

Summary
Hydrogen Sulfide (H2S) is a gasotransmitter with broad physiological activities, including protecting cells against stress, but little is known about the regulation of cellular H2S homeostasis. We have performed a high-content small molecule screen and identified genotoxic agents, including cancer chemotherapy drugs, as activators of intracellular H2Slevels. DNA damage induced H2S in vitro and in vivo. Mechanistically, DNA damage elevated autophagy and upregulated H2S-generating enzyme CGL; chemical or genetic disruption of autophagy or CGL impaired H2S induction. Importantly, exogenous H2S partially rescued autophagy-deficient cells from genotoxic stress. Furthermore, stressors that are not primarily genotoxic (growth factor depletion and mitochondrial uncoupler FCCP) increased intracellular H2S in an autophagy-dependent manner. Our findings highlight the role of autophagy in H2S production and suggests that H2S generation may be a common adaptive response to DNA damage and other stressors.
In Brief
Jiang et al. developed a high-content screen and identified genotoxic agents as activators of intracellular H2S levels. H2S induction is a physiological DNA damage response and is regulated by autophagy and CGL enzyme. Autophagy dependent H2S induction may be a general adaptive response to stress.
Graphical Abstract
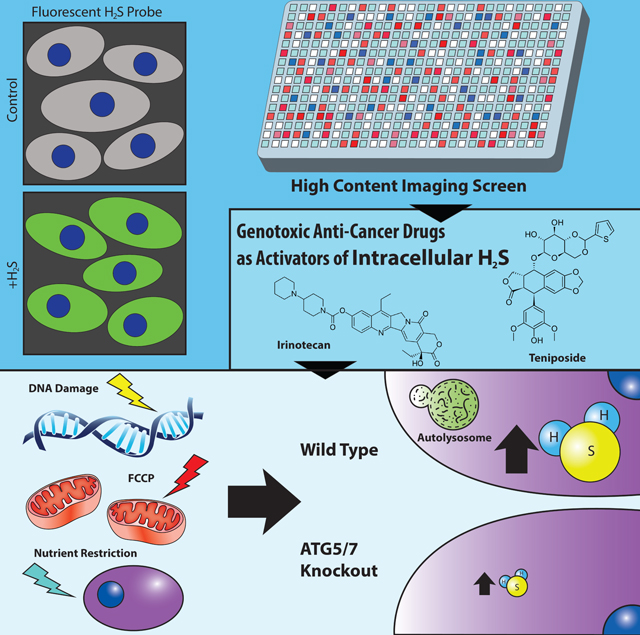
Introduction
Endogenous H2S is a gasotransmitter with broad physiological activities in cardiovascular, neuronal, immune and respiratory systems through pleiotropic mechanisms including protein post-translational modification via sulfhydration and mitochondrial electron transport (Filipovic et al., 2018; Wang, 2012) (Murphy et al., 2019). While high levels of H2S are toxic and induce DNA damage and apoptosis (Baskar et al., 2007), physiological levels of H2S can protect cells from hypoxia, neurological damage, ischemia reperfusion injury and oxidative stress (Kimura, 2014) (Murphy et al., 2019), and the lack of H2S is associated with hypertension, neurovegetative disorders and diabetes (Hine et al., 2018). Exogenous H2S addition has proven beneficial in numerous preclinical and cellular models, while increased endogenous H2S has been implicated in the cytoprotective effects of dietary restriction, including longevity (Hine et al., 2015) (Zivanovic et al., 2019).
Intracellular H2S exists in at least 3 potentially inter-convertible cellular pools, free H2S (in equilibrium with its hydrosulfide anion HS−), sulfane sulfur (bound pool existing mostly as protein thiol persulfides or polysulfides ) and in iron-sulfur clusters (acid-labile pool) (Nagy, 2015). At least three enzymes, cystathionine β-synthase (CBS), cystathionine γ-lyase (CGL) and 3-mercaptopyruvate sulfurtransferease (3MST), participate in cysteine-dependent intracellular H2S generation, while sulfide oxidation is catalyzed by sulfide quinone oxidoreductase (SQR) (Hine et al., 2018). Expression of H2S generating enzymes and cellular H2S production capacity are regulated by endogenous and exogeneous factors, including redox conditions, endoplasmic reticulum (ER) stress, inflammation, nutrient deprivation, transcription factors, hormones and protein post-translational modifications (Hine et al., 2018)(Kabil et al., 2011; Murphy et al., 2019). However, detailed mechanisms of cellular H2S homeostasis are poorly understood.
Macro-autophagy (hereafter termed autophagy) is a homeostatic cellular process essential for protein quality control in which proteins and organelles are delivered in autophagosomes to lysosomes, degraded and amino acids recycled (Eliopoulos et al., 2016). Autophagy is also part of a general cytoprotective response to stressors ranging from nutrient deprivation to genotoxic stress (Murrow and Debnath, 2013). There is abundant evidence linking H2S to autophagy, but the nature and directionality of this relationship remains opaque. H2S can positively or negatively regulate autophagy depending on concentration and context (Wu et al., 2018). On the other hand, autophagy is required for maximal endogenous H2S induction by growth factor deprivation (Hine et al., 2017), possibly through providing cysteine as a substrate for enzymatic H2S generation.
Cell-based high-throughput screening is a powerful unbiased discovery tool, however its use in understanding H2S biology has not been reported, probably due to the lack of sensitive and well-validated cellular H2S assays. Here, we developed quantitative image- and flow cytometry-based methods using existing fluorescence probes for primary screening and validation of small molecule modulators of endogenous H2S levels. Our high-throughput screen revealed genotoxic agents as a major class of activators of endogenous H2S levels. We further demonstrated a requirement for autophagy for maximal H2S induction upon DNA damage, as well as a number of non-genotoxic stressors, suggesting increased autophagy-dependent H2S production as a general adaptive response to cellular stress.
Results
A high-content screen identifies genotoxic agents as activators of intracellular H2S levels
To screen for small molecules modulating intracellular H2S levels, we developed a quantitative cell-based H2S assay using the chemical probe, P3 (Figure S1A). P3 reacts with HS− to generate fluorescence signal in a selective, sensitive and rapid manner (Singha et al., 2015). While originally developed for visualization by two photon fluorescence imaging, we adapted it to conventional one photon imaging in HeLa cells with serum depletion as a positive control for endogenous H2S induction (Figure S1B and S1C). For high-content screening, HeLa cells were grown in 384 well plates and treated with individual small molecule compounds for 20 h, stained with P3, washed, stained with the DNA probe DRAQ5 (for cell counting) and fixed. Fluorescence intensity and cell number were measured in treated cells relative to vehicle (DMSO) controls by high-content imaging (Figure 1A). The screening assay was validated using sildenafil (Fusco et al., 2012), a PDE5 inhibitor and H2S-inducing compound (Figure S1D), and AOAA, an inhibitor of H2S-generating enzymes CBS and CGL (Asimakopoulou et al., 2013) that may also limit the concerted production of H2S from the CAT-3MST system (Miyamoto et al., 2014) (Figure S1E). The Z-factor was determined to be 0.44 (Table S1), indicating a robust screening assay.
Figure 1. A high-content screen identifies genotoxic agents as activators of intracellular H2S levels.

(A) Schematic of high-content screen for small molecule modulators of intracellular H2S levels. (B) Scatter plot of the primary screening data. The dotted line indicates the cut-off for activator hits. Genotoxic hits are highlighted in red. (C) Intracellular H2S levels in HeLa cells treated with representative genotoxic hits for 20 h (n=3). (D) Representative P3 fluorescence images of HeLa cells treated with 30 μM teniposide (Ten) for 6h prior to staining with P3. Scale bar, 100 μm.
We screened a small molecule library of ∼ 12,000 compounds with diversified structures and molecular targets. Activator hits were defined as compounds that exhibited >1.4-fold increase in H2S level with >50% cell survival compared to DMSO controls (Figure 1B). Amongst the 123 putative activator hits identified (Datasheet S1) were the calorie restriction mimetic acarbose (Harrison et al., 2014) and resveratrol previously associated with vasodilation via increased H2S (Yetik-Anacak et al., 2016).
Surprisingly, the single largest class (n=39) of activating compounds was genotoxic agents (Datasheet S1). Amongst the strongest activator hits identified in multiple screening plates were four genotoxic compounds used in cancer chemotherapy that induce DNA damage through different mechanisms, including the topoisomerase I and II inhibitors irinotecan and teniposide (Ten), respectively, the nucleoside analogue trifluridine, and the free radical generator bleomycin (Cheung-Ong et al., 2013) (Figure S1F).
