

Abstract
Aneurysmal subarachnoid hemorrhage (aSAH) is associated with high socio-economic burden. Prothrombotic states of early brain injury (EBI) and delayed cerebral ischemia (DCI) after aSAH determine morbidity and mortality. To understand how activated platelets might contribute to such prothrombotic states, we studied trends in coated-platelets during EBI and DCI periods. Serial blood samples from a prospective cohort of aSAH patients were collected and assayed for coated-platelet levels. Patient’s coated-platelet level during post-hospital discharge follow-up served as an estimate of baseline. Occurrence of DCI, Montreal cognitive assessment (MOCA) score of < 26, and modified Rankin scale (mRS) of 3–6 were considered poor clinical outcomes. Non-linear regression analysis detected a transition between periods of rising and declining coated-platelet levels at day 4. Additional regression analyses of coated-platelet trends before day 4 showed differences among patients with modified Fisher 3–4 [4.2% per day (95% CI 2.4, 6.1) vs. − 0.8% per day (95% CI − 3.4, 1.8); p = 0.0023] and those developing DCI [4.6% per day (95% CI 2.8, 6.5) vs. − 1.9% per day (95% CI − 4.5, 0.5); p < 0.001]. Differences between peak coated-platelet levels and baseline levels were larger, on average for those with DCI [18.1 ± 9.6 vs. 10.6 ± 8.0; p = 0.03], MOCA < 26 [17.0 ± 7.8 vs. 10.7 ± 7.4; p = 0.05] and mRS 3–6 [24.8 ± 10.5 vs. 11.9 ± 7.6; p = 0.01]. Coated-platelet trends after aSAH predict DCI and short-term clinical outcomes. The degree of rise in coated-platelets is also associated with adverse clinical outcomes.
Keywords: Acute brain injury, Hemorrhagic stroke, Hypercoagulability, Reactive platelets, Stroke morbidity
Introduction
Aneurysmal subarachnoid hemorrhage (aSAH) affects middle-aged individuals and is associated with significant morbidity among survivors [1, 2]. For a long time, cerebral vasospasm (VSM) was postulated to be a primary cause for such morbidity. However, the absence of significant effect on clinical outcome in the CONSCIOUS-2 and CONSCIOUS-3 trials has brought into question the role of VSM [3, 4]. Moreover, brain perfusion studies have demonstrated cerebral hypo-perfusion occurs in brain regions without VSM [5]. Hence, newer mechanistic theories that distinguish early brain injury (EBI) during the first 3 days and subsequent delayed cerebral ischemia (DCI) during days 4–14 have replaced VSM as the leading mechanism for both short- and long-term clinical outcome after aSAH [6].
Microthrombosis following aSAH is one of the several mechanisms that are proposed to explain EBI and DCI [6–8]. Such microcirculatory ischemic events are likely contributors to global cerebral atrophy and cognitive deficits observed during long-term follow-up of aSAH patients [9, 10]. Radiological, serological, and clinical effects of such thrombotic events in the brain after aSAH have been demonstrated [11–13]. Derangement in thrombotic and fibrinolysis pathways after aSAH is also reported and is frequently dependent on presenting acuity of aSAH as measured by Hunt and Hess (H&H), World Federation of Neurological Surgeons (WFNS) scale, and Fisher/modified Fisher grades [14, 15]. Most of these studies investigated serological prothrombotic factors and does not report platelet-related thrombogenicity [12, 14–16].
Apart from its role in thrombosis, activated platelets participate in several other pathophysiological mechanisms including neuroinflammation [17, 18]. Coated-platelets are a subpopulation of platelets that retains prothrombotic substances on their surfaces, when appropriately stimulated, and can be used as a surrogate marker for platelet-related thrombogenicity. In normal populations, coated-platelet levels in individuals vary minimally over time [19]. Altered coated-platelet levels are noted in different subcategories of stroke [20–22]. Our group has previously reported higher coated-platelet levels during the first 4 days after aSAH as compared to age- and sex-matched controls and a possible association between lower levels and higher 30-day mortality [21].
Current practice for management of aSAH patients involves prolonged hospitalization to prevent and treat complications during the post-hemorrhage period. The extended hospital stay provides an opportunity to study disease evolution. In this hypothesis-driven study, our objective is to determine if coated-platelet levels change during different phases of the disease and whether the severity of aSAH at presentation has a “dose-effect” on these changes. While exploring the hypothesis, we were able to show not only “change in coated-platelet levels” but also a pattern with a specific “shape.” An understanding of coated-platelet trends would provide insight regarding role of activated platelets in pathophysiology of DCI and related clinical outcomes.
Materials and Methods
The present study was approved by Institutional Review Board (IRB) at the University of Oklahoma Health Sciences Centre in accordance with ethical standards for medical research laid down in the 1964 Declaration of Helsinki and its later amendments.
Patient Screening, Enrolment, and Characteristics
All patients presenting to Oklahoma University Medical Centre (OUMC), Oklahoma City, with diagnosis of spontaneous aSAH between July 1, 2014, and April 30, 2016, were screened for enrolment in the present study. Informed consent was obtained from each patient (if determined to be competent) or his/her next-of-kin as per local IRB guidelines. Patients included in the study were age > 18 years, presented within 48 h of ictus and had the first blood sample collected within 72 h. Of note, all patients included in the study presented within 24 h of symptom onset and underwent aneurysm securing procedure within 24 h of hospital presentation. This study reports only data from patients with aSAH as it is the most common and clinically relevant etiology of non-traumatic spontaneous SAH. Exclusion criteria consisted of: non-aneurysmal SAH, pregnancy, age > 85 years, family deciding on early withdrawal of care, patients with severe grade SAH with early comfort care in whom serial sample collections were not possible, patients on serotonin-reuptake inhibitors or antiplatelet agents preadmission or patients with SAH due to arterio-venous malformations or other vascular malformations and tumor.
After admission, all aSAH patients included in this study were managed in OUMC Neurosciences Intensive Care Unit (NSICU). As per our NSICU protocol, before securing ruptured aneurysms with coil embolization or clip ligation, systolic blood pressure (SBP) is kept below 140 mmHg with Nicardipine infusion and/or intermittent doses of parenteral labetalol. Consultant neurosurgeon and neurointerventionalist mutually determined the aneurysm securing modality. As per local clinical protocol, Nimodipine is administered for 21 days and Levetiracetam used for seizure prophylaxis for 7 days or longer (if seizure was present at time of admission or later during hospitalization). Statins were continued for patients already using them but never initiated. Prophylactic antithrombotic therapy, using Heparin, was started 24–48 h after clip ligation or coil embolization. Euvolemia was maintained with crystalloids and/or colloids as per discretion of treating physician. Such management was continued until daily transcranial Doppler studies show evidence of down-trending velocities in intracranial vessels, indicating VSM resolution. Blood pressure goal after diagnosis of symptomatic VSM was maintained between SBP of 160 and 200 mmHg.
