

Summary
Invasion of hepatocytes by sporozoites is essential for Plasmodium to initiate infection of the mammalian host. The parasite’s subsequent intracellular differentiation in the liver is the first developmental step of its mammalian cycle. Despite their biological significance, surprisingly little is known of the signalling pathways required for sporozoite invasion. We report that sporozoite invasion of hepatocytes requires signalling through two second-messengers – cGMP mediated by the parasite’s cGMP-dependent protein kinase (PKG), and Ca2+, mediated by the parasite’s calcium-dependent protein kinase 4 (CDPK4). Sporozoites expressing a mutated form of Plasmodium berghei PKG or carrying a deletion of the CDPK4 gene are defective in invasion of hepatocytes. Using specific and potent inhibitors of Plasmodium PKG and CDPK4, we demonstrate that PKG and CDPK4 are required for sporozoite motility, and that PKG regulates the secretion of TRAP, an adhesin that is essential for motility. Chemical inhibition of PKG decreases parasite egress from hepatocytes by inhibiting either the formation or release of merosomes. In contrast, genetic inhibition of CDPK4 does not significantly decrease the number of merosomes. By revealing the requirement for PKG and CDPK4 in Plasmodium sporozoite invasion, our work enables a better understanding of kinase pathways that act in different Plasmodium stages.
Introduction
Malaria infection begins with the infection by Plasmodium ‘sporozoites’ of the liver. After asexual replication in the liver to form ‘liver stages’, the parasites invade erythrocytes and replicate to produce merozoites and gametocytes. Merozoites initiate repeated rounds of erythrocytic invasion and asexual development that cause disease. Gametocytes initiate the sexual cycle in the mosquito that is essential for continued parasite transmission. Sexual development in the mosquito leads to stages known as ookinetes. Ookinetes undergo differentiation in the mosquito midgut to form sporozoites. Sporozoites are carried from the mosquito midgut to the salivary glands, from where they can commence another round of parasite transmission to the mammalian host.
The first obligate step of malaria in the mammalian cycle is the infection of hepatocytes by Plasmodium sporozoites. The increase in parasite numbers at this stage is essential for the parasite to establish a niche in the mammalian host (Graewe et al., 2012). Decreasing sporozoite infection of the liver reduces the incidence and severity of disease (Alonso et al., 2005). A mechanistic understanding of sporozoite invasion could reveal pathways that may be targeted for preventing malaria.
In addition to its clinical relevance, invasion by Plasmodium sporozoites is biologically significant because it displays a unique combination of features (Meissner et al., 2013). Sporozoites are motile over a large distance as they leave the site of inoculation in the skin to reach the liver. During this process, they migrate through several cells, breaching the host cell plasma membrane in the process. Once an appropriate host hepatocyte is encountered, they switch their mode of cell entry to one accompanied by the formation of a vacuole that serves as the site for further development (Coppi et al., 2007; Risco-Castillo et al., 2015). While Plasmodium ookinetes, like sporozoites, are motile and migrate through the mosquito midgut epithelium, their invasion does not involve the formation of a parasitophorous vacuole. Plasmodium merozoites, like sporozoites, invade forming a parasitophorous vacuole but they are not motile and do not migrate through cells. Therefore, Plasmodium sporozoites are an excellent model for studying the complexity of Apicomplexan invasion.
Sporozoite invasion of hepatocytes is triggered through a cascade of signalling events initiated by interaction between the circumsporozoite (CS) protein on the sporozoite surface and the highly negatively charged heparan sulfate proteoglycans on the hepatocyte surface (Coppi et al., 2007). These signalling events regulate diverse processes in the sporozoite, such as protein secretion from specialized organelles, Ca2+ mediated signalling and processing of surface adhesins (Ejigiri and Sinnis, 2009). How these diverse pathways are regulated in sporozoites is unknown. Evidence from other life cycle stages of Plasmodium falciparum and Plasmodium berghei has shown that the parasite’s cyclic GMP dependent protein kinase (PKG) plays an essential role as an upstream regulator of Ca2+ signals during both the mammalian and the mosquito cycle (Brochet et al., 2014). During the mammalian cycle, PKG is required for merosome formation and/or release in the liver (Falae et al., 2010), erythrocytic stage schizogony, merozoite invasion and egress (Taylor et al., 2010; Collins et al., 2013). During the mosquito cycle, PKG is required for gametogenesis and ookinete motility (McRobert et al., 2008; Moon et al., 2009). Here we study the role of PKG in P. berghei sporozoite biology.
We show that P. berghei PKG (PbPKG) is a key regulator of sporozoite motility that is a pre-requisite for sporozoite invasion of hepatocytes. In addition, P. berghei calcium dependent protein kinase 4 (PbCDPK4) contributes to sporozoite motility and invasion. Indirectly our data implicate cGMP and Ca2+ as important second messengers for regulating sporozoite invasion. In addition, we demonstrate that PbPKG but not PbCDPK4 is required for the formation and/or release of merosomes that allow parasites to exit the infected hepatocyte. Our results have implications for understanding the network of kinase interactions at different parasite stages and for therapies aimed at multiple parasite stages.
Results
Hepatocyte invasion by sporozoites requires PbPKG
To determine if PKG is expressed in sporozoites, we examined expression of HA-tagged PbPKG under the control of its endogenous promoter using immunofluorescence assays (IFA) (Fig. 1A, Supporting Information Fig. 1A [8]). We find PbPKG distributed throughout the cytoplasm of sporozoites and in liver stages, suggesting a functional role for PbPKG in these stages. A function for PKG in these stages is further supported by the ability of a selective inhibitor of Apicomplexan PKG, a trisubstituted pyrolle (TSP) known as Compound 1 (Gurnett et al., 2002) to potently block sporozoite infection in vitro and in vivo (Panchal and Bhanot, 2010).
Fig. 1.
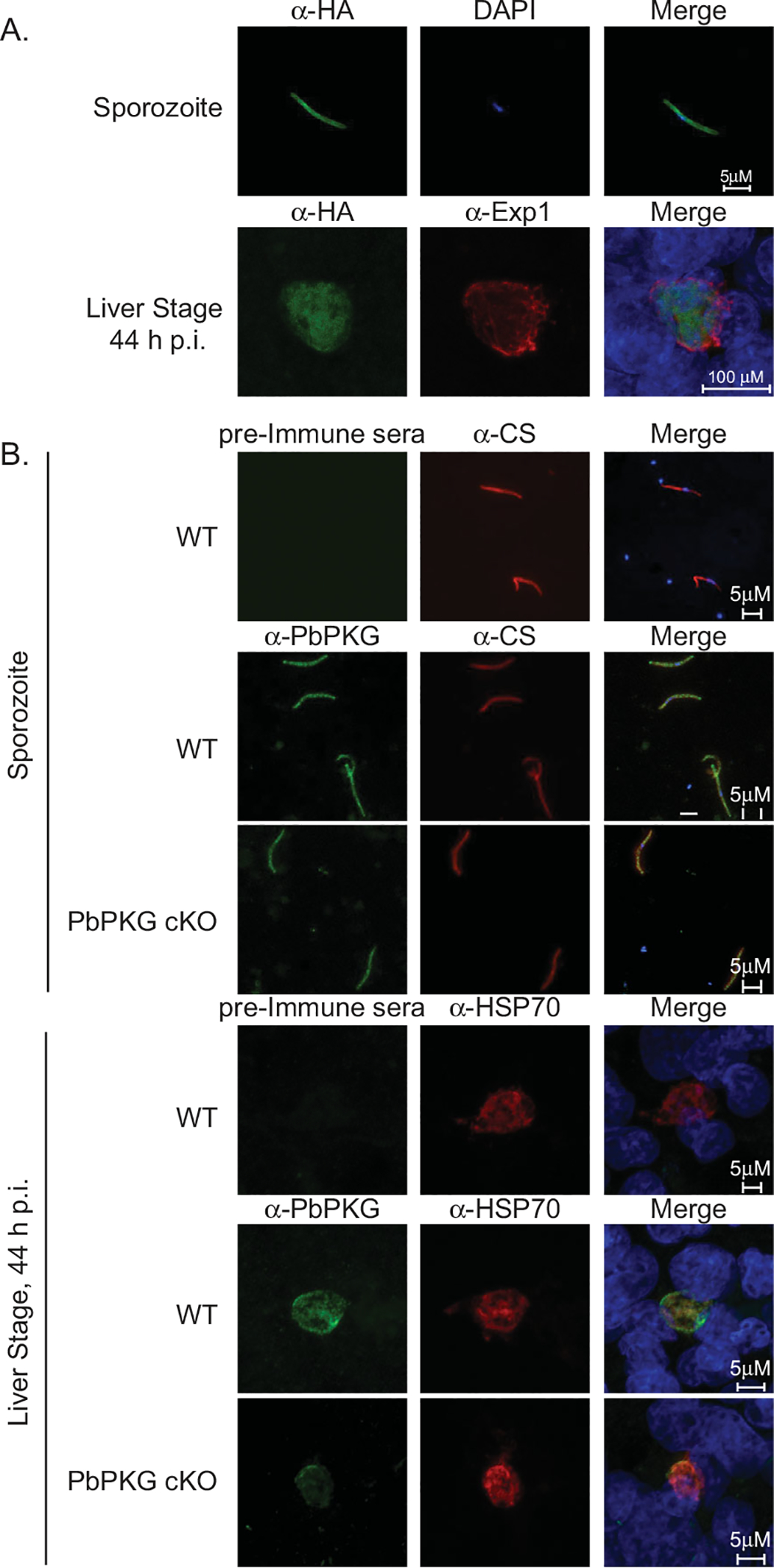
PbPKG is expressed in pre-erythrocytic stages.
A. HA-tagged PbPKG (PKG-HA) was localized in sporozoites and liver stages using immunostaining with an anti-HA antibody. All sporozoites and liver stages express HA-tagged PKG in the cytoplasm. The anti-Exp1 antibody recognizes Exp1, a resident protein of the parasitophorous vacuole membrane in liver stage parasites. Merged panels include DAPI for nuclear localization.
