

Summary
Tumors with defective mismatch repair (dMMR) are responsive to immunotherapy because of dMMR-induced neoantigens and activation of the cGAS-STING pathway. While neoantigens result from the hypermutable nature of dMMR, it is unknown how dMMR activates the cGAS-STING pathway. We show here that loss of the MutLα subunit MLH1, whose defect is responsible for ~50% of dMMR cancers, results in loss of MutLα-specific regulation of exonuclease 1 (Exo1) during DNA repair. This leads to unrestrained DNA excision by Exo1, which causes increased ssDNA formation, RPA exhaustion, DNA breaks, and aberrant DNA repair intermediates. Ultimately, this generates chromosomal abnormalities and the release of nuclear DNA into the cytoplasm, activating the cGAS-STING pathway. In this study, we discovered a hitherto unknown MMR mechanism that modulates genome stability and has implications for cancer therapy.
eTOC Blurb
The mechanism by which mismatch repair deficiency benefits immunotherapy is unclear. Guan et al. show that mismatch repair protein MLH1 controls Exo1 nuclease activity during DNA repair, and loss of MLH1 causes DNA hyper-excision, leading to chromosomal instability and cytosolic DNA accumulation. This activates the cGAS-STING pathway to facilitate immunotherapy.
Graphical Abstract
Introduction
DNA mismatch repair (MMR) maintains genome stability by correcting base mismatches generated during DNA replication. MMR is nick-directed and specifically targets the newly synthesized DNA strand. MMR in human cells is initiated when a mismatch recognition protein, MutSα (MSH2-MSH6) or MutSβ (MSH2-MSH3), identifies a mis-incorporated nucleotide, which induces coordinated interactions between MutSα/MutSβ, MutLα (MLH1-PMS2), and PCNA (proliferating cell nuclear antigen). These interactions lead recruitment of the 5′ to 3′ exonuclease 1 (Exo1) to a single strand break 5′ to the mismatch via its physical interactions with MLH1, MSH2, and/or PCNA (Schmutte et al., 1998; Tishkoff et al., 1998; Tran et al., 2001). Upon removing the mispaired base, the Exo1-catalyzed excision is promptly terminated, possibly through interactions with MutLα. The excision-generated ssDNA gap is then filled by DNA polymerase δ, and repair is concluded by ligase I-catalyzed nick ligation (Constantin et al., 2005; Zhang et al., 2005).
Consistent with MMR’s role in maintaining replication fidelity, defects in MMR genes, particularly in MLH1 and MSH2, lead to genome-wide mutations and cancer development (Kolodner and Marsischky, 1999; Li, 2008; Modrich and Lahue, 1996). Tumors defective in MMR (dMMR) exhibit an elevated mutation frequency and numerous alterations in microsatellite sequence, a phenomenon called microsatellite instability (MSI) (Li, 2008). Interestingly, recent studies have revealed that dMMR tumors are responsive to checkpoint blockade immunotherapy (Le et al., 2017; Le et al., 2015). The dMMR-triggered responses to immunotherapy have been attributed to the high mutation rate in dMMR tumors, which allows production of a large quantity of neoantigens that are subsequently recognized by the immune system as non-self to elicit immune responses and kill dMMR tumor cells. However, ∼50% of dMMR tumors do not respond to immune-checkpoint blockade (Cristescu et al., 2018; Kim et al., 2018; Le et al., 2017), suggesting that neoantigen-generated mutation burden found in dMMR is not sufficient to drive responsiveness to immunotherapy.
The cytosolic DNA sensor cyclic GMP-AMP synthase (cGAS), a critical factor in the innate immune response pathway, is essential for checkpoint blockade immunotherapy in animal models (Wang et al., 2017). Upon binding cytosolic DNA, cGAS synthesizes the second messenger molecule cyclic GMP-AMP (cGAMP), which, in turn, binds and activates the adaptor stimulator of interferon genes (STING). The activated STING then triggers a signaling cascade that induces type I interferons (IFNs) and other immune molecules (Ablasser and Chen, 2019; Li and Chen, 2018). Recent studies have also shown that cGAS activation is associated with DNA damage-induced genome instability, which leads to the formation of micronuclei, the cGAS-STING activation platform (Harding et al., 2017; Mackenzie et al., 2017). Interestingly, 5-fluorouracil treated MLH1-deficient tumor cells exhibit double strand breaks (DSBs), micronuclei, and chromosomal instability when they express no or reduced ATR or Chk1 (Jardim et al., 2009), suggesting that MLH1 deficiency triggers activation of the cGAS-STING pathway. Indeed, we have recently shown that tumor cells defective in MLH1 accumulate cytosolic DNA to activate the cGAS-STING pathway, which sensitizes MLH1-deficient tumors to immunotherapy by promoting the priming and infiltration of anti-tumor CD8+ T cells. Impairing the DNA sensing function of cGAS or STING renders MLH1-deficient tumors resistant to immunotherapy (Lu et al., 2021). However, the mechanism by which MLH1 deficiency activates the innate immune sensing pathway is unknown.
MLH1 has been shown to play a role in repairing DNA strand breaks. For example, it was shown that MLH1 is immediately recruited to DNA strand breaks induced by various DNA damaging agents (Hong et al., 2008). Exo1 is known to conduct the DNA end resection required for double-strand break (DSB) repair via the homologous recombination (HR) pathway (Cejka, 2015; Niu et al., 2009; Tsubouchi and Ogawa, 2000). Because DNA hyper-resection is mutagenic and toxic to the genome (Ochs et al., 2016), Exo1-catalyzed excision during end resection must be properly regulated, as it is in MMR (Zhang et al., 2005). Collectively, these data drove our hypothesis that MutLα regulates Exo1 activity and that loss of MLH1 causes Exo1-catalyzed hyper-excision, which leads to genome instability and activation of the cGAS-STING pathway to facilitate immune checkpoint inhibitor therapy.
We show here that MutLα modulates Exo1’s activity through a mutual physical interaction, and depleting MLH1 or disrupting the MLH1–Exo1 interaction allows Exo1 to conduct hyperactive excision in both MMR and DNA end resection. Exo1-mediated hyper-resection causes increased ssDNA, RPA exhaustion, increased ssDNA breaks. These aberrant intermediates eventually result in chromosomal abnormalities and accumulation of cytosolic DNA, which activates the cGAS-STING pathway.
Results
MLH1-deficiency activates the cGAS-STING pathway
To determine whether dMMR-mediated immunotherapy responsiveness occurs via activation of the cGAS-STING pathway, we generated Mlh1-knockout (Mlh1−/−) and Mlh1-rescued mouse 4T1 tumor cells, and the resulting cells were analyzed for DNA breaks and cytosolic DNA accumulation, events required for cGAS activation (Ablasser and Chen, 2019; Li and Chen, 2018). We measured cytosolic DNA using the PicoGreen dye, a widely used fluorescent stain that selectively binds to double-stranded DNA (Shen et al., 2015), and found that without any treatment, the percentage of cells accumulating cytosolic DNA in Mlh1−/− 4T1 cells was significantly higher than that in wild-type (WT) 4T1 and Mlh1-rescued Mlh1−/− 4T1 cells (Figures 1A and 1B). This number increased significantly (Figure 1B) when cells were treated with 10 Gy of ionizing radiation (IR). These results suggest that MLH1 deficiency causes DNA release from the nucleus to the cytoplasm, and IR stimulates this process.
Figure 1. MLH1 deficiency activates innate immune signaling pathway.

(A) Detection of cytosolic DNA in WT, Mlh1−/− 4T1 and Mlh1-rescued (Rescd) 4T1 cells treated with or without IR, as indicated. DNA was detected using the PicoGreen fluorescence dye selectively binding dsDNA. Arrows point to cytosolic DNA. (B) Percentage of cells displaying cytosolic DNA with and without IR treatment. (C) Western blot analysis showing prolonged γH2AX in Mlh1−/−, but not in WT and Mlh1-rescued 4T1 cells after IR treatment. (D) Quantification of relative γH2AX levels in various 4T1 cells. (E) Increased production of cGAMP in Mlh1−/− 4T1 cells. (F) Western blots showing enhanced phosphorylation of STING (pSTING) and STAT1 (pSTAT1) induced by IR in Mlh1−/− cells. (G) and (H) Quantification of relative levels of pSTING and pSTAT1, respectively. (I) qRT-PCR analysis showing increased production of Isg15 in Mlh1−/− cells. (J) and (K) Western blots and qRT-PCR, respectively, showing that immune signaling induced by MLH1 deficiency depends on cGAS. When present, “−” indicates untreated cells. Data represent the mean ± SEM of 3 independent experiments (B, D, G and H) or 3 replicates (E, I and K). P value was calculated using one-way ANOVA. **, p<0.01; ***, p<0.001; and ****, p<0.0001. See also Figure S1.
Because the accumulation of cytoplasmic DNA is a consequence of nuclear DNA damage (Li and Chen, 2018), we next assessed the phosphorylation of histone H2AX (γH2AX), an indirect marker of DNA double-strand breaks (DSBs) in the cell lines. Comparable levels of γH2AX were found 1-hour after IR in all cell lines (Figure 1C). Interestingly, γH2AX levels persisted in Mlh1−/− 4T1 cells, as indicated by γH2AX still being present 12 hours after IR in Mlh1−/− 4T1 cells, but not in WT 4T1 cells (Figures 1C and 1D). The same results were also obtained in the MLH1-deficient colorectal cancer cell line HCT116, and its MLH1-rescued derivative (Supplemental Figures 1A–1C). These results strongly suggest that MLH1−/− cells have delayed repair and/or persistent generation of DSBs.