To validate activator hits, we developed a flow cytometry-based assay using another H2S-selective fluorescent probe, SF7-AM (Lin et al., 2013) (Figure S1A) and validated the assay using sulfur amino acid depletion to induce intracellular H2S (Longchamp et al., 2018) (Figure S1G and S1H) and H2S donor Na2S (Figure S1I). The SF7-AM assay had better sensitivity than the assay with P3 (Figure S1J–M) and was thus used for most subsequent measurements. Each of the above 4 representative genotoxic compounds exhibited dose-dependent ability to enhance intracellular H2S levels in HeLa cells (Figure 1C), as measured using SF7-AM. Among the 4 hits, teniposide was the strongest one, and also significantly increased H2S/P3 fluorescence intensity in imaging assays (Figure 1D and Figure S1P).
DNA damage increases sulfide levels in vitro and in vivo
In immortalized mouse embryonic fibroblast (MEF) cells, Ten treatment increased intracellular H2S levels in a dose and time-dependent manner (Figure 2A and B). Both ultraviolet-C (UVC) irradiation, which induces DNA base damage (Rastogi et al., 2010), and ionizing radiation (IR), which induces a variety of oxidative DNA lesions including strand breaks (Santivasi and Xia, 2014), increased intracellular H2S (Figure 2C, D and S2A). Together these results suggest that several different primary DNA lesions can induce intracellular H2S.
Figure 2. DNA damage increases sulfide levels in vitro and in vivo.

(A-D) Intracellular H2S in immortalized MEFs treated with teniposide (Ten) for 6 h (A) or up to 24 h (B), or 24 h after ultraviolet-C (UVC) (C) or ionizing radiation (IR) (D) exposure (n=3). (E, F) Intracellular H2S levels in primary MEFs (E; n=3) or primary MDFs (F; n= 3) treated with Ten for 6 h. (G) Intracellular H2S levels in bone marrow cells from mice treated by intraperitoneal injection of etoposide (Eto) (n=3 or 4/group). (H) Intracellular H2S levels in circulating leukocytes from Csa−/−/Xpa−/− (Cx) mice (n=3). (I, J) Intracellular sulfane sulfur (I) and medium H2S (J) levels in primary MEFs treated with Ten for 6h (n=3). (K) Intracellular H2S levels in immortalized WT and PARP-1 knockout (KO) MEFs treated with Ten for 6 h (n=3). (L) Intracellular H2S in immortalized MEFs treated with 5 μM pan-caspase inhibitor Z-VAD-FMK (Z-VAD) and Ten for 24 h (n=3). Probe SF7-AM was used for intracellular and medium H2S measurements. Probe SSP4 was used for intracellular sulfane sulfur assay. Error bars indicate SD. ns, not significant; *P < 0.05; **P < 0.01; **** P < 0.0001. One-way ANOVA (A-E, I, J); Two-way ANOVA (K, L); Student’s t-test (F-H).
We next measured the effects of genotoxic stress on H2S levels in primary cells. In primary MEFs, Ten treatment increased intracellular H2S levels as measured by SF7-AM (Figure 2E) and confirmed with an additional H2S probe, HSip-1 DA (Sasakura et al., 2011) (Figure S1A and S2B). UVC irradiation increased H2S as measured using SF7-AM or HSip-1 DA (Figure S2C and S2D) concomitant with induction of the DNA damage marker γH2AX (Jackson and Bartek, 2009) (Figure S2E and S2F). Ten treatment also induced H2S in cultured mouse dermal fibroblast (MDF) cells (Figure 2F) and circulating leukocytes treated ex vivo (Figure S2G). Importantly, i.p. injection of etoposide (Cheung-Ong et al., 2013), an analogous compound of teniposide that also induces DNA damage and is used in cancer chemotherapy, in mice increased H2S level in bone marrow cells (Figure 2G and S2H).
We further used a genetic mouse model of nucleotide excision DNA repair deficiency (Csa−/−/Xpa−/−, hereafter Cx) (Brace et al., 2016; Brace et al., 2013) to test the role of unrepaired endogenous DNA damage. Intracellular H2S levels were higher in circulating leukocytes (Figure 2H and S2I–L) and lung endothelial cells (Trocha et al., 2020) (Figure S2M) from Cx mice relative to WT controls, concomitant with increased hepatic H2S production capacity (Figure S2N). These results suggest that H2S induction is a physiological response to exogenous or endogenous genotoxic stress.
Free intracellular H2S can be converted to sulfane sulfur, including protein S-sulfhydration, and/or diffuse outside of cells in a highly dynamic process (Nagy, 2015). We detected an increase in intracellular sulfane sulfur levels upon treatment of primary MEFs with either Ten (Figure 2I) or UVC (Figure S2O) using the sulfane sulfur-selective probe, SSP4 (Bibli et al., 2018). We also detected an increase in free H2S in the culture media of primary MEFs upon Ten treatment (Figure 2J) using SF7-AM fluorescence in the media (Figure S2P and S2Q). DNA damage thus appears to have a broad impact on cellular sulfide metabolism, increasing intracellular H2S, sulfane sulfur, and extracellular H2S release.
PARP-1, not apoptosis, mediates intracellular H2S levels upon DNA damage
The DNA damage response (DDR) is controlled by a network of sensor, mediator, transducer and effector proteins (Jackson and Bartek, 2009). PARP-1 is a sensor protein involved in initiation of the DDR that is recruited to damaged DNA (Olaussen et al., 2013). In immortalized MEFs lacking PARP-1, induction of H2S by Ten or UVC was significantly decreased relative to WT controls (Figure 2K, Figure S3A and S3B). Apoptosis, a terminal DDR event, is activated by caspases (Haupt et al., 2003). Treatment with the pancaspase/apoptosis inhibitor Z-VAD-FMK significantly impaired activation of caspase 3/7 upon DNA damage (Figure S3C) but had little effect on baseline or Ten induced intracellular H2S (Figure 2L). This dependence on PARP-1 but not apoptosis indicates that genotoxin-induced H2S is mediated by early DDR events.
Autophagy regulates H2S induction upon DNA damage
Activation of autophagy is another characteristic DDR response to multiple forms of genotoxic stress (Eliopoulos et al., 2016) including Ten treatment, which induced formation of autophagic puncta, a marker of autophagy, in MEFs expressing GFP-LC3 (Mizushima et al., 2010) (Figure 3A). Chloroquine (Figure 3B) and another lysosome/autophagy inhibitor NH4Cl (Figure S3D), dampened H2S induction upon Ten exposure, consistent with the ability of autophagy to potentiate intracellular H2S induction.
Figure 3. DNA damage induced H2S generation is an autophagy dependent adaptive response.

(A) Representative images of autophagic puncta (green) formation in MEF cells expressing GFP-LC3 treated with 30 μM Ten or vehicle (DMSO) for 6 h. Scale bar, 20 μm. (B) Intracellular H2S in immortalized MEFs treated with Ten and 50 μM chloroquine (CQ) or DMSO for 6 h (n=3). (C) Immunoblots of the indicated protein from immortalized WT and ATG5 knockout (KO) MEFs treated with 30 μM Ten for 6 h. (D, E) Intracellular H2S in immortalized WT vs. ATG5 KO (D) or AMPKα1/α2 KO (E) MEFs treated with Ten for 6 h (n=3). (F) Intracellular H2S in immortalized WT and ATG5 KO MEFs treated with Ten and 2.5 μM brefeldin A (BFA) or DMSO for 6 h (n=3). (G) Intracellular H2S levels in immortalized MEFs treated with rapamycin for 24 h (n=3). (H) Intracellular H2S in immortalized WT and ATG5 knockout MEFs treated with Ten and 100 nM MG132, 100 nM bortezomib (Btz) or DMSO for 6 h (n=3). (I) Gene expression of immortalized MEF cells treated with 30 μM Ten for 3 h (n=3). (J) Immunoblots of the indicated protein from immortalized MEFs treated with 30 μM Ten for the indicated time. Relative intensities of the blots are shown. (K) Intracellular H2S levels in WT and CGL knockout primary MDFs treated with Ten for 6 h (n=3). (L) Intracellular H2S in immortalized WT and ATF4 KO MEFs treated with Ten for 6 h (n=3). (M) Intracellular H2S in immortalized WT and ATG5 KO MEFs treated with Ten and 150 μM PAG or DMSO for 6 h (n=3). (N, O) Percent survival of immortalized WT and ATG5 KO MEFs treated with the indicated dose of Ten for 24 h (N; n=3) or 30 μM Ten and the indicated dose of AP39 (AP) for 24 h (O; n=3). All intracellular H2S measurements were performed using probe SF7-AM. Error bars indicate SD. ns, not significant; *P < 0.05; **P < 0.01; *** P < 0.001; **** P < 0.0001. One-way ANOVA (G, H, O); Two-way ANOVA (B, D-F, K-N); Student’s t-test (I, O).
