

Abstract
Rationale:
Phosphorylation of sarcomeric proteins has been implicated in heart failure with preserved ejection fraction (HFpEF); such changes may contribute to diastolic dysfunction by altering contractility, cardiac stiffness, Ca2+-sensitivity and mechanosensing. Treatment with cardiosphere-derived cells (CDCs) restores normal diastolic function, attenuates fibrosis and inflammation, and improves survival in a rat HFpEF model.
Objective:
Phosphorylation changes that underlie HFpEF and those reversed by CDC therapy, with a focus on the sarcomeric subproteome were analyzed.
Methods and Results:
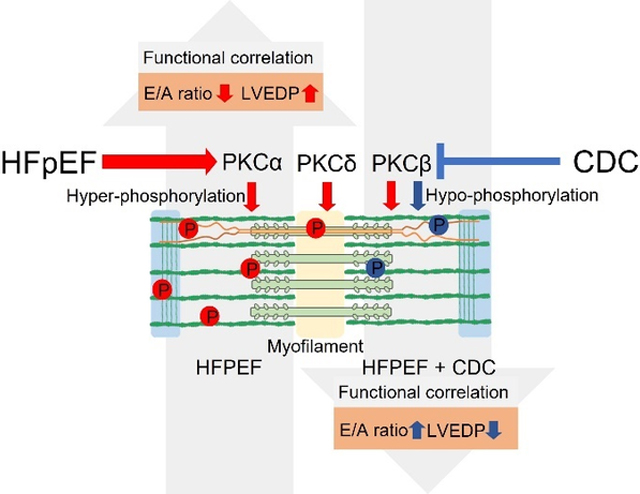
Dahl salt-sensitive rats fed a high-salt diet, with echocardiographically-verified diastolic dysfunction, were randomly assigned to either intracoronary CDCs or placebo. Dahl salt-sensitive rats receiving low salt diet served as controls. Protein, and phosphorylated Ser, Thr and Tyr residues from left ventricular tissue, were quantified by mass spectrometry. HFpEF hearts exhibited extensive hyperphosphorylation with 98% of the 529 significantly changed phospho-sites increased compared to control. Of those 39% were located within the sarcomeric subproteome, with a large group of proteins located or associated with the Z-disk. CDC treatment partially reverted the hyperphosphorylation, with 85% of the significantly altered 76 residues hypophosphorylated. Bioinformatic upstream analysis of the differentially phosphorylated protein residues revealed PKC as the dominant putative regulatory kinase. PKC isoform analysis indicated increases in PKC α, β and δ concentration, whereas CDC treatment led to a reversion of PKCβ. Use of PKC isoform specific inhibition and overexpression of various PKC isoforms strongly suggests PKCβ is the dominant kinase involved in hyperphosphorylation in HFpEF and is altered with CDC treatment.
Conclusion:
Increased protein phosphorylation at the Z-disk is associated with diastolic dysfunction, with PKC isoforms driving most quantified phosphorylation changes. Because CDCs reverse the key abnormalities in HFpEF and selectively reverse PKCβ upregulation, PKCβ merits being classified as a potential therapeutic target in HFpEF, a disease notoriously refractory to medical intervention.
Keywords: Heart Failure
Graphical Abstract
INTRODUCTION
Heart failure with preserved ejection fraction (HFpEF) is characterized by diastolic dysfunction leading to insufficient filling of the heart and increased myocardial stiffness (1, 2). Hypertrophy, fibrosis (3), abnormal Ca2+-handling (4) and/or titin phosphorylation and isoform expression have all been implicated in HFpEF associated cardiomyocyte stiffness (5–8). Sarcomeric proteins are involved in regulating contraction in response to cardiac stiffness and a main focus of today’s research identifying therapy targets for HFpEF has been titin. Titin is a mechanosensing component of sarcomere extensibility, translating mechanical information to appropriate intracellular signaling pathways which could affect other sarcomere proteins (6, 9). In HFpEF, changes in titin phosphorylation, particularly involving residues within the titin isoform elements N2B and N2A, are associated with stiffness changes in HFpEF and alter passive force in HF (5, 10). The phosphorylation changes in other sarcomere proteins occurring in HFpEF and their functional impact are poorly understood including identification of the kinases and or phosphatases that drive the phospho-signaling (11–13).
Cell based therapies for HFpEF are an alternative to small molecules (14, 15), the latter having generally failed in clinical trials. A very promising approach uses cardiosphere derived cells (CDCs), whose therapeutic bioactivity has been verified in several clinical trials for treatment of mild forms of heart failure with reduced ejection fraction (HFrEF) and following myocardial infarction (16–23). Recently, CDC treatment of Dahl salt sensitive (SS) rats with diastolic dysfunction and signs of heart failure (HF) (i.e., HFpEF), improved diastolic function while suppressing myocardial fibrosis, inflammation and apoptosis (24). The driving concept of CDC therapy postulates a paracrine effect arising from the transplanted cells secreting factors, notably exosomes, but the underlying driving signaling pathways remain to be elucidated (25).
We hypothesize that dysregulated kinase/phosphatase signaling results in hyperphosphorylation of sarcomeric and myofilament regulating protein residues in the HFpEF Dahl SS rat model of diastolic dysfunction and is reversed with CDC treatment. This, at least partially, mediates the beneficial impact of this therapy on cardiac myocyte performance. To address this hypothesis, and to narrow down the potential signaling pathways of interest, we: i) determined the phosphorylation sites of the myofilament proteins, including titin, that were altered in hearts with diastolic dysfunction compared to control hearts and reversed with CDC treatment, ii) determined which kinases have a known consensus sequence that could be assigned to the majority of observed myofilament phosphorylation sites and iii) determined which of those kinases had MS data supporting a change in their quantity or their activity in heart failure with diastolic dysfunction and a reversal following CDC treatment.
METHODS
Data Availability.
Please see the major resources table in the supplemental materials. The methods are described in detail in the online supplements. The authors declare that all supporting data are available within the article and its online supplementary files.
RESULTS
Hyperphosphorylation of sarcomeric proteins in HFpEF.
MS analysis of the LV (n=6 per experimental group) was able to quantify 3058 unambiguous proteins and 3396 phosphorylated Ser, Thr and Tyr residues based on the use of annotated and nonannotated rat and annotated mouse databases. The MS-analysis was carried out in discovery mode set up with the intention to maximize coverage of the quantifiable cardiac proteome. Since myofilaments are the most abundant subproteome in cardiac tissue, each sample were fractionated mechanically into cytosolic, myofilament and an in-soluble fraction in order to increase the depth. Even so, due to the high complexity of the samples, some low abundant protein or phosphorylated protein signals were out of the quantifiable linear range. Phosphorylation was altered at 529 sites (p≤0.05), of which 98.8% had an increase in phosphorylation in HFpEF compared to control (high salt fed compared to low salt). The subproteome that had the largest number of phosphorylated residue changes between HFpEF and control was the sarcomere with 208 modified residues (p≤0.05) (and additional 36 phospho-sites trending to be changed (p≤0.1). The listed p-values were calculated using the linear mixed effects model built into MSSTATs (Supplemental table I A). Within the sarcomere, the dominant functions affected by changed phosphorylation were actin-filament binding (18% for HFpEF compared to control) and actin-filament organization (22% for HFpEF compared to control) (Figure 1A). Further cellular functions that can be regulated by phosphorylation and are of special relevance for the pathophysiology of diastolic dysfunction are titin and actinin regulation, myofilament anchoring, extracellular matrix generated tension and mechanosensing (Figure 1A and 2).
Figure 1:

Shown are the represented GO functions in percent for proteins significantly changed in HFpEF vs control left and for CDC vs placebo right within the subproteome of sarcomeric proteins A. With a bioinformatic approach using GPS 2.0, consensus sequences with potential upstream kinases were identified and the graph shows the kinases that potentially could target the most consensus sequences of changed phosphorylation-sites for HFpEF vs control left and CDC vs placebo right. The color of the bar of a given kinase indicates if there was supporting MS data indicating an upregulation by increased concentration of the kinase itself or by changes of a known regulator suggesting an upregulation (red), downregulation by decreased concentration of the kinase itself or by changes of a known regulator suggesting a downregulation (blue), or no supporting MS data (gray).
Figure 2:

The graphic shows sarcomeric proteins with changes in phosphorylation sites relevant for myofilament structure and organization, whereas on the left are proteins with significant changed phosphorylation for comparison HFpEF versus control and on the right comparison of CDC versus placebo. The shown proteins were assigned to the dominant primary functions, e.g., titin regulation, myofilament regulation, structural component of the myofilament, or myofilament anchoring (A). The network figures show potential upstream kinases for altered phosphorylated sarcomere protein residues in HFpEF compared to control (B) and CDC compared to placebo (C) and was generated based on consensus sequence analysis using GPS2.0. Only kinases with MS data support for their changed activity concentration, phosphorylation or known regulators were included in the figure. The blue color indicates a downregulation and the red color an upregulation. The intensity of the node color represents the fold change intensity for HFpEF compared to control or CDC compared to placebo. Edge color shows PKC phosphorylation sites that were confirmed by kinase inhibition, kinase overexpression or recombinant kinase assay. Blue edges indicate a confirmed reduction and red a confirmed increase in phosphorylation confirmation experiments. Purple arrows show increased PKC kinase interactions with target proteins which were identified by PKC pull down experiments.
In LVs with diastolic dysfunction compared to control, we observed extensive phosphorylation across titin (a total of 83 residues), but with significant quantity changes of only a few modified residues, and even fewer residues were sensitive to CDC treatment. Most of the titin phosphorylation sites were located within the N-terminal (25 phosphorylated Ser or Thr residues) and C-terminal region (36 phosphorylated residues) close to the immunoglobulin-like (Ig) domain, the C-terminal Ig domain bound to the M-band and the titin kinase domain of titin (Supplemental figure I). Four residues at the C-terminus, S33927, S33961, T33962 and the residue S20869 were significantly increased in this HFpEF model (p≤0.05) compared to control. Phosphorylation within titin isoform elements N2B and N2A-PEVK was identified for 8 (S3479, S3751, S3888, T4117) and 2 (T11971 and S12022) residues, respectively. Note, the titin isoforms N2B and N2BA were identified by 5 peptides and 4 peptides respectively based on isoform-specific amino acid sequences for the N2B and N2A-PEVK elements (Supplemental figure I). As with total titin, there were no significant changes in overall expression of the different isoforms between any experimental group and control (Supplemental figure I B and I C). Only phosphorylation of residue T4117 within the N2B domain of titin was increased (p≤0.05) in the HFpEF rats compared to control animals (Supplemental table I A).