To determine whether the observed cytosolic DNA in Mlh1−/− cells leads to cGAS activation, we measured the production of cGAMP in WT and Mlh1−/− 4T1 cells with or without IR treatment, as described previously (Gui et al., 2019). The results show that Mlh1−/− cells, regardless of IR treatment, contained a cGAMP level significantly higher than that in WT cells (Figures 1E and S1D), and that IR treatment significantly stimulated the production of cGAMP (Figure 1E). Consistent with the fact that cGAMP activates STING and its downstream factors, the increase in cGMAP in Mlh1−/− 4T1 cells is associated with increased phosphorylation of STING (Figures 1F and 1G) and STAT1 (Figures 1F and 1H), as well as upregulation of interferon-stimulated gene 15 (Isg15) (Figure 1I) and interferon regulatory factor 7 (Irf7) (Figure S1E). The innate immune signaling triggered by MLH1 deficiency depends on cGAS and STING, as knocking out either abolished this signaling (Figures 1J, 1K, and S1F–S1I). Collectively, the data indicate that MLH1 deficiency decreases the repair of IR-induced DSBs, which increases leaking of nuclear DNA into the cytoplasm, thereby inducing innate immune signaling.
Exo1 is essential for MLH1−/−-mediated DNA breaks and innate sensing signaling
Previous studies have shown that MLH1 physically interacts with Exo1 (Schmutte et al., 1998; Tishkoff et al., 1998; Tran et al., 2001) and that MutLα modulates Exo1 nuclease activity during mismatch-provoked excision (Zhang et al., 2005). This drove us to postulate that the increased presence of DSBs in Mlh1−/− cells is because MLH1 also modulates Exo1 activity in DSB repair and loss of MLH1 results in excessive Exo1-mediated DNA end resection and dysfunctional DSB repair (Cejka, 2015; Niu et al., 2009; Tsubouchi and Ogawa, 2000). To explore this, we knocked out Exo1 in both 4T1 and Mlh1−/− 4T1 cells, treated the resulting Exo1−/− and Mlh1−/−-Exo1−/− cells with IR, and analyzed for cytosolic DNA and innate immune signaling. Depleting Exo1 in Mlh1−/− cells significantly reduced the number of cells exhibiting cytosolic DNA both in IR-treated and untreated cells, and the double knockout (Mlh1−/−-Exo1−/−) cells, as well as Exo1−/− cells, behaved like WT 4T1 cells (Figures 2A and S2A). Correspondingly, we did not observe the prolonged γH2AX detected in Mlh1−/− cells after IR treatment in Mlh1−/−-Exo1−/− cells (Figures 2B and 2C), which suggests that Exo1 is directly responsible for the prolonged DNA breaks in Mlh1-deficient cells, and that DSBs in Mlh1−/−-Exo1−/− cells could be repaired via the non-homologous end-joining pathway or processed by other end resection enzymes (e.g., DNA2) in the absence of Exo1 before being repaired via HR (Liu and Huang, 2016). The lower γH2AX levels in Mlh1−/−-Exo1−/− double knockout cells were also associated with reduced phosphorylation of STAT1 (Figures 2B and 2C) and STING (Figures 2D and 2E), as well as reduced expression of Isg15 (Figure 2F), Irf7 (Figure S2B), and type I interferon Ifn-β (Figure S2C). The results strongly suggest that Exo1 is essential for generating persistent, prolonged DNA damage in Mlh1−/− cells, which leads to cytosolic DNA, followed by cGAS-STING pathway activation.
Figure 2. Exo1 is essential for innate sensing signaling in Mlh1−/− 4T1 cells.

(A) Depletion of Exo1 reduces cytosolic DNA accumulation in Mlh1−/− cells regardless of IR treatment. (B) Western blots showing reduced DNA breaks and pSTAT1 when Exo1 was depleted from Mlh1−/− cells. A non-specific band detected by an Exo1 antibody is indicated by an asterisk. (C) Quantification of the relative γH2AX levels in Mlh1 knockout and Mlh1-Exo1 double knockout (Dbl KO) cells. (D) Western blots showing that Exo1 knockout abolishes IR-induced STING activation. (E) Quantification of relative pSTING levels in Mlh1-knockout and Mlh1-Exo1 Dbl KO cells. (F) qRT-PCR analysis showing that Exo1 depletion suppressed expression of Isg15. Data represent the mean ± SEM of 3 independent experiments (A, C and E) or 3 replicates (F). P value was calculated using one-way ANOVA. ****, p<0.0001. See also Figure S2.
MutLα–Exo1 interaction controls Exo1’s nuclease activity
To determine whether the MutLα–Exo1 interaction regulates Exo1’s nuclease activity in MMR and DNA end resection, we generated several Exo1 mutants that disrupt Exo1’s MLH1-interaction domains (Tran et al., 2004) (also see Figure 3A). We selected mutants that retained Exo1 nuclease activity but reduced or ablated the Exo1–MLH1 interaction. The three mutants selected were substitution of Phe at residues 506 and 507 with Ala (Exo1-FF-AA, or FF-AA), deletion of the C terminal 244 (603–846) residues (Exo1-ΔC or ΔC), and combined FF-AA and ΔC (FF-AAΔC) mutations (Figure 3A). Co-immunoprecipitation (Co-IP) experiments using purified proteins (Figure 3B, left) revealed that each of the three Exo1 mutants interacted less with MutLα than WT Exo1 did (Figure 3B, right).
Figure 3. MutLα regulates Exo1 nuclease activity.

(A) Diagram of major functional domains in Exo1. (B) Co-immunoprecipitation–Western analysis of MutLα interactions with WT and mutant Exo1 (right) using purified proteins (left). (C) Southern blot analysis determining the impact of the MutLα–Exo1 interaction on mismatch-provoked excision in a purified MMR system. The excision products were digested with SspI and processed for Southern blot analysis, as described in the Methods section. Schematic representation of the 5′ G-T heteroduplex after SspI digestion is shown on the right side of the gel. Positions of the nick and mismatch (red asterisk) are 544-bp and 416-bp away, respectively, from the bottom SspI site. Red bar indicates the 32P-labeled oligonucleotide probe, which is complementary to the nicked strand near the bottom SspI site. Red bracket shows mismatch-provoked excision products terminated upon mismatch removal in reactions with WT Exo1, but in those with Exo1-FF-AA. (D) In vitro end resection assay to determine the impact of the MutLα–Exo1 interaction on Exo1-catalyzed resection using purified proteins and a linearized 2.7-kb pUC19 plasmid DNA. (E) Percentage of end resection product II shown in D in three independent assays. (F) In vitro end resection assay to determine the role of RPA in Exo1-catalyzed resection. (G) Principle of in vivo end resection assay. (H) qPCR analysis determining the amount of ssDNA generated at a specific DBS site (AsiSI) in WT and MLH1−/− U2OS cells. Data represent the mean ± SEM of 3 independent experiments (E) or 3 replicates (H). P value was calculated using one-way ANOVA. **, p<0.01; ***, p<0.001; and ****, p<0.0001.
First, we tested the MutLα–Exo1 interaction on the mismatch-provoked excision. We performed a reconstituted excision reaction using purified proteins and a circular heteroduplex DNA containing a single G-T mismatch and a nick 128 bp 5’ to the mismatch, as previously described (Zhang et al., 2005). As shown in Figure 3C, higher levels of MutLα were associated with higher levels of excision intermediates (see red bracket), which are ~25-nt and 60-nt downstream of the mismatch site (red asterisk, the 416-nt marker), in reactions with WT Exo1 (lanes 4–6). However, the same conditions did not generate higher levels of excision intermediates immediately downstream of the mismatch in reactions with the Exo1-FF-AA mutant (lanes 9–11), which suggests that appropriate excision termination in MMR relies on the proper interaction between MutLα and Exo1.
We next determined whether MutLα modulates Exo1 activity in DNA end resection, using previously described in vitro end resection assays (Cannavo et al., 2013; Cejka et al., 2010). The results show that Exo1 alone digested a 2.7-kb linear dsDNA into a series of molecules much smaller than the 2.7-kb ssDNA (Figure 3D, lane 3, Product I). Addition of MutLα to the reaction made the excision products migrate more slowly (Figure 3D, lanes 4 and 5), and this was concentration dependent. In the presence of 4 pmol MutLα, all excision products (Product II) became bigger than the 2.7-kb ssDNA (Figure 3D, lane 5). However, MutLα essentially failed to do the same when Exo1 mutants FF-AA and FF-AAΔC were used in the DNA end resection assays (Figure 3D, lanes 7, 8, 10 and 11). The amount of Product II is apparently determined by how strongly MutLα interacts with Exo1, as the reaction containing WT Exo1, which has the strongest interaction with MutLα generated the most Product II, and the reaction containing FF-AA•ΔC, which has the weakest interaction with MutLα produced the least Product II (Figure 3D, compare lane 5 with lanes 8 and 11). Quantitative analysis revealed that the amount of product II in the WT Exo1-containing reaction is significantly higher than that in reactions containing either Exo1 mutant (Figure 3E). Taken together, these results strongly support the hypothesis that MutLα regulates Exo1’s excision activity during DNA end resection through its physical interaction with Exo1.
RPA has also been reported to suppress Exo1’s nuclease activity during end resection (Cannavo et al., 2013). We performed the DNA end resection assay in the presence of RPA, as described previously (Cannavo et al., 2013), and we found that, under experimental conditions, RPA by itself did not suppress DNA end resection conducted either by Exo1 or FF-AA (Figure 3F, lanes 3 and 8). However, RPA appears to facilitate MutLα in effectively controlling Exo1’s excision activity. Reactions with RPA generated Product II using less MutLα than reactions without RPA (Figure 3F, lane 5), and the highest MutLα concentration (4 pmol) essentially inhibited Exo1 activity (Figure 3F, lane 6). We did not observe this phenomenon when Exo1-FF-AA replaced Exo1 in the DNA end resection reactions (Figure 3F, lanes 7–11).
