

Abstract
Impaired apoptosis is one of the hallmarks of cancer, and almost all of the non‐surgical approaches of eradicating tumour cells somehow promote induction of apoptosis. Indeed, numerous studies have stated that non‐ionizing non‐thermal extremely low‐frequency magnetic fields (ELF‐MF) can modulate the induction of apoptosis in exposed cells; however, much controversy exists in observations. When cells are exposed to ELF‐EMF alone, very low or no statistically significant changes in apoptosis are observed. Contrarily, exposure to ELF‐EMF in the presence of a co‐stressor, including a chemotherapeutic agent or ionizing radiation, can either potentiate or inhibit apoptotic effects of the co‐stressor. In our idea, the main point neglected in interpreting these discrepancies is “the cellular stress responses” of cells following ELF‐EMF exposure and its interplay with apoptosis. The main purpose of the current review was to outline the triangle of ELF‐EMF, the cellular stress response of cells and apoptosis and to interpret and unify discrepancies in results based on it. Therefore, initially, we will describe studies performed on identifying the effect of ELF‐EMF on induction/inhibition of apoptosis and enumerate proposed pathways through which ELF‐EMF exposure may affect apoptosis; then, we will explain cellular stress response and cues for its induction in response to ELF‐EMF exposure; and finally, we will explain why such controversies have been observed by different investigators.
Keywords: apoptosis, cellular stress response, controversy, ELF‐EMF, reactive oxygen species
The outcome of cancer cells exposure to ELF‐EMF, as cellular stress response, is closely related to ER stress, NF‐κB inflammatory, Heat Shock Protein, Sirtuin, and DNA repair responses. The controversial evidence of ELF‐EMF‐induced apoptosis or autophagy in cancer cells has been hypothetically described by the way these cellular feedback happen.
![]()
1. INTRODUCTION
In modern world, electromagnetic fields (EMFs) have become an inseparable part of routine life. Numerous electric power‐generating human‐made devices are now producing EMFs which are overlaid on those of earth's magnetic field. EMFs are usually identified with a 50 or 60 Hz frequency and therefore are classified under the extremely low‐frequency, non‐ionizing span of electromagnetic spectrum. 1 Due to these physical characteristics, ELF‐EMFs are not capable of breaking molecular bond or inducing thermal effects on tissue. However, it is now proven that they can interact with human tissues and induce some weak electrical currents. 2 In addition, it is not completely understood whether biological effects induced by EMFs are hazardous for human or environment. During last few decades, a number of studies have reported beneficial effects of ELF‐EMFs in treatment of cancer both in vitro and in vivo. 3 , 4 , 5 , 6 , 7 Despite this, the exact mechanism of these anti‐neoplastic effects has not been confirmed yet.
So far, the most probable mechanism proposed for explaining anticancer effects of ELF‐EMF is induction of apoptosis through upregulation of intracellular reactive oxygen species (ROS) which has also been confirmed by different experimental studies. In the study performed by Ding et al., 8 it was demonstrated that 24‐h exposure to 60 Hz, 5 mT ELF‐EMF could potentiate apoptosis induced by H2O2 in HL‐60 leukaemia cell lines. Similarly, in the study performed by Jian et al., 9 exposure to an intermittent 100 Hz, 0.7 mT EMF significantly enhanced rate of apoptosis in human hepatoma cell lines pretreated with low‐dose X‐ray radiation. Kaszuba‐Zwoinska et al. 10 also showed that short‐term exposure of human acute monocytic leukaemia cell line exposure to 50 Hz, 45 ± 5 mT pulsed EMF, significantly potentiated rate of apoptosis induced by cyclophosphamide and colchicine. Benassi et al. reported that co‐treatment of human ovarian adenocarcinoma cell lines with cisplatin, a chemotherapeutic agent with DNA‐damaging and ROS‐promoting activity, significantly enhanced sensitivity to apoptosis through increasing both caspases 3 and 9 activity. This is in accordance with previous studies demonstrating an enhancement in 1‐methyl‐4‐phenylpyridinium (MPP +) induced caspase‐dependent apoptosis following 24‐h exposure to 50 Hz, 1 mT ELF‐MF in SH‐SY5Y neuroblastoma cell lines. 11
One of the main mechanisms proposed for defining anticancer effects of ELF‐EMF is induction of apoptosis through upregulation of reactive oxygen species (ROS) which has also been confirmed by different experimental studies.
Contrary to above‐mentioned studies, several reports propose an anti‐apoptotic activity for ELF‐EMF. Pirozzoli et al. 12 reported that 24‐h exposure to 50 Hz, 1 mT ELF field significantly attenuated apoptosis induced by camptothecin in LAN‐5 neuroblastoma cell lines. De Nicola et al reported that puromycin‐induced apoptosis in human lymphoblasts was significantly weakened in response to 2‐h exposure to a 0.1 mT ELF field. 13 They reported that reduced glutathione (GSH) was the key mediator of the observed effect. In addition, based on the study performed by Palumbo et al., 14 pretreatment of Jurkat leukaemic cell lines with 50 Hz, 1 mT EMF resulted in 22% reduction in caspase 3‐dependent apoptosis induced by anti‐Fas therapy. Moreover, based on Cid et al., 15 the anti‐apoptotic activity of melatonin on HepG2 cell lines was completely abrogated in response to 42‐h intermittent exposure with a 50 Hz, 10 µT EMF. Similarly, bleomycin‐induced apoptotic activity in K562 erythroleukaemia cell line was significantly reduced in response to a short‐term (~10 min) exposure period to a 217 Hz, 120 µT ELF‐MF. 16 Based on Brisdelli et al., 17 concurrent treatment of K562 cell lines with ELF‐EMF and quercetin for 24 h significantly increased expression of Bcl2, a protein with anti‐apoptotic activity, compared with quercetin alone treated and control groups. They also reported that extending ELF‐EMF exposure for 1–3 days results in attenuation of growth inhibitory effects of quercetin in leukaemia cell lines which was in association with reduced level of caspase 3 activity, along with inhibition of quercetin induced reduction in expression of Bcl‐xL and Mcl‐1 anti‐apoptotic proteins.
Still, some reports have stated no statistically significant cytotoxic or cytostatic activity for ELF‐EMF. Laqué‐Rupérez et al. 18 reported no statistically significant changes in methotrexate‐induced cytotoxicity in MCF‐7 breast cancer cell lines after exposing them to 25 Hz, 1.5 mT pulsed EMF. Similarly, in the study performed by Mizuno et al., 19 no statistically significant changes in survival rates of SV40 cells were observed between cells which were subjected to UV radiation alone and group subjected to concurrent administration of 24‐h 60 Hz, 5 mT EMF and UV radiation. Finally, Höytö et al. 20 reported no statistically significant enhancement in anti‐proliferative and cytotoxic activities of menadione on SH‐SY5Y neuroblastoma cells when combined with 24 h exposure to ELF‐MF of 100 µT intensity.
This discrepancy in observations has made it difficult to come into a unit conclusion, and therefore, application of ELF‐EMF in clinic for treatment of cancer still remains a big dilemma. In our idea, the main point neglected in interpreting the discrepancies observed in results is consideration of cellular stress responses induced by ELF‐EMF exposure and its interplay with the molecular mechanisms underlying apoptosis. The main purpose of current review was to outline the triangle of ELF‐EMF, cellular stress response of cells and apoptosis, and interpret and unify the discrepancies in results based on this theory. Therefore, initially we will explain studies performed on identifying the effect of ELF‐EMF on induction/inhibition of apoptosis, enumerate proposed pathways through which ELF‐EMF exposure may affect apoptosis; then, we will explain cellular stress response, cues for activation of this phenomenon in response to ELF‐EMF exposure and finally under a separate “discussion” section we will try to explain why such controversy has been obtained by different investigators.
2. APOPTOSIS AND ELF‐EMF EXPOSURE
Considering hallmarks of cancer, aberrant cellular survival is an important characteristic of malignant cells which is usually attributed to a mis‐regulated apoptotic state in cells. Apoptosis is a type of programmed cell death which is abundantly observed under both physiological and pathological conditions, upon interaction of cells with specific stimulators, capable of activating either of intrinsic and extrinsic pathways. Moreover, failure in induction of apoptosis, as a consequence of aberrant expression of antigens, secreted angiogenic growth factors, or their receptors has already been linked to an elevated risk of metastasis, promotion of angiogenesis and an accelerated risk of resistance development to anti‐angiogenic cancer therapies. 21 , 22 , 23 , 24 , 25 , 26 , 27 Either mediated by the extrinsic (mediated by FASL, TNFα and so on) or intrinsic pathway (most importantly, accumulation of ROS and development of oxidative stress), the rest of the process will be followed by modulation of specific sets of procaspase molecules cleavage (caspase 8 and caspase 9 for extrinsic and intrinsic pathways respectively), ending in degradation of numerous intracellular target proteins, blebbing of cellular membrane, cleavage and degradation of chromosomal DNA, and finally, getting phagocytosed and scavenged by polymorphonuclear cells. 28 Apoptosis can be triggered upon activation of two main pathways which are broadly referred as “intrinsic” and “extrinsic” pathways.
The most prevalent mechanism through which several chemotherapeutic agents trigger apoptosis is induction of mitochondrial membrane permeabilization, the intrinsic apoptosis pathway, which is mainly controlled by Bcl‐2 proteins family. This process results in leakage of several pro‐apoptotic molecules such as cytochrome c, Smac/DIABLO, apoptosis‐inducing factor (AIF) and endonuclease G (Endo G) into the cytoplasm. 29 Released Endo G and AIF initiate nuclear modifications while the others activate caspases. Cytochrome c promotes formation of apoptosis protease activating factor‐1 (Apaf‐1) oligomers using ATP or dATP. 30 , 31 This complex in next place recruits procaspase 9 and forms “apoptosome” which in turn induces autoactivation of procaspase 9. 32 , 33 Matured caspase 9 further activates caspase 3 and 7 which in turn results in initiation of downstream caspase cascades 34 and induction of apoptotic cell death. In parallel, Smac/DIABLO antagonize suppressing effects of inhibitors of apoptosis proteins (IAPs) on activated caspases. 35 , 36
In some cell types however, chemotherapeutic‐induced apoptotic cell death may be initiated through the death receptor Fas (APO‐1/CD95), the extrinsic apoptosis pathway. Ligation of Fas with its natural ligand, FasL, promotes Fas clustering, which in next place attracts FADD 37 and procaspase 8, 38 totally forming a complex referred as death‐inducing signalling complex (DISC). The mature caspase 8 would be exhausted from the DISC after oligomerization andautoactivation of procaspase 8. 39 Based on the cell type, mature caspase 8 initiates apoptosis by two distinct pathways. 40 In first pathway, high quantities of mature caspase 8 induce direct cleavage and activation of procaspase 3 without enrolment of mitochondrial pathway. In second pathway however, low quantities of mature caspase 8 are formed which is not capable of directly inducing activation of procaspase 3. Alternatively, herein, caspase 8 promotes cleavage of the “BH3‐only protein” Bid and formation of truncated Bid which, in turn, triggers mitochondrial apoptosis pathway. 41 , 42
Different groups of anticancer agents are capable of activating death receptor pathway through enhancement of Fas or FasL expression. 43 This process is transcription‐dependent and involves p53 activity. 44 The activated signalling pathway following Fas/FasL complexation outlines an autocrine/paracrine pathway like that happening during activation‐induced cell death in T lymphocytes. Nevertheless, FasL plays minimal role in chemotherapy‐induced apoptosis, as administration of antagonist antibodies or any small molecule preventing from FasL/Fas interaction does not suppress apoptosis. 45 Likewise, the pro‐apoptotic effects of chemotherapeutic agents on embryonic fibroblasts from FADD and caspase 8 knockout mice remained unaltered. 46 , 47
Although apoptosis is usually induced upon overproduction of ROS and development of oxidative stress, a mild‐to‐moderate level of ROS is required for maintenance and regulation of physiological function of cells including growth, proliferation, differentiation and migration 52 ; regulation of immune system's function and maintaining redox balance 48 ; and promotion of autophagy through activation of different signalling pathways including phosphoinositide 3‐kinase (PI3K)/Akt, mitogen‐activated protein kinases (MAPK), nuclear factor (erythroid‐derived 2)‐like 2 (Nrf2)/Kelch‐like ECH‐associated protein 1 (Keap1), nuclear factor‐κB (NF‐κB) and the tumour suppressor p53. 48 , 49 , 50 , 51 Hence, manipulation of ROS level in cells is a good strategy for cancer therapy.