We further examined the relationship between autophagy and genotoxic stress-induced H2S using genetic approaches. ATG5 and ATG7 are two key proteins that control autophagosome formation (Eliopoulos et al., 2016). The conversion of LC3I to LC3II, another autophagy marker, was induced by Ten treatment in WT but less efficiently in ATG5 (Figure 3C) or ATG7 knockout (KO) MEFs (Figure S3E) concomitant with reduced H2S induction (Figure 3D and Figure S3F). The metabolic stress sensor AMPK is another DDR mediator that is activated by PARP-1 and promotes autophagosome formation (Eliopoulos et al., 2016) (Garcia and Shaw, 2017). In AMPK α1/α2 catalytic subunits double KO MEFs, induction of autophagy (Figure S3G) and H2S (Figure 3E) upon Ten treatment were blunted, consistent with the requirement for autophagy for maximal H2S induction.
An alternative autophagy pathway has been reported in genotoxic stressor-treated MEFs that is regulated by AMPK but independent of ATG5 and lacking LC3I to LC3II conversion (Nishida et al., 2009) (Shimizu, 2018). Using an inhibitor of this alternative autophagy, Brefeldin A (Ma et al., 2015; Nishida et al., 2009), we found a decrease in Ten-induced H2S in both wild type and ATG5 KO MEFs (Figure 3F), suggesting a contribution of both autophagy pathways to H2S induction upon genotoxic stress.
To examine whether autophagy is sufficient to induce H2S, we treated immortalized MEFs with rapamycin, an inhibitor of the autophagy repressor mTOR (Laplante and Sabatini, 2012). While rapamycin induced autophagic puncta formation (Figure S3H) and increased the degradation of GFP-LC3 (Figure S3I), it did not induce detectable changes in intracellular H2S levels (Figure 3G). Together, these results suggest the necessity but not the sufficiency for autophagy in H2S induction upon DNA damage.
DNA damage also increases protein turn-over via the ubiquitin-proteasome system (UPS) that tags substrate proteins with ubiquitin for proteasomal degradation (Rousseau and Bertolotti, 2018) (Kouranti and Peyroche, 2012). Two proteasome catalytic inhibitors, MG132 and bortezomib (Btz), did not change H2S levels upon genotoxic stress either in WT or ATG5 KO cells (Figure 3H), arguing against a general role of the UPS in induction of H2S levels upon DNA damage.
CGL partially contributes to H2S induction upon DNA damage
To better understand the source of increased intracellular H2S upon genotoxic stress, we examined expression levels of H2S generating enzymes CGL, CBS and 3MST, as well as the enzyme responsible for H2S removal, SQR. Of these, only CGL was significantly upregulated in both mRNA and protein levels by Ten treatment (Figure 3I and 3J). Genetic ablation of CGL in primary MDFs (Figure S3J) resulted in a small but significant decrease in intracellular H2S upon Ten treatment (Figure 3K), supporting a role for CGL in H2S induction by DNA damage.
CGL mRNA expression is activated by the transcription factor ATF4 under stress conditions (Gao et al., 2015) (Longchamp et al., 2018) including amino acid deprivation, ER stress or DNA damage as shown here in immortalized MEFs (Figure S3K). In the absence of ATF4, induction of CGL expression (Figure S3K) and intracellular H2S (Figure 3L) by Ten were both attenuated. Furthermore, the CGL-specific inhibitor PAG decreased H2S induction by Ten in WT as well as ATG5 KO MEFs (Figure 3M). These results suggest the additive roles of ATF4-mediated CGL expression and autophagy in H2S induction upon genotoxic stress.
Autophagy-dependent H2S production is an adaptive response to DNA damage
Like autophagy, which can promote genotoxic stress resistance and cell survival but also contribute to autophagic cell death (Eliopoulos et al., 2016), H2S can enhance the DDR (Szczesny et al., 2014; Zhao et al., 2014) (Szczesny et al., 2016) but also induces DNA damage and apoptosis at higher concentrations (Baskar et al., 2007). To assess whether increased endogenous H2S contributes to a beneficial adaptive response to genotoxic stress similar to autophagy, we analyzed sensitivity to Ten in WT and ATG5 KO MEFs with or without the mitochondrial H2S donor AP39 (Sasakura et al., 2011). Cells lacking ATG5 were more sensitive to Ten than WT cells (Figure 3N), and this hypersensitivity was partially rescued by AP39 (Figure 3O). Together these data suggest that H2S induction, as an adaptive response to DNA damage, contributes to genotoxic stress resistance and cell survival induced by autophagy.
Autophagy dependent H2S generation may be a general stress response
Because transient nutrient or energy restriction induce autophagy (Bagherniya et al., 2018; Murrow and Debnath, 2013) and H2S in vitro and in vivo (Hine et al., 2015)(Hine et al., 2017; Longchamp et al., 2018) without overtly causing genotoxic stress, we next tested the possibility that autophagy-dependent H2S induction is a more general response to cellular stressors beyond DNA damage. To this end we challenged WT and autophagy-deficient cells with diversified stressors that are not primary genotoxins and also induce autophagy (Georgakopoulos et al., 2017; Hine et al., 2017; Wu et al., 2009)(Hoyer-Hansen and Jaattela, 2007; Swerdlow et al., 2008), including growth factor deprivation via serum withdrawal, energy stress via the glycolysis inhibitor 2-deoxyglucose (2DG), mitochondrial stress via the uncoupler FCCP or the ATP synthesis inhibitor oligomycin, ER stress with tunicamycin, or physiological stress via the glucocorticoid receptor agonist dexamethasone (Dex). Each of these treatments increased intracellular H2S in primary MEFs (Figure 4A–F). Furthermore, serum depletion, FCCP and dexamethasone (Figure 4G–I) were all less efficient at H2S induction in ATG5 or ATG7 KO cells than WT cells. It is noted that the treatments did not significantly induced the DNA damage marker γH2AX (Figure S4). Together these results suggest that autophagy-dependent H2S generation may be a general response to cellular stress.
Figure 4. Autophagy-dependent H2S generation is a general response to stress.

(A-F) Primary MEF cells were treated with serum (FBS) depletion (A), 2DG (B), FCCP (C), oligomycin (D), tunicamycin(E), dexamethasone (Dex) (F) for 6h, and intracellular H2S level was measured (n=3). (G-I) Immortalized WT, ATG5 knockout and ATG7 knockout MEF cells were treated with serum depletion (G), 20 μM FCCP (H), 150 μM Dex (I) for 6 h, and intracellular H2S level was measured. The relative H2S levels to the WT cells vehicle control are shown (n=3). All intracellular H2S measurements were performed using probe SF7-AM. Error bars indicate SD. *P < 0.05; **P < 0.01; *** P < 0.001; **** P < 0.0001. Student’s t-test (A, B, F); One way ANOVA (C-E, G-I).
Discussion
The volatility and reactivity of endogenous hydrogen sulfide species have complicated their utility in unbiased discovery platforms such as high-throughput small molecule screening. We adapted the use of an existing H2S-selective fluorescence probe P3 (Singha et al., 2015) to measure relative changes in endogenous intracellular H2S in the context of a high-content image screen. Using this approach, we identified DNA damage agents with varied mechanisms of action, including cancer chemotherapy drugs, as the largest class of inducers of endogenous intracellular H2S in our small molecule library. We also identified other potential activator hits (Datasheet S1) suggestive of unidentified pathways regulating endogenous H2S beyond the scope of the current study. In addition to validation of such potential targets, this approach paves the way for further unbiased small molecule screens for inhibitory compounds, as well as genetic screens for endogenous cellular H2S regulators.