B. PbPKG cKO sporozoites contain PbPKG protein. Polyclonal antisera against PbPKG (amino acids 988–1001) were used to localize PbPKG in sporozoites and liver stages from wildtype and PbPKG cKO parasites. Sporozoites were co-stained with an antibody against the circumsporozoite protein (CS). Liver stages were co-stained with an antibody against Heat Shock Protein 70 (HSP70). Merged images include DAPI as a nuclear marker. PKG protein is readily detected in PbPKG cKO sporozoites but is decreased significantly in PbPKG cKO liver stages.
TSP’s efficacy against sporozoites would suggest that its primary target, PKG is required for sporozoite infection of hepatocytes (Donald et al., 2002; McRobert et al., 2008). However, this is at odds with our previous genetic data demonstrating that salivary gland sporozoites generated using excision of the PbPKG open reading frame in developing midgut sporozoites (PbPKG cKO) did not display a significant decrease in sporozoite infection (Falae et al., 2010). The failure of stage-specific PbPKG gene excision to reveal a phenotype in sporozoite infectivity could be explained by the carryover of PbPKG protein from oocysts, which contain an intact PbPKG locus, into sporozoites that develop from them. Carryover of PbPKG protein is highly likely since stable isotope labelling of ookinete cultures has demonstrated that 87% of PbPKG protein in ookinetes is inherited from the preceding gamete stages, suggesting that the protein can turnover very slowly (Sebastian et al., 2012).
Using antisera raised against a carboxy terminal peptide of PbPKG, we readily detected PKG protein in PbPKG cKO sporozoites by IFA (Fig. 1B). In contrast, PKG protein expression in PbPKG cKO liver stages was significantly reduced (Fig. 1B). These results support our hypothesis that cKO sporozoites retain sufficient PbPKG protein. They suggest that PKG function in sporozoites is best examined using the available fast-acting and specific chemical inhibitors. Indeed this approach was useful for functional ablation of PKG orthologs in Toxoplasma gondii (Donald et al., 2002) and in Plasmodium schizonts and sexual stages (McRobert et al., 2008; Taylor et al., 2010; Brochet et al., 2014).
To rule out off-target effects of TSP on hepatocyte invasion, we generated a transgenic P. berghei line expressing a 3xHA-tagged, TSP-resistant allele of PbPKG using a strategy validated previously (Brochet et al., 2014) but with a GFP-expressing reference line to facilitate monitoring of parasite movement and development (Supporting Information Fig. 1A–C). The modified PbPKG allele carries a substitution of the 'gatekeeper' residue, Thr619 T619Q-HA) that prevents TSP from accessing its binding pocket. Therefore, PKG T619Q is resistant to TSP while maintaining normal catalytic efficiency (Donald et al., 2002). As a control, we used the line expressing 3xHA-tagged wildtype, inhibitor-sensitive PbPKG (PKG-HA) that was integrated in an identical manner to the PKG T619Q-HA allele (Supporting Information Fig. 1A–C). Asexual stage parasites expressing PKG T619Q-HA contain normal amounts of PbPKG protein (Supporting Information Fig. 1D), and undergo grossly normal asexual and sexual development (Supporting Information Fig. 1E, Brochet et al., 2014). However, calcium mobilization in gametocytes and ookinetes expressing PKG T619Q-HA, as well as motility in ookinetes expressing PKG T619Q-HA, are significantly less sensitive to inhibition by TSP (Brochet et al., 2014).
To determine if PbPKG is essential for sporozoite infection of hepatocytes, we tested the sensitivity of PKG T619Q-HA sporozoites to TSP. Sporozoites were allowed to infect HepG2 cells in the presence of TSP and compound exposure was maintained for 14 h post infection (p.i.) before the number of infected cells was quantified at 44 h p.i. Infection of HepG2 cells by PKG T619Q-HA sporozoites was about 19-fold less sensitive to TSP compared to infection by PKG-HA sporozoites (Fig. 2A, Supporting Information Table S1A). TSP has an IC50 of about 2.12 μM against PKG T619Q-HA sporozoites and about 0.11 μM against PKG-HA sporozoites. The refractoriness of PKG T619Q-HA sporozoites to TSP demonstrates clearly that TSP inhibits sporozoite infection by acting on PKG and that sporozoite infection of hepatocytes requires PKG.
Fig. 2.
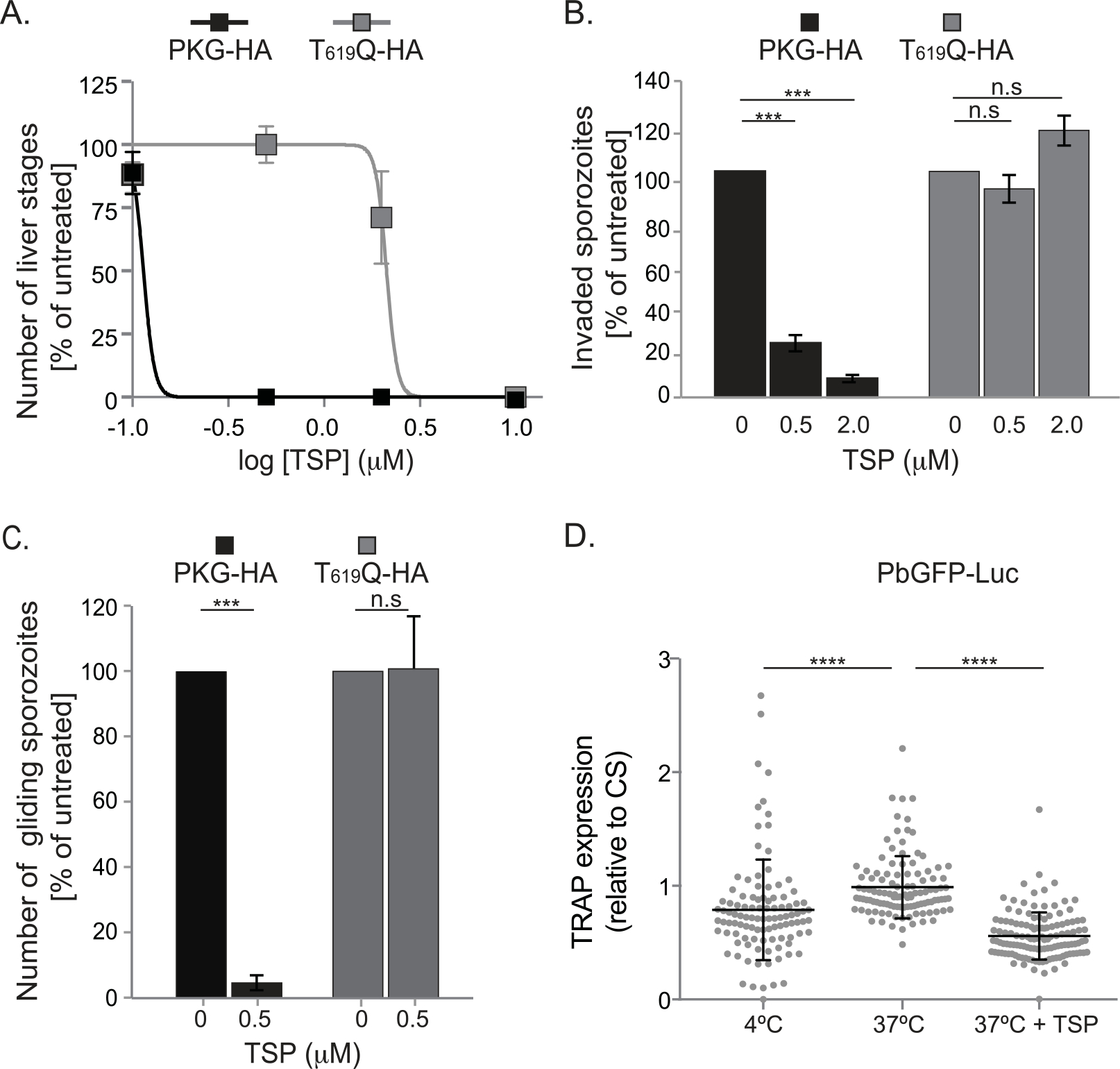
PbPKG is required for sporozoite motility, invasion and merosome formation.
A. Inhibition of PKG activity blocks sporozoite infectivity. HepG2 cells were infected with PKG-HA or PKG T619Q-HA sporozoites in the presence of TSP and compound exposure was maintained for 0–14 h p.i. Liver stages were quantified at 44 h p.i. IC50 values were calculated using curve-fitting software from Graphpad Prism. Shown are results from a representative experiment (mean of 4 replicates ± standard deviation). The experiment was repeated twice.
B. Inhibition of PKG activity blocks sporozoite invasion. PKG-HA or PKG T619Q-HA sporozoites were added to HepG2 cells for 2 h in the presence of TSP. Invasion was determined by quantifying the percentage of sporozoites that became intracellular within 2 h. Shown are results from two independent experiments (mean ± standard deviation) each performed with 3–4 replicates.
C. PKG is required for sporozoite motility. Motility of PKG-HA or PKG T619Q-HA sporozoites was examined in the presence of TSP using live imaging. Movement patterns of individual sporozoites were assigned manually. The percentage (± standard error, n = 2 experiments) of gliding sporozoites was determined. Data were analyzed using chi-square tests, ***P < 0.005. For both PKG-HA and PKG T619Q-HA sporozoites, 100% is the proportion of sporozoites that glide in the absence of TSP.
D. Inhibition of PKG activity decreases expression of TRAP on the sporozoite surface. The ratio of TRAP and CS expression on the surface of PbGFP-Luc sporozoites, incubated at 37°C in the presence or absence of TSP (2 μM), was determined. Sporozoites incubated at 4°C served as controls. Shown are results from a representative experiment (mean ± standard deviation, n = 104–127). Data were analyzed using a log transformed unpaired t-test, **** P value < 0.0001. The experiment was repeated twice.
