

Abstract
Hypertrophic cardiomyopathy (HCM) is often caused by pathogenic variants in sarcomeric genes and characterized by left ventricular (LV) hypertrophy, myocardial fibrosis and increased risk of heart failure and arrhythmias. There are no existing therapies to modify disease progression. In this study, we conducted a multi-center, double-blind, placebo-controlled phase 2 clinical trial to assess the safety and efficacy of the angiotensin II receptor blocker valsartan in attenuating disease evolution in early HCM. In total, 178 participants with early-stage sarcomeric HCM were randomized (1:1) to receive valsartan (320 mg daily in adults; 80–160 mg daily in children) or placebo for 2 years (NCT01912534). Standardized changes from baseline to year 2 in LV wall thickness, mass and volumes; left atrial volume; tissue Doppler diastolic and systolic velocities; and serum levels of high-sensitivity troponin T and N-terminal pro-B-type natriuretic protein were integrated into a single composite z-score as the primary outcome. Valsartan (n = 88) improved cardiac structure and function compared to placebo (n = 90), as reflected by an increase in the composite z-score (between-group difference +0.231, 95% confidence interval (+0.098, +0.364); P = 0.001), which met the primary endpoint of the study. Treatment was well-tolerated. These results indicate a key opportunity to attenuate disease progression in early-stage sarcomeric HCM with an accessible and safe medication.
HCM is a primary heart muscle disorder defined by left ventricular hypertrophy (LVH) that is not attributable to extrinsic factors, such as pressure overload1. HCM is often familial, and it was the first heritable cardiovascular disorder to have genetic etiology determined. Seminal studies in the 1980s and 1990s established the paradigm that HCM is a disease of the sarcomere, most commonly caused by pathogenic variants in genes encoding the contractile apparatus2. Patients with HCM are at increased risk of atrial fibrillation, heart failure and sudden cardiac death. Additionally, HCM is progressive, and disease burden increases over an individual’s lifetime, particularly in patients with sarcomeric disease3. Thus, developing therapies to counteract the pathobiology of sarcomeric variants, thereby slowing progression or preventing emergence of disease, is an important unmet need.
Studies in genetically modified mouse models of sarcomeric HCM indicated that activation of transforming growth factor-beta (TGF-β) is centrally involved in triggering the development of myocardial hypertrophy and fibrosis4–6. Angiotensin receptor blockers (ARBs) can inhibit TGF-β activation7, and administration of the ARB losartan to HCM mice abrogated development of LVH and fibrosis if administered early in life, when cardiac morphology was normal. However, ARB treatment did not appear to be beneficial if administered after hypertrophy was established6. Similarly, human clinical trials using ARBs failed to show substantive clinical benefit in adults with well-established HCM8–13. The Valsartan for Attenuating Disease Evolution in Early Sarcomeric Hypertrophic Cardiomyopathy (VANISH) trial was designed to test a novel strategy of disease modification by assessing the efficacy and safety of the ARB valsartan in individuals with early-stage HCM, based on young age and absence of severe LVH or limiting symptoms14,15.
Results
Study participants.
From April 2014 to May 2017, a total of 219 participants entered active run-in and were randomly assigned to study treatment at 17 sites in four countries. Participating sites, investigators, core laboratory directors and executive committee members are listed in Supplementary Table 1 in the Supplementary Appendix. Participant flow is shown in Fig. 1. Seven participants withdrew during active run-in, and 34 qualified for a parallel exploratory cohort of preclinical sarcomeric variant carriers with normal LV wall thickness and no diagnosis of HCM (to be reported separately). A total of 178 participants were randomized to the primary analysis cohort reported in this study. Of these, 88 were assigned to receive valsartan, and 90 were assigned to receive placebo. The mean (standard deviation) age was 23.3 (10.1) years, and 39% of participants were female. The groups were relatively balanced with respect to baseline characteristics (Tables 1 and 2). The valsartan group had slightly greater maximal LV wall thickness and N-terminal pro-B-type natriuretic protein (NTproBNP) levels. As pre-specified in the statistical analysis plan, models were adjusted for baseline values of these and other variables, indicated in the Methods and Tables 1 and 2.
Fig. 1 |.
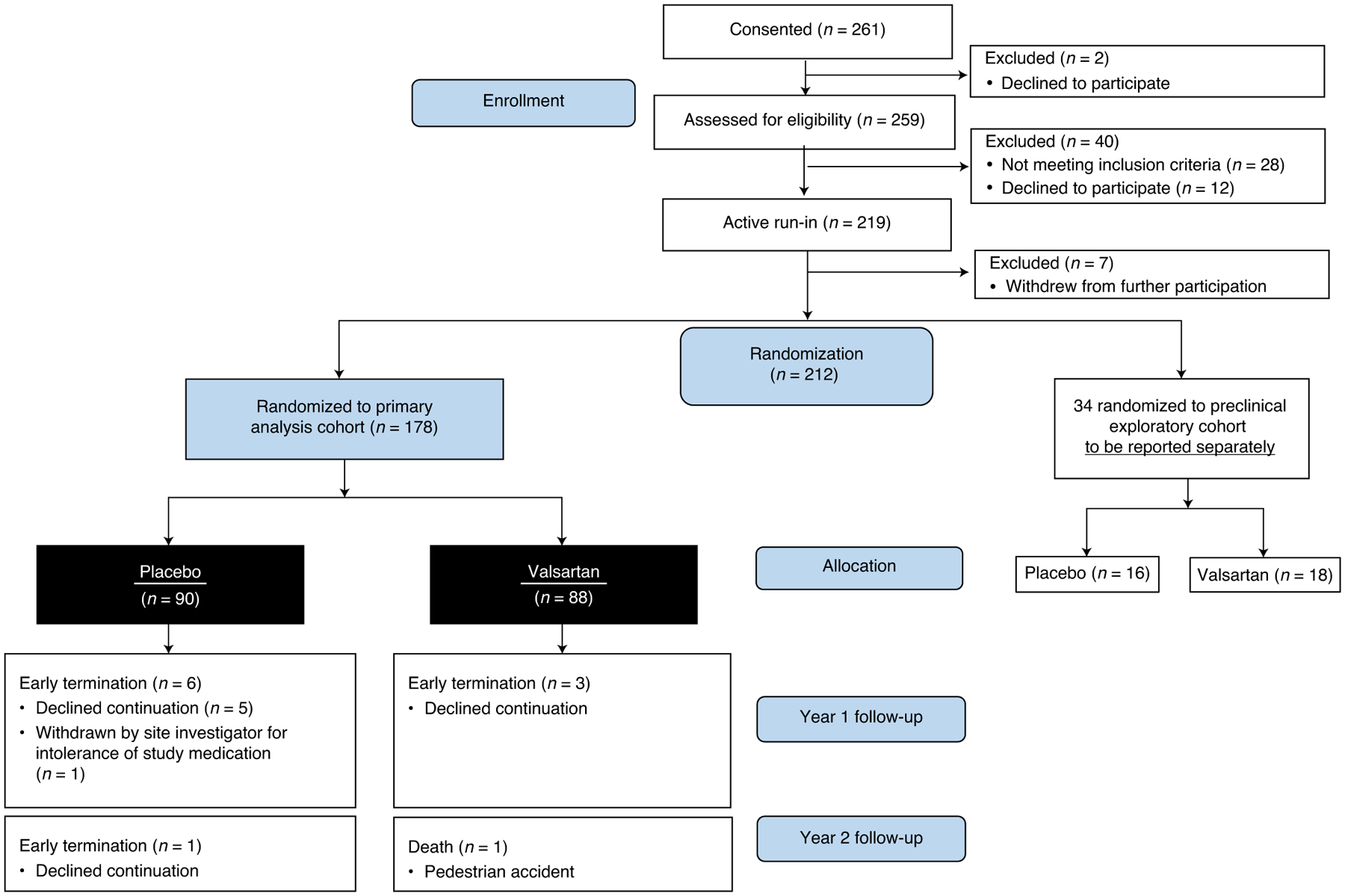
Participant enrollment, allocation and follow-up.
