affect fumarate or aspartate concentrations. The New Zealand patient’s gastrointestinal tract showed a large amount of variation in inflammation plus variation in the microbiota composition and these may be influencing the concentration of succinate and possibly other metabolites. The concentration of calprotectin was negatively associated with the proportion of isolates made up of the lineage missing dcuB, suggesting that the two lineages could have emerged in response to the variation in gastrointestinal inflammation and microbiota disruptions. The dcuA gene is a fumarate/aspartate:H+ symporter and 90% of isolates from the dcuB-negative clade contained the L212F mutation in the dcuA gene, whilst all isolates belonging to the dcuB-containing clade contained the S148F mutation in this gene. However, it is unclear how these mutations affect the function of this transporter. DcuA and DcuB work in close association with the cytoplasmic-facing fumarate reductase FrdABC, but no mutations were found in the frdA, frdB or frdC genes [30]. Nevertheless, the absence of DcuB would prevent correct stoichiometric exchange of succinate and fumarate that allow FrdABC to function. Campylobacter also has a unidirectional fumarate reductase that cannot oxidize succinate, MfrABE, that operates independently of DcuA and DcuB, as it is periplasm-facing [31]. For the clade with an intact dcuB gene, three out eighteen of the isolates contained a truncated mfrA gene due to a frameshift. It has been shown that FrdABC is the major contributor to fumarate reduction but that MfrABE is required for full fitness when the bacteria rely on fumarate respiration to conserve energy [31]. Taken together, these observations suggest that different clades will have distinct contributions of Dcu/Frd versus Mfr mediated fumarate respiration during colonization. The role of DcuA and DcuB in aspartate uptake in low oxygen intestinal niches may also be physiologically important. Further work on the concentrations of gastrointestinal aspartate, succinate and fumarate in the patient and how this relates to the different lineages is required to determine if gastrointestinal inflammation is creating distinct niches where certain metabolites are not required. However, multiple other mutations were found amongst the two lineages and we cannot rule out the possibility that the mutations associated with dcuB were genetic hitchhikers.
C. jejuni ST45 is one of the most frequently isolated strains of Campylobacter collected from humans, domestic animals and the environment [32–34]. It is regarded as a ‘generalist’ strain, as it has been isolated from multiple host species and environments [35, 36], and has demonstrated frequent host switching [37]. We found clades of ST45 that consisted of isolates collected from different sources, supporting ST45 as a generalist strain, but also found some clades that only consisted of isolates from single sources and many clades that only consisted of a single non-human source. Further analysis of non-human ST45 isolates is required to determine the extent that ST45 is a “generalist” strain.
The human microbiota is affected by disease [38], diet [39] and genetics [40]. Youmans et al. [38] investigated the microbiota of individuals with traveler’s diarrhea and found that regardless of cause, diarrheic samples contained a high Prevotella-to-Bacteroides (P/B) ratio. This observation is supported in this study, where the third diarrheal sample collected from the patient was higher on the Bristol scale (softer) and had a higher P/B ratio. Braun et al. [41] investigated the microbiota of healthy individuals and hospitalized patients suspected of infectious diarrhea, and found diarrhea patients were associated with an increased abundance of Proteobacteria. The first diarrheal sample collected from the patient had the highest Proteobacteria proportion, but this is because it has the highest Campylobacter proportion which made up most of the Proteobacteria in the microbiota of this sample. Zhuang et al. [42] found that diarrhea brought on by irritable bowel syndrome resulted in increased Bacteroidetes and decreased Firmicutes, such as with the first two samples. However, the amount of Prevotella and Proteobacteria in the microbiota is also affected by diet [39, 43]. Further microbiota samples and dietary information from the time of collection are required to investigate the role of disease and diet on the patient’s microbiota.
The sampled ancestor model identified multiple isolates from the United Kingdom patient that represented ancestors to other isolates collected from this patient, but it detected no likely ancestors amongst the New Zealand patient’s isolates. This suggests that the population of Campylobacter was more diverse within the New Zealand patient than within the United Kingdom patient, and therefore sampled isolates were less likely to represent an ancestral state. However, Campylobacter within these patients have likely undergone multiple bottlenecks, especially with antimicrobial therapies [8, 9], and we cannot rule out the possibility that the United Kingdom patient was sampled during several of these bottlenecks when the genetic diversity was smaller, whilst the New Zealand patient was not.
Campylobacter collected from the New Zealand patient had a substitution rate twice that of those collected from the United Kingdom patient, with no overlap between 95% HPD intervals. Multiple differences were found in genes involved in DNA replication and repair between isolates collected from these patients. The mutS gene had the largest number of differences, but knockouts in this gene in the closely related bacterium, Helicobacter pylori, are not associated with increased substitution rates [44]. Mutations in mutY have been associated with faster substitution rates in Campylobacter [45, 46], but isolates collected from the long-term patients had identical mutY genes. Further work is required to determine the effects of other mutations in DNA replication and repair genes on the substitution rate of Campylobacter.
The New Zealand patient’s gastrointestinal health, amount of inflammation and immunosuppression varied significantly between the three samples obtained, as indicated by the variation in serum IgA, IgG, IgM and CRP concentrations, and fecal calprotectin concentration. Apart from the association between the proportion of each clade isolated and fecal calprotectin concentrations, no other associations were identified between these markers, the fecal Campylobacter concentration, microbiota constitution, and the proportion of each clade isolated. The variation in these biochemical tests over these five months does suggest that the patient was undergoing changes in gastrointestinal health and immunosuppression.