Outcome Measures
Clinical outcome measures included DCI during the hospitalization and physical (modified Rankin scale, mRS) and cognitive (Montreal cognitive assessment, MOCA) outcomes during first post-hospitalization follow-up. We defined DCI as a new hypodensity (excluding periprocedural hypodensities) on computed tomography (CT) scans located in a vascular or other territories and/or associated symptoms, including a decrease of consciousness and focal deficits, due to cerebral vasospasm and not explained by other causes (e.g., re-bleeding, hydrocephalus, cardio-embolic sources of emboli, hypoxia, electrolyte disturbances, or seizures). This definition of DCI is in accordance to that proposed by Vergouwen et al. [23], i.e., “The occurrence of focal neurological impairment (such as hemiparesis, aphasia, apraxia, hemianopia, or neglect), or a decrease of at least 2 points on the Glasgow Coma Scale (either on the total score or on one of its individual components [eye, motor on either side, verbal]). This should last for at least 1 hour, is not apparent immediately after aneurysm occlusion, and cannot be attributed to other causes by means of clinical assessment, by CT or MRI scanning of the brain, or by appropriate laboratory studies.”. Investigators prospectively reviewed personal interview and medical records to document patient’s baseline clinical, laboratory, and imaging information (Table 1).
Table 1.
Baseline demographic and clinical characteristics of patients (n = 45)
| Characteristics | n (%) |
|---|---|
| No. of women (%) | 31 (68.9) |
| Age | |
| Mean ± SD (min. 26; max 83) | 54.3 ± 13.0 |
| Median (IQR) | 54.0 (46.0–83.0) |
| Hypertension | 27 (60.0) |
| Diabetes mellitus | 4 (8.9) |
| Hyperlipidemia | 10 (22.2) |
| Smoking | 32 (71.1) |
| Location of aneurysm | |
| Anterior circulation | 37 (82.2) |
| Posterior circulation | 8 (17.8) |
| Procedure for securing ruptured aneurysms | |
| Clip ligation | 33 (73.3) |
| Coil embolization | 12 (26.7) |
| Clinical characteristics | |
| Modified Fisher | |
| 1, 2 | 16 (35.6) |
| 3, 4 | 29 (64.4) |
| Hunt and Hess | |
| Grade I–III | 36 (80.0) |
| Grade IV, V | 9 (20.0) |
| World Federation of Neurological Surgeons Scale | |
| Grade 1–3 | 27 (60.0) |
| Grade 4, 5 | 18 (40.0) |
IQR interquartile range, SAH subarachnoid hemorrhage, SD standard deviation
Sample Collection and Analysis
After obtaining informed consent, blood samples were collected on alternate days on an average following admission till patient was discharged or 21 days whichever was earlier (blood collection varied from 9 to 21 days post-ictus) and also during follow-up clinic visit. Coated-platelet levels (expressed as percentages of total platelet count) were measured using flow cytometry as previously described [21]. Individuals performing the coated-platelet assay were not aware of patients’ clinical diagnoses. Similarly, the neuroradiologist reading the imaging studies and the neurologist establishing the diagnosis of DCI were not aware of the coated-platelet measurements.
Follow-up Assessment
Clinic follow-up was conducted 6 to 20 weeks after the initial ictus. As per study protocol, we planned clinic follow-up post-hospital discharge at 12 weeks (i.e., ~90 days post-ictus). However, follow-up was delayed for some patients with prolonged stays in long-term acute care facilities, or early for others due to surgeon’s decisions or health insurance requirements. Even so, mean follow-up was 12.8 ± 6.3 weeks and median 12 weeks with interquartile range, IQR of 6.5 weeks (8, 14.5). During this visit, investigators assessed physical status using mRS and cognitive status using MOCA test. A score of 3–6 on mRS and less than 26 on MOCA were considered as poor clinical outcomes. A blood sample was collected to determine coated-platelet level. Since, we obtained this sample at a time remote from ictus; we considered its value to reflect the patient’s baseline coated-platelet level.
Statistical Analysis
We hypothesized that a period of EBI and a subsequent period of DCI could be reflected in the trends that coated-platelet levels exhibit after aSAH. To explore these trends, including the possibility that the trend might change or even reverse itself over the first 21 days, we constructed non-linear mixed regression models (SAS PROC NLMIXED v9.4) that included patient-specific random effects. The models’ parameters were designed to estimate the slope of the two linear trends that intersected at a day of transition. The model also estimated the day of transition, along with a 95% confidence interval for that estimate.
When the non-linear regression models found consistent evidence of a transition or change in trend at around 4 days, we constructed a set of segmented mixed linear regression models (SAS PROC MIXED v9.4) that permitted more precise estimation of the rate of change in coated-platelet levels (in percent per day) during both the “acute phase” from 0 to 4 days, and during the “post-acute phase” from 5 to 21 days. The segmented linear mixed models, by assuming a change in trend at 4 days, had better power and precision to estimate rates of change than did the non-linear models. To avoid overfitting in this sample, we constructed models that included time since aSAH (in days) and a single covariate. Covariates included measures of disease severity (H&H, WFNS, modified Fisher scales), clinical outcomes like the presence or absence of DCI, and surgical treatment (clip ligation vs. coil embolization). Each linear mixed regression models also included an interaction term, which combined the covariate and time, to explore whether patterns of change in coated-platelet levels differed between patient groups.
For each patient who attended clinic follow-up visit, we calculated the maximum change in coated-platelet levels as the difference between presumed baseline, recorded at the time of follow-up visit, and the highest level recorded over the first 21 days. Between-group differences in mean maximum change in coated-platelet levels were assessed using non-parametric exact permutation tests.
Results
Forty-five aSAH patients were enrolled during the study period (Table 1). A total of 289 blood samples were collected and analyzed for coated-platelet levels during the first 21 days of hospitalization. Blood samples were also collected from 33 patients during their post-hospital discharge follow-up visit which was 12.8 ± 6.3 weeks. Six patients (13.3%) died during hospitalization and six patients were lost to follow-up. We could not perform MOCA testing during the follow-up visit on three patients, two of who were in a minimal conscious state and one of whom was aphasic (Fig. 1). During our initial sample collection phase, we observed in some patients that coated-platelet levels acutely increased up to certain post-bleed day (PBD) and then trended downward (Fig. 2a, b).
Fig. 1.