Table 1 |.
Baseline participant characteristics, by arm
| Placebo n = 90 | Valsartan n = 88 | |
|---|---|---|
| Mean age*, years | 23.5 (10.1) | 23.1 (10.1) |
| 18 years of age or younger, n (%) | 39 (43%) | 38 (43%) |
| Pre-puberty*, n (%) | 20 (22%) | 19 (22%) |
| Female*, n (%) | 35 (39%) | 34 (39%) |
| White*, n (%) | 88 (98%) | 85 (97%) |
| Black, n (%) | 0 | 3 (3%) |
| Unknown, n (%) | 2 (2%) | 0 |
| Hispanic, n (%) | 21 (23%) | 15 (17%) |
| Sarcomeric gene*, n (%) | ||
| MYH7 | 36 (40%) | 25 (28%) |
| MYBPC3 | 44 (49%) | 47 (53%) |
| TNNT2 | 3 (3%) | 5 (6%) |
| TNNI3 | 2 (2%) | 3 (3%) |
| TPM1 | 1 (1%) | 4 (5%) |
| MYL2 | 1 (1%) | 1 (1%) |
| MYL3 | 2 (2%) | 1 (1%) |
| ACTC | 1 (1%) | 2 (2%) |
| Mean BSA, m2 | 1.85 (0.35) | 1.82 (0.34) |
| Mean BMI* | 25.6 (5.9) | 25.0 (5.6) |
| Systolic blood pressure*, mmHg | 118 (13) | 118 (10) |
| Diastolic blood pressure, mmHg | 69 (11) | 68 (10) |
| NYHA*: Class I, n (%) | 84 (93%) | 80 (91%) |
| Class II, n (%) | 6 (7%) | 8 (9%) |
| BSA-adjusted z-score for maximum LV wall thickness (participants ≤18 years) | 8.1 (4.6) | 8.2 (5.1) |
| Maximum LV wall thickness, mm (participants >18 years) | 16.4 (3.4) | 17.9 (4.7) |
| LVEF*, % | 66.3 (7.2) | 66.1 (5.8) |
| Beta blocker use*, n (%) | 14 (16%) | 18 (20%) |
| Calcium channel blocker use*, n (%) | 1 (1%) | 4 (5%) |
Numbers are mean and standard deviation, unless otherwise indicated.
Variables pre-specified to be adjusted in the analysis model. Because of sparse categories, race was dichotomized to white versus non-white, and genotype was dichotomized to thick filament genes (MYBPC3, MYH7, MYL2 or MYL3) versus thin filament genes (ACTC, TNNT2, TNNI3 or TPM1). One participant in the placebo group was missing LVEF.
Table 2 |.
Baseline values for outcome components, by arm
| Placebo n = 90 | Valsartan n = 88 | |
|---|---|---|
| Troponin T, ng l−1 (99th percentile reference limit <19 ng l−1) | ||
| Geometric mean (IQR) | 7.3 (5.0, 8.7) | 7.8 (5.0, 10) |
| NTproBNP, pg ml−1 | ||
| Geometric mean (IQR) | 86 (36, 200) | 124 (45, 420) |
| LV mass index, g m−2 | 72 (25) | 74 (23) |
| Left atrial volume index, ml m−2 | 39 (16) | 38 (14) |
| LV end diastolic volume index, ml m−2 | 74 (16) | 74 (17) |
| LV end systolic volume index, ml m−2 | 25 (9.2) | 24 (7.3) |
| Max LV wall thickness, mm | ||
| Mean | 15.5 (3.6) | 16.5 (4.6) |
| BSA-adjusted z-score* | 8.2 (4.1) | 9.5 (5.1) |
| E′ velocity, cm s−1 | ||
| Mean | 9.54 (2.60) | 9.38 (3.67) |
| Age-adjusted z-score | −1.8 (1.0) | −1.8 (1.5) |
| S′ velocity, cm s−1 | ||
| Mean | 8.03 (1.42) | 7.68 (1.46) |
| Age-adjusted z-score | −0.14 (1.1) | −0.35 (1.2) |
Numbers are mean and s.d., unless otherwise indicated.
Variables pre-specified to be adjusted in the analysis model. Two participants in the placebo group were missing troponin T and NTproBNP. Troponin T values less than 6 were set = 5; NTproBNP values less than 5 were set = 4. One person in the valsartan group was missing LV mass, and one was missing S′ velocity. IQR, interquartile range.
Study drug administration and follow-up.
Of the 88 participants on valsartan, one died (pedestrian accident), and three withdrew from the trial before completion of follow-up. Of the 90 participants on placebo, zero died, six withdrew from the trial and one was withdrawn by the site investigator for study drug intolerance (Fig. 1). Imputation was used to provide year 2 data for these 11 participants. Twenty-six participants (13 in each arm, including the 11 participants above who withdrew early) stopped the study drug before the end of the treatment period. The remaining 152 participants completed the study on target dose with an average adherence of ~86% for valsartan and ~87% for placebo.
Study outcomes.
The complex alterations in cardiac structure and function that occur in response to growth and disease progression were interrogated by standardizing and integrating nine individual metrics into a composite z-score that assessed the change from baseline to year 2 (Methods). These metrics included: body surface area (BSA)-indexed LV mass and BSA-indexed left atrial (LA) volume (decrease considered improvement); BSA-indexed LV end diastolic and end systolic volumes (increase considered improvement) determined by the cardiac magnetic resonance (CMR) imaging core laboratory (echocardiographic core laboratory measures substituted if CMR studies could not be performed); BSA-adjusted maximal LV wall thickness (decrease considered improvement); age-adjusted tissue Doppler diastolic (E′) and systolic (S′) velocities (increase considered improvement) determined by the echocardiographic core laboratory; and log-transformed serum high-sensitivity cardiac troponin T (TnT) and NTproBNP levels (decrease considered improvement). For each patient, the nine individual z-scores for change in each metric from baseline to end of study at year 2 were averaged to produce the composite z-score, which was the pre-specified primary outcome16. An increase (positive change) in the composite z-score indicates a greater than average improvement from baseline to end of study.