If ROS generation and accumulation can be considered the first cellular event of ELF‐EMFs exposure, the modification of intracellular Ca2+ levels could be one of the most important mechanisms by which ROS have their multiple actions in cells. 52 Over the past few years, lots of data have shown that ELF‐EMF exposure regulates intracellular Ca2+ level which can, in turn, activate multiple physiological mechanisms such as differentiation of chromaffin cells into neuronal‐like cells (ELF‐MF, 60 Hz, 0.7 mT, 2 h/day twice a day 53 ); cell death by apoptosis (ELF‐MF, 50/60 Hz, 0.2–5 mT, 2–3 consecutive days 54 , 55 ); functional modification of the immune system's cells through involvement of P2Y membrane receptors (sinusoidal electric fields, 0.3 or 30 kV/m, 50 Hz, for 24 h 56 ), activation of mechanically operated stretch‐activated Ca2+ channels (noninvasive electrical stimulus, 0.1‐V/cm direct current 57 ); and the enhancement of the expression of voltage‐gated Ca2+ channels in different human cell systems (static magnetic fields, 0.15 and 66 mT 58 ). In this context, Kapri‐Pardes et al. examined responses of cells (both transformed and non‐transformed) to ELF‐EMFs across a broad range of field strengths by examining activation of ERK1/2 and other signalling pathways. They reported that all cell lines could sense and respond to ELF‐EMFs. Nevertheless, the extent to which transformed cells responded to EMFs was significantly lower compared to non‐transformed ones, and interestingly, in MDA‐MB‐231 cells, exposure decreased phosphorylation of ERK1/2. Perhaps the more important finding of their study was that contrary to what previously was though, cells can sense magnetic field strengths as low as 0.15 µT which is at least partly mediated through activation of NADH oxidase. 59
So far, multiple signalling pathways have identified to be affected under acute or short‐term exposure to ELF‐EMF. Indeed, exposure to ELF‐EMF promotes tyrosine phosphorylation of specific protein components of signalling pathways in cells. For instance, it has been shown that 1‐ to 30‐min exposure to 60 Hz, 0.1 mT ELF‐EMFs results in activation of Lyn, a protein tyrosine kinase and serine/threonine kinase protein kinase C (PKC) in B lymphocytes. 60 Likewise, acute exposure of Jurkat cell line (~5 min) to 50 Hz, 0.1 mT ELF‐EMFs activates Lck, which in turn promotes complexation of T cell receptors. 61 Similar result was also reported in adherent cells, where 5 min exposure to 50 Hz, 04 mT ELF‐EMFs promoted epidermal growth factor receptor (EGFR) clustering and subsequent stimulation of Ras GTPases in long fibroblast cells of Chinese hamster. 62 In addition, cyclic AMP/protein kinase A (cAMP/PKA) is another pathway which is activated in response to exposure to ELF‐EMF in rat's cerebellar granule cells and human skin fibroblasts. 63 , 64
Mitogen‐activated protein kinase (MAPK) cascades are among the other important signalling cascades which are stimulated upon exposure to ELF‐EMF in several types of examined cells. 65 MAPK pathways consisting from four main cascades, including extracellular signal regulated kinase 1 and 2 (ERK1/2), ERK5, p38 and c‐Jun N‐terminal kinase (JNK), are central in regulation of almost all stimulated cellular events such as differentiation, proliferation, stress responses and apoptosis. 65 , 66 After initial stimulation, these cascades function by serially activating specific protein kinases in each level of cascade with ultimate result of phosphorylation of thousands of target proteins and modulation of related cellular processes. Akt is another protein kinase with responsibilities similar to the MAPKs. 67 Akt becomes activated in response to extracellular stimuli upon phosphorylation of its two activatory moieties following interaction with PI3K‐phosphorylated phospholipids. Any dysregulation or abnormalities in mentioned five signalling pathways is associated with certain disorders including cancer. 67 , 68 Interestingly, both acute/short‐term and chronic/long‐term exposure to ELF‐EMF has shown to induce activation of Akt and MAPK. 69 , 70 For instance, 30‐min exposure to ELF‐EMF results in activation of ERKs and Akt in several cancer cell lines including MCF7, HaCaT, NB69, HL‐60 and so on. 69 , 70 , 71 , 72 Furthermore, 3‐ to 15‐min exposure to 50 Hz in CHL cells and 15, 30, or 60 min in NB69 cells results in activation of stress‐associated MAPKs, p38 and JNK. 70 , 73 , 74
In most of these studies, the strengths of applied ELF‐EMFs were more than 100 µT, and none has investigated the relation between the changes in strength of EMFs and induction of cell signalling cascades. Recently, Kapri‐Pardes et al. examined responses of cells (both transformed and non‐transformed) to ELF‐EMFs across a broad range of field strengths by examining activation of ERK1/2 and other signalling pathways. They reported that all cell lines could sense and respond to ELF‐EMFs. However, the extent to which transformed cells responded to EMFs was significantly lower compared to non‐transformed ones, and interestingly, in MDA‐MB‐231 cells, exposure decreased phosphorylation of ERK1/2. Perhaps the more important finding of their study was that contrary to what previously was though, cells can sense magnetic field strengths as low as 0.15 µT which is at least partly mediated through activation of NADH oxidase. 59
Although effects of EMF exposure on TGF‐β/BMP signalling pathway have been studied during the process of bone repair, same pathway is a key player in pathophysiology of cancer and its modulators demonstrate statistically significant anti‐metastatic activities. 75 Different studies have shown that exposure to pulsed EMF results in a statistically significant increase in TGF‐β, in osteoblastic cells and both atrophic and non‐hypertrophic cells. 76 , 77 In addition, based on a recent study, exposing differentiating osteoblasts to pulsed EMF, promotes activation of TGF‐β signalling pathway through Smad2 and increases expression of osteoblastic differentiation markers such as ALP and type I collagen. 78 BMP expression during osteogenesis was also increased after exposure to pulsed EMFs. 79 , 80 , 81 Moreover, it has been shown that exposure to pulsed EMFs, stimulates osteogenic differentiation and maturation through the activation of BMP‐Smad1/5/8 signalling. In this case, BMP receptor II, BMPRII, regulates differentiation in a cilium‐dependent manner. 82 Considering the separate effects of BMP and pulsed EMFs on differentiation and maturity of osteoblasts, many studies have shown that concurrent treatment with BMP and pulsed EMF enhances bone formation to a much greater degree compared to each treatment alone. 83 , 84 , 85 , 86
In addition to the mentioned signalling pathways, electromagnetic fields can also affect pathways underlying VEGF and FGF signalling molecules. 87 , 88 Based on a recent report, exposure to pulsed EMF significantly increases expression of IGF‐1 at mRNA level and promotes bone formation. 89 In addition, pulsed EMF (1.5 mT, 75 Hz) can also increase synthesis of proteoglycans and protect human articular cartilage from further damage. 90 Finally, it has been shown that exposure to pulsed EMF reverses osteoporotic effect of dexamethasone. 91
Notch signalling is a highly conserved pathway that regulates cellular fate and skeletal development. Recent reports have shown that exposure to pulsed EMF can regulate expression levels of Notch4 receptor, as well as DLL4 ligands and target genes (Hey1, Hes1 and Hes5) during the osteogenic differentiation of human mesenchymal stem cells. Interestingly, expression of osteogenic markers, including Runx2, Dlx5, Osterix, Hes1 and Hes5, after pulsed EMF treatment was reversed following treatment of cells with notch pathway inhibitors. 92 Furthermore, exposure to pulsed EMF significantly increases the level of cAMP, protein kinase A activity and accelerates osteogenic differentiation of MSCs. 93 , 94 Anti‐inflammatory effects of pulsed EMFs have also been reported both in vitro 95 , 96 and in vivo, 97 , 98 , 99 as well as in clinical settings. 100
3. ELF‐EMF AND INDUCTION OF CELLULAR STRESS RESPONSE
Numerous studies have shown that cells are physiologically well buffered against negative effects of ELF‐EMF alone. However, in the presence of stressful condition, including exposure to toxins, viruses, DNA damage and proteotoxic, hypoxic, metabolic and oxidative stress, an additional weak stressor like ELF‐EMF might produce large effects. 101 Based on Mattsson and Simko who extensively investigated the oxidative response of cells following ELF‐EMF exposure, ROS levels can be consistently altered in different cell types or experimental conditions following exposure to magnetic fields. These effects were prominent for fields with intensities more than 1 mT, but were also documented at or below 100 mT. Despite this, all observed effects where moderate and majority of changes were below 50%. 102 Consequently, the produced amounts of ROS by ELF‐EMF are not high enough to induce major DNA damage. Although this mild elevation in ROS levels in response to acute or chronic exposure to ELF‐EMF cannot trigger cell death, it may induce cellular resistance against oxidative damage through upregulation of antioxidant pathways and induction of cellular stress response. Small change in ROS levels stated above is capable of promoting different cell signalling pathways especially by means of superoxide ions. 20 , 103 , 104 , 105 , 106 This phenomenon requires a certain time to develop and promotes several other time‐dependent changes. 105
As discussed earlier, antioxidant defence capacity of cells can be changed following exposure to ELF‐EMF. For example, it has been shown that exposure to ELF‐EMF can significantly increase SOD levels in cells. 107 Furthermore, ELF‐EMF can enhance activity of both glutathione‐S‐transferase and ‐reductase enzymes in malignant cells. 108 Also, based on Cichon et al., 109 ELF‐EMF exposure can upregulate expression of different antioxidant target genes including CAT, SOD1, SOD2, GPx1 and GPx4. In addition, multiple pathways involved in orchestrating cellular stress response of cells to stressful condition can also become activated following ELF‐EMF exposure. Based on literature, generation of mitochondrial ROS at the time of ELF‐EMF exposure is pivotal for activation of signalling pathways involved in cellular adaption. 110 Activation and upregulation of Nrf2 expression, the master redox‐sensing transcription factor may be the most prominent example in this regard which has been confirmed in a Huntington's disease‐like rat model. 111 Another cellular stress response to ELF‐EMF involves activation of MAPK and NF‐κB which, in turn, may upregulate expression of peroxisome proliferator‐activated receptor‐γ coactivator‐1α (PGC‐1α) and enhance mitochondrial biogenesis. 112 Activation of autophagy, ER stress, heat‐shock response and sirtuin 3 expression are among the other identified cellular stress responses to ELF‐EMF exposure, all of which have been discussed earlier.
This cellular stress response is very important when ELF‐EMF exposure is applied before chemotherapy. The main mechanism through which several chemotherapeutic agents induce apoptosis in cancer cells is elevation of ROS and induction of apoptosis. However, as antioxidant defence after ELF‐EMF exposure is enhanced, cells become more resistant to these agents. Such effects have also been reported during radiation therapy and are responsible for development of resistance to radiotherapy. Contrarily, when chemotherapy and ELF‐EMF exposure are performed simultaneously, this increase in ROS levels potentiates the oxidative stress induced by chemotherapeutic agents, as the ROS levels become excessively high and cells do not have time for adaption. Therefore, the result is enhancement of apoptosis. Differences between extent of apoptosis induced or when no significant differences are observed in combination are mostly dependent on the nature of the cell (ie the antioxidant defence), type and dose of the chemotherapeutic agent applied and the number of cells seeded in the plate.
A number of other harmful agents or conditions, such as thermal stress, 113 exposure to alkylating agents, 114 heavy metals 115 and ionizing radiation 116 have shown to initiate a similar response. Generally, cellular stress response is characterized by modulation of expression of various genes. The main outcome of this alteration in pattern of gene expression is protection of cells from cytotoxic doses of a harmful agent. This response represents that following exposure to a toxin, cells expect or at least prepare themselves for a lethal concentration of the agent. In addition to mild exposure to toxic agents or stressful conditions, physiological conditions may also promote development of an cellular stress response. 117 For instance, exercise training reduces the extension of lipid peroxidation during acute exercise which has been attributed to induction of oxidative stress. 118 , 119 Likewise, an enhanced repairing capacity was observed in lymphocytes of workers which were occupationally become exposed with low levels of ionizing radiation. 120
It is now clear that sub‐lethal doses of oxidants are capable of inducing cellular stress responses in cells. This phenomenon was initially discovered in bacteria, but now it has also been documented in eukaryotic cells. 121 , 122 , 123 , 124 The protective responses induced in cells during challenge with sub‐lethal doses of oxidants have been identified in three major systems. These include hydrogen peroxide (H2O2) and superoxide anion (O2−)‐induced reactions in bacteria, protective responses induced by sub‐lethal doses of oxidants in eukaryotic cells which render them resistant to lethal doses of the same or a related oxidant and finally protective responses induced by sub‐lethal doses of oxidants in eukaryotic cells which render them resistant to lethal doses of other toxic agents. Overall, cells possess two primary defence mechanisms against oxidative stress. The first includes cellular molecules or enzymes that directly participate in scavenging free radicals and preventing oxidative stress‐induced damage to cells such as catalase, superoxide dismutases (SOD), glutathione peroxidases, ascorbate and glutathione. The second line, however, consists of enzymes involved in repairing or scavenging oxidatively damaged macromolecules such as DNA and proteins. Typical examples of such enzymes are DNA nucleases and glycosylases. 117
4. MECHANISMS UNDERLYING ELF‐EMF‐MEDIATED CELLULAR STRESS RESPONSE
The cellular stress response to oxidative stress in mammalian cells consists of seven main pathways including unfolded protein response (UPR), antioxidant response, heat‐shock response, autophagic response, NF‐kB inflammatory response, sirtuin response and DNA repair response. Numerous studies in literature have reported that exposure to ELF‐EMF can activate most of these pathways without inducing significant increase in cell death or apoptosis both in normal and in cancer cells (Table 1). Here, we will comprehensively review the ways through which cells respond to elevated ROS following exposure to ELF‐EMF and orchestrate cellular stress response (Figures 1 and 2).
TABLE 1.
Different cellular stress responses affected by ELF‐EMF exposure
| Experiment performed | ELF‐EMF treatment | Cell line | Observed effects |
|---|---|---|---|
| 1. Heat‐shock protein response | |||
| Corallo et al. 140 | 100 Hz | Primary osteoarthritic chondrocytes | Increased Mn‐superoxide‐dismutase and heat‐shock proteins expression |
| Alfieri et al. 141 | 50 Hz, 0.68 mT | Endothelial cells | A poor and transient activation of HSF1 |
| Frahm et al. 142 | 50 Hz, 1 mT | Mouse macrophages | Hsp70 and Hsp110 exhibited increased levels at certain time point |
| Wei et al. 143 | 15 Hz, 2 mT | Hypoxic cardiomyocytes | Significantly increased HSP70 mRNA expression |
| Bernardini 144 | 50 Hz | Porcine aortic endothelial cells |
Increase in the mRNA levels of HSP70 No increase in Hsp27, Hsp70 and Hsp90 protein levels |
| Akan et al. 145 | 50 Hz, 1 mT | THP‐1 cells | Increased hsp70 levels in a time‐dependent manner |
| 2. Unfold protein response | |||
| Chen et al. 161 | Picosecond pulsed electric fields | HeLa cells | Affected the phosphorylation levels of endoplasmic reticulum sensors and upregulated the expression of GRP78, GRP94 and CHOP |
| Keczan et al. 162 | PEMF | HEK263T | No remarkable effect |
| HepG2 | No remarkable effect | ||
| HeLa | Increased BiP, Grp94 and CHOP expression | ||
| 3. Autophagy | |||
| Chen et al. 173 | Pulsed electromagnetic fields (2 mT, 50 Hz) | Embryonic fibroblasts (MEF) | A significant increase in autophagic biomarkers including LC3‐II and formation of GFP‐LC3 puncta was observed |
| 4. NF‐kB activation | |||
| Kim et al. 204 | RAW264.7 cells | Enhanced translocation of phosphorylated NF‐κB in to the nucleus and induction of inflammatory responses | |
| 5. SIRT3 activation | |||
| Falone et al. 196 |
ELF‐EMF 1 mT, 50 Hz |
SH‐SY5Y | Upregulation of the major sirtuins, increased signalling activity of the NRF2 |
FIGURE 1.