For target validation and mechanistic studies, we further developed sensitive and reproducible flow cytometry-based methods with existing H2S- or sulfane sulfur-selective fluorescent probes. We found that various form of genotoxic stress that induce DNA damage, from Ten-induced strand breaks to UVC induced base lesions, increased sulfide in a dose- and time-dependent manner. Notably this did not require continuous exposure, as both UVC and IR are delivered acutely. Given the lag in detection of intracellular H2S and the ability of even homogenous DNA damaging agents such as UVC to give rise to a range of subsequent lesions including single- or double-strand breaks, future studies will be required to determine which lesion or lesions trigger the response. However, the finding that PARP-1, which participates in single-strand break repair and is implicated in multiple different DNA repair pathways, is required for maximal H2S generation is consistent with multiple different upstream lesions signaling through a common downstream signal.
Although a large number of reaction-based H2S fluorescence probes have been developed for H2S detection, only a few are suitable for endogenous cellular H2S measurement (Takano et al., 2017). To achieve high-throughput screening for small molecular modulators of intracellular H2S levels and the subsequent validation, we selected three probes with distinctive reaction mechanisms, P3 (Singha et al., 2015), SF7-AM (Lin et al., 2013) and HSip-1 DA (Sasakura et al., 2011), for their sensitivity, selectivity, fast response and biocompatibility. In imaging based or flow cytometry based assays, all these three probes detected significant induction of intracellular H2S upon DNA damage. The selectivity of the probes over other biological thiols have been tested and verified, however, the potential non-specific responses of the probes would be hard to completely rule out. The application of multiple probes with distinctive reaction mechanisms, along with the proper negative controls, would help to overcome the limitations of individual probes in terms of non-specific responses and thus strengthens the conclusion that DNA damage induces H2S.
Maximal H2S induction upon genotoxic stress required the transcription factor ATF4, its target, the H2S-producing enzyme CGL, AMPK activation and ATG5-dependent and -independent autophagy, all of which were also increased by genotoxic stress. Autophagy activation by mTORC1 inhibition was insufficient to induce cellular H2S production, whereas constitutive mTORC1 activation can both suppress autophagy and prevent induction of hepatic H2S production capacity via dietary restriction (Hine et al., 2015). Future studies are required to determine the mechanism by which autophagy contributes to H2S induction. One possibility would be that protein turnover via autophagy (but not the UPS) upon DNA damage provides cysteine as a substrate for CGL or other H2S generating enzymes to catalyze the generation of H2S. This hypothesis was previously suggested by cellular models of H2S induction by growth factor withdrawal, which is also partially dependent on ATG5 and ATG7 (Hine et al., 2017). Another possibility would be that the fusion of autophagosome, which may provide H2S-generating enzymes, and lysosome, which contains high level of cysteine (Abu-Remaileh et al., 2017), facilitates enzymatic H2S generation in autolysosome. In addition, it would also be possible that non-enzymatic H2S generation partially contributes to H2S induction. Notably,our proposed mechanisms of increased intracellular H2S are fundamentally different from H2S induction by hypoxia, which prevents oxidation of H2S by SQR and ETHE1 and occurs on a more rapid time frame than observed here (Olson, 2015).
While CGL was the only enzyme whose expression was significantly increased upon genotoxic stress, its ablation only partially reduced overall H2S induction. This observation may suggest a potential redundant role of H2S-producing enzymes in H2S induction upon genotoxic stress. Other stressors, including ER stress (Gao et al., 2015), amino acid restriction (Longchamp et al., 2018) and Golgi stress (Sbodio et al., 2018) also upregulate CGL expression in an ATF4-dependent manner, although whether H2S induction by the stressors is mediated by CGL and/or other enzymes remains to be examined. The combination of gene knockout of individual or multiple H2S-generating enzyme gene(s) in cells, and inhibition of enzyme activities using AOAA, PAG or other inhibitors, would help to determine the enzyme(s) responsible for H2S induction in response to DNA damage and other stressors.
Our results also highlight autophagy-dependent H2S induction as a potential general adaptive response to cellular stress. The ability of the mitochondrial H2S donor AP39 to partially rescue hypersensitivity of ATG5 KO cells to Ten treatment is consistent with H2S generation as a beneficial adaptive response to genotoxic stress. In addition, amino acid deprivation, growth factor depletion, energy/mitchondrial stress in the form of 2DG, oligomycin or FCCP, and ER stress, none of which lead directly to DNA damage, all increased intracellular H2S. Given that both autophagy and low levels of H2S are cytoprotective (Murphy et al., 2019; Murrow and Debnath, 2013), our observations imply that autophagy-dependent H2S induction may be a general adaptive response to cellular stress. Future studies are required to determine the downstream cellular mechanism of action, including the potential for cell autonomous effects on energy metabolism and/or protein S-sulfhydration, as well as paracrine/endocrine effects on the organ/organismal level due to the observed increase in H2S released into the culture media upon genotoxic stress.
Finally, our results reveal a previously unidentified connection between DNA damage and hydrogen sulfide biology with implications for genotoxic chemotherapeutics. Increased endogenous hydrogen sulfide production, in part due to CBS overexpression, is associated with cancer progression and drug resistance in colorectal (Hellmich et al., 2015), ovarian (Bhattacharyya et al., 2013), breast and lung (Szczesny et al., 2016) cancers. Inhibition of H2S biosynthesis, which can sensitize cancer cells to chemotherapeutic drugs (Szczesny et al., 2016), may thus improve the efficacy of chemotherapeutic agents by preventing adaptive H2S production induced by the agents themselves.
Star* Methods
Resource availability
Lead Contact
Further information and requests for resources and reagents should be directed and will be fulfilled by Lead Contact, Sarah J. Mitchell (smitchell@ethz.ch).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
The datasheet of activator hits identified in the primary screen is included in the supplemental information. The datasheet of activator hits identified in the primary screen is included in the supplemental information as Datasheet S1.
Experimental models and subject details
Primary cells and cell lines
HeLa and MEF cell linesHeLa and immortalized mouse embryonic fibroblast cells (MEFs) were maintained in DMEM with 25 mM glucose and GlutaMAX (Gibco) supplemented with 10% FBS (Thermo) and 1% penicillin–streptomycin (Thermo) in 20% O2, 5% CO2 at 37°C. Immortalized WT and Atf4−/− MEFs were additionally supplemented with 55 μM mercaptoethanol (Gibco) and 1X non-essential amino acids (Sigma-Aldrich). Information relating to the origin of immortalized MEF cells can be found in the STAR methods (Cheong et al., 2011; de Murcia et al., 1997; Han et al., 2013; Yang et al., 2010; Zadra et al., 2014).,
Primary cells
Primary cells were cultured in the same DMEM media but with 20% FBS and maintained in 3% O2, 5% CO2 at 37°C. For cell-based assays including the screen and subsequent validation and mechanistic studies, cells were grown in DMEM with 25 mM glucose, 4 mM glutamine and 25 mM HEPES, without sodium pyruvate and phenol red dye (Gibco), supplemented with 10% dialyzed FBS (Thermo) and 1% penicillin–streptomycin. For sulfur amino acid depletion, cells were cultured in DMEM without L-methionine/L-cystine (Gibco) supplemented with 10% dialyzed FBS and 1% penicillin–streptomycin. For sulfide measurement, cell were grown to 60–80% confluence.
Mouse experiments
All mouse experiments were performed with the approval of the Harvard Medical Area Institutional Animal Care and Use Committee. Mice carrying Xpa and Csa knockout alleles on a C57BL/6 background, and CGL knockout mice (derived from the International Knockout Mouse Consortium construct http://www.informatics.jax.org/allele/MGI:5435787) on a C57BL/6 background were maintained under standard laboratory conditions (temperature 20–24°C, relative humidity 50–60%, 12 h light/12 h dark) and allowed free access to water and standard chow pellets (PicoLab 5053, Purina). For in vivo etoposide treatment, 16-week-old male C57BL6 mice were dosed with 90 mg/kg of etoposide via intraperitoneal injection, and euthanized after 8 h. Primary MEFs were isolated from embryos of pregnant females harvested at E13.5 stage using a primary mouse embryonic fibroblast isolation kit (Thermo). Primary mouse dermal fibroblast cells (MDFs) were generated from tail using collagenase II (Thermo) digestion. Mouse circulating leukocytes were prepared from whole blood following lysis of red blood cells using ammonium-chloride-potassium (ACK) lysis buffer (Thermo). Total bone marrow cells were flushed out from bone followed by the lysis of red blood cells using ammonium-chloride-potassium (ACK) lysis buffer (Thermo). For lung endothelial cell preparation, minced lung tissue was digested in DMEM (Gibco) supplemented with 200 U/mL collagenase types II and IV (Gibco), 1 U/mL dispase (Sigma-Aldrich) and 0.1 mg/mL DNase I (Sigma-Aldrich) for 30 min at 37°C, and passed through a 100-micron filter (Denville Scientific). The cells were washed twice with PBS, stained with anti-CD31-APC (Miltenyi Biotec, 1:10 dilution) for 30 min, fixed with 2% PFA (Chemcruz) for 10 min and measured by FACS. Endothelial cells were determined via CD31+ gating.
