

Abstract
The larval zebrafish is an increasingly popular host model for the study of Aspergillosis. The visual accessibility, genetic resources, small size, and ease of handling make zebrafish larvae compatible with higher throughput investigation of fungal virulence and host resistance mechanisms. This article provides the protocols needed to prepare Aspergillus fumigatus spore inocula and use microinjection to infect the hindbrain ventricle of zebrafish larvae. Furthermore, we include protocols for analyzing host survival, immobilizing larvae for live imaging, and suggestions for image analysis.
Keywords: Zebrafish, Aspergillus, Aspergillosis, Infection model, pathogenesis
INTRODUCTION
Aspergillus infections remain a major health threat to the expanding immunocompromised population. Increased use of immunosuppressive therapies and growing antifungal drug resistance underline our need for greater understanding of fungal virulence determinants and host resistance mechanisms in Aspergillosis (Friedman & Schwartz, 2019; Latge & Chamilos, 2019; Sharma, Nelson-Sathi, Singh, Radhakrishna Pillai, & Chowdhary, 2019). The larval zebrafish model of Aspergillus infection recapitulates relevant features of human Aspergillosis, in which phagocyte recruitment and fungal clearance provide resistance to infection in healthy individuals, but immunosuppression largely increases risk of mortality (Knox et al., 2014). Genetic tractability and optical transparency make the larval zebrafish a unique, practical model to investigate Aspergillus infection biology in vivo and to visualize the specific functions and interactions of innate immune cells with Aspergillus (Munoz et al., 2021; Rosowski et al., 2018; Thrikawala & Rosowski, 2020).
In this article, we describe methods to prepare Aspergillus fumigatus spores and larval zebrafish for infection, microinjection of the larval hindbrain ventricle, larval survival analysis, and confocal imaging of fungal growth and phagocyte recruitment. These protocols provide a basic in vivo toolbox for querying host-pathogen interactions in Aspergillosis.
Biosafety Considerations
CAUTION: Aspergillus fumigatus is a Biosafety Level 2 (BSL-2) pathogen and should be handled in accordance with institutional guidelines for working with pathogenic microorganisms.
Animal Care and Use
Zebrafish larvae are considered live, vertebrate animals. Always adhere to appropriate national and institutional guidelines for proper care and use of animals in research and experimentation.
STRATEGIC PLANNING
For help planning zebrafish infection experiments, please review Figure 1 prior to starting protocols described in this article.
Figure 1.
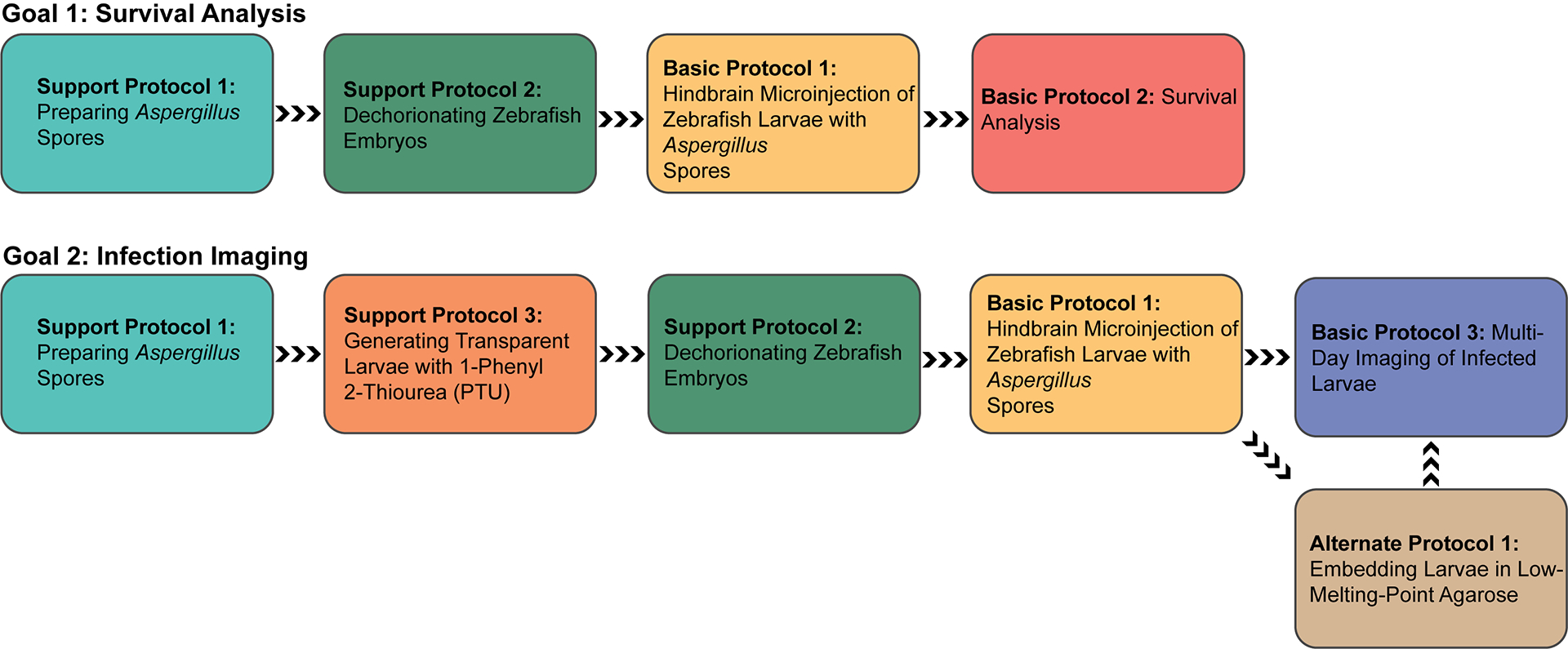
Flow chart guide to navigating the protocols in this article. Protocols are organized in chronological order for the purpose of 1) Survival Analysis or 2) Multi-Day Imaging of Infected Larvae. Colors are used to identify individual protocols.
SUPPORT PROTOCOL 1: PREPARATION OF ASPERGILLUS SPORES
Asexual spores, or conidia, are the infectious propagules derived from Aspergillus asexual reproduction. This protocol describes the growth, collection, and preparation of Aspergillus spores for hindbrain injection.
Materials
Aspergillus spores in glycerol suspension
The Aspergillus strain used for experimentation is at the discretion of the researcher. For an overview of commonly used A. fumigatus isolates, their lineages, and commercial availability, please refer to Bertuzzi et al. 2021.
Glucose Minimal Media (GMM) plates (see recipe in Reagents and Solutions)
50 ml 0.01% Tween-water (see recipe in Reagents and Solutions)
1X Phosphate Buffered Saline (PBS) (Thermo Fisher #14190144)
Fungal incubator (37°C)
50 ml conical tubes
Sterile Miracloth - cut in ~12×12 cm squares prior to autoclaving (Millipore Sigma # 475855)
Disposable L-shaped cell spreaders (Fisher Scientific #14-665-230)
25 ml serological pipettes (optional)
1.5 ml microcentrifuge tubes
Upright light microscope with 40X objective (Olympus X43 or equivalent)
Centrifuge such as Thermo Sorvall Legend XTR Centrifuge (Thermo Fisher #75004521)
Bright-Line Hemacytometer (Millipore Sigma #Z359629)
Part 1: Activate Aspergillus strains from glycerol stock
NOTE: Complete all spore preparations in a biosafety cabinet to prevent contamination. Always wear gloves when handling spores.
-
1Pipet a total volume of 10 μl of glycerol spore suspension (stored at −80°C) in 3 streaks on 2 GMM plates.
- Other methods of storing Aspergillus strains (e.g. silica, sterile water) can also be used to activate strains.
-
2Incubate plates at 37°C for 3–4 days.
- Sporulation should be evident by bold stripes of melanized spores. Please note that Aspergillus species can have different light requirements for asexual reproduction; ensure that the light cycle in the incubator is appropriate.
Part 2: Prepare short-term working water stock of Aspergillus spores
-
3
Assemble the plates from part 1, 1–50 ml conical tube, 1 square sterile Miracloth, 1 bottle 0.01% Tween-water, and L-shaped cell spreaders in the biosafety cabinet.
-
4
Tuck Miracloth into conical tube and replace the cap. This will serve as a filter to catch undesired hyphal debris in later steps.