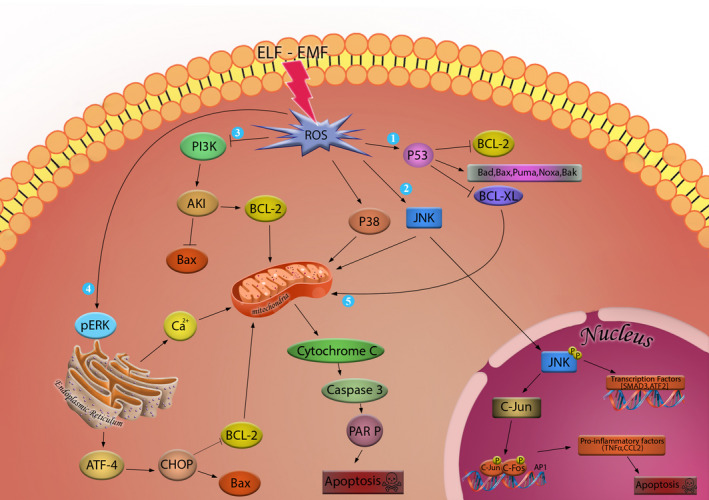
ROS‐mediated apoptosis signalling pathways: (1) Accumulation of ROS affects p53 protein which in turn inhibits Bcl‐2 and Bcl‐XL proteins function and promotes the activity of Bad, Bax, Bak, Puma and Noxa proteins. (2) ROS can induce phosphorylation of JNK. Phosphorylated JNK can activate transcription factors such as SMAD3 and ATF2. Phosphorylated JNK can also translocate to the nucleus and activate C‐Jun phosphorylation which in turn can activate transcription of several pro‐apoptotic factors. (3) Accumulation of ROS inhibits PI3K‐mediated activation of AKT. (4) Accumulated ROS promotes ER stress and expression of CHOP through activation of ATF‐4 which in turn can promote Bax activity and inhibit Bcl‐2. (5) All these pathways end in the release of cytochrome c which in turn can activate caspase 9 and caspase 3 and result in cleavage of PARP and induction of apoptosis
FIGURE 2.
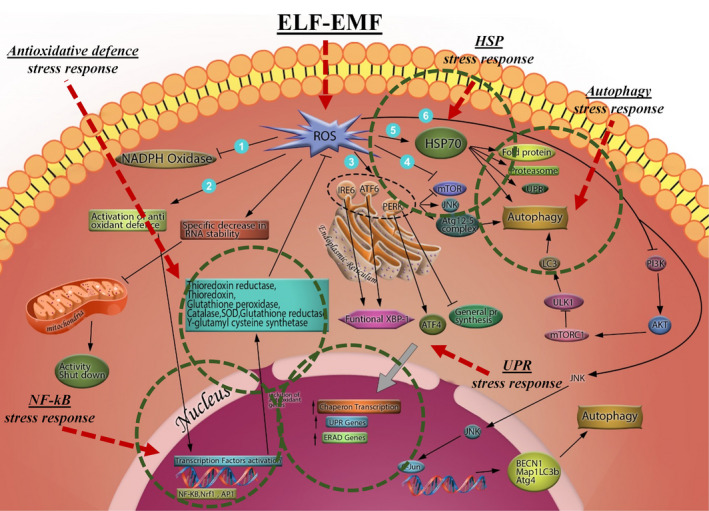
ROS‐mediated cellular stress response: (1) mild accumulation of ROS inhibits NADPH oxidase activity. (2) Mild accumulation of ROS activates antioxidant defence system which involves activation of transcription factors including NF‐kB, Nrf‐1 and AP‐1 which in turn upregulates expression of thioredoxin reductase, glutathione peroxidase, SOD, etc., which can suppress further accumulation of ROS. (3) Mild accumulation of ROS activates ER stress through affecting IRE6, ATF6 and PERK. PERK in turn inhibits general protein synthesis and ATF4 and functional XBP‐1 promote chaperon transcription, UPR genes and ERAD genes which can protect cells against accumulated ROS. (4) Mild accumulation of ROS can directly induce autophagy through inhibition of mTORC. (5) Mild accumulation of ROS can upregulate expression of HSP70 which can affect protein folding, proteasome activation and induction of autophagy. (6) Mild accumulation of ROS can also activate JNK and after that c‐JUN which can in turn activate BECN1, Atg4 and MAP1LC3B genes expressions, most important proteins involved in autophagy. ROS can also inhibit PI3K pathway and modulate autophagy. Finally, mild accumulation of ROS can induce specific decrease in RNA stability and result in mitochondrial activity shut down
4.1. Heat‐shock response
In most eukaryotes, heat‐shock factors (HSF) [ie transcription factors that regulate expression of heat‐shock proteins (HSPs)] are located in cytoplasm in bond with HSP70, HSP90 or other proteins which renders them to be inactive during normal condition. 125 , 126 During stressful condition however, cells are exposed to a much higher extent of denatured proteins. In this condition, as HSPs prefer to act more like a molecular chaperone instead of a regulatory protein, they become detached from HSF and undergo oligomerization. In next step, oligomerized HSFs translocate to the nucleus where they promote expression of HSP and related heat‐responsive genes. 127 , 128 Different studies have shown that treatment of cells with H2O2 and induced ROS can increase expression of heat‐responsive genes. 129 , 130 , 131 In the study performed by Volkov et al. 128 for example, it was shown that heat treatment and H2O2 elevate expression of AtHSP17.6 and AtHSP18.6 genes up to a similar level. In this regard, it has been hypothesized that heat may also activate HSFs by elevation of ROS in an indirect manner. Consistent with this finding, it has been shown that sub‐lethal amounts of ROS induced by thermal stress can enhance segregation of HSP‐HSF complexes. 126 In addition, certain HSFs have shown to play as a sensor for H2O2. 132 , 133
Among different ROS, H2O2 is the main player in modulating signalling pathways partly owing to its moderate reactivity and consequently long half‐life and stability. 134 Furthermore, produced H2O2 can also easily pass through membrane and therefore take role of a signalling molecule. 135 Based on Miller and Mittler, H2O2 may also trigger HSF’s trimerization through direct modification of HSFs. In addition, MAPK is another pathway through which ROS and HSFs may communicate with each other since HSF phosphorylation has been identified both in mammals and in yeasts. 136 , 137 , 138 Finally, oxidative stress is capable of promoting assembling and formation of high molecular weight HSE‐binding complexes which are the hallmarks of early HSFA1a/A1b‐dependent gene expression in thermal stressed leaves of Arabidopsis. 128 , 139
Exposing several primary or primary‐like cell lines to ELF‐EMF has shown to change HSP levels. These effects were identified both by upregulation of HSP genes mRNA levels and protein amounts in different cell types including human chondrocytes, 140 fibroblasts (HuDe, WI‐38) 141 and endothelial cells (SPAE, HUVECs), as well as mouse macrophages, 142 rat neonatal cardiomyocyte 143 and porcine aortic endothelial cells. 144 HSP expression was also induced following exposure of several human lymphoma and leukaemia cell lines including K562, HL‐60, U937, CEM and THP‐1 to ELF‐EMF. 106 , 141 , 145 , 146 , 147 As the energy required for partially unfolding of a protein is about 14 orders of magnitude higher than those possessed by magnetic fields energy, it is very unlikely that ELF‐EMF exposure cause (partial) unfolding in proteins. 101 Therefore, indirect pathways including promotion of ROS accumulation may be the main pathway through which ELF‐EMFs may affect folding of proteins and induce cellular stress responses.
4.2. Unfold protein response
In order to function properly, proteins require a specific three‐dimensional folding. 148 , 149 This unique structural folding is mainly stabilized through intramolecular disulphide bonds particularly for membranous and secretory proteins. 150 Endoplasmic reticulum (ER) is the specific place where nascent proteins synthesized by cytoplasmic ribosomes translocate and become folded and functional. 151 The main by‐product of the process of protein folding is H2O2 which results in maintenance of a high level of ROS. 152 , 153 Therefore, ER redox state is in close relation with correct ER functioning and maintenance of ER protein homeostasis. One of the main sources of ROS generation in cells is the electron transport chain in mitochondria. Different types of ROS are capable of disturbing protein folding in ER and inducing ER stress. Different studies have shown that ER and mitochondria are in close association via the mitochondrial‐associated membranes (MAM). 154 , 155 Diffusion of ROS produced in mitochondria through these membranes enable them to take part in ER redox homeostasis. Therefore, any stressful condition which results in overexpression of mitochondrial ROS can theoretically induce perturbation in ER redox homeostasis and trigger ER stress, a condition which is characterized by accumulation of unfolded proteins in the ER. In consequence of ER stress, cells begin to initiate UPR to counteract stressful condition. The main consequence of UPR is potentiation of protein folding capacity and decreasing protein folding overload. 156
During normal condition, the ER stress sensors, namely PERK, ATF6 and IRE1, are bound with Bip/GRP78 chaperones and are in an inactive state. However, accumulated unfolded protein during stressful condition promotes separation of chaperones from these three ER stress sensors and makes them active. Activated IRE1α and PERK become oligomerized and transphosphorylated in their cytosolic effector region. Activated ATF6, however, is transported to the Golgi apparatus, where it becomes cleaved to S1P and S2P. The signalling pathways underlying these sensors in next plate promotes activation of a number of transcription factors including Nrf2, NF‐κ B, CHOP, ATF4 and XBP1 as well as several protein kinases including JNK and AKT which, in turn, promotes cellular stress response consisting of induction of chaperons, proteasome degradation pathway, autophagy, ER expansion and finally enhancement of antioxidant defence capacity. In addition, PERK can phosphorylate elF2α which, in turn, can suppress total mRNA translation in stressed cells. 157
PERK is the main component of UPR in maintaining redox homeostasis. It has been shown that PERK can act as an upstream activator of Nrf2, the most well‐known transcription factor activating expression of a vast variety of genes encoding antioxidant factors and repressing the pro‐oxidant ones. Activated PERK during stress condition phosphorylates Nrf2 and dissociates from Keap1. Released Nrf2 in next place translocate to the nuclease where it binds with promoter region of genes encompassing antioxidant response element and subsequently induces alterations in gene transcription. 158 , 159 Furthermore, Nrf2 and ATF4 can form heterodimers which can bind with stress response element and trigger expression of haem oxidase‐1 gene. 160 Furthermore, IRE1α can also induce autophagy through modulating JNK pathway. JNK phosphorylates Bcl‐2 which results in dissociation of Beclin1 from Beclin1/Bcl‐2 complex. Released Beclin1 in next place initiates formation of Vps34‐Beclin1 complex which accelerates isolated membrane nucleation and formation of autophagosomes. Also, activated XBP1 by IRE1α RNase domain can also induce autophagy by activating transcription of Beclin1. Finally, PERK pathway can also regulate autophagy by means of activating a number of transcription factors including CHOP, ATF4 and Nrf2. 157
Different studies have depicted putative effects of ELF‐EMF exposure on induction of ER stress. For instance, Chen et al. 161 have shown that exposure to a picosecond pulsed electric field not only promote mitochondrial apoptosis pathway, but also increases expression of ER chaperons including Grp78 and Grp94, and CHOP. Further studies by Keczan et al. demonstrated that ELF‐EMF exposure effects on ER stress are also cell type dependent. They reported that exposure to pulsed EMF did not affect ER stress markers in HepG2 liver carcinoma and HEK 293T human embryonic kidney cell lines while similar exposure condition promoted expression of ER stress markers in HeLa human cervical cancer cell line. 162
4.3. Autophagic response
Observed during nutrition starvation or related stressful metabolic condition, autophagy is defined as a catabolic event through which cellular components are degraded and recycled following transportation in to lysosomes via specific bilayer structures named autophagosomes. 163 It has been shown that other stressful conditions such as hypoxia or oxidative stress are also capable of inducing autophagy. Likewise, several chemotherapy agents including arsenic trioxide and oxaliplatin are capable of inducing autophagy. 164 , 165 PI3K type III‐Atg6/Beclin 1 complex is responsible for initiation of autophagosomes nucleation while Atg12‐Atg5 and Atg8/LC3‐phosphatidylethanolamine conjugates monitor the process of autophagosomes elongation. These two processes are considered as the main characteristics of autophagy. 163 , 166 , 167 It is noteworthy to mention that while accumulation of a large body of autophagic vacuoles may result in initiation of autophagic cell death, a controlled autophagic response guarantees physiological recycling of damaged organelles and biomacromolecules to cope with energy demands following exposure to cytotoxic drugs or a stressful condition. 168 , 169 , 170
As mentioned above, ROS accumulation can trigger initiation of autophagy which in turn, can promote clearance of extra cellular ROS and prevent oxidative damage occurrence in cells. 171 , 172 Since exposure to ELF‐EMF can alter concentrations of cellular ROS, autophagy may also become activated in response to exposure. With this in mind, Chen et al. exposed mouse embryonic fibroblasts (MEF) to PFMF with an intensity of 2 mT and frequency of 50 Hz for different time periods (0.5, 2, 6, 12 and 24 h) and examined alterations in autophagy biomarkers. Based on their results, at 6 h time point, a statistically significant increase in autophagic biomarkers including LC3‐II and formation of GFP‐LC3 puncta was observed. Furthermore, examining cells at this time point with transmission electron microscope demonstrated a statistically significant increase in number of autophagic vacuoles. Using chloroquine, they further confirmed that these alterations in markers of autophagy were resulted from enhancement in autophagic flux and did not take place as a consequence of inhibition of lysosomal function. Investigating the molecular pathway underlying PFMF‐induced autophagy, they observe that the process was totally independent from mammalian target of rapamycin (mTOR) signalling pathway and was largely mediated by cellular ROS. 173
4.4. NF‐kB inflammatory response
Based on different studies, during oxidative stress, activation of NF‐κB transcription factor can protect cells from injury through inhibition of ROS accumulation. Suppressing activation of NF‐κB has shown to be together with an enhancement in TNFα‐induced ROS generation, oxidation of proteins and peroxidation of lipids. 174 In addition, NF‐κB can also modulate activation of autophagy which is another efficient protective mechanism against oxidative stress. Exposing retinal pigment epithelial cells to different concentrations of H2O2, a potent inducer of oxidative stress, resulted in phosphorylation of p65 subunit of NF‐κB which, in turn, promoted upregulation of p62 which is a potent promoter of autophagy. 175 ROS can also delay inactivation of JNK pathway by inhibiting phosphatases responsible for inactivating JNKs. This is mainly mediated through conversion of the catalytic cysteine of these enzymes to sulfenic acid. 176 Studies have shown an increase in TNF‐alpha‐mediated apoptosis in response to a decline in NF‐κB‐mediated inhibitory effect on JNK activation. 177 Therefore, it has been proposed that the anti‐apoptotic effect of NF‐κB may be partly mediated through suppression of JNK pathway activation, in which ROS maybe the bridging molecule. 178 Reported by Wu et al., 179 suppressing activation of NF‐κB during recovery period from a temporarily induced oxidative stress condition resulted in a statistically significant decline in cell viability which further confirms the vital role of NF‐κB activation in cell recovery.