We previously observed that TSP did not significantly decrease the number of liver stages when added 3 h p.i. (Panchal and Bhanot, 2010), suggesting that PKG’s critical functions are during early steps of sporozoite infection. To further investigate the steps at which PKG is important, PKG-HA and PKG T619Q-HA sporozoites were pre-treated with TSP for 30 min prior to infection. Upon subsequent addition to HepG2 cells, compound was diluted to levels ineffective when tested alone. At 2 h p.i., existing media was replaced with compound-free media. The exposure of sporozoites to TSP prior to addition to HepG2 cells was sufficient to significantly decrease the number of liver stages formed by PKG-HA but not PKG T619Q-HA sporozoites at 44 h p.i. (Supporting Information Table S1A). These results raised the possibility that PbPKG’s major role in sporozoite infection is around the point of host-cell invasion rather than subsequent trophic growth. Previous work has demonstrated that P. falciparum PKG functions in merozoite invasion (Alam et al., 2015) and its homolog in T. gondii, is required for tachyzoite invasion (Wiersma et al., 2004). To investigate PbPKG’s role in sporozoite invasion, we compared TSP’s effect on the fraction of PKG-HA and PKG T619Q-HA sporozoites that become intracellular within 2 h post-addition to cells. TSP significantly inhibited entry by PKG-HA, but not PKG T619Q-HA sporozoites (Fig. 2B, Supporting Information Table S1A). The refractoriness of invasion by PKG T619Q-HA sporozoites to TSP demonstrates that PKG is required for sporozoite invasion.
Sporozoite motility requires PKG
Sporozoite invasion consists of three loosely defined steps – attachment to the substrate, motility to reach the target cells and entry into the host cell (Meissner et al., 2013). PKG is required for secretion of micronemal adhesins and motility in T. gondii tachyzoites (Wiersma et al., 2004, Brown et al., 2016) and for motility in Plasmodium ookinetes (Moon et al., 2009). By analogy to its roles in these zoites, we hypothesized that PKG’s role around the time of sporozoite invasion reflects its function in sporozoite motility.
We tested PKG’s role in sporozoite motility by filming PKG-HA and PKG T619Q-HA sporozoites for 120 sec at 1Hz. Sporozoite movement patterns in vitro were categorized as previously described (Hegge et al., 2009) – (i) ‘gliding’ which describes sporozoites moving in circular tracks for the entire observation period of 120 sec (ii) ‘adherent’ which describes sporozoites adhering to the substrate with minor displacement (iii) ‘waving’ which describes sporozoites attached to the substrate at one end with the other end moving freely in the media (iv) ‘complex’ which describes sporozoites that display a combination of these simple patterns, for example those that glide for part of the time, detach and move out of the field of observation. Sporozoites that attached weakly to the substrate moving in the direction of media flow were categorized as ‘drifting’.
Motility of PKG-HA, but not PKG T619Q-HA, sporozoites was highly sensitive to TSP. Treatment with 0.5 μM TSP significantly decreased the percentage of PKG-HA sporozoites that glide (Fig. 2C, Supporting Information Fig. 2, Table S1B) and the number of circles executed during the observation period by the gliding sporozoites (from an average of 15.7 ± 1.7 circles/sporozoite to 12.7 ± 1.5 circles/sporozoite in 180 s). The number of circles made by sporozoites in unit time is an accurate proxy for their speed of movement (Hegge et al., 2010). In addition, in the absence of TSP, a smaller percentage (± standard error, n = 2 experiments) of PKG T619Q-HA sporozoites glide compared to PKG-HA sporozoites: 6.45 ± 0.99% versus 19.1 ± 1.65%, respectively, (Supporting Information Table S1B).
To rule out the possibility that HA-tagged PKG may have subtle functional differences from untagged PKG, we also examined motility in PbGFP-Luc sporozoites (Franke-Fayard et al., 2005), which contain an unmodified PKG locus. We found that PbGFP-Luc sporozoites were also robustly inhibited by TSP, although they were less sensitive to the lower doses of TSP compared to PKG-HA sporozoites (Supporting Information Table S1B). This difference may be due to different genetic backgrounds, or could reflect subtle effects of the HA-tag and generic 3' UTR on PKG-HA, leading to the sensitivation of PKG-HA to TSP. At 2 μM, TSP reduced not only the percentage of gliding sporozoites but also the number of circles they make – 13.7 ± 4.7 circles/sporozoite for vehicle-treated to 7.0 ± 1.7 for 2.0 μM-treated sporozoites (Supporting Information Table S1B).
We noted that in addition to decreased motility, TSP-treated PbGFP-Luc sporozoites were unable to attach strongly to the substrate and showed a linear displacement of 25–50 μm in the direction of medium flow (Supporting Information Table S1B, Fig. 2). These sporozoites were characterized as ‘drifting’. Weak initial attachment to the substrate would severely impair motility. Therefore, PKG could also be required for subsequent cycles of attachment-detachment from the substrate that occur during translocation or for generating the force needed for movement (Munter et al., 2009; Hegge et al., 2010).
Together, these data demonstrate PKG’s key role in sporozoite motility and suggest that inhibition of PKG may also decrease sporozoite adhesion to the surface. Since motility is required for invasion, PKG most likely regulates sporozoite invasion by controlling motility.
PKG regulates secretion of micronemal proteins
Sporozoite motility requires exocytosis of micronemal proteins. Previous studies in T. gondii tachyzoites and P. falciparum merozoites have demonstrated that PKG is required for the secretion of micronemal proteins and associated processes (Wiersma et al., 2004; Collins et al., 2013; Brown et al., 2016). We hypothesized that PbPKG could similarly regulate sporozoite motility by stimulating micronemal secretion. To examine PbPKG’s role in regulating micronemal exocytosis in sporozoites, we quantified the expression of TRAP, a canonical micronemal protein whose secretion onto the sporozoite surface and subsequent cleavage is essential for motility (Ejigiri et al., 2012; Takala-Harrison et al., 2015).
The surface expression of TRAP in non-permeabilized PbGFP-Luc sporozoites was quantified using immunofluorescence intensities (Fig. 2D). Expression of the CS protein, a constitutively expressed membrane protein of sporozoites, was used as an internal reference control. As previously reported (Gantt et al., 2000; Silvie et al., 2004), incubation of sporozoites at 37°C increased TRAP expression on the surface (p value < 0.0001, unpaired t-test of log transformed ratios). The increase in TRAP expression was blocked by TSP (P value < 0.0001, unpaired t-test of log transformed ratios) (Fig. 2D). The inhibition of TRAP surface expression by TSP demonstrates that PKG regulates micronemal exocytosis in sporozoites.
Merosome formation and/or release requires PKG
We previously showed that PKG cKO sporozoites do not form merosomes, suggesting that PKG is required for parasite egress from hepatocytes (Falae et al., 2010). These results were confirmed by testing TSP’s effect on merosome formation by PKG-HA or PKG T619Q-HA parasites. Addition of TSP to HepG2 cells infected with PKG-HA sporozoites decreased the number of merosomes found in the media at 65 h p.i., in a dose-dependent manner (Supporting Information Table S1A). In contrast, merosome formation and/or release by PKG T619Q-HA sporozoites was less sensitive to TSP treatment. Therefore, genetic and chemical inhibition confirm PKG’s essential role in merosome formation and/or release.
The T619Q mutation has subtle effects in the absence of inhibitor
Despite undergoing normal intraerythrocytic development (Supporting Information Fig. 1E, Brochet et al., 2014) and sexual development in vitro (Brochet et al., 2014), PKG T619Q-HA parasites consistently produced only about half as many liver stages as the isogenic PKG-HA control clone (Supporting Information Fig. 1F, Table S1C). This loss of infectivity occurs at the point of invasion since we observed that PKG T619Q-HA parasites displayed an approximately two-fold decrease in the fraction of sporozoites that were intracellular 2 h after addition to HepG2 cells (Supporting Information Fig. 1F, Table S1C). Following invasion, the T619Q mutation did not further impact the number of intracellular liver stages that develop at 24 h p.i. and 48 h p.i. (Supporting Information Fig. 1F, Table S1C). These differences could be a result of unknown genetic differences amongst the nominally isogenic PKG T619Q-HA and PKG-HA clones, although we find it more likely that PKG T619Q-HA is a hypomorphic allele in sporozoites.
We attempted to compare PKG protein levels amongst PKG T619Q-HA and PKG-HA sporozoites by Western blotting but were unable to collect sufficient material. As an alternative, we quantified anti-HA immunofluorescence intensities with anti-GFP as an internal reference control in IFA. The average ratio of anti-HA to anti-GFP fluorescence intensity in PKG T619Q-HA sporozoites was 0.17 ± 0.04 (n = 78 sporozoites) and in PKG-HA sporozoites was 0.24 ± 0.04 (n = 70 sporozoites). The difference in the sporozoite populations was statistically significant (P value < 0.0001, unpaired t-test of log transformed ratios, Supporting Information Fig. 1G), and similar results were obtained in three independent experiments. In contrast, levels of PKG protein in erythrocytic stage parasites from the two lines were very similar (Supporting Information Fig. 1D), consistent with the normal erythrocytic cycle of the PKG T619Q-HA parasites (Supporting Information Fig. 1E). We conclude that decreased PKG expression in PKG T619Q-HA sporozoites is a possible cause of their reduced infectivity. In addition, PKG T619Q-HA enzyme could have reduced activity in vivo as seen in ‘gatekeeper’ mutants of some kinases (Zhang et al., 2005). Since PKG T619Q-HA sporozoites have significantly decreased infectivity whereas PbPKG cKO sporozoites do not (Falae et al., 2010), we hypothesize that PKG enzymatic activity is closer to wildtype levels in PbPKG cKO sporozoites because of the presence of a significant amount of PKG protein in the cKO sporozoites. (Fig. 1B)
PbCDPK4 plays a crucial role in sporozoite invasion of hepatocytes
PKG-dependent pathways include phosphoinositide metabolism, protein secretion, vesicular trafficking, proteolysis, gene regulation and cellular signalling (Brochet et al., 2014; Alam et al., 2015). Many of PKG’s pleiotropic roles in different parasite stages are likely explained by its regulation of critical Ca2+ signals that control merozoite invasion and egress, gametocyte activation and ookinete motility (Brochet et al., 2014; Alam et al., 2015). In a model developed in P. berghei ookinetes, gametocytes and P. falciparum schizonts, PKG-dependent phosphatidylinositol (4,5)-biphosphate production releases internal Ca2+ in the parasite (Brochet et al., 2014). The resulting Ca2+ flux is transduced by Ca2+ effectors, including a family of calcium dependent protein kinases (CDPK). Different CDPKs act downstream of PKG at various steps of the parasite life-cycle – CDPK1 during merozoite invasion (Alam et al., 2015), CDPK5 during merozoite egress (Dvorin et al., 2010), CDPK3 during ookinete but not sporozoite motility (Siden-Kiamos et al., 2006) and CDPK4 in microgametogenesis (Billker et al., 2004). The specific CDPK that acts downstream of PKG in sporozoite and liver stages has not been identified, although efficient invasion of hepatocytes requires CDPK6 (Coppi et al., 2007).