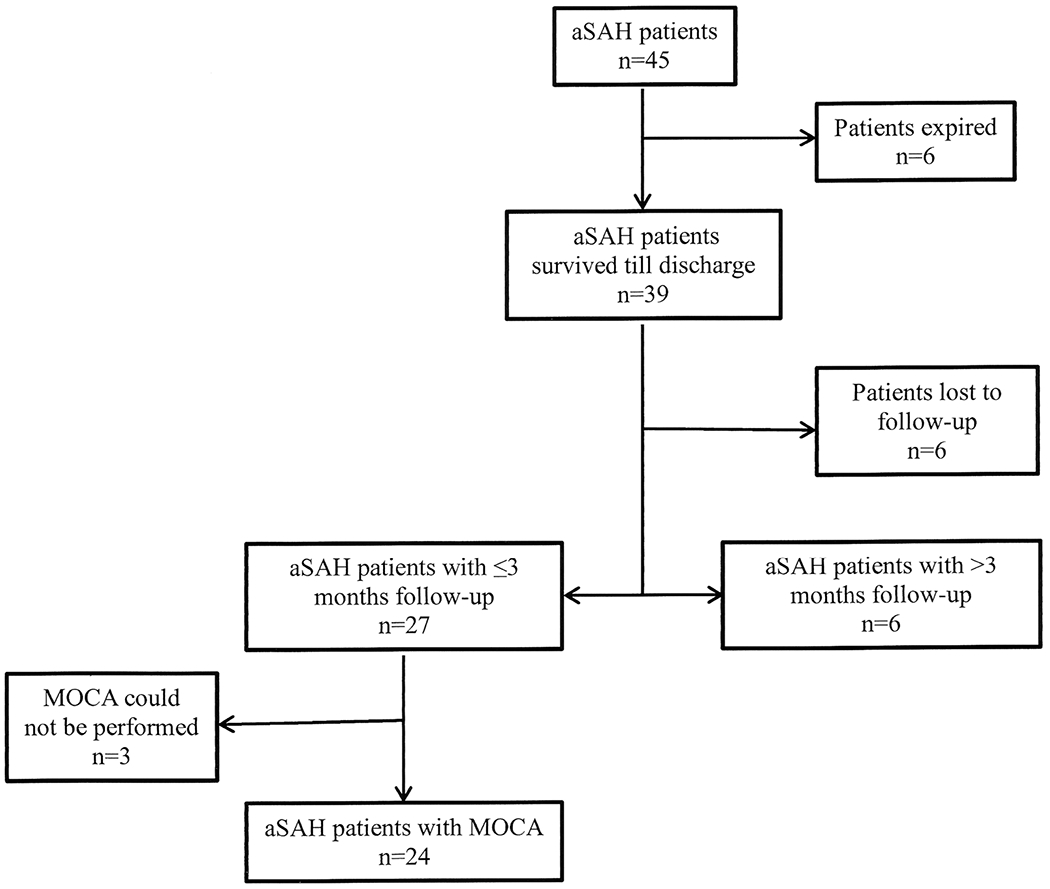
Flowchart illustrating patients enrolled for the study
Fig. 2.
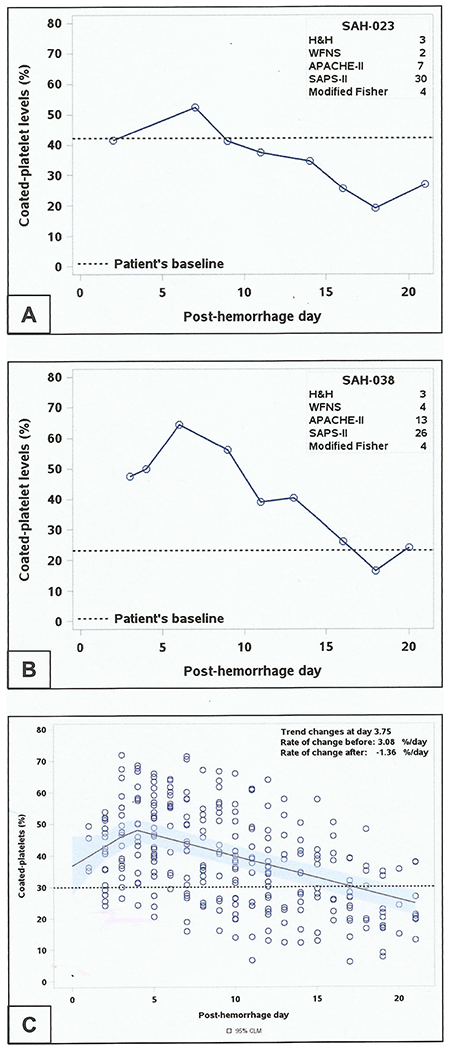
Trend of coated-platelet levels after aneurysmal subarachnoid hemorrhage. Individual trends of coated-platelets during first 21 days of aSAH in two patients (a, b). c Scatter plot depicting coated-platelet trend of all patients with aSAH (n = 45) over 21 days. Line shows the best fit for the trend with transition point at ~4 days. Dashed line indicates population average of coated-platelets
A non-linear regression model estimated that, in the entire cohort (n = 45), rising coated-platelet levels reversed at 3.8 days after SAH (95% confidence interval, CI 2.5 days, 5.1 days) [Fig. 2c]. Similar non-linear regression models, applied to patient groups defined by clinical (H&H and WFNS) and radiological (modified Fisher) severity and by the incidence of adverse clinical outcome (DCI), estimated similar times of transition (Table 2). Changes in trend were detected in all groups except for those with modified Fisher 1–2 and those without DCI. The non-linear regression analysis estimated coated-platelet levels at PBD 0 to be 36.7% (95% CI, 27.2, 46.2) of total platelet population, a value that is similar to averages previously reported normal population averages.
Table 2.
Transition day in coated-platelet trend after aSAH
| Clinical severity/outcome | n | Transition day | Lower limit 95% CI | Upper limit 95% CI |
|---|---|---|---|---|
| H&H | ||||
| IV–V | 9 | 3.1 | 2.0 | 4.3 |
| I–III | 36 | 4.1 | 2.8 | 5.5 |
| WFNS | ||||
| 4–5 | 18 | 3.0 | 2.996 | 3.004 |
| 1–3 | 27 | 4.2 | 2.5 | 5.8 |
| Modified Fisher | ||||
| 3–4 | 29 | 4.3 | 3.4 | 5.1 |
| 0–2 | 16 | na | na | na |
| DCI | ||||
| Yes | 30 | 4.3 | 3.4 | 5.1 |
| No | 15 | na | na | na |
| Procedure | ||||
| Clip | 33 | na | na | na |
| Coil | 12 | 4.3 | 2.5 | 6.2 |
aSAH aneurysmal subarachnoid hemorrhage, DCI delayed cerebral ischemia, H&H Hunt and Hess, na not applicable, WFNS World Federation of Neurological Surgeons Scale
Having found evidence that coated-platelet levels rose and peaked at day 4, we then used segmented linear regression, constructing models that assumed the joining of two linear trends at day 4, to more precisely estimate rates of change in coated-platelet levels (in percent per day) during the early post-hemorrhage (days 0–4) and later post-hemorrhage (days 5–21) periods. These models found that patients developing DCI had steeper rise and fall in coated-platelet levels compared to those without DCI (Table 3; Fig. 3a). Trends of coated-platelet levels did not differ depending on short-term clinical outcome determined by MOCA and mRS (Table 3; Fig. 3b, c). Similarly, although rise in coated-platelet levels did not differ among those undergoing clip ligation from those undergoing coil ligation but rate of decline was more prominent among those undergoing coil embolization (Table 3). While patients who present with more severe clinical grades (H&H 4–5, WFNS 4–5) had statistically significant rise and fall in coated-platelet levels from their estimated baseline during the abovementioned periods (analysis not shown), they did not reach significance when compared to patients in the corresponding clinically lower grade. However, when aSAH patients with the worst clinical outcome of mRS 5–6 (n = 8) were compared with those who had best clinical outcome of mRS 0–1 (n = 12), a trend of steeper rise and relative persistence of elevated coated-platelet levels was detected (Fig. 3d). On the contrary, patients with higher radiological severity (as determined by modified Fisher grade of 3–4) showed significant rate of rise in coated-platelets [4.2% per day (95% CI, 2.4, 6.1)] as compared to those with modified Fisher grade 0–2 [−0.8% per day (95% CI – 3.4, 1.8); p = 0.0023]. Similar persistence of coated-platelet levels was also observed among those undergoing clip ligation as compared to coil embolization (Fig. 3e). However, there was no significant difference in regard to DCI occurrence as per aneurysm securing procedure performed in these patients [DCI in clip vs. coil, 22/33 (66.7%) vs. 8/12 (66.7%), p = 1.0].