Activation of NF‐κB has also been linked to upregulation of expression of several antioxidant targets. One of these prominent targets is manganese superoxide dismutase (MnSOD). 180 NF‐κB activation following treatment of Ewing's sarcoma cells with TNF‐α resulted in a statistically significant increase in amounts of both thioredoxin and MnSOD. 174 In addition, ferritin heavy chain is another prominent antioxidant which is significantly upregulated by NF‐κB after treatment with TNF‐α. This protein is mainly responsible for suppressing reaction of iron with H2O2 and subsequently, inhibiting formation of highly reactive hydroxyl radicals. 181 Other antioxidant targets affected by NF‐κB consist of glutathione S‐transferase, NAD(P)H dehydrogenase, metallothionein‐3 and glutathione peroxidase‐1. 178
Examining putative role of ELF‐EMF on NF‐κB, Kim et al. 182 demonstrated that exposing RAW264.7 cells results in enhanced translocation of phosphorylated NF‐κB in to the nucleus and induction of inflammatory responses. Contrarily, however, Vianale et al. 183 reported a statistically significant decrease in RANTES, MCP‐1, MIP‐1α and IL‐8 production following ELF‐EMF exposure in HaCaT human keratinocyte cells which was attributed to the inhibition of NF‐κB pathway. These controversial results may be in part due to the different nature of cells used in these studies.
5. ANTI‐/PRO‐APOPTOTIC EFFECTS OF ELF‐EMF: STATEMENT OF THE CONTROVERSY
As discussed above, a vast variety of densities ranging from a few micro Tesla up to tens of milli tesla have been applied in studies examining effects of ELF‐EMF on apoptosis. 184 , 185 , 186 In addition, in some cases, promotion of apoptosis by ELF‐EMF exposure was examined in the presence of a co‐stressor (eg chemotherapeutic agents). One obvious result of these studies is that contrary to chemotherapeutic agents, ELF‐EMF exposure does not demonstrate a clear dose–response pattern. In another words, increase in intensity of magnetic fields does not necessarily result in enhancement of apoptosis or other biological effects. Also, no threshold can be considered for induction of ELF‐EMF biological effects. Despite this, another conclusion from reported data is that very low magnetic flux densities and exposure time are also enough for induction of biological responses. Based on these facts, the differential responses to magnetic fields cannot be attributed to the exposure conditions. Instead, biological state of the experiment including the studied cell type or animal tissue, time point after exposure which the assays are performed, density of cells seeded in plate, or other experimental conditions of the study may determine whether ELF‐EMF exposure can induce biological effects. Interestingly, all these factors can significantly affect the extension of ROS produced in response to exposure to ELF‐EMF. Trying to explain these controversies, McCreary et al. 187 studied fluctuations in cytosolic Ca2+ concentrations in Jurkat E6.1 cells following exposure to ELF‐EMF and stated that magnetic fields effects on Ca2+ can be only consistently detected when biological characteristics of system including pH of the environment, cell cycle phase and response to a Ca2+ agonist were specifically determined. Likewise, several other investigators have also listed some specific criteria necessary to be fulfilled in order to record a consistent response to ELF‐EMF exposure. 188 , 189 , 190 In situations where a co‐stressor such as treatment with a chemotherapeutic agent is also exist, prediction of results becomes more complex. One of the main points, which must be considered additionally in this context, is that how the study has been scheduled. In another words, how is the order of treating with ELF‐EMF exposure and the co‐stressor. In order to make discussion easier, in this section, we only focus on the controversies in apoptosis induced by ELF‐EMF exposure at the presence of another stressor and will not consider the discrepancies related to the biological nature of the materials in the study. In next few paragraphs, we will classify studies based on their experimental design, propose our theory and enumerate the cues in support of the study.
Based on the experimental design of studies existing in literature, we broadly classified treatments into three subclasses: the first group consisted of studies where ELF‐EMF exposure was performed prior to treatment with chemotherapeutic agent. Second group encompassed studies where ELF‐EMF and chemotherapy were performed simultaneously, and finally, the third group consisted of studies where ELF‐EMF exposure was performed after treatment with chemotherapeutic agent. Data regarding the characteristics of ELF‐EMF, duration of exposure, cell type and chemotherapeutic agents applied in the study have been summarized in Table 2. Interestingly, in majority of cases where ELF‐EMF exposure was performed prior to the administration of chemotherapy regimen, the pro‐apoptotic effects of chemotherapeutic agents were reduced. However, if exposure to ELF‐EMF was performed either simultaneously or after pretreatment with chemotherapeutic agent, the pro‐apoptotic effect of regimen was significantly increased. Few exceptions from this pattern however exist in second group. In this manner, simultaneous exposure to ELF‐EMF and chemotherapy with puromycin, 10 camptothecin (only for 24‐h exposure experiment) 191 and melatonin 15 protected cancer cells from pro‐apoptotic effects of these agents. Other important finding of these studies was that exposure to ELF‐EMF per se is not enough for induction of apoptosis.
TABLE 2.
Consequence of different sequential ELF‐EMF/stressor exposure on induction of apoptosis
| Classification of studies | ELF‐EMF treatment | Cell line | Agent co‐used (co‐stressor) | Interaction | |
|---|---|---|---|---|---|
| ELF‐EMF exposure prior to co‐stressor | |||||
| Kaszuba‐Zwoinska et al. 10 | Pulsed electromagnetic field | 50 Hz, 45 ± 5 mT, 4 h/stimulation, 3 times in 24 h | Monocytic cell line MonoMac6 | Minocycline puromycin, colchicine, cyclophosphamide, hydrogen peroxide | Diminished amount of apoptotic and necrotic cells; enhanced expression of gene belonging to pro‐apoptotic family of Bcl‐2 and AIF agent (antagonism) |
| Harland et al. 210 | Environmental‐level magnetic fields |
60 Hz, 1.2 µT 6 days |
MCF‐7 |
Tamoxifen Melatonin |
Significantly block the growth inhibitory action (antagonism) |
| Palumbo et al. 14 | ELF‐EMF | Intermittent 50 Hz, 1 mT; 1 h | Jurkat cells | Anti‐Fas | Significant decrease of anti‐Fas‐induced apoptosis (antagonism) |
| Mansourian et al. 16 | Static (DC) magnetic fields | 93.25–159.4 µT; 10 min | Erythroleukaemia K562 | Electrochemotherapy | Can incur resistance of the cells in response to electric pulses (antagonist) |
| Falone et al. 196 | ELF‐EMF |
75 Hz, 1 mT 5–10 days |
SH‐SY5Y human neuroblastoma |
H2O2 Doxorubicin |
Reduced vulnerability against both H2O2 and ROS‐generating doxorubicin (antagonism) |
| De Nicola et al. 13 | ELF‐EMF | 100 mT,N/A; 4 h | U937 cells | Puromycin | Protect U937 from apoptosis (antagonist) |
| Osera et al. 107 | Pulsed EMF |
75 Hz, 2 mT 40 min |
SH‐SY5Y cell line | H2O2 | Protected SH‐SY5Y cell line (antagonist) |
| Falone et al. 193 | ELF‐EMF | 75 Hz, 2 mT | SK‐N‐BE(2) neuroblastoma | H2O2 | Reduced vulnerability against H2O2 (antagonist) |
| Simultaneous exposure to ELF‐EMF and co‐stressor | |||||
| Marcantonio et al. 211 | ELF‐EMF |
50 Hz, 1 mT 24–72 h |
Neuroblastoma BE(2)C | All trans retinoic acid (ATRA) | Decreased cellular proliferation and increased proportion of G0/G1 phase cells (potentiation) |
| Kaszuba‐Zwoinska et al. 10 | Pulsed EMF |
50 Hz, 45 ± 5 mT 12 h |
Neuroblastoma (U937) |
Puromycin Cyclophosphamide H2O2 Colchicine |
PEMF protects U937 cells against puromycin‐induced cell death (antagonism) |
| Baharara et al. 10 | ELF‐EMF |
50 Hz, 20 mT 2 h |
A2780 ovarian cancer cells | Cisplatin | Increased apoptotic as well as necrotic cells (potentiation) |
| Ding et al. 8 | ELF‐EMF |
60 Hz, 5 mT 24 h |
Leukaemia HL‐60 | H2O2 | Changes generated by the ELF‐EMF can make resistant cells sensitive (potentiation) |
| Liburdy et al. 191 | DC Fields | 12–50 Hz, 6.5 mT | MCF‐7 | Melatonin | Increased the number of apoptotic and necrotic cells (potentiation) |
| Cid et al. 15 | ELF‐EMF |
50 Hz, 10 µT 90 h |
HepG2 | Melatonin | Enhancement of proliferation by blocking melatonin's oncostatic action (antagonism) |
| Pirozzoli et al. 12 | ELF‐EMF |
50 Hz, 1 mT 3 days |
Neuroblastoma cell line LAN‐5 | Camptothecin | Enhancement of proliferation by blocking melatonin's oncostatic action (antagonism) |
| Brisdelli et al. 17 | ELF‐EMF |
50 Hz, 1 mT 72 h |
K562 cells | Quercetin | Protective effect towards apoptosis only at 24 h exposure (antagonism) |
| ELF‐EMF exposure following co‐stressor | |||||
| Jian et al. 9 | Intermittent |
100 Hz, 0.7 mT 1–3 h |
BEL‐7402 | X‐ray radiotherapy | Significantly higher apoptosis rates (potentiation) |
| Ruiz‐Gomez et al. 212 | Pulsed EMF | 1–25 Hz, 1.5 mT; 1 h | Human colon adenocarcinoma (HCA) |
Vincristine Mitomycin Cisplatin |
Increased cytotoxicity (potentiation) |
5.1. Theory: Cellular stress response to ELF‐EMF protects cells from chemotherapy‐induced apoptosis
As mentioned in previous sections, studies in the literature have demonstrated that ELF‐EMF exposure can effectively activate adaptive response and underlying pathways in cells without significantly affecting cellular viability. Consistently, numerous studies have shown that cells are physiologically well buffered against negative effects of ELF‐EMF in monotherapies. Nevertheless, upon addition of even a weak co‐stimulator, (eg upon exposure to a toxin, viruses, or a DNA‐alkylating agents, as well as stressful environmental conditions including a hypoxic incubation area, hyperthermic microenvironment, or under oxidative stressful condition), tolerable ELF‐EMF exposure will meaningfully increase the number of apoptotic cell deaths. 101 Finally, sequential pretreatment with ELF‐EMF and then exposure to apoptotic agents could meaningfully increase the tolerability of the cells to anti‐neoplastic agents and reduce cell deaths (Figure 3). Clearly, considering the sequential events in this scenario, ELF‐EMF pretreatment had induced a series of protective events which could have an antagonizing effect on pro‐apoptotic effects of the anti‐neoplastic agent. Since the only proven effects of ELF‐EMF exposure on cells are cellular adaptive responses, ROS overproduction and intracellular calcium overload, from which the first one is only protective and the other two can be significantly deleterious upon co‐treatment and pretreatment, we hypothesized that the nature of studies (ie the relation between the time of ELF‐EMF exposure and treatment with pro‐apoptotic agent) may be the result beneath this controversy.
FIGURE 3.
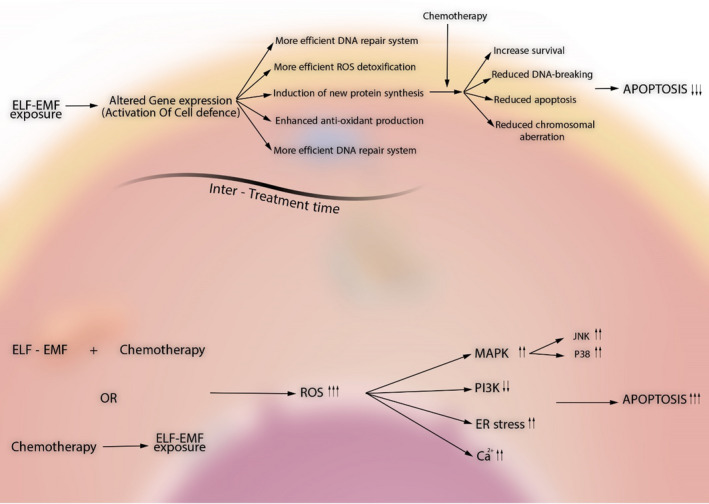
A schematic illustration of the hypothesis for explanation of controversial effects of ELF‐EMF on apoptosis. Upper side: ELF‐EMF exposure prior to treatment with the apoptosis‐inducing agent will result in activation of cellular defence system and alteration in expression of a number of genes which, in next place, will end in promotion of DNA repair system, ROS detoxification system and Ca2+ homeostasis through production of new protective proteins and antioxidative enzymes or restoration of antioxidative stress molecule reservoirs such as glutathione and so on. In next place, upon introduction of the apoptosis‐inducing agent, cells will defend themselves with robust protective system and consequently, lower rate of apoptosis will occur. Lower side: Contrarily, ELF‐EMF co‐treatment with or immediately after chemotherapeutic agent will enhance the rate of injury by ROS overproduction or unbalancing Ca2+ homeostasis which will end in promotion of apoptosis
Numerous data exist in support of this theory. For instance, when SH‐SY5Y neuroblastoma cells were concurrently exposed to H2O2 and ELF‐EMF with intensity of 1 mT and frequency of 50 Hz for 24 h, the increase in catalase was significantly restricted. 192 Contrarily, reports have shown that exposure to magnetic fields with above‐mentioned characteristics alone can induce expression of cytochrome P450 (CYP450) and glutathione S‐transferase (GST), both of which play key roles in cellular detoxification process. 192 , 193 Reported by Patruno et al., 194 administration of phorbol 12‐myristate 13‐acetate (PMA) to human erythro‐leukaemic cells following exposure to ELF‐EMF with intensity of 1 mT and frequency of 50 Hz more effectively enhanced the activity of CYP450. Recently, it has been shown that long‐term exposure of SH‐SY5Y cells to 50 Hz, 1 mT ELF‐MF significantly enhances CAT and GPX free radical scavenging activity and improves reduced glutathione's availability. 195 , 196 This is important, as GSH is a vital co‐factor for GPX and several other enzymes involved in phase II drug metabolization. 197 , 198 , 199 Recently, it has been shown that long‐term exposure to ELF‐EMF before exogenous treatment with methylglyoxal (MG) significantly reduces susceptibility of cancer cells to cytotoxic effects of this agent. 195 This has been mainly attributed to the enhanced accessibility of GSH following ELF‐EMF exposure which is an important co‐factor for directing MG into the glyoxalase‐mediated detoxifying system. 200 Recently, it has also been shown that ELF‐EMF is capable of inducing sirtuin 3 (SIRT3) expression. 196 The signalling cascade mediated by SIRT3 is capable of improving mitochondrial integrity and fitness following exposure to oxidative proteotoxic stress. 201 Finally, activation of Nrf‐2 following exposure to 50 Hz, 1 mT ELF‐MF has shown to be in association with development of resistance to ROS‐producing chemotherapeutic agents in different types of cancer cells. 202 , 203 , 204 , 205
5.2. Possible explanation for exceptional results
As stated above, few exceptions from this pattern however exist in second group. In this manner, simultaneous exposure to ELF‐EMF and chemotherapy with puromycin, camptothecin (only for 24‐h exposure experiment) and melatonin, protected cancer cells from pro‐apoptotic effects of these agents. These observations, however, can be easily explained based on the nature of the anticancer agent applied. For instance, melatonin is a potent free radical scavenger and several points of evidence exist that it can balance upregulated ROS content of cells. In cancer cells, upregulated ROS levels result in activation of tyrosine kinase pathway inhibitors including transcription factor and promote cellular growth. Administration of melatonin in cancer cells modulates ROS levels, inhibits NF‐kB activation and suppresses tumour growth. 206 Contrarily, when cells are exposed to ELF‐EMF, a new source of ROS production is introduced in cells which can at least partially reverse anticancer effects observed with cell's treatment with melatonin.