An original objective of this research was to determine if Campylobacter was contributing to the patient’s diarrheal episodes or simply colonizing the patient. The lack of evidence for anti-Campylobacter antibodies suggests that the patient had not mounted an immune response against the Campylobacter. However, most studies on Campylobacter serology have focused on acute infections, rather than the possible chronic infection described here. In addition, most serological tests have a high false-negative rate and for those individuals that do seroconvert the antibody titer quickly decreases after a few months [47, 48]. Studies on acute Guillain-Barré syndrome, a disease often triggered by Campylobacter infections have found up to 80% of cases display serological evidence of Campylobacter, but it is unclear whether the negative cases were triggered by Campylobacter or other infections [49], and false positives have been observed [50]. Regarding the New Zealand patient, this could be explained by a number of scenarios including: the Campylobacter was not the cause of any pathology and had not been presented to the immune system or triggered an immune response; the Campylobacter contributed to the diarrheal episodes but the patient was unable to form an immune response sufficient to remove the bacteria or be detected using the serological method described.
Conclusions
In this study we investigated the genomes of Campylobacter from two patients that were both colonized for over a decade and found that multiple selective pressures were imposed on the Campylobacter. There was some evidence of genome degradation, but more evidence that Campylobacter adapted through the accumulation of non-synonymous SNPs and frameshifts in genes involved in cell motility, signal transduction and the major outer membrane protein, possibly to evade host defenses. However, we also found that the Campylobacter collected from the patients differed in substitution rates and diversity as evidenced by sampled ancestral states, although the reason for these differences remains unclear. Through 16S rRNA metabarcoding and biochemical markers we demonstrated that the New Zealand patient displayed a large amount of variation in their microbiome, inflammation and immunosuppression over five months, and that the Campylobacter collected from the patient could be divided into two subpopulations, the proportion of which correlated with the amount of gastrointestinal inflammation. This suggests that subpopulations of Campylobacter evolve within the gastrointestinal tract to adapt to changing environments. Overall, this study demonstrates how genomics, phylogenetics, 16S rRNA metabarcoding and biochemical markers can provide insight into how Campylobacter adapts to changing environments within human hosts.
Methods
Sample collection and storage
The previously described New Zealand patient [8] was invited to take part in a year-long study to investigate the effect of long-term Campylobacter colonization. Every month serum and fecal samples were collected from the patient. The Bristol fecal chart was used to grade the consistency of the fecal sample [51]. Approximately 100 mg of fecal sample was set aside for immediate Campylobacter culturing. The rest of the fecal sample and the serum sample were stored at −80 °C until all samples were collected and were then further analyzed in a single batch.
Campylobacter quantification
Approximately 100 mg of each fecal sample was resuspended in 9.9 ml of phosphate-buffered saline (PBS) (prepared in house). The reconstituted solution was serially diluted three times by adding 1 ml of sample to 9 ml of PBS, before 100 µl of each dilution was spirally plated onto modified charcoal-cefoperazone-deoxycholate agar (mCCDA) (Fort Richard Laboratories, Auckland, New Zealand) in duplicate. For plates that contained 50–500 colonies, the number of colonies was quantified and used to calculate the concentration of Campylobacter in the fecal samples. From each sample, 3–6 Campylobacter colonies underwent genomic DNA extractions and were whole genome sequenced as previously described [8] using the Illumina MiSeq platform (Illumina, San Diego, California, United States).
Serum biochemistry and serology
The SERION ELISA classic Campylobacter jejuni IgA, IgG and IgM tests (Institut Virion\Serion GmbH, Wurzburg, Germany) were used to calculate the amount of anti-Campylobacter immunoglobulin A (IgA), immunoglobulin G (IgG) and immunoglobulin M (IgM) in the serum according to the manufacturer’s instructions. An aliquot of each serum sample was sent to MedLab Central (Palmerston North, New Zealand), who quantified the total serum IgA, IgG and IgM, and the serum inflammation marker, C-reactive protein (CRP), on a Cobas 8000 modular analyzer with the c702 module (Roche, Basel, Switzerland).
Fecal biochemistry
An aliquot of each fecal sample was sent to Canterbury Health Laboratories to quantify the amount of the gastrointestinal inflammation marker, calprotectin, using the Calpro Calprotecin ELISA (Calpro AS, Lysaker, Norway).
16S rRNA gene-based microbiota analysis
The DNeasy PowerSoil Kit (Qiagen, Hilden, Germany) was used to extract DNA from an aliquot of each fecal sample. PCR was used to amplify the 16S V3-V4 region of bacterial DNA in the extracts and libraries were made from the 16S rRNA gene amplicons using a single-step PCR with barcoded primers [52]. The libraries were sequenced on an Illumina MiSeq as 2 × 250 paired-end reads. The reads were investigated with the MG-RAST pipeline [53].
ST45 pan genome analysis
The accession numbers of available C. jejuni ST45 genome sequences were identified by searching the PATRIC database [54] and the Sequence Read Archive (SRA; https://www.ncbi.nlm.nih.gov/sra) for “Campylobacter”, “ST45” and “Paired-end reads”. The raw reads of these sequences were downloaded and trimmed along with those collected from the patient using Trimmomatic v0.39 [55]. ARIBA v2.14.4 [56] was used to determine the sequence type of these reads using the C. jejuni multi-locus sequence type (MLST) [57], and all isolates that were identified as ST45 were taken forward for further analysis (Additional file 1).
The trimmed reads of all ST45 isolates were assembled using Spades v3.14.0 [58], and the assemblies produced were analyzed using Quast v5.0.2 [59] and CheckM v1.1.2 [60]. All assemblies that contained more than 300 contigs greater than 500 bp or had more than 50 duplicate genes were regarded as potentially contaminated or not sequenced at a high enough read depth and were not analyzed further.