Method details
Sulfide species measurement
Intracellular H2S P3 imaging assay
Cells grown in a 24-well glass bottom plate (Cellvis). DMSO stock solution of P3 (Calbiochem, ( E)-2-(3-(6-(2-Hydroxyethylamino)naphthalen-2-yl)-3-oxoprop-1-enyl)-3,5-dimethoxybenzaldehyde, 10 μM as a final concentration) was added to the medium and cells were stained for 30 min in a CO2 incubator at 37°C, washed with PBS and fixed with 2% PFA. Images were acquired under an Axio A1 fluorescence microscopy (Zeiss, Ex/Em=365/524 nm). Cells treated with DMSO were used as the blank control for the calculation of fluorescence intensity.
Intracellular H2S flow cytometry assay
Cells were grown in a 24-well plate (Corning). DMSO stock solution of P3 (Calbiochem, 10 μM as a final concentration), SF7-AM (Tocris, 0.25 μM as a final concentration) or HSip-1 DA (Dojindo, 0.25 μM as a final concentration) was added to the cell culture media followed by a 30 min incubation in the same CO2 incubator for cell growth at 37°C. Cells were washed with PBS. For adherent cells, cells were incubated with Accumax Cell Dissociation Solution (Innovative Cell Technologies) at room temperature for 10 min to disassociate cells from plate, and harvested in DMEM medium supplemented with 1% dialyzed FBS for the analysis on a Fortessa flow cytometer (BD Biosciences, Ex/Em=378/524 nm for P3, Ex/Em=480/520 nm for SF-7AM and HSip-1 DA). FlowJo software was used to analyze mean values of the population. Most intracellular H2S measurements were performed by flow cytometry using SF7-AM unless otherwise specified.
Intracellular polysulfide flow cytometry assay
Cell were grown in a 24-well plate. Detergent CTAB (Sigma-Aldrich, 32.5 μM as a final concentration) and sulfane sulfide probe SSP4 (Dojindo, 1.25 μM as a final concentration) were added to the cell culture media followed by a 15 min incubation in a CO2 incubator at 37°C. Cells were washed with PBS prepared for analysis by flow cytometer (Ex/Em=480/520 nm) as described above. Cells without probe treatment were used as the blank control for the calculation of fluorescence intensity.
Medium H2S assay
Cells were grown in a 24-well plate. SF7-AM was added to the cell culture media to 0.25 μM as a final concentration. After 6 h incubation in a CO2 incubator at 37°C, 100 μL of media was transferred to a 96 well clear-bottom, black-sided plate (Corning) and measured on a fluorescence plate reader (BioTek, Ex/Em=480/520 nm). Culture media without cells incubated with SF7-AM was used as the blank control to calculate relative fluorescence intensity.
Lead sulfide assay
H2S production capacity in liver lysates was measured as previously described (Hine et al., 2015). Briefly, equal amounts of protein were added to a reaction master mix containing PBS, 10 mM Cys (Sigma-Aldrich) and 1 mM PLP (Sigma-Aldrich) in 96 well plate. A lead acetate H2S detection paper was put above the liquid and incubated for 2–5 h at 37°C until lead sulfide darkening of the paper occurred. The dark blots were scanned and quantitated using ImageJ software.
High-content compound screening
The screen was carried out against the Harvard ICCB Known Bioactive collection (https://iccb.med.harvard.edu/known-bioactives-collection) of ∼ 12,000 small molecule compounds including FDA approved drugs, NIH clinical collections and other bioactive molecules whose targets have been identified and which were selected to maximize chemical structure and biological pathway diversity. All compounds were dissolved in DMSO.
HeLa cells were dispensed into 384-well clear-bottom, black-sided plates (Corning) at 1200 cells per well in 30 μL of DMEM with 25 mM glucose, 4 mM glutamine and 25 mM HEPES, without sodium pyruvate and phenol red dye supplemented with 10% dialyzed FBS and 1% penicillin–streptomycin. After 4 h incubation, 100 nL of compound (0.1–30 μM as a final concentration) was pin transferred using a transfer robot (Seiko) to individual wells. After 20 h treatment, 2.5 μL of DMSO containing 130 μM P3 was added into wells using a Vprep automated pipettors (Agilent) for a final concentration of 10 μM P3. The cells were then returned to the 37°C 5% CO2 incubator for 60 min and washed with PBS, and then 30 μL of PBS containing 3% PFA and 5 μM DRAQ5 (Thermo) was dispensed into wells to fix cells and stain DNA at room temperature for 30 min. After washing with PBS, 30 μL of PBS containing 0.02% NaN3 (Sigma-Aldrich) was dispensed to wells. Plates were automatically imaged on Image Xpress Micro fluorescence microscope (Molecular Devices) at 378/524 nm (Ex/Em) for P3 and at 647/681 nm (Ex/Em) for DRAQ5. Each compound was tested in duplicated plates at the same concentration. DMSO and 1.3 mM AOAA served as the controls, respectively. The images were processed and analyzed using MetaXpress software. DRAQ5 images were used for cell counting and calculation of cell survival (total cell number in test well divided by the mean number of cells in DMSO control wells). The pixel-intensity of the P3 image for each well was divided by the cell number, and the resulting value divided by the mean value of the DMSO control wells for calculation of the relative intensity value. For quality control of the screening assay, the Z-factor was determined to be 0.44 for 1.3 mM AOAA. The average values of cell survival and the relative P3 staining intensity of the compound across duplicate plates was used for identification of activator hits, defined as compounds with the survival > 50% and average P3 intensity >1.4.
Immunoblotting
Cells were washed with PBS and lysed with RIPR buffer (Thermo) supplemented with protease and phosphatase inhibitor cocktail (Thermo). Protein was resolved on 4–20% or 10% Tris-glycine gel (Bio-rad) and transferred to PVDF membrane (Millipore). Primary antibodies against the following proteins were purchased from Cell Signaling Technology: phospho-H2AX (Ser139) (2577, 1:1000 dilution), vinculin (13901, 1:16000 dilution), PARP-1 (9542, 1:1000 dilution), β-tubulin (2128, 1:2000 dilution), ATG5 (12994, 1:1000 dilution), ATG7 (8558, 1:1000 dilution), AMPKα1 (2532, 1:500 dilution), phosphor-AMPKα1 (Thr172) (2535, 1:500 dilution) and ATF4 (11815, 1:1000 dilution). Anti-CBS (ab135626, 1:1000 dilution) and anti-SQR (ab118772, 1:1000 dilution) were purchased from Abcam. Anti-CGL (12217–1-AP, 1:300 dilution) was purchased from Proteintech. Anti-3MST (HPA001240, 1:1000 dilution) and anti-LC3B (NB100–2220, 1:4000 dilution) were purchased from Atlas antibodies and Novus Biologicals, respectively. Secondary HRP conjugated goat antibodies against rabbit was obtained from Dako.
Autophagic puncta assay
Immortalized MEFs expressing GFP-LC3 were fixed with 2% PFA for 15 min and stained with Hoechst 33342 (Invitrogen, 1:1000). Images were acquired under an Eclipse Ti fluorescence confocal microscope (Nikon).
Gene expression analysis
Total RNA was extracted from cells and purified using RNeasy kit (Qiagen). Reverse transcription was performed using SuperScript IV VILO MasterMix (Thermo). Quantitative PCR was carried out with PowerUp SYBR Green Master Mix (Thermo). Fold change was calculated by the ΔΔCt method using β-actin gene as the internal control. The following primers for mouse genes were used:
GGGACAAGGATCGAGTCTGGA and AGCACTGTGTGATAATGTGGG for CBS, TTGGATCGAAACACCCACAAA and AGCCGACTATTGAGGTCATCA for CGL, TCACAGCCGCTGAAGTTACTG and CAGCATGTGGTCGTAGGGG for 3MST, CCCGGCTCTTTGCCTGTTT and CCAGCACCTCATAGTGGTTCTT for SQR, GTGACGTTGACATCCGTAAAGA and GCCGGACTCATCGTACTCC for β-actin.