Elevated Ca2+ levels in gliding sporozoites suggest that sporozoite motility requires Ca2+ signalling pathways (Carey et al., 2014). However, the identity of these pathways is as yet unknown. Since CDPKs are major mediators of Ca2+ signalling in Plasmodium and distinct CDPKs act downstream of PKG throughout the parasite life-cycle (Billker et al., 2004; Siden-Kiamos et al., 2006; Dvorin et al., 2010; Sebastian et al., 2012), we hypothesized that sporozoite motility and invasion is likely to require CDPK activity. We focused on CDPK4 whose function in sporozoites has not yet been examined and whose homolog in T. gondii, T. gondii CDPK1 (TgCDPK1) is required for tachyzoite invasion and egress from host cells (Lourido et al., 2010; Lourido et al., 2012). Using antisera against TgCDPK1 which cross-reacts with P. berghei CDPK4 (Billker et al., 2004), we determined that CDPK4 is present in sporozoites (Fig. 3A). Since P. berghei CDPK4 (PbCDPK4) is essential for male gametogenesis, CDPK4 knockout parasites do not infect mosquitoes and consequently, cannot produce sporozoites (Billker et al., 2004). Therefore, we generated a stage-specific knockout (cKO) allele using the FlpL/FRT system (Lacroix et al., 2011) (Fig. 3B). In the CDPK4 cKO line, the CDPK4 open reading frame is excised during development in the mosquito midgut, generating a sporozoite population in which CDPK4 expression was below the level of detection (Fig. 3A). CDPK4 expression in liver stages of CDPK4 cKO line was similarly significantly reduced (Fig. 3A). Immunofluorescence assays were utilized since sufficient numbers of sporozoites for Western blot analysis could not be obtained. TRAP/FlpL parasites (Panchal et al., 2012), the FlpL-expressing parent line used to modify the CDPK4 locus, served as controls. CDPK4 cKO sporozoites developed normally (8175 ± 1247 sporozoites/mosquito for TRAP/FlpL-infected mosquitoes and 6887 ± 765 sporozoites/mosquito for CDPK4 cKO-infected mosquitoes) and were used to assess the role of CDPK4 in sporozoite infection and the liver cycle.
Fig. 3.
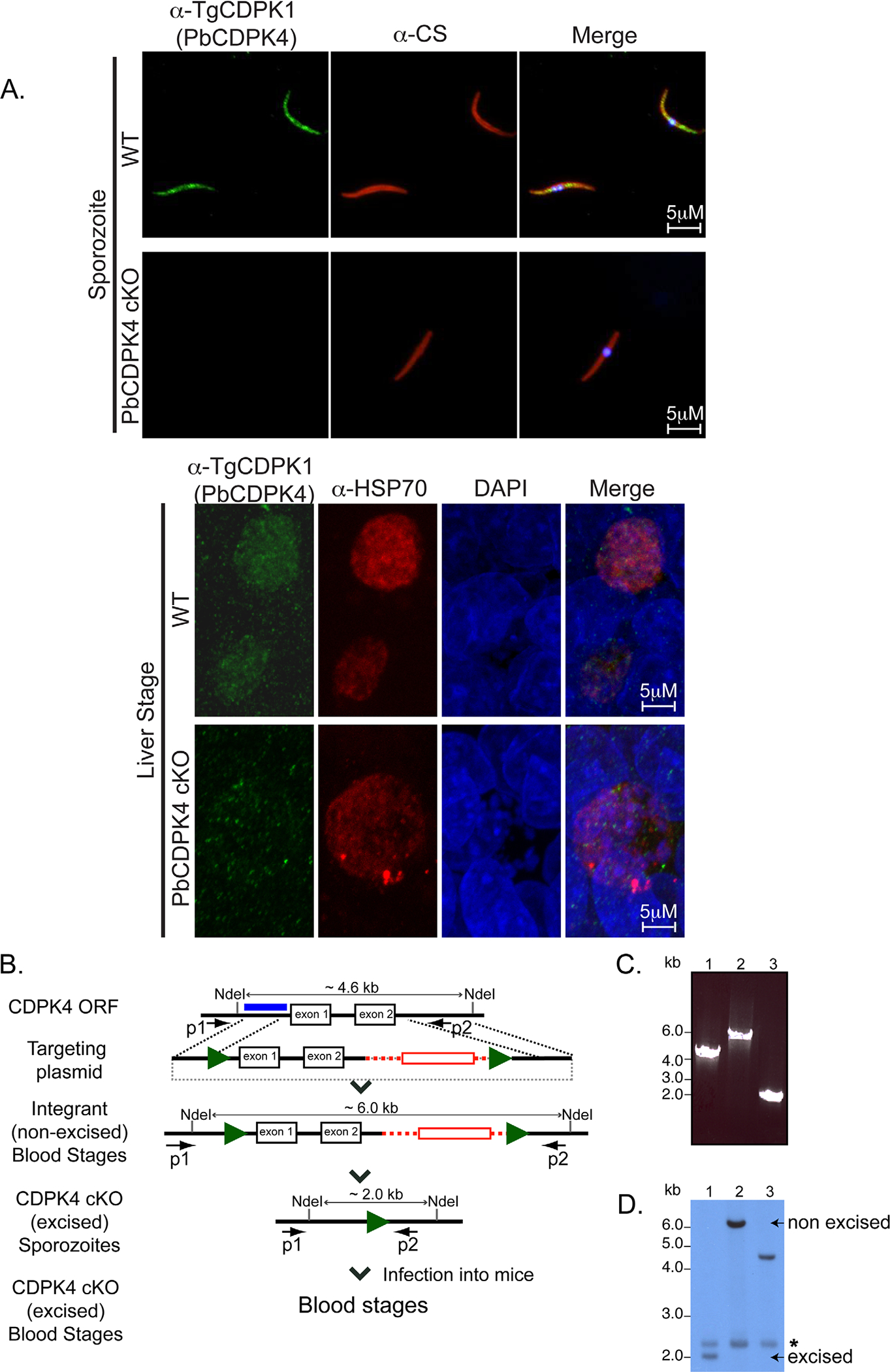
Conditional mutagenesis of PbCDPK4 using the FlpL-FRT system.
A. PbCDPK4 is expressed in the cytoplasm of pre-erythrocytic stages. Immunofluorescence assays using anti-TgCDPK1 antibodies were used to determine PbCDPK4’s subcellular localization in sporozoites and liver stages. The anti-CS antibody recognizes CS, a sporozoite membrane protein. Anti-HSP70 recognizes HSP70, a cytoplasmic protein. PbCDPK4 co-localizes with HSP70, a cytoplasmic marker in liver stage parasites. Merged panels include DAPI for nuclear localization.
B. Modification of CDPK4 open reading frame through addition of two FRT sites (green arrows) and a hDHFR expression cassette (red box) in FlpL-expressing parasites. The CDPK4 ORF is excised during sporogony in the mosquito midgut generating CDPK4 cKO sporozoites in the mosquito salivary glands.
C. PCR analysis demonstrates excision of CDPK4 in blood stage parasites obtained from cKO sporozoite infection. Amplification products of primers P1 and P2 from genomic DNA of (1) WT blood stages, (2) parasites obtained after integration of the targeting plasmid, (3) blood stages resulting from infection with CDPK4 cKO sporozoites.
D. Southern blot analysis confirms loss of CDPK4 in blood stage parasites obtained from cKO sporozoites. NdeI-digested genomic DNA obtained from (1) blood stages resulting from infection by CDPK4 cKO sporozoites (2) blood stages obtained after integration of the targeting construct (3) WT parasites. The probe demonstrates non-specific hybridization to a 2.3kb fragment (indicated by an asterisk) in genomic DNA from all parasite populations.
Hepatocyte invasion by CDPK4 cKO sporozoites was examined by quantifying the fraction of sporozoites that are intracellular 2 h after addition to host cells. In CDPK4 cKO infected cells, there was a two-fold decrease in the fraction of intracellular sporozoites compared to control sporozoites (Fig. 4A, Supporting Information Table S2A), suggesting an important role for CDPK4 in sporozoite entry into host cells. To determine if CDPK4 has additional functions during intrahepatic development, we quantified the number of liver stages present in infected HepG2 cultures at 24 h p.i. and 48 h p.i. CDPK4 cKO sporozoites form half as many liver stages compared to control sporozoites (Fig. 4A, Supporting Information Table S2A), a decrease equivalent to the reduction in sporozoites that invade hepatocytes. These data suggest that PbCDPK4 does not have an additional role in intracellular development after the sporozoite has successfully invaded the hepatocyte.
Fig. 4.