Participants assigned to valsartan demonstrated significant improvement in the primary outcome. The change in the composite z-score from baseline to year 2 with valsartan was +0.136, 95% confidence interval (+0.049, +0.223), compared to −0.095 (−0.192, +0.002) with placebo; between-group difference was +0.231 (+0.098, +0.364), P = 0.001 (Table 3). Pre-specified exploratory secondary analyses of the individual components of the composite outcome indicated that the largest contributors to the improvement in the composite outcome in favor of valsartan administration were E′ velocity, LV end diastolic volume and NTproBNP level. Sensitivity analyses excluding the 11 participants who withdrew from the trial early gave consistent results (Supplementary Table 2 in the Supplementary Appendix). Early during the enrollment phase of the trial, eligibility criteria were revised to increase the upper age limit from 30 years to 45 years and allowable maximal LV wall thickness from 20 mm (BSA-adjusted z-score 10) to 25 mm (BSA-adjusted z-score 18). Sensitivity analyses performed on participants meeting the original criteria (n = 104; most of the decrease in sample size was due to the change in the age limit) demonstrated increased responsiveness in this group of younger participants with more modest LVH. Results are provided in Supplementary Table 3 of the Supplementary Appendix. Owing to concerns that the revised eligibility criteria allowed inclusion of participants with severe LVH, sensitivity analyses excluding participants with maximal LV wall thickness >20 mm were also performed and showed consistent results. Owing to potential ambiguity regarding the long-term clinical implications of an increase in LV cavity size, sensitivity analyses were performed omitting LV volumes as components of the composite outcome and showed similar findings (Supplementary Table 3). Pre-specified exploratory analysis of the number of components of the composite outcome that showed improvement demonstrated a greater improvement with valsartan than placebo (Supplementary Table 4 in the Supplementary Appendix).
Table 3 |.
Primary and secondary efficacy outcomes, intention-to-treat
| Adjusted* mean change in raw measure | Between-group differences | Adjusted* mean change in normalized measure | Between-group differences | Covariate-adjusted* P value | |||
|---|---|---|---|---|---|---|---|
| Placebo (n = 90) | Valsartan (n = 88) | Placebo (n = 90) | Valsartan (n = 88) | ||||
| Primary outcome | NA | NA | −0.095 | +0.136 | +0.231 | 0.001 | |
| Composite z-score | [−0.192, +0.002] | [+0.049, +0.223] | [+0.098, +0.364] | ||||
| Components of the primary outcome | |||||||
| Decrease in value indicates improvement | |||||||
| Maximum LV wall | +1.57 | +0.40 | −1.17 | +1.32 | +0.16 | −1.16 | 0.06 |
| Thicknessa, mm | [+0.80, +2.35] | [−0.32, +1.12] | [−2.21, −0.14] | [+0.42, +2.21] | [−0.69, +1.00] | [−2.37, +0.06] | |
| LV mass indexb, g m−2 | NA | NA | NA | −1.11 | −4.32 | −3.20 | 0.12 |
| [−4.04, +1.81] | [−7.32, −1.32] | [−0.81, +7.22] | |||||
| LA volume indexb, ml m−2 | NA | NA | NA | +1.45 | +2.47 | +1.02 | 0.59 |
| [−1.27, +4.17] | [−0.31, +5.25] | [−2.70, +4.74] | |||||
| NTproBNPc, pg ml−1 | +84.2 | +23.1 | −61.0 | +0.27 | +0.02 | −0.25 | 0.03 |
| [+23.1, 1+45.2] | [−36.6, +82.9] | [−122.9, +0.9] | [+0.11, +0.43] | [−0.14, +0.18] | [−0.47, −0.03] | ||
| Troponin Tc, ng l−1 | +1.18 | +1.44 | −0.27 | +0.11 | +0.06 | −0.05 | 0.37 |
| [−0.70, +3.05] | [−0.42, +3.30] | [−2.37, +1.84] | [+0.02, +0.20] | [−0.02, +0.14] | [−0.16, +0.06] | ||
| Increase in value indicates improvement | |||||||
| LV end diastolic volume | NA | NA | +0.41 | +4.51 | +4.10 | 0.047 | |
| Indexd, ml m−2 | [−2.77, +3.59] | [+1.41, +7.61] | [+0.06, +8.14] | ||||
| LV end systolic volume | NA | NA | +2.20 | +3.69 | +1.49 | 0.24 | |
| Indexb, ml m−2 | [+0.28, +4.13] | [+1.77, +5.62] | [−1.00, +3.97] | ||||
| E′ velocityd, cm s−1 | −0.83 | +0.09 | +0.92 | −0.33 | +0.016 | +0.35 | 0.02 |
| [−1.37, −0.29] | [−0.39, +0.57] | [0.24, +1.61] | [−0.57, −0.10] | [−0.19, +0.22] | [+0.06, +0.63] | ||
| S′ velocityd, cm s−1 | −0.15 | +0.02 | 0.17 | −0.165 | −0.031 | +0.13 | 0.45 |
| [−0.48, +0.17] | [−0.29, +0.33] | [−0.26, +0.61] | [−0.43, +0.10] | [−0.28, +0.21] | [−0.21, +0.48] | ||
| Improvement on any componente | NA | NA | 91% | 95% | Unadjusted odds ratio | 0.37 | |
| 2.05 | |||||||
| [0.59, 7.07] | |||||||
Results of mixed model linear regressions comparing changes in the outcome between treatment groups, adjusting through random effects for clustering within sites and within families. Data are presented as marginal mean change at year 2 compared to baseline, with pre-specified adjustment for *: baseline value of the outcome (normalized), NYHA class, maximum LV wall thickness, sex, age, pubertal status, body mass index, race (white versus non-white), Hispanic ethnicity, genotype (thick versus thin filament variants), LVEF, systolic blood pressure, beta blocker use and calcium channel blocker use. Between-group difference: valsartan–placebo. P values for secondary outcomes were not adjusted for multiple testing.
Normalized measure is adjusted for BSA, but no adjustment is made in the raw measure.
Normalized measure is indexed to BSA.
Normalized measure was log-transformed, but no adjustment is made in the raw measure.
Normalized measure is adjusted for age, but no adjustment is made in the raw measure.
Unadjusted and based on unimputed data; too few people had no improvements to model. Not applicable (NA) is indicated because only BSA-indexed values of LV mass, LA volume, LV end diastolic volume and LV end systolic volume can be validly combined into the composite z-score, owing to the strong influence of body size on these measures.
In pre-specified exploratory subgroup analyses (Fig. 2 and Supplementary Table 5 in the Supplementary Appendix), the benefit of valsartan was most striking in participants with less hypertrophic remodeling. Participants with baseline maximal LV wall thickness less than or equal to the median value for the cohort (BSA-adjusted z-score of 7.3) showed a between-group difference in the composite z-score of +0.368 (+0.169, +0.567). By contrast, participants with baseline LV wall thickness greater than the median had a between-group difference of +0.069 (−0.115, +0.249) (P for interaction = 0.04). Otherwise, the effect of valsartan was consistent across the remaining pre-specified subgroups. Relative improvement in the primary composite outcome was seen with valsartan in males and females, with participants older and younger than 18 years and with disease caused by variants in MYH7 or MYBPC3 genes (Fig. 2).
Fig. 2 |. Forest plots of pre-specified subgroups and secondary outcomes.