The assemblies that passed QC were annotated using Prokka v1.12 [61] and the pan-genome evaluated using Roary v3.11.2 [62] with a 95% identity cut-off. Genes that were found in over 95% of the isolates were classified as part of the core genome. A maximum likelihood tree was constructed from single nucleotide polymorphisms (SNPs) in the core gene alignment using RAxML v8.2.11 [63]. Annotation, pan-genome evaluation and phylogenetics were repeated with reference genome NC_002163 [64] to root the tree. TreeCluster v1.0.1 [65] was used to predict clades in the maximum likelihood tree.
RFPlasmid v0.0.16 [66] was run on all assemblies with the “Campylobacter” database. Contigs with > 0.6 votes for plasmid were classified as “plasmid-associated”. Abricate v1.0.1 (https://github.com/tseemann/abricate) with the ResFinder [67] and virulence finder database (VFDB) [68] were used to search plasmid-associated contigs for AMR and virulence genes. Mob-typer [69] v3.0.0 was used to determine the mobility of the plasmid-associated contigs.
The ResFinder database, VFDB database, PlasmidFinder database [70], and a custom database consisting of all genes clustered by Roary were used with ARIBA to identify the presence of acquired AMR genes, virulence genes, plasmids and pseudogenes, respectively, in the ST45 genomes. Pseudogenes were identified from the custom database using the method described by Mather et al. [71]. Pseudogenes were analyzed if the original gene or pseudogene were found in 95% of isolates. To identify mutations associated with macrolide or fluoroquinolone resistance, an ARIBA database was formed from the 23S rRNA and gyrA genes of C. jejuni NCTC 11168 (NC_002163).
Genes and pseudogenes were classified as “long-term patient-specific” if they were found in 95% of isolates collected from the New Zealand and United Kingdom patients and fewer than 5% of ST45 isolates collected from other sources or other human sources. EggNOG v4.5.1 [72] was used to predict the function of the genes. Linear regression models were used to model the total number of genes and pseudogenes with source and country as the explanatory variables. Isolates where the country or source were unknown were excluded from the model, as were singletons. The long-term patients contained multiple isolates for a sample, so the mean number of genes and pseudogenes for these patients was modeled. Partial-F tests were used to determine if these variables had a significant effect on the model.
For the long-term New Zealand patient, linear regression models were used to model the total number of genes and pseudogenes with date of collection and clade as the explanatory variables. For samples where multiple isolates were collected, the mean number of genes and pseudogenes for each clade was calculated. Partial-F tests were used to determine if inclusion of clades had a significant effect on the model.
The trimmed reads of the genome sequences collected from the New Zealand patient and those belonging to the most closely related clade, were aligned to the ST45 reference genome NC_022529 using Snippy v4.4.5 (https://github.com/tseemann/snippy). Areas of putative recombination were removed from the full alignments produced using Gubbins [73]. RAxML was used to form a maximum likelihood tree from the non-recombinant SNPs identified.
Within-patient ST45 phylogenetics
The trimmed reads of all genomes that were collected from the New Zealand patient were aligned to the ST45 reference genome NC_022529 using Snippy and areas of putative recombination were removed from the full alignments produced using Gubbins. To determine if there was a clock signal, IQ-TREE v1.6.12 [74] was used to form a maximum likelihood tree from the identified non-recombinant SNPs and TempEst v1.5.3 [75] was used to test for temporal signal.
The non-recombinant SNPs were exported into BEAUti v2.5 to create an Extensive Markup Language (XML) file for BEAST v2.5 [76]. The ST45 reference genome NC_022529, consists of 584,548 adenine, 254,724 cytosine, 253,882 guanine and 576,175 thymine nucleotides; these nucleotide proportions (adjusted for the SNPs) were added as non-varying sites to keep the model representative of ST45 genomes and to calculate the substitution rate. bModelTest [77] was used to choose the substitution model (Supplementary Material). Multiple molecular clock (strict, random [78] and uncorrelated relaxed [79]) and tree models (constant and Extended Bayesian Skyline) [80]) were trialled for 100 million steps. Nested sampling [81] was used to select the model. A Generalized Time Reversible (GTR) [82] model was used to model nucleotide substitutions, an Extended Bayesian Skyline model was used to model the effective population size, and a strict clock model was used to model the molecular clock and was calibrated by the tip dates. A uniform prior was placed on the substitution rate that it would not exceed 10–4 substitutions site−1 year−1 nor fall below 10–8 substitutions site−1 year−1 [83], and due to the large number of constant sites added, the proportion of invariant sites was assumed to be zero. The XML file was run in BEAST in three separate chains with different starting seeds of 50 million steps each, before LogCombiner v2.5 was used to combine the runs with a 10% burn-in removed. Tracer v1.6 [84] was used to visualize the results.
TreeAnnotator v2.5 was used to form a maximum clade credibility tree of the SNP dataset. Evolview v2 [85] was used to visualize and edit the tree. The Sampled Ancestor (SA) v2.02 model [86] was also run in BEAST2 to determine the likelihood that any of the isolates collected from the patient represented ancestral states.
Second long-term Campylobacter patient
The ST45 isolates downloaded included 22 genomes from an immunosuppressed long-term Campylobacter patient from the United Kingdom [9]. The phylogentic analyses outlined above were also performed on these isolates.
Phase variation and non-synonymous SNPs
To investigate phase variation, Tatajuba v1.0.2 [87] was used to align the trimmed reads of all the ST45 isolates to the reference genome, NC_022529, and identify tracts that differed in size between the isolates. A cut-off of 0.95 was used when comparing frameshifts between different groups of ST45 isolates to account for small proportions of sequencing errors. To investigate non-synonymous SNPs, Snippy was used to align the trimmed reads of all the ST45 isolates to the same reference genome and identify SNPs that resulted in amino acid changes. Frameshifts and SNPs from long-term patient isolates were compared to ST45 isolates from other sources or other human sources as above to determine if they were associated with long-term patients.