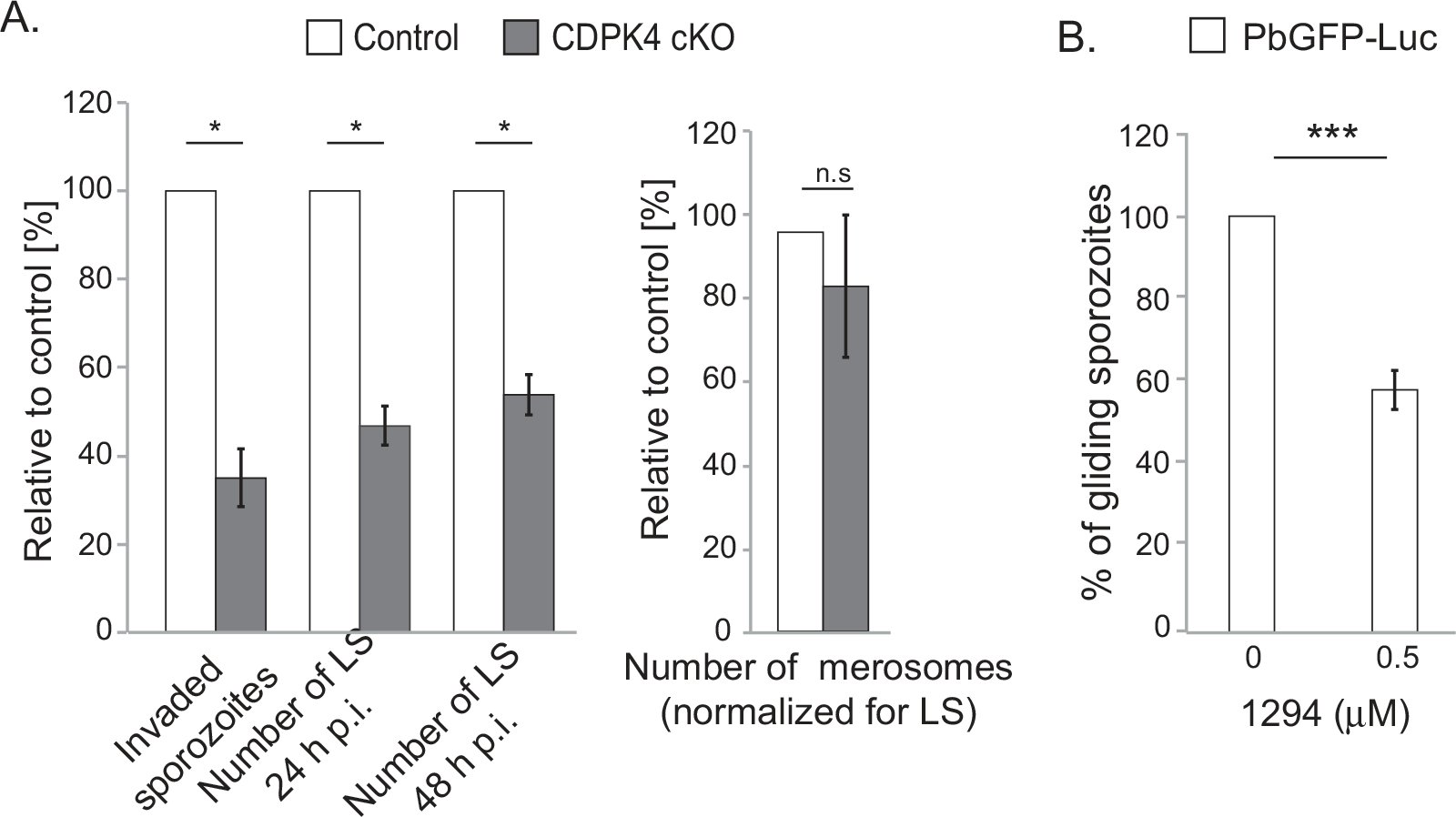
CDPK4 plays a role in sporozoite invasion.
A. Invasion by CDPK4 cKO sporozoites of HepG2 cells and intracellular development of CDPK4 cKO liver stages (LS) relative to FlpL-expressing sporozoites. Equal numbers of control (FlpL-expressing parent line) and CDPK4 cKO sporozoites were used to assay invasion and intracellular development. Sporozoite invasion was quantified by determining the fraction of sporozoites that are intracellular 2 h after addition to HepG2 cells. Intracelluar development of sporozoites was quantified by determining the number LS at 24 h p.i and 48 h p.i. Shown are results from a representative experiment (average of 4 replicates ± standard deviation). The experiment was performed four times. Parasite egress was examined by quantifying merosomes released at 65–72 h p.i. relative to the number of LS formed at 48 h p.i. To compensate for their decreased invasion, twice as many CDPK4 cKO sporozoites as FlpL-expressing sporozoites were used to infect HepG2 cells. Shown are results from a representative experiment (mean of 3–4 replicates ± standard deviation). The experiment was performed thrice. Data were analyzed using Kruskal–Wallis test, *P < 0.05.
B. Inhibition of CDPK4 activity decreases sporozoite motility. PbGFP-Luc sporozoites were filmed in the presence of 1294. The percentage of sporozoites that glide was determined. The percentage (± standard error, n = 2 experiments) of gliding sporozoites was determined. Data were analyzed using chi-square tests, ***P < 0.005. 100% is the proportion of sporozoites that glide in the absence of 1294.
A potential role for CDPK4 during egress from hepatocytes was examined by quantifying merosome formation in cKO and control cultures. To compensate for the two-fold lower infectivity of CDPK4 cKO sporozoites and ensure similar numbers of liver stages leading up to merosome formation and/release, we infected HepG2 cells with twice as many cKO sporozoites compared to control. The numbers of intracellular liver stages and merosomes in the media were quantified at 65–72 h p.i. CDPK4 cKO sporozoites were not significantly affected in their ability to form merosomes (Fig. 4A, Supporting Information Table S2A). Our data suggest that CDPK4’s most important role in pre-erythrocytic stages is during sporozoite invasion of hepatocytes and that it does not have a major role in parasite egress from hepatocytes.
CDPK4’s effect on parasite infection in mice was examined by determining the pre-patent period in mice (days to appearance of erythrocytic stage parasites in Giemsa-stained blood smears) following intravenous injection of 1×104 sporozoites/mouse. In the first experiment, deletion of CDPK4 increased the pre-patent period from 4.2 ± 0.22 days (n = 5, 5/5 control-infected mice developed blood stage parasitemia) to 4.75 ± 0.25 days (n = 4, 4/4 CDPK4 cKO-infected mice developed blood stage parasitemia). Average parasitemias at days 4 and 6 post-infection were not significantly different in the two groups (Supporting Information Table S2B). In a repeat experiment, the pre-patent periods of control sporozoites was 4.0 ± 0.46 days (n = 10, 8/10 mice control-infected mice developed blood stage parasitemia) and of cKO sporozoites was 4.66 ± 0.25 days (n = 10, 6/10 CDPK4 cKO-infected mice developed blood stage parasitemia). While at day 4 in the second experiment, blood parasitemias of the two groups were significantly different (p value < 0.05, unpaired t-test), there was only a trend towards delay in patency of CDPK4 cKO sporozoites that did not reach significance (Supporting Information Table S2B). The effect size in both replicates is however, entirely consistent with the two-fold decrease in hepatocyte infection by cKO sporozoites observed in vitro since a 1-day delay in patency determined by microscopic examination of blood smears would require a 10-fold decrease in liver parasitemia.
Next we examined if CDPK4’s role in invasion could result from its function in sporozoite motility. We utilized a bumped kinase inhibitor compound 1294 that is relatively specific for CDPK4 in vitro since it exploits the small ‘gatekeeper’ residue of the kinase that is absent in most Plasmodium kinases (Ojo et al., 2012; Ojo et al., 2014). In P. falciparum, treatment of gametocytes with 1294 phenocopies the effects of deleting CDPK4 by inhibiting male gametogenesis, thus blocking oocyst development. Importantly, inhibition of oocyst development by 1294 was reversed by introducing a larger amino acid in the ‘gatekeeper’ position of P. falciparum CDPK4, demonstrating that CDPK4 is the primary in vivo target of 1294 (Ojo et al., 2012; Ojo et al., 2014). Although PKG and CDPK1 also have small ‘gatekeeper’ residues, the IC50 of 1294 against PfCDPK4 (10 nM) is a log lower than against PfPKG (200 nM) or PfCDPK1 (100nM) (Ojo et al., 2012; Ojo et al., 2014). Since gametocytes and sporozoites express PKG, CDPK4 and CDPK1 (Billker et al., 2004; McRobert et al., 2008; Sebastian et al., 2012; Philip and Waters, 2015), 1294’s relative specificity for CDPK4 in gametocytes suggests that it can be used to probe CDPK4 function in sporozoites.
Using the same gliding assay as before, we found that 0.5 μM 1294 significantly decreased the percentage (± standard error, n = 2 experiments) of sporozoites that glide from 29.67 ± 1.91% to 17.29 ± 1.4% (Fig. 4B, Supporting Information Table S2C). These data suggest that CDPK4 may be one of a number of effectors that mediate the Ca2+ fluxes observed in gliding sporozoites, and which are associated with micronemal secretion and motility (Carey et al., 2014). We cannot rule out the possibility that 1294’s inhibition of sporozoite motility is mediated in part through inhibitory effects on other protein kinases.
Discussion
Our work adds to a comprehensive understanding of the role of cGMP and Ca2+ signalling in apicomplexans. It extends the role of parasite cGMP and Ca2+ signalling pathways, as mediated by PKG and CDPK4, to sporozoites in addition to their previously recognized roles in asexual and liver stages (PKG) as well as sexual stages (PKG and CDPK4). We clearly demonstrate an essential role for PKG and an important role for CDPK4 in invasion of hepatocytes by Plasmodium sporozoites, that has not been previously appreciated (Figs 2B and 4A). The function of the two enzymes in sporozoite invasion may be explained in part by their roles in sporozoite motility (Figs 2C and 4B). Sporozoites expressing a 1294-refractory allele of CDPK4 and/or fluorescently labeled CDPK4 cKO sporozoites can be used in the future to address the possibility of 1294 having off-target effects and to verify CDPK4’s role in sporozoite motility. While PKG is additionally required for the formation of merosomes through which parasites exit the hepatocyte (Fig. 2D, Falae et al., 2010), the same knockout strategy has not produced evidence linking CDPK4 to liver stage egress (Fig. 4A). It is possible that CDPK4 has a subtle role in merosome formation and release, which is not detected in the current assay.