To determine the impact of MLH1 on DNA end resection in vivo, we used the ER-AsiSI system established in U2OS cells, in which the AsiSI restriction enzyme is fused to the estrogen receptor hormone-binding domain and induced when cells are treated with 4-hydroxytamoxifen (4-OHT) (Iacovoni et al., 2010; Zhou et al., 2014). Upon expression, AsiSI can generate DSBs at an AsiSI sequence (5′-GCGATCGC-3′) located in chromosome 1 (Figure 3G). Since ssDNA resists restriction digestion, the amount and length of ssDNA generated during end resection at the AsiSI-created DSB can be determined using three pairs of quantitative PCR (qPCR) primers, i.e., near each of the three BsrGI sites, which are located 335, 1618, and 3500 bp away, respectively, from the AsiSI site (see Figure 3G). As expected, the amount of ssDNA generated in WT U2OS cells is inversely correlated to the distance between the DSB and BsrGI sites (Figure 3H), i.e., resection at the DSB site is more frequently terminated near the strand break, which is consistent with previous findings (Iacovoni et al., 2010; Zhou et al., 2014). However, MLH1-deficent cells generated significantly higher amounts of ssDNA than WT cells, and we did not observe any differences in the amount of ssDNA produced in the three different qPCR reactions (Figure 3H ). This suggests that end resection at the AsiSI site can go at least 3500 bp and likely beyond in the absence of MLH1. Collectively, these observations suggest that MLH1 regulates Exo1’s nuclease activity during DNA end resection.
Exo1 is more stable and abundant in MLH1-deficient cells
To directly visualize the recruitment of Exo1 to IR-induced strand breaks, we micro-irradiated WT and MLH1-deficient HeLa cells transiently expressing GFP-tagged Exo1 using a laser microscope, as previously described (Lu et al., 2019). We observed that similar amounts of Exo1 were recruited to the damage sites at early time points in WT and MLH1-deficient cells. However, the level of Exo1 in WT cells reached a plateau at ~2 min before gradually declining. In contrast, the fluorescence intensity in MLH1-deficient cells did not reach the peak until 4 min and stayed at that level for at least several hours (Figures 4A, 4B and 4C). Similar results were also obtained in MLH1-deficient HCT116 and MLH1-rescued HCT116 cells (Figures S3A and S3B). These observations suggest that, in MLH1-deficient cells, not only does more Exo1 interact with damaged DNA, but Exo1 is retained longer at DNA damage sites. This supports the idea that, in the absence of MLH1, Exo1 excessively excises damaged DNA.
Figure 4. Exo1 recruitment, abundance and stability in MLH1−/− cells.

(A) Live cell imaging showing real-time recruitment and retention dynamics of GFP-tagged Exo1 after laser micro-irradiation in WT and MLH1−/− HeLa cells. (B) Quantification of GFP-tagged Exo1 levels from the indicated number of cells. (C) Western blots showing whole cell lysate (WCL) and chromatin-bound levels of Exo1 and phosphorylated Exo1 (pExo1) in WT and MLH1−/− HeLa cells. (D) Quantification of relative total Exo1 levels in WT and MLH1−/− HeLa cells. (E) Western blots showing WCL levels of Exo1 in WT and MLH1−/− U2OS cells. (F) RNA-seq data from the TCGA database showing significantly higher Exo1 expression in dMLH1 tumors than in MSS tumors. (G) Quantification of relative pExo1 levels in whole cell lysate (upper) and on chromatin (lower) in WT and MLH1−/− HeLa cells. Data represent the mean ± SEM of 3 independent experiments. P value was calculated using one-way ANOVA. ****, p<0.0001. See also Figure S3.
To determine whether the higher levels of Exo1 in MLH1-deficient cells are related to Exo1 expression and/or stability, we measured Exo1 levels in MLH1-proficient and deficient HeLa cells. We found that the overall level of Exo1 is higher in MLH1-deficient cells than in WT HeLa cells both before and after IR treatment (Figures 4C upper and 4D). We also observed a higher level of Exo1 in U2OS-MLH1−/− cells than in WT U2OS cells both before and after inducing DSBs by 4-OHT (Figure 4E). Similar results were found in MLH1-deficient HCT116 cells, in comparison with MLH1-rescued HCT116 cells (Figures S3C–S3F). To confirm this finding in human cancers, we analyzed Exo1 expression in microsatellite stable (MSS) and MLH1-deficient (dMLH1) colorectal tumors from the Cancer Genome Atlas (https://gdac.broadinstitute.org/). Consistent with the results in cell lines, we found that the level of Exo1 in dMLH1 tumors is significantly higher than that in MSS tumors (Figure 4F). These results suggest that MLH1 may have a role in modulating Exo1 expression. Although the exact mechanism(s) by which MLH1 regulates Exo1 expression is unknown, previous studies have suggested that MutLα plays an important role in regulating miRNA biogenesis (Mao et al., 2012). It is possible that MutLα regulates the production of Exo1-specific miRNAs. We would like to mention that since we could not obtain a specific antibody for mouse Exo1, we used the human Exo1 antibody to detect Exo1 expression in 4T1 cells (Figure 2B). Although the human Exo1 antibody can recognize a specific band with the correct size, we did not observe IR-enriched Exo1 in 4T1 cells, which could be due to the fact that the human antibody cannot detect modified (e.g., phosphorylated) Exo1 in mouse cells (see below).
Recent studies have shown that the phosphorylation of Exo1 promotes its recruitment to DSBs and is required for appropriate DNA end resection (Bolderson et al., 2010; Tomimatsu et al., 2014). We examined Exo1 phosphorylation in WT and MLH1-deficient cells and found that, as with Exo1 expression, the level of phosphorylated Exo1 was significantly higher in MLH1-deficient HeLa cells than in WT HeLa cells, regardless of IR treatment (Figure 4C). Interestingly, we found that more Exo1, as well as pExo1, was associated with chromatin in MLH1-deficient cells than in WT HeLa cells (Figures 4C and 4G). These results are consistent with the fact that Exo1 is retained at laser-generated DSBs longer in MLH1-deficient cells than in WT cells (Figure 4B), suggesting that higher levels of Exo1 on DNA results in excessive DNA end resection (Figure 3H) and more DSBs (Figure 1C). The current literature reports contradictory roles for pExo1 in the DNA damage response, with two studies showing that Exo1 phosphorylation activates its activity (Doerfler and Schmidt, 2014; Tomimatsu et al., 2014), one showing Exo1 inactivation (Morafraile et al., 2020), and another showing that pExo1 results in protein degradation (El-Shemerly et al., 2005). Our data suggest that chromatin-bound pExo1 is active in DNA end resection.
RPA exhaustion and phosphorylation in MLH1-deficient cells
RPA protects ssDNA from nicking during DNA replication, repair, and recombination (Bhat and Cortez, 2018; Marechal and Zou, 2015). However, generating excess ssDNA causes RPA exhaustion and genomic instability (Chen et al., 2013; Ibler et al., 2019). We hypothesized that Exo1-catalyzed hyper-resection in MLH1-deficient cells increases the production of ssDNA and subsequently, RPA exhaustion. To examine this, we performed RPA-protection assays, as described previously (Ibler et al., 2019), in U2OS cells that carry the ER-AsiSI system. In this assay (see Figure S4A), hyper-resection-generated ssDNA fragments, which are not protected by RPA, can serve as a template for DNA synthesis using nucleotide analogue 5-bromo-2’-deoxyuridine (BrdU) and Taq DNA polymerase, and an antibody specific for BrdU can be used to identify sites of unprotected ssDNA (see STAR Methods for details). Upon treatment with 4-OHT, a BrdU-specific antibody detected few foci in WT cells, but significantly more in MLH1-deficient U2OS cells (Figures 5A and 5B), which is consistent with the fact that there are more than 150 AsiSI restriction sites in the human genome. In addition to the number of foci per cell (Figure 5B), the percentage of cells displaying BrdU foci is significantly lower in WT U2OS cells than in MLH1-deficient cells (Figure 5C). These RPA exhaustion assay results strongly suggest that MLH1-deficient cells contain high levels of RPA-free ssDNA, which supports the idea that Exo1 conducts extensive end resection in the absence of MLH1.
Figure 5. RPA exhaustion and aberrant resection intermediates in MLH1−/− cells.

(A) Microscope imaging showing BrdU incorporation by DNA polymerase using hyperesection-generated unprotected RPA as a template for DNA synthesis in the RPA exhaustion assay. ssDNA binding by phosphorylated RPA (pRPA) is also shown. (B) and (C) Quantification of BrdU foci/cell and percentage of cells exhibiting BrdU foci, respectively, in WT and MLH1−/− U2OS cells. (D) Quantification of pRPA foci per cell. (E) Western blots detecting pRPA and its association with DNA break marker γH2AX in the indicated cells before and after IR. (F) Quantification of relative pRPA levels shown in E, with 3 independent assays. (G) Immunoflurescence confocal analysis showing large RPA foci in HeLa MLH1−/− cells. (H) Quantification and comparison of the percentage of WT and MLH1−/− cells displaying RPA foci. (I) Immunofluorescence confocal analysis showing large Rad51 foci in MLH1−/− HeLa cells. (J) Quantification of RAD51 foci/nucleus in various HeLa cells, as indicated. (K) Immunofluorescence confocal analysis showing large Rad51 foci in HCT116 and MLH1-rescued HCT116 cells. Data represent the mean ± SEM of 3 independent experiments (C, F and H) or the indicated number of cells (B, D and J). P value was calculated using one-way ANOVA. **, p<0.01; ***, p<0.001; and ****, p<0.0001.