Interestingly, several proteins that bind titin or play a role in anchoring titin within the Z-disk (i.e, actinin binding proteins) exhibited increased phosphorylation in HFpEF, e.g., Telethonin (Tcap; S161 increased by 350 ± 72.9%, p≤0.05), Myopalladin (Mypn; S622 increased by 51.6 ± 19.2%, S623 increased by 53.5 ± 18.4%, and S737 increased by 76.7 ± 25.8%, p≤0.05), Nebulin-related-anchoring protein (Nrap, S275 increased by 194 ± 54.2%, and S1693 increased by 186.6 ± 53%, p≤0.05), Synaptopodin-2 (Synpo2; S895 increased by 72.3 ± 29.7%, p≤0.05), LIM domain-binding 3 (Ldb3; S44 increased by 81.9 ± 33.1%, S98 increased by 155 ± 41.1%, S1221 increased by 114.9 ± 33%, and S123 increased by 116.3 ± 33.5%, p≤0.05) and actinin (ACTN2; increased by 127.7 ± 71.6%, p≤0.05). Two additional titin binding proteins with increased phosphorylation in the LV of HFpEF rats were Myomesin-2 (Myom2; S535 increased phosphorylation by 113.5 ± 38.6%, p≤0.05) and Obscurin (Obscn; S6609 increased phosphorylation by 183.6 ± 39.4%, p≤0.05). Most of the proteins with changed phosphorylation in HFpEF were structural and regulatory components of the actin-myosin filaments. In addition, wide-ranging phosphorylation changes in proteins affecting myofilament anchoring to the extracellular matrix (ECM), ECM generated tension and mechonosensing were identified, e.g., Vinculin (Vcl) (significantly increased phosphorylation at S97, S290, T602, T719, S721, S579, S795), Desmin (Des) (significantly increased phosphorylation at S300, S303 and trending increased phosphorylation at S25, S28, and S32) and Paxillin (significantly increased phosphorylation at T313, S317, S320, S321, S323, S327, and S328) (Supplemental table I, Figures 2–4).
Figure 4:

The graphic shows sarcomeric proteins with changes in phosphorylation sites relevant for actin stability and actin bundle assembly. On the left are proteins with significantly changed phosphorylation in HFpEF versus control and on the right, a comparison of CDC versus placebo (A). The network figures show potential upstream kinases for altered phosphorylated sarcomere protein residues in HFpEF compared to control (B) and CDC compared to placebo (C) and was generated based on consensus sequence analysis using GPS2.0. Only kinases with MS data support for their changed activity concentration, phosphorylation or known regulators were included in the figure. The blue color indicates a downregulation and the red color an upregulation. The intensity of the nodelor represents the fold change intensity for HFpEF compared to control or CDC compared to placebo. Edge color shows PKC phosphorylation sites that were confirmed by kinase inhibition, kinase overexpression or recombinant kinase assay. Blue edges indicate a confirmed reduction and red a confirmed increase in phosphorylation confirmation experiments.
Sarcomeric phosphorylation events following CDC therapy.
Following CDC treatment of HFpEF rats, 76 phosphorylated residues were significantly altered compared to placebo, 84.2% of which were decreased following CDC treatment. Supplemental table I B lists all significant or trending changed phosphorylated peptides, the fold change, if any, of the corresponding protein and the ratio of phosphorylation/protein quantity. Phosphorylation changes are presented in log2 foldchanges (log2FC). The listed p-values were calculated using the linear mixed effects model built into MSSTATs. This provides insight into whether the observed phosphorylation change is an alteration in site occupancy or due to a change in protein concentration with little or no change in stoichiometry of phosphorylation.
With CDC treatment, there were 35 significantly changed phospho-sites (p≤0.05) and an additional 37 phospho-sites trending to be changed (p≤0.1) compared to placebo. Within the sarcomeres the dominant functions were actin-filament binding (27% for CDC compared to placebo treatment of HFpEF rats) and actin-filament organization (18% for CDC compared to placebo treatment of HFpEF rats) (Figure 1). Further cellular functions that showed phosphorylation changes (mainly hypophosphorylation) following CDC treatment with relevance for diastolic function were titin and actinin regulation, myofilament anchoring, extracellular matrix generated tension and mechanosensing (Figures 1–4). Titin phosphorylation changes following CDC treatment were observed at the N-terminal residue S322. Phosphorylation of the C-terminal residues of titin, T34067, Y34068 and S34488, were reduced (p≤0.05) compared to placebo treatment. However, these sites were not statistical different in rat LVs with high salt induced diastolic dysfunction relative to control rats receiving a low salt diet. The C-terminal residues T34067 and Y34068 were significantly hypophosphorylated in their mono- as well in the di-phosphorylated state of the same tryptic peptide which contains both modifiable amino acid residues. While most CDC-associated changes indicated trends toward hypophosphorylation, particularly in the N-terminal domain, there was a single residue within the fibronectin type III domain of titin, S16620, that was significantly hyperphosphorylated exclusively with CDC treatment (Supplemental table I B). Only the phosphorylation of residue S20869 was significantly reversed with CDC treatment relative to diastolic dysfunction. Following CDC treatment, a few of the titin binding proteins showed altered phospho-modifications, e.g., Obscn (reduced phosphorylation at S454 by 77.1 ± 40.2%, p=0.069), Myosin-binding protein C (Mybpc3; reduced phosphorylation at S302 by 79.3 ± 35%, p≤0.05), Synpo2 (reduced phosphorylation at residues S546 and 549 by 83.6 ± 36.3%, p≤0.05), and Myomesin-1 (Myom1; increased phosphorylated at Y118 by 262 ± 87.3%, p≤0.05) (Figure 2). The most extensive changes in protein phosphorylation following treatment with CDCs occurred in structural and regulating components of actin-myosin filaments. These include Sorbin and SH3 domain-containing protein 2 (Sorbs2; T136 and S143 reduced by 62.8 ± 25.12%, p=0.081, S340 and S343 were reduced by 63.6 ± 69.3%, p=0.077, and S394 increased by 275.6 ± 69.6%, p≤0.05), Filamin-C (Flnc; increased phosphorylation at S2663 by 92.5 33.4%, p=0.057), and Actin-binding LIM protein 1 (Ablim1; S115 increased phosphorylated by 146.2 ± 49.9%, p≤0.05, T495 reduced phosphorylated by 75.1 ± 32.3%, p≤0.05, S496 and S499 were reduced phosphorylated by 55.2 ± 18.2%, p≤0.05) (Figures 2 and 4). In addition, wide phosphorylation changes in proteins affecting myofilament anchoring to the ECM, ECM generated tension and mechanosensing were identified. Some proteins regulating those cellular functions showed changed phosphorylation modifications following CDC treatment, e.g., Ctnna1(reduced phosphorylation at S643 and T647 by 71.3 ± 37.9%, p=0.062), Vcl (reduced phosphorylation at S290 by 25.9 ± 4.12%, p=0.066 and S346 by 32.6 ± 5.9%, p=0.67), and Des (reduced phosphorylation at S25 and S28 by 84.1 ± 48.8%, p=0.051, at S25 and S31 by 68.5 ± 25.7%, p≤0.05, and at S31 and S32 by 78.5 ± 37.7%, p≤0.05) (Figures 2 and 3).