-
5Open 1 plate at a time and pour enough Tween-water to cover ~75% of the fungal growth, do not exceed 25 ml. Gently tilt the plate until liquid covers most of the plate.
- Aspergillus spores are hydrophobic, and the Tween-water will likely not cover the plate evenly.
-
6
Gently scrape the liquid-covered portions of the plate with an L-spreader until nearly all fungal material is homogenized in the Tween-water. Change gloves if they are noticeably covered in spores.
-
7Open conical tube, take out Miracloth and fold it in half. Insert the folded Miracloth into the top of the conical tube to create a filter. Hold the Miracloth filter tightly around the rim of the tube and pour in the fungal homogenate from each plate. Dispose of used Miracloth and recap tube.
- If this method leads to excessive spilling, instead use a 25 ml serological pipette to transfer fungal homogenate into the tube.
-
8
In 1.5 ml microcentrifuge tubes, prepare 10-fold dilutions (10X, 100X, 1000X) of the spore suspension with Tween-water in a final volume of 100 μl.
-
9
To determine the concentration of the spore suspension, select dilution in which spores are not visible by eye, and load 8–10 μl into each chamber of the hemacytometer. Wait 1 minute to allow spores to settle in a single plane.
-
10
Using an upright microscope, locate the central counting square of each hemacytometer chamber and count spores contained within the 25 boxes at 40X magnification. Calculate the average number of spores from the 2 chambers and use this number in step 11.
-
11Spore concentration (spores/ml) = average spore count × dilution factor × 104.
- This calculation is accurate for the Bright-Line Hemacytometer (0.1 mm depth). If using a different hemacytometer, refer to the manufacturer instructions.
-
12Record concentration of spore suspension and store at 4°C up to 1 month.
- Do not store spores longer than one month as this can affect germination, particularly of certain genetic mutants.
Part 3: Prepare Aspergillus spores in PBS for injection
-
13Using the spore suspension prepared in Part 2, calculate the volume needed to have 106 spores.
- This volume should be between 20–100 μl to ensure even plating.
-
14
Pipet and spread 106 spores on 2 GMM plates using an L-spreader.
-
15
Incubate plates at 37°C for 3–4 days.
-
16
Repeat steps 3–7 of Part 2.
-
17
Centrifuge spore suspension 10 min at 900 × g, room temperature.
-
18
Pour off supernatant into appropriate liquid biohazardous waste container.
-
19
Add 50 ml sterile PBS, vortex or shake to resuspend pellet. Repeat centrifugation.
-
20
Pour off supernatant and resuspend in 5 ml sterile PBS.
-
21
Prepare another 50 ml conical tube with a Miracloth filter and pour the 5 ml spore suspension into new tube. Dispose of used Miracloth and recap tube.
-
22
Repeat steps 8–12 of Part 2.
-
23
Prepare 1 ml dilution of spores in sterile PBS at concentration of 1.5 × 108 spores/ml. Store at 4°C up to 1 month.
SUPPORT PROTOCOL 2: DECHORIONATING ZEBRAFISH EMBRYOS
During early development, zebrafish embryos are surrounded by an acellular membrane known as the chorion. At 28.5°C, larvae will hatch (dechorionate) at 3 days post fertilization (dpf). For hindbrain injection, larvae must be physically removed from the chorion at 2 dpf (Figure 2). When embryos are raised in E3 media, dechorionation should not impact larval development and can be done as early as 1 dpf, however, the larvae are especially fragile at this time (Westerfield, 1993).
Figure 2.
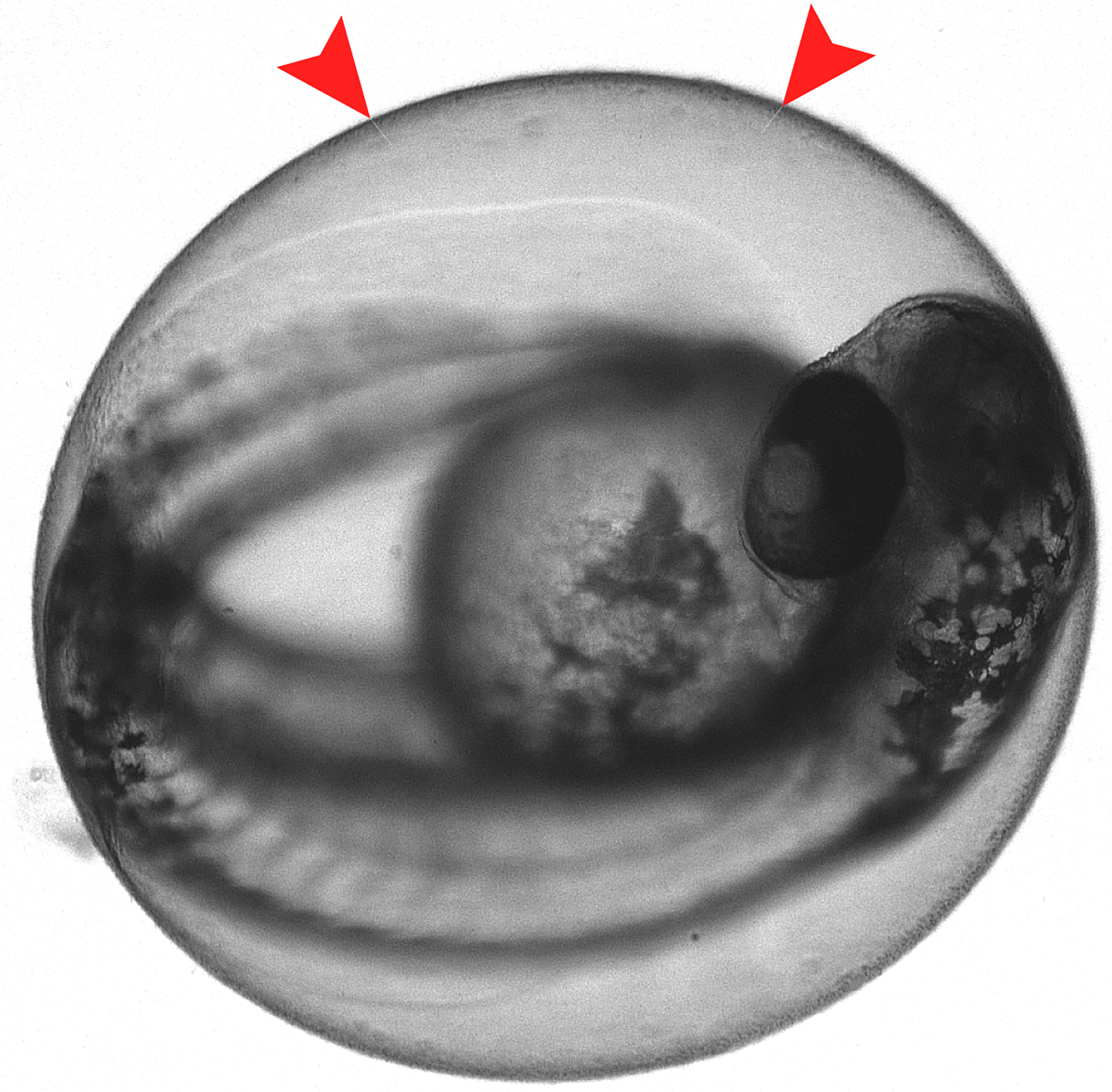
2 dpf larva inside the chorion. Red arrows provide an example of where to place forceps while dechorionating to avoid damaging larvae.
Materials
E3 media (see recipe in Reagents and Solutions)
1–2-day post fertilization (dpf) embryos in 10 cm petri dish
2 pairs of fine-tipped forceps (Dumont forceps #5, Fine Science Tools # 11251-20)
Stereo Zoom Microscope (Nikon SMZ745 or equivalent) with 10 mm reticle (MicroscopeWorld # RETR10)
Plastic transfer pipets (Fisher Scientific #S304673)
- Gloves
- Place dish of embryos in E3 onto microscope stage and gently swirl in a circular motion to collect embryos in center of dish.
- Zoom until the larvae and the surrounding chorion are clearly distinguishable.
- With one pair of forceps, pinch a small portion of the chorion (Figure 2).
- Select a part of the chorion that is not directly covering the larva to avoid harming them.
- With the other pair of forceps, pinch the chorion adjacent to the piece that is already being held.
- Gently pull the forceps away from each other to tear the chorion away from the larva.
- After dechorionation, remove chorions from dish using transfer pipet and replace E3 as needed.
- Return larvae to 28.5°C incubator until it is time to inject.