RPA is phosphorylated in response to DNA damage (Bhat and Cortez, 2018; Marechal and Zou, 2015). As expected, we observed phosphorylated RPA (pRPA) foci in U2OS cells treated with 4-OHT (Figure 5A). However, the number of pRPA foci per nucleus was significantly higher in mutant cells than in WT cells (Figures 5A and 5D). We also observed similar results in HeLa cells, as we detected higher levels of pRPA in MLH1-deficient cells, which correlated to the levels of γH2AX (Figure 5E, lanes 5–8). This effect appears to depend on MLH1 deficiency because rescuing MLH1 in MLH1−/− HeLa cells has largely reduced the production of both pRPA and γH2AX (Figure 5E, lanes 9–12, and Figure 5F). Although the rescued MLH1 level is lower than that in WT cells (Figure 5E, compare MLH1 expression in lanes 1–4 with lanes 9–12), the intermediate MLH1 expression in the rescued cells seems to be able to restore most of the MLH1 activity in controlling Exo1 nuclease activity, because the RPA/RAD51 foci number and size in MLH1-rescued cells are closer to those observed in WT cells. Therefore, these data further support the idea that MLH1 regulates Exo1 nuclease activity.
RPA and Rad51 form large foci in MLH1−/− cells
Because Exo1-catalyzed hyper-resection in MLH1-deficient cells generated excess ssDNA and increased RPA binding, we postulated that RPA foci formed in MLH1-deficient cells should be larger than those in WT cells. We tested this hypothesis in MLH1-proficient and deficient HeLa cells treated with IR. We found that RPA foci in MLH1-deficient HeLa cells are significantly larger and brighter than those in WT HeLa cells, as judged by focus size and fluorescence intensity (Figure 5G). In addition, both focus number per nucleus and percentage of cells displaying RPA foci were significantly higher in MLH1-deficient cells than in WT HeLa cells (Figures 5H and S4B). In support of these data, we observed that pRPA foci were larger and more numerous in U2OS-MLH1−/− cells than in WT U2OS cells (Figure 5A). These data strongly support the hypothesis that the hyper-resection by Exo1 in MLH1-deficient cells increases the formation of ssDNA, which leads to more RPA loading, larger RPA foci, and RPA exhaustion.
The next step in DSB repair by HR is the replacement of RPA by Rad51 and the formation of Rad51 nucleofilament on the ssDNA, which is required for strand invasion. We hypothesized that the increase in DNA end resection in MLH1-deficient cells would result in increased and longer Rad51 nucleofilaments. To test this, we examined IR-induced Rad51 focus formation in MLH1-proficient and -deficient HeLa cells. The results revealed that, like RPA, Rad51 forms much larger foci in MLH1-deficient than in WT and HeLa cells (Figure 5I). In addition, we observed more Rad51 foci per nucleus in MLH1−/− cells than in WT cells (Figure 5J). We also observed a few relatively large Rad51 foci (Figure 5I), as well as relatively higher levels of pRPA, particularly 8 h after IR (Figure 5E) in MLH1-rescued HeLa-MLH1−/− cells than those found in WT HeLa cells. We attributed this to the relatively lower expression of MLH1 in the MLH1-rescued cells (Figure 5E, compare MLH1 expression between WT and MLH1-rescued cells). These results further support the idea that MLH1 regulates Exo1 nuclease activity during end resection. Similar results were also obtained in HCT116 and MLH1-rescued HCT116 cells (Figures 5K, S4C–4E). Taken together, we conclude that in the absence of MLH1, Exo1-catalyzed end resection generates a large quantity of ssDNA, which results in RPA exhaustion and genomic instability.
DNA hyper-excision causes various chromosomal abnormalities
We hypothesized that RPA exhaustion results in SSBs that lead to abnormal HR and chromosome breaks. We performed a metaphase chromosome spreading experiment to visualize chromosomal abnormalities associated with hyper-resection in WT and Mlh1−/− 4T1 cells. Representative metaphase chromosome spreads are shown in Figure 6 (A–D). In the absence of IR, about 20% of WT 4T1 cells showed chromosome abnormalities, mainly 1 chromosome aberration (Figure 6E, column 1); but most (70%) Mlh1−/− 4T1 cells exhibited this low number of chromosome abnormalities (Figure 6E, column 2). These numbers changed dramatically after we delivered a single low dose (2 Gy) of radiation. More than 92% of the Mlh1−/− cells displayed chromosomal instability (Figure 6E, column 4), as compared with ~50% of WT cells showing instability (Figure 6E, column 3). MLH1−/− HeLa cells also display a higher percentage of chromosome instability than WT HeLa cells (Figures S5E and S5F). In addition to chromosomal breakages (see green arrows in Figures 6D and S5D), we also observed unseparated chromosomes (see blue arrows in Figures 6D and S5D), which could be derived from unresolved Holliday junctions as a result of defective HR.
Figure 6. Chromosomal abnormalities in MLH1−/− cells.

(A-D) Chromosomal spreading analysis to determine metaphase chromosomal breaks and other aberrations in 4T1 cells (A and C) and Mlh1−/− 4T1 cells (B and D) with (C and D) and without (A and B) IR treatment. Chromosome breaks are indicated by green arrows, while unresolved chromosomes are indicated by blue arrows. (E) Percentage of WT and Mlh1−/− 4T1 cells containing the indicated number of chromosome abnormalities. (F) and (G) Average number of chromosomal abnormalities in WT and Mlh1−/− 4T1 (F) and HeLa (G) cells, respectively. (H) Average number of chromosomal abnormalities in HCT116 and MLH1-rescued HCT116 cells. *** and **** represent p<0.001 and p<0.0001, respectively.
We then analyzed the average number of chromosomal abnormalities in our cohort of 4T1 cells, and found that cells with an MLH1 knockout exhibited a significant increase in chromosomal abnormalities with or without IR treatment, in comparison with their corresponding WT cells (Figure 6F). The same experiments were performed in MLH1-proficient and MLH1-deficient HeLa cells (Figure 6G), as well as in HCT116 and MLH1-rescued HCT116 cells (Figure 6H). The results are essentially the same, i.e., all MLH1-deficient cells (HCT116 and MLH1−/− HeLa) with or without IR treatment show a rate of chromosomal instability that is 3–5-fold higher than that in their corresponding WT cells (MLH1-rescued HCT116 and HeLa). These results are consistent with the fact that MLH1-deficient tumor cells expressing reduced ATR or Chek1, essential DNA damage response factors, display chromosomal instability when treated with chemotherapy drugs (Jardim et al., 2009), and that Mlh1-knockout mice are associated with abnormal crossover in meiosis I and severely damaged chromosomes (Baker et al., 1996). Taken together, we conclude that MLH1 deficiency-triggered hyper-excision by Exo1 induces chromosome instability.
Discussion
We demonstrate here that MLH1 deficiency induces cytosolic DNA and activates the cGAS-STING pathway. This is because MutLα regulates Exo1’s nuclease activity in DNA end resection via the physical interaction between these two proteins. Depleting MLH1 or disrupting the MLH1-Exo1 interaction leads to hyper-excision during DNA end resection, which causes RPA exhaustion, DNA breaks, and eventually chromosome abnormalities. This leads to the release of nuclear DNA into the cytoplasm to activate the cGAS-STING pathway.
Interestingly, we observed chromosomal abnormalities in MLH1-deficient cells, especially when they were treated with IR. MMR deficiency is known to cause base-base substitution and small insertion-deletion mutations. However, increasing evidence suggests that dMMR, particularly MLH1 deficiency, can cause chromosomal instability (CIN) under certain circumstances. It is known that Mlh1-knockout mice are infertile because of prematurely separated chromosomes and unsuccessful completion of recombination, leading to abnormal crossover in meiosis and severely damaged chromosomes (Baker et al., 1996). Partial knockdown ATR or Chk1 expression in multiple MLH1-deficient colorectal cancer cell lines resulted in cells displaying a CIN phenotype, including chromosomal breaks and gaps, chromosome bridge, and micronuclei formation (Jardim et al., 2009). Strikingly, the chromosomal abnormalities disappeared when the MLH1 gene was restored to these tumor cells (Jardim et al., 2009). It is possible that the observed phenomena in these MLH1-deficient cells and animals are caused, at least in part, by Exo1-catalyzed hyper-resection. The CIN phenotype in MLH1-deficient cells could be related to dMMR-caused mutations that impair critical DNA damage response genes, such as ATR and Chk1. Thus, cells cannot appropriately deal with damaged DNA, which induces an abnormal repair process, e.g., hyper-resection by Exo1, and augments the DNA damage, leading to chromosomal instability.