Figure 3:
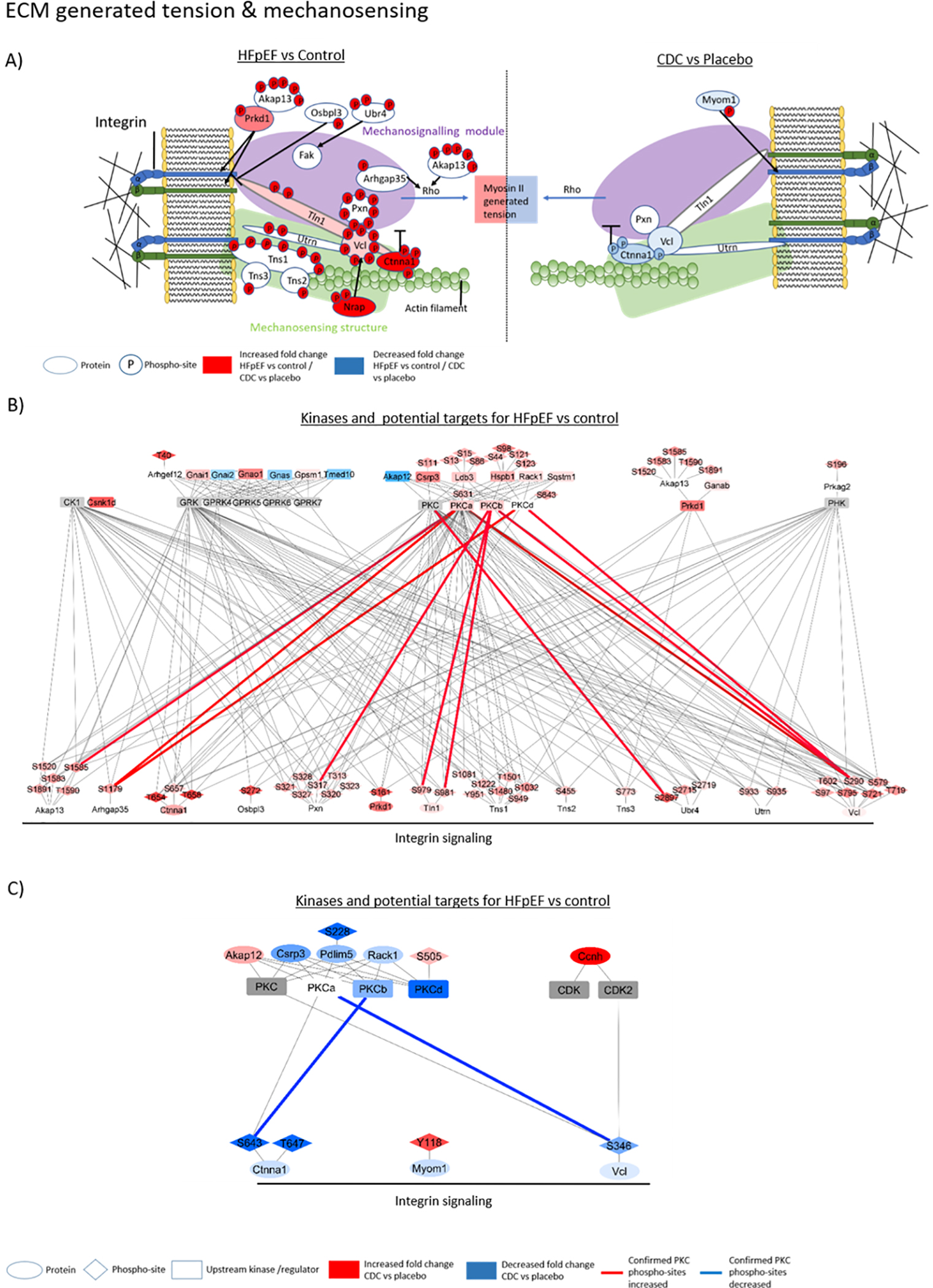
The graphic shows sarcomeric proteins with changes in phosphorylation sites relevant for ECM generated tension and mechanosensing. On the left are proteins with significantly changed phosphorylation of HFpEF versus control and on the right, a comparison of CDC versus placebo (A). The network figures show potential upstream kinases for altered phosphorylated sarcomere protein residues in HFpEF compared to control (B) and CDC compared to placebo (C) and was generated based on consensus sequence analysis using GPS2.0. Only kinases with MS data support for their changed activity concentration, phosphorylation or known regulators were included in the figure. The blue color indicates a downregulation and the red color an upregulation. The intensity of the node color represents the fold change intensity for HFpEF compared to control or CDC compared to placebo. Edge color shows PKC phosphorylation sites that were confirmed by kinase inhibition, kinase overexpression or recombinant kinase assay. Blue edges indicate a confirmed reduction and red a confirmed increase in phosphorylation confirmation experiments.
Identification of Kinases driving phosphorylation events in HFpEF and following CDC treatment.
To facilitate interpretation of sarcomeric phosphorylation patterns, we sought to define which specific kinases were driving the identified phosphorylation changes of protein residues associated with myofilament regulation and the status of those kinases in diastolic dysfunction with or without CDC treatment. A well-established algorithm (GPS 3.0) was used to identify a list of kinases ranked by their likelihood of targeting the altered phosphorylated residues (Supplemental table V). There were 366 potential kinases that share a phosphorylation consensus sequence with at least one changed phosphorylated residue from HFpEF versus control comparison. We filtered this large list of potentially important kinases with additional informatics analysis of the MS data from the total or phospho-enriched peptide samples across treatment groups, specifically prioritizing candidates based on evidence of altered enzyme concentration, phosphorylation changes or alterations in concentration of known kinase regulatory proteins in HFpEF compared to control (left) and HFpEF treated with CDCs compared to placebo (right) (Figure 2). The MS data for total protein concentration of the kinases that met these criteria and their regulators are listed in supplemental table II A and the quantitative data for phosphorylation changes of upstream kinases and their regulators are listed in supplemental table II B. Using these stringent criteria, 4 kinases (Casein kinase 1 alpha [CK1], Protein kinase C [PKC], Phosphoinositide-dependent kinase [PKD] and Phosphorylase kinase [PHK]) were identified within our proteomic dataset to be upregulated in LVs from the HFpEF vs control group and which were designated by the GPS analysis to have the potential to drive the observed hyperphosphorylation of greater than 100 residues known to be involved in of interest for myofilament regulation. The same ranking strategy was then used to identify potential kinases upstream of CDC-induced, hypo- and hyperphosphorylated residues. In total, 334 kinases were predicted by the GPS algorithm as able to phosphorylate the identified residues concerning the myofilament and MS data supported a CDC-associated significant downregulation of PKC kinase and CDK kinase upregulation that also share consensus sequences with more than 20 of the changed phosphorylation sites with CDC treatment compared to placebo. Thus, the data indicate that CK1, Phosphoinositide-dependent kinase-1 (PDK1), PHK and PKC are the potential driving kinases responsible for the observed phosphorylation changes in HFpEF animals compared to control. PKC is the most dominant potential driving kinase targeting the majority of changed phospho-sites (86.9%). Protein kinase A (PKA), which previous studies associated with HFpEF, had relatively low amounts of shared consensus sequences (105 shared sequences) with changed phosphorylation sites in this hypertensive HFpEF rat model. Several PKA subunits were altered in the HFpEF rats, e.g., PKA C-alpha (Prkaca; reduced by 26.6 ± 6.8%, p≤0.05), PKA type I-alpha regulatory subunit (Prkar1a; reduced by 19.5 ± 4.9%, p≤0.05), PKA type I-beta regulatory subunit (Prkar1b; reduced by 30.4 ± 8.4%, p≤0.05), PKA type II-beta regulatory subunit (Prkar2b; increased by 86 ± 19.8%, p≤0.05). Additionally, cAMP-dependent protein kinase inhibitor alpha (PKIA) was increased 14.5fold ± 49% (p≤0.05). Taken together the MS data indicates a reduction of PKA activity in HFpEF rats. Cyclic GMP–dependent protein kinase (PKG) was also associated with HFpEF in previous studies. In this study, PKG was increased by 64.7 ± 16.6% (p≤0.05), but not statistical different following CDC treatment. Unfortunately, PKG is lacking the characteristics of a consensus sequence and thus it is not possible to draw any conclusions regarding its potential contribution to the hyperphosphorylation in HFpEF rats. With CDC treatment, the dominant predicted upstream kinases were PKC (including all isoforms) and G protein-coupled receptor kinase (GRK), potentially targeting 74.7% and 66.2% of following CDC treatment changed phospho-sites (Figure 1B). MS data supports change in these kinases, either change in the quantity or phosphorylation status of the kinase or their regulatory proteins.
PKC has multiple isoforms (26) and among these, PKCα and PKCβ were predominantly predicted to target the changed phospho-sites. First, quantification of each PKC isoform was carried out based on i) analysis of tryptic peptides from the mass spectrometry data that are composed of amino acid sequences that are unique to each isoform and ii) western blot data using isoform specific antibodies. Secondly, the phosphorylation consensus sequence of PKCα was assigned to 86.9% of the phosphorylated residues that changed between the LV from the rats with diastolic dysfunction vs control. In addition, PKCβ could be assigned to 17.6%, whereas 50.5% of the changed phospho-sites were lying within a general PKC phosphorylation consensus site. Interestingly, PKCα was also the dominant kinase potentially targeting 71.8% of phospho-sites changed following CDC treatment compared to placebo, whereas only 16.9% of the phospho-sites were within specific consensus sequences for PKCβ. Furthermore, 43.7% of changed phospho-sites for CDC versus placebo were lying within a general PKC consensus site, thus, leaving only 23.9% of the PKC consensus that couldn’t be affected by the PKCβ downregulation following CDC treatment. All other PKC isoforms have either none or very few of the sarcomeric phosphorylation sites assigned to the known consensus sequences (Figure 2–4).
Western blots were performed to quantify changes in specific PKC isoforms and statistical significance was calculated using the non-parametric Krustal-Wallis one-way ANOVA. There was an increase in the quantity of PKCα by 61.8 ± 21.3% (p≤0.05) (Figure 5A), PKCβ by 80.2 ± 38.3% (p≤0.05) (Figure 5C) and PKCδ by 133.1 ± 46.7% (p≤0.05) (Figure 5D). Phosphorylation sites known to activate PKC (27–30) weren’t significantly changed in this HFpEF model compared to control. However, one analyzed activity inducing phosphorylation site, S505 of PKCδ, was increased following CDC treatment compared to placebo by 92.1 ± 12.6% (p≤0.05) (Figure 5E). Taken together these data support both quantity and kinase activity increase in HFpEF compared to control for PKC isoforms α, β and δ with PKCβ and δ isoforms exclusively reverting towards control levels with CDC treatment. The protein quantity and phosphorylation status changed also for several PKC regulators in HFpEF: A-kinase anchor protein 12 (Akap12) was reduced 66.2 ± 18.6% (p≤0.05), Ldb3 increased by 63.6 ± 10.4% (p≤0.05) and its phospho-sites S44, increased by 81.5 ± 33.1% (p≤0.05), S98 increased by 192.8 ± 41% (p≤0.05), S121 increased by 114.4 ± 33% (p≤0.05) and S123 increased by 115.9 ± 33.5% (p≤0.05). Pdlim5 was increased by 128.2 ± 16.2% (p≤0.05), Rack1 40.4 ± 8.3% (p≤0.05), Csrp3 increased by 119.1 ± 9.1% (p≤0.05) and its phosphorylation site S111 increased by 169.5 ± 44.6% (p≤0.05). Hspb1 increased by 165.7 ± 12.4% (p≤0.05) and its phosphorylation sites S13 107 ± 36.4% (p≤0.05), S86 126.6 ± 24.3% (p≤0.05), S15 129.7 ± 30% (p≤0.05), and Sqstm1 increased by 35.7 ± 15.8% (p≤0.05) in diastolic dysfunction compared to control (Supplemental table II A). Many of these changed towards control values following CDC treatment, specifically Csrp3 was reduced by 35.4 ± 7.9% (p≤0.05), Pdlim5 was reduced by 25.8 ±14.9% (p≤0.05) and its phosphorylation site S228 was trending to be reduced by 83.9 ± 59.9% (p=0.059). Rack1 was reduced by 18.2 ± 7.1% (p≤0.05), but Akap12 was increased by 81.5 ± 21.1% (p≤0.05) (Supplemental table II B).
