

Abstract
Maturity‐onset diabetes of young (MODY) is an autosomal dominant genetic disorder that causes insulin deficiency without autoimmunity. We present the first family with pancreatic duodenal homeobox 1 (PDX1) mutation causing diabetes from Qatar. Routine genetic screening of all antibody‐negative diabetic patients with diabetes should be offered to avoid misdiagnosis.
Keywords: MODY, PDX1, pediatric diabetes mellitus
We report the first family with PDX1 mutation causing diabetes in children from Qatar and present a complex pedigree which is contributing to the disease condition. Efforts to implement precision medicine for all patients with antibody‐negative diabetes will bring better clinical outcomes.

1. INTRODUCTION
Maturity‐onset diabetes of the young (MODY) is an autosomal dominant genetic disorder characterized by impaired insulin secretion causing hyperglycemia at an early age, most commonly before 25 years of age. There is minimal or no defect in insulin action, absence of autoimmunity or insulin resistance. 1 Serum insulin and/or c‐peptide with some residual pancreatic function is usually present. In the family history, typically multiple members will have diabetes. 2 MODY is the most common form of monogenic diabetes affecting 1%–5% of all patients with diabetes mellitus (DM). 3 However, these figures are based on studies in European and other western countries, with limited information about MODY in Middle Eastern countries. 2 , 3 MODY subjects are often misdiagnosed as type 1 or type 2 diabetes; however, they have different treatment modalities and prognosis. Diagnosis of MODY should be considered in subjects with autoantibody negative atypical diabetes with multiple affected family members. 1
14 subtypes of MODY have been described in the literature, the most common being MODY due to glucokinase (GCK), hepatocyte nuclear factor 1A (HNF1A), and hepatocyte nuclear factor 4A (HNF4A) gene mutations. 4 Some types of MODY need no treatment whereas others can be treated with diet or oral sulphonylureas rather than insulin injections; therefore, an accurate diagnosis is important for clinical management.
2. CASE REPORT
We present a family of three children and both parents with diabetes. They are of Arab ethnicity and products of non‐consanguineous marriage. The index case (Patient 1) is a 14‐year‐old boy born at term by normal vaginal delivery. There was a history of gestational diabetes (GDM) during the pregnancy but was otherwise uneventful. He was developmentally normal with normal body mass index (BMI) and eating patterns. At the age of 10 years, he presented to the clinic with increased frequency of urination and the investigations confirmed hyperglycemia with blood glucose level of 12.5 mmol/L. He was diagnosed as type 1 diabetes initially. Patient 2 is a 12‐year‐old boy and patient 3 is an 8‐year‐old boy, both are products of full‐term normal delivery after a pregnancy complicated by DM in the mother. They were physically and developmentally within normal limits. Patient 2 presented to the clinic with dysglycaemia with blood glucose level of 16.7 mmol/L at 8 years of age while patient 3 presented at 4 years with blood glucose level of 14.4 mmol/L. These last 2 siblings reported no history of polydipsia, polyuria, weight loss, or fatigue. Autoantibody testing was conducted and found to be negative. The autoantibodies tested were glutamic acid decarboxylase 65 (GAD65), insulin autoantibody (IAA), islet antigen‐2 autoantibody (IA‐2A), and zinc transporter 8 (ZnT8A). The patients were given diabetes education sessions to achieve target blood sugar levels and maintain a healthy lifestyle. Advice was given regarding carbohydrate counting and exercise routine and was followed up 3 monthly initially to monitor the blood sugar levels closely. Currently, all three patients are managed with diet control and lifestyle modifications only, insulin supplementation is not required and is being followed up 6 monthly. HbA1c after diabetes education and management is 33, 37, and 32 mmol/mol in patient 1, 2, and 3, respectively. Age of onset of diabetes in father was 55 years and in mother was 32 years.
The salient clinical and biochemical features are described in Table 1.
TABLE 1.