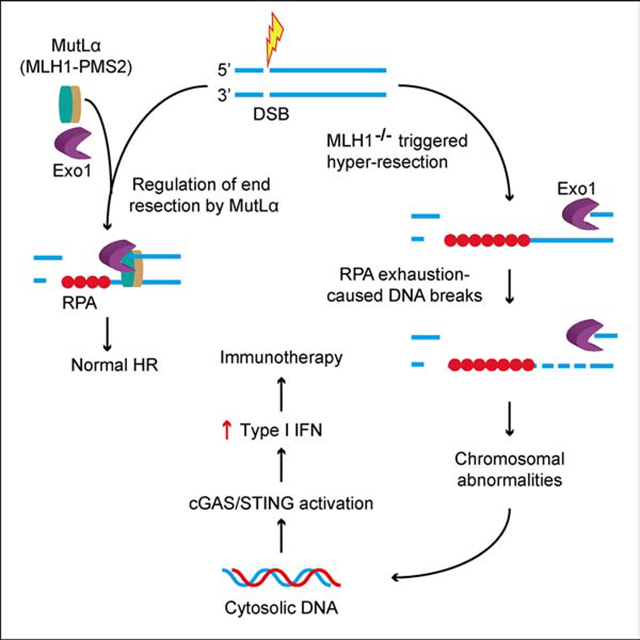
Based on published data and the results presented here, we propose a model that elucidates the mechanism through which MLH1 deficiency triggers the activation of the cGAS-STING pathway to facilitate immunotherapy. Under normal circumstances, DSBs repaired via HR undergo end resection by Exo1. The excision reaction is appropriately terminated through the MutLα-Exo1 interaction, which generates appropriate length of ssDNA essential for normal HR (Figure 7, left). However, MLH1 deficiency depletes Exo1’s regulator, MutLα from cells. Also, Exo1 appears to be more abundant and more stable in MLH1-deficient cells (Figure 4). Thus, a stable and highly concentrated Exo1 can digest DNA essentially at will, and generate a large quantity of ssDNA to use up cellular RPA stores, which leaves the remaining but continuously growing ssDNA chain unprotected. The unprotected ssDNA can be easily digested or nicked by various nucleases. These could impair HR by blocking DNA invasion and Holliday junction formation and/or resolution. These abnormal HR intermediates can provoke cells to degrade a part or all of the damaged chromosome, which leads to the release of nuclear DNA into the cytoplasm. The latter then activates the cGAS-STING pathway and downstream immune responses. Together with dMMR-generated large number of neoantigens, the immune signaling activated by Exo1 hyper-resection facilitates immunotherapy. Because IR stimulates the accumulation of cytosolic DNA and immune signaling, the data presented suggest that a radio-immunotherapy combination could be a more effective treatment for dMMR tumors, especially those defective in MLH1. This appears to be consistent with a recent study reporting that adjuvant radiotherapy improves survival in patients with dMMR cancers (Reijnen et al., 2019). However, further clinical trials are required to confirm this prediction.
Figure 7. Model for MLH1−/−-mediated cGAS activation and immunotherapy.

MutLα (MLH1-PMS2) properly terminates Exo1-catalyzed end resection, which facilitates DSB repair by HR (left). However, depleting MLH1 deprives cells of MutLα, allowing Exo1 to conduct uncontrolled excision. This hyper-resection generates a large quantity of ssDNA that exhausts the RPA pool, leaving ssDNA chain unprotected. The unprotected ssDNA can be digested or nicked by various nucleases in the nucleus, which leads to abnormal recombination intermediates and chromosome breaks. The latter can trigger cells to degrade a part or all of the damaged chromosome to release nuclear DNA into the cytoplasm, activating the cGAS-STING pathway and the downstream immune responses. Together with the large number of neoantigens generated from mutations caused by MLH1 deficiency, the immune signaling activated by Exo1 hyper-resection facilitates immunotherapy.
While MLH1 deficiency-caused chromosome instability benefits immunotherapy via activating the cGAS-STING pathway, merging evidence suggests that MLH1 deficiency can cause genome instability that leads cancer cells to evade immunotherapy. A study by von Loga et al. (von Loga et al., 2020) indicated that MLH1-deficient gastro-oesophageal adenocarcinomas (GOAs) display unusually high intratumor heterogeneity (>20-fold found in other cancers), a tumor phenotype mainly driven by chromosome instability (Bakhoum and Landau, 2017) and characterized by a diverse population of cancer cells with high numbers of both mutations and copy number alterations (Gerlinger and Swanton, 2010; Raynaud et al., 2018). This tumor heterogeneity has been shown to introduce significant challenges in treatment (Gerlinger and Swanton, 2010). In addition, the MLH1-deficient GOAs also have a gain of chromosomes (von Loga et al., 2020). Because tumor heterogeneity in dMMR cancers is usually associated with mutations in antigen presentation genes (Cancer Genome Atlas Research, 2014) and interferon signaling pathway genes (Albacker et al., 2017; Shin et al., 2017; von Loga et al., 2020), it has been postulated that this type of dMMR tumors are refractory to checkpoint blockade immunotherapy (von Loga et al., 2020). Therefore, although dMMR benefits immunotherapy, the hypermutable nature of dMMR will likely introduce mutations in the pathways (e.g., the cGAS-STING-IFN pathway) that are required for dMMR-mediated immunotherapy. As a result, these tumors will not respond to checkpoint blockade treatment, which may explain why ~50% of dMMR tumors refractory to checkpoint blockade immunotherapy.
STAR★Methods
RESOURCE AVAILABILITY
Lead Contact:
Guo-Min Li (Guo-Min.Li@UTSouthwestern.edu).
Material Availability
All materials generated in this study are being made available upon request.
Data and Code Availability
Source data for figures in the paper is available [Mendeley Data Reserved DOI: 10.17632/mrg4443z7m.1].
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
Human cell lines HeLa, HCT116, ER-AsiSI U2OS (a gift from Tanya T. Paull and Gaelle Leube), and mouse breast cancer cell line 4T1 were used in this study. HCT116 was grown in McCoy’s 5A medium and other cell lines were grown in DMEM medium supplemented with 10% FBS at 37 °C in a 5% CO2 incubator. MLH1-deficient cell lines 4T1-Mlh1−/−, HeLa-MLH1−/−, and ER-AsiSI U2OS-MLH1−/− were created by CRISPR-Cas9 technologies using sgRNA sequences targeting mouse Mlh1 (sg1: CAACCAGGGCACCCTGATCA; sg2: CTAATTCAGATCCAAGACAA) or human MLH1 (TGATAGCATTAGCTGGCCGC), as described (Shalem et al., 2014). 4T1-cGas−/−, 4T1-Sting−/−, and 4T1-Exo1−/− were generated using sgRNA sequences targeting mouse cGas (sg: CGCAAAGGGGGGCTCGATCG), Sting (sg: GTGCCCAGGGCGTCTCCTTG), and Exo1 (sg: TAGAGCGAGCGAAGCAGTCT), respectively. 4T1-Mlh1−/−cGas−/−, 4T1-Mlh1−/−Sting−/−, and 4T1-Mlh1−/−Exo1−/− double knock cells were generated by targeting specific sequences based on 4T1-Mlh1−/− cells. Positive clones were confirmed by DNA sequencing and Western blotting using corresponding antibodies. HeLa-MLH1−/−+MLH1 and HCT116+MLH1-rescued cell lines were created by transfecting plasmid pLVX-CMV-hMLH1 and sorted by FACS. The 4T1-Mlh1−/−+Mlh1 cell line was generated by transfecting pCMV6-Entry-mouse tagged Mlh1 and screening with 400 μg/ml G418.
METHOD DETAILS
Preparation of cell extracts, proteins, and Western blotting
Whole cell lysates were prepared using radioimmunoprecipitation assay (RIPA) lysis and extraction buffer, as instructed by the manufacturer (ThermoFisher Scientific, Waltham, MA). Chromatin fractionation was performed, as described previously (Daikoku et al., 2006). Briefly, cells were resuspended and incubated on ice for 20 min in hypotonic buffer (10 mM KCl, 2 mM MgCl2, 20 mM Hepes (pH 7.9), 1 mM DTT, 1 mM EDTA, 0.3% Nonidet P-40, phosphatase inhibitor cocktail). Samples were centrifuged at 14000 rpm for 15 min, and the supernatants were collected as cytosolic extracts. The pellets were washed once with PBS and once with ddH2O, centrifuged at 14000 rpm for 5 min. The pellets were resuspended in 0.2 N HCl and incubated on ice for 10 min, followed by centrifugation at 14000 rpm for 5 min. The supernatants were neutralized with 1.0 M Tris buffer (pH 8.5) and mixed with 6xSDS protein loading buffer. Proteins were separated in SDS-PAGE and transferred onto a nitrocellulose membrane, followed by Western blotting analysis using the indicated antibodies. Human MMR proteins MutLα, MutSα, Exo1, Exo1(FF-AA), Exo1(ΔC244), Exo1(FF-AAΔC244), and RPA were expressed and purified as described previously (Zhang et al., 2005). Protein concentrations were determined using the Bradford reagent (Bio-Rad, Hercules, CA).
cGAMP Extraction and Quantification
cGAMP extraction was essentially performed as described previously (Gui et al., 2019). In brief, WT 4T1 or Mlh1−/− 4T1 cells (3.3 ×106) were treated with or without 20 Gy IR, and harvested 36 hr post IR treatment. After washing with PBS, cells were quickly frozen with liquid nitrogen. Cells were then lysed in 80% (vol/vol) methanol and 2% (vol/vol) acetic acid solution before the addition of an internal standard and an equal volume of 2% acetic acid. Cell lysates were fractionated by centrifugation (10,000 g for 5 min), and pellets were extracted (with 2% acetic acid) and fractionated (by centrifugation) two more times. The supernatants from all three centrifugations were combined and applied to a HyperSep Aminopropyl SPE column for cGAMP purification. The purified cGAMP was then loaded onto a tandem mass spectrometer for quantification.
RNA Extraction and Quantitative Real-Time PCR
Total mRNAs were isolated from 4T1 cells and reversely transcribed to cDNA by using M-MuLV Reverse Transcriptase (NEB). Quantitative Real-Time PCR (qRT-PCR) was performed by using SsoAdv Univer SYBR GRN SMX (Bio-Rad) with specific primers for mouse Isg15 (Primer F: GAGCTAGAGCCTGCAGCAAT, Primer-R: TCACGGACACCAGGAAATCG) and mouse Irf7 (Primer F: TTGGGCAAGACTTGTCAGCA; Primer R: ATACCCATGGCTCCAGCTTC). Mouse Gapdh (Primer F: CAACTGCTTAGCCCCCCTGG; Primer R: GCAGGGTAAGATAAGAAATG) was used for normalization.