SUPPORT PROTOCOL 3: GENERATING TRANSPARENT LARVAE WITH 1-PHENYL 2-THIOUREA (PTU)
SAFETY NOTE: PTU is toxic when inhaled, ingested, or absorbed through the skin. Always wear gloves and avoid inhaling or making skin contact with PTU or media containing PTU. Refer to appropriate institutional guidelines for correct disposal of PTU liquid waste.
This protocol describes how to prevent pigment formation in zebrafish embryos to facilitate live-imaging experiments (Basic Protocol 3). Pigment formation begins at approximately 24 hours post fertilization (hpf) and may interfere with detection of fluorescent proteins or probes during microscopy. PTU is commonly used in zebrafish research to prevent formation of melanophores and generate transparent larvae for experimentation (Karlsson, von Hofsten, & Olsson, 2001). PTU treatment should not impair larval development.
Materials
1 dpf zebrafish embryos in 10 cm petri dish
- E3 media with PTU (see recipe in Reagents and Solutions)
- Remove E3 from plate containing 1 dpf embryos.
- Replace E3 with E3 containing 0.2 mM PTU and return plate to 28.5°C incubator.
- The effects of PTU are reversible therefore larvae should remain in PTU for the entirety of an experiment. It is not necessary to exchange E3 and PTU throughout an experiment unless the media appears dirty.
BASIC PROTOCOL 1: HINDBRAIN MICROINJECTION OF ZEBRAFISH LARVAE WITH ASPERGILLUS SPORES
This protocol has been adapted from the hindbrain injection method originally described in Knox et al. 2014. The hindbrain ventricle is a fluid-filled cavity separated from other parts of the body by an epithelial layer, which allows for localized infection with Aspergillus spores. In this technique, Aspergillus spores are microinjected into the hindbrain ventricle of 2 dpf larvae. Infected larvae can be subsequently used for survival analyses (Basic Protocol 2) and live-imaging experiments (Support Protocol 3, Basic Protocol 3). For a review of other infection locations in zebrafish, refer to Rosowski et al. 2018a. Note that this is a technically challenging protocol for new users and will likely require multiple practice experiments before the user feels confident with the technique.
Materials
E3 media without methylene blue (E3-MB) (see recipe in Reagents and Solutions)
1X Tricaine (see recipe in Reagents and Solutions)
2% bovine serum albumin (BSA) (Sigma A7906)
1% phenol red (Ricca Chemical R5725000)
1X Phosphate Buffered Saline (PBS) (Thermo Fisher #14190144)
E3-Agarose injection plate (see recipe in Reagents and Solutions)
Aspergillus inoculum (see Support Protocol 1)
2-day post fertilization zebrafish larvae – dechorionated (see Support Protocol 2)
Stereo Zoom Microscope (Nikon SMZ745 or equivalent) with 10 mm reticle (MicroscopeWorld # RETR10)
Intracellular Microinjection Dispense System (Picospritzer III or equivalent, Parker # 052-0500-900)
Micromanipulator (Tritech M-152 Three-Axis Direct-Drive Coarse Micromanipulator)
Magnetic stand for micromanipulator (Tritech GJ-1 or equivalent)
CO2 tank or other source of compressed air
Thin wall glass capillaries with no filament (WPI #TW100-3)
Flaming/Brown micropipette puller (Sutter Instruments P-97 or equivalent)
Settings for creating microinjection needles from glass capillaries: Pressure setting = 200, Heat = 502, Pull = 90, Velocity = 80, Time = 70, Delay =5.
Mini-beadbeater (Mini-beadbeater-16, Biospec Cat. No. 607 or equivalent)
Micropipette (p10 or p20)
Microloader pipette tips 0.5–20 μl (Eppendorf #930001007)
35 mm milk-treated or tissue culture treated dishes (see recipe for milk treatment)
50 ml conical tubes
1.5 ml microcentrifuge tubes
2 pairs of fine-tipped forceps (Dumont forceps #5, Fine Science Tools # 11251-20)
Hair loop (a piece of human hair or fine fiber inserted into a micropipette tip, used to move zebrafish larvae)
Plastic transfer pipets (Fisher Scientific #S304673)
Disposable L-shaped cell spreaders (Fisher Scientific #14-665-230)
Gloves
Part 1: Injection preparation
- Prepare injection plate.
- Pre-warm E3-agarose plate at RT for ~1 h before injection.
- Coat plate with 1 ml 2% BSA. Allow BSA to soak in for ~ 1 min.
- Wash plate with E3.
- Wash plate with 1X Tricaine, leave Tricaine on plate until it is time to inject.
- Using a transfer pipet, aliquot dechorionated larvae (see Support Protocol 2) into milk-treated petri dishes. Separate groups of larvae by condition and replace E3 with 1X Tricaine for anesthesia. Larvae must be in Tricaine for at least 1 min prior to injection and can remain in Tricaine up to 2–2.5 hours.
- For survival analysis, aim to inject 12–24 larvae per condition (plus 8 larvae for CFU counting). For imaging analysis, aim to inject 8–16 larvae per condition.
- Milk-treating dishes prevents larvae from sticking to the bottom of the dish, which can damage the skin of the animal. Tissue culture-treated dishes can be used as a substitute.
- Prepare Aspergillus inoculum.
- In microcentrifuge tube, dilute 20 μl of PBS spore stock (see Support Protocol 1) with 10 μl 1% phenol red. Mix well by pipetting. Return PBS spore stock to 4°C.
- Phenol red is added to visualize the inoculum in the hindbrain.
- For survival analyses, also prepare an injection control of 20 μl 1X PBS + 10 μl 1% phenol red.
- This serves as a control for any larval death that may result from rough handling during injection. None, or very few, of PBS-injected larvae should die following injection.
- Open CO2 tank and turn on Picospritzer. Adjust pressure to 20 psi and set time to 30 ms.
- Pressure and timing of the pulse may need to be adjusted later depending on injection needle.
- Arrange larvae for 1 condition on E3-agarose injection plate.
- Pour off Tricaine from injection plate.
- Take 1 dish of larvae and gently swirl to cluster larvae in the center of dish.
- Using a transfer pipet, pick up larvae in smallest possible volume of liquid. Gently squeeze the bulb of the pipet to collect larvae near the pipet opening.
- Pipet larvae onto center of injection plate in small amount of liquid. Too much liquid will make injection more difficult as larvae will slide when touched with needle.
- If not already, move injection plate onto microscope.
- With the hairloop, gently arrange larvae into 2 rows, 1 row facing right and 1 row facing left (Figure 3). Re-cover the plate and remove from microscope.
- Prepare microinjection needle.
-
1Draw up 5 μl of well-mixed inoculum using a microloader pipette tip.
-
2Using fingers or forceps, carefully pick up a pulled glass needle, always facing the tip away from hands.
-
3Insert the microloader tip into the needle until it reaches the area where the needle begins to narrow.
-
4Slowly pipet into the needle, simultaneously pulling the tip out as the needle fills.
- Be careful to not create air bubbles, as the injection will not work if there are air pockets.
-
5Insert the needle into the micromanipulator and tighten to secure it in place.
- Safety note: To prevent injury, keep the needle pointing away from hands while not injecting, and avoid moving hands across the stage of the microscope.
-
6With the microscope at the lowest magnification, carefully angle the micromanipulator down so the tip of the needle is in the center of the field of view. The needle tip should be close to, but not touching, the stage plate of the microscope.
-
7Increase zoom to 4X and bring needle into focus.
-
8Using forceps, begin cutting back the needle point in small sections while periodically pressing the injection pedal. Using a calibrated reticle (ocular micrometer), measure the diameter of the droplet after each press of the pedal. Stop cutting when the droplet has a diameter that corresponds to approximately 3 nl in volume (V= 4/3πr3). The diameter needed for a 3 nl droplet will vary depending on the microscope and reticle being used to measure.
- Example with the following equipment:
- Nikon SMZ745 stereo zoom microscope, Nikon C-W10xB/22 eyepieces fitted with a single 10 mm reticle with 100 divisions.
- At 4X zoom, 1 division = 37.5 um. A droplet diameter of 5 divisions = 178.5 um. Using the equation for spherical volume, the droplet volume is 2.98 nl. On the same microscope at 5X zoom, 1 division = 30 um. Use 6 divisions at 5X to inject 3.06 nl.