Salient clinical features observed
| Feature | Patient 1 | Patient 2 | Patient 3 |
|---|---|---|---|
| Age of onset | 10 years | 8 years | 4 years |
| HbA1c at diagnosis (mmol/mol) | 55 | 80 | 65 |
| HbA1c after diet change (mmol/mol) | 33 | 37 | 32 |
| C‐peptide (nmol/L) | 0.28 | 0.28 | 0.12 |
| Insulin (pmol/L) | 20 | 40 | 15 |
| Autoantibody status | Negative | Negative | Negative |
| Thyroid Peroxidase | Negative | Negative | Negative |
| Celiac autoantibodies | Negative | Negative | Negative |
3. FAMILY HISTORY
There is a strong positive family history of diabetes. Maternal and paternal grandparents as well as one maternal uncle and one paternal aunty suffered from diabetes. Detailed family pedigree is shown in Figure 1. Age of onset in grandparents and uncles are unknown.
FIGURE 1.

Pedigree chart for the family
4. GENETIC ANALYSIS
DNA samples were extracted from peripheral blood specimen of all individuals and whole‐exome sequencing was performed on Illumina HiSeq platform using a 150‐base paired‐end single‐index‐read format. Genomic variants belonging to genes already known to be implicated in MODY were extracted. Sequencing in the three subjects and mother revealed a heterozygous missense variant on chr13:28494372 C>A, NM_000209 and c.97C>A (p. Pro33Thr) of PDX1 gene, minor allele frequency (MAF < 0.01) with conflicting interpretation of pathogenicity. It was absent in the father. To confirm the variant identified by WGS, DNA was used for Sanger sequencing. Specific Primers (Forward Primer 5′‐ CACGCAGCTTTACAAGGACC 3′ and Reverse Primer 5′ GGAGGTGGTGGTGAAGGTG 3′) were designed using primer3 software (http://primer3.ut.ee/) to amplify the region of interest using polymerase chain reaction (PCR). The amplified PCR products were purified with PCR purification kit (Qiagen), followed by cycle sequencing PCR using a BigDye Terminator Sequencing Kit v.3.1 (Thermo Fisher Scientific, Inc.). The product was analyzed using a 96‐capillary ABI 3500xl automated genetic Analyzer (Applied Biosystems; Thermo Fisher Scientific). See Figures 2 and 3 for pictorial representation.
FIGURE 2.

IGV analysis showing the heterozygous variant
FIGURE 3.
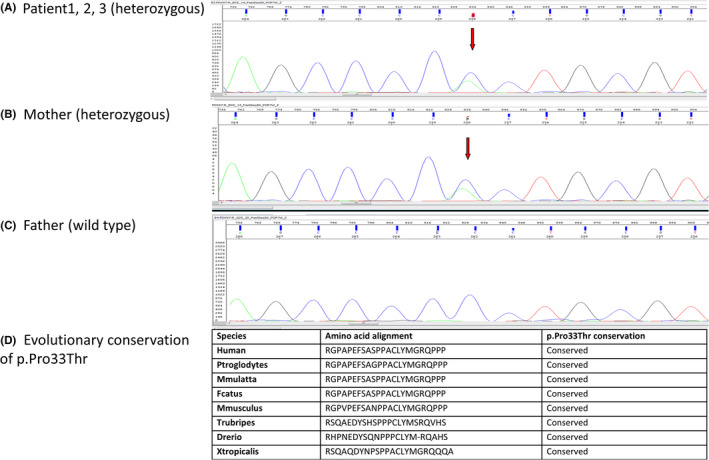
Sanger sequencing and evolutionary conservation of the PDX1 mutation. (A) Patient1, 2, 3 (heterozygous). (B) Mother (heterozygous). (C) Father (wild type). (D) Evolutionary conservation of p. Pro33Thr
5. DISCUSSION
The homeodomain‐containing transcription factor pancreatic duodenal homeobox 1 is a nuclear transcription factor consisting of 283 amino acids. It has a dual role initially in contributing to pancreatic development during embryogenesis and subsequently regulating pancreatic islet cell physiology in mature islet cells. It is a key transcriptional regulator of the INS, GCK, GLUT2 genes in the pancreatic beta cell. Homozygous mutations in the PDX1 gene lead to pancreatic agenesis/hypoplasia and neonatal diabetes mellitus with exocrine insufficiency whereas heterozygous PDX1 mutations lead to MODY 4 and late‐onset type 2 DM. 5 Caetano et al. 6 described two Brazilian patients with MODY4 with dorsal pancreatic agenesis and one of them also had exocrine insufficiency. Previous studies indicate that heterozygous PDX1 mutations result in reduction in secretion of insulin and a relatively mild glucose intolerance. A study from China reported MODY 4 in a family consisting of multiple members with diabetes and obesity including a 15‐year‐old boy, where, after diagnosis of MODY insulin supplementation was completely stopped and treated with oral metformin and healthy lifestyle alone to maintain glycemic control. 7 Another study from Japan reported PDX1 MODY in a patient at 16 years of age. He was lean his entire life and developed early‐onset diabetes that required oral anti‐diabetic treatment. However, the proband's father with the same mutation was obese with later‐onset diabetes and did not need any medication. 8 There is heterogeneity in the clinical phenotype observed in PDX1 MODY cases in the literature.