In vitro and in vivo excision assays
Unless otherwise mentioned, in vitro mismatch-provoked excision assays were performed in 20-μL reactions containing 5 fmol Exo1 or its derivatives, 400 fmol MutSα, 260 fmol MutLα, 800 fmol RPA, 24 fmol of a nicked single G-T mismatch-containing circular heteroduplex DNA, 10 mM Tris-HCl (pH 7.5), 5 mM MgCl2, 1.5 mM ATP, and 110 mM KCl, as described (Zhang et al., 2005). The amount of each protein used, except for Exo1, whose amount is difficult to detect in cells or extracts, was essentially based on its amount in 50 μg of HeLa nuclear extracts (Dzantiev et al., 2004). The reactions were incubated at 37°C for 10 min and terminated by Proteinase K digestion. Excision products were digested with SspI and separated on a 6% denaturing polyacrylamide gel, followed by Southern hybridization analysis using a 32P-labeled oligonucleotide probe (5′-ATTGTTCTGGATATTACC-3′) near the 3′ SspI site. Reaction products were visualized by autoradiography.
In vitro end resection assays were performed as described previously (Cannavo et al., 2013). The end resection assays were assembled in 20 μL reactions containing 18 ng of 3’-end 32P-labeled linearized pUC19 plasmid DNA (2.7 kb), 1 pmol Exo1 WT or its derivatives, 1 pmol, 2 pmol or 4 pmol MutLα and 4 pmol RPA (if present), 25 mM MOPS (pH 7.0), 2 mM DTT, 60 mM KCl, 1% Tween 20, 2 mM ATP, 5 mM MgCl2, terminating with 5 μl of Proteinase K buffer (2% SDS, 150 mM EDTA, 1 mg/ml proteinase K). After separation through agarose gel electrophoresis, the 32P signal was detected by using a Typhoon phosphor imaging system.
For in vivo end resection assays, WT and MLH1−/− ER-AsiSI U2OS cells were treated with 900 nM 4-OHT for 4 h before harvest. Genomic DNA was carefully prepared, and the resection assay was performed as described (Zhou et al., 2014). Briefly, 1.5 μL of genomic DNA (~10 ng) were mixed with 10 μL of the Taqman qPCR reaction reagent (ThermoFisher), and primers and probes specific for DSB1–335 (primer-F GAATCGGATGTATGCGACTGATC and primer-R TTCCAAAGTTATTCCAACCCGAT, probe 6FAM CACAGCTTGCCCATCCTTGCAAACC-TAMRA), DSB1–1618 (primer-F TGAGGAGGTGACATTAGAACTCAGA and primer-R AGGACTCACTTACACGGCCTTT, probe 6FAM TTGCAAGGCTGCTTCCTTACCATTCAA-TAMRA), and DSB1–3500 (primer-F TCCTAGCCAGATAATAATAGCTATACAAACA and primer-R TGAATAGACAGACAACAG ATAAATGAGACA, probe 6FAM ACCCTGATCAGCCTTTCCATGGGTTAAG-TAMRA). The percentage of resection-generated ssDNA was calculated using the formula ssDNA%=1/(2^(ΔCt-1)+0.5)*100. For each condition, ΔCt means subtracting the Ct value of undigested DNA from the Ct value of digested DNA.
RPA exhaustion assay
The RPA exhaustion assay was performed as described (Ibler et al., 2019) (also see Supplementary Materials, Figure S4A). Briefly, WT and MLH1−/− ER-AsiSI U2OS cultures on glass coverslips were treated with 300 nM 4-OHT to induce DSBs. The cells were then inverted onto Parafilm placed in a sealed chamber and incubated with Taq DNA polymerase, dNTP, and BrdU at 72 °C for 5 min. After being permeabilized with 0.25% TritonX-100 in PBS for 20 min, the slides were incubated with a specific antibody against BrdU (BDB347580, Fisher Scientific) at 4 °C overnight, followed by incubation with a secondary anti-mouse IgG Alexa Fluor 555 for 1 h. The slides were mounted using Prolong Diamond Antifade Mountant with DAPI (P36962, Fisher Scientific), then imaged using a Leica TCS SP8 confocal microscope. Phosphorylated RPA was similarly detected using an antibody against serine residues of the RPA2 subunit (A300–245A, Bethyl laboratories, Montgomery, TX).
Microscopy and immunofluorescence analysis
Cells were cultured on glass coverslips, treated with IR for indicated times, and fixed for 10 min with 4% paraformaldehyde in PBS. After washing with PBS, the cells were permeabilized with 0.25% TritonX-100 in PBS for 20 min and blocked with 1% BSA in PBS. To detect cytosolic DNA, cells were incubated with Pico488 dsDNA quantification reagent (Lumiprobe, Hunt Valley, Maryland). For immunofluorescence analysis, cells were incubated with the indicated primary antibodies (i.e., those against RPA from Millipore, cyclin A from AB clonal, Rad51 from Cell Signaling Technology) and then with a corresponding secondary antibody before mounting with DAPI and imaging via Leica TCS SP8 confocal microscope.
Real-time recruitment and retention dynamics of GFP-tagged Exo1 were performed as previously described (Lu et al, NAR, 2019). Briefly, 0.3×106 WT or MLH1−/− HeLa cells that had been plated in a 35 mm glass-bottomed dish (MatTek) were transiently transfected utilizing 2 μg of GFP-Exo1 plasmid via JetPrime® (Polyplus). Twenty-four hours after transfection, GFP-Exo1 expression was verified, and the cells were placed and maintained in CO2-independent medium (Invitrogen) at 37°C in a heated chamber. The fluorescence signal of GFP-Exo1 was monitored by using a Carl Zeiss Axiovert 200M microscope with a Plan-Apochromat 63X/NA 1.40 oil immersion objective (Carl Zeiss, Inc). A 365-nm pulsed nitrogen laser (Spectra-Physics) was applied to generate DNA double-stranded breaks (DSBs), and time-lapse images were taken via a Carl Zeiss AxioCam HRm camera. Fluorescence intensities of the micro-irradiated area and control area were determined by Carl Zeiss Axiovision software, v4.91, and the intensity of irradiated was normalized to non-irradiated control area.
Metaphase chromosome spreading
Cells were cultured to 70% confluence and treated with or without 2 Gy IR. Two hours after treatment, the cells were incubated with Colcemid (Sigma) for 4 h before dissolving in 75 mM KCl solution. After incubation at 37°C for 15 min, the cells were fixed with fresh fixation buffer (Methanol : Acetic acid 3:1), and chromosome spreading was performed onto the pre-cleaned slide and stained with 5% Giemsa solution. Images were captured by using a Zeiss AxioImager microscope (×100 objective lens). Chromosome aberration was analyzed by using Zeiss ZEN lite software.
QUANTIFICATION AND STATISTICAL ANALYSIS
Western blotting was repeated 3 times independently and blots were quantified using Image J software. For whole cell lysates, the relative amounts of γH2AX, pSTING, pSTAT1, Exo1, and pRPA were normalized with that of tubulin. For chromatin-bound Exo1, the relative Exo1 level was normalized with that of histone H3. Quantitative data were shown as mean ± SEM.
For in vitro end resection assay, three independent experiments were performed and quantified by Image J software. The amount of resection product II generated in each reaction was divided by the total amount of DNA substrates, and a percentage of product II was shown as mean ± SEM. Statistical analysis was performed using GraphPad Prism 8.
To quantify cells with cytosolic DNA, 50–100 cells were used to calculate BrdU and RPA foci in three independent experiments. Quantitative data are shown as mean ± SEM. For Exo1 recruitment, immunofluorescence assaying, and chromosomal aberration experiments, the indicated number of cells were counted and calculated. For real-time PCR results, three independent experiments were performed and the data are shown as means ± SEM. All data were analyzed using GraphPad Prism with one-way ANOVA multiple comparisons, and Tukey’s Multiple Comparison Test was used to compare variations. P values <0.05 were considered significant (*p<0.05, **p<0.01, ***p<0.001, ****p<0.0001).