Figure 5:
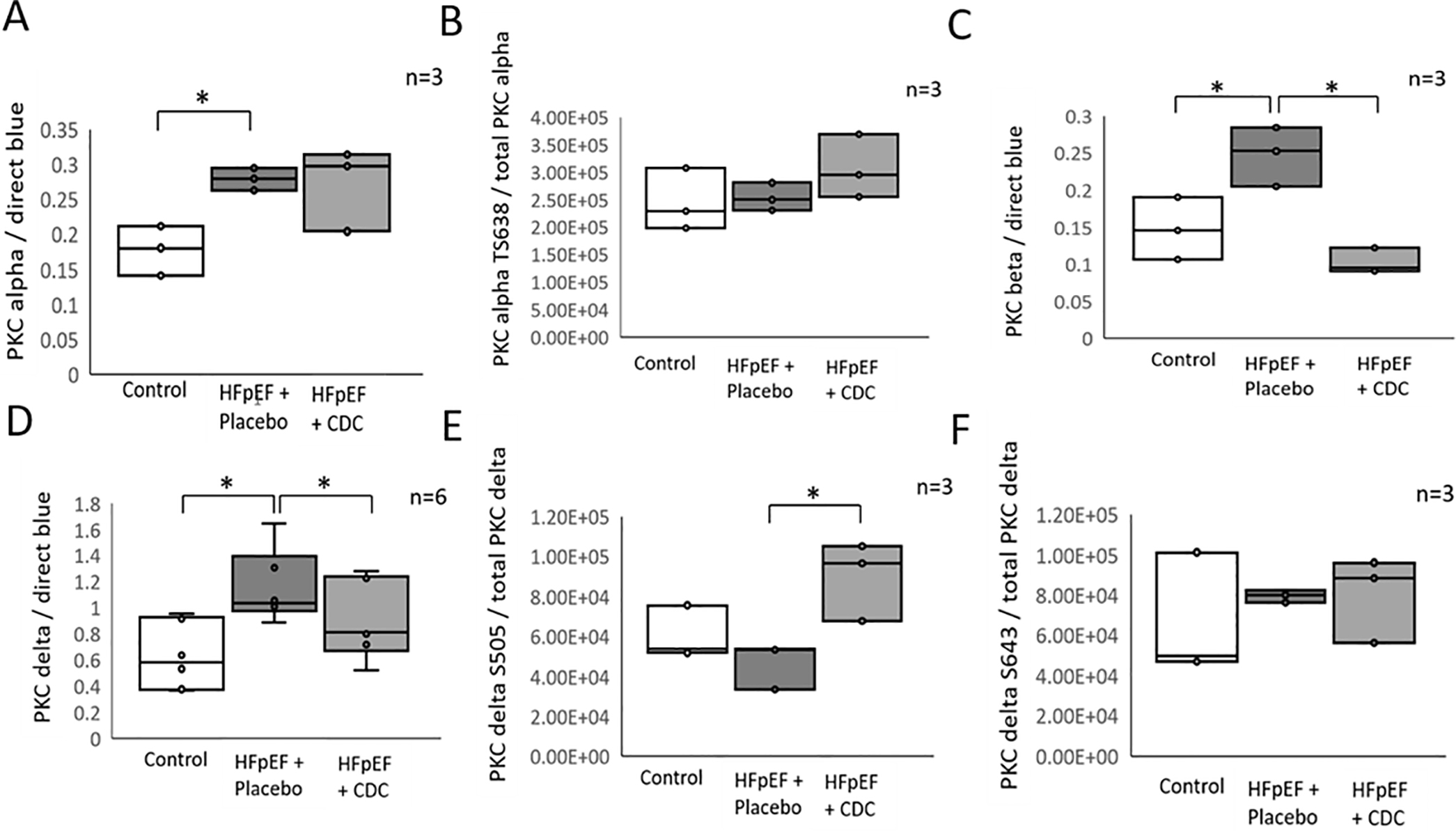
Relative quantification of PKC kinase isoforms and their regulators. Western blots were performed for PKC α (A), β (C), and δ (D) the phosphorylation sites T638 of PKCα (B), S505 (E), and S643 (F) of PKCδ from the identical LV rat samples used for MS analysis. All intensities were normalized to total protein staining with direct blue. * indicates significance p≤0.05 among treatment groups tested with non-parametric Krustal-Wallis one-way ANOVA (n=6 for D, n=3 for A, B, C, E and F).
Additional experiments were performed to confirm PKC is the upstream kinase driving the observed phosphorylation changes in the sarcomere: i) recombinant PKCα, PKCβ, or PKCδ were added to cardiac lysate of control rats, ii) individual PKC isoforms were inhibited with 1,2,3,4-Tetrahydro Staurosporine (PKCα inhibitor), PKCβ inhibitor (CAS 257879–35-9), Rottlerin (PKCδ inhibitor), and H-89 dihydrochloride (PKC inhibitor) in hypertrophic and control H9C2 cells, and iii) each PKC isoform was overexpressed in H9C2 cells. The protein phosphorylation was quantified by MS for each experimental group and validated for a subset of the phosphorylation sites that were altered between groups. Collectively, 47 phosphorylation sites on the myofilament proteins were phosphorylated by addition of PKC to cell lysate, specifically 27 for PKCα, 16 for PKCβ and 24 PKCδ (p≤0.05) (Supplemental table III A). Conversely, small molecule inhibition of PKC isoforms in hypertrophic H9C2 cells led to a reduction in phosphorylation of 11 residues of myofilament proteins, all having the PKC consensus sequences (Supplemental table III B). Finally, the overexpression of PKC isoforms or PKC regulators Ldb3 and Pdlim5 confirmed 2 additional sites which were also increased in HFpEF animals (Supplemental table III C). Finally, co-immunoprecipitation analysis revealed significantly increased interactions between PKC kinases and myofilament associated proteins in HFpEF rats compared to control, e.g., Ldb3, titin, actin (Acta2), alpha-crystallin B chain (Cryab), myosin-7 (Myh7), myosin-11 (Myh11), and Des.
To address the potential importance of particular PKC isoforms to specific heart functions we correlated the cellular concentration and phosphorylation status of each PKC isoform (based on WB) for each individual animal with the functional data for E/A ratio, Tau and/or LVEDP obtained for the same corresponding animal (functional data is listed in Soetkamp et al. 2021) (31) (Figure 6). Although PKCα, PKCβ, and PKCδ cellular concentrations were each negatively correlating with the E/A ratio (R2= − 0.233, −0.28, and −0.718, respectively), the PKCδ isoform was highly correlative to the E/A ratio. As well, the PKCβ cellular concertation also correlated strongly with LVEDP (R2=0.712). The phosphorylation status but not the cellular concentrations of PKCα T638 (R2= −0.285) and PKCδ S505 (R2= −0.205) correlated to LVEDP. The cellular concentrations of the other PKC isoforms did not correlate with LVEDP and none of the isoforms correlated well with Tau (R2=0.162 – 0.113).
Figure 6:

The graphs are showing the correlation between cardiac functional data, E/A ratio, TAU, and LV end-diastolic pressure (LVEDP) and PKC isoform concentrations (A, C, D) or PKC phosphorylation sites T638 of PKCα (B), S505 (E), and S643 (F) of PKCδ of specific animals (HFpEF + placebo, HFpEF + CDC, and control) used in this study.
Taken together, ~30% of the myofilament and myofilament regulating proteins within PKC consensus sequences were affected downstream by PKC manipulations (Supplemental table III A, B and C). Interestingly, there are 22 residues that were changed in the opposed direction of the kinase manipulation indicating phosphate regulation downstream of PKC. Equally important is the potential role of phosphatases in mediating the identified phosphorylation changes. Twenty-five phosphatases not including subunits or isoforms were found to have changed abundances and 11 residues had increased phosphorylation in rat LVs with diastolic dysfunction compared to control (Supplemental table IV A). Following CDC treatment, 17 of the phosphatases that were changed in HFpEF rats reverted towards control as did two phosphorylation-sites on these phosphatases. In addition, CDC treatment uniquely altered an additional 3 phosphatases (Supplemental table IV B).
DISCUSSION
Here we have dissected the proteomic changes underlying HFpEF, a common but poorly understood disease which has proven refractory to treatment. Recent studies have implicated inflammation and fibrosis as major factors in the HFpEF phenotype (1, 32, 33). Increased inflammatory cells in the heart could lead to increased LV fibrosis; the resulting increase in collagen would result in increased passive stiffness leading to impaired diastolic function (32). The Dahl salt sensitive rat, a well-established model of HFpEF, has been widely used to test new treatments for HFpEF, reproducing some of the typical features of HFpEF (34). The animals used in this study were verified to have HFpEF by echocardiography (hypertrophy, reduced early to late ventricular filling velocities (E/A) ratio, slowed LV relaxation); histology revealed increased fibrosis and increased proinflammatory and profibrotic cytokines (tumor necrosis factor-α, interleukin-6, monocyte chemotactic protein-1 and tissue inhibitor of metalloproteinase) (24). Although the DS model does not fully recapitulate human HFpEF, it does have comorbidities (hypertension, insulin resistance and hyperlipidemia) that are clinically relevant. In a previous study, sarcomeric phosphorylation changes in heart failure were reversed after a stem cell therapy (35). In this study, we take advantage of our earlier finding that cell therapy with CDCs reverses the key phenotypic abnormalities of HFpEF in Dahl salt-sensitive rats: diastolic dysfunction is ameliorated, fibrosis and inflammation are blunted, and survival is enhanced (24).