PKG was known to regulate micronemal secretion in P. falciparum merozoites (Collins et al., 2013) and in T. gondiii tachyzoites (Wiersma et al., 2004; Brown et al., 2016). Our work extends its role in regulating exocytosis to sporozoites. PKG’s regulation of sporozoite motility occurs through regulation of TRAP secretion (Fig. 1D). Future work will have to elucidate whether PKG regulates the release of additional sporozoite adhesins such as TRAP-like protein and S6 (Sultan et al., 1997; Heiss et al., 2008; Moreira et al., 2008; Combe et al., 2009; Steinbuechel and Matuschewski, 2009; Hegge et al., 2010). The exact contribution of each adhesin to the formation of the primary attachment site between the sporozoite and the substrate, turnover of adhesion sites and the generation of the force needed to propel sporozoites remains to be established using reflection interference contrast, traction force and/or total internal reflection fluorescence microscopy (Hegge et al., 2010).
By regulating protein secretion in sporozoites, PKG could also regulate cell traversal since proteins, like SPECT2 and PLP1, that are required for passage of sporozoites through cells are localized to micronemes (Ishino et al., 2004; Ishino et al., 2005; Risco-Castillo et al., 2015). Indeed, our unpublished data suggest that TSP treatment blocks cell traversal by sporozoites. However, since cell traversal is an active process that is blocked by treatments that inhibit gliding motility (Risco-Castillo et al., 2015), TSP’s inhibition of cell traversal could be explained entirely by its inhibition of sporozoite motility. To gain a better understanding of PKG’s regulation of cell traversal independent of its regulation of motility, future work will determine TSP’s effect on secretion of SPECT2 and PLP1. Another process that could be affected by micronemal secretion is the formation of the ‘tight junction’ between the zoite and the host cell that serves as an anchor point during sporozoite entry into hepatocytes (Giovannini et al., 2011). The ‘tight junction’ is thought to require micronemal proteins whose secretion may be regulated by PKG activity. In this way, PKG could contribute to multiple steps of cell invasion by sporozoites.
Our results suggest that a previously published model for microgamete activation in which PKG acts upstream of CDPK4 to transduce effects of Ca2+ released through PKG signalling (Brochet et al., 2014) likely extends to sporozoite motility. However, since CDPK4 inhibition produces a relatively less severe phenotype compared to PKG inhibition, we propose that additional Ca2+ mediators are involved. Another possible explanation for the reduced reliance of sporozoites on CDPK4 during invasion is that PbPKG compensates for the loss of PbCDPK4. Co-operativity between PKG and CDPKs has been demonstrated in T. gondii tachyzoites where activation of PKG rescues the egress defect caused by inhibition of T. gondii CDPK1 (Lourido et al., 2012). We recognize that an additional possibility - small amounts of CDPK4 protein in cKO sporozoites, that would have to be below the level of detection by IFA, cannot be ruled out. Residual CDPK4 protein may be sufficient to allow the reduced invasion demonstrated by CDPK4 cKO sporozoites.
Together with previous work (Wiersma et al., 2004; McRobert et al., 2008; Moon et al., 2009; Dvorin et al., 2010; Taylor et al., 2010) these studies provide an opportunity to trace the evolution of orthologous parasite kinases across parasite stages and species. This is the first time we identify a stage-transcending requirement for a Plasmodium CDPK. Our data suggest that CDPK4 has an important but likely non-essential role in sporozoite invasion (although the possibility of residual protein cannot be ruled out). This is in contrast to the same enzyme’s essential role in male gametogenesis (Billker et al., 2004) or to CDPK5’s essential role in merozoite egress (Dvorin et al., 2010). PbCDPK4 shares with its T. gondii ortholog, TgCDPK1, a role in regulating zoite gliding and invasion, which in T. gondii was shown to be due to its requirement for microneme secretion (Lourido et al., 2010). However, unlike TgCDPK1 (Lourido et al., 2012), PbCDPK4 does not have a major role in parasite egress at any life cycle stage.
We were intrigued to find that PbPKG T619Q-HA sporozoites have a phenotypic effect in the parasite that has not been previously observed in erythrocytic stages and ookinetes carrying the same mutation (Brochet et al., 2014) (Supporting Information Fig. 2F). Similarly, the equivalent substitutions in PKG in other species did not impact gametogenesis and schizogony of P. falciparum (McRobert et al., 2008; Taylor et al., 2010) or growth of T. gondii tachyzoites (Donald et al., 2002). Compared to PbPKG T619Q-HA, PbPKG cKO sporozoites have normal infectivity because they contain wildtype enzyme. We propose that the T619Q substitution causes subtle differences in enzyme stability in vivo as is reported for ‘gatekeeper’ mutations in several other kinases, including PfPKG (Zhang et al., 2005; McRobert et al., 2008). P. falciparum PKG in which the gatekeeper Thr is substituted by Met is unexpectedly more sensitive to TSP implying a subtle change in enzymatic function (McRobert et al., 2008).The sporozoite-specific effect of the T619Q mutation raises the intriguing possibility that PKG has stage-specific interactions that affect its stability in sporozoites and have functional consequences in the form of decreased sporozoite motility and invasion.
Our work has implications for therapies aimed at preventing liver infection by Plasmodium. Inhibitors of PKG and CDPK4 with greater potency could significantly decrease the parasite burden in the liver. Another possibility is to use inhibition of PKG and CDPK4 to block multiple parasite stages. Work presented here together with previous reports [8,23] demonstrate that parasite PKG and CDPK4 are required for both steps of Plasmodium transmission – mosquito to mammalian host and mammalian host to mosquito. The current work on sporozoite infection demonstrates the role of PKG and CDPK4 during transmission of parasites from mosquito to mammalian host. Previous reports demonstrate that PKG is required for male and female gametogenesis (McRobert et al., 2008) and CDPK4 is required for male specific gametogenesis events in the mosquito (Billker et al., 2004). Since both PKG and CDPK4 can be inhibited with small molecule inhibitors that are selective over most host protein kinases (Collins et al., 2013; Brochet et al., 2014; Ojo et al., 2014; Vidadala et al., 2014), they could be attractive targets for both prophylaxis and transmission blocking approaches.
Experimental procedures
Ethics statement
All experiments were approved by the Institutional Animal Care and Use Committee (IACUC) at the Rutgers New Jersey Medical School, under protocol number 13086D1016, following guidelines of the Animal Welfare Act, The Institute of Laboratory Animal Resources Guide for the Care and Use of Laboratory Animals, and Public Health Service Policy. Swiss-webster mice (6–8 weeks, female, Taconic Biosciences) were utilized for all experiments.
Conditional mutagenesis and parasite transfection
The CDPK4 cKO targeting vector was constructed by cloning (In-Fusion, Clontech) three PCR products into a vector that carries two FRT sites and the human dihydrofolate reductase expression cassette (Falae et al., 2010). Primers CTTGCATGCGCGGCCGCGTCTTTTACCATTTCTAC AAT and TCGCCCTTATGCGGCCGCCTTTAACTTTCCTATA TTTTATGC were used to amplify an approximately 1.0 kB fragment upstream of the 5'UTR of PbCDPK4 (PBANKA_061520). The product was cloned into the vector linearized with NotI. Primers TAGGAACTTCCTCGAGTACA TATGTTCATATTAAGAAA and CTGGGCTGCACTCGAGAA TAAATGAGTATTTAAAATATATAGG were used to amplify a 2.6 kB fragment encompassing the 5’UTR, exons 1–2 and 3'UTR of PbCDPK4. It was inserted into the previously generated plasmid using XhoI. Primers CTAGAGGATCCCC GGGTACCAATTATATATATGTATATAGTGTACGTTG and CC ATGATTACGAATTCTTGTATCATGTATATTCATGTTA were used to amplify a 0.5 kB fragment that was inserted into the previously derived plasmid digested with KpnI and EcoRI. The final insert was released from the targeting construct using NdeI and EcoRI. Transfections of TRAP/FlpL parasites (Panchal et al., 2012) were carried out using standard methodology. Transfected parasites were selected using pyrimethamine and cloned by limiting dilution. Integration into the genome was assayed using primers P1 (CTTGCATGCGCGGCCGCGTCTTTTACCATTTCTACAAT) and P2 (CCATGATTACGAATTCTTGTATCATGTATATTCAT GTTA), which amplify a 5.64 kB product in the presence of integration and a 4.14 kB product in the absence of integration. Integration was verified using Southern blotting of NdeI-digested genomic DNA followed by hybridization with a dioxygenin-labeled probe (DIG High Prime DNA labelling and detection kit, Roche Applied Sciences) following the manufacturer’s protocol.
Transgenic PKG-HA and PKG T619Q parasites were constructed in ANKA strain 507cl1 which expresses GFP under the control of the strong constitutive eef1α promoter that is active throughout the P. berghei life-cycle. Transfections and genotypic analysis were carried out essentially as described previously [8].
Mosquito infections
Anopheles stephensi mosquitoes were fed on infected Swiss-Webster mice. Mosquitoes infected with PKG-HA and PKG T619Q-HA parasites were maintained at 20°C. Sporozoites were obtained at days 18–21 post-feeding through dissections of salivary glands. Mosquitoes infected with CDPK4 cKO and TRAP/FlpL parasites were maintained at 20°C until day 11 post bloodmeal and transferred to 25°C thereafter. Sporozoites were obtained at days 21–26 post-bloodmeal.
Sporozoite invasion, liver stage infection and merosome formation
HepG2 cells (obtained from ATCC) were cultured in Dulbecco’s Modified Eagle Medium (high glucose) supplemented with 10% FCS. Cells were seeded on collagen-coated multi-chambered slides for overnight growth at 37°C prior to addition of sporozoites. Invasion assays were performed using cells at 90% confluency and infection assays were perfomed with cells at 50% confluency, essentially as previously described (Sinnis et al., 2013). For invasion assays, cells were fixed in 4% paraformaldehyde 2 h after addition of sporozoites (1–2 × 104). Cells were blocked in 1% BSA/PBS before incubation with anti-CS antibody (3D11, 1 μg/ mL) and anti-mouse Alexa594. Cells were then permeabilized with cold methanol, blocked, incubated with 3D11 and anti-mouse Alexa488. Extracellular sporozoites were quantified by determining the number of sporozoites that stained exclusively with anti-mouse Alexa594. The total sporozoite number was quantified by determining the number of sporozoites stained with both Alexa488 and Alexa594. For infection assays, cells infected with sporozoites (1–2 × 104) were fixed in 4% paraformaldehyde 24–48 h p.i. Liver stages were detected in IFA using a monoclonal antibody against PbHSP70 (10 μg/mL). For merosome assays, cells were seeded on collagen-coated coverslips for overnight growth before infection with 5–8 × 104 sporozoites. Media was changed every 12 h. The number of merosomes released in the media was quantified at 66–72 h p.i. using a hemocytometer. To determine the pre-patent period of infection was Swiss-webster mice (female, 6–8 weeks, Taconic Biosciences) were injected intravenously with sporozoites. Parasitemia was determined daily either through microscopic counting of Giemsa-stained blood smears or FACS analysis.