-
1
Inspect shape of the needle point, a beveled or straight cut is best (Figure 3). If needle point has a jagged edge, re-cut and re-calibrate, or begin with a new needle.
Figure 3.
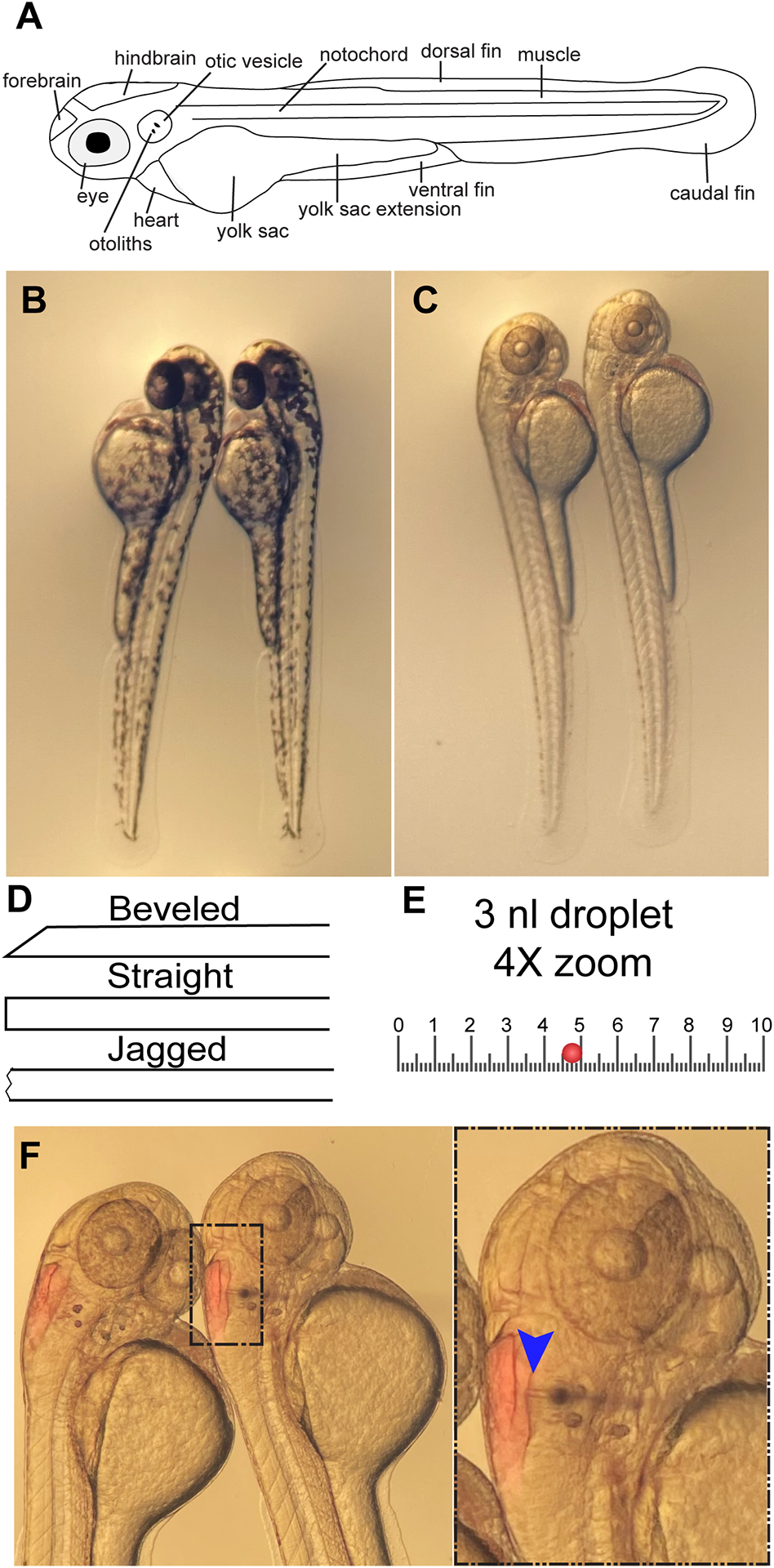
Hindbrain microinjection. A) Basic anatomy of 2 dpf larva. B) 2 dpf wild-type larvae facing left on agarose injection plate. C) 2 dpf wild-type larvae treated with PTU facing right on agarose injection plate. D) Illustration of possible needle points. E) Illustration of 10 mm eyepiece reticle at 4X zoom with a 3 nl droplet (red circle). F) Phenol red is visible in the hindbrains of injected larvae. Inset shows needle entering hindbrain via the otic vesicle. Blue arrow indicates needle point of entry.
Part 2: Hindbrain ventricle microinjection
-
9
Lift the micromanipulator and injection needle away from the stage and place the injection plate with larvae on the microscope.
-
10
Zoom out to 1X.
-
11
Angle the micromanipulator down until the needle is again in the center of the field of view. The needle point should be close to, but not touching, the agarose surface.
-
12
Move injection plate to align the needle point next to the right-most larvae in the right-facing row.
-
13
Zoom to 4 or 5X and re-focus such that the larvae and needle point are in focus.
-
14Use controls on micromanipulator to bring needle in contact with liquid surrounding larvae and press the injection pedal.
- Aspergillus spores should now be visible through the microscope, often looking like small, dark specks.
-
15Continue injecting onto plate until the concentration of spores in each drop is consistent.
- Aim to inject 30–75 spores per larvae. It is difficult, if not impossible, to count the number of spores ejected. Therefore, completing the colony forming unit plating in Part 3 is essential for learning how to estimate spore number by eye. It is most important that the concentration appears consistent throughout injection.
-
16Aim the needle in the direction of the hindbrain ventricle, placing the needle point at the ridge of tissue along the edge of the otic vesicle.
- Needle placement can change depending on larval position, but the otic vesicle should always be the point of entry.
-
17
Holding the injection plate with one hand, adjust the micromanipulator controls to move the needle up/down or forward/backward until the needle pierces the tissue.
-
18Still using the micromanipulator, guide the needle point into the hindbrain ventricle. Once positioned in the hindbrain, press the foot pedal (Figure 3).
- The hindbrain should be almost entirely filled with red inoculum, with little overflow into the midbrain or other cavities. If there is overflow, adjust the volume of the injection droplet by changing the pressure and/or timing of the burst. If red has leaked outside the body, the hindbrain has likely been pierced through and the larvae should be discarded.
-
19Retract the needle from the first larvae, and repeat steps 8–10 for other larvae in this row. Remember to stop every 3–5 larvae and inject onto plate to ensure that the spore concentration remains consistent.
- The concentration of spores will change over the course of injection. The concentration can be adjusted by changing the timing and pressure of the burst. If this does not work, make a new needle.
-
20
Turn the plate, and similarly inject left-facing larvae.
-
21
When finished, angle the micromanipulator up and away to prevent accidental injury.
-
22
Look carefully at injected larvae and use a hairloop to move any that do not have red in the hindbrain away from the successfully injected larvae. Use a small amount of 1X Tricaine and a transfer pipet to remove from plate and set aside for euthanasia.
-
23
Hold the plate vertically and use 1X Tricaine and a transfer pipet to gently rinse the successfully injected larvae to the bottom of the plate. Carefully transfer larvae back into the milk-treated dish and label with the experimental condition and time.
-
24
Repeat injections for other conditions including a PBS-injected control for survival analyses. Be mindful to maintain a similar spore concentration for every strain and larval background.
-
25
When injections are finished and larvae are returned to milk-treated dishes, collect 8 larvae/condition and aliquot larvae into individual microcentrifuge tubes. Euthanize larvae as these will be used for colony forming unit (CFU) plating in Part 3.
-
26
Wash remaining larvae 3X with E3-MB. For survival analysis, continue to Basic protocol 2. For live imaging, continue to Basic protocol 3. Complete Part 3 for both survival and imaging experiments.
Part 3: Colony forming unit plating
Following injection, a subset of larvae from each condition are homogenized and plated on fungal growth media to count colony forming units (CFUs). CFU plating is an important quality control step to ensure that the number of spores injected was consistent throughout the experiment.
-
27
Pipet off most of the liquid from larvae in microcentrifuge tubes, up to 10 μl can remain in tube if needed.
-
28
Add 90 μl of 1X PBS with kanamycin (f.c. 500 μg/ml) and gentamycin (f.c. 500 μg/ml) to each larva.