Cell survival assay
Cells were seeded in 96 well clear-bottom, black-sided plates (Corning; 10,000 cells/well), grown overnight, and treated for 24 h. Cell survival was measured using CyQUANT Direct Cell Proliferation Assay Kit (Invitrogen) on a Synergy2 fluorescence plate reader (BioTek).
Caspase 3/7 activity assay
Cells were grown in 96 well clear-bottom, black-sided plates (Corning; 16,000 cells/well) and caspase 3/7 activity was assayed using Apo-ONE Homogeneous Caspase-3/7 kit (Promega).
Quantification and statistical analysis
GraphPad Prism was used for statistical analysis. The replicates shown in all figures are biologically independent samples. Data are displayed as means ± standard deviation (SD). Student’s t-test (unpaired, two-tailed) was used to compare the values between two specific groups, and ordinary one-way ANOVA with Dunnett’s multiple comparisons test was used to compare multiple groups to a common control. Ordinary two-way ANOVA with Sidak’s multiple comparisons test was used to compare groups with two factors. A P value of 0.05 or less was determined to be statistically significant. Please note that statistical details are found in the figure legends.
Supplementary Material
Datasheet S1. List of activator hits identified in the primary screen. Relative to Figure 1 Genotoxic hits are highlighted in red.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti- phospho-H12AX (Ser139) | Cell Signaling Technology | Cat# 2577 |
| Anti- vinculin | Cell Signaling Technology | Cat# 13901 |
| Anti- PARP-1 | Cell Signaling Technology | Cat# 9542 |
| Anti- β-tubulin | Cell Signaling Technology | Cat# 2128 |
| Anti- ATG5 | Cell Signaling Technology | Cat# 12994 |
| Anti- ATG7 | Cell Signaling Technology | Cat# 8558 |
| Anti- AMPKα1 | Cell Signaling Technology | Cat# 2532 |
| Anti- phospho-AMPKα1 (Thr172) | Cell Signaling Technology | Cat# 2535 |
| Anti- ATF4 | Cell Signaling Technology | Cat# 11815 |
| Anti- CBS | Abcam | Cat# ab135626 |
| Anti- SQR | Abcam | Cat# ab118772 |
| Anti- CGL | Proteintech | Cat# 12217–1-AP |
| Anti- 3MST | Atlas antibodies | Cat# HPA001240 |
| Anti- LC3B | Novus Biologicals | Cat# B100–2220 |
| HRP conjugated goat antibodies against rabbit | Dako | Cat# P044801–2 |
| Bacterial and Virus Strains | ||
| N/A | ||
| Biological Samples | ||
| N/A | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| Compound library: Harvard ICCB Known Bioactive collection | ICCB-Longwood | https://iccb.med.harvard.edu/known-bioactives-collection |
| P3 | Calbiochem | Cat# 534329 |
| SF-7 AM | Tocris | Cat# 4943 |
| HSip-1 DA | Dojindo | Cat# SB22–10 |
| SSP4 | Dojindo | Cat# SB10–10 |
| Na2S | Sigma-Aldrich | Cat# 407410 |
| AOAA | Cayman | Cat# 28298 |
| Etoposide | Sigma-Aldrich | Cat# E1383 |
| NH4Cl | Sigma-Aldrich | Cat# 254134 |
| Oligomycin | Sigma-Aldrich | Cat# 75351 |
| FCCP | Sigma-Aldrich | Cat# C2920 |
| Tunicamycin | Sigma-Aldrich | Cat# T7765 |
| 2-Deoxy-D-glucose (2DG) | Sigma-Aldrich | Cat# D8375 |
| Dexamethasone | Sigma-Aldrich | Cat# D4902 |
| Irinotecan | Selleckchem | Cat# S1198 |
| Trifluridine | Selleckchem | Cat# S1778 |
| Teniposide | Selleckchem | Cat# S1787 |
| Bleomycin | Selleckchem | Cat# S1214 |
| DL-Propargyl Glycine (PAG) | Cayman | Cat# 10010948 |
| AP39 | Cayman | Cat# 17100 |
| Sildenafil | Tocris | Cat# 3784 |
| Z-VAD-FMK | Tocris | Cat# 2163/1 |
| brefeldin A | Tocris | Cat# 1231/5 |
| MG132 | Tocris | Cat# 1748/5 |
| Chloroquine | Invivogen | Cat# tlrl-chq |
| Bortezomib | LC laboratories | Cat# B-1408 |
| Rapamycin | EMD Millipore | Cat# 553210 |
| Critical Commercial Assays | ||
| CyQUANT Direct Cell Proliferation Assay Kit | Invitrogen | Cat# C7026 |
| Apo-ONE Homogeneous Caspase-3/7 kit | Promega | Cat# G7792 |
| Deposited Data | ||
| N/A | ||
| Experimental Models: Cell Lines | ||
| HeLa cells | ATCC | Cat # ATCC CCL-2 |
| Primary mouse embryonic fibroblast (MEF) cells from C57/BL6 mice | This study | N/A |
| Primary CGL WT and KO mouse tail dermal fibroblast cells | This study | N/A |
| Mouse circulating leukocytes | This study | N/A |
| Mouse bone marrow cells | This study | N/A |
| WT and Parp-1−/− immortalized MEF cells | From the lab of Dr. Samuel Wilson | (de Murcia et al., 1997) |
| WT and Atg5−/− immortalized MEF cells | From the lab of Gokhan Hotamisligil | (Yang et al., 2010) |
| WT and Atg7−/− immortalized MEF cells | From the lab of Gokhan Hotamisligil | (Yang et al., 2010) |
| WT and Atf4−/− immortalized MEF cells | From the lab of Dr. Craig Thompson | (Han et al., 2013) |
| Immortalized MEF cells expressing GFP-LC3 | From the lab of Dr. Craig Thompson | (Cheong et al., 2011) |
| WT and AMPKα1−/−/α2−/− immortalized MEF cells | From the lab of Dr. Massimo Loda | (Zadra et al., 2014) |
| Experimental Models: Organisms/Strains | ||
| Cx mouse | Generated in the lab of James Mitchell | (Brace et al., 2013) |
| CGL WT and KO mouse | International Knockout Mouse Consortium | Cat #MGI:5435787 |
| C57BL6 mice | Jackson Lab | Cat #000664 |
| Oligonucleotides | ||
| Mouse CBS FP: GGGACAAGGATCGAGTCTGGA | N/A | N/A |
| Mouse RP: AGCACTGTGTGATAATGTGGG | N/A | N/A |
| Mouse CGL FP: TTGGATCGAAACACCCACAAA | N/A | N/A |
| Mouse CGL RP: AGCCGACTATTGAGGTCATCA | N/A | N/A |
| Mouse 3MST FP: TCACAGCCGCTGAAGTTACTG | N/A | N/A |
| Mouse 3MST RP: CAGCATGTGGTCGTAGGGG | N/A | N/A |
| Mouse SQR FP: CCCGGCTCTTTGCCTGTTT | N/A | N/A |
| Mouse SQR RP: CCAGCACCTCATAGTGGTTCTT | N/A | N/A |
| Mouse β-actin FP: GTGACGTTGACATCCGTAAAGA | N/A | N/A |
| Mouse β-actin RP: GCCGGACTCATCGTACTCC | N/A | N/A |
| Recombinant DNA | ||
| N/A | ||
| Software and Algorithms | ||
| Image J | National Institute of Health | https://imagej.nih.gov |
| GraphPad Prism v 8.4.1 | GraphPad Software | https://www.graphpad.com |
| MetaXpress 6 | Molecular Devices | https://mdc.custhelp.com |
| FlowJo | BD Biosciences | https://www.flowjo.com |
| Other | ||
| N/A | ||
Significance
Biological activities of the gasotransmitter hydrogen sulfide (H2S) are highly dependent on its concentration. While high levels of H2S are cytotoxic, and low levels are associated with neurodegeneration and hypertension, supplementation with exogenous H2S or mildly increasing endogenous H2S can protect cells against stress. The mechanisms regulating endogenous cellular H2S homeostasis, however, remain poorly understood. We developed a high-content fluorescence image based screening assay, screened ∼ 12,000 diversified compounds, and identified a wide range of genotoxic agents, including cancer chemotherapy drugs, as potent inducers of H2S. DNA damage increased the levels of intracellular H2S, sulfane sulfide and extracellular H2S. This response occurred in both primary and transformed cells in vitro and in vivo and required both autophagy and the H2S-producing enzyme CGL, which were elevated upon DNA damage, for maximal production. Importantly, exogenous H2S partially rescued autophagy-deficient cells from genotoxic stress, supporting H2S production as a beneficial adaptive response to DNA damage. We also identified non-genotoxic stressors that induced H2S in an autophagy dependent manner. Our findings provide broadly applicable methods for screening and validating small molecules and genes regulators of cellular sulfide levels, highlight the importance of autophagy in H2S production, and suggest increased endogenous H2S production as a common beneficial adaptive response to a variety of cellular stressors. The results also suggest that inhibition of adaptive H2S production may improve the efficacy of chemotherapeutic agents.