Replication genes
To identify possible reasons for variation in substitution rates between isolates from each long-term patient, KEGG (https://www.genome.jp/kegg/) was searched for all genes involved in Campylobacter DNA replication and repair. An ARIBA database was created from these genes and used to search all the ST45 isolates for their presence and mutations in these genes. Mutations found in > 95% of isolates in one patient and < 5% of isolates in the other were classified as associated with a patient.
Stably-inherited genetic changes
To identify genetic changes that were stably-inherited, non-synonymous SNPs, frameshifts and gene loss were identified in the Campylobacter colonizing the long-term patients that were found in all isolates collected after a time point and not in any prior to this time point. Genetic changes that occurred between the collection of the first and second isolates, and last and second-to-last isolates were ignored as their stability was based on one isolate.
Supplementary Information
Additional file 1. Genomic information and metadata on ST45 isolates investigated.
Additional file 2. ST45 pangenome linear regression modeling.
Additional file 3. ST45 clade 18 analysis.
Additional file 4. New Zealand patient phylogenetic analysis.
Additional file 5. New Zealand patient clade gene analysis.
Additional file 6. New Zealand patient gene number modeling.
Additional file 7. United Kingdom patient phylogenetic analysis.
Additional file 8. United Kingdom patient gene number modeling.
Additional file 9. Tables of the proportion of DNA replication genes found in long-term Campylobacter patients and other ST45 isolates (Table S4), and the mutations in Campylobacter DNA replication genes associated with isolates collected from long-term patients (Table S5).
Additional file 10. Long-term patients’ pangenome function analysis.
Additional file 11. Long-term patients’ gene variation function analysis.
Acknowledgements
We thank the New Zealand patient and their partner, for their participation; MidCentral District Health Board and MedLab Central, for their help with obtaining samples and serum biochemical tests; Tui Shadbolt, for bringing the New Zealand case to our attention; Rudyard Yap for providing infectious disease expertise; Lynn Rogers of mEpiLab for technical assistance; Canterbury Health Laboratory for fecal biochemical tests; and the staff at Massey Genome Service (part of New Zealand Genomics), Massey University, Palmerston North, New Zealand, for their help and advice with the genome sequencing for this study.
Abbreviations
- AMR
Antimicrobial resistance
- C. jejuni
Campylobacter jejuni
- CF
Cystic fibrosis
- COG
Clusters of orthologous groups
- CRP
C-reactive protein
- CVID
Common variable immune deficiency
- E. faecium
Enterococcus faecium
- HPD
Highest posterior density
- IgA
Immunoglobulin A
- IgG
Immunoglobulin G
- IgM
Immunoglobulin M
- GTR
Generalized time reversible
- mCCDA
Modified charcoal cefoperazone deoxycholate agar
- MLST
Multi-locus sequence typing
- MOMP
Major outer membrane protein
- P. aeruginosa
Pseudomonas aeruginosa
- P/B
Prevotella-To-Bacteroides
- PBS
Phosphate buffered saline
- S. enterica
Salmonella enterica
- SA
Sampled ancestor
- SNP
Single nucleotide polymorphism
- SRA
Sequence read archive
- ST
Sequence type
- VFDB
Virulence finder database
- XML
Extensible markup language
Authors' contributions
SJB drafted the work and contributed to the design of the laboratory work and analysis of genomic data; ACM contributed to the design of the laboratory work and substantially revised the manuscript; PJB contributed to the analysis and interpretation of genomic and metagenomic data; NPF contributed to the design of the genomic analyses and interpretation of statistical data; JCM contributed to the analysis and interpretation of statistical data; DTSH contributed to the analysis of statistical and medical data; PEC contributed to the bacteriology analysis; AEM contributed to the pangenome analysis; AF contributed to the acquisition of genomic data; CF contributed to the design of the sampling; DJK contributed to the analysis and interpretation of mutations involved in fumarate transport and metabolism; JB contributed to the conception of the study, design of the sampling and analysis. All authors read and approved the final manuscript.
Funding
This work was supported by the Palmerston North Medical Research Fund and the Massey University Institute of Veterinary and Biological Science postgraduate fund. DTSH is funded by the Royal Society Te Apārangi Rutherford Discovery Fellowship RDF-MAU1701. SJB and AEM are supported by the Biotechnology and Biological Sciences Research Council Institute Strategic Programme Microbes in the Food Chain BB/R012504/1 and its constituent project BBS/E/F/000PR10348 (Theme 1, Epidemiology and Evolution of Pathogens in the Food Chain).
Availability of data and materials
The reads of the Campylobacter genomes and the 16S rRNA sequences were uploaded to NCBI under the Accession Numbers PRJEB24941 and PRJNA605845, respectively.
Declarations
Ethics approval and consent to participate
This study was approved by the Central Health Disease Ethics Committee (16/CEN/13) and consent was obtained by the patient before the study began.