These salutary effects occur despite persistent hypertension and hypertrophy in the rats, making CDCs an intriguing tool for selective dissection of pathways specifically related to the HFpEF phenotype. Combined phosphoproteomics and informatics reveal several novel findings in this HFpEF model, and the mechanism of CDC benefit: i) Hyperphosphorylation compared to control animals; ii) Selective changes in phosphorylation with CDC treatment; iii) Primary phosphorylation changed subproteome were sarcomeric proteins with a large fraction of proteins located or associated with the Z-disk or M-band; iv) Prediction of PKC as the primary kinase involved based on bioinformatic consensus sequence analysis; v) PKC isoforms and upstream regulator concentration alterations in the HFpEF rat model and a reversal of PKCβ and PKC regulators following CDC treatment; vi) Use of PKC isoform specific inhibition and overexpression of PKC isoforms confirming downstream sites strongly suggests that PKC isoforms α, β and δ are the dominant kinases involved in hyperphosphorylation in HFpEF and may be altered with CDC treatment. Of the large number of phosphorylation sites (653) that are induced in this model of HFpEF compared to controls, the majority of the hyperphosphorylated residues (205 significantly changed and further 38 trending, p≤0.05 and p≤0.1 respectively) were in the sarcomeric subproteome. It is notable that there were no hypophosphorylated residues indicating activation of kinases or inhibition of phosphatases. In this study we could not detect cardiac troponin phosphorylation due to the sample complexity analyzed. However, hyperphosphorylation of cardiac troponin I in association with an increased PKC expression was shown previously in a hypertensive rat model, supporting the importance of our findings here (36).
The majority of the sarcomeric proteins altered by HFpEF are involved in actin filament stability and actin bundle assembly, extracellular matrix generated tension and mechanosensing, or Z-disk organization which could act as a potential hub for regulating myofilament contraction and relaxation. Titin was the most phosphorylated protein with 83 modified residues. Interestingly, 65% percent of all phosphorylated titin residues were located near the N- and C-termini of titin. The N-terminus anchors to the Z-disk by binding to α-actinin (37, 38) which contains the C-terminus protein binding motif, tetratricopeptide repeat (TPR) (39–42). Only a small subset S20869, S33927, S33961, T33962 and T4117 were significantly p≤0.05 increased in the HFpEF model compared to control. The significantly changed phospho-sites located at titin’s C-terminus are located between the Ig-like 138 and TPR 14 domain of titin. Those titin domains are thought to form interactions with myosin, keeping the A-band in a central position within the sarcomere absorbing misbalances of active forces (43, 44). How phosphorylation changes within these titin domains affect the myofilament structure and contractility performance remains to be elucidated. Hypophosphorylation of titin was identified across multiple sites and was only associated with a significant reversal of phosphorylation on one of the 5 residues significantly increased in LVs with diastolic dysfunction following CDC treatment. This includes general hypophosphorylation of titin including residue S20869, which is conserved in mouse and human (S20007) located in the C-terminus within the myosin binding region. A previous study suggests this site is a target of β-adrenergic signaling (45). Titin phosphorylation is additionally discussed online.
In total, 23 proteins located within the Z-disk had increased phosphorylation with HFpEF, e.g., actinin (46), Tcap (47), Myom2 (48) and Obscn (49). For more detailed information see the online supplement. The HFpEF induced hyperphosphorylation of Tcap residue S161 is known to be a target of β-adrenergic signaling and is linked to cytoskeletal reorganization (50, 51). In our data, we determined that this site is also a PKCδ target (Table 2). The role of the Z-disk is not well understood, but there is increasing evidence that the sarcomere Z-disk function is sensing and regulating mechanical tension during cardiomyocyte hemodynamic adaptations (52, 53) and can respond to changes in systolic force and diastolic tension (54). This role of the Z-disk is supported by mouse genetic studies with the knockouts of a number of Z -disk proteins, including LIM protein (55), calsarcin-1 (56) or melusin (57), all developing heart failure phenotypes. This includes a rat model with titin Z-disk truncations showing diastolic heart failure phenotypes (58). Our study is the first to associate increased protein phosphorylation at the Z-disk with diastolic dysfunction. Taken together, this suggests a broader regulated role of the Z-disk in this model of HFpEF. This concept is further supported by the fact that CDC treatment reverted the HFpEF-induced phosphorylation of the majority of the myofilament and myofilament regulating proteins located within the Z-disk or M-band.
Another fundamental impact that could derivate from the identified altered myofilament phosphorylation is myofilament anchoring, ECM generated tension and mechanosensing, potentially affecting cardiac contraction and relaxation. Multiple proteins involved in these processes were hyperphosphorylated with HFpEF. CDC induced phosphorylation changes included Myom1, Mybpc3, Des, and Vcl. Several of these phosphorylation sites have been confirmed as PKC targets (Table 3). The identified phosphorylation changes could be part of CDC mediated signaling cascades that result in a downregulation of PKC. These changes could be a result of reduced fibrosis occurring with CDC treatment (24) and therefore could be an adaptation to reduced ECM generated stiffness.
Our data strongly supports that PKC is the predominant kinase driving the phosphorylation of the sarcomere. It is already documented, that PKC family members, PKCα and PKCβ, are both increased in human heart failure and numerous animal studies have implicated PKC activation in HF and other cardiac diseases (59–62). In this model of HFpEF, the quantity of PKC isoforms α, β and δ was increased based on MS and western blot and were hyperphosphorylated at residues T638 and S643, which are known to activate these kinases (28, 63) Interestingly, there is an increased quantity of two proteins known to be involved in the sarcomeric localization of PKC, Ldb3 and Pdlim5. Pdlim5 is an PKC interacting protein, which can bind to various PKC isoforms and regulate their activity (64, 65). The protein localizes to the Z-disk by binding to α-actinin (66). Like Pdlim5, the PKC binding protein Ldb3 is co-localized with α-actinin at the Z-line suggesting an adapter role to mediate localized PKC signaling within the sarcomeric Z-line (67–69). Further support for a Ldb3-induced increased PKCα activity in HFpEF rats arises from the hyperphosphorylation of residue S44 located in the PKCα binding PDZ domain. In addition, there is increased interaction of PKC with Ldb3 in co-immunoprecipitation studies. Taken together we hypothesize that increased quantities of both Ldb3 and Pdlim5 are contributing to the observed hyperphosphorylation located at or near the Z-line, by localizing PKC to this specific region. This hypothesis is backed by the increased interaction between PKC and myofilament proteins (Titin, Actin, Des, Myh7, Myh11 and Cryab). Furthermore, we find that CDC treatment results in phosphorylation patterns consistent with decreased PKC activity.
It is of interest if these phosphorylation events driven by PKC correlate with specific cardiac dysfunctions known to HFpEF and if those functional parameters are reversing in correlation with PKC concentrations following CDC treatment. The cellular quantity of each PKC isoforms correlated negatively with E/A ratio suggesting that the increased PKC concentrations are driving, at least in part, the reduced E/A ratio that is observed in HFpEF (31) and is supported by the reduction of PKC isoform concentrations following CDC treatment correlating to an increase in E/A ratio post-CDC treatment. Since LVEDP pressure specifically correlated with PKCβ suggests that concentration changes of this isoform could have the strongest impact on cardiac function in HFpEF and a reduction of PKCβ could be key to reduce diastolic pressure in HFpEF. The fact that only weaker correlations for Tau and PKC isoform concentrations were found could point out that there could be another key element regulation cardiac stiffness in HFpEF and CDC therapy.
There are a few limitations of our study that are noteworthy. First, the evidence we present here is correlational and indirect, and we have not demonstrated that any of the specific phosphorylated residues changing with chronic diastolic dysfunction and CDC treatment will alter myocardial function. The study should be viewed as hypothesis-generating, more than hypothesis-testing. Additional experiments will be required to reveal the impact of the phospho-sites changed in hearts with diastolic dysfunction with or without CDC treatment and integrate these data with those already accumulated for other PTMs. Besides PKC, phosphatases or other kinases could have a role in protein hyperphosphorylation in the HFpEF rat model and induced beneficial phosphorylation changes following CDC treatment. For instance, the identification of cGMP-dependent protein kinase 1 (PKGI) phosphorylation sites is highly problematic because the enzyme is lacking the characteristics of a PKGI consensus sequence. In this study PKG was increased by 145 ± 97% (p≤0.05) in HFpEF rats (not statistical different with CDC treatment) in contrast to previous studies where decreased PKG activity was correlated with HFpEF (70). Additional phosphorylation sites previously identified in HFpEF or CDC were possibly not represented here due to the non-targeted nature of this study and therefore could not be ruled out as mechanisticly relevant.
In conclusion, this study showed protein hyperphosphorylation in a Dahl salt sensitive rat model of HFpEF that reversed with CDC therapy, indicating an upregulation of kinase signaling that was reduced with the stem cell therapy. For the first time it is shown that the sarcomere is hyperphosphorylated at the Z-line and M-line, as well as at the myofilament anchoring and mechanosensing structures. These changes are correlated with pathology in a hypertension-induced diastolic dysfunction animal model. These same regions demonstrate hypophosphorylation following treatment with CDCs. Furthermore, our data suggests that these changes are most likely to mediate PKC isoforms α, β and δ, which represent potential new drug targets for treatment of HFpEF. In humans with type 2 diabetes, treatment with PKCβ-inhibitor ruboxistaurin mesylate improved kidney function (71). Since CDC treatment of this HFpEF model led to reduced PKCβ activity specifically, it would be worthwhile to test PKCβ-inhibition as a replacement therapy for CDC treatment given the difficulties of establishing a biological drug as an FDA approved therapy.
Supplementary Material
NOVELTY AND SIGNIFICANCE.
What Is Known?
In heart failure with preserved ejection fraction (HFpEF) phosphorylation of sarcomeric proteins contribute to diastolic dysfunction by altering contractility and cardiac stiffness.
Stem cell therapy (cardiosphere-derived cells, CDCs) has proven to be beneficial, restoring normal diastolic heart function in a rat HFpEF model.
What New Information Does This Article Contribute?
Extensive hyper-phosphorylation in myofilament proteins driven by increase in PKCα, β, and/or δ concentrations in correlation with reduced E/A ratio and increased LVEDP.