-
29
Example: Add 10 μl kanamycin (50 mg/ml) and 10 ul gentamicin (50 mg/ml) per 1 ml 1X PBS.
-
30Homogenize larvae using a mini bead-beater/tissue homogenizer for 10–20 sec, or until larvae are no longer visible.
- Not all bead-beaters are designed to hold 1.5 ml tubes, ensure that the samples are secure before running the machine.
-
31
Centrifuge samples at 15000 rpm (21130 rcf) for 30 sec.
-
32
Use a p200 pipette to resuspend pellet, making sure to collect any tissue that may be stuck to the side of the tube.
-
33Pipet the entirety of each sample onto individual GMM plates and spread evenly with an L-spreader.
- Perform in a biosafety cabinet or under flame.
-
34Incubate plates upside down at 37°C for 2–3 days.
- Check plates daily to prevent overgrowth in the case of fast-growing Aspergillus strains.
-
35
Count and record number of colonies. Calculate the mean for each condition and compare.
BASIC PROTOCOL 2: SURVIVAL ANALYSIS
Survival curves can be a useful tool to assess fungal virulence determinants and host factors important for survival. This protocol describes how to set up and monitor larval survival following infection.
Materials
Infected zebrafish larvae (Basic Protocol 1)
E3 media without methylene blue (E3-MB)
96-well plates
- Plastic transfer pipets (Fisher Scientific #S304673)
- Using a transfer pipet, move infected larvae into a 96-well plate with 1 larva/well. Each well should have ~200 μl of E3-MB.
- Media can evaporate over the course of the experiment, top off wells with E3-MB when needed.
- Label the plate lid such that each condition is easily identifiable.
- Ensure that PBS-injected larvae are included as a control.
- Keep plates in 28.5°C incubator for the entirety of the experiment.
- Using a stereo microscope, check each well daily for 7 days and mark dead larvae by writing the day of the experiment on the lid (1-day post infection= 1).
- Larval death is defined by the absence of heartbeat.
- On day 7, take a photo of the plate lid and euthanize surviving larvae.
- For euthanasia of larvae, please refer to the animal use and care protocol approved by your institution.
- Record and plot data using a Kaplan-Meier curve (Figure 4).
Figure 4.
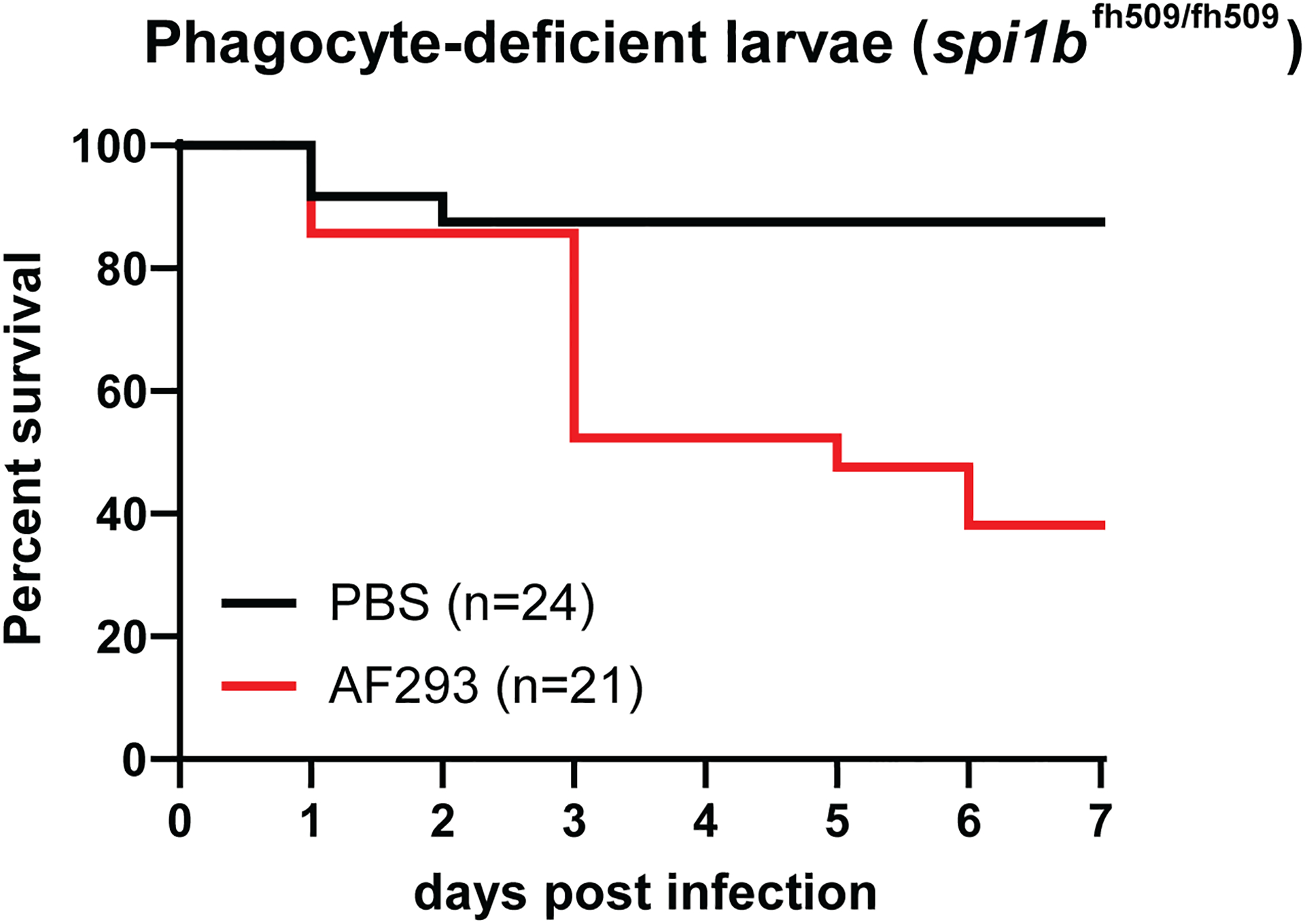
Kaplan-Meier curve. Results represent survival analysis of spi1b mutant larvae infected with A. fumigatus strain Af293. Larvae are deficient in macrophages and neutrophils as spi1b mutation impairs myeloid differentiation. Average spore dose of infected larvae = 48. Number of larvae injected indicated by n.
BASIC PROTOCOL 3: MULTI-DAY IMAGING OF INFECTED LARVAE
A primary advantage of the larval zebrafish-Aspergillus infection model is the ability to perform multi-day imaging experiments that provide longitudinal infection data from the same animal. In this protocol, infected larvae are mounted in the zWEDGI device and imaged using confocal microscopy at multiple time points (6, 24, 48, 72, 96 hpi). The zWEDGI (zebrafish Wounding and Entrapment Device for Growth and Imaging) device was specifically designed to accommodate long-term or repeated confocal imaging of zebrafish larvae. Instructions for device design and fabrication are provided in Huemer et al. 2017. If you do not have a zWEDGI device or are unable to fabricate a zWEDGI device at your institution, please refer to Alternate Protocol 1. This protocol is intended to be used with fluorescently labeled Aspergillus strains and transgenic zebrafish larvae with fluorescently labeled immune cells. Specific confocal microscopy guidelines for this protocol are not provided. See Figure 5 for further details.
Figure 5.