Camptothecin is a unique chemotherapeutic agent which induces apoptosis at S phase of cell cycle through inhibition of topoisomerase I. 207 Thus, depending on the doubling time of the cells, a specific time period is required for initiation of anticancer effects of camptothecin which is usually about 24 h. Thus, when cells are simultaneously exposed to ELF‐EMF and camptothecin, during the first 24 h, cells have enough time to undergo cellular stress response prior to initiation of action of camptothecin, and therefore, it is not surprising to observe protective effects against apoptosis. However, when the exposure time extends to 48 h, camptothecin is completely active and have produced excess ROS levels through which induced cellular stress response is not capable of coping with it, and therefore, extra ROS produced by ELF‐EMF can help in promotion of apoptosis.
Puromycin is a specific chemotherapeutic agent which is capable of inducing cell death through inhibition protein synthesis, accelerating accumulation of misfolded proteins and induction of apoptosis. 208 Grassi et al. 209 have shown that pre‐exposing cells to ELF‐EMF can significantly reduce apoptosis induced by puromycin which is consistent with the cellular stress response theory. However, the study by Kaszuba‐zwoinska et al. 10 also showed that simultaneous treatment with ELF‐EMF and puromycin can also protect from apoptotic effect. As protein synthesis process and accumulation of unfolded proteins in cells also requires a time period which is usually about 10–12 h, it can also be concluded that cells during concurrent treatment also have enough time for adaption. It is also noteworthy to mention that observed protective effect in this study was very low and about 5%–10% in its highest point. 10
6. CONCLUSION AND FUTURE PERSPECTIVE
As discussed herein, cellular stress response is a unique behaviour of cells following exposure to ELF‐EMF which helps them to cope with next more stressful encountered conditions. This response is mainly due to the mild increase in cellular ROS levels mediated by ELF‐EMF exposure which is not capable of inducing apoptosis alone. The statement discussed in present article, if proved to be true, will become very important in designation of new therapeutic schedules for treatment of cancer as concurrent ELF‐EMF exposure/chemotherapy or ELF‐EMF exposure immediately following chemotherapy can significantly improve pro‐apoptotic effects of chemotherapeutic agents. Furthermore, following this hypothesis, one can apply ELF‐EMF in treatment of resistant cancers as one of the main mechanisms of resistance to chemotherapeutic agents is high capacity of these cells in scavenging free radicals. Additive effects of ELF‐EMF to chemotherapeutic agents in induction of an oxidative stress condition may be helpful in this context. More importantly, ELF‐EMF exposure to normal cells in most cases has shown to be safe and un‐harmful. Therefore, ELF‐EMF therapy may not pose any other adverse effects except for those observed with potentiation of chemotherapeutic agents’ cytotoxic effects. As discussed herein, determination of ELF‐EMF’s intensity window is also very important as no response occurs outside this range, making it a highly personalized therapy. Finally, although this hypothesis apparently sounds rational, future studies comparing results of ELF‐EMF exposure before, during and immediately after chemotherapy is highly recommended for further confirmation of this theory.
CONFLICT OF INTEREST
Authors declare that they have no conflict of interest.
AUTHORS’ CONTRIBUTION
MB, MAJ and AM drafted the main body of the manuscript; BD and MRE modified the manuscript and extended some sections and also designed illustrations; SPS and AMA conceived the original idea of the manuscript; and AMA furthermore supervised the team.
ACKNOWLEDGEMENT
The authors highly acknowledge Prof. Mats‐Olof Mattsson and Prof. Myrtill Simko for their expert review and opinion about this manuscript and their valuable commentary which has been used to improve the consistency of this paper.
Barati M, Darvishi B, Javidi MA, et al. Cellular stress response to extremely low‐frequency electromagnetic fields (ELF‐EMF): An explanation for controversial effects of ELF‐EMF on apoptosis. Cell Prolif. 2021;54:e13154. 10.1111/cpr.13154
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Ahlbom A, Feychting M. Electromagnetic radiation: environmental pollution and health. Br Med Bull. 2003;68(1):157‐165. [DOI] [PubMed] [Google Scholar]
- 2. Feychting M, Ahlbom A, Kheifets L. EMF and health. Annu Rev Public Health. 2005;26:165‐189. [DOI] [PubMed] [Google Scholar]
- 3. Simkó M. Cell type specific redox status is responsible for diverse electromagnetic field effects. Curr Med Chem. 2007;14(10):1141‐1152. [DOI] [PubMed] [Google Scholar]
- 4. Rice‐Evans C, Miller NJ. Total antioxidant status in plasma and body fluids. Methods Enzymol. 1994;234:279‐293. [DOI] [PubMed] [Google Scholar]
- 5. Fanò G, Mecocci P, Vecchiet J, et al. Age and sex influence on oxidative damage and functional status in human skeletal muscle. J Muscle Res Cell Motil. 2001;22(4):345‐351. [DOI] [PubMed] [Google Scholar]
- 6. Lawrence RA, Burk RF. Glutathione peroxidase activity in selenium‐deficient rat liver. Biochem Biophys Res Comm. 1976;71(4):952‐958. [DOI] [PubMed] [Google Scholar]
- 7. Reznick AZ, Packer L. Oxidative damage to proteins: spectrophotometric method for carbonyl assay. Methods Enzymol. 1994;233:357‐363. [DOI] [PubMed] [Google Scholar]
- 8. Ding G‐R, Nakahara T, Hirose H, Koyama S, Takashima Y, Miyakoshi J. Extremely low frequency magnetic fields and the promotion of H2O2‐induced cell death in HL‐60 cells. Int J Radiat Biol. 2004;80(4):317‐324. [DOI] [PubMed] [Google Scholar]
- 9. Jian W, Wei Z, Zhiqiang C, Zheng F. X‐ray‐induced apoptosis of BEL‐7402 cell line enhanced by extremely low frequency electromagnetic field in vitro. Bioelectromagnetics. 2009;30(2):163‐165. [DOI] [PubMed] [Google Scholar]
- 10. Kaszuba‐Zwoinska J, Wojcik K, Bereta M, et al. Pulsating electromagnetic field stimulation prevents cell death of puromycin treated U937 cell line. J Physiol Pharmacol. 2010;61(2):201‐205. [PubMed] [Google Scholar]
- 11. Benassi B, Filomeni G, Montagna C, et al. Extremely Low Frequency Magnetic Field (ELF‐MF) exposure sensitizes SH‐SY5Y cells to the pro‐Parkinson's disease toxin MPP+. Mol Neurobiol. 2016;53(6):4247‐4260. [DOI] [PubMed] [Google Scholar]
- 12. Pirozzoli MC, Marino C, Lovisolo GA, Laconi C, Mosiello L, Negroni A. Effects of 50 Hz electromagnetic field exposure on apoptosis and differentiation in a neuroblastoma cell line. Bioelectromagnetics. 2003;24(7):510‐516. [DOI] [PubMed] [Google Scholar]
- 13. De Nicola M, Cordisco S, Cerella C, et al. Magnetic fields protect from apoptosis via redox alteration. Ann N Y Acad Sci. 2006;1090(1):59‐68. [DOI] [PubMed] [Google Scholar]
- 14. Palumbo R, Capasso D, Brescia F, et al. Effects on apoptosis and reactive oxygen species formation by Jurkat cells exposed to 50 Hz electromagnetic fields. Bioelectromagnetics. 2006;27(2):159‐162. [DOI] [PubMed] [Google Scholar]
- 15. Cid M, Úbeda A, Hernández‐Bule MA, Martínez MA, Trillo MÁ. Antagonistic effects of a 50 Hz magnetic field and melatonin in the proliferation and differentiation of hepatocarcinoma cells. Cell Physiol Biochem. 2012;30(6):1502‐1516. [DOI] [PubMed] [Google Scholar]
- 16. Mansourian M, Firoozabadi SMP, Shankayi Z, Hassan ZM. Magnetic fields with frequency of 217 Hz can reduce cell apoptosis caused by electrochemotherapy. Electromagn Biol Med. 2013;32(1):70‐78. [DOI] [PubMed] [Google Scholar]
- 17. Brisdelli F, Bennato F, Bozzi A, Cinque B, Mancini F, Iorio R. ELF‐MF attenuates quercetin‐induced apoptosis in K562 cells through modulating the expression of Bcl‐2 family proteins. Mol Cell Biochem. 2014;397(1‐2):33‐43. [DOI] [PubMed] [Google Scholar]
- 18. Laqué‐Rupérez E, Ruiz‐Gómez MJ, de la Peña L, Gil L, Martínez‐Morillo M. Methotrexate cytotoxicity on MCF‐7 breast cancer cells is not altered by exposure to 25 Hz, 1.5 mT magnetic field and iron (III) chloride hexahydrate. Bioelectrochemistry. 2003;60(1‐2):81‐86. [DOI] [PubMed] [Google Scholar]
- 19. Mizuno K, Narita E, Yamada M, Shinohara N, Miyakoshi J. ELF magnetic fields do not affect cell survival and DNA damage induced by ultraviolet B. Bioelectromagnetics. 2014;35(2):108‐115. [DOI] [PubMed] [Google Scholar]
- 20. Höytö A, Herrala M, Luukkonen J, Juutilainen J, Naarala J. Cellular detection of 50 Hz magnetic fields and weak blue light: effects on superoxide levels and genotoxicity. Int J Radiat Biol. 2017;93(6):646‐652. [DOI] [PubMed] [Google Scholar]
- 21. Frisch SM, Screaton RA. Anoikis mechanisms. Curr Opin Cell Biol. 2001;13(5):555‐562. [DOI] [PubMed] [Google Scholar]
- 22. Darvishi B, Boroumandieh S, Majidzadeh‐A K, Salehi M, Jafari F, Farahmand L. The role of activated leukocyte cell adhesion molecule (ALCAM) in cancer progression, invasion, metastasis and recurrence: a novel cancer stem cell marker and tumor‐specific prognostic marker. Exp Mol Pathol. 2020;115:104443. [DOI] [PubMed] [Google Scholar]
- 23. Farahmand L, Merikhian P, Jalili N, Darvishi B, Majidzadeh‐A K. Significant role of MUC1 in development of resistance to currently existing anti‐cancer therapeutic agents. Curr Cancer Drug Targets. 2018;18(8):737‐748. [DOI] [PubMed] [Google Scholar]
- 24. Mahdi A, Darvishi B, Majidzadeh‐A K, Salehi M, Farahmand L. Challenges facing antiangiogenesis therapy: the significant role of hypoxia‐inducible factor and MET in development of resistance to anti‐vascular endothelial growth factor‐targeted therapies. J Cell Physiol. 2019;234(5):5655‐5663. [DOI] [PubMed] [Google Scholar]
- 25. Darvishi B, Majidzadeh‐A K, Ghadirian R, Mosayebzadeh M, Farahmand L. Recruited bone marrow derived cells, local stromal cells and IL‐17 at the front line of resistance development to anti‐VEGF targeted therapies. Life Sci. 2019;217:34‐40. [DOI] [PubMed] [Google Scholar]
- 26. Sakhtianchi R, Darvishi B, Mirzaie Z, Dorkoosh F, Shanehsazzadeh S, Dinarvand R. Pegylated magnetic mesoporous silica nanoparticles decorated with AS1411 Aptamer as a targeting delivery system for cytotoxic agents. Pharm Dev Technol. 2019;24(9):1063‐1075. [DOI] [PubMed] [Google Scholar]
- 27. Barzaman K, Samadi M, Moradi‐Kalbolandi S, et al. Development of a recombinant anti‐VEGFR2‐EPCAM bispecific antibody to improve antiangiogenic efficiency. Exp Cell Res. 2021;405:112685. [DOI] [PubMed] [Google Scholar]
- 28. Carneiro BA, El‐Deiry WS. Targeting apoptosis in cancer therapy. Nat Rev Clin Oncol. 2020;17(7):395‐417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ravagnan L, Roumier T, Kroemer G. Mitochondria, the killer organelles and their weapons. J Cell Physiol. 2002;192(2):131‐137. [DOI] [PubMed] [Google Scholar]
- 30. Li P, Nijhawan D, Budihardjo I, et al. Cytochrome c and dATP‐dependent formation of Apaf‐1/caspase‐9 complex initiates an apoptotic protease cascade. Cell. 1997;91(4):479‐489. [DOI] [PubMed] [Google Scholar]
- 31. Hu Y. Role of cytochrome c and dATP/ATP hydrolysis in Apaf‐1‐mediated caspase‐9 activation and apoptosis. EMBO J. 1999;18(13):3586‐3595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Srinivasula SM, Ahmad M, Fernandes‐Alnemri T, Alnemri ES. Autoactivation of procaspase‐9 by Apaf‐1‐mediated oligomerization. Mol Cell. 1998;1(7):949‐957. [DOI] [PubMed] [Google Scholar]
- 33. Saleh A, Srinivasula SM, Acharya S, Fishel R, Alnemri ES. Cytochrome c and dATP‐mediated oligomerization of Apaf‐1 is a prerequisite for procaspase‐9 activation. J Biol Chem. 1999;274(25):17941‐17945. [DOI] [PubMed] [Google Scholar]
- 34. Slee EA, Harte MT, Kluck RM, et al. Ordering the cytochrome c‐initiated caspase cascade: hierarchical activation of caspases‐2, ‐3, ‐6, ‐7, ‐8, and ‐10 in a caspase‐9‐dependent manner. J Cell Biol. 1999;144(2):281‐292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Deveraux QL, Stennicke HR, Salvesen GS, Reed JC. Endogenous inhibitors of caspases. J Clin Immunol. 1999;19(6):388‐398. [DOI] [PubMed] [Google Scholar]
- 36. Vaux DL, Silke J. Mammalian mitochondrial IAP binding proteins. Biochem Biophys Res Comm. 2003;304(3):499‐504. [DOI] [PubMed] [Google Scholar]
- 37. Chinnaiyan AM, O'Rourke K, Tewari M, Dixit VM. FADD, a novel death domain‐containing protein, interacts with the death domain of fas and initiates apoptosis. Cell. 1995;81(4):505‐512. [DOI] [PubMed] [Google Scholar]