Consent for publication
Consent to publish the obtained results was obtained from the patient.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Kaakoush NO, Castano-Rodriguez N, Mitchell HM, Man SIM. Global epidemiology of Campylobacter infection. Clin Microbiol Rev. 2015;28:687–720. doi: 10.1128/CMR.00006-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.French N, Barrigas M, Brown P, Ribiero P, Williams N, Leatherbarrow H, et al. Spatial epidemiology and natural population structure of Campylobacter jejuni colonizing a farmland ecosystem. Environ Microbiol. 2005;7:1116–1126. doi: 10.1111/j.1462-2920.2005.00782.x. [DOI] [PubMed] [Google Scholar]
- 3.Gripp E, Hlahla D, Didelot X, Kops F, Maurischat S, Tedin K, et al. Closely related Campylobacter jejuni strains from different sources reveal a generalist rather than a specialist lifestyle. BMC Genomics. 2011;12:1–22. doi: 10.1186/1471-2164-12-584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sheppard SK, Colles F, Richardson J, Cody AJ, Elson R, Lawson A, et al. Host association of Campylobacter genotypes transcends geographic variation. Appl Environ Microbiol. 2010;76:5269–5277. doi: 10.1128/AEM.00124-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hedstrom OR, Sonn RJ, Lassen ED, Hultgren BD, Crisman RO, Smith BB, et al. Pathology of Campylobacter jejuni abortion in sheep. Vet Pathol. 1987;24:419–426. doi: 10.1177/030098588702400509. [DOI] [PubMed] [Google Scholar]
- 6.Thepault A, Meric G, Rivoal K, Pascoe B, Mageiros L, Touzain F, et al. Genome-wide identification of host-segregating epidemiological markers for source attribution in Campylobacter jejuni. Appl Environ Microbiol. 2017;83:1–13. doi: 10.1128/AEM.03085-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Crofts AA, Poly FM, Ewing CP, Kuroiwa JM, Rimmer JE, Harro C, et al. Campylobacter jejuni transcriptional and genetic adaptation during human infection. Nat Microbiol. 2018;3:494–502. doi: 10.1038/s41564-018-0133-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bloomfield SJ, Midwinter AC, Biggs PJ, French NP, Marshall JC, Hayman DTS, et al. Long-term colonization by Campylobacter jejuni within a human host: Evolution, antimicrobial resistance, and adaptation. J Infect Dis. 2018;217:103–111. doi: 10.1093/infdis/jix561. [DOI] [PubMed] [Google Scholar]
- 9.Barker CR, Painset A, Swift C, Jenkins C, Godbole G, Maiden MCJ, et al. Microevolution of Campylobacter jejuni during long-term infection in an immunocompromised host. Scientif. 2020;10:1–11. doi: 10.1038/s41598-020-66771-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Griggs DJ, Peake L, Johnson MM, Ghori S, Mott A, Piddock LJV. Beta-lactamase-mediated beta-lactam resistance in Campylobacter species: prevalence of Cj0299 (bla(OXA-61)) and evidence for a novel beta-lactamase in C. jejuni. Antimicrob Agents Chemother. 2009;53:3357–3364. doi: 10.1128/AAC.01655-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Espinoza N, Rojas J, Pollett S, Meza R, Patino L, Leiva M, et al. Validation of the T86I mutation in the gyrA gene as a highly reliable real time PCR target to detect Fluoroquinolone-resistant Campylobacter jejuni. BMC Infect Dis. 2020;20:1–7. doi: 10.1186/s12879-020-05202-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gibreel A, Taylor DE. Macrolide resistance in Campylobacter jejuni and Campylobacter coli. J Antimicrob Chemother. 2006;58:243–255. doi: 10.1093/jac/dkl210. [DOI] [PubMed] [Google Scholar]
- 13.Resnick ES, Moshier EL, Godbold JH, Cunningham-Rundles C. Morbidity and mortality in common variable immune deficiency over 4 decades. Blood. 2012;119:1650–1657. doi: 10.1182/blood-2011-09-377945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dunn SJ, Pascoe B, Turton J, Fleming V, Diggle M, Sheppard SK, et al. Genomic epidemiology of clinical Campylobacter spp. at a single health trust site. Microb Genomics. 2018;4:1–8. doi: 10.1099/mgen.0.000227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cody AJ, McCarthy ND, van Rensburg MJ, Isinkaye T, Bentley SD, Parkhill J, et al. Real-Time genomic epidemiological evaluation of human Campylobacter isolates by use of whole-genome multilocus sequence typing. J Clin Microbiol. 2013;51:2526–2534. doi: 10.1128/JCM.00066-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ramiro RS, Durao P, Bank C, Gordo I. Low mutational load and high mutation rate variation in gut commensal bacteria. PLoS Biol. 2020;18:1–34. doi: 10.1371/journal.pbio.3000617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smith JM, Haigh J. The hitch-hiking effect of a favourable gene. Genet Res (Camb) 1974;23:23–35. [PubMed] [Google Scholar]
- 18.Koskiniemi S, Sun S, Berg OG, Andersson DI. Selection-driven gene loss in bacteria. PLoS Genet. 2012;8:1–7. doi: 10.1371/journal.pgen.1002787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Branchu P, Charity OJ, Bawn M, Thilliez G, Dallman TJ, Petrovska L, et al. SGI-4 in monophasic Salmonella Typhimurium ST34 is a novel ICE that enhances resistance to copper. Front Microbiol. 2019;10:1–12. doi: 10.3389/fmicb.2019.01118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gabrielaite M, Johansen HK, Molin S, Nielsen FC, Marvig RL. Gene loss and acquisition in lineages of bacteria evolving in a human host environment. Rxiv. 2020;1:31. doi: 10.1128/mBio.02359-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Klemm EJ, Gkrania-Klotsas E, Hadfield J, Forbester JL, Harris SR, Hale C, et al. Emergence of host-adapted Salmonella Enteritidis through rapid evolution in an immunocompromised host. Nat Microbiol. 2016;1:1–15. doi: 10.1038/nmicrobiol.2015.23. [DOI] [PubMed] [Google Scholar]