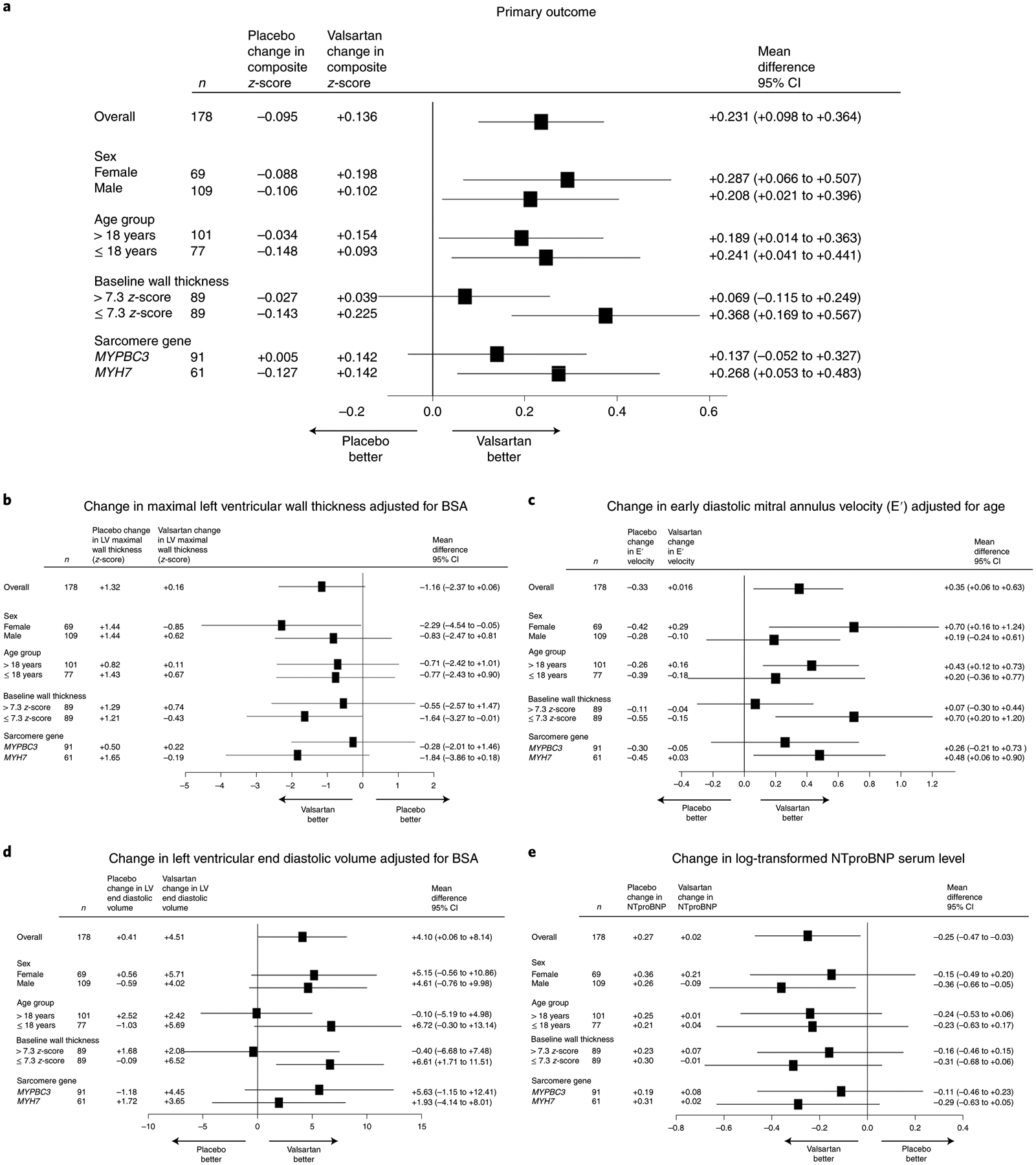
a, Mean difference in primary composite z-score between placebo- and valsartan-treated participants, along with 95% confidence interval, for pre-specified subgroups. b–e, Mean differences in z-score for the following key endpoint components: BSA-adjusted LV maximal wall thickness (b), age-adjusted tissue Doppler diastolic velocity (e′) (c), BSA-adjusted LV end diastolic volume (d) and log-transformed NTproBNP levels (e). CI, confidence interval.
Figure 3 displays values of key secondary endpoints over time during treatment. Maximal LV wall thickness, E′ velocity and NTproBNP levels remained stable or improved in the valsartan group but worsened progressively in the placebo group. LV end diastolic volume improved in the valsartan group while remaining stable in the placebo group.
Fig. 3 |. Longitudinal changes in key components of the primary composite outcome.
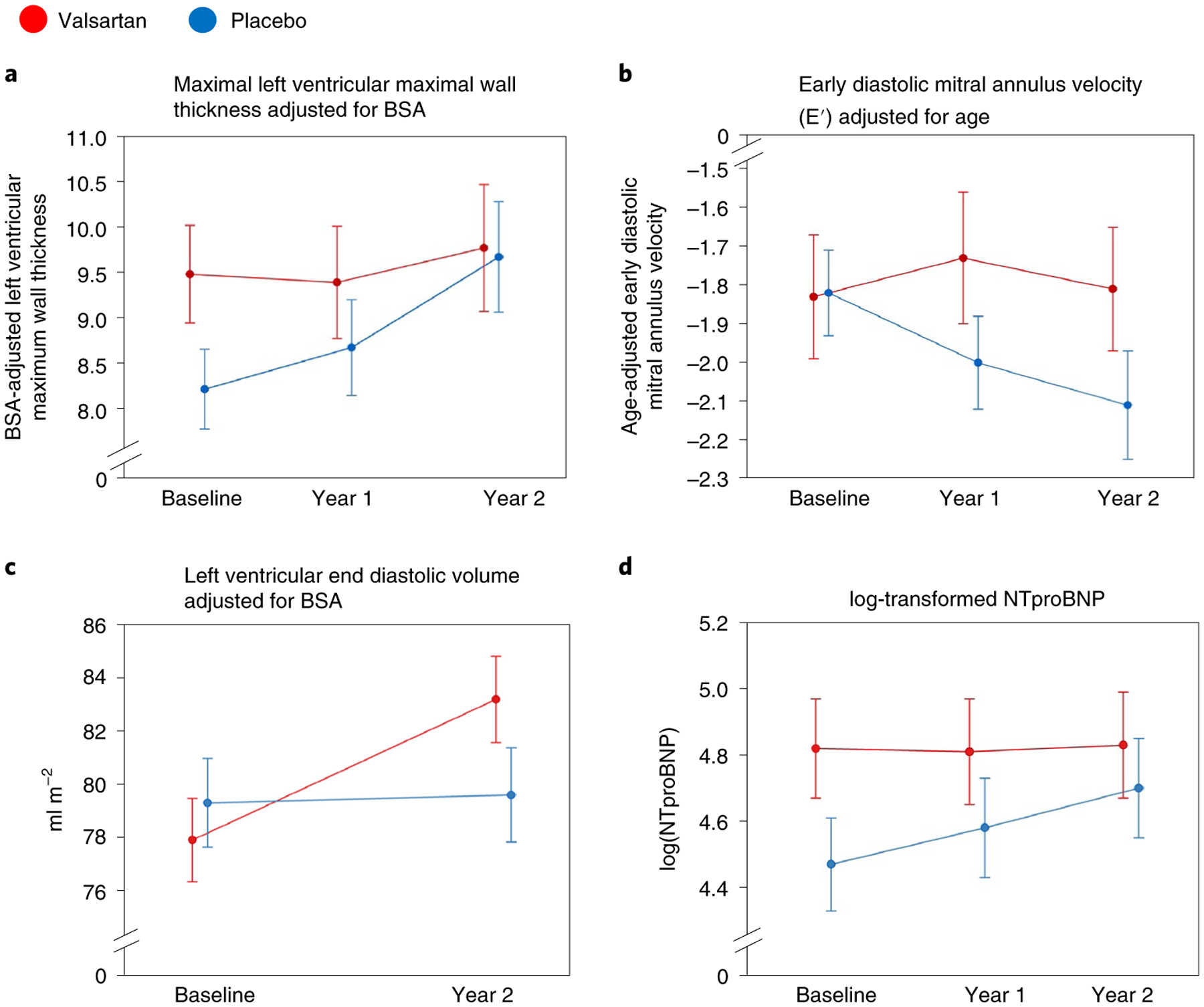
Values at baseline, year 1 and year 2 (end of study) for the valsartan (n = 88; red) and placebo (n = 90; blue) groups are presented as mean and standard error of the mean. a–d, Shown are data for BSA-adjusted z-score for maximal LV wall thickness (a), age-adjusted tissue Doppler diastolic velocity (E′) (b), BSA-indexed LV end diastolic volume, measured by CMR imaging performed only at baseline and year 2 (echo measures substituted for both time points if participants could not undergo either baseline or year 2 CMR imaging) (c) and log-transformed NTproBNP serum levels (d).