CDC therapy leads to hypo-phosphorylation of myofilament proteins and reduction of PKCβ, and/or δ in correlation with an increased E/A ratio and reduced LVEDP.
HFpEF, is a clinical syndrome associated with premature mortality. There is no clinical treatment for HFpEF and HFpEF is lacking molecular characterization to identify key regulatory elements that could lead to potential treatment targets. CDCs restore normal diastolic heart function in a rat HFpEF model. There are challenges associated with this therapeutic strategy for patients. Identifying stem cell derived-biologically active agents could be potential drug targets. By showing for the first time that CDC therapy reverses phosphorylation of sarcomeric proteins and linking the changes to PKC, we were able to identify a mechanism that could be beneficial for HFpEF patients.
SOURCES OF FUNDING
This work was generously supported by a U.S. Department of Defense Grant (E.M and J.E.V.E); National Institutes of Health/Heart, Lung and Blood Institute (J.E.V.E); the American Heart Association generously support this work with a Postdoctoral Fellowship Award (DS); Smidt Heart Institute (E.M.); the Erika J. Glazer chair in Women’s Heart Health (J.E.V.E.); the Barbra Streisand Women’s Heart Center J.E.V.E.; the Advanced Clinical Biosystems Institute (J.E.V.E.).
Nonstandard Abbreviations and Acronyms:
- Ca2+
Calcium
- CDC
Cardiosphere-derived cell
- ECM
Extracellular matrix
- FC
Fold change
- HF
Heart failure
- HFpEF
Heart failure with preserved ejection fraction
- HFrEF
Heart failure with reduced ejection fraction
- LC-MS/MS
Liquid chromatography-high-resolution mass spectrometry
- Ldb3
LIM domain-binding protein 3
- LV
Left ventricle
- MS
Mass spectrometry
- PKA
Protein kinase A
- PKC
Protein kinase C
- Pdlim5
PDZ and LIM domain protein 5
- Ser
Serine (S)
- SS
Salt sensitive
- Thr
Threonine (T)
- Tyr
Tyrosine (Y)
Footnotes
DISCLOSURE
Dr. Marbán owns founder’s equity in and serves as an unpaid advisor to Capricor Inc. All other authors declare no conflicts of interest.
SUPPLEMENTAL MATERIALS
Publisher's Disclaimer: This article is published in its accepted form. It has not been copyedited and has not appeared in an issue of the journal. Preparation for inclusion in an issue of Circulation Research involves copyediting, typesetting, proofreading, and author review, which may lead to differences between this accepted version of the manuscript and the final, published version.
REFERENCES
- 1.Paulus WJ, Tschope C. A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. Journal of the American College of Cardiology. 2013;62:263–71. [DOI] [PubMed] [Google Scholar]
- 2.Aurigemma GP, Zile MR, Gaasch WH. Contractile behavior of the left ventricle in diastolic heart failure: with emphasis on regional systolic function. Circulation. 2006;113:296–304. [DOI] [PubMed] [Google Scholar]
- 3.Martos R, Baugh J, Ledwidge M, O’Loughlin C, Conlon C, Patle A, Donnelly SC, McDonald K. Diastolic heart failure: evidence of increased myocardial collagen turnover linked to diastolic dysfunction. Circulation. 2007;115:888–95. [DOI] [PubMed] [Google Scholar]
- 4.Zile MR, Baicu CF, Gaasch WH. Diastolic heart failure--abnormalities in active relaxation and passive stiffness of the left ventricle. The New England journal of medicine. 2004;350:1953–9. [DOI] [PubMed] [Google Scholar]
- 5.Linke WA, Hamdani N. Gigantic business: titin properties and function through thick and thin. Circulation research. 2014;114:1052–68. [DOI] [PubMed] [Google Scholar]
- 6.Linke WA. Sense and stretchability: the role of titin and titin-associated proteins in myocardial stress-sensing and mechanical dysfunction. Cardiovascular research. 2008;77:637–48. [DOI] [PubMed] [Google Scholar]
- 7.van Heerebeek L, Borbely A, Niessen HW, Bronzwaer JG, van der Velden J, Stienen GJ, Linke WA, Laarman GJ, Pauluset WJ. Myocardial structure and function differ in systolic and diastolic heart failure. Circulation. 2006;113:1966–73. [DOI] [PubMed] [Google Scholar]
- 8.Borbely A, Falcao-Pires I, van Heerebeek L, Hamdani N, Edes I, Gavina C, Leite-Moreira AF, Bronzwaer JGF, Papp Z, van der Velden J, et al. Hypophosphorylation of the Stiff N2B titin isoform raises cardiomyocyte resting tension in failing human myocardium. Circulation research. 2009;104:780–6. [DOI] [PubMed] [Google Scholar]
- 9.LeWinter MM, Wu Y, Labeit S, Granzier H. Cardiac titin: structure, functions and role in disease. Clinica chimica acta; international journal of clinical chemistry. 2007;375:1–9. [DOI] [PubMed] [Google Scholar]
- 10.Gladden JD, Linke WA, Redfield MM. Heart failure with preserved ejection fraction. Pflugers Archiv : European journal of physiology. 2014;466:1037–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hamdani N, Kooij V, van Dijk S, Merkus D, Paulus WJ, Remedios CD, Duncker DJ, Stienen GJM, van der Velden J. Sarcomeric dysfunction in heart failure. Cardiovascular research. 2008;77:649–58. [DOI] [PubMed] [Google Scholar]
- 12.Solaro RJ, Kobayashi T. Protein phosphorylation and signal transduction in cardiac thin filaments. The Journal of biological chemistry. 2011;286:9935–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kobayashi M, Machida N, Tanaka R, Yamane Y. Effects of beta-blocker on left ventricular remodeling in rats with volume overload cardiac failure. The Journal of veterinary medical science. 2008;70:1231–7. [DOI] [PubMed] [Google Scholar]
- 14.Reddy YNV, Lewis GD, Shah SJ, LeWinter M, Semigran M, Davila-Roman VG, Anstrom K, Hernandez A, Braunwald E, Redfield MM, et al. INDIE-HFpEF (Inorganic Nitrite Delivery to Improve Exercise Capacity in Heart Failure With Preserved Ejection Fraction): Rationale and Design. Circulation Heart failure. 2017;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hwang IC, Kim YJ, Park JB, Yoon YE, Lee SP, Kim HK, Cho GY, Sohn DW. Pulmonary hemodynamics and effects of phosphodiesterase type 5 inhibition in heart failure: a meta-analysis of randomized trials. BMC cardiovascular disorders. 2017;17:150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aghila Rani KG, Kartha CC. Effects of epidermal growth factor on proliferation and migration of cardiosphere-derived cells expanded from adult human heart. Growth factors (Chur, Switzerland). 2010;28:157–65. [DOI] [PubMed] [Google Scholar]
- 17.Gaetani R, Ledda M, Barile L, Chimenti I, De Carlo F, Forte E, Ionta V, Giuliani L, D’Emilia E, Frati G, et al. Differentiation of human adult cardiac stem cells exposed to extremely low-frequency electromagnetic fields. Cardiovascular research. 2009;82:411–20. [DOI] [PubMed] [Google Scholar]
- 18.Mishra R, Vijayan K, Colletti EJ, Harrington DA, Matthiesen TS, Simpson D, Goh SK, Walker BL, Almeida-Porada G, Wang D, et al. Characterization and Functionality of Cardiac Progenitor Cells in Congenital Heart Patients. Circulation. 2011;123:364–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Takehara N, Tsutsumi Y, Tateishi K, Ogata T, Tanaka H, Ueyama T, Takahashi T, Takamatsu T, Fukushima M, Komeda M, et al. Controlled Delivery of Basic Fibroblast Growth Factor Promotes Human Cardiosphere-Derived Cell Engraftment to Enhance Cardiac Repair for Chronic Myocardial Infarction. Journal of the American College of Cardiology. 2008;52:1858–65. [DOI] [PubMed] [Google Scholar]
- 20.Tang YL, Zhu W, Cheng M, Chen L, Zhang J, Sun T, Kishore R, Phillips MI, Losordo DW, and Qin G. Hypoxic Preconditioning Enhances the Benefit of Cardiac Progenitor Cell Therapy for Treatment of Myocardial Infarction by Inducing CXCR4 Expression. Circulation research. 2009;104:1209–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zakharova L, Mastroeni D, Mutlu N, Molina M, Goldman S, Diethrich E, Gaballa MA. Transplantation of cardiac progenitor cell sheet onto infarcted heart promotes cardiogenesis and improves function. Cardiovascular research. 2010;87:40–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Makkar RR, Smith RR, Cheng K, Malliaras K, Thomson LE, Berman D, Czer LS, Marbán L, Mendizabal A, Johnston PV, et al. Intracoronary cardiosphere-derived cells for heart regeneration after myocardial infarction (CADUCEUS): a prospective, randomised phase 1 trial. Lancet. 2012;379:895–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Malliaras K, Makkar RR, Smith RR, Cheng K, Wu E, Bonow RO, Marbán L, Mendizabal A, Cingolani E, Johnston PV, et al. Intracoronary cardiosphere-derived cells after myocardial infarction: evidence for therapeutic regeneration in the final 1-year results of the CADUCEUS trial. Journal of the American College of Cardiology. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gallet R, de Couto G, Simsolo E, Valle J, Sun B, Liu W, Tseliou E, Zile MR, Marbán E. Cardiosphere-derived cells reverse heart failure with preserved ejection fraction (HFpEF) in rats by decreasing fibrosis and inflammation. JACC Basic to translational science. 2016;1:14–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gnecchi M, Zhang Z, Ni A, Dzau VJ. Paracrine mechanisms in adult stem cell signaling and therapy. Circulation research. 2008;103:1204–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kikkawa U The story of PKC: A discovery marked by unexpected twists and turns. IUBMB life. 2019;71:697–705. [DOI] [PubMed] [Google Scholar]