- 38. Medema JP, Scaffidi C, Kischkel FC, et al. FLICE is activated by association with the CD95 death‐inducing signaling complex (DISC). EMBO J. 1997;16(10):2794‐2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Salvesen GS, Dixit VM. Caspase activation: the induced‐proximity model. Proc Natl Acad Sci USA. 1999;96(20):10964‐10967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Scaffidi C, Fulda S, Srinivasan A, et al. Two CD95 (APO‐1/Fas) signaling pathways. EMBO J. 1998;17(6):1675‐1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Li H, Zhu H, Xu Cj, Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998;94(4):491‐501. [DOI] [PubMed] [Google Scholar]
- 42. Luo X, Budihardjo I, Zou H, Slaughter C, Wang X. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell. 1998;94(4):481‐490. [DOI] [PubMed] [Google Scholar]
- 43. Sordet O, Khan Q, Kohn K, Pommier Y. Apoptosis induced by topoisomerase inhibitors. Curr Med Chem Anticancer Agents. 2003;3(4):271‐290. [DOI] [PubMed] [Google Scholar]
- 44. Müller M, Wilder S, Bannasch D, et al. p53 activates the CD95 (APO‐1/Fas) gene in response to DNA damage by anticancer drugs. J Exp Med. 1998;188(11):2033‐2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Micheau O, Solary E, Hammann A, Martin F, Dimanche‐Boitrel MT. Sensitization of cancer cells treated with cytotoxic drugs to fas‐ediated cytotoxicity. J Natl Cancer Inst. 1997;89(11):783‐789. [DOI] [PubMed] [Google Scholar]
- 46. Yeh WC, Pompa JL, McCurrach ME, et al. FADD: essential for embryo development and signaling from some, but not all, inducers of apoptosis. Science. 1998;279(5358):1954‐1958. [DOI] [PubMed] [Google Scholar]
- 47. Varfolomeev EE, Schuchmann M, Luria V, et al. Targeted disruption of the mouse Caspase 8 gene ablates cell death induction by the TNF receptors, Fas/Apo1, and DR3 and is lethal prenatally. Immunity. 1998;9(2):267‐276. [DOI] [PubMed] [Google Scholar]
- 48. Zhang J, Wang X, Vikash V, et al. ROS and ROS‐mediated cellular signaling. Oxid Med Cell Longev. 2016;2016:4350965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Covarrubias L, Hernández‐García D, Schnabel D, Salas‐Vidal E, Castro‐Obregón S. Function of reactive oxygen species during animal development: passive or active? Dev Biol. 2008;320(1):1‐11. [DOI] [PubMed] [Google Scholar]
- 50. Bae YS, Oh H, Rhee SG, Yoo YD. Regulation of reactive oxygen species generation in cell signaling. Mol Cells. 2011;32(6):491‐509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kaminskyy VO, Zhivotovsky B. Free radicals in cross talk between autophagy and apoptosis. Antioxid Redox Signal. 2014;21(1):86‐102. [DOI] [PubMed] [Google Scholar]
- 52. Camello‐Almaraz C, Gomez‐Pinilla PJ, Pozo MJ, Camello PJ. Mitochondrial reactive oxygen species and Ca2+signaling. Am J Physiol Cell Physiol. 2006;291(5):C1082‐C1088. [DOI] [PubMed] [Google Scholar]
- 53. Morgado‐Valle C, Verdugo‐Díaz L, García DE, Morales‐Orozco C, Drucker‐Colín R. The role of voltage‐gated Ca2+ channels in neurite growth of cultured chromaffin cells induced by extremely low frequency (ELF) magnetic field stimulation. Cell Tissue Res. 1998;291(2):217‐230. [DOI] [PubMed] [Google Scholar]
- 54. Walleczek J. Electromagnetic field effects on cells of the immune system: the role of calcium signaling. FASEB J. 1992;6(13):3177‐3185. [DOI] [PubMed] [Google Scholar]
- 55. Walleczek J, Budinger TF. Pulsed magnetic field effects on calcium signaling in lymphocytes: dependence on cell status and field intensity. FEBS Lett. 1992;314(3):351‐355. [DOI] [PubMed] [Google Scholar]
- 56. Takahashi K, Doge F, Yoshioka M. Prolonged Ca2+ transients in ATP‐stimulated endothelial cells exposed to 50 Hz electric fields. Cell Biol Int. 2005;29(3):237‐243. [DOI] [PubMed] [Google Scholar]
- 57. Sun S, Cho M. Human fibroblast migration in three‐dimensional collagen gel in response to noninvasive electrical stimulus. II. Identification of electrocoupling molecular mechanisms. Tissue Eng. 2004;10(9–10):1558‐1565. [DOI] [PubMed] [Google Scholar]
- 58. Fanelli C, Coppola S, Barone R, et al. Magnetic fields increase cell survival by inhibiting apoptosis via modulation of Ca2+ influx. FASEB J. 1999;13(1):95‐102. [DOI] [PubMed] [Google Scholar]
- 59. Kapri‐Pardes E, Hanoch T, Maik‐Rachline G, et al. Activation of signaling cascades by weak extremely low frequency electromagnetic fields. Cell Physiol Biochem. 2017;43(4):1533‐1546. [DOI] [PubMed] [Google Scholar]
- 60. Uckun FM, Kurosaki T, Jin J, et al. Exposure of B‐lineage lymphoid cells to low energy electromagnetic fields stimulates Lyn kinase. J Biol Chem. 1995;270(46):27666‐27670. [DOI] [PubMed] [Google Scholar]
- 61. Lindström E, Still M, Mattsson MO, Hansson Mild K, Luben RA. ELF magnetic fields initiate protein tyrosine phosphorylation of the T cell receptor complex. Bioelectrochemistry. 2001;53(1):73‐78. [DOI] [PubMed] [Google Scholar]
- 62. Ke XQ, Sun WJ, Lu DQ, Fu YT, Chiang H. 50‐Hz magnetic field induces EGF‐receptor clustering and activates RAS. Int J Radiat Biol. 2008;84(5):413‐420. [DOI] [PubMed] [Google Scholar]
- 63. Thumm S, Löschinger M, Glock S, Hämmerle H, Rodemann HP. Induction of cAMP‐dependent protein kinase A activity in human skin fibroblasts and rat osteoblasts by extremely low‐frequency electromagnetic fields. Radiat Environ Biophys. 1999;38(3):195‐199. [DOI] [PubMed] [Google Scholar]
- 64. He YL, Liu DD, Fang YJ, Zhan XQ, Yao JJ, Mei YA. Exposure to extremely low‐frequency electromagnetic fields modulates Na+ currents in rat cerebellar granule cells through increase of AA/PGE2 and EP receptor‐mediated cAMP/PKA pathway. PLoS One. 2013;8(1):e54376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Keshet Y, Seger R. The MAP kinase signaling cascades: a system of hundreds of components regulates a diverse array of physiological functions. Methods Mol Biol. 2010;661:3‐38. [DOI] [PubMed] [Google Scholar]
- 66. Piala AT, Humphreys JM, Goldsmith EJ. MAP kinase modules: the excursion model and the steps that count. Biophys J. 2014;107(9):2006‐2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Risso G, Blaustein M, Pozzi B, Mammi P, Srebrow A. Akt/PKB: one kinase, many modifications. Biochem J. 2015;468(2):203‐214. [DOI] [PubMed] [Google Scholar]
- 68. Toker A, Marmiroli S. Signaling specificity in the Akt pathway in biology and disease. Adv Biol Regul. 2014;55:28‐38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Patruno A, Pesce M, Grilli A, et al. mTOR activation by PI3K/Akt and ERK signaling in short ELF‐EMF exposed human keratinocytes. PLoS One. 2015;10(10):e0139644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Martínez M, Úbeda A, Moreno J, Trillo M. Power frequency magnetic fields affect the p38 MAPK‐mediated regulation of NB69 cell proliferation implication of free radicals. Int J Mol Sci. 2016;17(4):510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Nie K, Henderson A. MAP kinase activation in cells exposed to a 60 Hz electromagnetic field. J Cell Biochem. 2003;90(6):1197‐1206. [DOI] [PubMed] [Google Scholar]
- 72. Jin M, Blank M, Goodman R. ERK1/2 phosphorylation, induced by electromagnetic fields, diminishes during neoplastic transformation. J Cell Biochem. 2000;78(3):371‐379. [DOI] [PubMed] [Google Scholar]
- 73. Sun W, Chiang H, Fu Y, Lu D, Xu Z. Effects of electromagnetic noise on the enhancement of stress‐activated protein kinase(SAPK) phosphorylation induced by 50 Hz magnetic fields. Zhonghua Lao Dong Wei Sheng Zhi Ye Bing Za Zhi. 2002;20(4):246‐248. [PubMed] [Google Scholar]
- 74. Sun W, Yu Y, Chiang H, Fu Y, Lu D. Exposure to power‐frequency magnetic fields can induce activation of P38 mitogen‐activated protein kinase. Zhonghua Lao Dong Wei Sheng Zhi Ye Bing Za Zhi. 2002;20(4):252‐255. [PubMed] [Google Scholar]
- 75. Farahmand L, Darvishi B, Majidzadeh‐A K, Madjid Ansari A. Naturally occurring compounds acting as potent anti‐metastatic agents and their suppressing effects on Hedgehog and WNT/β‐catenin signalling pathways. Cell Prolif. 2017;50(1):e12299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Kuzyk PR, Schemitsch EH. The science of electrical stimulation therapy for fracture healing. Indian J Orthop. 2009;43(2):127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Lohmann C, Schwartz Z, Liu Y, et al. Pulsed electromagnetic field stimulation of MG63 osteoblast‐like cells affects differentiation and local factor production. J Orthop Res. 2000;18(4):637‐646. [DOI] [PubMed] [Google Scholar]
- 78. Selvamurugan N, He Z, Rifkin D, Dabovic B, Partridge NC. Pulsed electromagnetic field regulates MicroRNA 21 expression to activate TGF‐β signaling in human bone marrow stromal cells to enhance osteoblast differentiation. Stem Cells Int. 2017;2017:1‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Streit A, Watson BC, Granata JD, et al. Effect on clinical outcome and growth factor synthesis with adjunctive use of pulsed electromagnetic fields for fifth metatarsal nonunion fracture: a double‐blind randomized study. Foot Ankle Int. 2016;37(9):919‐923. [DOI] [PubMed] [Google Scholar]
- 80. Bodamyali T, Bhatt B, Hughes FJ, et al. Pulsed electromagnetic fields simultaneously induce osteogenesis and upregulate transcription of bone morphogenetic proteins 2 and 4 in rat osteoblastsin vitro. Biochem Biophys Res Comm. 1998;250(2):458‐461. [DOI] [PubMed] [Google Scholar]
- 81. Zhou J, Ming LG, Ge BF, et al. Effects of 50 Hz sinusoidal electromagnetic fields of different intensities on proliferation, differentiation and mineralization potentials of rat osteoblasts. Bone. 2011;49(4):753‐761. [DOI] [PubMed] [Google Scholar]
- 82. Xie YF, Shi WG, Zhou J, et al. Pulsed electromagnetic fields stimulate osteogenic differentiation and maturation of osteoblasts by upregulating the expression of BMPRII localized at the base of primary cilium. Bone. 2016;93:22‐32. [DOI] [PubMed] [Google Scholar]
- 83. Selvamurugan N, Kwok S, Vasilov A, Jefcoat SC, Partridge NC. Effects of BMP‐2 and pulsed electromagnetic field (PEMF) on rat primary osteoblastic cell proliferation and gene expression. J Orthop Res. 2007;25(9):1213‐1220. [DOI] [PubMed] [Google Scholar]
- 84. Schwartz Z, Simon BJ, Duran MA, Barabino G, Chaudhri R, Boyan BD. Pulsed electromagnetic fields enhance BMP‐2 dependent osteoblastic differentiation of human mesenchymal stem cells. J Orthop Res. 2008;26(9):1250‐1255. [DOI] [PubMed] [Google Scholar]
- 85. Ongaro A, Pellati A, Bagheri L, Fortini C, Setti S, De Mattei M. Pulsed electromagnetic fields stimulate osteogenic differentiation in human bone marrow and adipose tissue derived mesenchymal stem cells. Bioelectromagnetics. 2014;35(6):426‐436. [DOI] [PubMed] [Google Scholar]
- 86. Yang HJ, Kim RY, Hwang SJ. Pulsed electromagnetic fields enhance bone morphogenetic protein‐2 dependent‐bone regeneration. Tissue Eng Part A. 2015;21(19–20):2629‐2637. [DOI] [PubMed] [Google Scholar]
- 87. Tepper OM, Callaghan MJ, Chang EI, et al. Electromagnetic fields increase in vitro and in vivo angiogenesis through endothelial release of FGF‐2. FASEB J. 2004;18(11):1231‐1233. [DOI] [PubMed] [Google Scholar]
- 88. Li RL, Huang JJ, Shi YQ, et al. Pulsed electromagnetic field improves postnatal neovascularization in response to hindlimb ischemia. Am J Transl Res. 2015;7(3):430. [PMC free article] [PubMed] [Google Scholar]
- 89. Zhou J, Ma XN, Gao YH, et al. Sinusoidal electromagnetic fields promote bone formation and inhibit bone resorption in rat femoral tissues in vitro. Electromagn Biol Med. 2016;35(1):75‐83. [DOI] [PubMed] [Google Scholar]
- 90. Ongaro A, Pellati A, Masieri FF, et al. Chondroprotective effects of pulsed electromagnetic fields on human cartilage explants. Bioelectromagnetics. 2011;32(7):543‐551. [DOI] [PubMed] [Google Scholar]
- 91. Esmail MY, Sun L, Yu L, Xu H, Shi L, Zhang J. Effects of PEMF and glucocorticoids on proliferation and differentiation of osteoblasts. Electromagn Biol Med. 2012;31(4):375‐381. [DOI] [PubMed] [Google Scholar]
- 92. Bagheri L, Pellati A, Rizzo P, et al. Notch pathway is active during osteogenic differentiation of human bone marrow mesenchymal stem cells induced by pulsed electromagnetic fields. J Tissue Eng Regen Med. 2018;12(2):304‐315. [DOI] [PubMed] [Google Scholar]
- 93. Yong Y, Ming ZD, Feng L, Chun ZW, Hua W. Electromagnetic fields promote osteogenesis of rat mesenchymal stem cells through the PKA and ERK1/2 pathways. J Tissue Eng Regen Med. 2016;10(10):E537‐E545. [DOI] [PubMed] [Google Scholar]
- 94. Xie LW, Huang F, Chen AM, Feng LI. Differentiation of rat adipose tissue‐derived mesenchymal stem cells towards a nucleus pulposus‐like phenotype in vitro. Chin J Traumatol. 2009;12(2):98‐103. [PubMed] [Google Scholar]
- 95. Ongaro A, Varani K, Masieri FF, et al. Electromagnetic fields (EMFs) and adenosine receptors modulate prostaglandin E2 and cytokine release in human osteoarthritic synovial fibroblasts. J Cell Physiol. 2012;227(6):2461‐2469. [DOI] [PubMed] [Google Scholar]
- 96. De Mattei M, Varani K, Masieri FF, et al. Adenosine analogs and electromagnetic fields inhibit prostaglandin E2 release in bovine synovial fibroblasts. Osteoarthritis Cartilage. 2009;17(2):252‐262. [DOI] [PubMed] [Google Scholar]