Safety and adverse events.
The safety and tolerability of valsartan were similar to placebo. Compared to placebo, average systolic blood pressure decreased by 3 mmHg (P = 0.07) and diastolic blood pressure decreased by 4 mmHg (P = 0.004) in the valsartan group. Serum potassium and creatinine levels were stable over time and similar in both groups (Extended Data Figs. 1 and 2 and Supplementary Table 6 in the Supplementary Appendix). There was one permanent treatment discontinuation for study drug intolerance in the placebo group but none in the valsartan group. There were no instances of hyperkalemia (serum K >5.2 mmol L−1), renal insufficiency (eGFR <75 ml/min/1.73m2) or development of obstructive HCM physiology. The incidence and severity of adverse events were similar in the treatment arms (Supplementary Table 7 in the Supplementary Appendix). There were eight serious adverse events in eight participants on valsartan (one considered possibly related to the study drug) and 14 in ten participants on placebo.
Discussion
The VANISH trial tested a novel strategy of disease modification in sarcomeric HCM, specifically whether administering ARBs early in the course of disease could attenuate progression of this genetic cardiomyopathy. Compared to placebo, we found that valsartan improved the primary composite outcome, which integrated nine measures of cardiac structure/function and remodeling. Analysis of pre-specified secondary outcomes showed that the largest effects of valsartan were on tissue Doppler diastolic velocity, LV end diastolic volume and serum NTproBNP levels. These measures were stable or improved over 2 years of treatment in the valsartan group, but diastolic velocity and NTproBNP worsened in the placebo group, along with LV wall thickness and cardiac troponin T levels. These results suggest that valsartan might not only stabilize disease progression but might also promote improvement. Additionally, valsartan treatment was safe and well-tolerated with no excess of adverse events and no instances of treatment withdrawal for drug intolerance, including hypotension, hyperkalemia or renal insufficiency.
The rationale for VANISH drew from studies on mouse models of sarcomeric HCM where treatment with either diltiazem or losartan appeared to attenuate disease progression but only if started before the development of LVH6,17. If treatment was started after an overt HCM phenotype developed, no significant benefit was detected. Similarly, previous clinical trials of angiotensin-converting enzyme inhibitors and ARBs in human HCM have not shown convincing benefit in older adult patients with well-established disease and undefined or heterogeneous genetic etiology8–13. By contrast, VANISH enrolled young individuals (mean age ~20–30 years younger than previous studies) with confirmed sarcomeric HCM (thus, homogeneous and defined disease etiology) and more modest disease expression (average maximal LV wall thickness 16 mm in VANISH versus 21 mm in other trials)15. Therefore, VANISH participants were anticipated to be more likely to respond to disease-modifying therapy. Indeed, treatment benefit in VANISH was more striking in participants with LV wall thickness below the median value. This finding supports the hypothesis that disease-modifying therapy might be most effective when started early in the disease course or in the absence of marked hypertrophic remodeling. Collectively, these experiences underscore the importance of precision in translating studies from bench to bedside, indicate that both the target population and timing of therapy are likely to be important and highlight the potential utility of genetic testing in guiding management.
It is important to recognize the substantial challenges to developing effective disease-modifying therapeutic strategies. In particular, it is difficult to demonstrate treatment benefit in participants who are healthy at baseline, have extremely low clinical event rates and have conditions whose phenotypic manifestations and natural history evolve over decades. In VANISH, we addressed these challenges by developing a primary composite outcome designed to interrogate disease biology rather than traditional long-term clinical outcomes, such as mortality and major adverse cardiac events. This composite outcome integrated multiple complementary objective metrics to capture dynamic changes in cardiac remodeling in response to growth and disease progression. Although this approach was necessary to maximize opportunity to detect treatment effect and impact on cardiac structure/function, the relationship between the composite endpoint and long-term clinical outcomes has not yet been determined. However, determining these relationships will require studying a large number of patients with a relatively rare disease for many years, potentially decades. Conducting such a trial might not be feasible. Recognizing the absence of alternative strategies for disease modification, physicians and patients might consider the findings from this trial and the low-risk nature of valsartan therapy to make individualized decisions to use valsartan with the intent to modify disease progression. Based on this trial, it might be most effective to start treatment in younger patients with more modest LVH early after HCM is diagnosed, but it is important to recognize that the effect of valsartan on long-term clinical outcomes is unknown. Determining how the composite primary endpoint relates to clinical outcomes and identifying additional robust, biologically relevant and clinically translatable metrics to track the evolution of disease and response to therapy are essential to continue developing effective disease-modifying treatment strategies.
We also recognize that maximal LV wall thickness and NTproBNP levels were lower at baseline in participants randomized to placebo despite stratifying randomization by baseline LV wall thickness. Although models were adjusted for baseline values, it remains possible that some of the overall favorable effect on the primary composite endpoint for these two measures individually was partly driven by regression to the mean in the placebo group. Additionally, the exclusive focus on sarcomeric HCM in VANISH might have facilitated detection of favorable effects of valsartan on cardiac remodeling. Future studies in patients with early-stage, non-sarcomeric HCM will be needed to address the broader generalizability of these results. Further studies are also needed to better characterize long-term treatment effects and to determine optimal timing of medication administration.
In conclusion, the VANISH trial leveraged insights gained in elucidating disease mechanism and targeted a genetically characterized cohort with HCM. Valsartan improved a composite score integrating measures of cardiac structure/function and remodeling in patients with early-stage sarcomeric HCM. Treatment with valsartan had beneficial effects on cardiac dimensions, tissue Doppler diastolic velocity and NTproBNP levels compared to placebo. These findings suggest an important opportunity to attenuate disease progression in sarcomeric HCM with a widely available and well-tolerated pharmacological intervention.
Methods
Study oversight.
VANISH was funded by the National Institutes of Health/National Heart, Lung, and Blood Institute and is registered at ClinicalTrials.gov (NCT01912534). An executive committee (E.B., C.Y.H., C.A.M., J.J.V.M., E.J.O. and S.D.S.) designed and oversaw the conduct of the trial and performed data analysis. Study medication was donated by Novartis, which played no role in the design, conduct or analysis of the trial. The trial was conducted and reported in accordance with the protocol and the statistical analysis plan, both of which are available in the Supplementary Note. The trial was approved by the ethics committee at each center, and all participants provided written informed consent and youth assent as appropriate. The safety of patients in the trial was overseen by an independent data and safety monitoring committee. An independent clinical events committee, unaware of treatment group assignments, adjudicated predefined clinical events.
Participants.