- 27.Kitatani K, Idkowiak-Baldys J, Hannun YA. Mechanism of inhibition of sequestration of protein kinase C alpha/betaII by ceramide. Roles of ceramide-activated protein phosphatases and phosphorylation/dephosphorylation of protein kinase C alpha/betaII on threonine 638/641. The Journal of biological chemistry. 2007;282:20647–56. [DOI] [PubMed] [Google Scholar]
- 28.Liu Y, Belkina NV, Graham C, Shaw S. Independence of protein kinase C-delta activity from activation loop phosphorylation: structural basis and altered functions in cells. The Journal of biological chemistry. 2006;281:12102–11. [DOI] [PubMed] [Google Scholar]
- 29.Gong J, Park M, Steinberg SF. Cleavage Alters the Molecular Determinants of Protein Kinase C-delta Catalytic Activity. Molecular and cellular biology. 2017;37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ringshausen I, Schneller F, Bogner C, Hipp S, Duyster J, Peschel C, Decker T. Constitutively activated phosphatidylinositol-3 kinase (PI-3K) is involved in the defect of apoptosis in B-CLL: association with protein kinase Cdelta. Blood. 2002;100:3741–8. [DOI] [PubMed] [Google Scholar]
- 31.Soetkamp D, Binek A, Gallet R, de Couto G, Kilfoil P, Mostafa R, Venkatraman V, Holewinski R, Bradshaw AD, Goldhaber J, et al. Mechanistic dissection of global proteomic changes in rats with heart failure and preserved ejection fraction. bioRxiv. 2021:2021.09.06.456608. [Google Scholar]
- 32.Zile MR, Baicu CF, Ikonomidis JS, Stroud RE, Nietert PJ, Bradshaw AD, Slater R, Palmer BM, Van Buren P, Meyer M, et al. Myocardial stiffness in patients with heart failure and a preserved ejection fraction: contributions of collagen and titin. Circulation. 2015;131:1247–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Westermann D, Lindner D, Kasner M, Zietsch C, Savvatis K, Escher F, von Schlippenbach J, Skurk C, Steendijk P, Riad A, et al. Cardiac inflammation contributes to changes in the extracellular matrix in patients with heart failure and normal ejection fraction. Circulation Heart failure. 2011;4:44–52. [DOI] [PubMed] [Google Scholar]
- 34.Valero-Muñoz M, Backman W, Sam F. Murine Models of Heart Failure with Preserved Ejection Fraction: a “Fishing Expedition”. JACC Basic to translational science. 2017;2:770–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gao L, Gregorich ZR, Zhu W, Mattapally S, Oduk Y, Lou X, Kannappan R, Borovjagin AV, Walcott GP, Pollard AE, et al. Large Cardiac Muscle Patches Engineered From Human Induced-Pluripotent Stem Cell-Derived Cardiac Cells Improve Recovery From Myocardial Infarction in Swine. Circulation. 2018;137:1712–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dong X, Sumandea CA, Chen YC, Garcia-Cazarin ML, Zhang J, Balke CW, Sumandea MP, Ge Y. Augmented phosphorylation of cardiac troponin I in hypertensive heart failure. The Journal of biological chemistry. 2012;287:848–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sorimachi H, Freiburg A, Kolmerer B, Ishiura S, Stier G, Gregorio CC, Labeit D, Linke WA, Suzuki K, Labeit S. Tissue-specific expression and alpha-actinin binding properties of the Z-disc titin: implications for the nature of vertebrate Z-discs. Journal of molecular biology. 1997;270:688–95. [DOI] [PubMed] [Google Scholar]
- 38.Young P, Ferguson C, Banuelos S, Gautel M. Molecular structure of the sarcomeric Z-disk: two types of titin interactions lead to an asymmetrical sorting of alpha-actinin. The EMBO journal. 1998;17:1614–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lamb JR, Tugendreich S, Hieter P. Tetratrico peptide repeat interactions: to TPR or not to TPR? Trends in biochemical sciences. 1995;20:257–9. [DOI] [PubMed] [Google Scholar]
- 40.Das AK, Cohen PW, Barford D. The structure of the tetratricopeptide repeats of protein phosphatase 5: implications for TPR-mediated protein-protein interactions. The EMBO journal. 1998;17:1192–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Goebl M, Yanagida M. The TPR snap helix: a novel protein repeat motif from mitosis to transcription. Trends in biochemical sciences. 1991;16:173–7. [DOI] [PubMed] [Google Scholar]
- 42.D’Andrea LD, Regan L. TPR proteins: the versatile helix. Trends in biochemical sciences. 2003;28:655–62. [DOI] [PubMed] [Google Scholar]
- 43.Horowits R, Podolsky RJ. The positional stability of thick filaments in activated skeletal muscle depends on sarcomere length: evidence for the role of titin filaments. The Journal of cell biology. 1987;105:2217–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Labeit S, Gautel M, Lakey A, Trinick J. Towards a molecular understanding of titin. The EMBO journal. 1992;11:1711–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lundby A, Secher A, Lage K, Nordsborg NB, Dmytriyev A, Lundby C, Olsen JV. Quantitative maps of protein phosphorylation sites across 14 different rat organs and tissues. Nature communications. 2012;3:876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ribeiro Ede A Jr., Pinotsis N, Ghisleni A, Salmazo A, Konarev PV, Kostan J, Sjöblom B, Schreiner C, Polyansky AA, Gkougkoulia EA, et al. The structure and regulation of human muscle alpha-actinin. Cell. 2014;159:1447–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Itoh-Satoh M, Hayashi T, Nishi H, Koga Y, Arimura T, Koyanagi T, Takahashi M, Hohda S, Ueda K, Nouchi T, et al. Titin mutations as the molecular basis for dilated cardiomyopathy. Biochemical and biophysical research communications. 2002;291:385–93. [DOI] [PubMed] [Google Scholar]
- 48.Gaudet P, Livstone MS, Lewis SE, Thomas PD. Phylogenetic-based propagation of functional annotations within the Gene Ontology consortium. Briefings in bioinformatics. 2011;12:449–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bang ML, Centner T, Fornoff F, Geach AJ, Gotthardt M, McNabb M, Witt CC, Labeit D, Gregorio CC, Granzier H, et al. The complete gene sequence of titin, expression of an unusual approximately 700-kDa titin isoform, and its interaction with obscurin identify a novel Z-line to I-band linking system. Circulation research. 2001;89:1065–72. [DOI] [PubMed] [Google Scholar]
- 50.Lundby A, Andersen MN, Steffensen AB, Horn H, Kelstrup CD, Francavilla C, Schmitt N, Thomsen MB, Olsen JV. In vivo phosphoproteomics analysis reveals the cardiac targets of beta-adrenergic receptor signaling. Science signaling. 2013;6:rs11. [DOI] [PubMed] [Google Scholar]
- 51.Candasamy AJ, Haworth RS, Cuello F, Ibrahim M, Aravamudhan S, Kruger M, Holt MR, Terracciano CMN, Mayr M, Gautel M, et al. Phosphoregulation of the titin-cap protein telethonin in cardiac myocytes. The Journal of biological chemistry. 2014;289:1282–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pyle WG, Solaro RJ. At the crossroads of myocardial signaling: the role of Z-discs in intracellular signaling and cardiac function. Circulation research. 2004;94:296–305. [DOI] [PubMed] [Google Scholar]
- 53.Hoshijima M, Pashmforoush M, Knoll R, Chien KR. The MLP family of cytoskeletal Z disc proteins and dilated cardiomyopathy: a stress pathway model for heart failure progression. Cold Spring Harbor symposia on quantitative biology. 2002;67:399–408. [DOI] [PubMed] [Google Scholar]
- 54.Cazorla O, Vassort G, Garnier D, Le Guennec JY. Length modulation of active force in rat cardiac myocytes: is titin the sensor? Journal of molecular and cellular cardiology. 1999;31:1215–27. [DOI] [PubMed] [Google Scholar]
- 55.Knoll R, Hoshijima M, Hoffman HM, Person V, Lorenzen-Schmidt I, Bang ML, Hayashi T, Shiga N, Yasukawa H, Schaper W, et al. The cardiac mechanical stretch sensor machinery involves a Z disc complex that is defective in a subset of human dilated cardiomyopathy. Cell. 2002;111:943–55. [DOI] [PubMed] [Google Scholar]
- 56.Frey N, Barrientos T, Shelton JM, Frank D, Rutten H, Gehring D, Kuhn C, Lutz M, Rothermel B, Bassel-Duby R, et al. Mice lacking calsarcin-1 are sensitized to calcineurin signaling and show accelerated cardiomyopathy in response to pathological biomechanical stress. Nature medicine. 2004;10:1336–43. [DOI] [PubMed] [Google Scholar]
- 57.Brancaccio M, Fratta L, Notte A, Hirsch E, Poulet R, Guazzone S, De Acetis M, Vecchione C, Marino G, Altruda F, et al. Melusin, a muscle-specific integrin beta1-interacting protein, is required to prevent cardiac failure in response to chronic pressure overload. Nature medicine. 2003;9:68–75. [DOI] [PubMed] [Google Scholar]
- 58.Ye L, Su L, Wang C, Loo S, Tee G, Tan S, Khin SW, Ko S, Su B, Cook SA, et al. Truncations of the titin Z-disc predispose to a heart failure with preserved ejection phenotype in the context of pressure overload. PloS one. 2018;13:e0201498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Goldberg M, Steinberg SF. Tissue-specific developmental regulation of protein kinase C isoforms. Biochemical pharmacology. 1996;51:1089–93. [DOI] [PubMed] [Google Scholar]
- 60.Rouet-Benzineb P, Mohammadi K, Perennec J, Poyard M, Bouanani Nel H, Crozatier B. Protein kinase C isoform expression in normal and failing rabbit hearts. Circulation research. 1996;79:153–61. [DOI] [PubMed] [Google Scholar]
- 61.Sugden PH, Bogoyevitch MA. Intracellular signalling through protein kinases in the heart. Cardiovascular research. 1995;30:478–92. [PubMed] [Google Scholar]