- 97. Zhou J, Liao Y, Xie H, et al. Pulsed electromagnetic field ameliorates cartilage degeneration by inhibiting mitogen‐activated protein kinases in a rat model of osteoarthritis. Phys Ther Sport. 2017;24:32‐38. [DOI] [PubMed] [Google Scholar]
- 98. Fini M, Torricelli P, Giavaresi G, et al. Effect of pulsed electromagnetic field stimulation on knee cartilage, subchondral and epiphyseal trabecular bone of aged Dunkin Hartley guinea pigs. Biomed Pharmacother. 2008;62(10):709‐715. [DOI] [PubMed] [Google Scholar]
- 99. Benazzo F, Cadossi M, Cavani F, et al. Cartilage repair with osteochondral autografts in sheep: effect of biophysical stimulation with pulsed electromagnetic fields. J Orthop Res. 2008;26(5):631‐642. [DOI] [PubMed] [Google Scholar]
- 100. Krath A, Klüter T, Stukenberg M, et al. Electromagnetic transduction therapy in non‐specific low back pain: a prospective randomised controlled trial. J Orthop. 2017;14(3):410‐415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Gutzeit HO. Interaction of stressors and the limits of cellular homeostasis. Biochem Biophys Res Commun. 2001;283(4):721‐725. [DOI] [PubMed] [Google Scholar]
- 102. Simko M, Mattsson MO. Extremely low frequency electromagnetic fields as effectors of cellular responses in vitro: possible immune cell activation. J Cell Biochem. 2004;93(1):83‐92. [DOI] [PubMed] [Google Scholar]
- 103. Luukkonen J, Liimatainen A, Juutilainen J, Naarala J. Induction of genomic instability, oxidative processes, and mitochondrial activity by 50Hz magnetic fields in human SH‐SY5Y neuroblastoma cells. Mutat Res. 2014;760:33‐41. [DOI] [PubMed] [Google Scholar]
- 104. Kesari KK, Juutilainen J, Luukkonen J, Naarala J. Induction of micronuclei and superoxide production in neuroblastoma and glioma cell lines exposed to weak 50 Hz magnetic fields. J R Soc Interface. 2016;13(114):20150995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Feng B, Ye C, Qiu L, Chen L, Fu Y, Sun W. Mitochondrial ROS release and subsequent Akt activation potentially mediated the anti‐apoptotic effect of a 50‐Hz magnetic field on FL cells. Cell Physiol Biochem. 2016;38(6):2489‐2499. [DOI] [PubMed] [Google Scholar]
- 106. Mannerling AC, Simkó M, Mild KH, Mattsson MO. Effects of 50‐Hz magnetic field exposure on superoxide radical anion formation and HSP70 induction in human K562 cells. Radiat Environ Biophys. 2010;49(4):731‐741. [DOI] [PubMed] [Google Scholar]
- 107. Osera C, Amadio M, Falone S, et al. Pre‐exposure of neuroblastoma cell line to pulsed electromagnetic field prevents H2 O2 ‐induced ROS production by increasing MnSOD activity. Bioelectromagnetics. 2015;36(3):219‐232. [DOI] [PubMed] [Google Scholar]
- 108. Srdjenovic B, Mrdjanovic J, Galovic AJ, et al. Effect of ELF‐EMF on antioxidant status and micronuclei in K562 cells and normal lymphocytes. Open Life Sci. 2014;9:931‐940. [Google Scholar]
- 109. Cichon N, Bijak M, Synowiec E, Miller E, Sliwinski T, Saluk‐Bijak J. Modulation of antioxidant enzyme gene expression by extremely low frequency electromagnetic field in post‐stroke patients. Scand J Clin Lab Invest. 2018;78(7‐8):626‐631. [DOI] [PubMed] [Google Scholar]
- 110. Morabito C, Rovetta F, Bizzarri M, Mazzoleni G, Fanò G, Mariggiò MA. Modulation of redox status and calcium handling by extremely low frequency electromagnetic fields in C2C12 muscle cells: a real‐time, single‐cell approach. Free Radic Biol Med. 2010;48(4):579‐589. [DOI] [PubMed] [Google Scholar]
- 111. Tasset I, Pérez‐Herrera A, Medina FJ, Arias‐Carrión Ó, Drucker‐Colín R, Túnez I. Extremely low‐frequency electromagnetic fields activate the antioxidant pathway Nrf2 in a Huntington's disease‐like rat model. Brain Stimul. 2013;6(1):84‐86. [DOI] [PubMed] [Google Scholar]
- 112. Akbarnejad Z, Eskandary H, Vergallo C, et al. Effects of extremely low‐frequency pulsed electromagnetic fields (ELF‐PEMFs) on glioblastoma cells (U87). Electromagn Biol Med. 2017;36(3):238‐247. [DOI] [PubMed] [Google Scholar]
- 113. Somero GN. The cellular stress response and temperature: Function, regulation, and evolution. J Exp Zool A Ecol Integr Physiol. 2020;333(6):379‐397. [DOI] [PubMed] [Google Scholar]
- 114. Mohanan G, Das A, Rajyaguru PI. Genotoxic stress response: What is the role of cytoplasmic mRNA fate? BioEssays. 2021;43:2000311. [DOI] [PubMed] [Google Scholar]
- 115. Chakrabarti M, Mukherjee A. Metallo‐adaptive response: a unique survival strategy of plants under genotoxic stress. Nucleus. 2021;1‐8.33211621 [Google Scholar]
- 116. Payne KK. Cellular stress responses and metabolic reprogramming in cancer progression and dormancy. Semin Cancer Biol. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Crawford DR, Davies KJ. Adaptive response and oxidative stress. Environ Health Perspect. 1994;102(Suppl 10):25‐28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Davies KJA, Quintanilha AT, Brooks GA, Packer L. Free radicals and tissue damage produced by exercise. Biochem Biophys Res Commun. 1982;107(4):1198‐1205. [DOI] [PubMed] [Google Scholar]
- 119. Alessio HM, Goldfarb AH. Lipid peroxidation and scavenger enzymes during exercise: adaptive response to training. J Appl Physiol (1985). 1988;64(4):1333‐1336. [DOI] [PubMed] [Google Scholar]
- 120. Tuschl H, Kovac R, Altmann H. UDS and SCE in lymphocytes of persons occupationally exposed to low levels of ionizing radiation. Health Phys. 1983;45(1):1‐7. [DOI] [PubMed] [Google Scholar]
- 121. Spitz DR, Dewey WC, Li GC. Hydrogen peroxide or heat shock induces resistance to hydrogen peroxide in Chinese hamster fibroblasts. J Cell Physiol. 1987;131(3):364‐373. [DOI] [PubMed] [Google Scholar]
- 122. Laval F. Pretreatment with oxygen species increases the resistance of mammalian cells to hydrogen peroxide and gamma‐rays. Mutat Res. 1988;201(1):73‐79. [DOI] [PubMed] [Google Scholar]
- 123. Lu D, Maulik N, Moraru II, Kreutzer DL, Das DK. Molecular adaptation of vascular endothelial cells to oxidative stress. Am J Physiol. 1993;264(3):C715‐C722. [DOI] [PubMed] [Google Scholar]
- 124. Pacifici RE, Davies KJ. Protein, lipid and DNA repair systems in oxidative stress: the free‐radical theory of aging revisited. Gerontology. 1991;37(1–3):166‐180. [DOI] [PubMed] [Google Scholar]
- 125. Morimoto RI. Regulation of the heat shock transcriptional response: cross talk between a family of heat shock factors, molecular chaperones, and negative regulators. Genes Dev. 1998;12(24):3788‐3796. [DOI] [PubMed] [Google Scholar]
- 126. Schoffl F, Prandl R, Reindl A. Regulation of the heat‐shock response. Plant Physiol. 1998;117(4):1135‐1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Zou J, Guo Y, Guettouche T, Smith DF, Voellmy R. Repression of heat shock transcription factor HSF1 activation by HSP90 (HSP90 complex) that forms a stress‐sensitive complex with HSF1. Cell. 1998;94(4):471‐480. [DOI] [PubMed] [Google Scholar]
- 128. Volkov RA, Panchuk II, Mullineaux PM, Schöffl F. Heat stress‐induced H2O2 is required for effective expression of heat shock genes in Arabidopsis. Plant Mol Biol. 2006;61(4‐5):733‐746. [DOI] [PubMed] [Google Scholar]
- 129. Watanuki M, Sakai A, Sakata T, et al. Role of inducible nitric oxide synthase in skeletal adaptation to acute increases in mechanical loading. J Bone Miner Res. 2002;17(6):1015‐1025. [DOI] [PubMed] [Google Scholar]
- 130. Wahid A, Perveen M, Gelani S, Basra SMA. Pretreatment of seed with H2O2 improves salt tolerance of wheat seedlings by alleviation of oxidative damage and expression of stress proteins. J Plant Physiol. 2007;164(3):283‐294. [DOI] [PubMed] [Google Scholar]
- 131. Banti V, Loreti E, Novi G, Santaniello A, Alpi A, Perata P. Heat acclimation and cross‐tolerance against anoxia in Arabidopsis. Plant Cell Environ. 2008;31(7):1029‐1037. [DOI] [PubMed] [Google Scholar]
- 132. Ahn SG, Thiele DJ. Redox regulation of mammalian heat shock factor 1 is essential for Hsp gene activation and protection from stress. Genes Dev. 2003;17(4):516‐528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Miller G, Mittler R. Could heat shock transcription factors function as hydrogen peroxide sensors in plants? Ann Bot. 2006;98(2):279‐288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Vranova E, Inze D, Van Breusegem F. Signal transduction during oxidative stress. J Exp Bot. 2002;53(372):1227‐1236. [PubMed] [Google Scholar]
- 135. Petrov VD, Van Breusegem F. Hydrogen peroxide‐a central hub for information flow in plant cells. AoB Plants. 2012;2012:pls014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Chu B, Soncin F, Price BD, Stevenson MA, Calderwood SK. Sequential phosphorylation by mitogen‐activated protein kinase and glycogen synthase kinase 3 represses transcriptional activation by heat shock factor‐1. J Biol Chem. 1996;271(48):30847‐30857. [DOI] [PubMed] [Google Scholar]
- 137. Knauf U, Newton EM, Kyriakis J, Kingston RE. Repression of human heat shock factor 1 activity at control temperature by phosphorylation. Genes Dev. 1996;10(21):2782‐2793. [DOI] [PubMed] [Google Scholar]
- 138. Kim D, Kim SH, Li GC. Proteasome inhibitors MG132 and lactacystin hyperphosphorylate HSF1 and induce hsp70 and hsp27 expression. Biochem Biophys Res Commun. 1999;254(1):264‐268. [DOI] [PubMed] [Google Scholar]
- 139. Lohmann C, Eggers‐Schumacher G, Wunderlich M, Schöffl F. Two different heat shock transcription factors regulate immediate early expression of stress genes in Arabidopsis. Mol Genet Genomics. 2004;271(1):11‐21. [DOI] [PubMed] [Google Scholar]
- 140. Corallo C, Battisti E, Albanese A, et al. Proteomics of human primary osteoarthritic chondrocytes exposed to extremely low‐frequency electromagnetic fields (ELF EMFs) and to therapeutic application of musically modulated electromagnetic fields (TAMMEF). Electromagn Biol Med. 2014;33(1):3‐10. [DOI] [PubMed] [Google Scholar]
- 141. Alfieri RR, Bonelli MA, Pedrazzi G, et al. Increased levels of inducible HSP70 in cells exposed to electromagnetic fields. Radiat Res. 2006;165(1):95‐104. [DOI] [PubMed] [Google Scholar]
- 142. Frahm J, Mattsson MO, Simko M. Exposure to ELF magnetic fields modulate redox related protein expression in mouse macrophages. Toxicol Lett. 2010;192(3):330‐336. [DOI] [PubMed] [Google Scholar]
- 143. Wei J, Tong J, Yu L, Zhang J. EMF protects cardiomyocytes against hypoxia‐induced injury via heat shock protein 70 activation. Chem Biol Interact. 2016;248:8‐17. [DOI] [PubMed] [Google Scholar]
- 144. Bernardini C, Zannoni A, Turba ME, et al. Effects of 50 Hz sinusoidal magnetic fields on Hsp27, Hsp70, Hsp90 expression in porcine aortic endothelial cells (PAEC). Bioelectromagnetics. 2007;28(3):231‐237. [DOI] [PubMed] [Google Scholar]
- 145. Akan Z, Aksu B, Tulunay A, Bilsel S, Inhan‐Garip A. Extremely low‐frequency electromagnetic fields affect the immune response of monocyte‐derived macrophages to pathogens. Bioelectromagnetics. 2010;31(8):603‐612. [DOI] [PubMed] [Google Scholar]
- 146. Garip AI, Akan Z. Effect of ELF‐EMF on number of apoptotic cells; correlation with reactive oxygen species and HSP. Acta Biol Hung. 2010;61(2):158‐167. [DOI] [PubMed] [Google Scholar]
- 147. Tokalov SV, Gutzeit HO. Weak electromagnetic fields (50 Hz) elicit a stress response in human cells. Environ Res. 2004;94(2):145‐151. [DOI] [PubMed] [Google Scholar]
- 148. Kosuri P, Alegre‐Cebollada J, Feng J, et al. Protein folding drives disulfide formation. Cell. 2012;151(4):794‐806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Qin M, Wang W, Thirumalai D. Protein folding guides disulfide bond formation. Proc Natl Acad Sci USA. 2015;112(36):11241‐11246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Berridge MJ. The endoplasmic reticulum: a multifunctional signaling organelle. Cell Calcium. 2002;32(5–6):235‐249. [DOI] [PubMed] [Google Scholar]
- 151. Lakkaraju AKK, Mary C, Scherrer A, Johnson AE, Strub K. SRP keeps polypeptides translocation‐competent by slowing translation to match limiting ER‐targeting sites. Cell. 2008;133(3):440‐451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Araki K, Iemura SI, Kamiya Y, et al. Ero1‐alpha and PDIs constitute a hierarchical electron transfer network of endoplasmic reticulum oxidoreductases. J Cell Biol. 2013;202(6):861‐874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Enyedi B, Varnai P, Geiszt M. Redox state of the endoplasmic reticulum is controlled by Ero1L‐alpha and intraluminal calcium. Antioxid Redox Signal. 2010;13(6):721‐729. [DOI] [PubMed] [Google Scholar]
- 154. Araki K, Nagata K. Protein folding and quality control in the ER. Cold Spring Harb Perspect Biol. 2012;4(8):a015438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Gilady SY, Bui M, Lynes EM, et al. Ero1alpha requires oxidizing and normoxic conditions to localize to the mitochondria‐associated membrane (MAM). Cell Stress Chaperones. 2010;15(5):619‐629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334(6059):1081‐1086. [DOI] [PubMed] [Google Scholar]