Highlights
A high-content screen identifies genotoxic agents as activators of cellular H2S levels
DNA damage increases intracellular H2S, sulfane sulfide and extracellular H2S levels
Autophagy and CGL enzyme regulate H2S induction upon DNA damage
Autophagy dependent H2S induction may be a general adaptive response to stress
Acknowledgements
We are grateful for K. Ahn, S. Wilson, G. Hotamisligil, C. Thompson, M. Loda, G. Yang, J. Petrini and B. Paul for providing reagents or cell lines. We thank L. Brace, J. Miljkovic, C. Mann, C. Hine and other members of J.R.M lab for discussions and valuable suggestions. The compound screening was performed at Harvard ICCB-Longwood Screening Facility. The work was funded by grants from the NIH (R56AG036712, P01AG034906 to J.R.M).
Footnotes
Declaration of interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abu-Remaileh M, Wyant GA, Kim C, Laqtom NN, Abbasi M, Chan SH, Freinkman E, and Sabatini DM (2017). Lysosomal metabolomics reveals V-ATPase- and mTOR-dependent regulation of amino acid efflux from lysosomes. Science 358, 807–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asimakopoulou A, Panopoulos P, Chasapis CT, Coletta C, Zhou Z, Cirino G, Giannis A, Szabo C, Spyroulias GA, and Papapetropoulos A. (2013). Selectivity of commonly used pharmacological inhibitors for cystathionine beta synthase (CBS) and cystathionine gamma lyase (CSE). Br J Pharmacol 169, 922–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagherniya M, Butler AE, Barreto GE, and Sahebkar A. (2018). The effect of fasting or calorie restriction on autophagy induction: A review of the literature. Ageing Res Rev 47, 183–197. [DOI] [PubMed] [Google Scholar]
- Baskar R, Li L, and Moore PK (2007). Hydrogen sulfide-induces DNA damage and changes in apoptotic gene expression in human lung fibroblast cells. FASEB J 21, 247–255. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya S, Saha S, Giri K, Lanza IR, Nair KS, Jennings NB, Rodriguez-Aguayo C, Lopez-Berestein G, Basal E, Weaver AL, et al. (2013). Cystathionine beta-synthase (CBS) contributes to advanced ovarian cancer progression and drug resistance. PLoS One 8, e79167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bibli SI, Luck B, Zukunft S, Wittig J, Chen W, Xian M, Papapetropoulos A, Hu J, and Fleming I. (2018). A selective and sensitive method for quantification of endogenous polysulfide production in biological samples. Redox Biol 18, 295–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brace LE, Vose SC, Stanya K, Gathungu RM, Marur VR, Longchamp A, Trevino-Villarreal H, Mejia P, Vargas D, Inouye K, et al. (2016). Increased oxidative phosphorylation in response to acute and chronic DNA damage. NPJ Aging Mech Dis 2, 16022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brace LE, Vose SC, Vargas DF, Zhao S, Wang XP, and Mitchell JR (2013). Lifespan extension by dietary intervention in a mouse model of Cockayne syndrome uncouples early postnatal development from segmental progeria. Aging Cell 12, 1144–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheong H, Lindsten T, Wu J, Lu C, and Thompson CB (2011). Ammonia-induced autophagy is independent of ULK1/ULK2 kinases. Proc Natl Acad Sci U S A 108, 11121–11126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung-Ong K, Giaever G, and Nislow C. (2013). DNA-damaging agents in cancer chemotherapy: serendipity and chemical biology. Chem Biol 20, 648–659. [DOI] [PubMed] [Google Scholar]
- de Murcia JM, Niedergang C, Trucco C, Ricoul M, Dutrillaux B, Mark M, Oliver FJ, Masson M, Dierich A, LeMeur M, et al. (1997). Requirement of poly(ADP-ribose) polymerase in recovery from DNA damage in mice and in cells. Proc Natl Acad Sci U S A 94, 7303–7307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eliopoulos AG, Havaki S, and Gorgoulis VG (2016). DNA Damage Response and Autophagy: A Meaningful Partnership. Front Genet 7, 204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filipovic MR, Zivanovic J, Alvarez B, and Banerjee R. (2018). Chemical Biology of H2S Signaling through Persulfidation. Chem Rev 118, 1253–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fusco F, di Villa Bianca R, Mitidieri E, Cirino G, Sorrentino R, and Mirone V. (2012). Sildenafil effect on the human bladder involves the L-cysteine/hydrogen sulfide pathway: a novel mechanism of action of phosphodiesterase type 5 inhibitors. Eur Urol 62, 1174–1180. [DOI] [PubMed] [Google Scholar]
- Gao XH, Krokowski D, Guan BJ, Bederman I, Majumder M, Parisien M, Diatchenko L, Kabil O, Willard B, Banerjee R, et al. (2015). Quantitative H2S-mediated protein sulfhydration reveals metabolic reprogramming during the integrated stress response. Elife 4, e10067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia D, and Shaw RJ (2017). AMPK: Mechanisms of Cellular Energy Sensing and Restoration of Metabolic Balance. Mol Cell 66, 789–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgakopoulos ND, Wells G, and Campanella M. (2017). The pharmacological regulation of cellular mitophagy. Nat Chem Biol 13, 136–146. [DOI] [PubMed] [Google Scholar]
- Han J, Back SH, Hur J, Lin YH, Gildersleeve R, Shan J, Yuan CL, Krokowski D, Wang S, Hatzoglou M, et al. (2013). ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat Cell Biol 15, 481–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison DE, Strong R, Allison DB, Ames BN, Astle CM, Atamna H, Fernandez E, Flurkey K, Javors MA, Nadon NL, et al. (2014). Acarbose, 17-alpha-estradiol, and nordihydroguaiaretic acid extend mouse lifespan preferentially in males. Aging Cell 13, 273–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haupt S, Berger M, Goldberg Z, and Haupt Y. (2003). Apoptosis - the p53 network. J Cell Sci 116, 4077–4085. [DOI] [PubMed] [Google Scholar]
- Hellmich MR, Coletta C, Chao C, and Szabo C. (2015). The therapeutic potential of cystathionine beta-synthetase/hydrogen sulfide inhibition in cancer. Antioxid Redox Signal 22, 424–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hine C, Harputlugil E, Zhang Y, Ruckenstuhl C, Lee BC, Brace L, Longchamp A, Trevino-Villarreal JH, Mejia P, Ozaki CK, et al. (2015). Endogenous hydrogen sulfide production is essential for dietary restriction benefits. Cell 160, 132–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hine C, Kim HJ, Zhu Y, Harputlugil E, Longchamp A, Matos MS, Ramadoss P, Bauerle K, Brace L, Asara JM, et al. (2017). Hypothalamic-Pituitary Axis Regulates Hydrogen Sulfide Production. Cell Metab 25, 1320–1333 e1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hine C, Zhu Y, Hollenberg AN, and Mitchell JR (2018). Dietary and Endocrine Regulation of Endogenous Hydrogen Sulfide Production: Implications for Longevity. Antioxid Redox Signal 28, 1483–1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyer-Hansen M, and Jaattela M. (2007). Connecting endoplasmic reticulum stress to autophagy by unfolded protein response and calcium. Cell Death Differ 14, 1576–1582. [DOI] [PubMed] [Google Scholar]
- Jackson SP, and Bartek J. (2009). The DNA-damage response in human biology and disease. Nature 461, 1071–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabil O, Weeks CL, Carballal S, Gherasim C, Alvarez B, Spiro TG, and Banerjee R. (2011). Reversible heme-dependent regulation of human cystathionine beta-synthase by a flavoprotein oxidoreductase. Biochemistry 50, 8261–8263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura H. (2014). Production and physiological effects of hydrogen sulfide. Antioxid Redox Signal 20, 783–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouranti I, and Peyroche A. (2012). Protein degradation in DNA damage response. Semin Cell Dev Biol 23, 538–545. [DOI] [PubMed] [Google Scholar]