Table 3.
Rate of rise and fall in coated-platelets after aSAH
| Clinical response | Days 0–4 |
Days 4–21 |
||||||
|---|---|---|---|---|---|---|---|---|
| Estimated rate of change (%/day) | 95% CI on rate of change (%/day) | P value | Estimated rate of change (%/day) | 95% CI on rate of change (%/day) | P value | |||
| DCI (n = 45) | ||||||||
| Yes | 4.6 | 2.8 | 6.5 | < 0.0001* | − 1.6 | − 1.8 | − 1.4 | < 0.0001* |
| No | − 2.0 | − 4.5 | 0.5 | − 0.6 | − 1.0 | − 0.2 | ||
| MOCA (n = 24) | ||||||||
| < 26 | 2.9 | − 0.2 | 5.9 | 0.499 | − 1.3 | − 1.7 | − 1.0 | 0.360 |
| ≥ 26 | 1.4 | − 1.8 | 4.5 | − 1.6 | − 2.1 | − 1.2 | ||
| mRS (n = 27) | ||||||||
| 3–6 | 3.4 | 0.0 | 6.0 | 0.298 | − 1.3 | − 1.6 | − 1.0 | 0.421 |
| 0–2 | 1.5 | − 1.0 | 4.0 | − 1.5 | − 1.8 | − 1.1 | ||
| Aneurysm securing procedure (n = 45) | ||||||||
| Clip | 2.5 | 0.9 | 4.2 | 0.753 | − 1.6 | − 1.8 | − 1.3 | 0.001* |
| Coil | 3.1 | − 0.3 | 6.6 | − 0.8 | − 1.2 | − 0.4 | ||
The asterisk indicates a significant difference between groups in the rate of change of coated-platelets before and after the transition day (at a significance level of 0.05)
aSAH aneurysmal subarachnoid hemorrhage, DCI delayed cerebral ischemia, MOCA Montreal cognitive assessment test, mRS modified Rankin scale
Fig. 3.
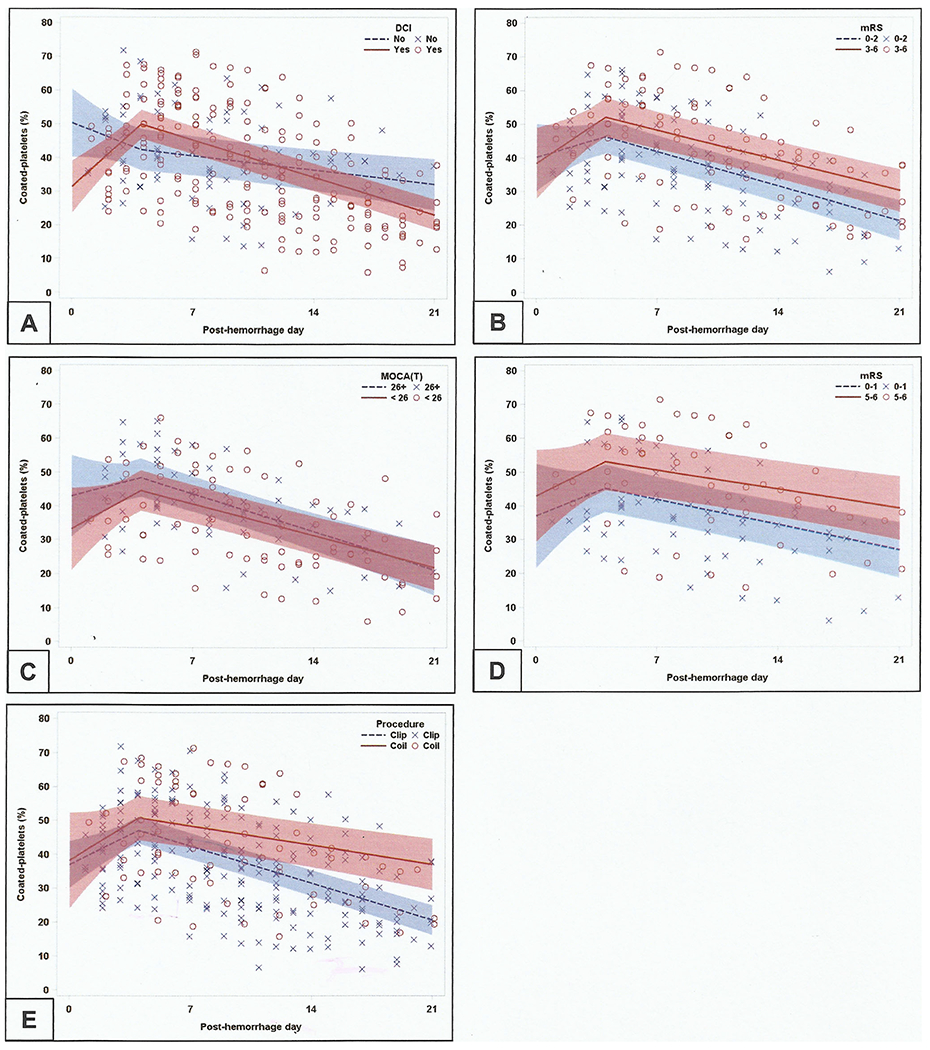
Trend of coated-platelets as delineated by various clinical outcomes. Segmented linear models segregating trends of coated-platelets among those with or without DCI (a), and good and poor mRS (b) and MOCA (c) scores during follow-up. N.B. Patients with worse clinical outcomes have steeper and early rise in coated-platelet levels with relative persistence of higher levels as compared to those with best clinical outcome (d). Similar persistence of higher levels of coated-platelets are also seen among those undergoing clip ligation (e)
Mean coated-platelet levels on the day of ictus were estimated using intercept terms from the same segmented regression models. Levels of coated-platelets at day 0 in patients who developed DCI were estimated to be 18.9% lower as compared to those without DCI [95% CI, – 31.7, – 6.1; p = 0.004]. Similarly, patients with higher radiological grades of modified Fisher 3–4 also had levels of coated-platelet at day 0 that were 13.8% lower [95% CI, – 27.0, – 0.7; p = 0.039].
Moreover, the mean overall change in coated-platelet levels, calculated for the 33 patients with follow-up visits as the difference between the presumed baseline level (recorded at follow-up) and the highest level recorded over the first 21 days, was greater in those with worse outcomes as determine by DCI incidence, MOCA scores, or mRS but was not related to manner in which aneurysm was secured (Table 4, Fig. 4).
Table 4.