- 157. Zhang Z, Zhang L, Zhou L, Lei Y, Zhang Y, Huang C. Redox signaling and unfolded protein response coordinate cell fate decisions under ER stress. Redox Biol. 2019;25:101047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Cullinan SB, Zhang D, Hannink M, Arvisais E, Kaufman RJ, Diehl JA. Nrf2 is a direct PERK substrate and effector of PERK‐dependent cell survival. Mol Cell Biol. 2003;23(20):7198‐7209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Cullinan SB, Diehl JA. PERK‐dependent activation of Nrf2 contributes to redox homeostasis and cell survival following endoplasmic reticulum stress. J Biol Chem. 2004;279(19):20108‐20117. [DOI] [PubMed] [Google Scholar]
- 160. He CH, Gong P, Hu B, Stewart D, Choi ME, Choi AMK, Alam J. Identification of activating transcription factor 4 (ATF4) as an Nrf2‐interacting protein. J Biol Chem. 2001;276(24):20858‐20865. [DOI] [PubMed] [Google Scholar]
- 161. Chen WJ, Xiong ZA, Zhang M, et al. Picosecond pulsed electric fields induce apoptosis in HeLa cells via the endoplasmic reticulum stress and caspase‐dependent signaling pathways. Int J Oncol. 2013;42(3):963‐970. [DOI] [PubMed] [Google Scholar]
- 162. Keczan E, Kéri G, Bánhegyi G, Stiller I. Effect of pulsed electromagnetic fields on endoplasmic reticulum stress. J Physiol Pharmacol. 2016;67(5):769‐775. [PubMed] [Google Scholar]
- 163. Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy. 2007;3(6):542‐545. [DOI] [PubMed] [Google Scholar]
- 164. O'Donovan TR, O'Sullivan GC, McKenna SL. Induction of autophagy by drug‐resistant esophageal cancer cells promotes their survival and recovery following treatment with chemotherapeutics. Autophagy. 2011;7(5):509‐524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Mujumdar N, Mackenzie TN, Dudeja V, et al. Triptolide induces cell death in pancreatic cancer cells by apoptotic and autophagic pathways. Gastroenterology. 2010;139(2):598‐608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol. 2010;22(2):124‐131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8(4):445‐544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Jin S, White E. Role of autophagy in cancer: management of metabolic stress. Autophagy. 2007;3(1):28‐31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Levine B, Kroemer G. Autophagy in aging, disease and death: the true identity of a cell death impostor. Cell Death Differ. 2009;16(1):1‐2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Scarlatti F, Granata R, Meijer AJ, Codogno P. Does autophagy have a license to kill mammalian cells? Cell Death Differ. 2009;16(1):12‐20. [DOI] [PubMed] [Google Scholar]
- 171. Filomeni G, Desideri E, Cardaci S, Rotilio G, Ciriolo MR. Under the ROS: thiol network is the principal suspect for autophagy commitment. Autophagy. 2010;6(7):999‐1005. [DOI] [PubMed] [Google Scholar]
- 172. Scherz‐Shouval R, Elazar Z. Regulation of autophagy by ROS: physiology and pathology. Trends Biochem Sci. 2011;36(1):30‐38. [DOI] [PubMed] [Google Scholar]
- 173. Chen Y, Hong L, Zeng Y, Shen Y, Zeng Q. Power frequency magnetic fields induced reactive oxygen species‐related autophagy in mouse embryonic fibroblasts. Int J Biochem Cell Biol. 2014;57:108‐114. [DOI] [PubMed] [Google Scholar]
- 174. Djavaheri‐Mergny M, Javelaud D, Wietzerbin J, Besançon F. NF‐κB activation prevents apoptotic oxidative stress via an increase of both thioredoxin and MnSOD levels in TNFα‐treated Ewing sarcoma cells. FEBS Lett. 2004;578(1‐2):111‐115. [DOI] [PubMed] [Google Scholar]
- 175. Song C, Mitter SK, Qi X, et al. Oxidative stress‐mediated NFkappaB phosphorylation upregulates p62/SQSTM1 and promotes retinal pigmented epithelial cell survival through increased autophagy. PLoS One. 2017;12(2):e0171940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Kamata H, Honda Si, Maeda S, Chang L, Hirata H, Karin M. Reactive oxygen species promote TNFα‐induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell. 2005;120(5):649‐661. [DOI] [PubMed] [Google Scholar]
- 177. Tang F, Tang G, Xiang J, Dai Q, Rosner MR, Lin A. The absence of NF‐κB‐mediated inhibition of c‐Jun N‐terminal kinase activation contributes to tumor necrosis factor alpha‐induced apoptosis. Mol Cell Biol. 2002;22(24):8571‐8579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Morgan MJ, Liu ZG. Crosstalk of reactive oxygen species and NF‐kappaB signaling. Cell Res. 2011;21(1):103‐115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Wu M, Bian Q, Liu Y, et al. Sustained oxidative stress inhibits NF‐kappaB activation partially via inactivating the proteasome. Free Radic Biol Med. 2009;46(1):62‐69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Kairisalo M, Korhonen L, Blomgren K, Lindholm D. X‐linked inhibitor of apoptosis protein increases mitochondrial antioxidants through NF‐κB activation. Biochem Biophys Res Commun. 2007;364(1):138‐144. [DOI] [PubMed] [Google Scholar]
- 181. Pham CG, Bubici C, Zazzeroni F, et al. Ferritin heavy chain upregulation by NF‐kappaB inhibits TNFalpha‐induced apoptosis by suppressing reactive oxygen species. Cell. 2004;119(4):529‐542. [DOI] [PubMed] [Google Scholar]
- 182. Kim SJ, Jang YW, Hyung KE, et al. Extremely low‐frequency electromagnetic field exposure enhances inflammatory response and inhibits effect of antioxidant in RAW 264.7 cells. Bioelectromagnetics. 2017;38(5):374‐385. [DOI] [PubMed] [Google Scholar]
- 183. Vianale G, Reale M, Amerio P, Stefanachi M, Di Luzio S, Muraro R. Extremely low frequency electromagnetic field enhances human keratinocyte cell growth and decreases proinflammatory chemokine production. Br J Dermatol. 2008;158(6):1189‐1196. [DOI] [PubMed] [Google Scholar]
- 184. Xu A, Wang Q, Lv X, Lin T. Progressive study on the non‐thermal effects of magnetic field therapy in oncology. Front Oncol. 2021;11. 10.3389/fonc.2021.638146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Barati M, Fahimi H, Farahmand L, Madjid Ansari A. 1Hz and 100mT electromagnetic field induces apoptosis in breast cancer cells through up‐regulation of P38 and P21. Multidiscip Cancer Investig. 2020;4(1):23‐29. [Google Scholar]
- 186. Cios A, Ciepielak M, Stankiewicz W, Szymański Ł. The influence of the extremely low frequency electromagnetic field on clear cell renal carcinoma. Int J Mol Sci. 2021;22(3):1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. McCreary CR, Thomas AW, Prato FS. Factors confounding cytosolic calcium measurements in Jurkat E6.1 cells during exposure to ELF magnetic fields. Bioelectromagnetics. 2002;23(4):315‐328. [DOI] [PubMed] [Google Scholar]
- 188. Lindström E, Lindström Pe, Berglund A, Lundgren E, Mild KH. Intracellular calcium oscillations in a T‐cell line after exposure to extremely‐low‐frequency magnetic fields with variable frequencies and flux densities. Bioelectromagnetics. 1995;16(1):41‐47. [DOI] [PubMed] [Google Scholar]
- 189. Löschinger M, Thumm S, Hämmerle H, Rodemann HP, Loschinger M, Hammerle H. Induction of intracellular calcium oscillations in human skin fibroblast populations by sinusoidal extremely low‐frequency magnetic fields (20 Hz, 8 mT) is dependent on the differentiation state of the single cell. Radiat Res. 1999;151(2):195. [PubMed] [Google Scholar]
- 190. Mattsson MO, Lindström E, Still M, Lindström P, Mild KH, Lundgren E. [Ca2+](i) rise in Jurkat E6‐1 cell lines from different sources as a response to 50 Hz magnetic field exposure as a reproducible effect and independent of poly‐L‐lysine treatment. Cell Biol Int. 2001;25(9):901‐907. [DOI] [PubMed] [Google Scholar]
- 191. Liburdy RP, Callahan DE, Harland J, Dunham E, Sloma TR, Yaswen P. Experimental evidence for 60 Hz magnetic fields operating through the signal transduction cascade. FEBS Lett. 1993;334(3):301‐308. [DOI] [PubMed] [Google Scholar]
- 192. Reale M, Kamal MA, Patruno A, et al. Neuronal cellular responses to extremely low frequency electromagnetic field exposure: implications regarding oxidative stress and neurodegeneration. PLoS One. 2014;9(8):e104973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Falone S, Grossi MR, Cinque B, et al. Fifty hertz extremely low‐frequency electromagnetic field causes changes in redox and differentiative status in neuroblastoma cells. Int J Biochem Cell Biol. 2007;39(11):2093‐2106. [DOI] [PubMed] [Google Scholar]
- 194. Patruno A, Tabrez S, Pesce M, Shakil S, Kamal MA, Reale M. Effects of extremely low frequency electromagnetic field (ELF‐EMF) on catalase, cytochrome P450 and nitric oxide synthase in erythro‐leukemic cells. Life Sci. 2015;121:117‐123. [DOI] [PubMed] [Google Scholar]
- 195. Falone S, Santini JS, Di Loreto S, et al. Improved mitochondrial and methylglyoxal‐related metabolisms support hyperproliferation induced by 50 Hz magnetic field in neuroblastoma cells. J Cell Physiol. 2016;231(9):2014‐2025. [DOI] [PubMed] [Google Scholar]
- 196. Falone S, Santini S, Cordone V, et al. Power frequency magnetic field promotes a more malignant phenotype in neuroblastoma cells via redox‐related mechanisms. Sci Rep. 2017;7(1):11470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Tew KD. Glutathione‐associated enzymes in anticancer drug resistance. Cancer Res. 1994;54(16):4313‐4320. [PubMed] [Google Scholar]
- 198. Fruehauf JP, Brem H, Brem S, et al. In vitro drug response and molecular markers associated with drug resistance in malignant gliomas. Clin Cancer Res. 2006;12(15):4523‐4532. [DOI] [PubMed] [Google Scholar]
- 199. Sau A, Pellizzari Tregno F, Valentino F, Federici G, Caccuri AM. Glutathione transferases and development of new principles to overcome drug resistance. Arch Biochem Biophys. 2010;500(2):116‐122. [DOI] [PubMed] [Google Scholar]
- 200. Thornalley PJ, Rabbani N. Glyoxalase in tumourigenesis and multidrug resistance. Semin Cell Dev Biol. 2011;22(3):318‐325. [DOI] [PubMed] [Google Scholar]
- 201. Kenny TC, Hart P, Ragazzi M, et al. Selected mitochondrial DNA landscapes activate the SIRT3 axis of the UPR(mt) to promote metastasis. Oncogene. 2017;36(31):4393‐4404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Rocha CR, Kajitani GS, Quinet A, Fortunato RS, Menck CFM. NRF2 and glutathione are key resistance mediators to temozolomide in glioma and melanoma cells. Oncotarget. 2016;7(30):48081‐48092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Ryoo IG, Kim G, Choi BH, Lee SH, Kwak MK. Involvement of NRF2 signaling in doxorubicin resistance of cancer stem cell‐enriched colonospheres. Biomol Ther (Seoul). 2016;24(5):482‐488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Kim EH, Jang H, Shin D, Baek SH, Roh JL. Targeting Nrf2 with wogonin overcomes cisplatin resistance in head and neck cancer. Apoptosis. 2016;21(11):1265‐1278. [DOI] [PubMed] [Google Scholar]
- 205. Zhu J, Wang H, Chen F, et al. An overview of chemical inhibitors of the Nrf2‐ARE signaling pathway and their potential applications in cancer therapy. Free Radic Biol Med. 2016;99:544‐556. [DOI] [PubMed] [Google Scholar]
- 206. Talib WH. Melatonin and cancer hallmarks. Molecules. 2018;23:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Mayer LD, Harasym TO, Tardi PG, et al. Ratiometric dosing of anticancer drug combinations: controlling drug ratios after systemic administration regulates therapeutic activity in tumor‐bearing mice. Mol Cancer Ther. 2006;5(7):1854‐1863. [DOI] [PubMed] [Google Scholar]
- 208. Croons V, Martinet W, Herman AG, De Meyer GRY. Differential effect of the protein synthesis inhibitors puromycin and cycloheximide on vascular smooth muscle cell viability. J Pharmacol Exp Ther. 2008;325(3):824‐832. [DOI] [PubMed] [Google Scholar]
- 209. Grassi C, D’Ascenzo M, Torsello A, et al. Effects of 50 Hz electromagnetic fields on voltage‐gated Ca2+ channels and their role in modulation of neuroendocrine cell proliferation and death. Cell Calcium. 2004;35(4):307‐315. [DOI] [PubMed] [Google Scholar]
- 210. Harland JD, Liburdy RP. Environmental magnetic fields inhibit the antiproliferative action of tamoxifen and melatonin in a human breast cancer cell line. Bioelectromagnetics. 1997;18:555‐562. [DOI] [PubMed] [Google Scholar]
- 211. Marcantonio P, Del Re B, Franceschini A, et al. Synergic effect of retinoic acid and extremely low frequency magnetic field exposure on human neuroblastoma cell line BE (2) C. Bioelectromagnetics. 2010;31(6):425‐433. [DOI] [PubMed] [Google Scholar]
- 212. Ruiz‐Gómez MJ, de la Peña L, Prieto‐Barcia MI, Pastor JM, Gil L, Martínez‐Morillo M. Influence of 1 and 25 Hz, 1.5 mT magnetic fields on antitumor drug potency in a human adenocarcinoma cell line. Bioelectromagnetics. 2002;23(8):578‐525. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.