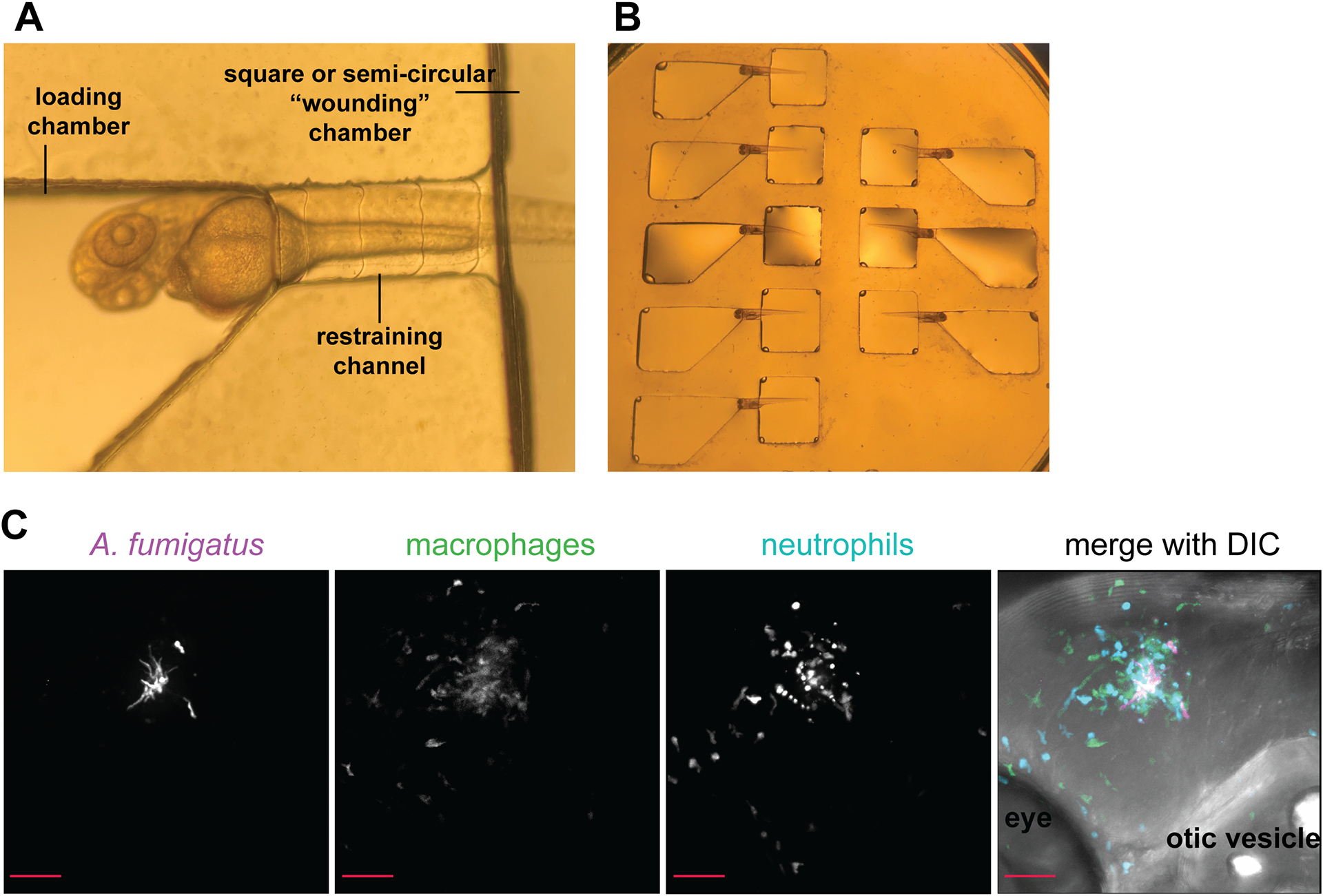
Immobilizing and imaging infected larvae. A) Dorsolateral positioning of 2 dpf larva in zWEDGI device. B) Multiple larvae may be positioned at the same time for time efficient imaging. C) Representative image A. fumigatus (magenta) infection in a wild-type larva at 72 hpi with labeled macrophages (green) and neutrophils (blue). Max intensity projection of z-stack, 20X magnification. Scale bar = 50 μm.
Materials
Infected zebrafish larvae (Basic Protocol 1)
E3 media without methylene blue (E3-MB)
E3-MB with 1-phenyl 2-thiourea (PTU)
1X Tricaine (see recipe in Reagents and Solutions)
35 mm milk-treated dishes (see recipe for milk treatment in Reagents and Solutions)
48-well plates
p200 pipette
Plastic transfer pipets (Fisher Scientific #S304673)
zWEDGI (see Huemer et al., 2017)
- Confocal microscope (CSU-X Yokogawa (spinning disk), Zeiss Observer Z.1 inverted confocal microscope with Plan-Apochromat NA 0.8/20x objective, and Photometrics Evolve EMCCD camera)
- Following infection, keep larvae at 28.5°C in 35 mm dishes (<25 larvae/dish) with E3-MB + PTU until 6 hpi.
- Fill 1 milk-treated 35 mm dish with 1X Tricaine. Fill another milk-treated dish with E3-MB + PTU.
- Using a transfer pipet, pick up larva in a small amount of liquid and drop into the Tricaine dish.
- On the first day of imaging, multiple larvae can be anesthetized simultaneously. After the first day, transfer only 1 larva at a time to keep track of individual animals.
- Place zWEDGI under the stereo microscope and fill the left side loading chambers with 1X Tricaine using a p200 pipette. Allow the Tricaine to pass through the restraining tunnels and into the semi-circular or square (shape dependent on model) chambers on the right side. Both sides of the zWEDGI channel should be filled with media.
- Using a p200 pipette, check for air bubbles by slowly drawing up media from one side of the chamber, the volume on the other side of the channel should decrease as well.
- If there is not fluid movement between the chambers, remove air bubbles from the restraining channel with a p200 pipette.
- Once zWEDGI channels are filled with Tricaine, use a transfer pipet to drop an anesthetized larva into the loading chamber.
- Pick up larva in a small volume of liquid, the media in the zWEDGI channels should not overflow.
- Using a p200 pipette, adjust the volume ratio of liquid in the chambers so that the larva is drawn into the restraining tunnel. The larva should be oriented such that the tail enters the semi-circular or square chamber, the head remains in loading chamber with the yolk sac resting on the angled portion, and the dorsal or dorsolateral side of the larva is positioned flat against the bottom of the zWEDGI (Figure 5).
- Dorsal or dorsolateral positioning is essential for imaging the hindbrain with an inverted objective lens. Aim for consistent positioning of all larvae.
- Move zWEDGI device to the confocal microscope and acquire z-stack images of larval hindbrains with 20X objective lens. Include each appropriate fluorescent channel and differential interference contrast (DIC) (Figure 5).
- The hindbrain is approximately 100 μm in depth. Recommended z-slice: 1–5 μm.
- Following imaging, return zWEDGI to stereo microscope. Add Tricaine to the semi-circular or square chamber to push larvae out of restraining chamber and into the loading chamber.
- Using a transfer pipet partially filled with tricaine, pick up larva and drop into Tricaine-filled dish. Pick up larva again in a small volume of liquid and transfer to the dish with E3-MB + PTU.
- This prevents excessive amounts of tricaine being added to the E3 dish.
- Wash larva in E3-MB + PTU and use transfer pipet to move larva to well of a 48-well plate.
- Repeat protocol at 24, 48, 72, and 96 hpi.
ALTERNATE PROTOCOL 1: EMBEDDING LARVAE IN LOW-MELTING-POINT AGAROSE
This protocol describes an alternate method for immobilizing zebrafish larval for confocal imaging. Note that this technique is not designed for multi-day imaging. Embedding larvae in agarose will limit you to observing Aspergillus infection at a single time point as it is difficult to safely remove larvae from agarose following microscopy.
Materials
Infected larvae (Basic Protocol 1)
p1000 pipette
Plastic transfer pipet (Fisher Scientific #S304673)
50 mm glass bottom dish (MatTek P50G-0-30-F)
E3 media without methylene blue (E3-MB) (see recipe in Reagents and Solutions)
1X Tricaine (see recipe in Reagents and Solutions)
2% low-melting-point agarose
Heat block or water bath
- Hairloop
- With a transfer pipet, move ~10 anesthetized, infected larvae to the center of glass bottom dish in small volume of liquid.
- Prepare a 1% low-melting-point agarose (LMA) solution.
- 1 ml 2% w/v LMA (Fisher Scientific BP16525) – melted at 95°C then cooled to 56°C
- 160 μl Tricaine stock (20X)
- 40 μl PTU stock (50X)
- 800 μl E3-MB
- Slowly pipet LMA solution over larvae. Aim to keep larvae in center of dish.
- Make sure agarose has sufficiently cooled. Hot agarose will kill larvae.
- Using a hairloop, arrange larvae in a dorsal or dorsolateral position such that the hindbrain is facing the bottom of dish.
- Allow LMA to harden for 10 min before moving the dish.
- Pipet enough 1X Tricaine into dish to cover hardened agarose. Larvae are ready for imaging.