Degree of rise in coated-platelets above patient’s baseline after aSAH
| Clinical outcome | n | Mean ± SD | Median (IQR) | P value |
|---|---|---|---|---|
| DCI (n = 33) | ||||
| Yes | 22 | 18.1 ± 9.6 | 17.0 (11.0–20.4) | 0.03* |
| No | 11 | 10.6 ± 8.0 | 9.1 (6.0–18.4) | |
| MOCA (n = 24) | ||||
| < 26 | 11 | 17.0 ± 7.8 | 15.6 (11.0–20.4) | 0.05 |
| ≥ 26 | 13 | 10.7 ± 7.4 | 9.1 (6.1–17.0) | |
| mRS (n = 27) | ||||
| 3–6 | 8 | 24.8 ± 10.5 | 20.9 (16.4–34.7) | 0.001* |
| 0–2 | 19 | 11.9 ± 7.6 | 11.0 (6.1–18.4) | |
| Surgical procedure (n = 33) | ||||
| Clip ligation | 25 | 15.4 ± 8.5 | 15.4 (9.1–20.0) | 0.88 |
| Coil embol. | 8 | 16.1 ± 13.4 | 13.4 (4.4–25.0) | |
The asterisk indicates a significant difference between patient’s follow-up baseline and peak level achieved during hospitalization (at a significance level of 0.05)
aSAH aneurysmal subarachnoid hemorrhage, DCI delayed cerebral ischemia, IQR interquartile range, MOCA Montreal cognitive assessment test, mRS modified Rankin scale, SD standard deviation
Fig. 4.
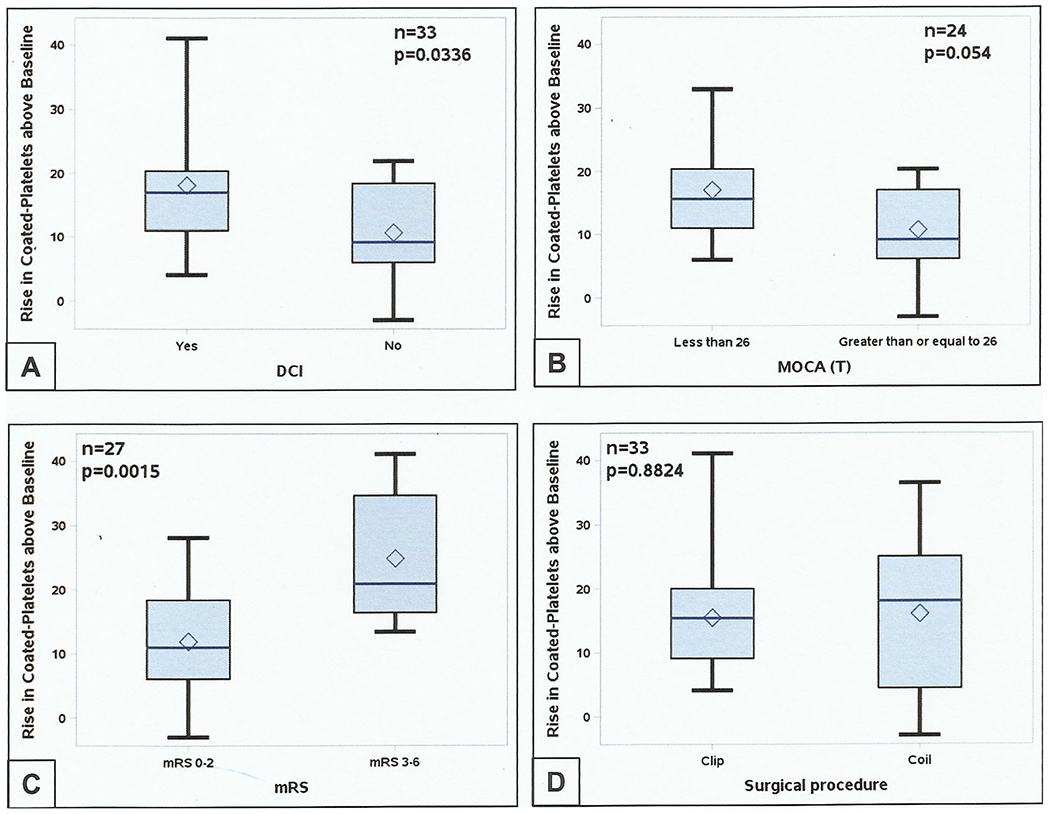
Rise in coated-platelets above patient’s post-ictal baseline. During the acute phase of subarachnoid hemorrhage degree of rise in coated-platelets predict DCI (a) and both cognitive (b) and physical (c) outcome at first post-hospital discharge clinic follow-up. However, rise was not specific for aneurysm securing procedure (d). DCI delayed cerebral ischemia, MOCA (T) Montreal cognitive assessment score (total), mRS modified Rankin score, VSM clinical cerebral vasospasm
Discussion
Our study found consistent evidence of a pattern of gradual increase, then decrease in coated-platelet potential following aSAH, with a change in trend approximately 4 days after hemorrhage. The distinctive pattern is most marked in subgroups with the most severe clinical presentation and adverse clinical outcomes. To the best of our knowledge, this is the first demonstration that coated-platelets (a marker for platelet-related systemic thrombogenicity) display temporal trends that differ between an acute and a subacute period following hemorrhage. The acute phase may relate to EBI while subacute phase may represent DCI stages following aSAH. Theoretically, EBI and DCI periods are demarcated in humans based on animal data [6, 24, 25]. Our study findings provide the first objective evidence in humans that there may be actually a transition time between a period (roughly PBD 0 to 4) related to acute brain injury (EBI), and a period when disease resolution starts (DCI). Unlike previous studies which used healthy controls to explore hypercoagulability after aSAH [16, 26], we used respective patient’s coated-platelet levels during follow-up as their own controls. Non-linear regression analysis, that does not make any presumption about the cohort’s trend, estimated coated-platelet levels on PBD 0 to be similar to previously reported population average [19]. Moreover, coated-platelet levels measured during first post-hospital discharge follow-up visit were similar to those measured at 1 year or later clinic visit levels (unpublished data).
Role of Coated-Platelets in Thrombogenicity After aSAH
Our study found a significant rise in coated-platelet levels after aSAH, which is especially marked in those presenting with higher clinical and radiological grades. Two recent articles also reported hypercoagulability using viscoelastic coagulation assays (VCA) [16, 26]. They noted that increased maximal amplitude (MA) indicates increased thrombogenicity to be associated with more severe clinical grades of aSAH during the EBI period [26]. The other study analyzing serial VCA showed similar increased MA values up to PBD 10. The VCA results were no different for aSAH patients on PBD 0 and for controls [16]. It is well known that prothrombotic proteins such as fibrinogen cause elevation in MA [27]. Hence, it may be inferred that various pro-inflammatory markers and other prothrombotic substances contributed to persistence of higher MA in these studies. Unlike our study, these studies used whole blood to determine systemic thrombogenic potency; they could not isolate platelet-related thrombogenicity from systemic thrombogenicity.
The present study estimated that coated-platelet levels rise progressively after aSAH, and that this rise persists, according to 95% confidence intervals for different subgroups, for between 2 and 6 days (Table 2). An early rise after aSAH has been similarly demonstrated using platelet aggregometry and elevated β-thromboglobulin levels [28]. Similar increase in prothrombotic factors in serum followed by fibrinolytic products that is more pronounced in those developing DCI has been described [15, 29]. Investigators in our group previously noted increased coated-platelets in samples collected during the first 4 days of ictus when compared to age matched controls [21]. These methods cannot ascertain if this rise is due to de novo activation of circulating platelets or to an increased production of coated-platelet subpopulation in the bone marrow. However, progressive rise in coated-platelet levels noted in previous studies on dogs would favor increased production rather than de novo activation, and alludes to immunological cross-talk with bone marrow in coated-platelet production [19]. SIRS response after aSAH as reported previously [13, 30] may also be responsible for a rise in coated-platelets. Coated-platelet trends among patients undergoing clip ligation and coil embolization did not differ statistically; however, early rise and persistence of higher levels among those undergoing clip ligation is noteworthy and may represent tissue injury response secondary to craniotomy. But associative role of coated-platelets in regard to manner of aneurysm secured was not evident in our cohort.