Supplementary Material
Key Resource Table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Purified mouse anti- human MLH1 Antibody | BD Bioscience | Cat#550838; RRID:AB_2297859 |
| cGAS (D3O8O) Rabbit mAb (Mouse specific) | Cell Signaling | Cat# 31659; RRID:AB_2799008 |
| STING (D2P2F) Rabbit mAb | Cell Signaling | Cat# 13647; RRID:AB_2732796 |
| Phospho-STAT1 (Tyr701) (58D6) Rabbit mAb | Cell Signaling | Cat# 9167; RRID:AB_561284 |
| Phospho-STING (Ser365) (D8F4W) Rabbit mAb | Cell Signaling | Cat# 72971; RRID:AB_2799831 |
| Anti-mouse IgG (H+L), F(ab’)2 Fragment (Alexa Fluor 555 Conjugate) antibody | Cell Signaling | Cat# 4409; RRID:AB_1904022 |
| Anti-rabbit IgG (H+L), F(ab’)2 Fragment (Alexa Fluor® 488 Conjugate) antibody | Cell Signaling | Cat# 4412; RRID:AB_1904025 |
| Anti-rabbit IgG (H+L), F(ab) 2 Fragment (Alexa Fluor 555 Conjugate) antibody | Cell Signaling | Cat# 4413; RRID:AB_10694110 |
| α Tubulin antibody (B-5-1-2) | Santa Cruz | Cat# sc-23948; RRID:AB_628410 |
| Beta Actin mAb (8H10D10) | Novus Biologicals | Cat# NBP1-47423; RRID:AB_10010376 |
| Phospho-RPA32 (S4/8) antibody (A300-245A) | Bethyl laboratories | Cat# A300-245A; RRID:AB_210547 |
| BrdU (5-bromo-2-deoxyuridine) antibody | Millipore Sigma | Cat# MAB3510; RRID:AB_2314031 |
| Anti-Replication Protein A, clone RPA34-20 antibody) | Millipore Sigma | Cat# MABE285; RRID:AB_11205561 |
| Phospho-Histone H2AX (Ser139) mouse mAb | Millipore Sigma | Cat# 05-636-I; RRID:AB_2755003 |
| Goat Anti-Rabbit IgG Antibody, (H+L) HRP conjugate | Millipore Sigma | Cat# AP307P; RRID:AB_92641 |
| Goat Anti-Mouse IgG & IgM Antibody, HRP conjugate | Millipore Sigma | Cat# AP130P; RRID:AB_91266 |
| Anti-human Exo1 Antibody | This paper | Custom-made |
| Phospho-Exo1 (S746) Antibody | (Li et al., 2019) | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| ProLong™ Diamond Antifade Mountant with DAPI | Fisher Scientific | Cat# P36962 |
| Pico488 dsDNA quantification reagent | Lumiprobe | Cat# 12010 |
| Critical Commercial Assays | ||
| Mouse IFN Beta ELISA Kit | PBL Assay Science | Cat# 42410 |
| M-MuLV Reverse Transcriptase | New England Biolabs | Cat# M0253S |
| SsoAdvanced Uni SYBR Grn Supmix | Bio-Rad | Cat# 1725272 |
| Taqman™ Fast Advanced master mix | Thermo Fisher | Cat# 4444557 |
| Bio-Rad protein assay dye reagent concentrate | Bio-Rad | Cat#5000006 |
| jetPRIME™ Transfection reagent | PolyPlus | #114-07 |
| Deposited Data | ||
| Raw and analyzed data | This paper | Mendeley data: Reserved DOI: 10.17632/mrg4443z7m.1 |
| Experimental Models: Cell Lines | ||
| 4T1 | ATCC | Cat# CRL-2539; RRID:CVCL_0125 |
| HeLa | ATCC | Cat# 60005; RRID:CVCL_0030 |
| HCT116 | ATCC | Cat# KCB 200706YJ; RRID:CVCL_0291 |
| ER-AsiSI-U2OS | (Zhou et al., 2014) | N/A |
| Oligonucleotides | ||
| Mouse Isg15 forward: 5’-GAGCTAGAGCCTGCAGCAAT-3’ | This paper | N/A |
| Mouse Isg15 reverse: 5’-TCACGGACACCAGGAAATCG-3’ | This paper | N/A |
| Mouse Irf7 forward: 5’-TTGGGCAAGACTTGTCAGCA-3’ | This paper | N/A |
| Mouse Irf7 reverse: 5’-ATACCCATGGCTCCAGCTTC-3’ | This paper | N/A |
| Mouse Gapdh forward: 5’-CAACTGCTTAGCCCCCCTGG-3’ | This paper | N/A |
| Mouse Gapdh reverse: 5’-GCAGGGTAAGATAAGAAATG-3’ | This paper | N/A |
| DSB1-335 forward: 5’-GAATCGGATGTATGCGACTGATC-3’ | This paper | N/A |
| DSB1-335 reverse: 5’-TTCCAAAGTTATTCCAACCCGAT-3’ | This paper | N/A |
| DSB1-335 probe: 6FAM-CACAGCTTGCCCATCCTTGCAAACC-TAMRA | This paper | N/A |
| DSB1-1618 forward: 5’-TGAGGAGGTGACATTAGAACTCAGA-3’ | This paper | N/A |
| DSB1-1618 reverse: 5’-AGGACTCACTTACACGGCCTTT-3’ | This paper | N/A |
| DSB1-1618 probe: 6FAM-TTGCAAGGCTGCTTCCTTACCATTCAA-TAMRA | This paper | N/A |
| DSB1-3500 forward: 5’-TCCTAGCCAGATAATAATAGCTATACAAACA-3’ | This paper | N/A |
| DSB1-3500 reverse: 5’-TGAATAGACAGACAACAG-3’ | This paper | N/A |
| DSB1-3500 probe: 6FAM-ACCCTGATCAGCCTTTCCATGGGTTAAG-TAMRA | This paper | N/A |
| Recombinant DNA | ||
| pLentiCRISPR v2 | (Sanjana et al., 2014) | Addgene Plasmid Cat#52961 |
| pSpCas9(BB)-2A-GFP (PX458) | (Hmelo et al., 2015) | Addgene Plasmid Cat #48138 |
| pCMV6-Entry-mouse Mlh1 | Origene | Cat#: MR210511 |
| pEGFP-N1-Exo1 | This paper | N/A |
| pLVX-CMV-human MLH1 | This paper | N/A |
| Software and Algorithms | ||
| GraphPad Prism software 8.0 | GraphPad Software | N/A |
| Carl Zeiss Axiovision software v4.91 | Carl Zeiss | N/A |
| Carl Zeiss ZEN lite software | Carl Zeiss | N/A |
| ImageJ software | NIH | N/A |
| the LAS X software | Leica | N/A |
Highlights.
MLH1 deficiency triggers cytosolic DNA release and activates the cGAS-STING pathway
MLH1 regulates Exo1 nuclease activity during DNA end resection
Loss of MLH1 leads to DNA hyper-excision, RPA exhaustion, and chromosomal instability
Acknowledgements
We thank Tanya T. Paull and Gaëlle Legube for providing ER-AsiSI-U2OS cells, Zhongsheng You for providing pExo1 antibody, Jonathan Feinberg and Damiana Chiavolini for editing the manuscript. This work was supported in part by the Cancer Prevention & Research Institute of Texas grants (CPRIT) RR160101 to G.-M.L. and RR150072 and RP180725 to Y.-X.F., and the National Institutes of Health grants CA092584, CA162804, and GM047251 to A.J.D. G.-M.L. and Y.-X.F. are CPRIT Scholars. G.-M.L. is the Reece A. Overcash, Jr. Distinguished Chair for Research on Conlon Cancer. Y.-X.F. is the Mary Nell and Ralph B. Rogers Professorship in Immunology. Z.J.C. is an investigator of the Howard Hughes Medical Institute. J.W.S. holds the Southland Financial Corporation Distinguished Chair in Geriatrics Research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ablasser A, and Chen ZJ (2019). cGAS in action: Expanding roles in immunity and inflammation. Science 363. [DOI] [PubMed] [Google Scholar]
- Albacker LA, Wu J, Smith P, Warmuth M, Stephens PJ, Zhu P, Yu L, and Chmielecki J (2017). Loss of function JAK1 mutations occur at high frequency in cancers with microsatellite instability and are suggestive of immune evasion. PLoS One 12, e0176181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker SM, Plug AW, Prolla TA, Bronner CE, Harris AC, Yao X, Christie DM, Monell C, Arnheim N, Bradley A, et al. (1996). Involvement of mouse Mlh1 in DNA mismatch repair and meiotic crossing over. Nat Genet 13, 336–342. [DOI] [PubMed] [Google Scholar]
- Bakhoum SF, and Landau DA (2017). Chromosomal Instability as a Driver of Tumor Heterogeneity and Evolution. Cold Spring Harb Perspect Med 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat KP, and Cortez D (2018). RPA and RAD51: fork reversal, fork protection, and genome stability. Nature structural & molecular biology 25, 446–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolderson E, Tomimatsu N, Richard DJ, Boucher D, Kumar R, Pandita TK, Burma S, and Khanna KK (2010). Phosphorylation of Exo1 modulates homologous recombination repair of DNA double-strand breaks. Nucleic acids research 38, 1821–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research, N. (2014). Comprehensive molecular characterization of gastric adenocarcinoma. Nature 513, 202–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannavo E, Cejka P, and Kowalczykowski SC (2013). Relationship of DNA degradation by Saccharomyces cerevisiae exonuclease 1 and its stimulation by RPA and Mre11-Rad50-Xrs2 to DNA end resection. Proceedings of the National Academy of Sciences of the United States of America 110, E1661–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cejka P (2015). DNA End Resection: Nucleases Team Up with the Right Partners to Initiate Homologous Recombination. The Journal of biological chemistry 290, 22931–22938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cejka P, Cannavo E, Polaczek P, Masuda-Sasa T, Pokharel S, Campbell JL, and Kowalczykowski SC (2010). DNA end resection by Dna2-Sgs1-RPA and its stimulation by Top3-Rmi1 and Mre11-Rad50-Xrs2. Nature 467, 112–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Lisby M, and Symington LS (2013). RPA coordinates DNA end resection and prevents formation of DNA hairpins. Molecular cell 50, 589–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constantin N, Dzantiev L, Kadyrov FA, and Modrich P (2005). Human mismatch repair: Reconstitution of a nick-directed bidirectional reaction. The Journal of biological chemistry 280, 39752–39761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cristescu R, Mogg R, Ayers M, Albright A, Murphy E, Yearley J, Sher X, Liu XQ, Lu H, Nebozhyn M, et al. (2018). Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science 362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daikoku T, Kudoh A, Sugaya Y, Iwahori S, Shirata N, Isomura H, and Tsurumi T (2006). Postreplicative mismatch repair factors are recruited to Epstein-Barr virus replication compartments. The Journal of biological chemistry 281, 11422–11430. [DOI] [PubMed] [Google Scholar]
- Doerfler L, and Schmidt KH (2014). Exo1 phosphorylation status controls the hydroxyurea sensitivity of cells lacking the Pol32 subunit of DNA polymerases delta and zeta. DNA repair 24, 26–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzantiev L, Constantin N, Genschel J, Iyer RR, Burgers PM, and Modrich P (2004). A defined human system that supports bidirectional mismatch-provoked excision. Molecular cell 15, 31–41. [DOI] [PubMed] [Google Scholar]
- El-Shemerly M, Janscak P, Hess D, Jiricny J, and Ferrari S (2005). Degradation of human exonuclease 1b upon DNA synthesis inhibition. Cancer Res 65, 3604–3609. [DOI] [PubMed] [Google Scholar]