- 62.Bowling N, Walsh RA, Song G, Estridge T, Sandusky GE, Fouts RL, Mintze K, Pickard T, Roden R, Bristow MR, et al. Increased protein kinase C activity and expression of Ca2+-sensitive isoforms in the failing human heart. Circulation. 1999;99:384–91. [DOI] [PubMed] [Google Scholar]
- 63.Li W, Zhang J, Bottaro DP, Pierce JH. Identification of serine 643 of protein kinase C-delta as an important autophosphorylation site for its enzymatic activity. The Journal of biological chemistry. 1997;272:24550–5. [DOI] [PubMed] [Google Scholar]
- 64.Maturana AD, Nakagawa N, Yoshimoto N, Tatematsu K, Hoshijima M, Tanizawa K, Kuroda S. LIM domains regulate protein kinase C activity: a novel molecular function. Cellular signalling. 2011;23:928–34. [DOI] [PubMed] [Google Scholar]
- 65.Kuroda S, Tokunaga C, Kiyohara Y, Higuchi O, Konishi H, Mizuno K, Kikkawa U. Protein-protein interaction of zinc finger LIM domains with protein kinase C. The Journal of biological chemistry. 1996;271:31029–32. [DOI] [PubMed] [Google Scholar]
- 66.Nakagawa N, Hoshijima M, Oyasu M, Saito N, Tanizawa K, Kuroda S. ENH, containing PDZ and LIM domains, heart/skeletal muscle-specific protein, associates with cytoskeletal proteins through the PDZ domain. Biochemical and biophysical research communications. 2000;272:505–12. [DOI] [PubMed] [Google Scholar]
- 67.Faulkner G, Pallavicini A, Formentin E, Comelli A, Ievolella C, Trevisan S, Bortoletto G, Scannapieco P, Salamon M, Mouly V, et al. ZASP: a new Z-band alternatively spliced PDZ-motif protein. The Journal of cell biology. 1999;146:465–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhou Q, Ruiz-Lozano P, Martone ME, Chen J. Cypher, a striated muscle-restricted PDZ and LIM domain-containing protein, binds to alpha-actinin-2 and protein kinase C. The Journal of biological chemistry. 1999;274:19807–13. [DOI] [PubMed] [Google Scholar]
- 69.Yamashita Y, Matsuura T, Kurosaki T, Amakusa Y, Kinoshita M, Ibi T, Sahashi K, Ohno K. LDB3 splicing abnormalities are specific to skeletal muscles of patients with myotonic dystrophy type 1 and alter its PKC binding affinity. Neurobiology of disease. 2014;69:200–5. [DOI] [PubMed] [Google Scholar]
- 70.van Heerebeek L, Hamdani N, Falcao-Pires I, Leite-Moreira AF, Begieneman MP, Bronzwaer JG, van der Velden J, Stienen GJ, Laarman GJ, Somsen A, et al. Low myocardial protein kinase G activity in heart failure with preserved ejection fraction. Circulation. 2012;126:830–9. [DOI] [PubMed] [Google Scholar]
- 71.Anderson PW, McGill JB, Tuttle KR. Protein kinase C beta inhibition: the promise for treatment of diabetic nephropathy. Current opinion in nephrology and hypertension. 2007;16:397–402. [DOI] [PubMed] [Google Scholar]
- 72.Kooij V, Venkatraman V, Kirk JA, Ubaida-Mohien C, Graham DR, Faber MJ, Van Eyk JE. Identification of cardiac myofilament protein isoforms using multiple mass spectrometry based approaches. Proteomics Clinical applications. 2014;8:578–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kessner D, Chambers M, Burke R, Agus D, Mallick P. ProteoWizard: open source software for rapid proteomics tools development. Bioinformatics. 2008;24:2534–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Craig R, Beavis RC. TANDEM: matching proteins with tandem mass spectra. Bioinformatics. 2004;20:1466–7. [DOI] [PubMed] [Google Scholar]
- 75.Eng JK, Jahan TA, Hoopmann MR. Comet: an open-source MS/MS sequence database search tool. Proteomics. 2013;13:22–4. [DOI] [PubMed] [Google Scholar]
- 76.Elias JE, Gygi SP. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nature methods. 2007;4:207–14. [DOI] [PubMed] [Google Scholar]
- 77.Apweiler R, Bairoch A, Wu CH, Barker WC, Boeckmann B, Ferro S, Gasteiger E, Huang H, Lopez R, Magrane M, et al. UniProt: the Universal Protein knowledgebase. Nucleic acids research. 2004;32:D115–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Perez-Riverol Y, Csordas A, Bai J, Bernal-Llinares M, Hewapathirana S, Kundu DJ, Inuganti A, Griss J, Mayer G, Eisenacher M, et al. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 2019;47:D442–d50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Choi M, Chang CY, Clough T, Broudy D, Killeen T, MacLean B, Vitek O. MSstats: an R package for statistical analysis of quantitative mass spectrometry-based proteomic experiments. Bioinformatics. 2014;30:2524–6. [DOI] [PubMed] [Google Scholar]
- 80.Benjamini Y, Hochberg Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society: Series B (Methodological). 1995;57:289–300. [Google Scholar]
- 81.Hamdani N, Franssen C, Lourenco A, Falcao-Pires I, Fontoura D, Leite S, Plettig L, López B, Ottenheijm CA, Becher PM, et al. Myocardial titin hypophosphorylation importantly contributes to heart failure with preserved ejection fraction in a rat metabolic risk model. Circ Heart Fail. 2013;6:1239–49. [DOI] [PubMed] [Google Scholar]
- 82.Schwarzl M, Hamdani N, Seiler S, Alogna A, Manninger M, Reilly S, Zirngast B, Kirsch A, Steendijk P, Verderber J, et al. A porcine model of hypertensive cardiomyopathy: implications for heart failure with preserved ejection fraction. Am J Physiol Heart Circ Physiol. 2015;309:H1407–18. [DOI] [PubMed] [Google Scholar]
- 83.Hamdani N, Bishu KG, von Frieling-Salewsky M, Redfield MM, Linke WA. Deranged myofilament phosphorylation and function in experimental heart failure with preserved ejection fraction. Cardiovascular research. 2013;97:464–71. [DOI] [PubMed] [Google Scholar]
- 84.Zakeri R, Moulay G, Chai Q, Ogut O, Hussain S, Takahama H, Lu T, Wang XL, Linke WA, Lee HC, et al. Left Atrial Remodeling and Atrioventricular Coupling in a Canine Model of Early Heart Failure With Preserved Ejection Fraction. Circ Heart Fail. 2016;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hudson B, Hidalgo C, Saripalli C, Granzier H. Hyperphosphorylation of mouse cardiac titin contributes to transverse aortic constriction-induced diastolic dysfunction. Circ Res. 2011;109:858–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kotter S, Gout L, Von Frieling-Salewsky M, Muller AE, Helling S, Marcus K, Dos Remedios CG, Linke WA, Krüger M. Differential changes in titin domain phosphorylation increase myofilament stiffness in failing human hearts. Cardiovasc Res. 2013;99:648–56. [DOI] [PubMed] [Google Scholar]
- 87.Granzier HL, Labeit S. The giant protein titin: a major player in myocardial mechanics, signaling, and disease. Circ Res. 2004;94:284–95. 2009;104:780–6. [DOI] [PubMed] [Google Scholar]
- 88.Hidalgo C, Hudson B, Bogomolovas J, Zhu Y, Anderson B, Greaser M, Granzier H. PKC phosphorylation of titin’s PEVK element: a novel and conserved pathway for modulating myocardial stiffness. Circ Res. 2009;105:631–8, 17 p following 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hamdani N, Krysiak J, Kreusser MM, Neef S, Dos Remedios CG, Maier LS, Krüger M, Backs J, Linke WA. Crucial role for Ca2(+)/calmodulin-dependent protein kinase-II in regulating diastolic stress of normal and failing hearts via titin phosphorylation. Circ Res. 2013;112:664–74. [DOI] [PubMed] [Google Scholar]
- 90.Kapoor N, Menon ST, Chauhan R, Sachdev P, Sakmar TP. Structural evidence for a sequential release mechanism for activation of heterotrimeric G proteins. Journal of molecular biology. 2009;393:882–97. [DOI] [PubMed] [Google Scholar]
- 91.Cho H, Kehrl JH. Localization of Gi alpha proteins in the centrosomes and at the midbody: implication for their role in cell division. The Journal of cell biology. 2007;178:245–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Fukuhara S, Chikumi H, Gutkind JS. Leukemia-associated Rho guanine nucleotide exchange factor (LARG) links heterotrimeric G proteins of the G(12) family to Rho. FEBS letters. 2000;485:183–8. [DOI] [PubMed] [Google Scholar]
- 93.Hodavance SY, Gareri C, Torok RD, Rockman HA. G Protein-coupled Receptor Biased Agonism. Journal of cardiovascular pharmacology. 2016;67:193–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sato PY, Chuprun JK, Ibetti J, Cannavo A, Drosatos K, Elrod JW, Koch WJ. GRK2 compromises cardiomyocyte mitochondrial function by diminishing fatty acid-mediated oxygen consumption and increasing superoxide levels. Journal of molecular and cellular cardiology. 2015;89:360–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Petrofski JA, Koch WJ. The beta-adrenergic receptor kinase in heart failure. Journal of molecular and cellular cardiology. 2003;35:1167–74. [DOI] [PubMed] [Google Scholar]
- 96.Park M, Steinberg SF. Carvedilol Prevents Redox Inactivation of Cardiomyocyte Beta1-Adrenergic Receptors. JACC Basic to translational science. 2018;3:521–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hahn HS, Marreez Y, Odley A, Sterbling A, Yussman MG, Hilty KC, Bodi I, Liggett SB, Schwartz A, Dorn GW 2nd. Protein kinase Calpha negatively regulates systolic and diastolic function in pathological hypertrophy. Circulation research. 2003;93:1111–9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Please see the major resources table in the supplemental materials. The methods are described in detail in the online supplements. The authors declare that all supporting data are available within the article and its online supplementary files.