The current study, while it identifies an association between coated-platelets and DCI, cannot establish causality. For example, it is possible that currently unknown factors in the serum of aSAH patients may have resulted in increase in coated-platelet levels as suggested by reports of increased oxygen consumption and presumed platelet activation in patients with sepsis [31, 32]. Hence, we propose that after aSAH, especially in those developing DCI related complications, neurohumoral activation of bone marrow and/or circulating platelets leads to increased availability of circulating activated platelets. Such readily available activated platelets could explain the occurrence of microinfarcts during the early phase following aSAH as previously reported with imaging studies [12] and autopsy findings [33].
Down-Trending Coated-Platelet Levels and aSAH Resolution
In our cohort, coated-platelet levels declined significantly after reaching peak levels, sometimes falling below baseline (Fig. 2). Decreased availability of circulating coated-platelets may be attributed to either decreased production in the bone marrow, increased peripheral removal of coated-platelets from the circulation or inability of the circulating platelets to be activated. To the best of our knowledge, this phenomenon of downtrend has not been described before in patients with aSAH. Most studies that investigated hypercoagulability associated with aSAH report increased platelet activation markers and rise in inflammatory cytokines and markers [12, 13, 16, 26] and their association with adverse outcome. Although coated-platelet levels increased in those developing DCI, the downtrend started prior to occurrence of any clinical symptoms. This pattern suggests that coated-platelets may be facilitating other prothrombotic factors in serum that in turn contribute to DCI. These prothrombotic substances in circulation may subsequently downregulate coated-platelet production in bone marrow, re-establish deranged homeostasis. However, our study was not designed to identify such serological mediators and cannot confirm this hypothesis. Activated platelets secreting chemoattractant mediators to facilitate inflammatory cells and its role in secretion of hepatic prothrombotic proteins are reported [34, 35]. Hence, abovementioned hypothesis maybe valid but cannot be established by our study.
It is also noteworthy that recent literature investigating resolution of acute inflammation describes release of several pro-resolution lipid mediators. These processes include active secretion of ω3 fatty acid, eicosapentaenoic acid (EPA), and docosahexaenoic acid (DHA) derivatives, like resolvins, protectins, and merasins (collectively known as specialized pro-resolution mediators, SPMs). Of these products, resolvin E1 selectively inhibits platelet aggregation and activation [36–38]. Serial lipid metabolomics in patients with sepsis reported temporal persistence of increased pro-inflammatory lipid mediators and some pro-resolving mediators among non-survivors [39]. Hence, we propose that after a period of increased thrombogenicity (that may vary depending on several disease and individual related factors), there is an active, programmed response that is “turned on” to signal disease resolution and determines down-trending coated-platelet levels. This resolution process might be delayed, prolonged or absent in certain patients, resulting in morbidity and adverse clinical outcomes. Future studies need to investigate this phenomenon. Therapies that facilitate the early initiation of inflammation resolution may hold a key to improving outcomes after aSAH.
Coated-Platelets and Clinical Outcome
Patients with lower levels of estimated coated-platelets were noted to have higher incidence of DCI during acute hospitalization period. Previous studies have found a similar association between low coated-platelet levels and 30-day mortality [21]. The present study indicates that a subgroup of patients with lower baseline coated-platelet levels is prone to have more severe initial course but not necessarily poor short-term outcomes, if they survive hospitalization. This morbidity is likely related to increased amount of subarachnoid blood products as patients with low coated-platelets also had higher modified Fisher grades and is consistent with previous research [14]. Patients with lower coated-platelet levels having worse clinical outcome may indicate opposing biological effect contrary to expected. However, it may be noted that similar derangement in homeostasis is also noted in other body systems after acute illness and brain injury. This homeostatic derangement may be related to the body’s inability to adequately respond to an unfamiliar state of increased thrombogenicity. As an analogy, in a similar manner, non-diabetic patients who experience stress induced hyperglycemia have worse outcomes than diabetic patients, who may be better equipped to handle hyperglycemic states [40].
Our study also suggests that patients who experience the largest transient increase in coated-platelet levels after aSAH have an increased burden of cerebral microinfarcts, which result in higher risk of DCI and worse cognitive (MOCA) and functional (mRS) scores at follow-up. A similar phenomenon of increased procoagulant platelet potential was seen with progression of minimal cognitive impairment to dementia and association of activated platelets with vascular dementia [41, 42]. The persistence of higher levels of coated-platelets among those undergoing clip ligation may provide explanation for reports of worse clinical outcome in this population of patients although our short-term follow-up did not show such results [43, 44]. However, if such morbidity persists during long-term follow-up, it needs further investigation.
Future Directions
Limitations of the present study include a small cohort of aSAH patients and a relatively short follow-up period, resulting in limited ability to identify mechanisms leading to the novel findings of temporal changes in coated-platelets. We also acknowledge that our study hypotheses were not established a priori but rather evolved, informed by knowledge of the disease process and by exploration of the data. Accordingly, the study has limitations that are characteristic of all exploratory studies, the most important of which is that its statistical estimates probably fit this particular patient cohort better than they will other cohorts. Despite these limitations, the results suggest possible link between coated-platelets and clinical outcomes after aSAH. Future studies should address how coated-platelets are generated and its relation with life-span of platelets after aSAH with plan for extended clinical follow-up. Coated-platelet milieu in blood in regard to pro-inflammatory cytokines and pro-resolution lipid mediators also needs further investigation.
Acknowledgments
The authors would like to acknowledge the contribution of research assistants, Ms. Brittany Karfonta, BS, and Ms. Chanel Seraphin, LPN, with Department of Neurology, OUHSC, for their help with sample and data collection during the study.
Sources of Funding
This study was supported in part by the US Department of Veterans Affairs Clinical Science Research and Development Service (CX000340) and Oklahoma Centre for Neuroscience and Oklahoma University Biomedical Engineering Centre Interdisciplinary Research Grant Program, 2016.
Conflict of Interest
Bappaditya Ray received funding from Oklahoma Centre for Neuroscience and Oklahoma University Biomedical Engineering Center for the present study. Vijay M Pandav, Eleanor A Mathews, David M Thompson, Lance Ford, Lori K Yearout, Bradley N Bohnstedt, Shuchi Chaudhary, and George L Dale declare that they have no conflict of interest. Calin I Prodan received funding from the US Department of Veterans Affairs Clinical Science Research and Development Service.