In our patients, there is an amino acid substitution from proline to threonine (p. Pro33Thr). The results of the analysis of this variant with different bioinformatics insilico prediction tools suggest that it is a deleterious variant:‐SIFT (deleterious, score 0), Polyphen‐2 (probably damaging, score 0.997), and MutationTaster (disease causing).
The variant Pro33Thr affects transactivation domain of the PDX1 gene in a highly conserved residues among several species, with genomic evolutionary rate profiling (GERP) value of 2.7 (Figure 3B). Gragnoli et al 9 reported this variant in an Italian extended family with many affected members. The clinical phenotypes observed in the family were GDM, MODY, and type 2 diabetes of the 22 member's genotypes, nine of them had the mutation. 9 In vitro studies showed that the mutant P33T protein had a reduction in DNA binding and transcription activation thus confirming pathogenicity of the variant. 9 Similar to previously reported cases of MODY 4, our patients only had mild dysglycemia, no ketoacidosis, and are not obese. Four type 1 diabetes autoantibodies namely GAD65, IAA, IA‐2A, and ZnT8A were negative and they are managed conservatively with healthy diet and exercise only. Many members of the extended family have DM, and accurate diagnoses of MODY were challenging since type 2 DM is also commonly inherited in families. We also observed that age of diagnosis also became earlier for subsequent siblings than patient 1. This could be due to the fact that parents are more aware of the signs and symptoms now. The c‐peptide and insulin level are just detectable in three siblings and indicate there is residual pancreatic function. It is interesting to note that the mother who also has the same mutation did not develop diabetes at a young age. She had GDM with her third pregnancy which later progressed to permanent type 2 diabetes. Previous studies have observed GDM in women with PDX1 mutation; however, the triggers for the different ages of onset of diabetes are not well understood. 10 This raises the question if the early onset of dysglycaemia in the children is a result of the combined effect of genetic triggers from paternal and maternal sides of the family or related to environmental and lifestyle factors in the Middle East.
We report the first family with PDX1 mutation causing diabetes in children from Qatar, and present a complex pedigree which is contributing to the disease condition. More efforts should be taken for genetic screening of families with multiple affected members and onset of DM in young age.
We suggest that screening for genetic causes should be considered in subjects based on their clinical history, Hba1c level, autoantibody negativity, detectable c‐peptide level, and positive family history. This will help to implement precision medicine for appropriate management and better clinical outcomes for all patients with MODY.
CONFLICT OF INTEREST
The authors declare that there is no conflict of interest regarding the publication of this article.
AUTHOR CONTRIBUTIONS
Basma Haris collected patient information, recruited the patients, analyzed and interpreted the data, and drafted the manuscript. Idris Mohammed analyzed genetic data, performed the laboratory confirmatory experiments, and drafted the manuscript. Najeeb Syed performed genetic analysis, drafted and reviewed the manuscript. Khalid Fakhro obtained funding and reviewed the manuscript. Khalid Hussain designed the study, obtained funding, collected patient information, reviewed and edited the manuscript.
ETHICAL APPROVAL
This study was approved by the Institutional Review Board (IRB) for the protection of human subjects in Sidra Medicine, Qatar (IRB reference number 1702007592) and as per the Declaration of Helsinki. Informed consent was taken from the subjects and parents as required.