All participants were required to carry a pathogenic or likely pathogenic HCM sarcomeric variant. Variant pathogenicity was determined using standard criteria accounting for segregation, conservation, published information and public databases and absence or very low frequency in appropriate control populations18,19. Investigators (C.Y.H., A.L.C. and C.E.S.) with expertise in genetics reviewed questionable variants to determine eligibility by consensus. Genetic variants are listed in Supplementary Table 8 in the Supplementary Appendix. Eligibility criteria included age 8–45 years, LV wall thickness 12–25 mm (or BSA-adjusted z-score 3–18 for pediatric participants), New York Heart Association (NYHA) Class I or II, absence of resting or provoked LV outflow gradients >30 mmHg, LV ejection fraction (LVEF) >55%, no contraindication to receiving ARB, no prior septal reduction and absence of appropriate implantable cardioverter–defibrillator (ICD) therapy or secondary prevention ICD. Full inclusion and exclusion criteria are listed in Supplementary Table 9 in the Supplementary Appendix. Early during the enrollment phase of the trial, eligibility criteria were revised to increase the upper age limit from 30 years to 45 years and maximal LV wall thickness from 20 mm (BSA-adjusted z-score 10) to 25 mm (BSA-adjusted z-score 18) to enhance enrollment. Sensitivity analyses were conducted to assess trial results using the original eligibility criteria. A parallel exploratory study enrolled familial sarcomeric variant carriers aged 10–25 years in the preclinical stage (normal LV wall thickness and no diagnosis with HCM) that will be analyzed and reported separately (Fig. 1).
Study design and procedures.
VANISH was a multi-center, placebo-controlled, double-blind, randomized clinical trial conducted in 17 HCM specialty centers in four countries. Participating sites, investigators, core laboratory directors and executive committee members are listed in Supplementary Table 1 in the Supplementary Appendix. Details of the study design and baseline characteristics were reported previously14,15. Eligible participants entered an active run-in period during which they received valsartan titrated to target dose (adults, 320 mg daily; children <18 years old weighing ≥35 kg, 160 mg daily; children <18 years old weighing <35 kg, 80 mg daily). Participants who successfully completed active run-in were randomly assigned in a 1:1 ratio to receive target dose valsartan or placebo for 2 years. Randomization was performed by the data coordinating center (New England Research Institutes/HealthCore). Because age, pubertal status (males ≥17 years and females ≥16 years were considered post-pubertal), NYHA class and LV wall thickness (<14 mm or ≥14 mm or BSA-adjusted z-score of 6) were anticipated to influence phenotypic expression and outcomes, randomization was stratified by each of these factors using permuted blocks.
Study visits occurred at baseline, year 1 and year 2, consisting of CMR and cardiopulmonary exercise testing (baseline and year 2 only), echocardiography, electrocardiography, safety laboratories and collection of blood for biomarker assessment, quality of life and activity questionnaires (all visits). Quarterly phone interviews were conducted to monitor for symptoms and adverse events.
The trial was double-blind, with participants, investigators, site staff and core laboratories (echocardiographic, CMR imaging, cardiopulmonary exercise testing and biomarker) masked to treatment assignment. Valsartan and matching placebo were identical in appearance. Study drug adherence was monitored by pill count.
Study outcomes.
The primary objective of VANISH was to investigate whether treatment with valsartan attenuated phenotypic progression in early-stage sarcomeric HCM by assessing changes in multiple metrics of cardiac structure and function from baseline to end of study (year 2). To capture the complex structural and functional remodeling and changes in response to cardiac growth and disease progression, we integrated individual metrics into a composite score. Based on previous studies that identified features that differentiated preclinical sarcomeric variant carriers from both healthy controls and clinically overt sarcomeric HCM20–23, we chose nine metrics to monitor treatment response/disease progression. Specifically, these included: BSA-indexed LV mass and BSA-indexed LA volume (decrease considered improvement); BSA-indexed LV end diastolic and end systolic volumes (increase considered improvement) determined by the CMR core laboratory (echocardiographic core laboratory measures substituted if CMR studies could not be performed); BSA-adjusted maximal LV wall thickness (decrease considered improvement); age-adjusted tissue Doppler diastolic (E′) and systolic (S′) velocities (increase considered improvement) determined by the echocardiographic core laboratory; and log-transformed serum high-sensitivity cardiac troponin T and NTproBNP levels (decrease considered improvement). Owing to pediatric and adolescent participation, echocardiographic measures were standardized to BSA- or age-adjusted z-scores, as appropriate, to account for differences in body size and age. For each patient, values were aligned so that a positive difference represented improvement for each metric when the value of each metric at year 2 was subtracted from its baseline value. These differences were converted to z-scores for change for each metric by subtracting the mean change for the cohort and dividing by the standard deviation of the change for the cohort. Finally, for each patient, the nine individual z-scores for change were averaged to produce the composite z-score, which was the pre-specified primary outcome16. A positive composite z-score indicates a greater than average improvement from baseline and end of study.
Statistical analysis.
Our primary efficacy analysis compared the composite z-scores (described above) between the study arms. We analyzed all randomized participants according to the intention-to-treat principle. Multiple imputation (MI Procedure in SAS (SAS Institute)) was used for missing data. Of the 3,204 variables measured for the nine components of the primary composite outcome in the cohort, fewer than 4% were missing and required imputation. The multiple imputation process used chained equations and a fully conditional specification to allow for non-normal variables. Logistic regression was used to impute binary variables, and skewed variables were log-transformed before imputation. The missing pattern was a combination of monotone missingness (due to early withdrawals) and arbitrary missingness (primarily due to insufficient blood samples or values outside of detection limits). Therefore, arbitrary missingness was allowed for the multiple imputation process with calculations carried out through chained equations.
We estimated that 150 participants (75 per treatment group) would provide 76–88% power to detect standardized effect sizes of 0.22 (moderate effect) to 0.25 (large effect) for the composite z-score outcome, respectively14. We also compared each of the nine components of the composite score as pre-specified secondary outcomes.
Our analyses of the composite z-score and its components were based on mixed model linear regressions, adjusting through random effects for clustering within sites and within families. Models compared changes in the outcome between the treatment groups and were adjusted for the following pre-specified patient characteristics: baseline value of the outcome, NYHA class, maximum LV wall thickness, sex, age, pubertal status, body mass index, race (white versus non-white), genotype (thick filament genes (myosin heavy chain (MYH7), myosin binding protein C (MYBPC3), myosin light chains (MYL2, MYL3)) or thin filament genes (actin (ACTC), tropomyosin (TPM1), cardiac troponin T (TNNT2), cardiac troponin I (TNNI3)), LVEF, systolic blood pressure, beta blocker use and calcium channel blocker use. Model details with covariate effects are shown in Supplementary Table 10 in the Supplementary Appendix. Results are presented as adjusted marginal means, 95% confidence intervals and adjusted P values from the model. In addition to the composite score outcomes, each participant was evaluated for improvement in each individual metric at year 2 as pre-specified secondary outcomes (for example, decrease in BSA-indexed LV mass, decrease in BSA-indexed LA volume, increase in BSA-indexed LV end diastolic volume, increase in BSA-indexed LV end systolic volume, decrease in BSA-adjusted maximal LV wall thickness, increase in age-adjusted E′ velocity, increase in age-adjusted S′ velocity, decrease in cardiac troponin T serum level and decrease in NTproBNP serum level, each analyzed individually). The proportion of patients with improvement in any of the nine components was also compared between treatment groups as a pre-specified secondary outcome, with adjustment for covariates and correlation, as described above, through a mixed effects logistic regression. To assess generalizability of the findings, exploratory analyses were also carried out for improvement in each of the nine components of the primary outcome and in pre-specified subgroups (sex, age dichotomized at 18 years, baseline maximum LV wall thickness dichotomized at the population median BSA-adjusted z-score of 7.3 and variants in the genes most commonly associated with HCM: MYBPC3 versus MYH7). P values for secondary outcomes were not adjusted for multiple testing, and exploratory analyses are presented with just effect estimates and unadjusted 95% confidence intervals.