- 22.Bayjanov JR, Baan J, Rogers MRC, Troelstra A, Willems RJL, van Schaik W. Enterococcus faecium genome dynamics during long-term asymptomatic patient gut colonization. Microb Genomics. 2019;5:1–11. doi: 10.1099/mgen.0.000277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Williams D, Evans B, Haldenby S, Walshaw MJ, Brockhurst MA, Winstanley C, et al. Divergent, coexisting Pseudomonas aeruginosa lineages in chronic cystic fibrosis lung infections. Am Jounral Respir Crit Care Med. 2015;191:775–785. doi: 10.1164/rccm.201409-1646OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Feliziani S, Marvig RL, Lujan AM, Moyano AJ, Di Rienzo JA, Johansen HK, et al. Coexistence and within-host evolution of diversified lineages of hypermutable Pseudomonas aeruginosa in long-term cystic fibrosis infections. PLoS Genet. 2014;10:1–17. doi: 10.1371/journal.pgen.1004651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gause GF. The struggle for existence. Baltimore: Williams & Wilkins; 1934. [Google Scholar]
- 26.Jelsbak L, Johansen HK, Frost A-L, Thogersen R, Thomsen LE, Ciofu O, et al. Molecular epidemiology and dynamics of Pseudomonas aeruginosa populations in lungs of cystic fibrosis patients. Infect Immun. 2007;75:2214–2224. doi: 10.1128/IAI.01282-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wosten MMSM, van de Lest CHA, van Dijk L, van Putten JPM. Function and regulation of the C4-dicarboxylate transporters in Campylobacter jejuni. Front Microbiol. 2017;8:1–13. doi: 10.3389/fmicb.2017.00174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Woodmansey EJ, McMurdo MET, Macfarlane GT, Macfarlane S. Comparison of compositions and metabolic activities of fecal microbiotas in young adults and in antibiotic-treated and non-antibiotic-treated elderly subjects. Appl Environ Microbiol. 2004;70:6113–6122. doi: 10.1128/AEM.70.10.6113-6122.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Macias-Ceja DC, Ortiz-Masia D, Salvador P, Gisbert-Ferrandiz L, Hernandez C, Hausmann M, et al. Succinate receptor mediates intestinal inflammation and fibrosis. Mucosal Immunol. 2019;12:178–187. doi: 10.1038/s41385-018-0087-3. [DOI] [PubMed] [Google Scholar]
- 30.Taylor AJ, Kelly DJ. The function, biogenesis and regulation of the electron transport chains in Campylobacter jejuni: new insights into the bioenergetics of a major food-borne pathogen. In: Poole R, editor. Adv Microb Physiol. 2019. p. 239–329. [DOI] [PubMed]
- 31.Guccione E, Hitchcock A, Hall SJ, Mulholland F, Shearer N, van Vliet AHM, et al. Reduction of fumarate, mesaconate and crotonate by Mfr, a novel oxygen-regulated periplasmic reductase in Campylobacter jejuni. Environ Microbiol. 2010;12:576–591. doi: 10.1111/j.1462-2920.2009.02096.x. [DOI] [PubMed] [Google Scholar]
- 32.Dingle KE, Colles FM, Ure R, Wagenaar JA, Duim B, Bolton FJ, et al. Molecular characterization of Campylobacter jejuni clones: A basis for epidemiologic investigation. Emerg Infect Dis. 2002;8:949–955. doi: 10.3201/eid0809.02-0122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sheppard SK, Dallas JF, MacRae M, McCarthy ND, Sproston EL, Gormley FJ, et al. Campylobacter genotypes from food animals, environmental sources and clinical disease in Scotland 2005/6. Int J Food Microbiol. 2009;134:96–103. doi: 10.1016/j.ijfoodmicro.2009.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lake RJ, Campbell DM, Hathaway SC, Ashmore E, Cressey PJ, Horn BJ, et al. Source attributed case-control study of campylobacteriosis in New Zealand. Int J Infect Dis. 2021;103:268–277. doi: 10.1016/j.ijid.2020.11.167. [DOI] [PubMed] [Google Scholar]
- 35.Revez J, Llarena A-K, Schott T, Kuusi M, Hakkinen M, Kivisto R, et al. Genome analysis of Campylobacter jejuni strains isolated from a waterborne outbreak. BMC Genomics. 2014;15:1–8. doi: 10.1186/1471-2164-15-768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sheppard SK, Cheng L, Méric G, De Haan CPA, Llarena A-K, Marttinen P, et al. Cryptic ecology among host generalist Campylobacter jejuni in domestic animals. Mol Ecol. 2014;23:2442–2451. doi: 10.1111/mec.12742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dearlove BL, Cody AJ, Pascoe B, Méric G, Wilson DJ, Sheppard SK. Rapid host switching in generalist Campylobacter strains erodes the signal for tracing human infections. ISME J. 2016;10:721–729. doi: 10.1038/ismej.2015.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Youmans BP, Ajami NJ, Jiang Z-D, Campbell F, Wadsworth WD, Petrosino JF, et al. Characterization of the human gut microbiome during travelers’ diarrhea. Gut Microbes. 2015;6:110–119. doi: 10.1080/19490976.2015.1019693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Senghor B, Sokhna C, Ruimy R, Lagier J-C. Gut microbiota diversity according to dietary habits and geographical provenance. Hum Microbiome J. 2018;7–8:1–9. [Google Scholar]
- 40.Bhute S, Pande P, Shetty SA, Shelar R, Mane S, Kumbhare SV, et al. Molecular characterization and meta-analysis of gut microbial communities illustrate enrichment of Prevotella and Megasphaera in Indian Subjects. Front Microbiol. 2016;7:1–14. doi: 10.3389/fmicb.2016.00660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Braun T, Di Segni A, BenShoshan M, Asaf R, Squires JE, Barhom SF, et al. Fecal microbial characterization of hospitalized patients with suspected infectious diarrhea shows significant dysbiosis. Sci Rep. 2017;7:1–9. doi: 10.1038/s41598-017-01217-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhuang X, Tian Z, Li L, Zeng Z, Chen M, Xiong L. Fecal microbiota alterations associated with diarrhea-predominant irritable bowel syndrome. Front Microbiol. 2018;9:1–11. doi: 10.3389/fmicb.2018.01600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Precup G, Vodnar D-C. Gut Prevotella as a possible biomarker of diet and its eubiotic versus dysbiotic roles: a comprehensive literature review. Br J Nutr. 2019;122:131–140. doi: 10.1017/S0007114519000680. [DOI] [PubMed] [Google Scholar]