- Laplante M, and Sabatini DM (2012). mTOR signaling in growth control and disease. Cell 149, 274–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin VS, Lippert AR, and Chang CJ (2013). Cell-trappable fluorescent probes for endogenous hydrogen sulfide signaling and imaging H2O2-dependent H2S production. Proc Natl Acad Sci U S A 110, 7131–7135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longchamp A, Mirabella T, Arduini A, MacArthur MR, Das A, Trevino-Villarreal JH, Hine C, Ben-Sahra I, Knudsen NH, Brace LE, et al. (2018). Amino Acid Restriction Triggers Angiogenesis via GCN2/ATF4 Regulation of VEGF and H2S Production. Cell 173, 117–129 e114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma T, Li J, Xu Y, Yu C, Xu T, Wang H, Liu K, Cao N, Nie BM, Zhu SY, et al. (2015). Atg5-independent autophagy regulates mitochondrial clearance and is essential for iPSC reprogramming. Nat Cell Biol 17, 1379–1387. [DOI] [PubMed] [Google Scholar]
- Miyamoto R, Otsuguro K, Yamaguchi S, and Ito S. (2014). Contribution of cysteine aminotransferase and mercaptopyruvate sulfurtransferase to hydrogen sulfide production in peripheral neurons. J Neurochem 130, 29–40. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Yoshimori T, and Levine B. (2010). Methods in mammalian autophagy research. Cell 140, 313–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy B, Bhattacharya R, and Mukherjee P. (2019). Hydrogen sulfide signaling in mitochondria and disease. FASEB J 33, 13098–13125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murrow L, and Debnath J. (2013). Autophagy as a stress-response and quality-control mechanism: implications for cell injury and human disease. Annu Rev Pathol 8, 105–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy P. (2015). Mechanistic chemical perspective of hydrogen sulfide signaling. Methods Enzymol 554, 3–29. [DOI] [PubMed] [Google Scholar]
- Nishida Y, Arakawa S, Fujitani K, Yamaguchi H, Mizuta T, Kanaseki T, Komatsu M, Otsu K, Tsujimoto Y, and Shimizu S. (2009). Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature 461, 654–658. [DOI] [PubMed] [Google Scholar]
- Olaussen KA, Adam J, Vanhecke E, Vielh P, Pirker R, Friboulet L, Popper H, Robin A, Commo F, Thomale J, et al. (2013). PARP1 impact on DNA repair of platinum adducts: preclinical and clinical read-outs. Lung Cancer 80, 216–222. [DOI] [PubMed] [Google Scholar]
- Olson KR (2015). Hydrogen sulfide as an oxygen sensor. Antioxid Redox Signal 22, 377–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rastogi RP, Richa, Kumar A, Tyagi MB, and Sinha RP (2010). Molecular mechanisms of ultraviolet radiation-induced DNA damage and repair. J Nucleic Acids 2010, 592980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousseau A, and Bertolotti A. (2018). Regulation of proteasome assembly and activity in health and disease. Nat Rev Mol Cell Biol 19, 697–712. [DOI] [PubMed] [Google Scholar]
- Santivasi WL, and Xia F. (2014). Ionizing radiation-induced DNA damage, response, and repair. Antioxid Redox Signal 21, 251–259. [DOI] [PubMed] [Google Scholar]
- Sasakura K, Hanaoka K, Shibuya N, Mikami Y, Kimura Y, Komatsu T, Ueno T, Terai T, Kimura H, and Nagano T. (2011). Development of a highly selective fluorescence probe for hydrogen sulfide. J Am Chem Soc 133, 18003–18005. [DOI] [PubMed] [Google Scholar]
- Sbodio JI, Snyder SH, and Paul BD (2018). Golgi stress response reprograms cysteine metabolism to confer cytoprotection in Huntington’s disease. Proc Natl Acad Sci U S A 115, 780–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu S. (2018). Biological Roles of Alternative Autophagy. Mol Cells 41, 50–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singha S, Kim D, Moon H, Wang T, Kim KH, Shin YH, Jung J, Seo E, Lee SJ, and Ahn KH (2015). Toward a selective, sensitive, fast-responsive, and biocompatible two-photon probe for hydrogen sulfide in live cells. Anal Chem 87, 1188–1195. [DOI] [PubMed] [Google Scholar]
- Swerdlow S, McColl K, Rong Y, Lam M, Gupta A, and Distelhorst CW (2008). Apoptosis inhibition by Bcl-2 gives way to autophagy in glucocorticoid-treated lymphocytes. Autophagy 4, 612–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szczesny B, Marcatti M, Zatarain JR, Druzhyna N, Wiktorowicz JE, Nagy P, Hellmich MR, and Szabo C. (2016). Inhibition of hydrogen sulfide biosynthesis sensitizes lung adenocarcinoma to chemotherapeutic drugs by inhibiting mitochondrial DNA repair and suppressing cellular bioenergetics. Sci Rep 6, 36125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szczesny B, Modis K, Yanagi K, Coletta C, Le Trionnaire S, Perry A, Wood ME, Whiteman M, and Szabo C. (2014). AP39, a novel mitochondria-targeted hydrogen sulfide donor, stimulates cellular bioenergetics, exerts cytoprotective effects and protects against the loss of mitochondrial DNA integrity in oxidatively stressed endothelial cells in vitro. Nitric Oxide 41, 120–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takano Y, Hanaoka K, Shimamoto K, Miyamoto R, Komatsu T, Ueno T, Terai T, Kimura H, Nagano T, and Urano Y. (2017). Development of a reversible fluorescent probe for reactive sulfur species, sulfane sulfur, and its biological application. Chem Commun (Camb) 53, 1064–1067. [DOI] [PubMed] [Google Scholar]
- Trocha KM, Kip P, Tao M, MacArthur MR, Trevino-Villarreal JH, Longchamp A, Toussaint W, Lambrecht BN, de Vries MR, Quax PHA, et al. (2020). Short-term preoperative protein restriction attenuates vein graft disease via induction of cystathionine gamma-lyase. Cardiovasc Res 116, 416–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R. (2012). Physiological implications of hydrogen sulfide: a whiff exploration that blossomed. Physiol Rev 92, 791–896. [DOI] [PubMed] [Google Scholar]
- Wu D, Wang H, Teng T, Duan S, Ji A, and Li Y. (2018). Hydrogen sulfide and autophagy: A double edged sword. Pharmacol Res 131, 120–127. [DOI] [PubMed] [Google Scholar]
- Wu H, Zhu H, Liu DX, Niu TK, Ren X, Patel R, Hait WN, and Yang JM (2009). Silencing of elongation factor-2 kinase potentiates the effect of 2-deoxy-D-glucose against human glioma cells through blunting of autophagy. Cancer Res 69, 2453–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Li P, Fu S, Calay ES, and Hotamisligil GS (2010). Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab 11, 467–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yetik-Anacak G, Sevin G, Ozzayim O, Dereli MV, and Ahmed A. (2016). Hydrogen sulfide: A novel mechanism for the vascular protection by resveratrol under oxidative stress in mouse aorta. Vascul Pharmacol 87, 76–82. [DOI] [PubMed] [Google Scholar]
- Zadra G, Photopoulos C, Tyekucheva S, Heidari P, Weng QP, Fedele G, Liu H, Scaglia N, Priolo C, Sicinska E, et al. (2014). A novel direct activator of AMPK inhibits prostate cancer growth by blocking lipogenesis. EMBO Mol Med 6, 519–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao K, Ju Y, Li S, Altaany Z, Wang R, and Yang G. (2014). S-sulfhydration of MEK1 leads to PARP-1 activation and DNA damage repair. EMBO Rep 15, 792–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zivanovic J, Kouroussis E, Kohl JB, Adhikari B, Bursac B, Schott-Roux S, Petrovic D, Miljkovic JL, Thomas-Lopez D, Jung Y, et al. (2019). Selective Persulfide Detection Reveals Evolutionarily Conserved Antiaging Effects of S-Sulfhydration. Cell Metab 30, 1152–1170 e1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Datasheet S1. List of activator hits identified in the primary screen. Relative to Figure 1 Genotoxic hits are highlighted in red.
Data Availability Statement
The datasheet of activator hits identified in the primary screen is included in the supplemental information. The datasheet of activator hits identified in the primary screen is included in the supplemental information as Datasheet S1.