Abbreviations
- aSAH
Aneurysmal subarachnoid hemorrhage
- CI
Confidence interval
- CONSCIOUS
Clazosentan to overcome neurological ischemia and infarction occurring after subarachnoid hemorrhage
- CT
Computed tomography
- DCI
Delayed cerebral ischemia
- DHA
Docosahexaenoic acid
- EBI
Early brain injury
- EPA
Eicosapentaenoic acid
- H&H
Hunt and Hess
- IQR
Interquartile range
- MA
Maximal amplitude
- MOCA
Montreal cognitive assessment
- mRS
Modified Rankin scale
- NSICU
Neurosciences intensive care unit
- OUMC
Oklahoma University Medical Center
- PBD
Post-bleed day
- SAH
Subarachnoid hemorrhage
- SBP
Systolic blood pressure
- SIRS
Systemic inflammatory response syndrome
- SPM
Specialized pro-resolution mediator
- VCA
Viscoelastic assay
- VSM
Cerebral vasospasm
- WFNS
World Federation of Neurological Surgeons
Footnotes
Ethical Approval All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed Consent Informed consent was obtained from all individual participants and/or their next-to-kin included in the study.
References
- 1.Brinjikji W, Kallmes DF, Lanzino G, Cloft HJ. Hospitalization costs for endovascular and surgical treatment of ruptured aneurysms in the United States are substantially higher than Medicare payments. AJNR Am J Neuroradiol. 2012;33(6):1037–40. 10.3174/ajnr.A2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Taylor TN, Davis PH, Torner JC, Holmes J, Meyer JW, Jacobson MF. Lifetime cost of stroke in the United States. Stroke. 1996;27(9):1459–66. 10.1161/01.STR.27.9.1459. [DOI] [PubMed] [Google Scholar]
- 3.Macdonald RL, Higashida RT, Keller E, Mayer SA, Molyneux A, Raabe A, et al. Randomized trial of clazosentan in patients with aneurysmal subarachnoid hemorrhage undergoing endovascular coiling. Stroke. 2012;43(6):1463–9. 10.1161/strokeaha.111.648980. [DOI] [PubMed] [Google Scholar]
- 4.Macdonald RL, Higashida RT, Keller E, Mayer SA, Molyneux A, Raabe A, et al. Clazosentan, an endothelin receptor antagonist, in patients with aneurysmal subarachnoid haemorrhage undergoing surgical clipping: a randomised, double-blind, placebo-controlled phase 3 trial (CONSCIOUS-2). Lancet Neurol. 2011;10(7):618–25. 10.1016/s1474-4422(11)70108-9. [DOI] [PubMed] [Google Scholar]
- 5.Dhar R, Diringer MN. Relationship between angiographic vasospasm, cerebral blood flow, and cerebral infarction after subarachnoid hemorrhage. Acta Neurochir Suppl 2015;120:161–5. 10.1007/978-3-319-04981-6_27. [DOI] [PubMed] [Google Scholar]
- 6.Macdonald RL. Delayed neurological deterioration after subarachnoid haemorrhage. Nat Rev Neurol. 2014;10(1):44–58. 10.1038/nrneurol.2013.246. [DOI] [PubMed] [Google Scholar]
- 7.Rowland MJ, Hadjipavlou G, Kelly M, Westbrook J, Pattinson KT. Delayed cerebral ischaemia after subarachnoid haemorrhage: looking beyond vasospasm. Br J Anaesth. 2012;109(3):315–29. 10.1093/bja/aes264. [DOI] [PubMed] [Google Scholar]
- 8.Vergouwen MD, Vermeulen M, Coert BA, Stroes ES, Roos YB. Microthrombosis after aneurysmal subarachnoid hemorrhage: an additional explanation for delayed cerebral ischemia. J Cereb Blood Flow Metab. 2008;28(11):1761–70. 10.1038/jcbfm.2008.74. [DOI] [PubMed] [Google Scholar]
- 9.Bendel P, Koivisto T, Aikia M, Niskanen E, Kononen M, Hanninen T et al. Atrophic enlargement of CSF volume after subarachnoid hemorrhage: correlation with neuropsychological outcome. AJNR Am J Neuroradiol. vol 2. United States 2010. p. 370–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tam AK, Kapadia A, Ilodigwe D, Li Z, Schweizer TA, Macdonald RL. Impact of global cerebral atrophy on clinical outcome after subarachnoid hemorrhage. J Neurosurg. 2013;119(1):198–206. 10.3171/2013.3jns121950. [DOI] [PubMed] [Google Scholar]
- 11.Romano JG, Forteza AM, Concha M, Koch S, Heros RC, Morcos JJ, et al. Detection of microemboli by transcranial Doppler ultrasonography in aneurysmal subarachnoid hemorrhage. Neurosurgery. 2002;50(5):1026–30. discussion 30-1 [DOI] [PubMed] [Google Scholar]
- 12.Frontera JA, Ahmed W, Zach V, Jovine M, Tanenbaum L, Sehba F, et al. Acute ischaemia after subarachnoid haemorrhage, relationship with early brain injury and impact on outcome: a prospective quantitative MRI study J Neurol Neurosurg Psychiatry. 2015;86(1):71–8. 10.1136/jnnp-2013-307313. [DOI] [PubMed] [Google Scholar]
- 13.Frontera JA, Aledort L, Gordon E, Egorova N, Moyle H, Patel A, et al. Early platelet activation, inflammation and acute brain injury after a subarachnoid hemorrhage: a pilot study J Thromb Haemost: JTH. 2012;10(4):711–3. 10.1111/j.1538-7836.2012.04651.x. [DOI] [PubMed] [Google Scholar]
- 14.Ikeda K, Asakura H, Futami K, Yamashita J. Coagulative and fibrinolytic activation in cerebrospinal fluid and plasma after subarachnoid hemorrhage. Neurosurgery. 1997;41(2):344–349; discussion 9-50. 10.1097/00006123-199708000-00002. [DOI] [PubMed] [Google Scholar]
- 15.Fujii Y, Takeuchi S, Sasaki O, Minakawa T, Koike T, Tanaka R. Hemostasis in spontaneous subarachnoid hemorrhage. Neurosurgery. 1995;37(2):226–34. 10.1227/00006123-199508000-00006. [DOI] [PubMed] [Google Scholar]
- 16.Ramchand P, Nyirjesy S, Frangos S, Doerfler S, Nawalinski K, Quattrone F, et al. Thromboelastography parameter predicts outcome after subarachnoid hemorrhage: an exploratory analysis. World Neurosurg. 96:215–21. 10.1016/j.wneu.2016.04.002. [DOI] [PubMed] [Google Scholar]
- 17.Leslie M Cell biology. Beyond clotting: the powers of platelets. Science. 2010;328(5978):562–4. 10.1126/science.328.5978.562. [DOI] [PubMed] [Google Scholar]
- 18.Langer HF, Chavakis T. Platelets and neurovascular inflammation. Thromb Haemost. 2013;110(5):888–93. 10.1160/th13-02-0096. [DOI] [PubMed] [Google Scholar]
- 19.Dale GL. Coated-platelets: an emerging component of the procoagulant response. J Thromb Haemost : JTH. 2005;3(10):2185–92. 10.1111/j.1538-7836.2005.01274.x. [DOI] [PubMed] [Google Scholar]
- 20.Prodan CI, Joseph PM, Vincent AS, Dale GL. Coated-platelets in ischemic stroke: differences between lacunar and cortical stroke. J Thromb Haemost : JTH. 2008;6(4):609–14. 10.1111/j.1538-7836.2008.02890.x. [DOI] [PubMed] [Google Scholar]
- 21.Prodan CI, Vincent AS, Kirkpatrick AC, Hoover SL, Dale GL. Higher levels of coated-platelets are observed in patients with subarachnoid hemorrhage but lower levels are associated with increased mortality at 30 days. J Neurol Sci. 2013;334(1–2):126–9. 10.1016/j.jns.2013.08.008. [DOI] [PubMed] [Google Scholar]