- 44.Bjorkholm B, Sjolund M, Falk PG, Berg OG, Engstrand L, Andersson DI. Mutation frequency and biological cost of antibiotic resistance in Helicobacter pylori. Proc Natl Acad Sci U S A. 2001;98:14607–14612. doi: 10.1073/pnas.241517298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dai L, Sahin O, Tang Y, Zhang Q. A mutator phenotype promoting the emergence of spontaneous oxidative stress-resistant mutants in Campylobacter jejuni. Appl Environ Microbiol. 2017;83:1–13. doi: 10.1128/AEM.01685-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dai L, Muraoka WT, Wu Z, Sahin O, Zhang Q. A single nucleotide change in mutY increases the emergence of antibiotic-resistant Campylobacter jejuni mutants. J Antimicrob Chemother. 2015;70:2739–2748. doi: 10.1093/jac/dkv190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Watson KC, Kerr EJC, McFadzean SM. Serology of human Campylobacter infections. J Infect. 1979;1:151–158. [Google Scholar]
- 48.Taylor BV, Williamson J, Luck J, Coleman D, Jones D, McGregor A. Sensitivity and specificity of serology in determining recent acute Campylobacter infection. Intern Med J. 2004;34:250–258. doi: 10.1111/j.1444-0903.2004.00517.x. [DOI] [PubMed] [Google Scholar]
- 49.Schmidt-Ott R, Schmidt H, Feldmann S, Brass F, Krone B, Gross U. Improved serological diagnosis stresses the major role of Campylobacter jejuni in triggering Guillain-Barre syndrome. Clin Vaccine Immunol. 2006;13:779–783. doi: 10.1128/CVI.00065-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Taylor BV, Williamson J, Jones D, Coleman D, Luck J, McGregor A. Utility of serum Campylobacter specific antibodies in determining prior Campylobacter infection in neurological disease. J Clin Neurosci. 2007;14:116–121. doi: 10.1016/j.jocn.2006.01.040. [DOI] [PubMed] [Google Scholar]
- 51.Lewis SJ, Heaton KW. Stool form scale as a useful guide to intestinal transit time. Scand J Gastroenterol. 1997;32:920–924. doi: 10.3109/00365529709011203. [DOI] [PubMed] [Google Scholar]
- 52.Kozich JJ, Westcott SL, Baxter NT, Highlander SK, Schloss PD. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl Environ Microbiol. 2013;79:5112–5120. doi: 10.1128/AEM.01043-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wilke A, Bischof J, Gerlach W, Glass E, Harrison T, Keegan KP, et al. The MG-RAST metagenomics database and portal in 2015. Nucleic Acids Res. 2016;44:D590–D594. doi: 10.1093/nar/gkv1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wattam AR, Abraham D, Dalay O, Disz TL, Driscoll T, Gabbard JL, et al. PATRIC, the bacterial bioinformatics database and analysis resource. Nucleic Acids Res. 2014;42:D581–D591. doi: 10.1093/nar/gkt1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hunt M, Mather AE, Sanchez-Buso L, Page AJ, Parkhill J, Keane JA, et al. ARIBA: rapid antimicrobial resistance genotyping directly from sequencing reads. Microb Genomics. 2017;3:1–11. doi: 10.1099/mgen.0.000131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dingle KE, Colles FM, Wareing DRA, Ure R, Fox AJ, Bolton FE, et al. Multilocus sequence typing system for Campylobacter jejuni. J Clin Microbiol. 2001;39:14–23. doi: 10.1128/JCM.39.1.14-23.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Compulational Biol. 2012;19:455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gurevich A, Saveliev V, Vyahhi N, Tesler G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics. 2013;29:1072–1075. doi: 10.1093/bioinformatics/btt086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015;25:1043–1055. doi: 10.1101/gr.186072.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Prokka ST. Rapid prokaryotic genome annotation. Bioinformatics. 2014;30:2068–2069. doi: 10.1093/bioinformatics/btu153. [DOI] [PubMed] [Google Scholar]
- 62.Page AJ, Cummins CA, Hunt M, Wong VK, Reuter S, Holden MTG, et al. Roary: Rapid large-scale prokaryote pan genome analysis. Bioinformatics. 2015;31:3691–3693. doi: 10.1093/bioinformatics/btv421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Stamatakis A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Parkhill J, Wren BW, Mungall K, Ketley JM, Churcher C, Basham D, et al. The genome sequence of the food-borne pathogen Campylobacter jejuni reveals hypervariable sequences. Nature. 2000;403:665–668. doi: 10.1038/35001088. [DOI] [PubMed] [Google Scholar]