REAGENTS AND SOLUTIONS
Bovine Serum Albumin (BSA)
2 % w/v BSA (Sigma A7906) in H2O
Filter sterilize
Store at −20°C
E3 media stock (60X): 1 L
17.2 g NaCl (Fisher 7647-14-5)
0.76 g KCl (Sigma DSP41000)
2.9 g CaCl2 (Thermo Fisher C5670)
4.9 g MgSO4 · 7H2O (Sigma M2773)
Add distilled H2O up to 1 L
Store at 4°C
E3 media with methylene blue (1X, working stock): 1 L
16.7 ml 60X E3
430 ul 0.05 M NaOH (MP Biomedical 153495)
3 ml 0.01% w/v methylene blue (Sigma 861243)
Add distilled H2O up to 1 L
Store at RT
*Exclude methylene blue to make E3-MB
E3-agarose injection plate
3 % w/v agarose (Promega v3125) in E3 with methylene blue
Heat in microwave until agarose is dissolved, pour into 10 cm petri dish, let cool
Can be made in advance, store at 4°C
Glucose minimal medium (GMM): 1 L
50 ml 20X Nitrate salts
1 ml Trace elements
10 g Glucose
Adjust pH to 6.5 with 10 M NaOH
16 g Agar
Add distilled water up to 1 L
Autoclave
Pour plates, store at 4°C or RT
Milk-treated dishes
Prepare 2% w/v skim milk (Thermo Fisher MP290288705) solution
Soak 35 mm dishes (exclude lids) in milk solution for at least 1 hr, then rinse and airdry
Store at RT
Nitrate salts (20X): 1 L
120 g NaNO3 (Sigma 221341)
10.4 g KCl (Sigma DSP41000)
10.4 g MgSO4 · 7H2O (Sigma M2773)
30.4 g KH2PO4 (Sigma 7778-77-0)
Add distilled H2O up to 1 L
Autoclave
Store at RT
PTU stock (10 mM, 50X): 500 ml
7.61 g 1-Phenyl-2-thiourea (Sigma P7629)
Add H2O up to 500 ml
Store at RT in darkness
*Dilute to 1X in E3 working stock (1X) to make E3 with PTU or E3-MB with PTU
Trace Elements: 100 ml
Add elements to 80 ml distilled H2O in order shown
2.2 g ZnSO4 · 7H2O (Sigma 7446-20-0)
1.1 g H3BO3 (Sigma 10043-35-3)
0.5 g MnCl2 · 4H2O (Sigma 221279)
0.5 g FeSO4 · 7H2O (Sigma 7782-63-0)
0.16 g CoCl2 · 6H2O (Sigma 7791-13-1)
0.16 g CuSO4 ·5H2O (Sigma 7758-99-8)
0.11 g (NH4)6Mo7O24 · 4H2O (Sigma 431346)
5 g Na4EDTA (Sigma EX0550)
Once dissolved bring volume to 100 ml with distilled H2O
Store at RT
Tricaine stock (20X): 500 ml
2 g Tricaine (Thermo Fisher NC0872873)
5g Na2HPO4 (Fisher AAA1181730)
4.2 ml 60X E3
Add H2O up to 500 ml
Store at 4°C up to 1 month
*Dilute to 1X in E3 or E3-MB to make working stock
Tween-water
0.01% v/v Tween-20 (Sigma P1379) in H2O
Autoclave
Store at RT
COMMENTARY
Background Information
Aspergillus species are saprophytic, filamentous fungi that thrive in a variety of ecological niches. Airborne conidia are frequently inhaled and can settle in the lower respiratory tract, causing pulmonary or invasive disease in immunocompromised individuals. Invasive Aspergillosis is one of the leading causes of infectious death in immunocompromised populations, such as those undergoing chemotherapy, iatrogenic immunosuppression, or those with inherited immune deficiencies (Latge & Chamilos, 2019). While various Aspergillus species have pathogenic potential, Aspergillosis cases are most often attributed to A. fumigatus; (Friedman & Schwartz, 2019; Latge & Chamilos, 2019).
Animal models are an essential resource for understanding fungal development and host response during A. fumigatus infection. Mammals have been long been favored for their similarity to humans but are often expensive and have limited visual accessibility for microscopy experiments. Larval zebrafish provide a complementary vertebrate platform for higher throughput screening of fungal virulence determinants and host resistance factors. Since its first publication in 2014, the larval zebrafish-Aspergillus infection model has continued to gain popularity as a practical, non-invasive solution to observe host-pathogen interactions in vivo (Knox et al., 2014; Koch, Hajdamowicz, Lagendijk, Ram, & Meijer, 2019; Rosowski et al., 2018).
A critical component of the larval zebrafish infection model is the conservation of a robust innate immune response to Aspergillus infection. The larval zebrafish shares essential features of the human innate immune system such as functional neutrophils and macrophages, pattern recognition receptors, and inflammatory signaling molecules like cytokines and chemokines (Henry, Loynes, Whyte, & Renshaw, 2013; Torraca, Masud, Spaink, & Meijer, 2014). The function of specific immune cell types and components can be readily assessed by utilizing gene editing techniques such as CRISPR, pharmacological immunosuppression, or the many available immunodeficient zebrafish lines (Rosowski et al., 2018). Furthermore, the optical transparency, small size, and availability of transgenic lines with fluorescently labeled immune cells make zebrafish larvae amenable to multi-day imaging experiments, allowing for observation of fungal growth and immune cell recruitment throughout disease progression (Knox, Huttenlocher, & Keller, 2017; Thrikawala & Rosowski, 2020).
This article describes infection of the hindbrain ventricle, a fluid-filled cavity bounded by an epithelial layer that separates the hindbrain from other structures. While the hindbrain ventricle lacks the specialized lung epithelium or alveolar macrophages that are present in mammals, it provides a visually accessible site for localized infection that is devoid of immune cells during early development. Importantly, injection into the hindbrain via the otic vesicle creates minimal tissue damage and inflammation. In response to infection, neutrophils and macrophages exit the vasculature to enter the hindbrain, which allows for quantification of immune cell migration dynamics and recruitment, specific to their interactions with Aspergillus (Knox et al., 2017; Rosowski et al., 2018).
In addition to investigating critical components of host immunity, larval zebrafish are being used to dissect fungal genes and pathways important for pathogenesis. Hindbrain infection is a useful tool to examine Aspergillus mutants for differences in in vivo germination rates, hyphal development, and resistance to host defenses. These techniques have been used primarily in studies of A. fumigatus; however, they can be easily adapted to work with other Aspergillus species (Koch et al., 2019; Schoen et al., 2019)
Critical Parameters
Support Protocol 1
At 4°C, Aspergillus spores will lose viability over time. Do not use spore stocks for injections that are older than 1 month.
Basic Protocols 1–3
It is essential that the number of spores injected is consistent among larvae of all conditions and between experimental replicates. Always homogenize and plate a subset of larvae for counting CFUs. Consistent spore dosage is critical for reproducibility.
Trouble Shooting
See Table 1 for troubleshooting suggestions.
Table 1:
Trouble shooting guide for common problems in zebrafish infection assays
| Problem | Possible Cause | Solution |
|---|---|---|
| Inconsistent CFU counts | Injecting inconsistent spore dose |
|
| Clogged injection needle |
|
|
| Unable to penetrate hindbrain with needle |
|
|
| Death of PBS-injected larvae in survival assay |
|
|
Understanding results
Survival curves (Basic Protocol 2)
One of the most important considerations while analyzing a survival experiment is the survival of the PBS-injected negative control larvae. PBS-injected larvae should not die over the course of the experiment, although it is not uncommon to see 1–2 larvae from each condition die on the day following injection (Figure 4). Day 1 larval deaths are most attributed to rough handling during injection, as fungal growth and inflammation are not likely to cause larval death at this time point.
The immune status of the larvae and the strain of A. fumigatus used will largely influence the outcome of survival analyses. Wild-type larvae typically do not succumb to infection; however, different A. fumigatus strains are known to have notable differences in rate of germination, growth, and virulence (Knox et al., 2016; Rosowski et al., 2018). If you observe no death of infected wild-type larvae, there are both pharmacological and genetic techniques to induce immunosuppression in zebrafish larvae, these have been recently reviewed in Rosowski et al., 2018a.
Confocal images (Basic Protocol 3)
Basic Protocol 3 produces confocal Z-stack images of individual, infected larvae at 6, 24, 48, 72, and 96 hpi (Figure 5). These images provide the information needed to measure fungal growth and immune cell recruitment during disease progression. Note that the selection of specific time points can be easily tailored to fit the needs of the researcher. See Table 2 for suggestions on measuring fungal development and immune cell recruitment.