Pre-specified safety assessments included the incidence of adverse events, study dropout, hypotension and hyperkalemia and increases in serum creatinine. Because the study had a single primary outcome analysis, a two-sided P value of 0.05 was used to determine significance of the composite outcome. Exploratory analyses are presented with effect estimates and unadjusted 95% confidence intervals.
Reporting Summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Extended Data
Extended Data Fig. 1 |. Longitudinal changes in blood pressure.
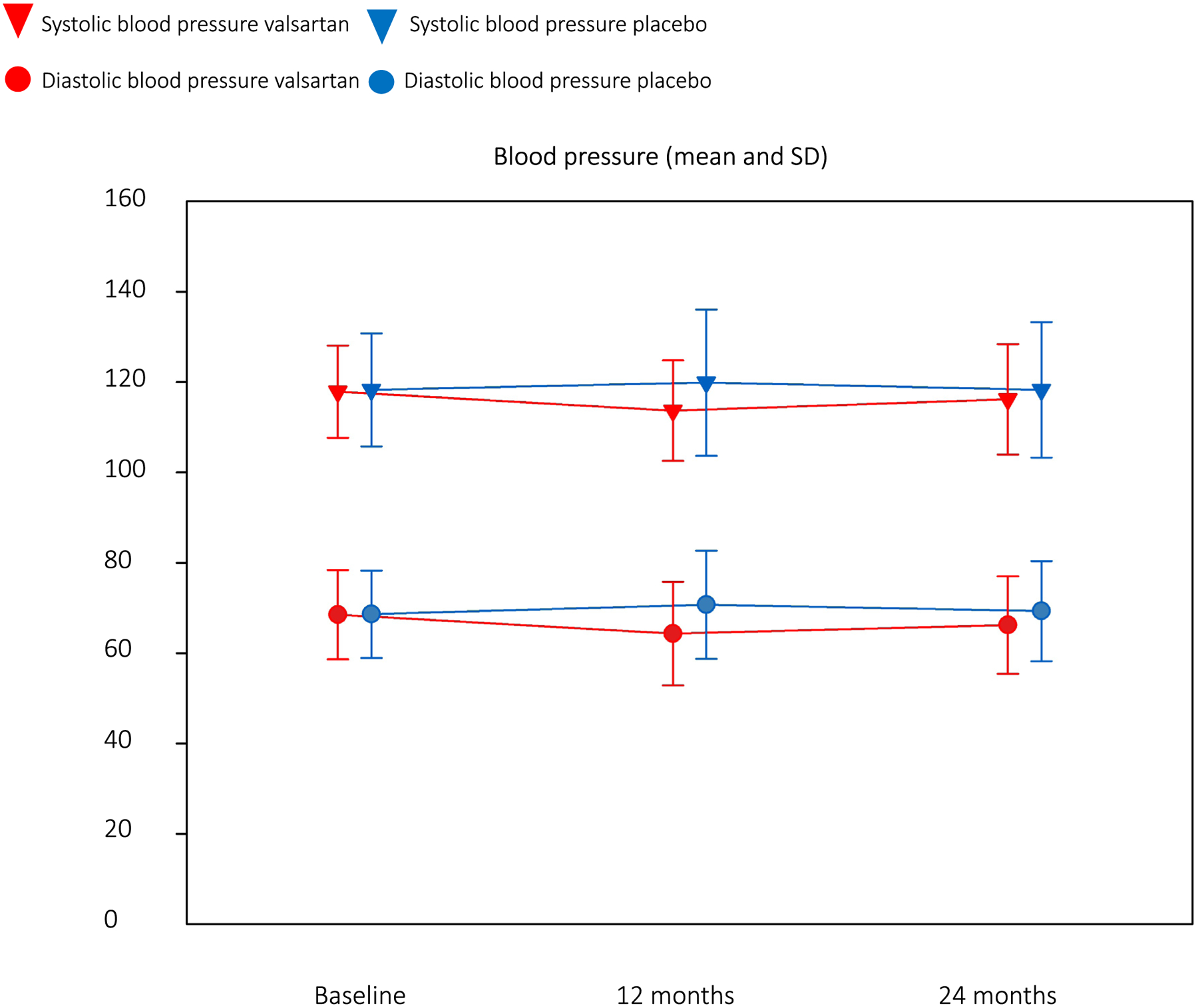
Measures at baseline, Year 1, and Year 2 (end of study) for systolic (a) and diastolic (b) blood pressure are shown for participants treated with valsartan (n = 88; red) and placebo (n = 90; blue). Values are presented as mean and standard deviation.
Extended Data Fig. 2 |. Longitudinal changes in serum creatinine and potassium levels.
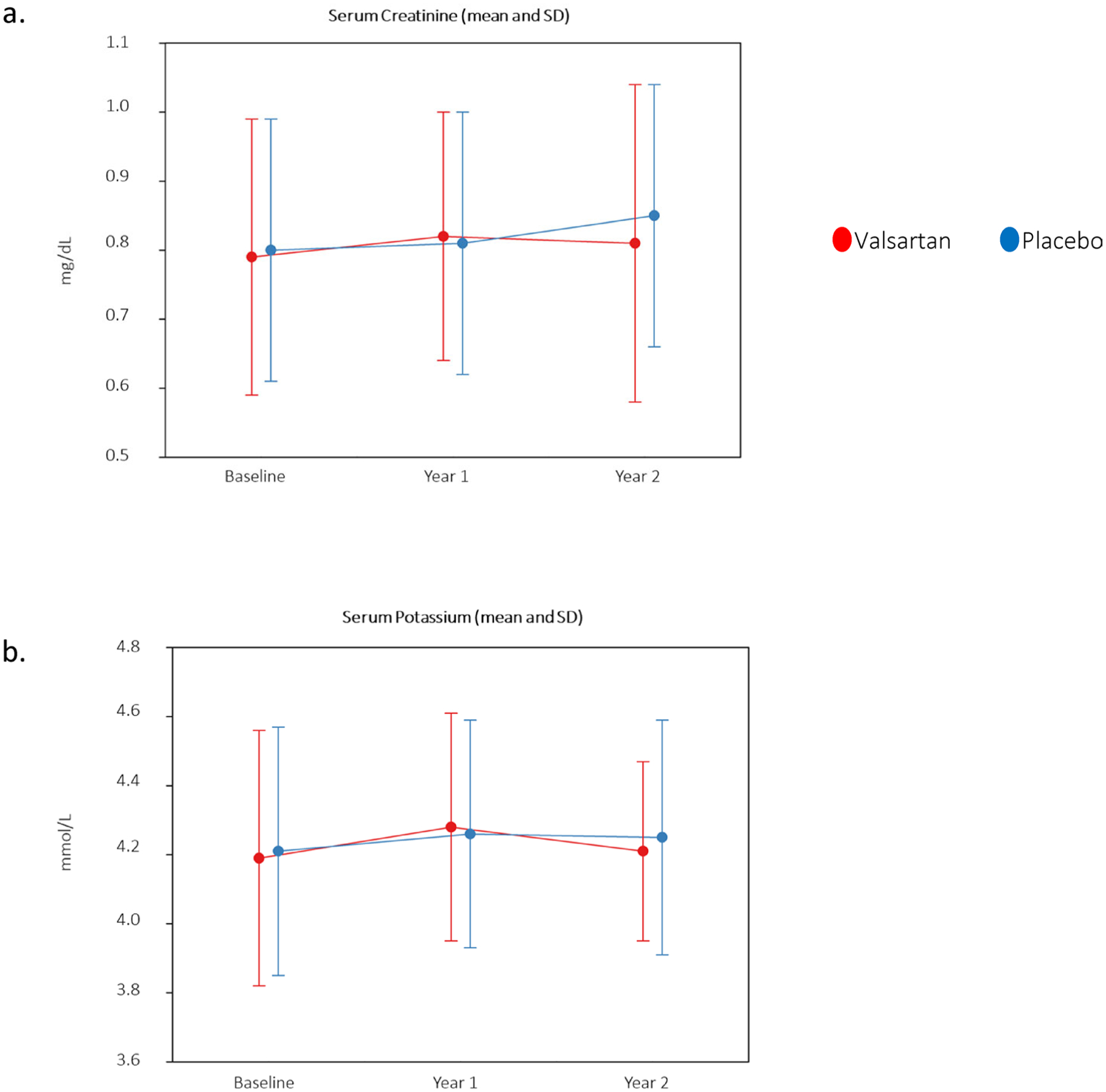
Mean values and standard deviation are shown for serum creatinine (a) and potassium (b) levels for participants treated with valsartan (n = 88; red) and placebo (n = 90; blue).