- Gerlinger M, and Swanton C (2010). How Darwinian models inform therapeutic failure initiated by clonal heterogeneity in cancer medicine. Br J Cancer 103, 1139–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gui X, Yang H, Li T, Tan X, Shi P, Li M, Du F, and Chen ZJ (2019). Autophagy induction via STING trafficking is a primordial function of the cGAS pathway. Nature 567, 262–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding SM, Benci JL, Irianto J, Discher DE, Minn AJ, and Greenberg RA (2017). Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 548, 466–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hmelo LR, Borlee BR, Almblad H, Love ME, Randall TE, Tseng BS, Lin C, Irie Y, Storek KM, Yang JJ, et al. (2015). Precision-engineering the Pseudomonas aeruginosa genome with two-step allelic exchange. Nat Protoc 10, 1820–1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong Z, Jiang J, Hashiguchi K, Hoshi M, Lan L, and Yasui A (2008). Recruitment of mismatch repair proteins to the site of DNA damage in human cells. Journal of cell science 121, 3146–3154. [DOI] [PubMed] [Google Scholar]
- Iacovoni JS, Caron P, Lassadi I, Nicolas E, Massip L, Trouche D, and Legube G (2010). High-resolution profiling of gammaH2AX around DNA double strand breaks in the mammalian genome. The EMBO journal 29, 1446–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibler AEM, ElGhazaly M, Naylor KL, Bulgakova NA, S, F.E.-K., and Humphreys, D. (2019). Typhoid toxin exhausts the RPA response to DNA replication stress driving senescence and Salmonella infection. Nat Commun 10, 4040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jardim MJ, Wang Q, Furumai R, Wakeman T, Goodman BK, and Wang XF (2009). Reduced ATR or Chk1 expression leads to chromosome instability and chemosensitization of mismatch repair-deficient colorectal cancer cells. Molecular biology of the cell 20, 3801–3809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim ST, Cristescu R, Bass AJ, Kim KM, Odegaard JI, Kim K, Liu XQ, Sher X, Jung H, Lee M, et al. (2018). Comprehensive molecular characterization of clinical responses to PD-1 inhibition in metastatic gastric cancer. Nat Med 24, 1449–1458. [DOI] [PubMed] [Google Scholar]
- Kolodner RD, and Marsischky GT (1999). Eukaryotic DNA mismatch repair. Current opinion in genetics & development 9, 89–96. [DOI] [PubMed] [Google Scholar]
- Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, Lu S, Kemberling H, Wilt C, Luber BS, et al. (2017). Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 357, 409–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D, et al. (2015). PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N Engl J Med 372, 2509–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li GM (2008). Mechanisms and functions of DNA mismatch repair. Cell research 18, 85–98. [DOI] [PubMed] [Google Scholar]
- Li S, Lavagnino Z, Lemacon D, Kong L, Ustione A, Ng X, Zhang Y, Wang Y, Zheng B, Piwnica-Worms H, et al. (2019). Ca(2+)-Stimulated AMPK-Dependent Phosphorylation of Exo1 Protects Stressed Replication Forks from Aberrant Resection. Mol Cell 74, 1123–1137 e1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, and Chen ZJ (2018). The cGAS-cGAMP-STING pathway connects DNA damage to inflammation, senescence, and cancer. J Exp Med 215, 1287–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, and Huang J (2016). DNA End Resection: Facts and Mechanisms. Genomics Proteomics Bioinformatics 14, 126–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C, Guan J, Lu S, Jin Q, Rousseau B, Lu T, Stephens D, Zhang H, Zhu J, Yang M, et al. (2021). DNA sensing in mismatch repair-deficient tumor cells is essential for anti-tumor immunity. Cancer cell in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H, Saha J, Beckmann PJ, Hendrickson EA, and Davis AJ (2019). DNA-PKcs promotes chromatin decondensation to facilitate initiation of the DNA damage response. Nucleic acids research 47, 9467–9479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie KJ, Carroll P, Martin CA, Murina O, Fluteau A, Simpson DJ, Olova N, Sutcliffe H, Rainger JK, Leitch A, et al. (2017). cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 548, 461–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao G, Lee S, Ortega J, Gu L, and Li GM (2012). Modulation of microRNA processing by mismatch repair protein MutLalpha. Cell research 22, 973–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marechal A, and Zou L (2015). RPA-coated single-stranded DNA as a platform for post-translational modifications in the DNA damage response. Cell research 25, 9–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modrich P, and Lahue R (1996). Mismatch repair in replication fidelity, genetic recombination, and cancer biology. Annual review of biochemistry 65, 101–133. [DOI] [PubMed] [Google Scholar]
- Morafraile EC, Bugallo A, Carreira R, Fernandez M, Martin-Castellanos C, Blanco MG, and Segurado M (2020). Exo1 phosphorylation inhibits exonuclease activity and prevents fork collapse in rad53 mutants independently of the 14–3-3 proteins. Nucleic acids research 48, 3053–3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu H, Raynard S, and Sung P (2009). Multiplicity of DNA end resection machineries in chromosome break repair. Genes & development 23, 1481–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochs F, Somyajit K, Altmeyer M, Rask MB, Lukas J, and Lukas C (2016). 53BP1 fosters fidelity of homology-directed DNA repair. Nature structural & molecular biology 23, 714–721. [DOI] [PubMed] [Google Scholar]
- Raynaud F, Mina M, Tavernari D, and Ciriello G (2018). Pan-cancer inference of intra-tumor heterogeneity reveals associations with different forms of genomic instability. PLoS genetics 14, e1007669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reijnen C, Kusters-Vandevelde HVN, Prinsen CF, Massuger L, Snijders M, Kommoss S, Brucker SY, Kwon JS, McAlpine JN, and Pijnenborg JMA (2019). Mismatch repair deficiency as a predictive marker for response to adjuvant radiotherapy in endometrial cancer. Gynecologic oncology 154, 124–130. [DOI] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, and Zhang F (2014). Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods 11, 783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmutte C, Marinescu RC, Sadoff MM, Guerrette S, Overhauser J, and Fishel R (1998). Human exonuclease I interacts with the mismatch repair protein hMSH2. Cancer Res 58, 4537–4542. [PubMed] [Google Scholar]
- Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelsen TS, Heckl D, Ebert BL, Root DE, Doench JG, et al. (2014). Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343, 84–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen YJ, Le Bert N, Chitre AA, Koo CX, Nga XH, Ho SS, Khatoo M, Tan NY, Ishii KJ, and Gasser S (2015). Genome-derived cytosolic DNA mediates type I interferon-dependent rejection of B cell lymphoma cells. Cell Rep 11, 460–473. [DOI] [PubMed] [Google Scholar]
- Shin DS, Zaretsky JM, Escuin-Ordinas H, Garcia-Diaz A, Hu-Lieskovan S, Kalbasi A, Grasso CS, Hugo W, Sandoval S, Torrejon DY, et al. (2017). Primary Resistance to PD-1 Blockade Mediated by JAK1/2 Mutations. Cancer discovery 7, 188–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tishkoff DX, Amin NS, Viars CS, Arden KC, and Kolodner RD (1998). Identification of a human gene encoding a homologue of Saccharomyces cerevisiae EXO1, an exonuclease implicated in mismatch repair and recombination. Cancer Res 58, 5027–5031. [PubMed] [Google Scholar]
- Tomimatsu N, Mukherjee B, Catherine Hardebeck M, Ilcheva M, Vanessa Camacho C, Louise Harris J, Porteus M, Llorente B, Khanna KK, and Burma S (2014). Phosphorylation of EXO1 by CDKs 1 and 2 regulates DNA end resection and repair pathway choice. Nat Commun 5, 3561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran PT, Erdeniz N, Symington LS, and Liskay RM (2004). EXO1-A multi-tasking eukaryotic nuclease. DNA repair 3, 1549–1559. [DOI] [PubMed] [Google Scholar]
- Tran PT, Simon JA, and Liskay RM (2001). Interactions of Exo1p with components of MutLalpha in Saccharomyces cerevisiae. Proceedings of the National Academy of Sciences of the United States of America 98, 9760–9765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsubouchi H, and Ogawa H (2000). Exo1 roles for repair of DNA double-strand breaks and meiotic crossing over in Saccharomyces cerevisiae. Molecular biology of the cell 11, 2221–2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Loga K, Woolston A, Punta M, Barber LJ, Griffiths B, Semiannikova M, Spain G, Challoner B, Fenwick K, Simon R, et al. (2020). Extreme intratumour heterogeneity and driver evolution in mismatch repair deficient gastro-oesophageal cancer. Nat Commun 11, 139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Hu S, Chen X, Shi H, Chen C, Sun L, and Chen ZJ (2017). cGAS is essential for the antitumor effect of immune checkpoint blockade. Proceedings of the National Academy of Sciences of the United States of America 114, 1637–1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Yuan F, Presnell SR, Tian K, Gao Y, Tomkinson AE, Gu L, and Li GM (2005). Reconstitution of 5’-directed human mismatch repair in a purified system. Cell 122, 693–705. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Caron P, Legube G, and Paull TT (2014). Quantitation of DNA double-strand break resection intermediates in human cells. Nucleic Acids Res 42, e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Source data for figures in the paper is available [Mendeley Data Reserved DOI: 10.17632/mrg4443z7m.1].