CONSENT
Written informed consent was obtained from the patient to publish this report in accordance with the journal's patient consent policy.
ACKNOWLEDGEMENTS
None.
Haris B, Mohammed I, Syed N, Fakhro K, Hussain K. Maturity‐onset diabetes of the young (MODY) due to PDX1 mutation in a sib‐pair diabetes family from Qatar. Clin Case Rep. 2021;9:e05141. doi: 10.1002/ccr3.5141
Funding information
This research was supported by the Qatar National Research Fund [QNRF‐NPRP 10‐6100017‐AXX] awarded to Professor Khalid Hussain.
DATA AVAILABILITY STATEMENT
The data generated/analyzed during this study is not available in a public repository to protect patient confidentiality but are available from the corresponding author on reasonable request.
REFERENCES
- 1. American Diabetes Association . 2. Classification and diagnosis of diabetes: standards of medical care in diabetes‐2021. Diabetes Care. 2021;44(Suppl 1):S15‐S33. 10.2337/dc21-S002. Erratum in: Diabetes Care. 2021 Sep;44(9):2182. PMID: 33298413. [DOI] [PubMed] [Google Scholar]
- 2. Nkonge KM, Nkonge DK, Nkonge TN. The epidemiology, molecular pathogenesis, diagnosis, and treatment of maturity‐onset diabetes of the young (MODY). Clin Diabetes Endocrinol. 2020;6(1):20. 10.1186/s40842-020-00112-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Urakami T. Maturity‐onset diabetes of the young (MODY): current perspectives on diagnosis and treatment. Diabetes Metab Syndr Obes. 2019;8(12):1047‐1056. 10.2147/DMSO.S179793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Anık A, Çatlı G, Abacı A, Böber E. Maturity‐onset diabetes of the young (MODY): an update. J Pediatr Endocrinol Metab. 2015;28(3–4):251‐263. 10.1515/jpem-2014-0384 [DOI] [PubMed] [Google Scholar]
- 5. Winter WE, Silverstein JH. Molecular and genetic bases for maturity onset diabetes of youth. Curr Opin Pediatr. 2000;12(4):388‐393. 10.1097/00008480-200008000-00019. Erratum. In: Curr Opin Pediatr 2001 Feb; 13(1):95‐6. PMID: 10943822. [DOI] [PubMed] [Google Scholar]
- 6. Caetano LA, Santana LS, Costa‐Riquetto AD, et al. PDX1 ‐MODY and dorsal pancreatic agenesis: new phenotype of a rare disease. Clin Genet. 2018;93(2):382‐386. 10.1111/cge.13044. Epub 2017 Jul 19 PMID: 28436541. [DOI] [PubMed] [Google Scholar]
- 7. Deng M, Xiao X, Zhou L, Wang T. First Case report of maturity‐onset diabetes of the young type 4 pedigree in a chinese family. Front Endocrinol (Lausanne). 2019;3(10):406. 10.3389/fendo.2019.00406. PMID: 31333579; PMCID: PMC6618295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Satoshi Y, Yukio H, Sodai K, et al. First Japanese family with PDX1‐MODY (MODY4): a novel PDX1 frameshift mutation, clinical characteristics, and implications. J Endocr Soc. 2021;bvab159. 10.1210/jendso/bvab159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gragnoli C, Stanojevic V, Gorini A, Von Preussenthal GM, Thomas MK, Habener JF. IPF‐1/MODY4 gene missense mutation in an Italian family with type 2 and gestational diabetes. Metabolism. 2005;54(8):983‐988. 10.1016/j.metabol.2005.01.037. PMID: 16092045. [DOI] [PubMed] [Google Scholar]
- 10. Wang X, Sterr M, Ansarullah BI, et al. Point mutations in the PDX1 transactivation domain impair human β‐cell development and function. Mol Metab. 2019;24:80‐97. 10.1016/j.molmet.2019.03.006. Epub 2019 Mar 20. PMID: 30930126; PMCID: PMC6531841. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data generated/analyzed during this study is not available in a public repository to protect patient confidentiality but are available from the corresponding author on reasonable request.