Supplementary Material
Acknowledgements
We are indebted to the families and individuals who partnered with us in conducing this trial. This study was funded by the National Heart, Lung, and Blood Institute (P50HL112349 to C.Y.H.). The views expressed in this manuscript are those of the authors and do not reflect official positions of the National Heart, Lung, and Blood Institute or the National Institutes of Health. Study medication (blinded valsartan and matching placebo) was provided by Novartis. Novartis was not involved in the design or conduct of the study; data collection, data management, data analysis or data interpretation; preparation, review or approval of the manuscript; or decision to submit the manuscript for publication.
Footnotes
Competing interests
The authors declare no competing interests.
Extended data is available for this paper at https://doi.org/10.1038/s41591-021-01505-4.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41591-021-01505-4.
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41591-021-01505-4.
Data availability
The final study protocol and statistical analysis plan are provided as Supplementary Note. The corresponding author will consider requests for collaborative study and analysis of de-identified individual participant data to further understanding of the trial results or genetic cardiomyopathies. Interested researchers should submit proposals and analytic plans to the corresponding author, and approval will be granted by consensus of the executive committee. A data use agreement will be issued and will be compliant with relevant patient confidentiality and privacy regulations.
References
- 1.Writing Committee M et al. 2020 AHA/ACC guideline for the diagnosis and treatment of patients with hypertrophic cardiomyopathy: a report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation 142, e558–e631 (2020). [DOI] [PubMed] [Google Scholar]
- 2.Seidman CE & Seidman JG Identifying sarcomere gene mutations in hypertrophic cardiomyopathy: a personal history. Circ. Res 108, 743–750 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ho CY et al. Genotype and lifetime burden of disease in hypertrophic cardiomyopathy: insights from the Sarcomeric Human Cardiomyopathy Registry (SHaRe). Circulation 138, 1387–1398 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kim JB et al. Polony multiplex analysis of gene expression (PMAGE) in mouse hypertrophic cardiomyopathy. Science 316, 1481–1484 (2007). [DOI] [PubMed] [Google Scholar]
- 5.Lopez B, Gonzalez A & Diez J Circulating biomarkers of collagen metabolism in cardiac diseases. Circulation 121, 1645–1654 (2010). [DOI] [PubMed] [Google Scholar]
- 6.Teekakirikul P et al. Cardiac fibrosis in mice with hypertrophic cardiomyopathy is mediated by non-myocyte proliferation and requires Tgf-β. J. Clin. Invest 120, 3520–3529 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Habashi JP et al. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science 312, 117–121 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kawano H et al. Valsartan decreases type I collagen synthesis in patients with hypertrophic cardiomyopathy. Circ. J 69, 1244–1248 (2005). [DOI] [PubMed] [Google Scholar]
- 9.Araujo AQ et al. Effect of losartan on left ventricular diastolic function in patients with nonobstructive hypertrophic cardiomyopathy. Am. J. Cardiol 96, 1563–1567 (2005). [DOI] [PubMed] [Google Scholar]
- 10.Yamazaki T et al. A new therapeutic strategy for hypertrophic nonobstructive cardiomyopathy in humans. A randomized and prospective study with an angiotensin II receptor blocker. Int. Heart J 48, 715–724 (2007). [DOI] [PubMed] [Google Scholar]
- 11.Penicka M et al. The effects of candesartan on left ventricular hypertrophy and function in nonobstructive hypertrophic cardiomyopathy: a pilot, randomized study. J. Mol. Diagn 11, 35–41 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shimada YJ et al. Effects of losartan on left ventricular hypertrophy and fibrosis in patients with nonobstructive hypertrophic cardiomyopathy. JACC Heart Fail. 1, 480–487 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Axelsson A et al. Efficacy and safety of the angiotensin II receptor blocker losartan for hypertrophic cardiomyopathy: the INHERIT randomised, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol. 3, 123–131 (2015). [DOI] [PubMed] [Google Scholar]
- 14.Ho CY et al. The design of the Valsartan for Attenuating Disease Evolution in Early Sarcomeric Hypertrophic Cardiomyopathy (VANISH) trial. Am. Heart J 187, 145–155 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Axelsson Raja A et al. Baseline characteristics of the VANISH cohort. Circ. Heart Fail 12, e006231 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sun H, Davison BA, Cotter G, Pencina MJ & Koch GG Evaluating treatment efficacy by multiple end points in phase II acute heart failure clinical trials: analyzing data using a global method. Circ. Heart Fail 5, 742–749 (2012). [DOI] [PubMed] [Google Scholar]
- 17.Semsarian C et al. The L-type calcium channel inhibitor diltiazem prevents cardiomyopathy in a mouse model. J. Clin. Invest 109, 1013–1020 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Richards S et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med 17, 405–424 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Landrum MJ et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 44, D862–D868 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ho CY et al. Echocardiographic strain imaging to assess early and late consequences of sarcomere mutations in hypertrophic cardiomyopathy. Circ. Cardiovasc. Genet 2, 314–321 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ho CY et al. Evolution of hypertrophic cardiomyopathy in sarcomere mutation carriers. Heart 102, 1805–1812 (2016). [DOI] [PubMed] [Google Scholar]
- 22.Ho CY et al. The burden of early phenotypes and the influence of wall thickness in hypertrophic cardiomyopathy mutation carriers: findings from the HCMNet study. JAMA Cardiol. 2, 419–428 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ho CY et al. Diltiazem treatment for pre-clinical hypertrophic cardiomyopathy sarcomere mutation carriers: a pilot randomized trial to modify disease expression. JACC Heart Fail. 3, 180–188 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The final study protocol and statistical analysis plan are provided as Supplementary Note. The corresponding author will consider requests for collaborative study and analysis of de-identified individual participant data to further understanding of the trial results or genetic cardiomyopathies. Interested researchers should submit proposals and analytic plans to the corresponding author, and approval will be granted by consensus of the executive committee. A data use agreement will be issued and will be compliant with relevant patient confidentiality and privacy regulations.