Immunofluorescence for protein detection
Purified sporozoites (Kennedy et al., 2012) were air-dried on poly-lysine coated slides, fixed in 4% paraformaldehyde for 15 min and permeabilized in 0.5% TritonX-100 for 5 min. They were blocked in 3% BSA/PBS for 1 h prior to incubation with antibodies. Liver stages were fixed with 4% paraformaldehyde for 15 min, permeabilized with cold methanol for 15 min, blocked in 3% BSA/PBS for 1 h prior to incubation with antibodies. Primary antibodies were anti-HA (mouse monoclonal, Covance, 10 μg/mL), anti-CS (mouse monoclonal 3D11, 1 μg/mL), anti-HSP70 (1 μg/mL, (Tsuji et al., 1994)), anti-PKG (polyclonal antisera raised against a peptide containing amino acids 989 to 1003 of PbPKG), anti-merosome antibody (rat, 1:100, a kind gift of Dr. Volker Heussler), anti-TgCDPK1 (rabbit polyclonal antisera, a kind gift of Dr. Conrad Beckers), which recognizes PbCDPK4 (Billker et al., 2004) and anti-TRAP (1:100, rabbit polyclonal sera raised against the repeat region of P. berghei TRAP, a kind gift of Dr. Rogerio Amino). Secondary antibodies were anti-mouse Alexa488, anti-mouse Alexa594, anti-rabbit Alexa594 and anti-rabbit Alexa488. All secondary antibodies were purchased from Santa Cruz Biotechnology and used at 0.7 μg/mL. Images were collected on a Nikon A1R laser scanning confocal microscope using 60X/NA1.4 oil objective. For quantification of signal intensities, all images in a given experiment were captured using the same excitation laser intensity and detector gain settings. A region-of-interest comprising a single sporozoite was automatically selected and the mean background corrected fluorescence intensity of Alexa488 and Alex594 within that region-of-interest was measured using the Nikon NIS Elements Advanced Research software. The average ratio of Alexa488 and Alexa594 signal intensities was determined for a given sporozoite population.
Western blot for protein detection
Protein lysates of schizont stage parasites were examined by SDS-PAGE using anti-HA (mouse monoclonal, Covance, 2 μg/mL) and anti-HSP-70 (0.4 μg/mL (Tsuji et al., 1994)) antibodies followed by detection using chemiluminescence (SuperSignal™ West Femto substrate, ThermoFisher Scientific) following the manufacturer’s protocol. Signal intensities were quantified using Image J software.
Detection of surface TRAP by indirect immunofluorescence
PbGFP-Luc sporozoites were dissected in DMEM, filtered through a 40 μM mesh filter (BD-Falcon) and treated with either TSP (2 μM) or vehicle for 20 min on ice. Sporozoites were activated by incubation at 37°C for 10 min after addition of 3% BSA. Following activation, sporozoites were deposited on glass coverslips, fixed in 4% paraformaldehyde for 15 min at room temperature, blocked for 30 min at room temperature with 1% BSA prior to labelling with anti-CS (3D11, 1 μg/mL) for 1 h at room temperature. Sporozoites were washed thrice with PBS, followed by labelling with anti-mouse Alexa594 (Santa Cruz Biotechnology, 0.7 μg/mL) for 1 h at 37°C. Sporozoites were washed twice with PBS prior to blocking with 1% BSA and labelling with anti-TRAP antisera (1:100, gift of Dr. Rogerio Amino) for 1 h at room temperature. Sporozoites were washed thrice with PBS, followed by labelling with anti-rabbit Alexa488 (Santa Cruz Biotechnology, 0.7 μg/mL) for 1h at 37°C. Sporozoites were washed thrice with PBS and mounted for analysis of the anti-TRAP and anti-CS fluorescence intensities. Images of randomly selected sporozoites in the Alexa594 channel were collected on a Nikon A1R laser scanning confocal microscope using 60X/NA1.4 oil objective. For quantification of signal intensities, all images in a given experiment were captured using the same excitation laser intensity and detector gain settings. A region-of-interest comprising a single sporozoite was automatically selected and the mean background corrected fluorescence intensity of Alexa488 and Alex594 within that region-of-interest was measured using the Nikon NIS Elements Advanced Research software. The average ratio of Alexa488 (TRAP) and Alexa594 (CS) signal intensities was determined for a given sporozoite population.
Compound treatment
The effect of TSP on sporozoite infectivity was determined by adding sporozoites to HepG2 cells in the presence of appropriate concentrations of TSP or vehicle alone. Compound-containing media was replaced with compound-free medium at 14 h p.i. Liver stages present at 44–48 h p.i. were detected as described above. The effect of compounds on sporozoite invasion was determined by treating sporozoites for 30 min on ice in a volume of 20 μL prior to addition to HepG2 cells in a volume of 200 μL. Cells were fixed 2 h later and processed as described above.
Imaging sporozoite motility
Sporozoites were filmed on a Nikon A1R laserscanning confocal microscope using a 20X/NA0.75 objective at 37°C in a 96-well plate with an optical bottom. Dissected sporozoites, in RPMI and 3% BSA, were incubated with appropriate compounds for 15 min on ice prior to centrifugation for 3 min at 4°C. Movies were recorded over 90 frames at 1 Hz. Image acquisition and analysis was performed using NIS Elements software from Nikon. Fluorescence intensity projections were processed using NIS Elements and movement patterns were determined through visual inspection of individual sporozoites.
Statistical analysis
Invasion and infection of HepG2 cells by sporozoites was examined using the Kruskal–Wallis test (Kruskal, 1952). The Kruskal–Wallis test is a non-parametric test appropriate for comparing a continuous outcome measured in two or more groups. It is the non-parametric analog of a ANOVA test when there are three or more groups; and analog of t-test when there are only two groups.
Supplementary Material
Acknowledgments
The authors gratefully acknowledge Dr. J. Hahn and L. Fritzky for assistance with imaging. This work was supported by grants from the National Science Foundation (IOS-1146221), National Institutes of Health (R21AI094167) and Department of Defense (W81XWH-13-1-0429) to P.B. Work at the Sanger Institute was supported by the Wellcome Trust (WT098051) and by a Marie Curie Fellowship (PIEF-GA-2009-253899) to M.B. M.B. is an INSERM investigator and is supported by the Swiss National Science Foundation (BSSGI0_155852).
Footnotes
Supporting information
Additional supporting information may be found in the online version of this article at the publisher’s web-site.
References
- Alam MM, Solyakov L, Bottrill AR, Flueck C, Siddiqui FA, Singh S, et al. (2015) Phosphoproteomics reveals malaria parasite Protein Kinase G as a signalling hub regulating egress and invasion. Nat Commun 6: 7285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso PL, Sacarlal J, Aponte JJ, Leach A, Macete E, Aide P, et al. (2005) Duration of protection with RTS, S/AS02A malaria vaccine in prevention of Plasmodium falciparum disease in Mozambican children: single-blind extended follow-up of a randomised controlled trial. Lancet 366: 2012–2018. [DOI] [PubMed] [Google Scholar]
- Billker O, Dechamps S, Tewari R, Wenig G, Franke-Fayard B, and Brinkmann V (2004) Calcium and a calcium-dependent protein kinase regulate gamete formation and mosquito transmission in a malaria parasite. Cell 117: 503–514. [DOI] [PubMed] [Google Scholar]
- Brochet M, Collins MO, Smith TK, Thompson E, Sebastian S, Volkmann K, et al. (2014) Phosphoinositide metabolism links cGMP-dependent protein kinase G to essential Ca(2)(+) signals at key decision points in the life cycle of malaria parasites. PLoS Biol 12: e1001806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown KM, Lourido S, and Sibley LD (2016) Serum albumin stimulates protein kinase G-dependent microneme secretion in Toxoplasma gondii. J Biol Chem 291: 9554–9565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey AF, Singer M, Bargieri D, Thiberge S, Frischknecht F, Menard R, and Amino R (2014) Calcium dynamics of Plasmodium berghei sporozoite motility. Cell Microbiol 16: 768–783. [DOI] [PubMed] [Google Scholar]
- Collins CR, Hackett F, Strath M, Penzo M, Withers-Martinez C, Baker DA, and Blackman MJ (2013) Malaria parasite cGMP-dependent protein kinase regulates blood stage merozoite secretory organelle discharge and egress. PLoS Pathog 9: e1003344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Combe A, Moreira C, Ackerman S, Thiberge S, Templeton TJ, and Menard R (2009) TREP, a novel protein necessary for gliding motility of the malaria sporozoite. Int J Parasitol 39: 489–496. [DOI] [PubMed] [Google Scholar]