- 65.Balaban M, Moshiri N, Mai U, Jia X, Mirarab S. TreeCluster: Clustering biological sequences using phylogenetic trees. PLoS ONE. 2019;14:1–20. doi: 10.1371/journal.pone.0221068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.van Bloois L, Wagenaar JA, Zomer AL. RFPlasmid: Predicting plasmid sequences from short read assembly data using machine learning. Cold Spring: Cold Spring Harbor Laboratory; 2020. pp. 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zankari E, Hasman H, Cosentino S, Vestergaard M, Rasmussen S, Lund O, et al. Identification of acquired antimicrobial resistance genes. J Antimicrob Chemother. 2012;67:2640–2644. doi: 10.1093/jac/dks261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chen L, Zheng D, Liu B, Yang J, Jin Q. VFDB 2016: hierarchical and refined dataset for big data analysis-10 years on. Nucleic Acids Res. 2016;44:D694–D697. doi: 10.1093/nar/gkv1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Robertson J, Nash JHE. MOB-suite: software tools for clustering, reconstruction and typing of plasmids from draft assemblies. Microb Genomics. 2018;4:1–7. doi: 10.1099/mgen.0.000206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Carattoli A, Zankari E, Garcia-Fernandez A, Larsen MV, Lund O, Villa L, et al. In silico detection and typing of plasmids using PlasmidFinder and plasmid multilocus sequence typing. Antimicrob agents chemotheropy. 2014;58:3895–3903. doi: 10.1128/AAC.02412-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mather AE, Phuong TLT, Gao Y, Clare S, Mukhopadhyay S, Goulding DA, et al. New variant of multidrug-resistant Salmonella enterica serovar Typhimurium associated with invasive disease in immunocompromised patients in Vietnam. MBio. 2018;9:1–11. doi: 10.1128/mBio.01056-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Huerta-Cepas J, Szklarczyk D, Forslund K, Cook H, Heller D, Walter MC, et al. eggNOG 4.5: a hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 2016;44:D286–D293. doi: 10.1093/nar/gkv1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Croucher NJ, Page AJ, Connor TR, Delaney AJ, Keane JA, Bentley SD, et al. Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using Gubbins. Nucleic Acids Res. 2015;43:1–13. doi: 10.1093/nar/gku1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nguyen L-T, Schmidt HA, von Haeseler A, Minh BQ. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 2015;32:268–274. doi: 10.1093/molbev/msu300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rambaut A, Lam TT, Carvalho LM, Pybus OG. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen) Virus Evol. 2016;2:1–7. doi: 10.1093/ve/vew007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bouckaert R, Heled J, Kühnert D, Vaughan T, Wu C-H, Xie D, et al. BEAST 2: A software platform for Bayesian evolutionary analysis. PLoS Comput Biol. 2014;10:1–6. doi: 10.1371/journal.pcbi.1003537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bouckaert RR, Drummond AJ. bModelTest: Bayesian phylogenetic site model averaging and model comparison. BMC Evol Biol. 2017;17:1–11. doi: 10.1186/s12862-017-0890-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Drummond AJ, Suchard MA. Bayesian random local clocks, or one rate to rule them all. BMC Biol. 2010;8:1–12. doi: 10.1186/1741-7007-8-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Drummond AJ, Ho SYW, Phillips MJ, Rambaut A. Relaxed phylogenetics and dating with confidence. PLoS Biol. 2006;4:699–710. doi: 10.1371/journal.pbio.0040088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Heled J, Drummond AJ. Bayesian inference of population size history from multiple loci. BMC Evol Biol. 2008;8:1–15. doi: 10.1186/1471-2148-8-289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Russel PM, Brewer BJ, Klaere S, Bouckaert RR. Model selection and parameter inference in phylogenetics using nested sampling. Syst Biol. 2018;68:219–233. doi: 10.1093/sysbio/syy050. [DOI] [PubMed] [Google Scholar]
- 82.Tavare S. Some probabilistic and statistical problems in the analysis of DNA sequences. Am Math Soc. 1986;17:57–86. [Google Scholar]
- 83.Biek R, Pybus OG, Lloyd-Smith JO, Didelot X. Measurably evolving pathogens in the genomic era. Trends Ecol Evol. 2015;30:306–313. doi: 10.1016/j.tree.2015.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rambaut A, Suchard MA, Xie D, Drummond AJ. Tracer. 2014;1(6):1–1. [Google Scholar]
- 85.He ZL, Zhang HK, Gao SH, Lercher MJ, Chen WH, Hu SN. Evolview v2: an online visualization and management tool for customized and annotated phylogenetic trees. Nucleic Acids Res. 2016;44:W236–W241. doi: 10.1093/nar/gkw370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gavryushkina A, Welch D, Stadler T, Drummond AJ. Bayesian inference of sampled ancestor trees for epidemiology and fossil calibration. PLOS Comput Biol. 2014;10:1–15. doi: 10.1371/journal.pcbi.1003919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.de Oliveira Martins L, Bloomfield S, Stoakes E, Grant A, Page AJ, Mather AE. Tatajuba-Exploring the distribution of homopolymer tracts. bioRxiv. 2021;1–12. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1. Genomic information and metadata on ST45 isolates investigated.
Additional file 2. ST45 pangenome linear regression modeling.
Additional file 3. ST45 clade 18 analysis.
Additional file 4. New Zealand patient phylogenetic analysis.
Additional file 5. New Zealand patient clade gene analysis.
Additional file 6. New Zealand patient gene number modeling.
Additional file 7. United Kingdom patient phylogenetic analysis.
Additional file 8. United Kingdom patient gene number modeling.
Additional file 9. Tables of the proportion of DNA replication genes found in long-term Campylobacter patients and other ST45 isolates (Table S4), and the mutations in Campylobacter DNA replication genes associated with isolates collected from long-term patients (Table S5).
Additional file 10. Long-term patients’ pangenome function analysis.
Additional file 11. Long-term patients’ gene variation function analysis.
Data Availability Statement
The reads of the Campylobacter genomes and the 16S rRNA sequences were uploaded to NCBI under the Accession Numbers PRJEB24941 and PRJNA605845, respectively.