- 22.Prodan CI, Vincent AS, Padmanabhan R, Dale GL. Coated-platelet levels are low in patients with spontaneous intracerebral hemorrhage. Stroke. 2009;40(7):2578–80. 10.1161/strokeaha.109.549014. [DOI] [PubMed] [Google Scholar]
- 23.Vergouwen MD, Vermeulen M, van Gijn J, Rinkel GJ, Wijdicks EF, Muizelaar JP, et al. Definition of delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage as an outcome event in clinical trials and observational studies: proposal of a multidisciplinary research group. Stroke. 2010;41(10):2391–5. 10.1161/strokeaha.110.589275. [DOI] [PubMed] [Google Scholar]
- 24.Ishikawa M, Kusaka G, Yamaguchi N, Sekizuka E, Nakadate H, Minamitani H, et al. Platelet and leukocyte adhesion in the micro-vasculature at the cerebral surface immediately after subarachnoid hemorrhage. Neurosurgery. 2009;64(3):546–553; discussion 53-4. 10.1227/01.neu.0000337579.05110.f4. [DOI] [PubMed] [Google Scholar]
- 25.Tso MK, Macdonald RL. Subarachnoid hemorrhage: a review of experimental studies on the microcirculation and the neurovascular unit. Transl Stroke Res. 2014;5(2):174–89. 10.1007/s12975-014-0323-4. [DOI] [PubMed] [Google Scholar]
- 26.Frontera JA, Provencio JJ, Sehba FA, McIntyre TM, Nowacki AS, Gordon E, et al. The role of platelet activation and inflammation in early brain injury following subarachnoid hemorrhage. Neurocrit Care. 2016;26(1):48–57. 10.1007/s12028-016-0292-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Salooja N, Perry DJ. Thrombelastography. Blood Coagul Fibrinolysis. 2001;12(5):327–37. 10.1097/00001721-200107000-00001. [DOI] [PubMed] [Google Scholar]
- 28.Ohkuma H, Suzuki S, Kimura M, Sobata E. Role of platelet function in symptomatic cerebral vasospasm following aneurysmal subarachnoid hemorrhage. Stroke. 1991;22(7):854–9. 10.1161/01.STR.22.7.854. [DOI] [PubMed] [Google Scholar]
- 29.Nina P, Schisano G, Chiappetta F, Luisa Papa M, Maddaloni E, Brunori A, et al. A study of blood coagulation and fibrinolytic system in spontaneous subarachnoid hemorrhage. Correlation with hunt-hess grade and outcome. Surg Neurol. 2001;55(4):197–203. 10.1016/S0090-3019(01)00402-5. [DOI] [PubMed] [Google Scholar]
- 30.Hirashima Y, Nakamura S, Endo S, Kuwayama N, Naruse Y, Takaku A. Elevation of platelet activating factor, inflammatory cytokines, and coagulation factors in the internal jugular vein of patients with subarachnoid hemorrhage. Neurochem Res. 1997;22(10):1249–55. 10.1023/A:1021985030331. [DOI] [PubMed] [Google Scholar]
- 31.Sjovall F, Morota S, Hansson MJ, Friberg H, Gnaiger E, Elmer E. Temporal increase of platelet mitochondrial respiration is negatively associated with clinical outcome in patients with sepsis. Crit Care. 2010;14(6):R214. 10.1186/cc9337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Puskarich MA, Kline JA, Watts JA, Shirey K, Hosler J, Jones AE. Early alterations in platelet mitochondrial function are associated with survival and organ failure in patients with septic shock. J Crit Care. 2016;31(1):63–7. 10.1016/j.jcrc.2015.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stein SC, Browne KD, Chen XH, Smith DH, Graham DI. Thromboembolism and delayed cerebral ischemia after subarachnoid hemorrhage: an autopsy study. Neurosurgery. 2006;59(4):781–787; discussion 7-8. 10.1227/01.neu.0000227519.27569.45. [DOI] [PubMed] [Google Scholar]
- 34.Coppinger JA, Cagney G, Toomey S, Kislinger T, Belton O, McRedmond JP, et al. Characterization of the proteins released from activated platelets leads to localization of novel platelet proteins in human atherosclerotic lesions. Blood. 2004;103(6):2096–104. 10.1182/blood-2003-08-2804. [DOI] [PubMed] [Google Scholar]
- 35.Levi M, van der Poll T, Schultz M. Systemic versus localized coagulation activation contributing to organ failure in critically ill patients. Semin Immunopathol. 2012;34(1):167–79. 10.1007/s00281-011-0283-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Serhan CN. Pro-resolving lipid mediators are leads for resolution physiology. Nature. 2014;510(7503):92–101. 10.1038/nature13479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fredman G, Van Dyke TE, Serhan CN. Resolvin E1 regulates adenosine diphosphate activation of human platelets. Arterioscler Thromb Vasc Biol. 2010;30(10):2005–13. 10.1161/atvbaha.110.209908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dona M, Fredman G, Schwab JM, Chiang N, Arita M, Goodarzi A, et al. Resolvin E1, an EPA-derived mediator in whole blood, selectively counter regulates leukocytes and platelets. Blood. 2008;112(3):848–55. 10.1182/blood-2007-11-122598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dalli J, Colas RA, Quintana C, Barragan-Bradford D, Hurwitz S, Levy BD, et al. Human sepsis eicosanoid and proresolving lipid mediator temporal profiles: correlations with survival and clinical outcomes. Crit Care Med. 2017;45(1):58–68. 10.1097/ccm.0000000000002014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smith FG, Sheehy AM, Vincent JL, Coursin DB. Critical illness-induced dysglycaemia: diabetes and beyond. Crit Care. 2010;14(6):327. 10.1186/cc9266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stellos K, Katsiki N, Tatsidou P, Bigalke B, Laske C. Association of platelet activation with vascular cognitive impairment: implications in dementia development? Curr Vasc Pharmacol. 2014;12(1):152–4. 10.2174/157016111201140327164641. [DOI] [PubMed] [Google Scholar]
- 42.Prodan CI, Ross ED, Stoner JA, Cowan LD, Vincent AS, Dale GL. Coated-platelet levels and progression from mild cognitive impairment to Alzheimer disease. Neurology. 2011;76(3):247–52. 10.1212/WNL.0b013e3182074bd2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McDougall CG, Spetzler RF, Zabramski JM, Partovi S, Hills NK, Nakaji P, et al. The barrow ruptured aneurysm trial. J Neurosurg. 2012;116(1):135–44. 10.3171/2011.8.jns101767. [DOI] [PubMed] [Google Scholar]
- 44.Molyneux A, Kerr R, Stratton I, Sandercock P, Clarke M, Shrimpton J, et al. International Subarachnoid Aneurysm Trial (ISAT) of neurosurgical clipping versus endovascular coiling in 2143 patients with ruptured intracranial aneurysms: a randomised trial. Lancet. 2002;360(9342):1267–74. 10.1016/S0140-6736(02)11314-6. [DOI] [PubMed] [Google Scholar]