- Coppi A, Tewari R, Bishop JR, Bennett BL, Lawrence R, Esko JD, et al. (2007) Heparan sulfate proteoglycans provide a signal to Plasmodium sporozoites to stop migrating and productively invade host cells. Cell Host Microbe 2: 316–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donald RG, Allocco J, Singh SB, Nare B, Salowe SP, Wiltsie J, and Liberator PA (2002) Toxoplasma gondii cyclic GMP-dependent kinase: chemotherapeutic targeting of an essential parasite protein kinase. Eukaryot Cell 1: 317–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvorin JD, Martyn DC, Patel SD, Grimley JS, Collins CR, Hopp CS, et al. (2010) A plant-like kinase in Plasmodium falciparum regulates parasite egress from erythrocytes. Science 328: 910–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ejigiri I, and Sinnis P (2009) Plasmodium sporozoite-host interactions from the dermis to the hepatocyte. Curr Opin Microbiol 12: 401–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ejigiri I, Ragheb DR, Pino P, Coppi A, Bennett BL, Soldati-Favre D, and Sinnis P (2012) Shedding of TRAP by a rhomboid protease from the malaria sporozoite surface is essential for gliding motility and sporozoite infectivity. PLoS Pathog 8: e1002725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falae A, Combe A, Amaladoss A, Carvalho T, Menard R, and Bhanot P (2010) Role of Plasmodium berghei cGMP-dependent protein kinase in late liver stage development. J Biol Chem 285: 3282–3288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke-Fayard B, Janse CJ, Cunha-Rodrigues M, Ramesar J, Buscher P, Que I, et al. (2005) Murine malaria parasite sequestration: CD36 is the major receptor, but cerebral pathology is unlinked to sequestration. Proc Natl Acad Sci U S A 102: 11468–11473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gantt S, Persson C, Rose K, Birkett AJ, Abagyan R, and Nussenzweig V (2000) Antibodies against thrombospondin-related anonymous protein do not inhibit Plasmodium sporozoite infectivity in vivo. Infect Immun 68: 3667–3673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giovannini D, Spath S, Lacroix C, Perazzi A, Bargieri D, Lagal V, et al. (2011) Independent roles of apical membrane antigen 1 and rhoptry neck proteins during host cell invasion by apicomplexa. Cell Host Microbe 10: 591–602. [DOI] [PubMed] [Google Scholar]
- Graewe S, Stanway RR, Rennenberg A, and Heussler VT (2012) Chronicle of a death foretold: Plasmodium liver stage parasites decide on the fate of the host cell. FEMS Microbiol Rev 36: 111–130. [DOI] [PubMed] [Google Scholar]
- Gurnett AM, Liberator PA, Dulski PM, Salowe SP, Donald RG, Anderson JW, et al. (2002) Purification and molecular characterization of cGMP-dependent protein kinase from Apicomplexan parasites. A novel chemotherapeutic target. J Biol Chem 277: 15913–15922. [DOI] [PubMed] [Google Scholar]
- Hegge S, Kudryashev M, Smith A, and Frischknecht F (2009) Automated classification of Plasmodium sporozoite movement patterns reveals a shift towards productive motility during salivary gland infection. Biotechnol J 4: 903–913. [DOI] [PubMed] [Google Scholar]
- Hegge S, Munter S, Steinbuchel M, Heiss K, Engel U, Matuschewski K, and Frischknecht F (2010) Multistep adhesion of Plasmodium sporozoites. FASEB J 24: 2222–2234. [DOI] [PubMed] [Google Scholar]
- Heiss K, Nie H, Kumar S, Daly TM, Bergman LW, and Matuschewski K (2008) Functional characterization of a redundant Plasmodium TRAP family invasin, TRAP-like protein, by aldolase binding and a genetic complementation test. Eukaryot Cell 7: 1062–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishino T, Yano K, Chinzei Y, and Yuda M (2004) Cell-passage activity is required for the malarial parasite to cross the liver sinusoidal cell layer. PLoS Biol 2: E4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishino T, Chinzei Y, and Yuda M (2005) A Plasmodium sporozoite protein with a membrane attack complex domain is required for breaching the liver sinusoidal cell layer prior to hepatocyte infection. Cell Microbiol 7: 199–208. [DOI] [PubMed] [Google Scholar]
- Kennedy M, Fishbaugher ME, Vaughan AM, Patrapuvich R, Boonhok R, Yimamnuaychok N, et al. (2012) A rapid and scalable density gradient purification method for Plasmodium sporozoites. Malar J 11: 421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruskal W (1952) Use of ranks in one-criterion variance analysis. J Am Stat Assoc 47: 583–621. [Google Scholar]
- Lacroix C, Giovannini D, Combe A, Bargieri DY, Spath S, Panchal D, et al. (2011) FLP/FRT-mediated conditional mutagenesis in pre-erythrocytic stages of Plasmodium berghei. Nat Protoc 6: 1412–1428. [DOI] [PubMed] [Google Scholar]
- Lourido S, Shuman J, Zhang C, Shokat KM, Hui R, and Sibley LD (2010) Calcium-dependent protein kinase 1 is an essential regulator of exocytosis in Toxoplasma. Nature 465: 359–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lourido S, Tang K, and Sibley LD (2012) Distinct signalling pathways control Toxoplasma egress and host-cell invasion. EMBO J 31: 4524–4534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McRobert L, Taylor CJ, Deng W, Fivelman QL, Cummings RM, Polley SD, et al. (2008) Gametogenesis in malaria parasites is mediated by the cGMP-dependent protein kinase. PLoS Biol 6: e139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meissner M, Ferguson DJ, and Frischknecht F (2013) Invasion factors of apicomplexan parasites: essential or redundant? Curr Opin Microbiol 16: 438–444. [DOI] [PubMed] [Google Scholar]
- Moon RW, Taylor CJ, Bex C, Schepers R, Goulding D, Janse CJ, et al. (2009) A cyclic GMP signalling module that regulates gliding motility in a malaria parasite. PLoS Pathog 5: e1000599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreira CK, Templeton TJ, Lavazec C, Hayward RE, Hobbs CV, Kroeze H, et al. (2008) The Plasmodium TRAP/MIC2 family member, TRAP-like Protein (TLP), is involved in tissue traversal by sporozoites. Cell Microbiol 10: 1505–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munter S, Sabass B, Selhuber-Unkel C, Kudryashev M, Hegge S, Engel U, et al. (2009) Plasmodium sporozoite motility is modulated by the turnover of discrete adhesion sites. Cell Host Microbe 6: 551–562. [DOI] [PubMed] [Google Scholar]
- Ojo KK, Pfander C, Mueller NR, Burstroem C, Larson ET, Bryan CM, et al. (2012) Transmission of malaria to mosquitoes blocked by bumped kinase inhibitors. J Clin Invest 122: 2301–2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojo KK, Eastman RT, Vidadala R, Zhang Z, Rivas KL, Choi R, et al. (2014) A specific inhibitor of PfCDPK4 blocks malaria transmission: chemical-genetic validation. J Infect Dis 209: 275–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panchal D, and Bhanot P (2010) Activity of a trisubstituted pyrrole in inhibiting sporozoite invasion and blocking malaria infection. Antimicrob Agents and Chemother 54: 4269–4274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panchal D, Govindasamy K, Rana A, and Bhanot P (2012) Improved Plasmodium berghei lines for conditional mutagenesis. Mol Biochem Parasitol 184: 52–54. [DOI] [PubMed] [Google Scholar]
- Philip N, and Waters AP (2015) Conditional degradation of Plasmodium calcineurin reveals functions in parasite colonization of both host and vector. Cell Host Microbe 18: 122–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risco-Castillo V, Topcu S, Marinach C, Manzoni G, Bigorgne AE, Briquet S, et al. (2015) Malaria sporozoites traverse host cells within transient vacuoles. Cell Host Microbe. 18: 593–603. [DOI] [PubMed] [Google Scholar]
- Sebastian S, Brochet M, Collins MO, Schwach F, Jones ML, Goulding D, et al. (2012) A Plasmodium calcium-dependent protein kinase controls zygote development and transmission by translationally activating repressed mRNAs. Cell Host Microbe 12: 9–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siden-Kiamos I, Ecker A, Nyback S, Louis C, Sinden RE, and Billker O (2006) Plasmodium berghei calcium-dependent protein kinase 3 is required for ookinete gliding motility and mosquito midgut invasion. Mol Microbiol 60: 1355–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvie O, Franetich JF, Charrin S, Mueller MS, Siau A, Bodescot M, et al. (2004) A role for apical membrane antigen 1 during invasion of hepatocytes by Plasmodium falciparum sporozoites. J Biol Chem 279: 9490–9496. [DOI] [PubMed] [Google Scholar]
- Sinnis P, De La Vega P, Coppi A, Krzych U, and Mota MM (2013) Quantification of sporozoite invasion, migration, and development by microscopy and flow cytometry. Methods Mol Biol 923: 385–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinbuechel M, and Matuschewski K (2009) Role for the Plasmodium sporozoite-specific transmembrane protein S6 in parasite motility and efficient malaria transmission. Cell Microbiol 11: 279–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sultan AA, Thathy V, Frevert U, Robson KJ, Crisanti A, Nussenzweig V, Nussenzweig RS, and Menard R (1997) TRAP is necessary for gliding motility and infectivity of Plasmodium sporozoites. Cell 90: 511–522. [DOI] [PubMed] [Google Scholar]
- Takala-Harrison S, Jacob CG, Arze C, Cummings MP, Silva JC, Dondorp AM, et al. (2015) Independent emergence of artemisinin resistance mutations among Plasmodium falciparum in Southeast Asia. J Infect Dis 211: 670–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor HM, McRobert L, Grainger M, Sicard A, Dluzewski AR, Hopp CS, et al. (2010) The malaria parasite cyclic GMP-dependent protein kinase plays a central role in blood-stage schizogony. Eukaryot Cell 9: 37–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuji M, Mattei D, Nussenzweig RS, Eichinger D, and Zavala F (1994) Demonstration of heat-shock protein 70 in the sporozoite stage of malaria parasites. Parasitol Res 80: 16–21. [DOI] [PubMed] [Google Scholar]
- Vidadala RS, Ojo KK, Johnson SM, Zhang Z, Leonard SE, Mitra A, et al. (2014) Development of potent and selective Plasmodium falciparum calcium-dependent protein kinase 4 (PfCDPK4) inhibitors that block the transmission of malaria to mosquitoes. Eur J Med Chem 74: 562–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiersma HI, Galuska SE, Tomley FM, Sibley LD, Liberator PA, and Donald RG (2004) A role for coccidian cGMP-dependent protein kinase in motility and invasion. Int J Parasitol 34: 369–380. [DOI] [PubMed] [Google Scholar]
- Zhang C, Kenski DM, Paulson JL, Bonshtien A, Sessa G, Cross JV, et al. (2005) A second-site suppressor strategy for chemical genetic analysis of diverse protein kinases. Nat Methods 2: 435–441. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.