Table 2:
Example parameters that can be measured from infection images.
| Parameter | Example measurement | Notes |
|---|---|---|
| Germination | % of larvae with germinated spores | Does larva contain at least one germinated spore? (Y/N) |
| Presence of hyphal branches | % of larvae with hyphal branches | Does larva contain hyphae with lateral branches? (Y/N) |
| Fungal burden | 2D area of fungal fluorescent signal | Using Fiji (ImageJ), generate a max intensity projection and manually threshold fluorescent signal of fungus, then measure area. |
| Immune cell recruitment | # of cells in the hindbrain OR 2D area of immune cell fluorescent signal in the hindbrain (if there are too many cells to accurately count) | Using Fiji (ImageJ), generate a max intensity projection and manually threshold fluorescent signal of immune cells in the hindbrain, then measure area. |
Time Considerations
See Table 3 for time considerations.
Table 3:
Time considerations for protocols described in this article. Please note that the time required for hindbrain injection and imaging experiments will vary depending on the number of larvae used and the level of experience of the researcher.
| Protocol | Total Time | Details |
|---|---|---|
| Support Protocol 1: Preparation of Aspergillus Spores | 6–8 days | Day 1: 20 min (Part 1) Day 3–4: 2 hr (Part 2) Day 6–8: 2 hr (Part 3) |
| Support Protocol 2: Dechorionating Zebrafish Embryos | 20 min | |
| Support Protocol 3: Generating Transparent Larvae with 1-Phenyl 2-Thiourea (PTU) | 5 min | |
| Basic Protocol 1: Hindbrain Microinjection of Zebrafish Larvae with Aspergillus Spores | 3–4 days | Day 1: 4 hr (Parts 1–3) Day 3–4: 1 hr (Part 3) |
| Basic Protocol 2: Survival Analysis | 7 days | 10 min daily |
| Basic Protocol 3: Multi-Day Imaging of Infected Larvae | 4 days | 4 hr daily |
| Alternate Protocol 1: Embedding Larvae in Low-Melting-Point Agarose | 30 min |
ACKNOWLEDGEMENTS:
This work was supported by R35GM118027-01 from the National Institute of General Medical Sciences (NIGMS) of the National Institutes of Health (NIH) to A.H. and 5R01AI065728-10 from the National Institute of Allergy and Infectious Diseases (NIAID) of the NIH to N.P.K. T.J.S. was supported by the National Institute on Aging of the National Institutes of Health under Award Number T32AG000213. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
CONFLICT OF INTEREST STATEMENT:
The authors declare no competing or financial interests.
DATA AVAILABILITY STATEMENT:
The data, tools and material (or their source) that support the protocol are available from the corresponding author upon reasonable request.
LITERATURE CITED:
- Bertuzzi M, van Rhijn N, Krappmann S, Bowyer P, Bromley MJ, & Bignell EM (2021). On the lineage of Aspergillus fumigatus isolates in common laboratory use. Med Mycol, 59(1), 7–13. doi: 10.1093/mmy/myaa075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman DZP, & Schwartz IS (2019). Emerging Fungal Infections: New Patients, New Patterns, and New Pathogens. J Fungi (Basel), 5(3). doi: 10.3390/jof5030067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry KM, Loynes CA, Whyte MK, & Renshaw SA (2013). Zebrafish as a model for the study of neutrophil biology. J Leukoc Biol, 94(4), 633–642. doi: 10.1189/jlb.1112594 [DOI] [PubMed] [Google Scholar]
- Huemer K, Squirrell JM, Swader R, LeBert DC, Huttenlocher A, & Eliceiri KW (2017). zWEDGI: Wounding and Entrapment Device for Imaging Live Zebrafish Larvae. Zebrafish, 14(1), 42–50. doi: 10.1089/zeb.2016.1323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson J, von Hofsten J, & Olsson PE (2001). Generating transparent zebrafish: a refined method to improve detection of gene expression during embryonic development. Mar Biotechnol (NY), 3(6), 522–527. doi: 10.1007/s1012601-0053-4 [DOI] [PubMed] [Google Scholar]
- Knox BP, Blachowicz A, Palmer JM, Romsdahl J, Huttenlocher A, Wang CC, … Venkateswaran K (2016). Characterization of Aspergillus fumigatus Isolates from Air and Surfaces of the International Space Station. mSphere, 1(5). doi: 10.1128/mSphere.00227-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knox BP, Deng Q, Rood M, Eickhoff JC, Keller NP, & Huttenlocher A (2014). Distinct innate immune phagocyte responses to Aspergillus fumigatus conidia and hyphae in zebrafish larvae. Eukaryot Cell, 13(10), 1266–1277. doi: 10.1128/EC.00080-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knox BP, Huttenlocher A, & Keller NP (2017). Real-time visualization of immune cell clearance of Aspergillus fumigatus spores and hyphae. Fungal Genet Biol, 105, 52–54. doi: 10.1016/j.fgb.2017.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch BEV, Hajdamowicz NH, Lagendijk E, Ram AFJ, & Meijer AH (2019). Aspergillus fumigatus establishes infection in zebrafish by germination of phagocytized conidia, while Aspergillus niger relies on extracellular germination. Sci Rep, 9(1), 12791. doi: 10.1038/s41598-019-49284-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latge JP, & Chamilos G (2019). Aspergillus fumigatus and Aspergillosis in 2019. Clin Microbiol Rev, 33(1). doi: 10.1128/CMR.00140-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz A, Bertuzzi M, Seidel C, Thomson D, Bignell EM, & Read ND (2021). Live-cell imaging of rapid calcium dynamics using fluorescent, genetically-encoded GCaMP probes with Aspergillus fumigatus. Fungal Genet Biol, 151, 103470. doi: 10.1016/j.fgb.2020.103470 [DOI] [PubMed] [Google Scholar]
- Rosowski EE, Raffa N, Knox BP, Golenberg N, Keller NP, & Huttenlocher A (2018). Macrophages inhibit Aspergillus fumigatus germination and neutrophil-mediated fungal killing. PLoS Pathog, 14(8), e1007229. doi: 10.1371/journal.ppat.1007229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoen TJ, Rosowski EE, Knox BP, Bennin D, Keller NP, & Huttenlocher A (2019). Neutrophil phagocyte oxidase activity controls invasive fungal growth and inflammation in zebrafish. J Cell Sci, 133(5). doi: 10.1242/jcs.236539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma C, Nelson-Sathi S, Singh A, Radhakrishna Pillai M, & Chowdhary A (2019). Genomic perspective of triazole resistance in clinical and environmental Aspergillus fumigatus isolates without cyp51A mutations. Fungal Genet Biol, 132, 103265. doi: 10.1016/j.fgb.2019.103265 [DOI] [PubMed] [Google Scholar]
- Thrikawala S, & Rosowski EE (2020). Infection of Zebrafish Larvae with Aspergillus Spores for Analysis of Host-Pathogen Interactions. JoVE(159), e61165. doi:doi: 10.3791/61165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torraca V, Masud S, Spaink HP, & Meijer AH (2014). Macrophage-pathogen interactions in infectious diseases: new therapeutic insights from the zebrafish host model. Dis Model Mech, 7(7), 785–797. doi: 10.1242/dmm.015594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerfield M (1993). The Zebrafish book : a guide for the laboratory use of zebrafish (Brachydanio rerio). Eugene. Or.: University of Oregon Press. [Google Scholar]
KEY REFERENCES/INTERNET RESOURCES:
- Thrikawala S, & Rosowski EE (2020). Infection of Zebrafish Larvae with Aspergillus Spores for Analysis of Host-Pathogen Interactions. JoVE(159), e61165. doi:doi: 10.3791/61165 URL: https://www.jove.com/t/61165/infection-zebrafish-larvae-with-aspergillus-spores-for-analysis-host [DOI] [PMC free article] [PubMed] [Google Scholar]; This reference provides excellent additional visual references for the hindbrain infection technique.
- Huemer K, Squirrell JM, Swader R, LeBert DC, Huttenlocher A, & Eliceiri KW (2017). zWEDGI: Wounding and Entrapment Device for Imaging Live Zebrafish Larvae. Zebrafish, 14(1), 42–50. doi: 10.1089/zeb.2016.1323 URL: https://morgridge.org/research/medical-engineering/fab-lab/designs/zwedgi/ [DOI] [PMC free article] [PubMed] [Google Scholar]; This URL provides information for downloading fabrication specifications for the zWEDGI device.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data, tools and material (or their source) that support the protocol are available from the corresponding author upon reasonable request.